Клинико-фармакологическое обоснование новых направлений патогенетической терапии диабетической полинейропатии
- Аннотация
- Статья
- Ссылки
- English
Диабетическая полинейропатия (ДПН) – частое осложнение сахарного диабета (СД) 1 и 2 типов. Однако оно развивается не у всех пациентов. Факторами риска развития ДПН при СД являются недостаточный гликемический контроль [1], сердечно-сосудистая коморбидность, в частности артериальная гипертензия, повышенный уровень триглицеридов, ожирение, курение [2], а также врожденная (генетическая) предрасположенность, заключающаяся в более высокой чувствительности периферических тканей к гипергликемии [3].
Понимание причин возникновения ДПН тем не менее не дает ответа на вопрос: почему у одних пациентов развивается это осложнение, а у других нет?
Другой не менее важный вопрос, не решенный до настоящего времени, – причины, по которым некоторые пациенты испытывают нейропатическую боль. Согласно эпидемиологическим данным, нейропатическая боль отмечается у 20–50% пациентов с СД и примерно у 60% больных ДПН [4].
Ключевыми факторами, приводящими к развитию ДПН, являются гипергликемия и дислипидемия. Влияние этих факторов опосредовано метаболическим и окислительным стрессом, приводящим к дегенерации аксонов [5, 6]. Повышенный уровень глюкозы способствует активации полиолового и гексозаминового путей, что ведет к образованию активных форм кислорода и развитию системного воспаления. Эти же процессы запускает дислипидемия, что в совокупности значительно усугубляет еще один крайне важный механизм поражения периферической нервной системы – повреждение митохондрий [7]. Крайне важно, что доминирующую роль в дебюте ДПН играет именно системное воспаление [8, 9].
При гипергликемии также наблюдается усиленное гликирование многочисленных структурных и функциональных белков нервной ткани с образованием конечных продуктов, которые тоже стимулируют высвобождение провоспалительных молекул и свободных радикалов. Изменение функции белков наблюдается и при естественном старении, поэтому у возрастных пациентов с диабетом модификация экспрессии генов и внутриклеточного (инсулинового) сигналинга выражена особенно ярко [10]. Аналогичные процессы происходят и в малых сосудах, изменения в которых вызывают нарушение перфузии нервов, провоцируя их гипоксию и дисфункцию [11].
Развитие ДПН связано также со снижением биодоступности тканей (чувствительности тканей и их рецепторов) к действию различных аутокоидов – нейромедиаторов, факторов роста, включая инсулиноподобный фактор роста, фактор роста эндотелия сосудов, а также газотрансмиттеров (оксида азота и углерода), обеспечивающих условия для нормального функционирования сосудов и нервов [12, 13].
Важно отметить, что строгий контроль уровня глюкозы способен снизить частоту развития ДПН только при СД 1 типа. При СД 2 типа данное направление лечения не сопровождается достоверными положительными результатами [14]. При СД 2 типа невелика значимость и коррекции гипергликемии инсулином в отличие от СД 1 типа, при котором подобная терапия достаточно эффективна в отношении ДПН [15].
Описанные механизмы приводят к возникновению различных соматосенсорных фенотипов ДПН. Клиническая симптоматика при этом определяется преимущественным поражением толстых или тонких волокон. При поражении толстых волокон у пациентов выявляются расстройства тактильной (онемение) и вибрационной чувствительности, нарушается восприятие тела в пространстве (проприоцепция), в связи с чем возрастает риск падений и переломов, снижаются миотатические рефлексы, прежде всего ахиллов, могут наблюдаться уменьшение мышечной силы и атрофия мышц стопы. Поражение тонких волокон сопровождается появлением расстройств температурной чувствительности. Пациенты предъявляют жалобы на чувство прохождения электрического тока, жжение. Кроме того, у них может наблюдаться вегетативная дисфункция.
Боль при ДПН встречается как при поражении толстых волокон (Аβ), так и при поражении тонких (С). При поражении первых боль имеет более глубокое распределение, а при поражении вторых – более поверхностное.
