Патогенетическое обоснование применения альфакальцидола для лечения остеопороза при некоторых эндокринных заболеваниях
- Аннотация
- Статья
- Ссылки
- English
Введение
Остеопороз, возникающий на фоне эндокринных заболеваний, относится к вторичным остеопорозам и рассматривается как осложнение основной патологии. Именно поэтому вопросами диагностики и лечения таких пациентов нередко занимаются эндокринологи. Остеопороз и остеопатии, возникающие на фоне различных эндокринных заболеваний, характеризуются патогенетическими и клиническими особенностями, знание которых играет важную роль при определении тактики лечения.
Глюкокортикоидный остеопороз
По распространенности и медико-социальной значимости первое место среди разных видов вторичного остеопороза занимает глюкокортикоидный остеопороз, который развивается вследствие длительного приема глюкокортикоидов (ГК) либо эндогенной гиперсекреции ГК надпочечниками.
Эндогенный гиперкортицизм встречается очень редко. Остеопороз отмечают у 60–90% пациентов с болезнью Иценко – Кушинга или синдромом Кушинга, а переломы позвонков – у 76% [1].
В настоящее время основной причиной развития вторичного остеопороза как у женщин, так и у мужчин считается прием системных ГК. Так, ятрогенный глюкокортикоидный остеопороз развивается в среднем у 30–50% пациентов [1]. ГК широко применяются для лечения различных заболеваний, в первую очередь обструктивных заболеваний легких и болезней костно-мышечной системы. В мире в среднем 2,7–4,6% женщин 55 лет и старше принимают пероральные ГК постоянно [2], среди жительниц Великобритании данного возраста – 1,4–1,7% [3].
Прием ГК приводит к быстрому снижению минеральной плотности кости (МПК), наиболее выраженному в первый год лечения. Уже в первые три – шесть месяцев терапии повышается риск переломов [4]. Кроме того, ГК негативно влияют на мышечную силу, что увеличивает риск падений [5–7]. Метаанализ исследований (порядка 42 000 пациентов) показал, что на фоне предшествующего или текущего приема ГК риск переломов у мужчин и женщин в возрасте 50 лет и старше повышается в равной степени: относительный риск (ОР) для любого перелома варьирует от 1,98 (в 50 лет) до 1,66 (в 85 лет), для любого остеопоротического перелома и перелома проксимального отдела бедра – 2,63–1,71 и 2,48–4,42 соответственно [8]. Получены данные, что на фоне приема пероральных ГК в период постменопаузы переломы происходят при более высоких значениях МПК, чем при постменопаузальном остеопорозе [8].
При этом установлен дозозависимый эффект ГК на риск переломов: у больных, принимавших ГК в дозе ≥ 7,5 мг/сут в преднизолоновом эквиваленте, ОР компрессионных переломов позвонков составил 5,18 при 95%-ном доверительном интервале (ДИ) 4,25–6,31, для непозвоночных переломов – 2,27 (95% ДИ 2,16–3,10) [6]. В период постменопаузы на фоне терапии ГК при снижении МПК на одно стандартное отклонение риск деформации позвонков возрастает на 85%, а в случае увеличения суточной дозы на каждые 10 мг – на 62% [4].
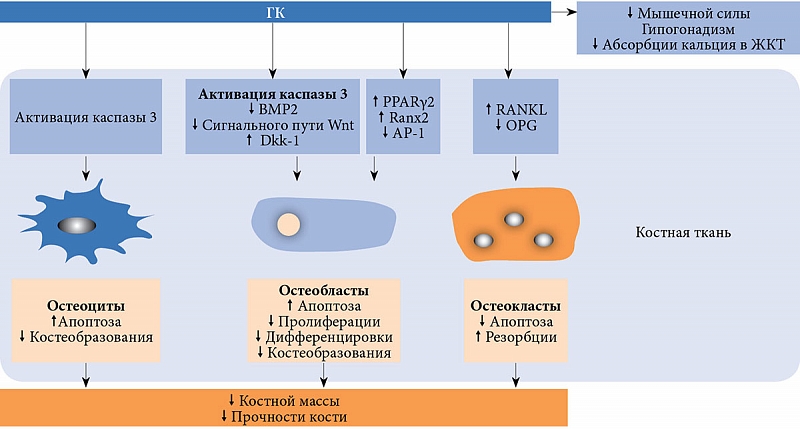
Основное влияние ГК на костное ремоделирование – подавление костеобразования в результате снижения показателей активности остеобластов, а также повышение апоптоза остеобластов и остеоцитов, что считается основной причиной развития остеопороза [5–7]. Кроме того, ГК способствуют активации каспазы 3 – важного триггера апоптоза остеобластов и остеоцитов [9]. Снижение функции остеобластов может быть связано и с активацией гликоген-синтазы-киназы-3β – протеина, вовлеченного в сигнальный путь Wnt и играющего ключевую роль в остеобластогенезе [10]. ГК активируют выработку сигнальных молекул Dickkopf-1 (Dkk-1) и склеростина – ингибиторов сигнального пути Wnt. Как следствие, повышается активность β-катепсина и замедляется продукция коллагена 1 за счет блокирования транскрипции соответствующих генов [5, 7, 10]. ГК могут также смещать направление дифференцировки стромальных клеток костного мозга (предшественниц остеобластов) в сторону образования адипоцитов путем угнетения АР-1 (Аctivator Protein 1 – активирующий протеин 1), повышения активности PPARγ2 (Peroxisome Proliferator-Activated Receptors – рецепторы, активируемые пероксисомными пролифераторами) и угнетения Ranx2 (рис. 1) [7, 10].
