Положительные эффекты гликлазида МВ в терапии сахарного диабета 2 типа
- Аннотация
- Статья
- Ссылки
Результаты проспективных клинических исследований, а также исследований по данным аутопсий показывают значительное снижение не только массы, но и функции бета-клеток в первые годы заболевания у пациентов с сахарным диабетом (СД) 2 типа. Основными патогенетическими звеньями СД 2 типа, как известно, являются наличие инсулинорезистентности (ИР) и развитие дисфункции бета-клеток поджелудочной железы [1–4]. Такие факторы, как гипергликемия, гиперинсулинемия, гиперлипидемия, наличие амилоида в островках поджелудочной железы, являются причиной развития дисфункции бета-клеток при СД 2 типа и способствуют ее дальнейшему прогрессированию [4–7]. Генетическая предрасположенность и факторы внешней среды (гиперкалорийная диета, сидячий образ жизни) оказывают влияние на степень развития и длительность поддержания этих патогенетических составляющих.
В развитии периферической ИР большое значение имеют дефекты инсулиновых рецепторов и патология транспортеров глюкозы. Нарушение синтеза инсулина может проявляться изменением последовательности аминокислот в молекуле инсулина и превращения проинсулина в инсулин. В обоих случаях инкретируемый инсулин будет иметь низкую аффинность к рецептору инсулина, что приведет к развитию гипергликемии. Нарушение инкреции инсулина может также являться следствием дефекта развития бета-клеток при неадекватном внутриутробном и постнатальном питании, при длительно существующей глюкозотоксичности. Возможны и генетические дефекты в механизме инкреции инсулина [8, 9].
На ранних стадиях заболевания ИР не сопровождается выраженной гипергликемией, поскольку бета-клетки инкретируют достаточное количество инсулина, что приводит к развитию гиперинсулинемии. Хроническая гиперинсулинемия, в свою очередь, уменьшает число рецепторов на клетках-мишенях, и бета-клетки постепенно теряют способность реагировать на повышение концентрации глюкозы. Таким образом, гипергликемия развивается вследствие снижения чувствительности тканей к инсулину, нарушения глюкозо-индуцированной секреции инсулина и истощения «немедленного» запаса инсулина [10, 11]. Поддержание гипергликемии уменьшает чувствительность клеток к инсулину, что усугубляет уже имеющиеся нарушения [12, 13].
В настоящее время в арсенале эндокринолога есть несколько классов сахароснижающих препаратов, а именно: бигуаниды, производные сульфонилмочевины, глиниды, ингибиторы альфа-глюкозидазы, тиазолидиндионы, ингибиторы дипептидилпептидазы-4, агонисты рецепторов глюкагоноподобного пептида-1 и инсулин. В клинической практике для лечения пациентов с СД 2 типа широко применяются производные сульфонилмочевины, как в монотерапии, так и в различных комбинациях, поскольку они эффективно снижают уровень глюкозы плазмы крови.
Гликлазид – препарат второго поколения производных сульфонилмочевины – помимо гипогликемизирующего действия положительно влияет на микроциркуляцию, реологические свойства крови и показатели оксидативного стресса. На сегодняшний день в клинической практике в основном используется форма препарата с модифицированным высвобождением – Диабетон МВ (компания «Сервье»), – что позволяет принимать его однократно в сутки. Связывание гликлазида с рецептором сульфонилмочевины SUR1 происходит быстро и обратимо, в отличие от других производных сульфонилмочевины (например, глибенкламида), которые обладают замедленным и стойким эффектом [14–17].
В частности, степень стимуляции инкреции инсулина Диабетоном МВ зависит от уровня глюкозы плазмы крови: препарат связывается с рецептором SUR1 и отделяется от него после достижения нормогликемии. Длительная стимуляция инкреции инсулина приводит к повышению риска развития гипогликемии и может способствовать прибавке массы тела, а также прогрессированию недостаточности функции бета-клеток.
