Амилоидоз: современные методы диагностики и лечения
- Аннотация
- Статья
- Ссылки
Термин «амилоидоз» объединяет заболевания, которые характеризуются внеклеточным отложением специфического нерастворимого фибриллярного белка амилоида. Немецкий патолог Рудольф Вирхов в 1853 г. предложил термин «амилоид» для обозначения вещества, откладывающегося в органах больных «сальной болезнью» (как называли амилоидоз в начале XIX века) при туберкулезе, сифилисе, лепре. Это вещество он ошибочно посчитал крахмалоподобным из-за характерной реакции с иодом. В настоящее время установлено, что полисахариды составляют не более 4% от массы амилоида, однако термины «амилоид» и «амилоидоз» закрепились.
Эпидемиология амилоидоза
Распространенность амилоидоза до настоящего времени изучена недостаточно. По данным S.Y. Tan и соавт. (1995) [1], реактивный АА-амилоидоз, один из наиболее частых вариантов амилоидоза, развивается у 5% больных с хроническими воспалительными заболеваниями в Европе; по данным других источников, АА-амилоидоз осложняет течение ревматоидного артрита в 6–10% случаев. В среднем доля АА-амилоидной нефропатии в структуре заболеваний почек в Европе составляет 2,5–2,8%, а в структуре болезней, приведших к хронической почечной недостаточности (ХПН), – 1% (в соответствии с данными Европейской ассоциации диализа и трансплантации).
В США частота амилоидоза варьирует от 5,1 до 12,8 случаев на 100 тыс. населения в год [2]. Эти данные касаются преимущественно распространенности первичного AL-амилоидоза, или амилоидоза в рамках миеломной болезни и других В-гемобластозов. В странах третьего мира, по мнению S.Y. Tan (1995), смертность от AL-амилоидоза составляет 1 на 2000 населения (0,05%). По-видимому, представленные данные о распространенности различных вариантов амилоидоза в целом могут быть экстраполированы и на другие страны.
Морфология амилоидоза
Основу тканевых отложений амилоида составляют амилоидные фибриллы – особые белковые структуры диаметром 5–10 нм и длиной до 800 нм, состоящие из 2 и более параллельных разнонаправленных (антипараллельных) филаментов, образующих кросс-бета-складчатую конформацию. Именно она определяет специфическое оптическое свойство амилоида – способность к двойному лучепреломлению. При микроскопии окрашенных конго красным препаратов в поляризованном свете амилоид изменяет красный цвет окраски на яблочно-зеленое свечение. Обнаружение этого свойства положено в основу диагностики амилоидоза.
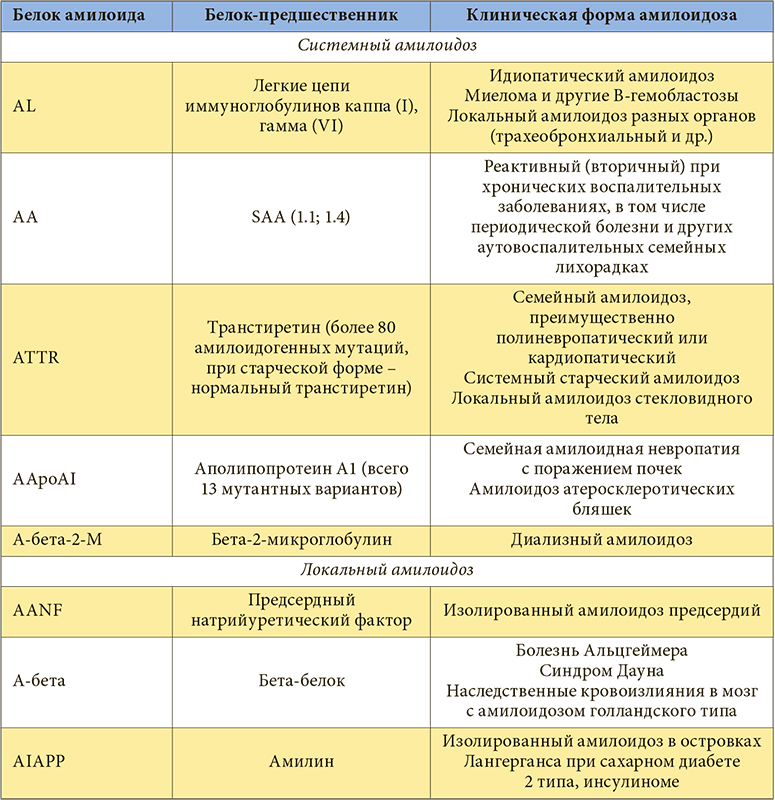
Структурные и химико-физические особенности амилоида определяются основным белком-предшественником, содержание которого в фибрилле достигает 80% и является специфичным признаком для каждого типа амилоидоза.
Характеристика основных типов амилоидоза
В группу АА-амилоидоза входят реактивный (вторичный) амилоидоз, амилоидоз при периодической болезни, криопиринопатиях (преимущественно при синдроме Макла – Уэллса – семейной периодической лихорадке в сочетании с глухотой и крапивницей). Наиболее частыми причинами вторичного амилоидоза в настоящее время служат ревматоидный артрит (30–50%), хронические гнойно-деструктивные болезни (остеомиелит, бронхоэктатическая болезнь), воспалительные заболевания кишечника (язвенный колит, болезнь Крона), туберкулез, опухоли (чаще лимфогранулематоз и рак почки) [3].
АА-амилоид образуется из сывороточного белка-предшественника SAA – острофазового белка, в норме синтезируемого гепатоцитами, нейтрофилами и фибробластами в следовых количествах. Его концентрация значительно возрастает под воздействием интерлейкинов 1 и 6, фактора некроза опухоли в ответ на воспаление, опухолевый рост. Повышение содержания SAA в крови играет основную роль в патогенезе АА-амилоидоза.