Механизм, усиливающий костную резорбцию в результате воздействия ГК, до конца не изучен. Однако известно, что ГК стимулируют созревание и активность остеокластов через повышение уровня RANKL (Receptor Activator of Nuclear Factor Kappa-B Ligand – лиганд рецептора активатора ядерного фактора каппа-В) к ОРG (osteoprotegerin – остеопротегерин) [5, 7]. Отмечается также снижение продукции половых гормонов, сопровождающееся активацией процессов костного ремоделирования [5, 6]. ГК снижают абсорбцию кальция в кишечнике и его тубулярную реабсорбцию, что приводит к повышению секреции паратиреоидного гормона (ПТГ) (рис. 1). Избыточное содержание ГК приводит к подавлению экспрессии VDR (Vitamin D Receptor – рецептор витамина D) и опосредованно негативно влияет на мышечную силу (следствие воздействия на клетки скелетных мышц повышенного уровня ПТГ, сниженного уровня IGF-1 (Insulinlike Growth Factor 1 – инсулиноподобный фактор роста 1) и дефицита VDR) [11].
Диабетическая остеопатия
Сахарный диабет (СД) – одна из важнейших проблем современного здравоохранения. За последние 20 лет численность больных СД в мире выросла более чем в два раза и достигла 366 млн (7% населения). При этом около 50% случаев приходится на возрастную категорию от 50 до 69 лет [12]. Медико-социальное значение СД обусловлено развитием системных сосудистых осложнений, которые являются основной причиной инвалидизации и смерти пациентов. Каждые 5 секунд в мире умирает один больной, за год эта цифра составляет 4,6 млн, в России – 66 тыс. [12]. Сегодня все больший интерес вызывает малоизученное осложнение СД – остеопороз, или диабетическая остеопатия, ассоциирующийся с повышенным риском переломов, и в первую очередь перелома проксимального отдела бедра.
Причина развития диабетической остеопатии – нарушение баланса процессов костного ремоделирования: скорость костной резорбции повышается, а костеобразования снижается. Увеличение количества остеокластов и усиление резорбции костной ткани происходят в результате повышения экспрессии RANK, синтеза RANKL – основного медиатора дифференцировки, созревания и функции остеокластов, фактора некроза опухоли α и колониестимулирующего фактора макрофагов [13].
Снижение активности костеобразования связано в первую очередь с хронической гипергликемией. Высокий уровень глюкозы в крови тормозит созревание остеобластов и процесс минерализации, что проявляется в снижении биохимических показателей костеобразования – остеокальцина и коллагена 1 [14]. Прочность костной ткани снижается также в результате деградации коллагена 1 AGE (Аssay of Advanced Glycation Endproducts – конечные продукты гликирования) [15]. Помимо прямого угнетающего воздействия на функцию остеобластов гипергликемия стимулирует накопление адипоцитов в костном мозге длинных трубчатых костей, что приводит к уменьшению количества остеобластов и, как следствие, истончению кортикального слоя [16]. Важным механизмом замедления костеобразования при СД типа 1 является снижение эндогенной секреции инсулина и IGF-1 [17].
Необходимо отметить, что при СД типа 2 снижение прочности костной ткани и повышение риска переломов не всегда ассоциируются с уменьшением МПК и остеопеническим синдромом [18, 19]. В то же время при СД типа 1, особенно в случае раннего дебюта заболевания, дефицит МПК и остеопороз встречаются чаще [20–22]. Причина – нарушение формирования адекватного пика костной массы в подростковом периоде [23]. У женщин с СД типа 1 риск перелома проксимального отдела бедра увеличивается в 12 раз по сравнению с женщинами без диабета [24]. При СД типа 2 таковой повышается в два раза [24, 25], а также несколько увеличивается риск переломов позвонков, дистального отдела предплечья, плечевой кости и костей стоп [26].