У пациентов с СД 2 типа в дебюте заболевания нарушается первая фаза секреции инсулина, что проявляется в снижении пика выброса последнего. Важным следствием этого расстройства является то, что производство глюкозы в печени больше не подавляется во время приема пищи, а также отсутствует подавление секреции глюкагона. Постпрандиальная гипергликемия не успевает возвратиться к нормальному уровню до следующего приема пищи, что вызывает хроническую гипергликемию в течение дня [18]. Несмотря на сохранную вторую фазу секреции инсулина, скорректировать гипергликемию не удается. Учитывая эти патогенетические особенности, очевидным подходом является восстановление ранней фазы секреции инсулина путем фармакологических вмешательств.
К препаратам, стимулирующим раннюю фазу секреции инсулина, относятся глиниды и гликлазид. Первые оказывают наибольшее влияние на скорость секреции инсулина в течение первых 30 минут после приема пищи [19]. C-Z. Wu и соавт. провели сравнительную оценку эффективности гликлазида и репаглинида у пациентов с впервые выявленным СД 2 типа [20]. Они обследовали 20 пациентов, которым провели оценку чувствительности к инсулину, глюкозе, изучили раннюю фазу секреции инсулина после 4 месяцев лечения указанными препаратами. В данном исследовании авторы не получили значимых различий в концентрации глюкозы плазмы натощак, инсулина, гликированного гемоглобина, показателях артериального давления, индекса массы тела, липидного спектра крови между двумя препаратами (гликлазид, репаглинид).
В другом исследовании также не было отмечено значимых различий между гликлазидом и репаглинидом в действии данных препаратов на гликемический контроль [21]. После 4-недельного лечения у пациентов обеих групп значительно улучшились показатели глюкозы плазмы натощак и после еды, увеличилась постпрандиальная концентрация инсулина (р < 0,05). Площадь под кривой (инсулин) увеличилась в обеих группах (р < 0,05), достоверных различий между группами выявлено не было. Индекс ранней фазы секреции инсулина (ΔI30/ΔG30) также увеличился в обеих группах (р < 0,05), особенно в группе лечения репаглинидом (p < 0,05). Авторы сделали вывод, что репаглинид и гликлазид оказывают сходное влияние на гликемический контроль и общую секрецию инсулина.
Хроническая гипергликемия приводит к избыточному образованию реактивных форм кислорода (РФК), оксидативному стрессу и стрессу эндоплазматического ретикулума в различных клетках. По сравнению с другими типами клеток, бета-клетки особенно чувствительны к такому повреждению, что повышает их чувствительность к апоптозу [22, 23]. Показано, что стойкое избыточное образование РФК приводит к подавлению экспрессии гена инсулина в результате дефицита факторов транскрипции [24], а также активации апоптоза бета-клеток [25]. Не следует забывать, что постоянное применение производных сульфонилмочевины у пациентов с СД 2 типа может также приводить к дисфункции и апоптозу бета-клеток.
В нескольких исследованиях отмечено, что причиной апоптоза бета-клеток может явиться стойкое увеличение поступления ионов Ca2+ в клетку, вызванное применением производных сульфонилмочевины [26, 27]. Данные экспериментальных исследований показали: инкубация островковых клеток человека с глибенкламидом значительно снижает содержание инсулина в бета-клетках, а также приблизительно в 2 раза увеличивает их чувствительность к апоптозу, преимущественно за счет повышения концентрации РФК [28, 29]. Однако длительное использование гликлазида МВ (Диабетона МВ) защищает панкреатические бета-клетки от апоптоза [30–32].
