Роль конечных продуктов гликирования и их рецепторов в развитии осложнений сахарного диабета
- Аннотация
- Статья
- Ссылки
- English
Установлено, что образование и накопление конечных продуктов гликирования (КПГ) негативно воздействует на функции внутри- и внеклеточных структур. В частности, КПГ нарушают образование поперечных связей между своими рецепторами и молекулами базальной мембраны внеклеточного матрикса. Подобные изменения приводят к прогрессированию атеросклероза, ускоренному росту атеросклеротических бляшек, патологическому фиброзу миокарда с последующим развитием сердечной недостаточности.
В статье рассматривается участие КПГ и их рецепторов в развитии сердечно-сосудистых заболеваний при сахарном диабете.
Установлено, что образование и накопление конечных продуктов гликирования (КПГ) негативно воздействует на функции внутри- и внеклеточных структур. В частности, КПГ нарушают образование поперечных связей между своими рецепторами и молекулами базальной мембраны внеклеточного матрикса. Подобные изменения приводят к прогрессированию атеросклероза, ускоренному росту атеросклеротических бляшек, патологическому фиброзу миокарда с последующим развитием сердечной недостаточности.
В статье рассматривается участие КПГ и их рецепторов в развитии сердечно-сосудистых заболеваний при сахарном диабете.
Конечные продукты гликирования (КПГ) – гетерогенная группа молекул, которые образуются в результате неферментативного гликирования и окисления белков, липидов и нуклеиновых кислот. К ним также относятся карбонильные соединения – продукты их деградации [1, 2]. Хорошо изученными КПГ являются пентосидин – производное перекрестного связывания белков и N-карбоксиметил-лизин (N-carboxymethyllysine – CML). Необходимо отметить, что именно флуоресценция пентосидина лежит в основе неинвазивных методов исследования уровня КПГ [3]. Однако чаще для определения уровня CML и КПГ используют иммуноферментный анализ.
В процессе образования КПГ выделяют несколько этапов. Сначала глюкоза связывается со свободными аминогруппами с формированием оснований Шиффа. Затем основания переходят в более стабильные продукты Амадори и в конечном итоге в разные по структуре КПГ – конечные продукты реакции Майяра. Образование КПГ в белках происходит в течение нескольких месяцев, поэтому их накопление больше характерно для медленно обменивающихся белков.
КПГ труднорастворимы, устойчивы к протеолитическому расщеплению, активны химически.
Данные молекулы способны менять функции и свойства тканей. Это достигается патологической сшивкой белков внутриклеточного и межклеточного матрикса [3, 4] путем связывания с рецептором КПГ (рКПГ).
С возрастом накопление КПГ в организме повышается. На это влияют как эндогенные, так и экзогенные факторы. Так, табачный дым и длительная термическая обработка стимулируют генерацию продуктов гликоокисления и липоокисления [5, 6]. Кроме того, при наличии определенных патологических состояний, например сахарного диабета (СД) или почечной недостаточности, скорость гликирования значительно увеличивается и количество КПГ достигает критических значений [7, 8].
Известно, что КПГ приводят к декомпенсации СД 2 типа. Кроме того, они признаны предикторами риска развития сердечно-сосудистых заболеваний. Избыток КПГ отвечает за такой феномен, как метаболическая память.
Образование КПГ – один из процессов, ассоциированных со старением клетки. Их воздействие преимущественно направлено на долгоживущие белки. Именно поэтому в настоящее время КПГ также рассматриваются как один из возможных биомаркеров старения.
Таким образом, изучение свойств и роли КПГ в патофизиологических процессах имеет важное значение, в том числе для разработки методов снижения риска развития сердечно-сосудистых заболеваний как основной причины смерти у пациентов с СД 2 типа [9].
Механизм действия конечных продуктов гликирования на ткани
Интерес к реакции Майяра, или взаимодействию глюкозы с белками, появился в середине 1990-х гг., после того как в условиях in vivo было установлено, что глюкоза способна модифицировать белки без участия ферментов [8]. Эффекты КПГ на ткани реализуются посредством трех основных механизмов:
- скрещивание внеклеточных (матричных) белков, влияющих на механические свойства тканей [7, 9];
- образование измененных поперечных межмолекулярных связей внутриклеточных белков, что приводит к их патологической функции [10, 11];
- связывание с рКПГ на клеточной поверхности для индуцирования множества внутриклеточных сигнальных каскадов [8, 9].
