Нарушение липидного обмена у детей с заболеваниями печени. XXIII конгресс детских гастроэнтерологов России и стран СНГ «Актуальные вопросы абдоминальной патологии у детей»
- Аннотация
- Статья
- Ссылки

Общие положения
О нарушении липидного обмена у пациентов любого возраста можно судить по содержанию фосфолипидов, неэстерифицированных жирных кислот, триглицеридов, свободного и эстерифицированного холестерина. Липиды находятся в плазме крови в связанной с белками форме и образуют следующие классы:
- хиломикроны;
- липопротеины очень низкой плотности (ЛПОНП);
- липопротеины низкой плотности (ЛПНП) – бета-липопротеины;
- липопротеины высокой плотности (ЛПВП) – альфа-липопротеины.
Разделение липопротеинов на классы обусловлено различием в плотности движения при электрофорезе.
В зависимости от показателей применяются формулы для вычисления индекса атерогенности. В педиатрической практике к подобному расчету прибегают нечасто, поскольку корреляции между уровнем указанного индекса и атеросклеротическим поражением у детей не установлено.
Под дислипидемией понимают самостоятельные заболевания и патогенетические состояния, обусловленные количественными и качественными нарушениями состава липидов и липопротеинов в крови.
Выделяют первичные и вторичные дислипидемии. Первичные генетически детерминированы. Выявляются стабильные изменения одного или нескольких показателей липидного обмена в отсутствие каких-либо заболеваний. Вторичные дислипидемии – следствие широкого спектра заболеваний: панкреатита, гепатита, холецистита, нефрита, гипотиреоза, наследственных болезней обмена и т.д.
Дислипидемии классифицируют на врожденные и приобретенные. Врожденные наблюдаются у ребенка с первых месяцев жизни, но не имеют наследственного или семейного характера и обусловлены неблагоприятными факторами воздействия на плод – хронической гипоксией, заболеваниями матери, в частности сахарным диабетом, нефропатией, поздним гестозом. Приобретенные дислипидемии развиваются в разные периоды жизни ребенка на фоне:
- нарушения режима питания;
- изменения химического состава рациона питания; гиподинамии;
- дисфункции желудочно-кишечного тракта;
- различных сопутствующих заболеваний.
Согласно классификации Всемирной организации здравоохранения, в зависимости от спектра в липидограмме различают пять типов гиперлипидемии. У детей чаще встречаются типы IIa и IIb.
Еще совсем недавно проведение молекулярно-генетических исследований для определения типа заболевания казалось невозможным. Сегодня у многих пациентов обнаружены генетические локусы, подтверждающие заболевание. Биохимическое исследование липидограммы считается основным методом определения типа дислипидемии.
При типе I гиперхиломикронемии клинические симптомы включают спленомегалию, абдоминальные колики, панкреатит. У детей раннего возраста достаточно быстро формируются атеросклеротические бляшки. Гиперхолестеринемия типа IIa предполагает наличие атеросклероза коронарных артерий и суставного синдрома. При типе IIb имеют место избыточная масса тела, неалкогольная жировая болезнь печени (НАЖБП), сахарный диабет и ранние формы атеросклероза. При типе III гиперлипидемии дебют происходит в раннем возрасте, появляются ксантомы на ладонях и местах давления одежды, формируется НАЖБП. При типе IV начало заболевания, как правило, приходится на молодой и средний возраст, развиваются гепатомегалия, сахарный диабет и ожирение. При типе V формируются атеросклероз сосудов, ожирение, панкреатит.
Не меньшую роль в структуре дислипидемий играют выявляемые у части пациентов различные формы гиполипидемии.
Гипо-альфа-липопротеинемия (болезнь Танжера) характеризуется отсутствием или снижением до 1–4% в плазме крови уровней ЛПВП и общего холестерина. Клинически наблюдаются увеличение миндалин яркого желто-оранжевого цвета (патогномоничный признак), спленомегалия, увеличение лимфатических узлов, мышечная слабость в верхних и нижних конечностях, ослабление рефлексов, потеря чувствительности. В пунктате костного мозга обнаруживаются «пенистые» клетки.
Семейная абеталипопротеинемия (синдром Бассена – Корнцвейга) представляет собой наследственное аутосомно-рецессивное нарушение образования Апо-В-содержащих липопротеинов. Заболевание характеризуется синдромом мальабсорбции, стеатореей, прогрессирующей дистрофией, задержкой развития, умственной отсталостью, пигментным ретинитом, наличием акантоцитоза, атаксической невропатией.