Таким образом, в клинической картине ДПН могут наблюдаться как симптомы выпадения (онемение, гипорефлексия), так и симптомы раздражения.
Болевые феномены могут быть спонтанными. При поражении толстых волокон это чаще ноющая боль или покалывание, а при поражении тонких – чаще жжение, острая или колющая боль [16]. Могут определяться повышенная чувствительность к болевым стимулам (гипералгезия) или болевые ощущения к стимулам различных неболевых модальностей (аллодиния – чаще при поражении тонких волокон). Механическая гипералгезия при ДПН имеет достаточно высокую распространенность [17–19]. Тепловая гипералгезия (обычно в сочетании с сохраненной функцией тонких волокон – фенотип раздраженного ноцицептора) встречается существенно реже. У большинства пациентов с ДПН наблюдается сочетанное поражение толстых и тонких волокон (фенотип деафферентации), при этом механическая боль в глубоких тканях (оценивается по порогу боли при давлении) отмечается редко [20].
В настоящее время не изучены клинико-патогенетические параллели, позволяющие определить ведущий механизм развития того или иного фенотипа ДПН. Для болевых форм ДПН описаны модифицируемые и немодифицируемые факторы риска. Нейропатическая боль при ДПН чаще встречается у женщин [21], в старших возрастных группах [22], у пациентов с ожирением [23], повышенным уровнем гликированного гемоглобина [17], значительным потреблением алкоголя, длительным течением СД и выраженными сенсорными расстройствами [24], наследственной предрасположенностью [25].
В патогенезе боли при ДПН немаловажную роль играют структурно-функциональные поражения сосудов нервной системы [26] и нарушения регуляции периферического кровотока [27]. Обнаружено, что при гипоксии, связанной с нарушением регуляции местного кровотока в коже, вырабатываются специфические белки – фактор, индуцируемый гипоксией, 1α [28], и фактор фон Виллебранда [29], концентрация которых прямо коррелирует с выраженностью боли. Было также установлено, что у пациентов с болевой формой ДПН снижена концентрация витамина D в плазме крови [30]. Наконец, результаты ряда исследований свидетельствуют о том, что в патогенезе нейропатической боли при СД участвует метилглиоксаль (альдегид пировиноградной кислоты) – побочный цитотоксический продукт гликолиза, который реализует свои эффекты через активацию С-ноцицепторов и тепловую гипералгезию [31].
У пациентов с болевой формой ДПН помимо периферических обнаруживаются центральные изменения невральных структур. Структурные и функциональные модификации определяются в спинальных, соматомоторных, лимбических, таламических, восходящих и нисходящих модуляционных системах [32]. Выявлены изменения в высших корковых центрах: атрофия и аномальная активность соматомоторной и островковой коры головного мозга [29, 33], усиление мозгового кровотока в передней поясной коре [34]. Эти модификации могут быть следствием первичного поражения, а также хронизации нейропатической боли.
Отсутствие единой четкой концепции, позволяющей установить в каждом конкретном случае вероятность запуска и релевантность механизмов возникновения и развития ДПН, в том числе ее болевых форм, послужило предпосылкой к продолжению исследований, значительная часть которых в настоящее время сконцентрирована на оценке роли глиальных клеток в патогенезе неврологических осложнений СД.
При ДПН в первую очередь страдают нейрональные структуры, однако клетки глии являются такой же мишенью СД, что подтверждается выявлением признаков демиелинизации у части пациентов [7, 35]. Очевидно, что глиальные клетки периферической нервной системы также реагируют на диабетические состояния, что в свою очередь влияет на их важную функцию – питание и содействие выживанию нейронов.
В экспериментах на животных с помощью стрептозотоцина, специфической диеты и генетической индукции были созданы модели СД 1 и 2 типов [36, 37]. В рамках исследований определялись не только поражения дистальных аксонов, но и изменение взаимодействия нейронов с глиальными клетками. Полученные результаты свидетельствуют о том, что при СД поражаются все структуры первичного сенсорного нейрона. Остается открытым вопрос о том, повреждаются ли в первую очередь периферические аксоны и связанные с ними шванновские клетки или доминирует поражение перикариона нейронов с сателлитными глиальными клетками, находящимися в дорсальных спинномозговых ганглиях.
