Дифференциальный диагноз хронической диареи у детей
- Аннотация
- Статья
- Ссылки










Хронической считается диарея продолжительностью более 3 недель с частотой стула три и более раз в день. Учитывая, что такая частота стула может быть нормой для детей первых месяцев жизни, необходимо также обращать внимание на характер стула и его объем. В норме у здорового ребенка объем стула обычно не превышает 10 г/кг массы тела в сутки. Водянистый или жирный характер стула, наличие зелени, слизи, крови, непереваренных остатков пищи являются признаками патологии.
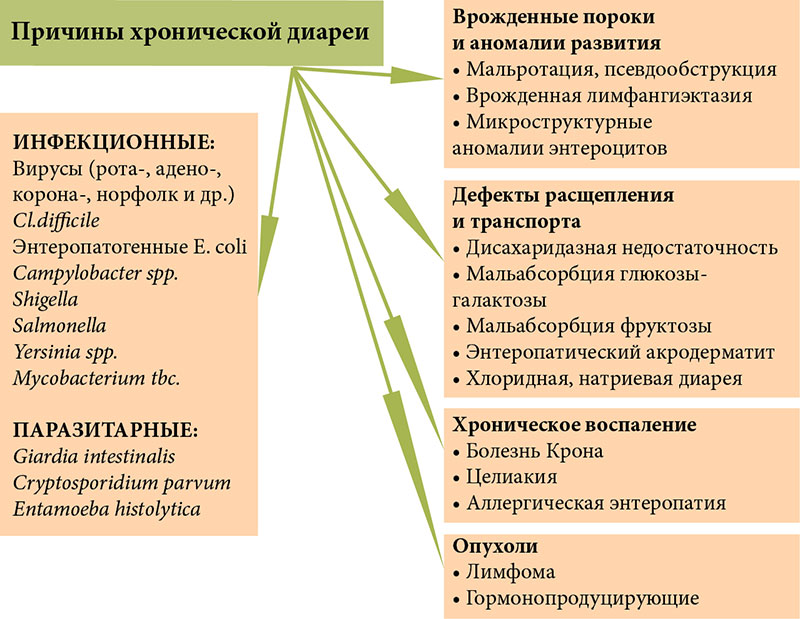
Причины хронической диареи (ХД) весьма разнообразны (рис. 1), наиболее частые из них: кишечные инфекции (чаще вирусные, реже бактериальные, вызванные условно-патогенной микрофлорой), а также паразитарные инвазии (лямблиоз, иногда криптоспоридиоз, амебиаз и др.). Трансформация острой диареи в хроническую может быть обусловлена неблагоприятным фоном, на котором протекает инфекция (гипотрофия, дефицит микронутриентов, иммунодефицит), а также неадекватной терапией. Как следствие длительного воспаления, развивается атрофия слизистой оболочки тонкой кишки (СОТК) с вторичным нарушением ее функций.
Наряду с инфекционными и паразитарными причинами, ХД может быть обусловлена врожденными генетически детерминированными дефектами пищеварения (лактазная недостаточность, сахаразо-изомаль-тазная недостаточность) и всасывания (мальабсорбция глюко-зы-галактозы, хлоридная, натриевая диарея, мальабсорбция желчных кислот, энтеропатический акродерматит и др.), врожденными аномалиями строения энтероцитов (атрофия микроворсин, интестинальная эпителиальная дисплазия) и иммунопатологическими заболеваниями, сопровождающимися хроническим воспалением СОТК (аллергическая энтеропатия, целиакия, аутоиммунная энтеропатия, болезнь Крона). Наиболее тяжелой формой ХД является так называемая трудноизлечимая младенческая диарея, обусловленная аномалиями энтероцитов или аутоиммунной энтеропатией.