Наследственная гипо-бета-липопротеинемия отличается аутосомно-доминантным типом наследования, дефектом Апо-В, приводящим к низкому уровню бета-липопротеинов в крови. У больных этой формой гиполипидемии значительно снижается уровень холестерина ЛПНП. Клиническая симптоматика почти такая же, как в предыдущем случае, но имеет более мягкое течение. Диагноз подтверждается при обнаружении в крови низкого содержания холестерина, триглицеридов, Апо-В. Концентрации хиломикронов, ЛПОНП и ЛПНП также низкие.
У детей с хроническими заболеваниями печени нередко регистрируются нарушения липидного метаболизма.
По данным исследований, из 2393 пациентов с хроническими заболеваниями печени нарушение липидного обмена зафиксировано в 15% случаев. Причем у большинства пациентов имели место гликогеновая болезнь, преимущественно типа I, хронические вирусные и хронические криптогенные гепатиты.
Частота выявления дислипидемий составила: при хроническом вирусном гепатите – 7%, аутоиммунных заболеваниях – 23%, хроническом криптогенном гепатите – 24%, гликогеновой болезни – 65%, фруктоземии – 23%, наследственном холестазе – 80%, болезни Вильсона – 10%. Более высокие уровни холестерина отмечались при наследственном холестазе – 9,3 ммоль/л, аномалии желчных протоков – 8,8 ммоль/л, гликогеновой болезни – 7,5 ммоль/л.
При гликогеновой болезни наиболее тяжелые изменения липидного спектра относятся к первому типу заболевания – уровень холестерина превышает 7 ммоль/л.
На сегодняшний день накоплен солидный клинический опыт ведения пациентов детского возраста с НАЖБП – свыше 700 человек. В основном это дети с избыточной массой тела и ожирением. Наибольший интерес представляют участившиеся случаи появления локального неалкогольного гепатоза. У названной категории больных часто выявляются нарушения липидного обмена.
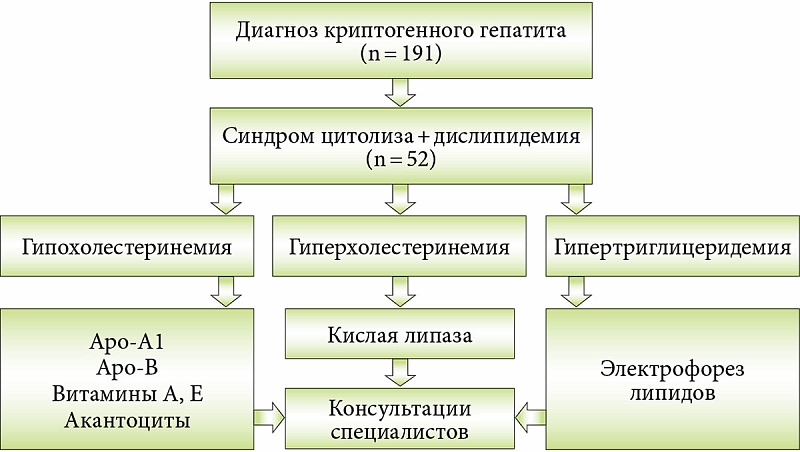
Определить наличие дислипидемии у детей с криптогенным гепатитом позволяет диагностический алгоритм. Докладчик привела данные собственного наблюдения за 191 ребенком с диагнозом хронического криптогенного гепатита. У 52 (27,2%) из них синдром цитолиза сочетался с дислипидемией. В соответствии с разработанным алгоритмом по результатам исследования липидограммы установлено наличие гипохолестеринемии, гиперхолестеринемии или гипертриглицеридемии. При наличии гипохолестеринемии проводили исследование уровня Апо-А1, Апо-В, содержания витаминов A, Е и исследование акантоцитов, поскольку акантоцитоз развивается у 30% пациентов с абеталипопротеинемией. При гиперхолестеринемии изучали активность кислой липазы. При гипертриглицеридемии необходимым считалось проведение электрофореза липидов. Важный момент: пациенты с выявленной дислипидемией требуют консультации ряда специалистов: офтальмолога, невропатолога, кардиолога и др. (рис. 1).
Таким образом, из 52 пациентов с синдромом дислипидемии 34 удалось поставить точный диагноз. Дефицит лизосомной кислой липазы (ДЛКЛ) обнаружен в 38% случаев, гиперхиломикронемия – в 6%, семейная гиперхолестеринемия – в 47%, болезнь Танжера и абеталипопротеинемия – в 3%.