Однако для развития амилоидоза недостаточно только высокой концентрации SAA, необходимо также наличие у белка-предшественника амилоидогенности. Развитие амилоидоза у человека связывают с депозицией SAA1; в настоящее время известно 5 изотипов SAA1, из которых наибольшую амилоидогенность приписывают изотипам 1.1 и 1.5 [4]. Конечный этап амилоидогенеза – образование фибрилл амилоида из белка-предшественника – осуществляется при неполном расщеплении протеазами моноцитов-макрофагов. Стабилизация амилоидной фибриллы и резкое снижение растворимости этого макромолекулярного комплекса во многом обусловлены взаимодействием с полисахаридами интерстиция. При АА-амилоидозе амилоид обнаруживают в различных органах, однако клиническая картина и прогноз обусловлены поражением почек.
AL-амилоидоз включает в себя первичный (идиопатический) амилоидоз и амилоидоз, ассоциированный с миеломной болезнью, развивающийся у 7–10% больных с множественной миеломой. По современным представлениям, первичный AL-амилоидоз и миеломную болезнь (как ассоциированную, так и не ассоциированную с амилоидозом) рассматривают в рамках единой В-лимфоцитарной дискразии, которая характеризуется пролиферацией аномального клона плазматических клеток или В-клеток в костном мозге с избыточной продукцией моноклонального иммуноглобулина, обладающего амилоидогенностью. Белком-предшественником при AL-амилоидозе считают моноклональные легкие цепи иммуноглобулинов (ЛЦИ), из названия которых происходит аббревиатура L [3]. AL-амилоидоз – это генерализованный процесс с преимущественным поражением сердца, почек, желудочно-кишечного тракта (ЖКТ), нервной системы и кожи.
К ATTR-амилоидозу относят семейную амилоидную полиневропатию, наследуемую по аутосомно-доминантному типу, и системный старческий амилоидоз. Белком-предшественником при этой форме амилоидоза служит транстиретин – компонент фракции преальбумина, синтезируемый печенью и выполняющий функции транспортного белка тироксина и ретинола. Установлено, что АTTR-амилоидоз бывает результатом мутации в гене, кодирующем синтез транстиретина, что приводит к замене аминокислот в молекуле TTR. В результате повышается амилоидогенность белка-предшественника и облегчается его полимеризация в амилоидные фибриллы. В настоящее время известно множество вариантных транстиретинов, чем и обусловлено разнообразие клинических форм наследственной невропатии [5]. Эта группа заболеваний характеризуется прогрессирующей периферической и вегетативной невропатией, которая сочетается с поражением сердца, почек и других органов различной степени.
Системный старческий амилоидоз развивается после 70 лет в результате возрастных изменений структуры нормального транстиретина, усиливающих его амилоидогенность. К органам-мишеням старческого амилоидоза относят сердце, сосуды головного мозга и аорту.
К семейным формам амилоидоза относятся также более редкие AGel-, AFib-, ALys-амилоидозы, при которых амилоидогенностью обладают мутантные формы соответственно гелсолина, фибриногена, лизоцима. При этих вариантах амилоидоза также отмечается преимущественное поражение почек, однако для гелсолинового амилоидоза характерно сочетание нефропатии с сетчатой дистрофией роговицы и периферической невропатией (преимущественно поражаются черепно-мозговые нервы).
В настоящее время известно более 20 амилоидогенных белков-предшественников и, соответственно, клинических вариантов амилоидоза. Так, А-бета-амилоид является морфологической основой болезни Альцгеймера, AIAPP-амилоид – сахарного диабета 2 типа и др. [3].
В нефрологической практике большое значение имеет А-бета-2-М-амилоидоз (ассоциированный с хроническим гемодиализом). Белком-предшественником при этой форме амилоидоза служит бета-2-микроглобулин, который в норме присутствует в крови, моче, спинномозговой и синовиальной жидкостях. При нормальной функции почек его концентрация в крови составляет 1–2 мг/л. Этот белок фильтруется в клубочках почек и метаболизируется после реабсорбции в проксимальных канальцах. У пациентов с ХПН концентрация этого белка в крови возрастает, коррелируя с содержанием креатинина, однако максимальных значений (в 20–70 раз превышающих норму) она достигает через несколько лет проведения регулярного гемодиализа. Бета-2-микроглобулин плохо удаляется при проведении стандартного гемодиализа, что неизбежно приводит к развитию амилоидоза [3]. Первые депозиты бета-2-М-амилоида обнаруживают в синовиальной оболочке суставов уже через 1–2 года заместительной почечной терапии, клинические симптомы появляются через 7 и более лет терапии [3]. У больных старше 60 лет диализный амилоидоз развивается быстрее. Кроме высокой концентрации белка-предшественника, в патогенезе диализного амилоидоза существенную роль играют и другие факторы. Амилоидогенность бета-2-микроглобулина возрастает при модификациях, обусловленных взаимодействием с продуктами неполного гликирования, а также при окислении бета-2-микроглобулина и ацидификации среды [3]. Одним из основных источников этих модификаций являются компоненты диализата, в том числе бактериальный липополисахарид; гликирование бета-2-микроглобулина возможно также при контакте с целлюлозными диализными мембранами. Таким образом, профилактика диализного амилоидоза во многом связана с отказом от целлюлозных диализных мембран и тщательностью контроля качества применяемого диализата. Модифицированный бета-2-микроглобулин является мощным индуктором образования цитокинов (интерлейкины 1 и 6, фактор некроза опухоли альфа). Результатом активации цитокинового каскада является тяжелое синовиальное воспаление с привлечением макрофагов [3]. Макрофагальное воспаление определяет высокий деструктивный потенциал воспаления в синовии с развитием костных эрозий и кист, рост которых обусловливает развитие переломов костей. Было установлено, что бета-2-микроглобулин обладает высокой коллагенсвязывающей активностью, возрастающей по мере увеличения его концентрации в крови. Кроме того, выявлена связь бета-2-микроглобулина с гликозоаминогликанами хряща. Этими фактами можно объяснить преимущественное отложение фибрилл амилоида в суставных тканях. Таким образом, при А-бета-2-М-амилоидозе отмечают поражение костей и периартикулярных тканей, реже – сосудов.