Диагностика заболеваний, проявляющихся ХД, должна основываться на тщательной оценке характера кишечного синдрома, симптомов со стороны других органов и систем, данных анамнеза и дополнительных исследований. В связи с широкой распространенностью кишечных инфекций и паразитозов у детей первым этапом диагностики является установление или исключение данных заболеваний. Для этого каждому ребенку с ХД необходимо провести:
- клинический анализ крови;
- посевы кала;
- исследование кала на яйца глистов и цисты лямблий методами микроскопии, ИФА и ПЦР;
- микроскопию мазка кала на криптоспоридиоз (с окраской по Цилю-Нильсену или обработкой 1% раствором HCl);
- исследование кала на токсин А и В Clostridium difficile методом ИФА;
- копрологическое исследование (не менее 3 копрограмм);
- исследование кала на скрытую кровь.
Даже в случае отрицательного результата бактериологического исследования при подозрении на инфекционный генез диареи показан курс антибактериальной терапии ex juvantibus. Обычно в этих случаях назначают нитрофураны (нифуратель 15 мг/кг или нифуроксазид 100 мг 2 раза в день), аминогликозиды (гентамицин 3–5 мг/кг, амикацин 10 мг/кг) или цефалоспорины III поколения (цефотаксим 50 мг/кг, цефтриаксон 50 мг/кг). Поскольку диарея в детском возрасте имеет преимущественно вирусную этиологию и характеризуется водянистым стулом со значительной потерей жидкости и солей, важно с первых же дней болезни проводить адекватную регидратацию и коррекцию электролитного обмена. Не менее важно обеспечить полноценное питание ребенка с учетом сниженных возможностей пищеварения.
Для коррекции микробиоценоза кишечника при постинфекционных диареях применяют препараты-пробиотики. При назначении пробиотического препарата необходимо убедиться в отсутствии лактозы в его составе. Для детей раннего неонатального периода выпускается безлактозный пробиотик Примадофилус Детский, который содержит подобранные в соответствии с возрастом штаммы лакто- и бифидобактерий в возрастной дозировке 2 х 10 9 КОЕ в суточной дозе.
Если ребенок находится на естественном вскармливании, его можно продолжить, но в каждое кормление рекомендуется добавлять фермент Лактаза Бэби.
При искусственном вскармливании с учетом вероятного развития вторичной лактазной недостаточности на фоне кишечной инфекции целесообразно использовать безлактозные или кисломолочные смеси: НАН кисломолочный, НАН безлактозный, Энфамил Лактофри.
Лактазная недостаточность у детей раннего возраста может быть не только вторичной, но и первичной: врожденной или транзиторной. Врожденная лактазная недостаточность – крайне редкая патология, при которой в результате генетических мутаций лактаза не транспортируется из аппарата Гольджи на поверхность мембраны энтероцита. Симптоматика врожденной лактазной недостаточности проявляется с первых дней жизни очень ярко в виде многократной водянистой диареи, сопровождающейся вздутием живота, потерей массы тела и дегидратацией. В абсолютном большинстве случаев лактазная недостаточность у новорожденного или недоношенного ребенка носит транзиторный характер и обусловлена незрелостью кишечных ворсинок и недостаточной активностью фермента лактазы на поверхности эпителия (гиполактазией).
Транзиторная лактазная недостаточность может быть причиной нетяжелой «кислой» водянистой диареи в первые месяцы жизни, которая нередко сопровождается кишечными коликами и беспокойством ребенка, но не влияет на его физическое развитие. Симптомы постепенно смягчаются на фоне продолжения грудного вскармливания. Это объясняется тем, что женское молоко обладает многочисленными факторами роста (EGF, IGF-1, TGF-β, спермин, спермидин, нуклеотиды, короткоцепочечные жирные кислоты и т.д.), которые способствуют росту и созреванию кишечного эпителия и повышению лактазной активности.
Поэтому при симптоматике гиполактазии грудное вскармливание следует продолжить, но для устранения симптомов целесо-образно применять фермент Лактаза Бэби по 700 ед. в начале каждого кормления. Детям с рождения назначается содержимое 1 капсулы Лактазы Бэби на 100 мл молока. Перед кормлением следует сцедить 20 мл молока и высыпать содержимое капсул. Кормление начинать через 10 минут с этой порции. В случае если ребенок получает только сцеженное грудное молоко, Лактазу Бэби добавляют в полный объем питания. При искусственном вскармливании необходимое количество фермента также добавляется в полный объем молочного питания. Время для ферментации составляет 10 минут.