Повышение риска переломов у пациентов с СД объясняется не только снижением костной массы и прочности костей, но и возрастанием риска падений вследствие гипогликемий и осложнений заболевания: диабетической ретинопатии, катаракты и т.д., из-за чего может снижаться периферическое сумеречное зрение [27]. Периферическая нейропатия также является значимым фактором риска падений [28]. На ее фоне отмечается повышение частоты периферических низкоэнергетических переломов у больных СД [29]. Следует отметить, что периферическая нейропатия при СД является самостоятельным фактором уменьшения МПК периферического скелета [30].
По сравнению со здоровой популяцией у больных СД часто наблюдается дефицит витамина D [31]. Кроме того, метаболизм витамина D до активного D-гормона нарушается вследствие развития диабетической нефропатии. Дефицит витамина D ассоциируется с мышечной слабостью, замедлением скорости и качества ходьбы, уменьшением массы и атрофией мышечной ткани [32]. Поэтому восполнение дефицита витамина D и дальнейшее применение его активных метаболитов являются важным звеном в профилактике падений и переломов у таких больных.
Почечная остеодистрофия
Почки, так же как и кишечник, паращитовидные железы и скелет, активно участвуют в регуляции минерального гомеостаза. Для регуляции кальций-фосфорного обмена важны как экскреторная, так и метаболические функции почек. Основная причина развития почечной остеодистрофии – тяжелая хроническая почечная недостаточность, тубулопатии, канальцевый ацидоз и другие заболевания, сопровождающиеся нарушением функции почечных канальцев. Особое место среди причин почечной остеодистрофии занимает диабетическая нефропатия. Ежегодно в мире 500 тыс. больных СД начинают получать заместительную почечную терапию вследствие развития почечной недостаточности до терминальной стадии [12].
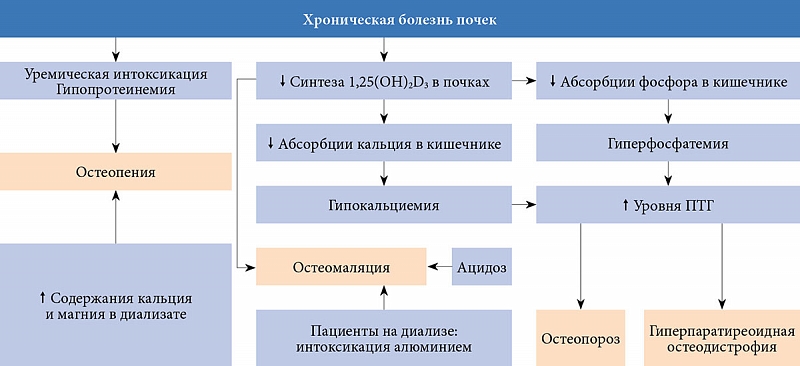
На терминальной стадии хронической болезни почек (ХБП) наблюдается комплекс метаболических нарушений, приводящих к повышению уровня ПТГ, изменению качества и прочности костной ткани. К ним относятся гипокальциемия, гиперфосфатемия, дефицит активного метаболита витамина D – 1,25(OH)2D3, нарушение обратной регуляции функции VDR, чувствительности и экспрессии CaR (кальций-чувствительных рецепторов) [13]. Главный патогенетический фактор почечной остеодистрофии –нарушение почечного синтеза и изменение концентрации в сыворотке крови активных метаболитов витамина D – 1,25(OH)2D3 и 24,25(OH)2D3 из-за выраженного дефицита ферментов почек – 1α-гидроксилазы и 24-гидроксилазы.
Хронический дефицит 1,25(OH)2D3 приводит к повышению секреции ПТГ и формированию симптомокомплекса вторичного гиперпаратиреоза. При уремии в паращитовидных железах может также происходить клональная экспансия клеток – носителей генных мутаций, способствующих росту этих желез и формированию в них узелковой гиперплазии [34]. Кроме того, при вторичном гиперпаратиреозе отмечаются снижение уровня кальций-связывающего протеина в сыворотке крови и экспрессия CaR, что ослабляет ингибирующее воздействие кальция на пролиферацию клеток паращитовидных желез [34]. Важным патогенетическим звеном изменений, происходящих в костной ткани у пациентов с терминальной стадией ХБП, является формирование фиброзно-кистозного остеита в результате избытка ПТГ, развитие остеомаляции на фоне выраженного дефицита 1,25(OH)2D3 и интоксикация алюминием при длительном нахождении на гемодиализе (рис. 2).