Патогенез амилоидоза
Несмотря на различие в типах амилоидного белка, существует общность патогенеза различных клинических форм амилоидоза. Основной причиной развития болезни служит наличие определенного, нередко повышенного количества амилоидогенного предшественника. Как уже указывалось, появление или усиление амилоидогенности может быть обусловлено циркуляцией вариантов белков с повышенной общей гидрофобностью молекулы, нарушенным соотношением поверхностных молекулярных зарядов, что приводит к нестабильности белковой молекулы и способствует ее агрегации в амилоидную фибриллу. Эти механизмы особенно ярко прослеживаются на примере белков, в функцию которых заложена необходимость физиологического изменения конформации. Так, практически все аполипопротеины, вынужденные разворачивать свою вторичную структуру в процессе транслокации холестерина через стенку сосуда, участвуют в патогенезе различных форм амилоидоза. На последнем этапе амилоидогенеза происходит взаимодействие амилоидного белка с белками плазмы крови и гликозоаминогликанами тканей. Кроме структурных особенностей, имеют значение также физико-химические свойства межклеточного матрикса, где происходит сборка амилоидной фибриллы. В практике экспериментального амилоидоза хорошо известна способность суспензии амилоидных масс (амилоидускоряющая субстанция), полученной из тканей животных, пораженных амилоидом, провоцировать амилоидоз при введении здоровым животным. В клинической практике у больных ATTR-амилоидозом, несмотря на прекращение циркуляции патологического транстиретина после трансплантации здоровой печени, продолжается нарастание массы амилоидных депозитов в сердце за счет захвата нормального неизмененного транстиретина [6]. Многие формы амилоидоза можно объединить также по признаку возникновения в пожилом и старческом возрасте (AL, ATTR, AIAPP, AApoA1, AFib, ALys, AANF, A-бета), что указывает на наличие механизмов возрастной эволюции структуры определенных белков в сторону повышения амилоидогенности и позволяет рассматривать амилоидоз как одну из моделей старения организма.
Классификация амилоидоза
Объяснение многообразия клинических форм амилоидоза наличием многочисленных сывороточных белков-предшественников привело к созданию современной классификации амилоидоза, в основу которой положен биохимический тип белка-предшественника (табл.). Все типы амилоидоза обозначают аббревиатурой, в которой первая буква А означает «амилоидоз», а последующие – сокращенное название основных фибриллярных белков амилоида: А – амилоидный белок А, L – легкие цепи иммуноглобулинов, TTR – транстиретин, бета-2-М – бета2-микроглобулин и др. С клинической точки зрения целесообразно выделять локальные и системные, или генерализованные, формы амилоидоза. Среди последних основными считают АА-, AL-, АTTR- и А-бета-2-М-амилоидоз.
Симптомы амилоидоза почек
В клинической практике наибольшее значение имеют АА- и AL-типы системного амилоидоза, которые протекают с вовлечением в патологический процесс многих органов, однако чаще манифестируют симптомами моноорганного поражения. В дальнейшем развивается, как правило, столь характерный для этих типов амилоидоза полиморфизм клинических проявлений. АА- и AL-амилоидоз у мужчин отмечают в 1,8 раза чаще, чем у женщин. Для вторичного амилоидоза характерно более раннее начало, чем для первичного (средний возраст заболевших – около 40 и 65 лет соответственно). Среди многочисленных клинических проявлений АА- и AL-амилоидоза наряду с общими для обоих типов симптомами существуют характерные лишь для AL-типа признаки (периорбитальные геморрагии, макроглоссия, поражение кожи и др.) [7–9]. Кроме того, клинические проявления, напоминающие признаки первичного амилоидоза, возможны при АTTR- и А-бета-2-М-амилоидозе (синдром запястного канала и др.).
Поражение почек – ведущий клинический признак АА- и AL-амилоидоза. При АА-амилоидозе почки вовлечены в патологический процесс практически у всех больных, при AL-типе частота нефропатии также высока и приближается к 80%. Поражение почек наблюдают и при АTTR-амилоидозе, однако у большинства больных семейной амилоидной невропатией отмечают несоответствие между клиническими и морфологическими признаками нарушений почек.
Амилоид при АА- и AL-типах амилоидоза локализуется преимущественно в клубочках, однако у 10% больных первичным амилоидозом и у значительной части больных наследственной невропатией отмечают только отложения вне клубочков. Амилоид откладывается также в других почечных структурах: в базальной мембране канальцев (преимущественно дистальных и петли Генле), интерстиции, стенках сосудов.
Клинически амилоидная нефропатия манифестирует, как правило, изолированно протеинурией и характеризуется неуклонно прогрессирующим течением с последовательной сменой стадий: протеинурическая, нефротическая, ХПН. Только у 20% больных АА-амилоидозом ХПН развивается без предшествующего нефротического синдрома. При AL-амилоидозе стадийность течения амилоидной нефропатии проявляется менее отчетливо [9].