В большей степени неферментативному гликированию подвергаются белки внеклеточного матрикса (ВМ) (особенно коллаген 4-го типа) [8–10]. Коллаген относится к долгоживущим белкам и является основным компонентом внеклеточного матрикса [11]. Коллагеновые нити образуют каркас для кожи, сухожилий, кровеносных сосудов, костной ткани, роговицы и стекловидного тела, а также являются основой большинства паренхиматозных органов. Гликирование белков внеклеточного матрикса – коллагена и эластина делает их более жесткими и менее восприимчивыми к протеолитическому расщеплению [5]. Это может способствовать увеличению жесткости сосудов, наблюдающейся у пациентов старшей возрастной группы и с хронической гипергликемией [8, 9].
Коллаген 1-го типа – основной органический компонент костной матрицы подвергается серии посттрансляционных модификаций, больше характерных для процессов старения. Это приводит к миграции миофибробластов и формированию фиброза [7]. Согласно результатам последних исследований, артериосклероз является следствием гликирования коллагеновых цепей в артериолах мышечного типа, вызванного образованием поперечных связей между коллагеновыми волокнами [11]. Ключевая роль КПГ в старении кожи подтверждена H. Pageon и соавт., проводивших эксперимент на модели восстановленной кожи, модифицированной гликированием коллагена [6]. S. Zeiman и соавт., а также R. Candido и соавт. показали, что под воздействием КПГ изменяются свойства миокардиального коллагена, что приводит к развитию диастолической дисфункции [5, 12]. Подобные изменения обусловливают утолщение базальной мембраны, например в мезангиальном матриксе почек, что вызывает развитие почечной недостаточности при СД [13].
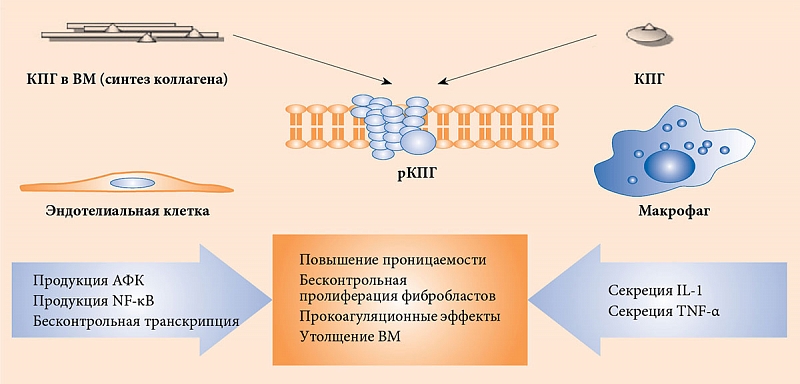
Гликирование влияет и на структуру липопротеинов низкой плотности (ЛПНП). Наиболее интенсивное разрушение ЛПНП и продуктов реакции Майяра происходит в макрофагах. При этом наблюдается активация эндоцитоза и синтеза многих регуляторных молекул, в том числе инсулиноподобного фактора роста 1 и фактора роста тромбоцитов, являющихся стимуляторами деления фибробластов, гладкомышечных и мезангиальных клеток [13]. Таким образом, создаются условия для образования большого количества пенистых клеток и последующего запуска атеросклеротических изменений в сосудистой стенке (рисунок) [14, 15].
Накопление КПГ приводит к бесконтрольному синтезу провоспалительных цитокинов и молекул адгезии, которые влияют на рост атеросклеротических бляшек [16, 17]. Речь, в частности, идет об интерлейкине (IL) 1α, IL-6, факторе некроза опухоли (Tumor Necrosis Factor – TNF) α, молекулах межклеточной адгезии 1, молекулах адгезии сосудистых клеток 1, факторах роста эндотелия сосудов, эндотелине 1, тканевом факторе, E-селектине, тромбомодулине [18, 19].
Запуск патогенетического каскада осуществляется при взаимодействии КПГ с их рецепторами и последующем фосфорилировании p21ras, митоген-активированных протеинкиназ, внеклеточной сигнально-регулируемой киназы 1/2, p38 и активации GTPases Cdc42 и Rac. Это в конечном итоге стимулирует миграцию транскрипционного фактора NF-κB к ядру, где он начинает транскрибировать собственный целевой набор генов [20].