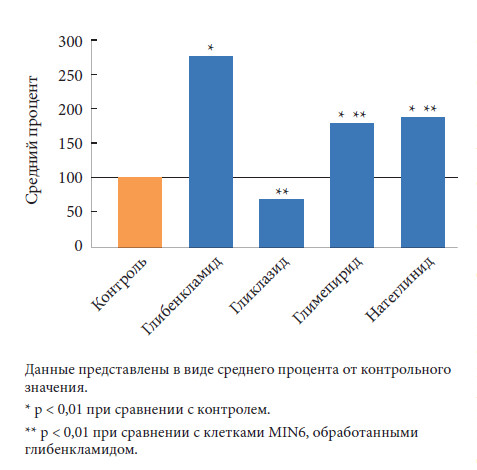
F. Sawada и соавт. оценили влияние различных секретагенов на апоптоз бета-клеток [32]. Авторы изучили дозозависимые эффекты глибенкламида, глимепирида, гликлазида, а также натеглинида на образование РФК и апоптоз в культуре панкреатических бета-клеток линии MIN6 и мышиных островках. В результате воздействия на бета-клетки глибенкламида, глимепирида и натеглинида отмечалось достоверное увеличение внутриклеточного синтеза РФК, которое зависело от концентрации препарата (0,1–10 мкмоль/л). Эти эффекты полностью блокировались ингибиторами никотинамидаденин-динуклеотидфосфат-[НАД(Ф)H]-оксидазы (апоцинин) или ингибитором протеинкиназы С (кальфостин C). Воздействие этих сахароснижающих препаратов в течение 48 часов также привело к достоверному увеличению числа апоптозных клеток (рис.) [32].
Данные эффекты значительно снижались под действием апоцинина и антиоксиданта N-ацетил-L-цистеина. Применение гликлазида в любых концентрациях не влияло на уровень внутриклеточного образования РФК и число апоптозных клеток. Препараты сульфонилмочевины (глибенкламид или глимепирид, но не гликлазид), а также натеглинид стимулировали образование РФК посредством зависимой от протеинкиназы С активации НАД(Ф)Н-оксидазы и, как следствие, вызывали апоптоз бета-клеток in vitro. Поскольку при использовании гликлазида подобных нежелательных эффектов не наблюдалось, данный препарат имеет преимущество в терапии пациентов с СД 2 типа в отношении сохранения массы функционирующих бета-клеток.
Аминоазабициклооктиловое кольцо, входящее в состав химической структуры гликлазида, обладает антиоксидантными свойствами [33, 34], что может обусловливать защитное действие Диабетона МВ в отношении бета-клеток поджелудочной железы [30, 31]. Кроме того, этот препарат защищает бета-клетки от окислительного стресса через увеличение активности супероксиддисмутазы и каталазы [15]. L. Chen и соавт. оценили влияние гликлазида на функцию эндотелия у больных с впервые выявленным СД 2 типа [16]. Пациенты получали гликлазид МВ в течение 12 недель. После лечения отмечено значительное улучшение функции эндотелия, увеличение количества циркулирующих эндотелиальных клеток-предшественников и активности супероксиддисмутазы (р < 0,05).
Уровень малонового диальдегида и уровень окиси азота в сыворотке крови снизились после терапии гликлазидом. Эти результаты показывают, что гликлазид МВ (Диабетон МВ) улучшает функцию эндотелия у больных СД за счет своих антиоксидантных свойств. Похожие результаты получены в ходе исследования, проведенного J. Drzewoski и соавт. [17]. Ученые обследовали 24 пациента в возрасте 61,2 ± 15,4 года с плохо контролируемым СД 2 типа (уровень HbA1c 7,6 ± 1,1%). Все пациенты в течение 12 недель получали гликлазид МВ. На фоне лечения значительно улучшились показатели углеводного обмена: снизилась концентрация глюкозы плазмы натощак (с 7,6 ± 1,4 до 6,6 ± 1,2 ммоль/л, р < 0,01), уровень HbA1c (с 7,6 ± 1,1 до 6,9 ± 0,8%, р < 0,01), а также отмечено снижение концентрации интерлейкина-6 (ИЛ-6) в плазме (с 2,5 ± 1,8 до 1,8 ± 1,2 пг/мл, р < 0,05). По завершении периода наблюдения было выявлено повышение концентрации адипонектина в плазме с 6,4 ± 3,3 до 7,6 ± 4,4 мкг/мл (р < 0,05). Снижение концентрации фактора некроза опухоли альфа (ФНО-альфа) в плазме и показателя ИР (HOMA-IR) оказалось незначительным.