Перспективы применения российских биологических препаратов в терапии злокачественных новообразований. Петербургский онкологический форум «Белые ночи – 2015». Сателлитный симпозиум компании BIOCAD
- Аннотация
- Статья
- Ссылки






Разработка отечественных высокотехнологичных препаратов как способ повышения доступности дорогостоящих лекарственных средств
Инновационная отечественная биофармацевтическая компания полного цикла BIOCAD (Biopharmaceutical Company) – одна из немногих компаний в России, которая обладает инфраструктурой, необходимой для полного цикла работ по созданию и производству первых в стране препаратов на основе моноклональных антител для лечения онкологических заболеваний. Вице-президент по биомедицинским исследованиям и развитию ЗАО «Биокад» Роман Алексеевич ИВАНОВ в своем выступлении кратко охарактеризовал основные отличия подходов к исследованиям воспроизведенных и оригинальных препаратов, а также биоаналогов.
Как известно, биологические лекарственные средства бывают оригинальными и воспроизведенными. Воспроизведенные биологические препараты с доказанной эквивалентностью оригинальному препарату называются биоаналогами, поскольку точную копию биологического лекарственного средства создать невозможно. В отношении биоаналогов применяются особые подходы к доказательству их эквивалентности оригинальным препаратам.
В настоящее время Всемирная организация здравоохранения (ВОЗ), Европейское агентство по лекарственным препаратам (European Medicines Agency – EMA) и Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США (Food and Drug Administration – FDA) разработали свод рекомендаций в отношении объема исследований биоаналогов для подтверждения их соответствия оригинальным лекарственным средствам. Рекомендации предусматривают прежде всего большой объем сравнительных исследований физико-химических и биологических свойств.
Основной акцент делается на сравнительном исследовании характеристик качества, способных оказывать влияние на биологические свойства молекул – структуру белка, посттрансляционные модификации, чистоту и профиль примесей, функциональные характеристики. Идентичность структуры – это не только последовательность аминокислот. Это еще и сложная пространственная ориентация белковых цепей и молекул, составляющая вторичную и третичную структуры белка. Это характеристики, связанные с зарядом белковой молекулы, ориентацией в пространстве и т.д. Не случайно для доказательства идентичности структуры применяется комплекс из десятков методов, дополняющих друг друга.
Молекулы терапевтических белков, синтезируемые клетками млекопитающих, имеют уникальный состав сахаров – так называемый профиль гликозилирования, характеристика которого во многом зависит от способа производства белка, условий культивирования клеток-продуцентов. Гликозилирование влияет на такие важные биологические свойства моноклонального антитела, как цитотоксичность, фармакокинетика, иммуногенность. Именно поэтому для подтверждения идентичности профиля гликозилирования применяется целый комплекс методов. Тщательным образом контролируются и примеси, как связанные с деградацией белковой молекулы, так и возникающие в процессе очистки.
Для выявления малейших различий в биологической активности между биоаналогом и оригинальным препаратом необходимо использовать данные многочисленных сравнительных исследований in vitro.
Только после доказанного полного соответствия всех характеристик биоаналога оригинальному препарату проводятся сравнительные доклинические исследования на релевантных животных, подробные сравнительные исследования фармакокинетики и фармакодинамики биоаналога. Затем выполняются сравнительные клинические исследования эффективности, безопасности и иммуногенности биоаналога по одному показанию, наиболее чувствительному в отношении выявления возможных различий. Если доказана эквивалентность по одному наиболее чувствительному показанию, считается, что биоаналог может применяться и по другим показаниям, одобренным для оригинального препарата (то есть возможна экстраполяция показаний к применению).
Основной целью сравнительных клинических исследований является демонстрация идентичности терапевтических эффектов биоаналога эффектам оригинального препарата.
Подход к выбору первичной конечной точки в исследованиях биоаналога отличается от такового в исследованиях оригинального препарата – в онкологии наиболее приемлема частота ответа. Точки, которые используются в исследованиях оригинальных препаратов (общая выживаемость, выживаемость, свободная от прогрессирования), менее чувствительны для выявления различий между биоаналогами и оригинальными препаратами.
Важным аспектом в исследованиях биоаналогов является изучение их профиля безопасности, который должен соответствовать таковому оригинального препарата и демонстрировать отсутствие нехарактерных для оригинального препарата побочных эффектов.
В России до 2010 г. отсутствовали четкие требования к клиническому исследованию биоаналогов. В результате на отечественном фармацевтическом рынке появились так называемые copy biologics – препараты, которые нельзя назвать биоаналогами из-за отсутствия данных сравнительных исследований. С сентября 2010 г. российским законодательством закреплено обязательное проведение сравнительных клинических исследований. В 2014 г. вышло в свет Руководство по экспертизе лекарственных средств ФГБУ «НЦЭСМП» Минздрава России, в котором требования к исследованиям биоаналогов соответствуют рекомендациям ВОЗ, EMA и FDA. С 1 января 2016 г. вступят в силу Правила исследований биологических препаратов Евразийского экономического союза, которые представляют аутентичный перевод всех рекомендаций Евросоюза.
По словам Р.А. Иванова, фактически завершена работа по приведению российских требований к исследованиям биоаналогов в соответствие с международными стандартами.