Для подтверждения лактазной недостаточности любого генеза у ребенка можно использовать различные тесты: исследование рН кала (ниже 5,5), гликемическую кривую с лактозной нагрузкой из расчета 2 г/кг (прирост уровня глюкозы не более 25% от исходного), водородный тест (прирост уровня водорода в выдыхаемом воздухе выше 20 ppm).
Если кишечная инфекция и лактазная недостаточность (наиболее частые причины хронической диареи) исключены, а диарея, несмотря на антибактериальную терапию и безлактозную диету, продолжается, необходимо продолжить обследование, обеспечивая адекватную нутриционную поддержку путем сочетания парентерального и энтерального питания смесями белковых гидролизатов без лактозы: Альфаре, Нутрилон Пепти ТСЦ, Нутрамиген, Фрисопеп АС, Прегестимил.
Дальнейший алгоритм диагностики ХД основывается на оценке характера стула (рис. 2). Следует различать водянистую диарею, диарею со стеатореей и диарею с колитическим синдромом (кровью и слизью в стуле).
Водянистая диарея
При сохраняющейся, несмотря на отмену лактозы, водянистой «кислой» диарее (рН кала < 5,5) у новорожденного ребенка вероятным диагнозом может быть мальабсорбция глюкозы-галактозы, для подтверждения которого проводится исследование экскреции сахаров с калом и пробы на толерантность к глюкозе и фруктозе. Поскольку при этом заболевании нарушена работа глюкозо-галактозного транспортера, в то время как функции фруктозного транспортера в СОТК сохраняются, нагрузку этими моносахаридами организм ребенка воспринимает по-разному. После приема глюкозы диарея усиливается, а гликемическая кривая имеет плоский вид. Фруктоза же является единственным углеводом, который хорошо переносится детьми с мальабсорбцией глюкозы-галактозы, поэтому при подтверждении диагноза вскармливание следует проводить артифициальной смесью, приготовленной из казеината кальция, растительного масла и фруктозы.
Все нарушения расщепления и всасывания углеводов – лактазная, сахаразо-изомальтазная недостаточность, мальабсорбция глюкозы-галактозы, мальабсорбция фруктозы – проявляются осмотической диареей, которая развивается в результате образования из невсосавшихся углеводов осмотически активных продуктов бактериального брожения. Временный перевод ребенка на парентеральное питание или исключение из рациона углевода, вызвавшего диарею, сопровождаются купированием симптомов.
Диарея, сохраняющаяся после перевода ребенка на полное парентеральное питание, свидетельствует о ее секреторном характере. Основным отличием секреторной диареи от осмотической является отсутствие эффекта от «голодания», то есть от отмены обычного питания и перевода ребенка на парентеральное (табл. 1). Для подтверждения секреторного характера диареи необходимо исследование уровня электролитов в крови и экскреции их с калом. При секреторной диарее осмоляльность кала зависит главным образом от содержания в нем электролитов и представляет собой сумму концентраций ионов Na+ и K+ (или Na+, K+ и Cl-), умноженную на два. При осмотической диарее осмоляльность кала обусловлена нерасщепленными или невсосавшимися частичками пищи, при этом концентрация электролитов в нем остается низкой, что объясняет большую осмотическую разницу, поэтому сумма электролитов кала (Na++K++Cl-) значительно меньше осмоляльности. Секреторная диарея чаще наблюдается при острых (так называемых водянистых) диареях, однако длительное упорное ее течение может наблюдаться при таких редких заболеваниях, как врожденная хлоридная или натриевая диарея, а также при мальабсорбции желчных кислот. Для хлоридной диареи характерно преобладание в кале ионов хлора над другими электролитами, для натриевой диареи – ионов натрия. При подтверждении селективной потери соответствующего электролита показана заместительная терапия в виде солевых растворов, сначала внутривенно, затем перорально.