Диагностика болезней нарушений липидного обмена основана на результатах клинического и параклинического обследования, а также данных семейного анамнеза. Обратите внимание: наследственные заболевания могут протекать по аналогии с ненаследственными. Нередко наследственное заболевание у детей сопутствует основному, ненаследственному заболеванию, с которым родители ребенка обратились за медицинской помощью. Поэтому выявленные нарушения липидного обмена требуют обязательного дополнительного обследования с целью своевременной постановки диагноза и начала этиопатогенетической терапии для предупреждения осложнений дислипидемий. Вместе с тем следует помнить, что дислипидемия нередко приводит к выраженному хилезу сыворотки и искажению лабораторных результатов исследования.
Клинический случай
Мальчик 2002 года рождения от третьей нормально протекавшей беременности (старшие дети здоровы). Наследственность по заболеваниям липидного обмена не отягощена. Вес при рождении – 4900 г, рост – 56 см. Профилактические прививки проведены согласно Национальному календарю вакцинации. Психомоторное развитие соответствует возрасту. Находился на грудном вскармливании до восьми месяцев. С трехмесячного возраста имели место проявления атопического дерматита. В настоящее время у ребенка наблюдаются бронхиальная астма, круглогодичный аллергический ринит.
В возрасте четырех лет в мае 2006 г. при подготовке к аденотомии у ребенка выявлены синдром цитолиза (уровень аланинаминотрансферазы, аспартатаминотрансферазы – 67 Ед/л), повышенное содержание триглицеридов, холестерина, ЛПНП в крови. При проведении ультразвукового исследования (УЗИ) брюшной полости выявлены гепатомегалия и диффузные изменения паренхимы печени. Исследование маркеров вирусных гепатитов дало отрицательный результат. После консультации генетика поставлен предварительный диагноз семейной гиперхолестеринемии типа IIа. В апреле 2007 г. при определении генетических полиморфизмов установлено наличие монозиготного варианта липопротеиновой липазы с повышенным уровнем триглицеридов, сниженным уровнем ЛПВП в крови, что свидетельствовало о повышенном риске развития атеросклеротических поражений сосудов. В том же году проведены генетические анализы.
В клинику ФИЦ питания и биотехнологии ребенок поступил с диагнозом хронического криптогенного гепатита. У него определялась гепатомегалия: печень выступала из-под края реберной дуги на 4 см, что подтверждалось данными УЗИ. При неоднократном обследовании у мальчика регистрировались сниженный уровень альфа-1-антитрипсина (на 30% от нормы), повышенное содержание ферритина (646 мг/л). Периодически отмечались гипогликемия (3,3–2,8 ммоль/л), которая определялась при исследовании биохимического анализа крови и в пробе с нагрузкой глюкозой, лактат-ацидоз. У пациента проводился дифференциальный диагноз между дефицитом альфа-1-антитрипсина, болезнями накопления гликогена, гемохроматозом, наследственными нарушениями липидного обмена.
При исследовании гена альфа-1-антитрипсина была обнаружена мутация Е34К (Z-аллель) в гетерозиготном состоянии, характерная для фенотипа PiMZ (риск развития заболевания – 20%). При дальнейших исследованиях уровень альфа-1-антитрипсина нормализовался и при генетическом анализе в альтернативной лаборатории в гене альфа-1-антитрипсина не обнаружено мутаций (фенотип PiMM – нормальная активность альфа-1-антитрипсина). Результаты молекулярно-генетического исследования гена HFE на выявление частых мутаций, ответственных за развитие гемохроматоза (C282Y, H63D), не подтвердили их наличия.
С целью верификации диагноза, а также определения стадии заболевания в сентябре 2010 г. ребенку провели пункционную биопсию печени. Исследование биоптата печени показало умеренный фиброз портальных трактов, фиброз стенок центральных вен, наличие порто-портальных септ и зоны перигепатоцеллюлярного фиброза. В гепатоцитах округлой формы со светлой цитоплазмой определялись множественные мелкие вакуоли, что указывало на микровезикулярный стеатоз. Согласно морфологическому заключению, признаков, характерных для дефицита альфа-1-антитрипсина, болезней накопления гликогена, нет. Индекс склероза по Desmet – 2–3 балла (умеренный).