К особенностям амилоидоза почек относят редкость гематурии и лейкоцитурии («скудный» мочевой осадок), а также артериальной гипертонии, которую даже при ХПН отмечают лишь у 20% больных АА-амилоидозом и еще реже при AL-амилоидозе. Нефротический синдром и большие размеры почек сохраняются даже при развитии и прогрессировании ХПН.
Величина протеинурии не коррелирует с выраженностью амилоидных отложений в почках (при преимущественно сосудистом поражении протеинурия может быть минимальной) и зависит от степени деструкции подоцитов. Максимальную потерю белка обнаруживают через участки базальной мембраны, которые пропитаны амилоидом и лишены эпителиального покрытия.
Амилоидоз почек у большинства больных диагностируют на стадии нефротического синдрома, у 33% – на стадии ХПН. В редких случаях амилоидная нефропатия может проявляться остронефротическим синдромом и макрогематурией. Описаны также синдром Фанкони и тромбоз почечных вен.
Поражение сердца отмечают у подавляющего большинства больных AL-амилоидозом и у части пациентов с АTTR-амилоидозом, в то время как для АА-амилоидоза этот симптом не характерен. В результате замещения миокарда амилоидными массами развивается рестриктивная кардиомиопатия [10]. Клинически определяют кардиомегалию, рано развивается сердечная недостаточность (у 22% больных уже в дебюте болезни), которая быстро прогрессирует и почти у 50% пациентов, наряду с аритмиями, бывает причиной смерти. Особенностью сердечной недостаточности при первичном амилоидозе служит ее рефрактерность к терапии, в первую очередь сердечными гликозидами [11].
Нарушения ритма и проводимости при AL-амилоидозе многообразны: мерцательная аритмия, наджелудочковая тахикардия, синдром преждевременного возбуждения желудочков, различные блокады и синдром слабости синусового узла. Вследствие отложения амилоида в коронарных артериях возможно развитие инфаркта миокарда, обнаруживаемого на аутопсии у 6% больных. Амилоидные отложения в клапанных структурах симулируют картину клапанного порока. Основным признаком амилоидоза сердца на электрокардиограмме бывает снижение вольтажа зубцов комплекса QRS. Описан инфарктоподобный тип электрокардиограммы. Наиболее адекватным методом диагностики амилоидной кардиомиопатии считают эхокардиографию, с помощью которой можно диагностировать симметричное утолщение стенок желудочков, дилатацию предсердий, нарушения гемодинамики.
Серьезным патологическим признаком при AL-амилоидозе служит ортостатическая артериальная гипотония, которую наблюдают у 11% больных в момент постановки диагноза. Обычно этот симптом связан с автономной дисфункцией (поражение вегетативной нервной системы) и в тяжелых случаях сопровождается синкопальными состояниями. Артериальная гипотония бывает также у больных АА-амилоидозом, но в этом случае ее причиной служит надпочечниковая недостаточность вследствие отложения амилоида в надпочечниках [3].
Поражение дыхательной системы возникает при первичном амилоидозе примерно у 50% больных, а при вторичном – у 10–14%. В большинстве случаев оно протекает бессимптомно или со скудной клинической симптоматикой [12]. При AL-амилоидозе одним из ранних признаков болезни может быть охриплость или изменение тембра голоса вследствие отложения амилоида в голосовых связках, опережающего его появление в дистальных отделах дыхательных путей. В легких амилоид откладывается преимущественно в альвеолярных перегородках (что приводит к развитию кашля и одышки) и стенках сосудов. Описаны также ателектазы и инфильтраты в легких. Рентгенологическая картина не специфична, смерть от прогрессирующей дыхательной недостаточности наступает редко [13].
Поражение органов пищеварения наблюдают при амилоидозе в 70% случаев, причем у больных с AL- и АА-типами амилоидоза частота поражения тех или иных отделов ЖКТ различна [3]. У 25% больных первичным амилоидозом отмечают амилоидное поражение пищевода, проявляющееся преимущественно дисфагией, которая может быть одним из ранних симптомов заболевания. О поражении желудка и кишечника могут свидетельствовать изъязвления и перфорации их стенок с возможным кровотечением, а также препилорическая непроходимость желудка или механическая кишечная непроходимость из-за отложения амилоидных масс. У больных с преимущественным поражением толстой кишки возможно появление клинических симптомов, имитирующих язвенный колит. Наиболее частым желудочно-кишечным проявлением AL-амилоидоза, отмечаемым почти у 25% пациентов, бывает тяжелая моторная диарея с вторичным нарушением всасывания. Возможной причиной тяжелой диареи при этом, наряду с инфильтрацией кишечной стенки, и в том числе ворсин, амилоидом, у больных AL-амилоидозом служит автономная (вегетативная) дисфункция. Истинный синдром нарушенного всасывания развивается приблизительно у 4–5% больных. При АА-амилоидозе эти симптомы иногда также возможны, в том числе как единственное клиническое проявление амилоидоза.
Поражение печени при АА- и AL-амилоидозе наблюдают практически в 100% случаев, при этом обычно отмечают небольшое увеличение печени и 3–4-кратное повышение активности гамма-глутамилтранспептидазы и щелочной фосфатазы [14]. Увеличению печени обычно сопутствует небольшое увеличение селезенки. Тяжелое поражение печени с выраженной гепатомегалией и развернутыми признаками тяжелого холестаза отмечается значительно реже (у 15–25% больных) и более характерно для AL-амилоидоза. Несмотря на выраженную гепатомегалию, функция печени обычно остается сохранной. Редким признаком амилоидоза печени бывает внутрипеченочная портальная гипертензия, которая чаще сочетается с выраженной желтухой, холестазом, печеночной недостаточностью и свидетельствует о далеко зашедшем поражении печени с риском пищеводного кровотечения, печеночной комы. При некоторых вариантах семейного ALys-амилоидоза описаны тяжелые спонтанные внутрипеченочные кровотечения [15]. Редким проявлением амилоидоза селезенки бывает ее спонтанный разрыв.