Сателлитные глиальные клетки локализуются в сенсорных и автономных ганглиях периферической нервной системы, образуя тонкую и плотную оболочку вокруг каждой отдельной нейрональной сомы. Их количество пропорционально размеру нейрона. Двунаправленная связь между нейронами и сателлитными глиальными клетками обнаружена в спинномозговых ганглиях и в значительной степени опосредована пуринергическими системами, особенно представленными классом P2 [38]. Сателлитные глиальные клетки экспрессируют рецепторы нейротрансмиттеров, транспортеры и ионные каналы, что позволяет контролировать метаболический и нейрональный гомеостаз. Кроме того, они высвобождают нейроактивные вещества, такие как аденозинтрифосфат и цитокины, которые способны самостоятельно передавать сигналы от глии к нейрону [39].
Сателлитные глиальные клетки, окружающие нейроны спинномозгового ганглия, реагируют на повреждение нерва, вызывая сопряженную активность нейронов, а блокирование этих контактов ослабляет гипервозбудимость нервных клеток и механическую гипералгезию [40]. Вместе с тем реакция сателлитных глиальных клеток на СД может быть первичной, и тогда их взаимосвязь с сомой нейронов, опосредованная пуринергическими рецепторами, определяет феномен нейронального раздражения, клиническими эквивалентами которого в том числе являются болевые феномены. Подтверждением этого тезиса служат результаты исследований, в которых у крыс с моделью диабета ингибировали пуринергическую связь пораженных сателлитных глиальных клеток с нейронами и получали клинический эффект в виде снижения выраженности механической и тепловой гипералгезии [41].
При СД также нарушается функция шванновских клеток, причем легкая сегментарная демиелинизация наблюдается уже в дебюте ДПН при сохранном аксоне. Это позволяет предположить, что миелинопатия может лежать в основе первичного повреждения нервных волокон и быть первым звеном в патогенезе ДПН [42]. Механизмы поражения нервных волокон при СД также актуальны для шванновских клеток, причем некоторые из них преобладают именно в данных глиальных элементах. В частности, это касается полиолового пути как наиболее изученного механизма развития ДПН. Доказано, что важнейший фермент этого метаболического процесса – альдозоредуктаза экспрессируется именно в шванновских клетках, что приводит к снижению выработки белков миелина и нейротрофических факторов [43, 44]. При этом также снижается экспрессия специфических белков клеточного сигналинга, что дополнительно способствует поражению нервных волокон, нарушению скорости проведения возбуждения, индуцирует эндотелиальную дисфункцию и в конечном счете способствует развитию ДПН [45, 46].
В шванновских клетках, так же как в нервных волокнах, при диабете наблюдаются ультраструктурные аномалии в митохондриях [47]. Кроме того, специфические рецепторы, на которые действуют конечные продукты усиленного гликирования, расположены не только на аксонах, но и на шванновских клетках. Это позволяет предполагать возможный запуск окислительного стресса и воспаления в этих структурах еще до манифестации ДПН [48].
При диабете усиливаются иммунные реакции. Шванновские клетки считаются иммунокомпетентными, поскольку экспрессируют основные молекулы комплекса гистосовместимости II и несколько Toll-подобных и воспалительных рецепторов, а также продуцируют ряд провоспалительных цитокинов, которые участвуют в патогенезе ДПН, могут сенсибилизировать Aβ- и С-волокна и способствовать развитию боли [49].
Таким образом, участие глиальных клеток в патогенезе ДПН очевидно. При этом морфофункциональные изменения в них имеют клинические эквиваленты лишь на поздних стадиях ДПН, однако могут стать значимым или даже пусковым механизмом поражения нервных волокон и появления боли уже в дебюте заболевания.