В стране завершилась и большая работа по созданию Правил организации производства и контроля качества лекарственных средств, которые соответствуют европейским правилам GMP. «Компания BIOCAD одной из первых в этом году получила заключение о соответствии производителя лекарственных средств требованиям стандарта надлежащей производственной практики», – констатировал Р.А. Иванов.
Между тем во многих странах пока полностью не решены вопросы взаимозаменяемости оригинальных препаратов и биоаналогов. Все чаще звучит мнение, что, если биоаналог прошел все необходимые сравнительные исследования, оригинальный биологический препарат может быть заменен биоаналогом.
В настоящее время во Франции законодательно закреплена возможность автоматической замены оригинальных препаратов биоаналогами у пациентов, начинающих лечение биологическими лекарственными средствами. Подобный порядок действует и в большинстве скандинавских стран.
В силу специфичности системы государственных закупок в России у отечественных врачей меньше свободы выбора в назначении препарата. Минздрав России разработал проект постановления о процедуре установления взаимозаменяемости препаратов. Документ вступит в силу в 2016 г. В ближайшие три года все ранее зарегистрированные препараты пройдут проверку на соответствие жестким современным требованиям. С 2018 г. у учреждений здравоохранения появится возможность закупать лекарственные средства только с доказанной взаимозаменяемостью референтному препарату.
Появление на отечественном фармацевтическом рынке качественных и безопасных биоаналогов – позитивная тенденция. Например, после появления первого биоаналога гранулоцитарного колониестимулирующего фактора (препарат Лейкостим® компании BIOCAD) средняя стоимость препаратов этой группы снизилась более чем в два раза, а потребление препаратов колониестимулирующего фактора возросло в три раза. То есть в три раза больше пациентов начали получать эффективное лечение.
Другой пример – биоаналог ритуксимаба – препарат Ацеллбия (ритуксимаб), произведенный компанией BIOCAD, – первый российский препарат моноклональных антител. Более низкая стоимость отечественного биоаналога по сравнению с оригинальным препаратом (на 23%) позволила государству сэкономить свыше 30 млн долларов США. В настоящее время более 1000 пациентов перешли на использование российского биоаналога ритуксимаба. При этом сигналов о различиях в профиле безопасности препаратов не зарегистрировано.
В ближайшем будущем на российском фармацевтическом рынке появится целый ряд произведенных компанией BIOCAD биоаналогов (рис. 1).
Завершается процедура регистрации биоаналога бевацизумаба, проходит процедуру регистрации биоаналог трастузумаба, осуществляются клинические исследования разных фаз биоаналогов интерферона бета-1а, пэгинтерферона альфа-2а, дарбэпоэтина, инфликсимаба, адалимумаба.
Вместе с тем, как отметил Р.А. Иванов, производство биоаналогов – только первый этап развития компании BIOCAD. Ее будущее связано с разработкой оригинальных лекарственных средств. Сейчас в разработке и на разных стадиях исследований находятся 11 инновационных лекарственных средств (рис. 2).
«В настоящее время мы завершили разработку антитела к PD-1, которое по функциональной активности не уступает ниволумабу. Во втором квартале 2016 г. начнется его клиническое исследование. На более ранних стадиях исследований находятся еще несколько препаратов, которые будут применяться в комбинации в области иммуноонкологии», – пояснил докладчик.
В 2016 г. компания планирует проведение клинических исследований таких инновационных лекарственных средств, как биспецифические антитела к HER2/HER3 и EGFR/HER3. Двойное блокирование этих мишеней позволит увеличить эффективность терапии рака молочной железы и колоректального рака соответственно. В начале 2016 г. предполагается старт клинических исследований ингибитора CDK8/19, который обладает огромным потенциалом как препарат первой линии терапии кастрационно-резистентного рака предстательной железы и ER-позитивного рака молочной железы.
Завершая выступление, Р.А. Иванов пообещал через год представить первые результаты исследований новых оригинальных лекарственных препаратов, разработанных компанией BIOCAD.
Итоги третьей фазы клинических исследований биоаналога трастузумаба компании BIOCAD
Во втором квартале 2015 г. был завершен анализ результатов международного сравнительного клинического исследования фазы III первого отечественного биоаналога трастузумаба – препарата BCD-022 и оригинального препарата трастузумаба Герцептин®. Трастузумаб – ключевой препарат таргетной терапии HER2-положительного рака молочной железы (РМЖ), однако его применение в рутинной клинической практике пока ограничено высокой стоимостью. Внедрение в клиническую практику воспроизведенного препарата трастузумаба будет способствовать значительному повышению доступности современной высокоэффективной терапии для больных РМЖ.
Научный сотрудник отделения клинической фармакологии и химиотерапии Российского онкологического научного центра им. Н.Н. Блохина, к.м.н. Екатерина Олеговна ИГНАТОВА ознакомила участников симпозиума с результатами международного многоцентрового двойного слепого рандомизированного клинического исследования фазы III, которое проводилось в более чем 30 исследовательских центрах на территории РФ, ближнего и дальнего зарубежья.