Поражение нервной системы, представленное симптомами периферической невропатии и вегетативной дисфункции, отмечают у 17% больных AL-амилоидозом и у пациентов с семейной амилоидной невропатией разных типов (ATTR, AApoA1 и др.). Клиническая картина невропатии при всех типах амилоидоза практически одинакова, поскольку обусловлена сходными процессами: в первую очередь, дегенерацией миелиновой оболочки нервов, а также компрессией нервных стволов отложениями амилоида и ишемией в результате амилоидных депозитов в стенках сосудов [3].
В большинстве случаев возникает симметричная дистальная невропатия с неуклонным прогрессированием. В дебюте поражения нервной системы наблюдают, главным образом, сенсорные нарушения, в первую очередь болевой и температурной чувствительности, позже присоединяются нарушения вибрационной и позиционной чувствительности, двигательные нарушения. Ранними симптомами невропатии бывают парестезии или мучительные дизестезии (чувство онемения). Нижние конечности вовлекаются в патологический процесс чаще, чем верхние.
Автономные дисфункции часто манифестируют ортостатической артериальной гипотонией (см. выше), иногда с обморочными состояниями, диареей, нарушением функции мочевого пузыря, импотенцией у мужчин.
У 20% больных AL-амилоидозом и у большинства пациентов с амилоидозом на фоне проведения гемодиализа выявляют синдром запястного канала, обусловленный сдавлением срединного нерва в запястном канале амилоидом, откладывающимся в связках запястья [2, 8, 9]. Клинически этот синдром проявляется интенсивными болями и парестезиями в I–III пальцах кисти с постепенной атрофией мышц тенара. К особенностям синдрома запястного канала при диализном амилоидозе относят его преимущественное развитие на той руке, где сформирована фистула, а также усиление болей во время процедуры гемодиализа, возможно, в результате развития феномена обкрадывания, индуцированного фистулой, что приводит к ишемии срединного нерва.
Поражение кожи наблюдают почти у 40% больных первичным амилоидозом. Для него характерно разнообразие проявлений, наиболее частыми из которых бывают параорбитальные геморрагии (патогномоничны для AL-амилоидоза), возникающие при малейшем напряжении. Описаны также папулы, бляшки, узелки, пузырьковые высыпания. Нередко наблюдают индурацию кожи, аналогичную склеродермической. Редким вариантом поражения кожи при AL-амилоидозе служат нарушения пигментации (от выраженного усиления до тотального альбинизма), алопеция, трофические нарушения.
Поражение опорно-двигательного аппарата характерно для пациентов с диализным амилоидозом и редко (в 5–10% случаев) возникает у больных AL-амилоидозом (исключая костные изменения при миеломной болезни). При этом характер тканевого отложения амилоида сходен при обоих этих типах амилоидоза: амилоид откладывается в костях, суставном хряще, синовии, связках и мышцах [16, 17].
При диализном амилоидозе наиболее часто отмечают триаду признаков – плече-лопаточный периартрит, синдром запястного канала и поражение сухожильных влагалищ сгибателей кисти, приводящее к развитию сгибательных контрактур пальцев. Кроме них, характерно развитие кистозного поражения костей из-за отложения амилоида. Типичны амилоидные кисты в костях запястья и головках трубчатых костей. Со временем эти отложения увеличиваются в размерах, становясь причиной патологических переломов.
Частым признаком диализного амилоидоза бывает также деструктивная спондилоартропатия в результате амилоидного поражения межпозвонковых дисков, преимущественно в шейном отделе позвоночника.
Амилоидные отложения в мышцах чаще наблюдают при первичном амилоидозе. Они проявляются псевдогипертрофией или атрофией мышц, затрудняющими движения, мышечными болями. Макроглоссия, обусловленная выраженной инфильтрацией мышц амилоидом, – это патогномоничный симптом AL-амилоидоза, который встречается примерно у 20% пациентов и нередко сочетается с псевдогипертрофией других групп поперечно-полосатой мускулатуры. В тяжелых случаях макроглоссия не только затрудняет прием пищи и речь, но и приводит к обструкции дыхательных путей [3]. При АА-амилоидозе она не развивается.
Среди других органных поражений при амилоидозе известны поражение щитовидной железы с развитием клинической картины гипотиреоза (AL-амилоидоз), надпочечников с появлением симптомов их недостаточности (чаще при АА-амилоидозе), экзокринных желез, приводящее к возникновению сухого синдрома, лимфаденопатия. Редким проявлением, описанным при AL- и АTTR- амилоидозе, бывает поражение глаз.
Диагностика амилоидоза
Предполагаемый на основании клинических и лабораторных данных амилоидоз необходимо подтвердить морфологически обнаружением амилоида в биоптатах тканей.
При подозрении на AL-тип амилоидоза рекомендуют производить пункцию костного мозга. Подсчет плазматических клеток и окраска пунктата на амилоид позволяют не только диагностировать амилоидоз, но и дифференцировать первичный и ассоциированный с миеломой варианты AL-амилоидоза. Положительный результат исследования костного мозга на амилоид отмечают у 60% больных AL-амилоидозом.
Простой и безопасной диагностической процедурой считают аспирационную биопсию подкожной жировой клетчатки, при которой выявляют амилоид в 80% случаев AL-амилоидоза. К преимуществам этой процедуры, кроме информативности, относят также редкость развития кровотечений, что позволяет использовать этот метод у больных с нарушениями свертывания крови (больные первичным амилоидозом нередко имеют дефицит Х фактора свертывания, при котором могут развиться геморрагии [8]).