Вторичный гиперпаратиреоз
Вторичный гиперпаратиреоз характеризуется гиперфункцией паращитовидных желез в ответ на хроническую гипокальциемию. Наиболее частая причина его развития – терминальная стадия ХБП. Вторичный гиперпаратиреоз также наблюдается при D-дефицитных и D-резистентных состояниях [33]:
-
остеомаляции;
-
дефиците витамина D, возникающем из-за его нехватки в пище или недостаточной инсоляции, синдрома мальабсорбции и др.;
-
приеме антирезорбтивных препаратов – бисфосфонатов, деносумаба;
-
нарушении метаболизма витамина D на фоне лечения антиконвульсантами, цитостатиками;
-
первичном дефиците 1α-гидроксилазы;
-
первичном билиарном циррозе и другой хронической патологии печени;
-
заболеваниях, сопровождающихся нарушением всасывания кальция в желудочно-кишечном тракте (ЖКТ), – резекция желудка или кишечника, снижение желудочной секреции и др.;
-
приеме алюминийсодержащих антацидов;
-
гиперфосфатемии;
-
первичной гиперкальциурии;
-
массивных гемотрансфузиях за счет добавления цитрата.
Все эти заболевания и состояния сопровождаются длительным хроническим дефицитом активного метаболита 1,25(OH)2D3, снижением абсорбции кальция в кишечнике и уровня кальция в крови. На уменьшение уровня ионизированного кальция реагируют рецепторы CaR паращитовидных желез, что в комплексе приводит к стойкому повышению секреции ПТГ, влияющего на три основные мишени – почки, кишечник и костную ткань. В почках ПТГ активирует 1α-гидроксилазу и выработку 1,25(OH)2D3. В результате повышается абсорбция кальция и фосфата в кишечнике. Кроме того, в почках ПТГ увеличивает реабсорбцию кальция и повышает экскрецию фосфата с мочой. В костной ткани он стимулирует функцию остеокластов и активность костной резорбции, благодаря чему кальций и фосфат костного гидроксиапатита попадают в кровь. Все эффекты ПТГ направлены на повышение поступления кальция во внеклеточную жидкость и поддержание его нормальной концентрации в крови [33, 34].
Длительная гиперстимуляция функции паращитовидных желез и повышенный уровень ПТГ способствуют формированию симптомокомплекса вторичного гиперпаратиреоза. Он характеризуется быстрой потерей костной массы, развитием остеопороза и переломов, остеомаляцией, проявляющейся мышечной слабостью, болями в костях, слабостью проксимальных мышечных групп, трудностями при подъеме по лестнице или вставании со стула, в тяжелых случаях – формированием внекостных кальцификатов [34].
Лечение остеопороза при эндокринных заболеваниях: роль альфакальцидола
В настоящее время для лечения остеопороза используется широкий арсенал препаратов, основной целью назначения которых является снижение риска переломов. В качестве самостоятельной терапии системного остеопороза применяются бисфосфонаты, деносумаб, терипаратид и стронция ранелат (вторая линия лечения). Все антиостеопоротические препараты назначаются на фоне базовой терапии солями кальция (в случае его недостаточного пищевого потребления), витамином D или его активным метаболитом – альфакальцидолом.
Альфакальцидол 1α(ОН)D3 (Альфа Д3-Тева®) – синтетический аналог естественных активных метаболитов витамина D. В отличие от нативного витамина D он превращается в D-гормон 1,25(ОН)2D3 за одну метаболическую стадию (минуя почки) при помощи печеночного фермента 25-гидроксилазы. Скорость образования 1,25(ОН)2D3 из альфакальцидола намного выше, чем из нативного витамина D, поэтому биологические эффекты первого проявляются быстрее и интенсивнее.
К основным биологическим и клиническим эффектам альфакальцидола относятся [35]:
1) усиление абсорбции кальция в ЖКТ и повышение кальциемии;
2) подавление гиперпродукции ПТГ паращитовидными железами и снижение благодаря этому активности костной резорбции;
3) стимуляция костного ремоделирования и синтеза костного матрикса путем прямого воздействия на VDR остеобластов, повышения их дифференцировки и функциональной активности (важный эффект при подавлении костеобразования у больных СД и принимающих ГК);
4) улучшение качества кости вследствие подавления перфорации трабекулярных пластин, усиления репарации костей, повышения синтеза костного матрикса и местных ростовых факторов;
5) снижение риска падений в результате повышения мышечной силы.