В исследовании приняли участие в общей сложности 126 пациенток с HER2-положительным метастатическим РМЖ. Все больные были стратифицированы согласно возрасту, предшествующей терапии, статусу гормональных рецепторов и рандомизированы на две группы в соотношении 1:1. Пациентки первой группы получали BCD-022 в нагрузочной дозе 8 мг/кг однократно (один цикл) с переходом на поддерживающую дозу 6 мг/кг каждые три недели (пять циклов). Пациентки второй группы получали Герцептин® по аналогичной схеме. Кроме того, в обеих группах вводился паклитаксел 175 мг/м2 внутривенно в течение трех часов в первый день каждого трехнедельного цикла. Лечение продолжалось до шести циклов или до прогрессирования заболевания либо до наступления непереносимой токсичности. По окончании шести циклов терапии больные с полным либо частичным ответом или стабилизацией заболевания по решению врача-исследователя переводились в период поддерживающего лечения и наблюдения.
Обе группы были сопоставимы по характеристикам основного заболевания и демографическим параметрам. Большинство пациенток имели инвазивный рак неспецифического типа с гиперэкспрессией HER2 3+, более чем у 50% больных отсутствовала экспрессия гормональных рецепторов. Метастазы в двух и более органах были зарегистрированы примерно у половины пациенток в обеих группах. Основными местами локализации отдаленных метастазов были печень, легкие, лимфатические узлы, кости. Большинству пациенток проводили хирургическое лечение и лучевую терапию. Менее половины из них получали адъювантную или неоадъювантную химиотерапию.
Эффективность терапии оценивалась по данным компьютерной томографии (КТ) с использованием критериев RECIST 1.1. КТ проводили на скрининге, после трех и шести циклов терапии. Если при КТ после третьего или шестого цикла терапии был выявлен полный или частичный ответ, выполняли КТ через четыре недели для подтверждения достигнутого ответа. Эффективность терапии (лучший ответ) оценивалась по результатам КТ независимым специалистом, заслепленным в отношении проводимого больным лечения.
Первичной конечной точкой анализа эффективности была общая частота ответов (полные ответы + частичные ответы), вторичными – частота полных и частичных ответов, частота стабилизации опухолевого ответа и частота прогрессирования заболевания. В анализ эффективности суммарно вошли 110 больных: 56 из группы BCD-022, 54 из группы Герцептина.
Общая частота ответа (ОЧО) в группе препарата BCD-022 составила 53,57% (95%-ный доверительный интервал (ДИ) 40,70–65,98%), а в группе препарата Герцептин® – 53,70% (95% ДИ 40,60–66,31%) соответственно. Разница ОЧО в группе исследуемой терапии и в группе препарата сравнения составила -0,13% (95% ДИ -19,83–18,35%) (р = 0,862, критерий хи-квадрат Пирсона с поправкой Йетса). Нижняя граница рассчитанного 95% ДИ (-19,83%) превышала установленную границу не меньшей эффективности, что позволило сделать вывод о не меньшей эффективности препарата BCD-022 по сравнению с препаратом Герцептин®. Отношение шансов для ОЧО составило 0,9947 (95% ДИ 0,470–2,105), что также указывало на отсутствие статистически значимых различий в эффективности между группами терапии. Статистически значимых различий не было и при сравнении прочих параметров оценки эффективности.
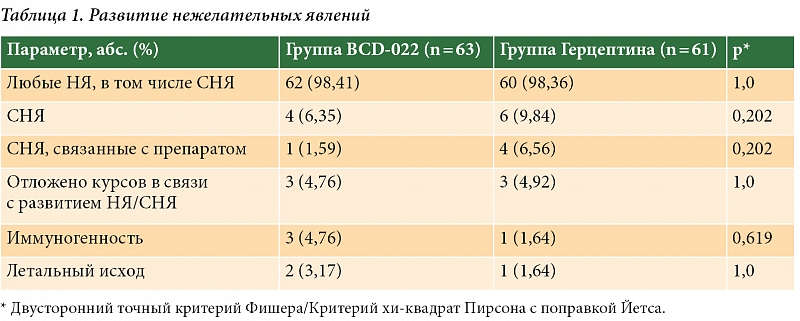
Анализ безопасности проводили стандартными методами – на основании данных о регистрации нежелательных явлений (НЯ) и серьезных нежелательных явлений (СНЯ), результатов клинико-лабораторных анализов, инструментальных обследований и данных физикального осмотра, которые осуществлялись согласно графику процедур. В анализ безопасности вошли все пациентки, получившие хотя бы одно введение исследуемого препарата или препарата сравнения. Суммарно 124 пациентки: 63 из группы BCD-022, 61 из группы Герцептина. В целом на протяжении исследования какие-либо НЯ были зарегистрированы у 62 (98,41%) пациенток группы препарата BCD-022 и у 60 (98,36%) больных группы препарата Герцептин®. СНЯ были выявлены в общей сложности у десяти пациенток: у четырех (6,35%) из группы BCD-022 и шести (9,84%) из группы препарата сравнения (причем у одной больной зарегистрировано два СНЯ). Таким образом, в группе сравнения насчитывалось семь (11,48%) СНЯ. В большинстве случаев СНЯ были обусловлены наличием сопутствующей патологии, воздействием химиопрепаратов, применяемых в рамках комбинированной химиотерапии, либо другими не связанными с исследуемой терапией причинами (табл. 1). По мнению исследователей, только у одной (1,59%) пациентки из группы BCD-022 и четырех (6,56%) из группы препарата сравнения связь с исследуемой терапией была расценена как «вероятная» или «возможная».