С этой точки зрения определенным прорывом в лечении ДПН могло бы стать применение препаратов, влияющих на процессы регенерации нервной ткани, скорость химического синтеза миелина и метаболизм глиальных элементов. Их традиционно относят к нестероидным анаболическим средствам. Перспективными соединениями этой группы являются вещества, обеспечивающие образование пиримидинов и пуринов, участвующие в синтезах ДНК и РНК. Среди них в большей степени изучено действие производных уридина, агониста Р2Y-медиаторных пуринергических систем, в которых он выполняет трансмиттерную функцию, столь важную, что за это открытие в 1972 г. была присуждена Нобелевская премия.
Пуринергические рецепторы относят к классу типичных 7ТМ-рецепторов. Это трансмембранные рецепторы с семью внешними петлями узнавания лиганда уридина, имеющие свою топику и четко очерченную функцию. Уридин участвует в системной нейротрансмиттерной регуляции жизненно важных функций организма, таких как промежуточный обмен, терморегуляция, кровоток, сократимость. Он оказывает ко-трансмиттерное действие прежде всего в отношении к ГАМК-, холин- и дофаминергических систем, каждая из которых несет антиноцицептивный потенциал. Нейрональная сеть пуринергических систем образует регулирующий ноцицепцию Hub-рецептор. Доказано, что на ранних стадиях поражения нервных стволов при сбое процессинга (созревание pre-mRNA) и сплайсинга (вырезание и соединение нуклеотидов) усиливается поглощение уридина и цитидина, а их назначение сопровождается нормальным синтезом и регенерацией компонентов нервного кластера, при этом повышаются болевой порог и проводимость возбуждения по нерву [50].
В исследованиях на клеточных культурах установлено, что внеклеточный уридинтрифосфат (УТФ) взаимодействует с рецепторами P2Y шванновских клеток и активирует молекулярные механизмы, которые вызывают изменения в их цитоскелете, улучшают межклеточные контакты и стимулируют их миграцию [51, 52]. Накопленные к настоящему времени данные позволяют констатировать, что уридин, включаясь в механизмы процессинга, по сути перепрограммирует шванновские клетки, результатом чего является активация восстановления нервных волокон [53]. Полученные на клеточных культурах результаты были подтверждены данными экспериментов на животных. В ходе их проведения зафиксирована значительная активация процессов ремиелинизации и регенерации аксонов травматически поврежденного нерва с восстановлением скорости проведения возбуждения при использовании уридинмонофосфата (УМФ) и цитидинмонофосфата (ЦМФ) в течение 60 дней [54].