Наиболее часто для диагностики разных типов амилоидоза проводят биопсию слизистой оболочки прямой кишки, почки, печени. Биопсия слизистого и подслизистого слоев прямой кишки позволяет выявить амилоид у 70% больных, а биопсия почки – практически в 100% случаев. У пациентов с синдромом запястного канала исследованию на амилоид необходимо подвергать ткань, удаленную при операции декомпрессии запястного канала.
Биопсийный материал для выявления амилоида необходимо окрашивать конго красным с последующей микроскопией в поляризованном свете для выявления способности к двойному лучепреломлению. Современная морфологическая диагностика амилоидоза включает не только обнаружение, но и типирование амилоида, поскольку тип амилоида определяет терапевтическую тактику. Для типирования часто применяют пробу с перманганатом калия. При обработке окрашенных конго красным препаратов 5%-ным раствором перманганата калия АА-тип амилоида теряет окраску и утрачивает свойство двойного лучепреломления, тогда как AL-тип амилоида сохраняет их. Использование щелочного гуанидина позволяет более точно дифференцировать АА- и AL-амилоидоз.
Наиболее эффективным методом типирования амилоида служит иммуногистохимическое исследование с применением антисывороток к основным типам амилоидного белка (специфические антитела против АА-белка, легких цепей иммуноглобулинов, транстиретина и бета-2-микроглобулина) [3, 18].
Лечение амилоидоза
Согласно современным представлениям, целью терапии любого типа амилоидоза является уменьшение количества (или, если возможно, удаление) белков-предшественников для того, чтобы замедлить или приостановить прогрессирование болезни. Неблагоприятный прогноз при естественном течении амилоидоза оправдывает применение некоторых агрессивных лекарственных режимов или других радикальных мер. Клиническое улучшение, достигаемое с помощью этих видов лечения, включает стабилизацию или восстановление функции жизненно важных органов, а также предотвращение функциональных нарушений, что увеличивает продолжительность жизни больных. Морфологическим критерием эффективности лечения считают уменьшение отложений амилоида в тканях, что в настоящее время можно оценить, применяя радиоизотопную сцинтиграфию с сывороточным Р-компонентом амилоида. Кроме основных терапевтических режимов, лечение амилоидоза должно включать симптоматические методы, направленные на уменьшение выраженности застойной недостаточности кровообращения, аритмии, отечного синдрома, коррекцию артериальной гипер- и гипотонии.
Целью терапии вторичного амилоидоза служит подавление продукции белка-предшественника SAA [19, 20], что достигается лечением хронического воспаления, в том числе и хирургическим (секвестрэктомия при остеомиелите, удаление доли легкого при бронхоэктатической болезни, опухоли, туберкулезе).
Особое значение в настоящее время придают терапии ревматоидного артрита, учитывая его лидирующее положение среди причин вторичного амилоидоза. Базисная терапия ревматоидного артрита цитостатическими лекарственными средствами (метотрексатом, циклофосфамидом, хлорамбуцилом) или современными антицитокиновыми средствами (блокаторы эффектов фактора некроза опухоли альфа, интерлейкины 1 и 6, блокаторы СD20 В-лимфоцитов и др.), назначаемая на длительный срок (более 12 мес.), уменьшает риск развития амилоидоза. У пациентов с уже развившимся амилоидозом это лечение позволяет в большинстве случаев уменьшить клинические проявления амилоидной нефропатии. В результате подобной терапии отмечают снижение выраженности протеинурии, купирование нефротического синдрома, стабилизацию функции почек. У части пациентов удается предотвратить развитие ХПН или замедлить ее прогрессирование, что существенно улучшает прогноз. Об эффективности лечения свидетельствует также нормализация концентрации С-реактивного белка в крови.
Средством выбора для лечения АА-амилоидоза при периодической болезни служит колхицин. При его постоянном приеме можно полностью избежать рецидивирования приступов у большинства больных, предотвращая у них также развитие амилоидоза. При развившемся амилоидозе длительный, возможно пожизненный, прием колхицина в дозе 1,8–2 мг/сут приводит к ремиссии, выражающейся в ликвидации нефротического синдрома, уменьшении или исчезновении протеинурии у больных с нормальной функцией почек. При наличии ХПН начальную дозу лекарственных средств уменьшают в зависимости от величины клубочковой фильтрации, хотя в случае снижения концентрации креатинина в крови возможно повышение дозы до стандартной. Колхицин также предотвращает рецидив амилоидоза в трансплантированной почке. Больные хорошо переносят данное лекарственное средство. Если эффективность колхицина при амилоидозе в рамках периодической болезни не вызывает сомнений, то имеются лишь единичные работы, свидетельствующие о его успешном применении у больных вторичным амилоидозом. Кроме колхицина, при АА-амилоидозе применяют диметилсульфоксид, вызывающий резорбцию амилоидных отложений. Однако использование его в высоких дозах (не менее 10 г/сут), необходимых для успешного лечения, ограничено из-за крайне неприятного запаха.
При AL-амилоидозе, как и при миеломной болезни, лечение должно быть направлено на подавление пролиферации клона плазматических клеток для уменьшения продукции ЛЦИ. Наибольший опыт лечения накоплен для мелфалана в сочетании с преднизолоном [21]. Лечение продолжают 7-дневными курсами с интервалом 4–6 недель на протяжении 12–24 месяцев. Доза мелфалана составляет 0,15 мг/кг массы тела в сутки, преднизолона – 0,8 мг/кг массы тела в сутки. У больных с ХПН (скорость клубочковой фильтрации менее 40 мл/мин) дозу мелфалана уменьшают на 50%. Эта схема лечения характеризуется низким риском осложнений химиотерапии и может применяться у абсолютного большинства больных, даже при наличии тяжелых висцеральных проявлений амилоидоза. Однако эффект этой терапии достигается только у 18–30% больных.