Среди НЯ наиболее часто встречались явления гематологической токсичности, включавшие нейтропению, лейкопению, лимфопению, анемию и тромбоцитопению. Несколько реже регистрировались отклонения ряда биохимических показателей: гипергликемия, гиперурикемия, повышение уровней лактатдегидрогеназы (ЛДГ), аспартатаминотрансферазы (АСТ), аланинаминотрансферазы (АЛТ), щелочной фосфатазы (ЩФ), мочевины. Среди прочих НЯ наиболее распространенными были алопеции, нарушения со стороны скелетно-мышечной системы (артралгии, миалгии, оссалгии, боль в спине), со стороны пищеварительной системы (тошнота, рвота, диарея, стоматит), со стороны сердечно-сосудистой системы (тахикардия, пароксизмальная фибрилляция предсердий, желудочковая экстрасистолия), слабость, головная боль, со стороны органов дыхания – острая пневмония, острые респираторные заболевания. Статистически значимых различий между группами не выявлено ни по одному из зарегистрированных НЯ.
Важным параметром безопасности при исследовании препаратов моноклональных антител является иммуногенность. В данном клиническом исследовании оценивали частоту образования и титр связывающих и нейтрализующих антител к трастузумабу. Исследование иммуногенности выявило три (4,76%) случая появления антител к трастузумабу в группе исследуемого препарата и один (1,64%) случай в группе препарата сравнения (р = 0,619). Из них нейтрализующая активность была выявлена у одной пациентки из группы исследуемого препарата и одной больной из группы сравнения (р = 1,0). Таким образом, статистически значимых различий между группами препарата BCD-022 и препарата Герцептин® по частоте образования как связывающих, так и нейтрализующих антител не было. Оба препарата характеризовались низкой частотой выявления антител к трастузумабу.
Фармакокинетику препаратов оценивали по результатам определения трастузумаба в сыворотке крови больных в конкретные временные точки. Проводили анализ стандартных фармакокинетических параметров после первого и шестого циклов терапии, а также определяли концентрации трастузумаба перед каждым введением с последующим расчетом Сtrough. Проведенный анализ показал, что как после однократного, так и после многократного введения исследуемого препарата и препарата сравнения концентрации трастузумаба в крови пациенток изменялись аналогичным образом.
Настоящее исследование выполнено с учетом европейских рекомендаций по оценке биоэквивалентности, а также руководства по изучению биоаналогов моноклональных антител. Статистическое сравнение по первичной фармакокинетической конечной точке (AUC(0–504)) позволило получить 90% ДИ для отношения средних геометрических AUC(0–504) – 83,31–113,55%. Данный интервал соответствует установленным пределам эквивалентности фармакокинетических показателей 80–125%. 90% ДИ для отношения средних геометрических Сmах исследуемого препарата и препарата сравнения составил 88,33–111,14%, что также соответствует интервалу 80–125%. Сравнение испытуемых групп по прочим вторичным конечным точкам (Тmax, T1/2) с помощью стандартных статистических критериев также показало отсутствие статистически значимых различий. При анализе на шестом цикле статистическое сравнение по всем основным фармакокинетическим параметрам (AUC(0-504), Сmах, Тmax, T1/2) с помощью стандартных статистических критериев продемонстрировало отсутствие статистически значимых различий между группами. Статистически значимых различий по показателю Ctrough между группами не зарегистрировано ни в одном из циклов. Следовательно, можно сделать вывод об эквивалентности фармакокинетических свойств препаратов BCD-022 и Герцептин® при внутривенном введении.
Резюмируя информацию, полученную в рамках крупномасштабного клинического исследования биоаналога трастузумаба, докладчик, которая также являлась исследователем в одном из ведущих клинических центров, отметила, что данные свидетельствуют о терапевтической эквивалентности исследуемого препарата BCD-022 и препарата сравнения Герцептин®.
Итоги клинического исследования фазы III биоаналога бевацизумаба в популяции пациентов с немелкоклеточным раком легкого
Ведущий научный сотрудник отдела хирургической пульмонологии Санкт-Петербургского государственного медицинского университета им. акад. И.П. Павлова, д.м.н. Сергей Владимирович ОРЛОВ начал свое выступление с краткого напоминания о том, какой серьезной проблемой для современной онкологии является лечение рака легкого, о достижениях в терапии этого заболевания и месте бевацизумаба в схеме лечения.
Рак легкого – одно из самых распространенных злокачественных новообразований в мире. Рак легкого занимает третье место по числу ежегодно выявляемых случаев заболеваемости и первое по ежегодному числу смертей среди всех злокачественных опухолей. При этом на долю немелкоклеточного рака легкого (НМРЛ) в структуре заболеваемости раком легкого приходится 80%. За последние десятилетия благодаря развитию знаний о молекулярных механизмах патогенеза НМРЛ достигнуты заметные успехи в разработке подходов к лечению этого заболевания, в первую очередь направленной, таргетной терапии. Внедрены в клиническую практику препараты, блокирующие фактор роста эндотелия сосудов (VEGF), рецептор эпидермального фактора роста (EGFR), киназу анапластической лимфомы (ALK).