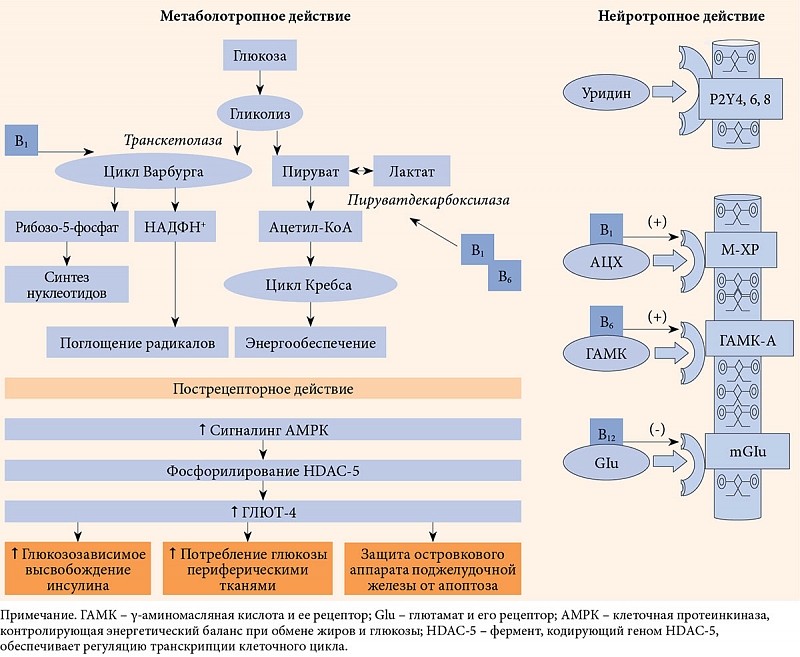
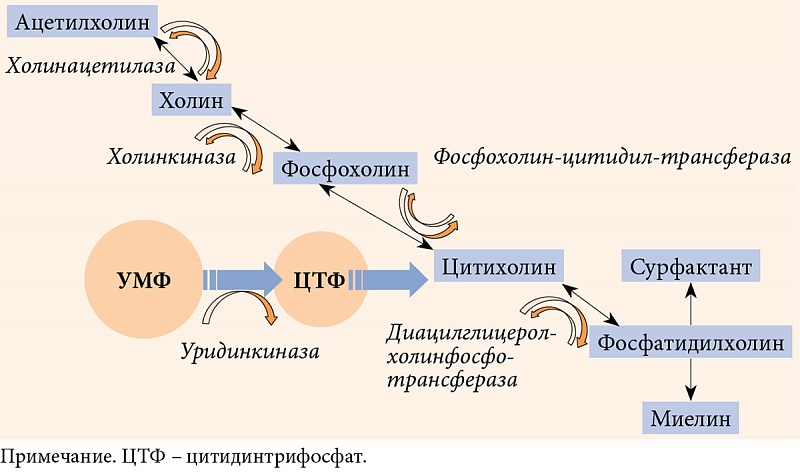
Следует различать действие уридина-медиатора и уридина-метаболита. Метаболотропные реакции промежуточного и липидного обменов регулирует уридин-мeтаболит, одной из метаболических ветвей которого является поддержание синтеза нуклеотидов, обеспечивающих синтез и кругооборот миелина. Помимо этого уридин-метаболит (в виде УМФ) кооперативно связан с цитидином и фосфохолином. В этой связке он регулирует синтез фосфатидилхолина, структурообразующего фосфолипида клеточных мембран любого типа. Фосфатидилхолин обеспечивает постоянство каркасной и матричной функций в мембранном цикле Кеннеди. В данном цикле финальными метаболитами фосфатидилхолина являются миелин и сурфактант – главные мишени диабетической нейропатии и различных болезней легких. Следовательно, снижение пула УМФ может сопровождаться не только нарушением целостности мембран, но и снижением образования миелина и сурфактанта [55].
Важной особенностью действия уридина-медиатора и препаратов, разработанных на его основе, является пострецепторное действие пуринергических систем, особенно реализуемое через Р2Y- и P4Y-рецепторы. Оно включает цепь молекулярных реакций, опосредуемых АМРК-системой – внутриклеточным белковым комплексом, который обеспечивает окисление глюкозы и жирных кислот в условиях низкой выработки клеточной энергии, что наблюдается при диабете, тем самым стимулируется митохондриальный биогенез за счет увеличения пула его ферментов, таких как цитохром С, сукцинатдегидрогеназа, малатдегидрогеназа [56].
Пуринергический сигналинг также затрагивает верхние этажи биотрансформации глюкозы, увеличивает активность ферментов ее массопереноса, таких как глюкозный транспортер 4 (ГЛЮТ-4) и гексокиназа [57].
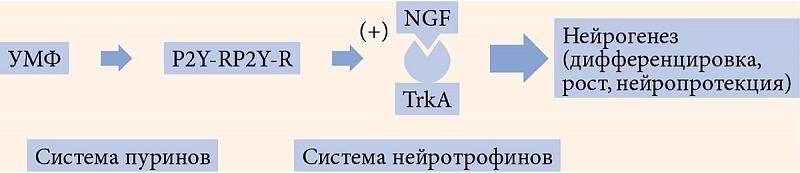
Помимо усиления активности АМРК-системы важной особенностью пострецепторного действия уридина является активация тирозинкиназных рецепторов, расположенных на мембранах ядер клеток, в частности TrkA-рецепторов. Эта взаимосвязь двух систем очень важна, так как лигандами Trk-рецепторов являются нейротрофины, один из которых – фактор роста нервов (NGF) поддерживает жизнеспособность нейронов и стимулирует их развитие и активность (рис. 1) [58].
Таким образом, пострецепторное действие пуринергических систем обеспечивает [59]:
- увеличение глюкозозависимого высвобождения инсулина;
- усиление потребления глюкозы периферическими тканями;
- защиту островкового аппарата поджелудочной железы от апоптоза.