Рецепторы конечных продуктов гликирования и их роль
В качестве специфических рКПГ рассматриваются различные мембранные белки. Это белки, принадлежащие к суперсемейству иммуноглобулинов, которые выполняют функцию рецепторов для гликозилированных молекул КПГ [21]. Однако были обнаружены и другие лиганды к рКПГ, включая семейство белков S100 [22], амилоид b [23, 24] и агрегаты фибриллярных белков [25, 26].
Рецепторы КПГ играют важную роль в развитии состояний, ассоциированных с участием перечисленных лигандов, например в повреждении сосудистой стенки, канцерогенезе, нейродегенерации и амилоидозах [25, 27–29]. Сообщалось, что ген рКПГ расположен на шестой хромосоме между генами, кодирующими основные комплексы гистосовместимости второго и третьего классов [30].
Связывание КПГ с их рецепторами приводит к эндотелиальной дисфункции вследствие активации ряда сигнальных путей, например никотинамидадениндинуклеотидфосфатоксидазы, которая усиливает образование активных форм кислорода (АФК) [31]. Последние образуются в результате митохондриального дыхания и клеточного метаболизма. В малых количествах, считающихся физиологичными, АФК задействованы в таких процессах, как индукция стрессорных белков и ферментов, синтез и распад цитокинов, рост, деление и дифференцировка клеток, антимикробный, противовирусный, противоопухолевый эффекты, старение и гибель клеток, разрушение поврежденных молекул, межклеточного вещества, регуляция репаративных процессов, продукция коллагена [32]. Необходимо отметить, что АФК, столь опасные согласно свободнорадикальной теории старения, вырабатываются организмом целенаправленно [33]. Было показано, что АФК играют ключевую роль в развитии сердечно-сосудистых осложнений за счет изменения структуры клеточных белков, липидов и нуклеиновых кислот и, следовательно, их физиологических функций [34].
В настоящее время известно несколько типов рКПГ. В частности, рКПГ-1 при связывании с КПГ инактивируется, что приводит к деградации лиганда. Снижение экспрессии рКПГ-1 ассоциируется с ускорением гломерулярной дисфункции при СД 2 типа [34] и активацией циркулирующих мононуклеарных клеток при высоких значениях КПГ у лиц с тяжелыми осложнениями СД 2 типа [35]. Функция рКПГ-3 (семейство углевод-связывающих белков) напрямую зависит от длительности и степени гипергликемии. При инактивированном рКПГ-3 достоверно чаще развивается диабетическая нефропатия [36].
Сердечно-сосудистая система
Образование КПГ в тканях ускоряет иммуновоспалительные реакции и перекисное окисление липидов, что в условиях хронической гипергликемии приводит к декомпенсации СД.
Кроме того, накопление КПГ связано не только с ранним развитием сердечно-сосудистых осложнений, но и с более негативным прогнозом в отношении выживаемости.
Так, E.Y. Choi и соавт., изучавшие в течение пяти лет уровень КПГ в сыворотке крови 203 пациентов, перенесших чрескожное коронарное вмешательство, установили, что высокие значения КПГ являются независимым фактором риска развития рестеноза при СД 2 типа (отношение шансов – 2,659 при 95%-ном доверительном интервале (ДИ) 1,431–4,940, p = 0,002) [37]. Уже через шесть месяцев у пациентов с высокими значениями КПГ (> 170 Ед/мл) частота рестенозирования была значительно выше (р < 0,001). Согласно результатам, полученным K. Kiuchi и соавт., повышение КПГ прямо коррелировало со степенью стенозирования коронарных артерий, обратно – с фракцией выброса левого желудочка, а также ассоциировалось с более длительным пребыванием в отделении интенсивной терапии [38]. A. Simm и соавт. показали, что значения КПГ влияли на риск летального исхода у пациентов после чрескожного коронарного вмешательства [39]. В работах R. Meerwaldt и соавт., а также V. Jakus и соавт. сообщалось, что снижение уровня КПГ служило доказательством эффективности проведенной реваскуляризации у больных СД 2 типа [40–42].