Один из первых и наиболее клинически изученных таргетных препаратов в онкологии – бевацизумаб, гуманизированное моноклональное антитело к VEGF. Препарат обладает доказанной в клинических исследованиях эффективностью в терапии поздних стадий немелкоклеточного рака легкого; его использование не только увеличивает частоту опухолевого ответа на химиотерапию и выживаемость без прогрессирования заболевания, но и продлевает жизнь пациентов. Согласно актуальным международным рекомендациям, применение бевацизумаба показано в первой линии лечения распространенного неплоскоклеточного НМРЛ. К сожалению, возможности использования бевацизумаба в клинике значительно ограничиваются высокой стоимостью оригинального препарата.
Перспективный путь повышения доступности современной терапии, активно развивающийся в наши дни во всем мире, в том числе в США и Европейском Союзе, – внедрение в клиническую практику биоаналогов. Компанией BIOCAD разработан первый в России биоаналог бевацизумаба. С.В. Орлов рассказал о его разработке, подробно остановившись на результатах проведенного клинического исследования фазы III, и поделился личным опытом применения препарата в клинической практике.
На доклиническом этапе разработки препарата был проведен весь необходимый объем сравнительных исследований с оригинальным препаратом бевацизумаба Авастин®, в ходе которых оценивали структурные, физико-химические и биологические свойства, специфическую активность, токсикологические характеристики, фармакокинетику. Проведенные исследования показали отсутствие значимых различий между биоаналогом бевацизумаба и оригинальным препаратом бевацизумаба по всем изучавшимся характеристикам, что определило возможность продолжения изучения препарата в клинических исследованиях. В конце 2014 г. были подведены итоги сравнительного клинического исследования фазы III биоаналога бевацизумаба производства компании BIOCAD (BCD-021) и оригинального препарата Авастин®, проводимого в 26 исследовательских центрах на территории России и стран ближнего зарубежья. Результаты данного исследования были представлены в июне 2015 г. в рамках постерной сессии ежегодного собрания Американского общества клинической онкологии (American Society of Clinical Oncology – ASCO).
В исследовании приняли участие 138 пациентов с впервые выявленным неоперабельным или метастатическим неплоскоклеточным НМРЛ, которые были рандомизированы в соотношении 1:1 в группу препарата BCD-021 и группу препарата Авастин®. Схема лечения была следующей: в обеих группах пациенты получали паклитаксел в дозе 175 мг/м2 внутривенно в течение трех часов и карбоплатин в дозе, достаточной для достижения AUC 6 мг/мл × мин, внутривенно в течение 15–30 минут, сразу после паклитаксела. Сразу после карбоплатина в зависимости от присвоенной группы пациенты получали BCD-021 или Авастин® в дозе 15 мг/кг внутривенно в виде 90-минутной инфузии. Все препараты вводились в первый день каждого трехнедельного цикла. Лечение продолжалось до шести циклов или до прогрессирования заболевания либо до наступления непереносимых явлений токсичности.
При оценке эффективности первичной конечной точкой была определена общая частота ответов (частичный ответ + полный ответ) после получения до шести курсов терапии, оценивавшаяся по данным компьютерной томографии с использованием критериев RECIST 1.1. Для оценки фармакокинетики в ходе исследования определяли сывороточные концентрации бевацизумаба у всех пациентов перед каждым введением препаратов, а также в конкретных временных точках на первом и шестом циклах химиотерапии. При оценке безопасности анализировали частоту НЯ, СНЯ, частоту отмены лечения вследствие НЯ. Дополнительно исследовали иммуногенность терапии.
Группы пациентов были сопоставимы по исходным демографическим параметрам (возраст, вес, рост, пол), общему состоянию по шкале ECOG и характеристикам основного заболевания (продолжительность времени с момента постановки диагноза, предшествующая терапия, морфологическая характеристика опухоли, число и локализация отдаленных метастазов). У большинства пациентов была диагностирована стадия IV НМРЛ (49 (90,74%) и 46 (82,14%) больных в группах BCD-021 и Авастина соответственно) (р > 0,05).
В анализ эффективности были включены все больные, которые получили хотя бы одно введение препарата BCD-021 или Авастина и у которых можно было оценить достигнутый ответ (n = 110, 54 (78,26%) пациента группы BCD-021 и 56 (81,20%) пациентов группы Авастина).
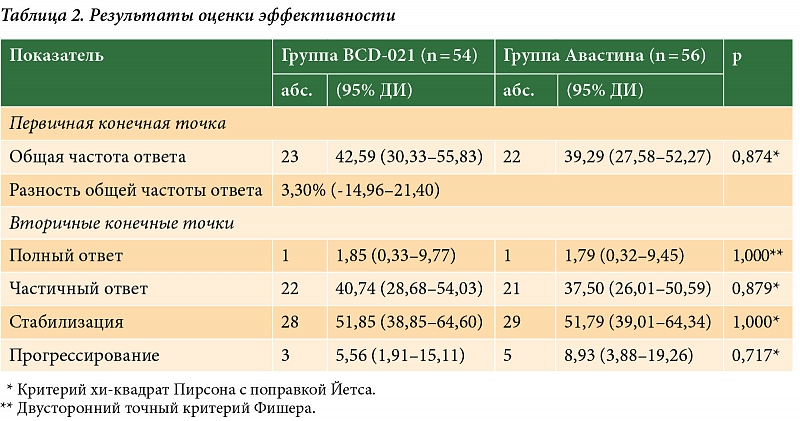
На основании данных КТ общая частота ответа в группе препарата BCD-021 составила 42,59% (95% ДИ 30,33–55,83%), а в группе препарата Авастин® – 39,29% (95% ДИ 27,58–52,27%).