Иными словами, в действии уридина и препаратов, созданных на его основе, в частности комплекса Нейроуридин, прослеживаются три важных компонента: метаболический, нейротропный и пострецепторный, каждый из которых вносит свой вклад в репаративные процессы, имеющие место в развитии диабетической нейропатии (рис. 2).
Накопленная доказательная база позволила провести ряд клинических исследований по оценке целесообразности применения уридина и его производных при ДПН. В одном из них эффективность пиримидиновых нуклеотидов оценивалась у пациентов с СД 2 типа и ДПН второй и третьей стадий. Обследование показало, что демиелинизация у больных имела не только вторичное, но и первичное происхождение, которое авторы связали с иммунными механизмами диабета. Терапия осуществлялась комплексом уридиндифосфата (УДФ), УТФ, УМФ, ЦМФ в течение 30 дней. Полученные результаты свидетельствовали о статистически значимом улучшении показателей ремиелинизации и реиннервации у принимавших пиримидиновые нуклеотиды. При этом достигнутая положительная динамика сохранялась в течение трех месяцев после начала терапии [60].
В другом двойном слепом плацебо-контролируемом исследовании эффективность уридина оценивалась у пациентов с СД 1 и 2 типов. Препарат назначали трехкратно в дозе 900 мг/сут в течение 180 дней. Авторы исследования констатировали повышение скорости распространения возбуждения по нерву у принимавших уридин в течение всего времени наблюдения. Примечательным является тот факт, что побочных эффектов и осложнений на фоне терапии выявлено не было [61].
Было также проведено клиническое исследование эффективности применения уридина при болевых формах ДПН. Пациентам с СД 1 и 2 типов в течение трех месяцев назначался комплекс пиримидиновых нуклеотидов (УТФ, УДФ, УМФ, ЦМФ). Наряду с улучшением клинических и инструментальных показателей, свидетельствующих об ускорении регенерации нервов и процессов ремиелинизации, отмечено выраженное снижение болевого синдрома – примерно на 30% [62].
Анальгетические эффекты уридина связаны с его прямым и опосредованным действием на механизмы боли. Прямое действие прежде всего подразумевает влияние на факторы системного воспаления – приоритетной составляющей развития ДПН, особенно на начальных стадиях поражения невральных структур. Опосредованное действие реализуется через восстановление структурной целостности и нормального функционирования невральных и глиальных элементов.
Как уже было сказано выше, любое поражение нервов сопровождается дефицитом уридина, являющегося важным компонентом ДНК и РНК. Дефицит возникает из-за возросшей потребности в этом нуклеотиде и невозможности его внутриклеточного синтеза. По-видимому, поэтому при нейропатической боли наблюдается экспрессия пуриновых рецепторов в различных структурах центральной и периферической нервной системы. Например, рецепторы P2Y2, которые расположены в нейронах тройничного ганглия, начинают инициировать и поддерживать аллодинии [63]. Рецепторы P2Y6 и P2Y11 экспрессируются в спинальной микроглии и регулируются в ответ на повреждение спинномозгового нерва, что проявляется гиперчувствительностью к механической боли и тактильной аллодинией [64, 65].
Таким образом, своевременное применение уридина предотвращает хронизацию болевых феноменов.
Представленный на отечественном рынке комплекс Нейроуридин содержит 150 мг УМФ, холин, фолиевую кислоту и витамины группы В (В1, В6 и В12). Нейроуридин относится к биологическим активным добавкам, что не умаляет его свойств. Компоненты Нейроуридина органично дополняют действие УМФ на нервную ткань. В частности, синтез холина, из которого образуется ацетилхолин-медиатор (АЦХ), поддерживается пулом уридина, без которого уровень АЦХ снижается. Субстрат холина был введен в рецептуру Нейроуридина еще по одной причине. При синтезе миелина, в котором участвует уридин, вектор цикла Кеннеди смещается в сторону образования фосфатидилхолина, вследствие чего синтез холина, из которого образуется АЦХ, снижается (рис. 3). Холин 82,5 мг при резорбции, распределении и поступлении в мембраны создает тот необходимый пул, при котором синтез АЦХ не страдает. Таким образом, продуманный состав Нейроуридина позволяет обеспечить синтез миелина и не допустить потери АЦХ.