В настоящее время разработан эффективный иммунохимический метод оценки ремиссии плазмоклеточной дискразии у больных амилоидозом – определение свободных ЛЦИ в сыворотке (Freelite). Ранее применявшиеся методы не позволяли отличить пул свободных ЛЦИ от пула связанных с иммуноглобулинами, что резко снижало чувствительность методов. Благодаря методу Freelite, который базируется на применении антител к скрытым эпитопам ЛЦИ, появилась возможность количественной оценки пула свободных ЛЦИ, впервые были сформулированы критерии нормальной продукции ЛЦИ. В настоящее время во всем мире метод Freelite стал стандартом оценки эффективности терапии и мониторирования течения болезни у больных амилоидозом [22, 23].
С целью повышения эффективности лечения широко применялись попытки лечения более агрессивными схемами химиотерапии с включением винкристина, доксорубицина, циклофосфана, мелфалана, дексаметазона в разных комбинациях. Наибольшая эффективность достигнута при химиотерапии с использованием высоких доз. Так, в 1996 г. были опубликованы предварительные результаты (R.L. Comenzo и соавт.) лечения 5 больных AL-амилоидозом внутривенными вливаниями мелфалана в дозе 200 мг/м² поверхности тела с последующим введением аутологичных стволовых клеток (CD34+) крови [24]. Аутологичные стволовые клетки получают методом лейкофереза крови больного после предварительной их мобилизации из костного мозга под влиянием введенного извне гранулоцитарного колониестимулирующего фактора. Однако тяжелый агранулоцитоз и другие осложнения этой терапии существенно ограничивают применение терапии сверхвысокими дозами мелфалана, в частности, у больных с недостаточностью кровообращения. В последние годы показана высокая эффективность при относительно удовлетворительной переносимости схемы «мелфалан (0,15 мг/кг 4 дня) в сочетании с дексаметазоном (20 мг/сут в 1–4-е и 9–12-е дни)» ежемесячными курсами [25]. В настоящее время эта схема стала первой линией терапии AL-амилоидоза. В качестве второй линии терапии обсуждаются схемы с включением бортезомиба и талидомида [26, 27]. Кардиотоксичность и нейротоксичность этих препаратов не позволяют рекомендовать их в качестве препаратов первой линии выбора, несмотря на высокую эффективность в отношении плазмоклеточной дискразии.
Основной целью лечения А-бета-2-М-амилоидоза является уменьшение поступления в ткани бета-2-микроглобулина. Поскольку способы подавления его продукции в настоящее время отсутствуют, необходимо увеличить клиренс бета-2-микроглобулина путем проведения современных методов очищения крови.
Широко распространено мнение о том, что наиболее эффективным методом выведения бета-2-микроглобулина является трансплантация почки. Эта точка зрения подкреплена высокой эффективностью трансплантации в отношении костных болей, характерных для диализного амилоидоза. В настоящее время установлено, что причиной костных болей, так же как и костных кист, является макрофагальное воспаление, характерное для этого типа амилоидоза. Быстрое исчезновение болей после трансплантации является следствием назначения стероидов и цитостатиков, необходимых для предотвращения отторжения трансплантата и одновременно подавляющих макрофагальное воспаление в зоне депозиции амилоида. На поздних этапах амилоидогенеза зависимость макрофагального воспаления от поставки бета-2-микроглобулина в ткани снижается, и улучшение клиренса этого белка после трансплантации не предупреждает дальнейшее прогрессирование костных кист и эрозий. Таким образом, трансплантация способна предотвратить прогрессирование диализного амилоидоза только на ранних, в основном доклинических, стадиях амилоидоза [28].
В этой связи в лечении диализного амилоидоза большое значение придают современным диализным технологиям увеличения клиренса бета-2-микроглобулина. При проведении стандартной процедуры гемодиализа с использованием низкопоточных мембран (диффузионная модель) клиренс бета-2-микроглобулина существенно ограничен, так как этот белок по своей массе (11 800 Да) приближается к классу так называемых средних молекул. Однако применение высокопоточных синтетических мембран и сочетание диффузионного механизма очищения крови с конвективным приводят к существенному повышению эффективности выведения бета-2-микроглобулина. Так, было показано, что лечение гемодиафильтрацией в режиме online (режим ГДФ ONLINE предусматривает использование в качестве замещающего раствора сверхчистого диализата, который готовится непосредственно в аппарате для гемодиализа) позволяет добиться значительного снижения постдиализного уровня бета-2-микроглобулина (на 70% и более) [29]. В этой связи наиболее предпочтительным методом гемодиализа с точки зрения профилактики и лечения диализного амилоидоза является высокоэффективная ГДФ ONLINE с использованием синтетических мембран и скоростью подачи замещающего раствора не менее 100 мл/мин. Пациенты, которые получали ГДФ ONLINE с объемом замещения не менее 24 л за 4 часа процедуры, имели достоверно более существенное (на 72,7%) снижение сывороточного уровня бета-2-микроглобулина по сравнению с пациентами, получавшими стандартный гемодиализ на низкопоточных мембранах (на 49,7%) [30].
Результаты недавно завершенного исследования CONTRAST (CONvective TRAnsport STudy), посвященного влиянию конвективного транспорта на сывороточный уровень бета-2-микроглобулина, свидетельствуют о том, что через 6 месяцев у пациентов, которым проводится гемодиафильтрация, уровень бета-2-микроглобулина в сыворотке достоверно ниже, чем у пациентов на низкопоточном гемодиализе [31].