Разница показателя в группе исследуемой терапии и в группе препарата сравнения составила 3,30% с 95% ДИ -14,96–21,40% (р = 0,874, критерий хи-квадрат Пирсона с поправкой Йетса). Нижняя граница рассчитанного 95% ДИ (-14,96%) превысила установленную границу не меньшей эффективности, а следовательно, препарат BCD-021 продемонстрировал не меньшую эффективность по сравнению с препаратом Авастин®. Отношение шансов для ОЧО составило 1,145 (95% ДИ 0,500–2,629), что также указывало на отсутствие статистически значимых различий в эффективности между группами терапии. При сравнении дополнительных параметров оценки эффективности также не выявлено статистически значимых различий (табл. 2).
В анализ безопасности были включены все больные, получившие хотя бы одно введение препарата BCD-021 или Авастина (n = 134). Оба препарата продемонстрировали приемлемый профиль токсичности и переносимости. Не выявлено значимых различий между группами препаратов как по общей частоте НЯ, так и по частоте каждого из зарегистрированных НЯ. Среди НЯ наиболее часто встречались явления гематологической токсичности, включавшие нейтропению, лейкопению, лимфопению, анемию и тромбоцитопению. Несколько реже регистрировались отклонения ряда биохимических показателей крови: гипергликемия, гиперурикемия, увеличение уровня мочевины, повышение активности ЛДГ, АСТ, АЛТ, ЩФ. Среди прочих НЯ наиболее часто наблюдались алопеции, нарушения со стороны скелетно-мышечной системы (артралгии, миалгии, оссалгии, боль в спине), со стороны пищеварительной системы (тошнота, рвота, диарея, стоматит), со стороны сердечно-сосудистой системы (тахикардия), слабость, головная боль, а также нарушения со стороны органов дыхания (легочное кровотечение, кровохарканье, одышка, кашель).
СНЯ выявлены в общей сложности у 22 пациентов: у 14 (20,59%) из группы BCD-021 (у двух из них зарегистрировано по два СНЯ) и восьми (12,12%) из группы сравнения. В большинстве случаев СНЯ были обусловлены наличием сопутствующей патологии, воздействием химиопрепаратов, применяемых в рамках комбинированной химиотерапии, либо другими причинами, не связанными с исследуемой терапией. Не установлено значимых различий между группами по частоте встречаемости любых СНЯ (р > 0,05).
Согласно международным рекомендациям по исследованию воспроизведенных препаратов моноклональных антител, клинические исследования эффективности и безопасности должны включать оценку иммуногенности препаратов. По результатам обследования пациентов установлено по одному случаю появления связывающих антител (с нейтрализующей активностью) к бевацизумабу в каждой группе. Таким образом, оба препарата характеризовались низкой иммуногенностью.
В целом анализ безопасности продемонстрировал отсутствие значимых различий между группами по основным параметрам безопасности.
Анализ показателей фармакокинетики проводили в соответствии с международными рекомендациями по исследованию биоэквивалентности. Результаты исследования показали, что как после однократного, так и после многократного введения препаратов BCD-021 и Авастин® концентрации бевацизумаба в крови пациентов изменялись аналогичным образом. Статистическое сравнение по первичной фармакокинетической конечной точке (AUC(0–504)) позволило получить 90% ДИ для отношения средних геометрических AUC(0–504) – 80,67–109,69%. Данный интервал соответствует установленным пределам эквивалентности фармакокинетических показателей 80–125%. 90% ДИ для отношения средних геометрических Сmах исследуемого препарата и препарата сравнения составил 89,12–111,35%, что также соответствует установленному интервалу 80–125%. При сравнении испытуемых групп по прочим вторичным конечным точкам (Тmax, T1/2) с помощью стандартных статистических критериев статистически значимые различия между группами также отсутствовали. При анализе на шестом цикле статистическое сравнение по всем основным фармакокинетическим параметрам (AUC(0–504), Сmах, Тmax, T1/2) с помощью стандартных статистических критериев показало отсутствие статистически значимых различий между группами. Статистически значимых различий по показателю Ctrough между группами не зарегистрировано ни в одном из циклов. Следовательно, можно говорить об эквивалентности фармакокинетических свойств препаратов BCD-021 и Авастин® при внутривенном введении.
Резюмируя сказанное, С.В. Орлов подчеркнул, что анализ данных клинического исследования подтвердил отсутствие значимых различий между препаратами BCD-021 и Авастин® по всем исследовавшимся показателям. Препарат BCD-021 может быть рекомендован к внедрению в клиническую практику, что позволит повысить доступность современной терапии для пациентов со многими злокачественными опухолями и значимо улучшить результаты терапии, одновременно уменьшив затраты на лечение.