Более того, в экспериментальных исследованиях были продемонстрированы анальгезирующий эффект холина, в том числе при нейропатической боли, улучшение функционального восстановления и регенерации нерва, сенсорных симптомов и скорости проведения возбуждения по моторному нерву, регенерации и восстановления аксонов при рассечении периферического нерва [66–68]. Преимущества анальгетического действия холина в комбинации с пиримидиновыми нуклеотидами представлены в ряде фундаментальных работ, на основании анализа результатов которых сделан вывод о возможном синергизме эффектов компонентов данной комбинации [69].
Фолиевая кислота и витамины группы В, входящие в состав Нейроуридина, содержатся в пределах суточной потребности, которая обеспечивает их участие как в метаболических процессах, регулируемых витаминопрепаратами, так и в синаптических системах, где они выполняют роль аллостерических эффекторов (см. рис. 2). Следует добавить, что такие дозы не обеспечивают самостоятельного анальгетического действия, но способствуют регенерации нервов, снижают риск нежелательных проявлений фармакотерапии и поддерживают Hub-рецептор в его взаимосвязях с другими синаптическими системами.
Следовательно, применение Нейроуридина уже на самых ранних этапах ДПН способно улучшить метаболические и регенеративные процессы в периферических нервах, а также функциональное состояние нервной ткани.
Проведенный обзор литературы не подразумевает применения уридиновых комплексов при ДПН в качестве монотерапии. Тем не менее уридинмонофосфат может быть использован в качестве дополнительного средства патогенетического лечения наряду с препаратами, позволяющими корректировать гипергликемию, дислипидемию, другие факторы риска развития ДПН, α-липоевой кислотой, бенфотиамином, ингибиторами альдозоредуктазы и протеинкиназы С, γ-линоленовой кислотой. Перспективным также видится применение уридинмонофосфата как ко-анальгетика при болевых формах ДПН в дополнение к антидепрессантам и габапентиноидам.
D.A. Iskra, MD, PhD, Prof., V.V. Afanasyev, MD, PhD, Prof., A.R. Volkova, MD, PhD, Prof.
St. Petersburg State Pediatric Medical University
North-Western State Medical University named after I.I. Mechnikov
Academician I.P. Pavlov First St. Petersburg State Medical University
Contact person: Dmitriy A. Iskra, iskradm@mail.ru
Analyzed modern conceptions about the etiopathogenesis of diabetic polyneuropathy. Thus, the main etiological factors leading to the development of the disease are hyperglycemia and dyslipidemia, which cause metabolic disorders in the neuronal cluster. The development of diabetic polyneuropathy is implemented through the mechanisms of impaired glycolysis, oxidative stress, systemic inflammation, mitochondrial dysfunction, and systemic inflammation plays a leading role in the onset of damage to the nervous system. The probability of neuropathic pain is associated with the presence of modifiable and unmodifiable risk factors: female gender, age, obesity, elevated levels of glycated hemoglobin, excessive alcohol consumption, prolonged diabetes, severity of sensory disorders, genetic predisposition. The clinical and pathogenetic relationship of pain with hypoxia of peripheral tissues, vitamin D deficiency, excessive methylglyoxal formation has been proven. The manifestation of clinical signs is caused not only by the defeat of the entire cluster, but also by its glial elements. Dysfunction of Schwann cells, sattelitic glial cells and mild segmental demyelination are observed already in the onset of diabetic polyneuropathy. These mechanisms lead to disruption of the neuroglial interactions underlying the trophic neurons and the formation of their genome. Metabotropic P2Y receptor agonists, pyrimidine nucleotides (uridine, cytidine), play an important role in the metabolism of glial elements. The prospects of using uridine-containing complexes in the treatment of diabetic polyneuropathy with the restoration of the structural integrity of the nerve fiber, regression of clinical symptoms, including the decrease in the severity of pain, have been confirmed by the results of laboratory and clinical studies.