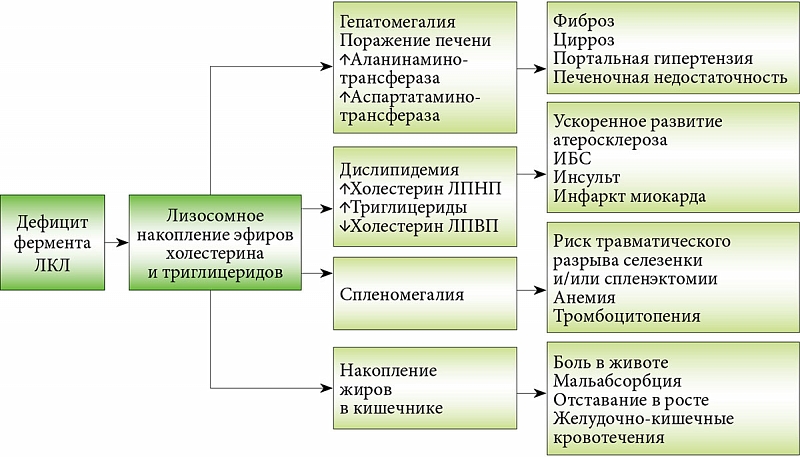
Ранее выявленные у пациента устойчивое повышение уровней трансаминаз, триглицеридов, холестерина и ЛПНП, прогрессирующая гепатомегалия с признаками фиброза, а также микровезикулярный стеатоз гепатоцитов говорили о высокой вероятности наличия болезни накопления эфиров холестерина (одна из фенотипических форм заболевания, которые в современной литературе объединены под названием ДЛКЛ).
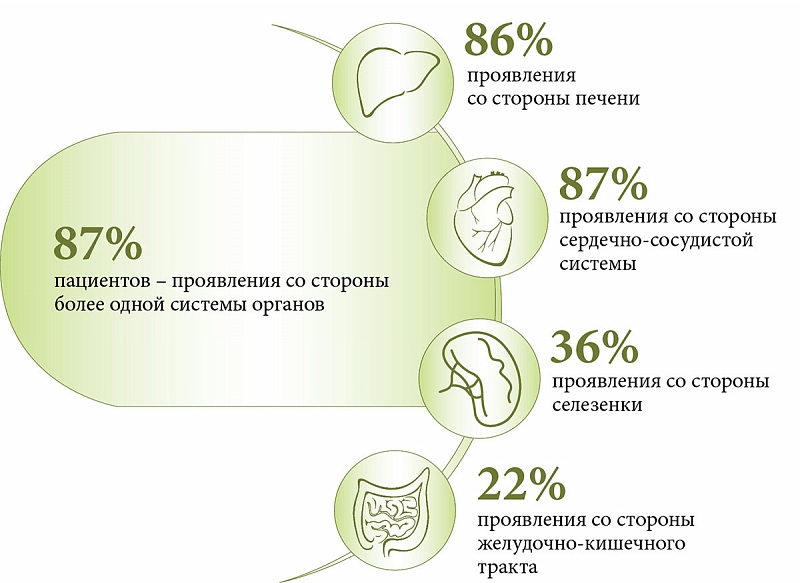
ДЛКЛ представляет собой моногенное нарушение, при котором мутации вызывают дефицит ЛКЛ, что способствует лизосомному накоплению эфиров холестерина и триглицеридов. Системное лизосомное накопление липидов приводит к прогрессирующему поражению различных органов. По данным литературы, среди детей и взрослых с ДЛКЛ клинические проявления со стороны более одной системы органов наблюдаются в 87% случаев (рис. 2 и 3).
В Медико-генетическом научном центре (МГНЦ) РАН впервые в России было налажено измерение активности пула липаз в лейкоцитах крови для диагностики ДЛКЛ. Данный метод позволяет определять общую липазную активность в лейкоцитах периферической крови, в том числе активность ЛКЛ. Не случайно образец крови пациента был исследован именно в этой лаборатории. Показатель активности составил 43,8 нМ/мг/ч, что превышало нижнюю границу нормы (30–118 нМ/мг/ч).
Несмотря на базисную терапию и строгое соблюдение диеты, при очередных обследованиях ребенка зафиксированы признаки нарастания гепатомегалии (до +7 см из-под края реберной дуги), синдрома цитолиза (до 180 Ед/л) и дислипидемии за счет гиперхолестеринемии (до 9,9 ммоль/л), повышения ЛПНП (до 8,14 ммоль/л).