Оригинальный отечественный препарат Г-КСФ пролонгированного действия эмпэгфилграстим: анализ результатов регистрационного исследования
Компанией BIOCAD разработан новый оригинальный препарат пегилированного филграстима – эмпэгфилграстим пролонгированного действия для профилактики таких тяжелых осложнений химиотерапии, как нейтропения. Препарат успешно прошел клинические испытания. Врач-онколог Архангельского клинического онкологического диспансера Марина Николаевна НЕЧАЕВА рассказала о ходе клинической разработки препарата и его уникальных особенностях.
Физико-химические свойства молекулы препарата эмпэгфилграстим имеют отличия от первого препарата пегилированного филграстима на рынке – Неуластима. Молекулярная масса остатка полиэтиленгликоля в составе эмпэгфилграстима составляет 30 кДа, в то время как в составе Неуластима содержится полиэтиленгликоль массой 20 кДа. Увеличение массы полиэтиленгликоля приводит к тому, что он стерически дальше располагается от активной части молекулы препарата – филграстима. Благодаря этому специфическая активность препарата эмпэгфилграстим превышает таковую Неуластима – 84–94 и 68% соответственно.
На сегодняшний день выполнен весь объем сравнительных физико-химических, доклинических и клинических исследований препарата эмпэгфилграстим. В конце 2014 г. завершен анализ данных исследования фазы III, в котором наряду с другими медицинскими центрами участвовали и специалисты Архангельского онкологического диспансера. М.Н. Нечаева представила участникам симпозиума основные результаты завершенного исследования.
В международном многоцентровом двойном слепом рандомизированном исследовании с двойным маскированием эффективность нового препарата BCD-017 (эмпэгфилграстим) оценивали на протяжении четырех циклов химиотерапии у больных РМЖ. Препаратом сравнения был непегилированный филграстим (Лейкостим®), производимый также компанией BIOCAD. В исследовании участвовали 135 пациенток с РМЖ стадий II–IV, рандомизированных в три группы в соотношении 1:1:1 (пациентки в двух группах получали две разных дозировки эмпэгфилграстима, в третьей группе – непегилированный филграстим). Все участницы исследования получали химиотерапию по схеме «доцетаксел 75 мг/м2 + доксорубицин 50 мг/м2». На второй день, не менее чем через 24 часа после введения химиотерапии, начиналось введение исследуемой терапии.
Пациенткам первой группы однократно подкожно вводили препарат BCD-017 в дозе 6 мг с последующим ежедневным введением плацебо (0,0083 мл/кг). Пациенткам второй группы на второй день однократно подкожно вводили препарат BCD-017 7,5 мг с последующим ежедневным введением плацебо (0,0083 мл/кг). Плацебо в первых двух группах вводили до достижения абсолютного числа нейтрофилов (АЧН) 10 × 109/л, но не более 14 дней. Пациенткам группы сравнения плацебо вводилось однократно, препарат сравнения вводился в дозе 5 мг/кг до тех пор, пока АЧН не достигало 10 × 109/л, но не более 14 дней. Использование двойного маскирования подразумевало применение двух видов плацебо – первый вид, внешне не отличимый от препарата сравнения, – в группах исследуемого препарата, второй, не отличимый от исследуемого препарата, – в группе сравнения.
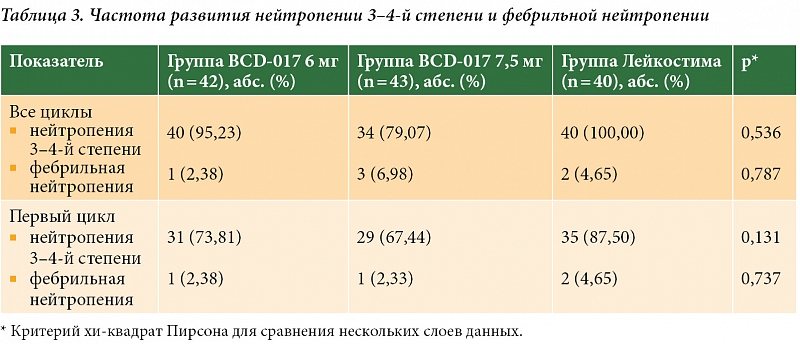
Основным показателем, по которому оценивали эффективность терапии, в данном исследовании была продолжительность нейтропении 4-й степени тяжести на первом цикле химиотерапии. Максимальные показатели продолжительности нейтропении 4-й степени отмечались в группе филграстима, различия с показателями в группах исследования препарата BCD-017 носили статистически значимый характер. Кроме того, согласно данным исследования, на протяжении всех четырех циклов тяжелая нейтропения (3–4-й степни) чаще имела место в группе филграстима, ее частота составляла 100%. Для сравнения: в группе, получавшей BCD-017 7,5 мг, частота тяжелой нейтропении была ниже – 79,07%. Тем не менее данные различия не достигали статистической значимости. Фебрильная нейтропения в ходе исследования встречалась редко, ее частота не отличалась в группах (табл. 3).
В течение первого цикла терапии тяжелые инфекции не регистрировались ни в одной группе, а со второго по четвертый цикл – только в группе Лейкостима (10%). К случаям тяжелой инфекции в ходе исследования относились по одному случаю пневмонии, рожистого воспаления правого предплечья, воспаления атеромы предплечья, фурункула области носа.