Дальнейшие перспективы в развитии гемодиализа во многом связаны с технологическим усовершенствованием и широким распространением методики ГДФ ONLINE в клинической практике. В частности, усовершенствованная техника ГДФ ONLINE – MIXED ГДФ – позволяет существенно увеличить эффективность конвективного транспорта, в том числе перенос средних молекул, в безопасном для пациента режиме [32–34].
Большое значение имеет применение современных синтетических диализных мембран, которые способны адсорбировать значительные количества бета-2-микроглобулина, в результате чего концентрация этого белка в крови в течение процедуры гемодиализа может снижаться на 30–40% [29]. Разработаны также специальные сорбционные колонки с высокими показателями очищения крови от бета-2-микроглобулина, однако их широкое применение сдерживается высокой себестоимостью [35–37].
Поскольку важным компонентом диализного амилоидоза является макрофагальное воспаление, большое значение имеет использование высокоочищенных растворов для приготовления диализата. G. Lonnemann и другие исследователи сообщают о том, что применение ультрачистых диализных жидкостей во время лечения конвективными методами позволяет снизить частоту синдрома запястного канала на 50% [37, 38].
В качестве симптоматической терапии больным диализным амилоидозом может быть рекомендовано местное применение стероидных мазей при болях, в литературе обсуждаются также показания к применению стероидов внутрь и в инъекциях, однако их использование ограничено из-за тяжелой ренальной остеодистрофии. В этой связи представляется перспективным применение у таких больных современных препаратов, подавляющих эффекты фактора некроза опухоли, интерлейкина-1 и интерлейкина-6.
Средством выбора для лечения АTTR-амилоидоза служит трансплантация печени, при которой удается удалить источник синтеза амилоидогенного предшественника. Трансплантация печени существенно задерживает прогрессирование амилоидоза, в первую очередь нервной системы [39]. В то же время накопленные депозиты амилоида в тканях, в особенности в сердце, способны адсорбировать также и нормальный транстиретин и тем самым способствовать дальнейшему прогрессированию амилоидоза [6]. В этой связи в настоящее время широко обсуждаются возможности стабилизации нормального транстиретина и предотвращения его амилоидогенности с помощью фармакологических препаратов (diflunisal) [40].
Поскольку ХПН служит одной из основных причин смерти больных системным амилоидозом, проведение гемодиализа или постоянного амбулаторного перитонеального диализа позволяет улучшить прогноз этих пациентов, в частности, выживаемость этих больных связывают с уровнем бета-2-микроглобулина [41–43].
Выживаемость больных амилоидозом при проведении гемодиализа, независимо от типа амилоидоза, сопоставима с выживаемостью больных другими системными заболеваниями и сахарным диабетом. Постоянный амбулаторный перитонеальный диализ имеет некоторые преимущества перед гемодиализом, поскольку нет необходимости в постоянном сосудистом доступе, отсутствует артериальная гипотония во время процедуры диализа, а у больных с AL-типом амилоидоза во время процедуры возможно удаление легких цепей иммуноглобулинов.
Трансплантация почки одинаково эффективна при обоих типах системного амилоидоза. Пятилетняя выживаемость больных и трансплантата составляет 65 и 62% соответственно и сопоставима с таковыми показателями других групп больных с ХПН. Трансплантация почки показана больным с медленным прогрессированием амилоидоза без поражения сердца и ЖКТ. Амилоидоз в трансплантированной почке возникает, по разным данным, примерно у 30% больных, однако он служит причиной потери трансплантата всего у 2–3% пациентов.
Прогноз при амилоидозе
Амилоидоз характеризуется неуклонно прогрессирующим течением. Прогноз заболевания зависит от типа амилоида, степени вовлечения различных органов, главным образом сердца и почек, наличия и характера предрасполагающего заболевания.
Наиболее серьезен прогноз при AL-амилоидозе. По данным клиники Мейо, средняя продолжительность жизни больных этим типом амилоидоза составляет лишь 13,2 мес., 5-летняя выживаемость – 7%, 10-летняя – всего 1%. При этом самая низкая продолжительность жизни отмечена у пациентов с застойной недостаточностью кровообращения (6 мес.) и ортостатической артериальной гипотонией (8 мес.) [2, 44, 45]. Наиболее частыми причинами смерти больных с AL-типом амилоидоза бывают сердечная недостаточность и нарушения ритма сердца (48%), уремия (15%), сепсис и инфекции (8%). Несмотря на то что смерть от уремии отмечают значительно реже, чем от кардиальных причин, ХПН разной степени выраженности регистрируют более чем у 60% умерших.
Прогноз при АА-амилоидозе более благоприятен и зависит, главным образом, от характера предрасполагающего заболевания и возможности его контроля. Средняя продолжительность жизни больных с этим типом амилоидоза от момента верификации диагноза составляет 30–60 мес. (большая при вторичном амилоидозе, меньшая при амилоидозе в рамках периодической болезни). Эффективное лечение предрасполагающих заболеваний, в том числе полное излечение туберкулеза или хронических нагноений, не исключает возможности развития амилоидоза в дальнейшем, однако замедляет его прогрессирование, улучшая прогноз. Эффективная терапия ревматоидного артрита позволяет продлить течение амилоидной нефропатии, замедляя наступление ХПН. Основной причиной смерти больных АА-типом амилоидоза служит почечная недостаточность.
Достижения последних лет в изучении проблемы амилоидоза, позволившие сформулировать четкие критерии классификации клинических форм амилоидоза и подходы к лечению, дали возможность существенно улучшить прогноз больных разными типами амилоидоза.