Увеличение уровня КПГ у пациентов с СД 1 типа влияло на частоту развития сердечно-сосудистых событий, независимо от наличия других факторов риска, таких как возраст, индекс массы тела, курение, артериальная гипертензия и гиперлипидемия [43].
Согласно результатам проспективного исследования EURODIAB, у больных СД 1 типа увеличение артериального давления было напрямую связано с высокими значениями КПГ [44]. У больных СД уровень рКПГ коррелировал с толщиной комплекса «интима – медиа» брахиоцефальных артерий [45]. В нескольких экспериментальных исследованиях установлено, что КПГ могут участвовать в образовании неоинтимы в месте повреждения. Z. Zhou и соавт. отметили, что у крыс, больных СД, при значительном увеличении уровня КПГ повышалась иммунореактивность рКПГ и S100/calgranulins в ответ на травматическое воздействие баллона в сонной артерии [46].
В условиях in vitro и in vivo предотвращение связывания рКПГ с лигандом снижало пролиферацию эндотелиоцитов.
КПГ также могут оказывать влияние на целостность структуры сосудистой стенки. В частности, чрезмерное гликирование молекул ВМ, таких как коллаген, может нарушать как клеточно-мембранные, так и межмембранные взаимодействия [47].
Для диабетической кардиомиопатии характерны гипертрофия и формирование патологического фиброза миокарда, что в конечном итоге приводит к диастолической дисфункции. Доказано, что эти процессы напрямую зависят от степени компенсации углеводного обмена, скорости снижения уровня гликированного гемоглобина. Однако в настоящее время в качестве основной причины ее развития рассматривают накопление КПГ [48]. Взаимодействие КПГ с внутриклеточными белками, в частности с основным фактором роста фибробластов (Basic Fibroblast Growth Factor – β-FGF), значительно стимулирует фиброзирование миокарда [49]. β-FGF – мощный модулятор клеточной дифференцировки, пролиферации и подвижности клеток [50].
Активация фибробластов в условиях гипергликемии обусловлена ускорением полиолового шунта, значительным повышением концентрации глюкозо-6-фосфата, фруктозы и фруктозо-3-фосфата, активацией С-протеинкиназы, окислительного стресса и гликирования факторов роста фибробластов [51, 52]. При неферментативном анаэробном гликолизе внутри клетки накапливаются дикарбонилы, которые признаются одними из основных участников сшивания белков [53]. Это приводит к патологической, бесконтрольной работе фибробластов. Они начинают активно пролиферировать, разрушать старый и синтезировать новый коллаген. Это приводит к перестройке стенки сосудов и, как следствие, к фиброзу.
На индукцию фиброза также влияют рКПГ – регулируют трансформирующий фактор роста β (Transforming Growth Factor β – TGF-β) [53]. Гликирование TGF-β увеличивает синтез коллагена 3, 4 (α-3), 5 и 6-го типов, а также ламинина и фибронектина в ВМ [54]. R. Petrova и соавт. обнаружили, что чрезмерная экспрессия рКПГ у трансгенных особей снижала внутриклеточную концентрацию кальция как во время систолы, так и во время диастолы [55]. Была также выявлена обратная связь между уровнем КПГ в перикардиальной жидкости и фракцией выброса левого желудочка [56, 57].
Метилгексилированные производные КПГ активируют мРНК кардиальных рКПГ, что стимулирует развитие сократительной дисфункции кардиомиоцитов. Накопление метилгексилированных производных КПГ приводит к деполяризации митохондриального мембранного потенциала, снижению инактивации гликоген синтазы киназы 3β в миокарде, что вызывает замедление регенерации (TR90) [58]. Стимуляция рецепторов γ, активируемых пероксисомными пролифераторами (Peroxisome Proliferator-Activated Receptors γ – PPAR-γ), способствует снижению уровня рКПГ.
В ряде работ были проанализированы эффекты агониста PPAR-γ (росиглитазон) у животных. Полученные данные свидетельствовали о важной роли рКПГ в инициации фиброза [59].