При оценке надира уровня нейтрофилов было выявлено, что в группе филграстима он был достоверно ниже, чем в группах BCD-017 6 и 7,5 мг. При этом в группе BCD-017 7,5 мг на первом цикле терапии надир нейтрофилов почти в два раза превышал показатели в группе филграстима и был близок к нижней границе нормы, в то время как в группе филграстима соответствовал нейтропении 4-й степени.
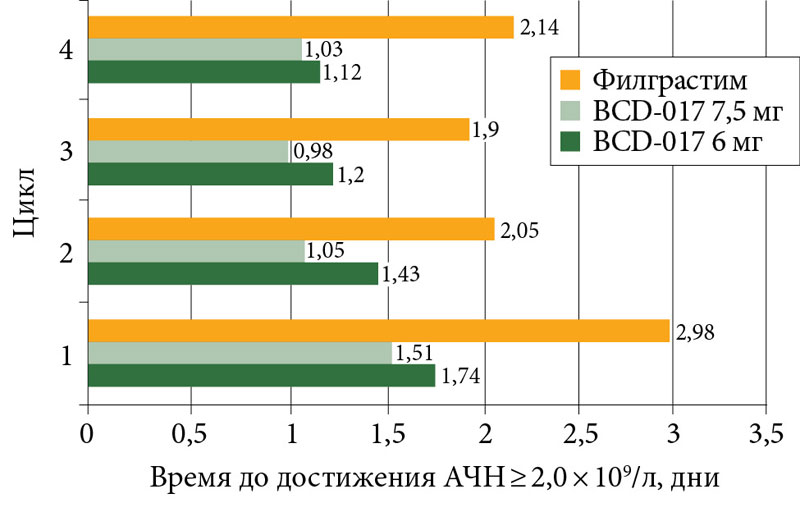
Время до восстановления нормального АЧН постепенно уменьшалось с первого по четвертый цикл (рис. 3).
Наиболее длительным процесс восстановления нейтрофилов в крови до нормального уровня был в группе Лейкостима. Так, во время четвертого цикла до достижения нормального числа нейтрофилов в группе Лейкостима потребовалось 2,14 дня. В группах BCD-017 6 мг и BCD-017 7,5 мг эти показатели составили 1,12 и 1,03 дня соответственно.
НЯ в ходе исследования наблюдались практически у всех пациенток, но в большинстве случаев были связаны с применявшейся схемой химиотерапии. Частота явлений гематологической токсичности не имела значимых различий во всех группах исследования, за исключением лейкопении 4-й степени, чаще наблюдавшейся в группе филграстима, что косвенно говорило в пользу большей эффективности препарата BCD-017. Не выявлено достоверных различий между группами по негематологическим явлениям и НЯ, в том числе свойственным препаратам гранулоцитарного колониестимулирующего фактора (Г-КСФ) и местным реакциям. СНЯ чаще отмечались в группе Лейкостима (11,63%), но различия между группами были недостоверны.
Таким образом, итоги клинического исследования фазы III продемонстрировали не меньшую эффективность нового пегилированного филграстима – препарата эмпэгфилграстим по сравнению с непегилированным филграстимом – препаратом Лейкостим®. При этом профиль безопасности препарата эмпэгфилграстим был аналогичен таковому препарата Лейкостим®.
На основании анализа данных об эффективности, безопасности и фармакокинетике доза препарата эмпэгфилграстим 7,5 мг была выбрана как наиболее предпочтительная для клинического применения, поскольку обеспечивала наибольшую эффективность без повышения токсичности.
В заключение М.Н. Нечаева рассказала об опыте применения препарата BCD-017 в рамках исследования фазы III в Архангельском областном онкологическом диспансере. В исследование было включено 36 пациенток. Данные, полученные в этом диспансере, соответствовали общим результатам исследования. По мнению М.Н. Нечаевой, внедрение эмпэгфилграстима в клиническую практику будет способствовать повышению безопасности терапии и улучшит результаты лечения больных, получающих миелосупрессивную химиотерапию по поводу злокачественных новообразований.
Заключение
Представленные на симпозиуме результаты клинических исследований подтверждают занимаемые компанией BIOCAD ведущие позиции среди отечественных фармацевтических компаний. Исследования фазы III с участием больных РМЖ и НМРЛ убедительно доказали терапевтическую эквивалентность биоаналога трастузумаба и биоаналога бевацизумаба производства компании BIOCAD оригинальным препаратам. Внедрение этих препаратов в клиническую практику может повысить доступность современной противоопухолевой терапии для пациентов с самыми распространенными в России злокачественными новообразованиями и снизить затраты на эффективное лечение.
Первый оригинальный отечественный препарат Г-КСФ пролонгированного действия, разработанный компанией BIOCAD, доказал свою эффективность и безопасность в сравнении с золотым стандартом профилактики нейтропении у больных, получающих химиотерапию препаратом филграстим. Удобный режим введения и высокая эффективность препарата эмпэгфилграстим позволяют рассматривать его применение как весьма перспективный метод профилактики таких тяжелых осложнений химиотерапевтического лечения, как фебрильная нейтропения и нейтропенические инфекции, в данной популяции пациентов.