Необходимо отметить, что клинически значимые эффекты нативного витамина D проявляются только у лиц с его исходным дефицитом при нормальной функции почек, в то время как альфакальцидол активно превращается в D-гормон вне зависимости от уровня 25(ОН)D3 и функции почек [36]. Однако при лабораторно подтвержденном снижении уровня 25(ОН)D3 назначать альфакальцидол для его восполнения нецелесообразно, так как он лишь незначительно повышает уровень 25(ОН)D3 в сыворотке крови за счет пассивного снижения расхода его запасов в жировой ткани для синтеза D-гормона. Несмотря на то что 25(ОН)D3 сам по себе не оказывает выраженных клинических эффектов, сначала следует нормализовать его концентрацию в крови (выше 30 нг/мл), а потом лечить пациента активным метаболитом витамина D – альфакальцидолом.
Альфакальцидол успешно применяется для лечения и профилактики глюкокортикоидного остеопороза. Патогенетическая основа – формирование отрицательного баланса кальция и повышение продукции ПТГ, уменьшение экспрессии VDR и развитие мышечной слабости на фоне приема ГК. В перечисленных ситуациях альфакальцидол оказывает более выраженный эффект на МПК и риск перелома позвонков, чем нативный витамин D. В исследовании J.D. Ringe и соавт. (2004) участвовало 204 больных, которые постоянно получали ГК и которым был поставлен диагноз «глюкокортикоидный остеопороз». В течение трех лет пациенты получали альфакальцидол в дозе 1 мкг/сут или витамин D3 в дозе 1000 МЕ/сут. Результаты исследования продемонстрировали более выраженное повышение МПК в группе альфакальцидола по сравнению с группой витамина D3: в позвоночнике – на 3,2% (р < 0,0001), в шейке бедра – на 1,8% (р < 0,01). Кроме того, у больных, получавших альфакальцидол, отмечено статистически более значимое снижение риска переломов позвонков – на 39% (р = 0,005) и риска всех типов переломов – на 48% (р = 0,001) [37].
В другом исследовании показано снижение риска переломов позвонков при глюкокортикоидном остеопорозе в 1,8 раза при лечении активными метаболитами витамина D по сравнению с плацебо, нативным витамином D и/или кальцием (ОР 0,56 (95% ДИ 0,34–0,92)) [38].
Еще один важный аспект лечения остеопороза при различных эндокринных заболеваниях – снижение риска падений, который, как указывалось ранее, значительно повышается на фоне СД, выраженной почечной недостаточности и при применении больших доз ГК. В ряде исследований продемонстрировано положительное влияние альфакальцидола на мышечную силу, равновесие и координацию тела и функциональные возможности пациентов [39]. Так, лечение альфакальцидолом значительно снижало риск падений у пожилых пациентов (ОР 0,45 (95% ДИ 0,21–0,97), p = 0,042) [40].
Для пациентов с вторичным гиперпаратиреозом или почечной остеодистрофией альфакальцидол является препаратом выбора вследствие целого ряда клинических эффектов: способности значительно повышать кишечную абсорбцию кальция, подавлять гиперпродукцию ПТГ и стимулировать активность костного ремоделирования. У пациентов с почечной недостаточностью, в том числе терминальной стадией ХБП на гемодиализе, терапия альфакальцидолом в дозах от 0,5 мкг в сутки до 4 мкг два-три раза в неделю повышает уровень сывороточного кальция, снижает уровень ПТГ и способствует увеличению МПК, уменьшая таким образом проявления остеопении и остеомаляции [41–43]. Самый существенный эффект альфакальцидола в профилактике падений – снижение риска последних на 71% при приеме препарата 1 мкг/сут – наблюдался также у пожилых больных со сниженной фильтрационной способностью почек [44]. При этом альфакальцидол продемонстрировал хороший профиль безопасности: частота гиперкальциемии – 1,1%, риск развития нефролитиаза отсутствовал [45].
Заключение
Остеопороз и остеопатии, возникающие на фоне ряда эндокринных заболеваний, характеризуются патогенетическими и клиническими особенностями, которые открывают широкие возможности и перспективы для назначения терапии активным метаболитом витамина D – альфакальцидолом.
L.A. Marchenkova
Russian Scientific Center of Medical Rehabilitation and Balneology
Contact person: Larisa Aleksandrovna Marchenkova, MarchenkovaLA@rncmrik.com
Benefits of active metabolite of vitamin D (alfacalcidol) over native vitamin D as well as its biological and clinical effects mediated via activity of 1,25(ОН)2D3 and discussed, which determine a rationale for its use in treatment of osteoporosis under various endocrine diseases. It is demonstrated that use of alfacalcidol (Alpha D3-Teva®) is among important links in prophylaxis of falls and fractures in such patients.