В исследовании, проведенном R.D. Semba и соавт., было подтверждено участие КПГ и их рецепторов в развитии сердечно-сосудистых заболеваний в период постменопаузы [60]. Высокий уровень КПГ (95% ДИ 1,08–3,48, р = 0,026) и рКПГ (95% ДИ 0,98–1,65, р = 0,07) также ассоциировался с высокой смертностью среди женщин старшей возрастной группы, имевших нарушения углеводного обмена. В другом исследовании, проведенном Y. Koyama и соавт., установлено, что сывороточные уровни рКПГ коррелировали с классами Функциональной классификации хронической сердечной недостаточности Нью-Йоркской ассоциации кардиологов и низкой фракцией выброса [61]. Выдвинуто предположение, что рКПГ являются независимым фактором развития диастолической дисфункции.
K. Sugaya и соавт. подтвердили участие КПГ в воспалительных процессах. В частности, в исследовании (длительность наблюдения – 18 лет) выявлена связь между высокими значениями сывороточного КПГ и увеличением количества летальных случаев у пациенток с СД 2 типа в исходе ишемической болезни сердца [30].
Особый интерес представляют результаты иммуногистохимического анализа и вестерн-блоттинга 60 атеросклеротических бляшек, полученных в результате каротидной эндартерэктомии. Так, при СД 2 типа уровень макрофагов, рКПГ, Т-лимфоцитов, HLA-DR+, NF-κB, COX-2/mPGES-1, липидов и MMP был достоверно выше (р < 0,0001). При этом экспрессия рКПГ, COX-2/mPGES-1 и MMP линейно коррелировала с уровнем гликированного гемоглобина в плазме [62]. В ходе интервенционного исследования установлено, что лечение статинами до каротидной эндартерэктомии снижает не только активность воспаления, но и экспрессию рКПГ.
В исследовании in vivo дезактивирование рКПГ ассоциировалось со снижением степени повреждения миоцитов, о чем свидетельствовало уменьшение уровня ЛПНП, продуктов с низким содержанием гликозидов – CML и пентосидина, улучшение скорости функционального восстановления и синтеза аденозинтрифосфата [63]. Более того, согласно результатам иммуногистохимического исследования, у мышей с модифицированным рКПН, несмотря на наличие СД 2 типа, активность ключевых маркеров апоптоза, таких как каспаза-3 и цитохром С, была снижена.
Отмечено негативное влияние КПГ на рецепторы рианодина [4] и SER-CA2a [5] в кардиомиоцитах. Нарушение их функции приводит к изменениям гомеостаза кальция и последующему развитию диабетической кардиомиопатии [35, 36]. Фибриноген состоит из трех пар неидентичных цепей, стабилизированных несколькими дисульфидными связями. Известно, что концентрация фибриногена и скорость образования сгустка не зависят от наличия нарушений углеводного обмена [58], однако при диабете высокие уровни КПГ обусловливают нарушение гомеостаза и активацию атеросклеротических процессов. Гликирование фибриногена, активного участника свертывающей системы крови, приводит к замедлению фибринолиза и образованию в дальнейшем тромбогенной фибриновой сети [57].
Заключение
Ускоренное гликирование белков и накопление КПГ играют важную роль в патогенезе сердечно-сосудистых заболеваний у пациентов с СД. КПГ следует рассматривать в качестве маркеров развития воспалительных процессов и окислительного стресса.
Изучение механизмов регуляции взаимодействия КПГ и их рецепторов позволит разработать методы предотвращения развития осложнений, связанных с хронической гипергликемией.
E.V. Ivannikova, PhD, O.M. Smirnova, DM, Prof.
National Medical Research Center for Endocrinology
Contact person: Ekaterina Vladimirovna Ivannikova, doc.ivannikova@gmail.com
Diabetic complications appear to be multifactorial in origin, but in particular, the biochemical process of advanced glycation, which is accelerated in diabetes as a result of chronic hyperglycemia and increased oxidative stress, has been postulated to play a central role in these disorders. The presence and accumulation of аdvanced glycation end-products (AGEs) in many different cell types affect extracellular and intracellular structure and function. AGEs contribute to a variety of microvascular and macrovascular complications through the formation of cross-links between molecules in the basement membrane of the extracellular matrix and by engaging the receptor for advanced glycation end products.
These biological effects translate to accelerated plaque formation in diabetes as well as increased cardiac fibrosis with consequent effects on cardiac function. The purpose of this review is to discuss the role of AGEs in cardiovascular disease and in particular in heart failure.