Позднее, с накоплением опыта диагностики такого редкого заболевания, как ДЛКЛ, выяснились некоторые особенности измерения активности ЛКЛ в лейкоцитах периферической крови. При высокой активности прочих липаз крови итоговая активность может маскировать активность ЛКЛ в случае ее патологического снижения. В такой ситуации у пациента с ДЛКЛ итоговая активность будет выше нормы (ложноотрицательный результат). Еще одна особенность метода – чувствительность к качеству образца. В ряде случаев, например при несоблюдении необходимых условий транспортировки, липазная активность в образце снижается, что может дать ложноположительный результат. Таким образом, несмотря на наличие клинических, лабораторных и морфологических данных, указывающих на высокую вероятность ДЛКЛ, данный диагноз не был исключен или подтвержден.
В 2016 г. в Лаборатории наследственных болезней обмена веществ МГНЦ РАН был внедрен современный метод измерения специфической активности ЛКЛ в сухих пятнах крови. Такой метод характеризуется очень высокой чувствительностью, поскольку применяется специфический ингибитор ЛКЛ. Ингибитор лалистат 2 практически полностью ингибирует активность ЛКЛ, при этом не влияет на другие липазные активности в сухих пятнах крови. Это позволяет практически однозначно классифицировать здоровых людей, носителей и больных ДЛКЛ.
В январе 2016 г. ребенку было проведено определение ЛКЛ в пятне крови. В результате исследования обнаружена очень низкая специфическая активность ЛКЛ, характерная только для ДЛКЛ, – 0,02 нмоль/мг/ч (норма 0,37–2,3 нмоль/мг/ч). Диагноз ДЛКЛ был подтвержден специфическим методом измерения активности ЛКЛ. Кроме того, в ходе дополнительных исследований пациенту провели секвенирование экзонов гена LIPA, кодирующего ЛКЛ. В кодирующей части гена мутации выявлена компаунд-гетерозиготная мутация, в экзоне 8 – мутация c.894G>A в гетерозиготном состоянии, приводящая к нарушению сплайсинга. Мутация описана S.A. Scott и соавт. у пациентов с болезнью Вольмана1. В экзоне 4 обнаружена делеция c.421del в гетерозиготном состоянии, приводящая к сдвигу рамки считывания p.Ala141Leufs*20. Выявленная делеция ранее не была описана и, по данным компьютерного анализа (Alamut Visual), является патогенной.
Таким образом, с 2006 по 2016 г. основным диагнозом пациента был «хронический криптогенный гепатит», сопровождавшийся гепатомегалией, повышением уровней трансаминаз, дислипидемией (увеличением содержания холестерина, ЛПНП). В 2010 г. были обнаружены характерные для редкого наследственного заболевания ДЛКЛ фибротические изменения и микровезикулярный стеатоз в образце печени пациента. Однако данный диагноз не удалось своевременно подтвердить или отвергнуть из-за ограничений метода диагностики ДЛКЛ в период исследования пациента. Только в 2016 г., благодаря внедрению наиболее высокочувствительного избирательного метода измерения ЛКЛ в пятнах крови, удалось однозначно подтвердить диагноз ДЛКЛ.
Анализ последовательности гена LIPA позволил определить генетическую поломку, которая стала причиной заболевания у данного пациента. При ДЛКЛ исследование гена стандартными методами может быть только подтверждающим диагностическим способом, поскольку известны случаи, когда мутация в кодирующих областях не выявлялась. Это может быть обусловлено наличием мутаций в некодирующих участках гена LIPA, наличием крупных перестроек, а также другими механизмами регулирования активности ЛКЛ, например эпигенетическими, которые не обнаруживаются при стандартных генетических исследованиях. Таким образом, основным критерием диагностики ДЛКЛ является снижение активности ЛКЛ в сухих пятнах крови.
Заключение
Данный клинический пример демонстрирует целесообразность индивидуального подхода к ведению пациентов с нарушениями липидного обмена, а также необходимость использования современных диагностических методов.
При дислипидемии у пациентов повышается риск развития НАЖБП, патологии сосудов, атеросклеротических изменений. Именно поэтому при нарушениях липидного метаболизма у детей необходим комплексный терапевтический подход врачей разных специальностей.
При наличии признаков гепатомегалии, синдрома цитолиза и дислипидемии различной степени выраженности пациентам следует проводить диагностику активности лизосомной кислой липазы в пятнах крови для исключения ДЛКЛ, опасного тяжелыми поражениями печени, селезенки, сосудов, а также инвалидизацией и снижением качества жизни пациентов.