Наблюдаемая с рождения тяжелая смешанная диарея с секреторным компонентом, потерей большого количества жидкости (более 300 мл в сутки) в сочетании с потерей плазменного белка характерна для врожденных аномалий энтероцитов (табл. 2). Этому типу диареи свойственно отсутствие эффекта от перевода ребенка на парентеральное питание и усиление поноса после еды. Осмотическая разница в кале отсутствует, как и при секреторной диарее. Такая диарея является показанием к проведению биопсии СОТК и электронно-микроскопического исследования биоптатов. Обнаружение феномена «включенных микроворсин» или «пучковой дисплазии» энтероцитов подтверждает факт врожденной аномалии их строения. Единственным способом лечения этих врожденных аномалий является пересадка тонкой кишки.
Диарея со стеатореей
Стеаторея сопровождается не столько учащенным, сколько жирным на вид, объемным и зловонным стулом, который обычно имеет кашицеобразный характер, плохо отмывается от горшка. При копрологическом исследовании важно различать два типа стеатореи: стеаторею нейтральным жиром, которая является симптомом недостаточности поджелудочной железы, и стеаторею жирными кислотами, которая характерна для патологии тонкой кишки.
Стеаторея панкреатического типа (нейтральным жиром) возможна при муковисцидозе, синдроме Швахмана-Даймонда, синдроме Пирсона, изолированном дефиците липазы (синдроме Шелдона-Рея). При ее обнаружении в серии копрограмм показана более точная оценка экзокринной функции поджелудочной железы с помощью теста на эластазу-1 в кале. Снижение уровня эластазы-1 ниже 200 мг/г подтверждает вероятность панкреатической недостаточности. Последующий диагностический алгоритм направлен на выявление конкретного заболевания поджелудочной железы. Диагноз муковисцидоза подтверждается на основании повышения уровня хлоридов в поте и обнаружения мутаций CFTR-гена.
Для синдрома Швахмана-Даймонда характерно сочетание панкреатической недостаточности и гематологических нарушений (нейтропении, реже тромбоцитопении и анемии), а также отставание в росте и наличие некоторых костных аномалий (дисхондроплазии тазобедренных суставов, несращение ребер, клиновидные пальцы и т.д.).
Синдром Пирсона тоже проявляется гематологическими нарушениями, но они, как правило, более тяжелые, характерно упорное течение анемии и тромбоцитопении. Костные аномалии при синдроме Пирсона отсутствуют.
Синдром Шелдона-Рея характеризуется отсутствием липазы в крови и моче, выраженной стеаторей; нарушений роста нет.
Основой лечения всех этих генетически детерминированных заболеваний является заместительная терапия панкреатическими ферментами. Доза ферментов подбирается индивидуально, обычно начинают с 2 000 ЕД/кг липазы в день, при необходимости дозу повышают. Предпочтительны препараты с микросферическими ферментами (Креон, Панцитрат), которые следует давать ребенку в начале каждого приема пищи и распределять в соответствии с ее количеством. При тяжелой гипотрофии в питании больных используют смеси со среднецепочечными триглицеридами (СЦТ): Альфаре, Нутрилон Пепти ТСЦ, Прегестимил, Пептамен, Портаген. Дополнительно назначают жирорастворимые витамины: А, Е, Д, К.
Стеаторея кишечного типа (жирными кислотами) не является специфическим признаком определенных заболеваний, но довольно часто отмечается при заболеваниях тонкой кишки. Для уточнения характера интестинальной патологии необходимо морфологическое исследование биоптатов СОТК, взятых в ходе эндоскопического исследования из дистального отдела двенадцатиперстной или проксимального отдела тощей кишки.
Наличие лимфоплазмоцитарной инфильтрации собственной пластинки СОТК свидетельствует о хроническом воспалении, которое в большинстве случаев сопровождается атрофией слизистой, проявляющейся укорочением ворсин и истончением эпителия. Оценка степени атрофии СОТК проводится в соответствии с классификацией Marsh, различают 3 степени. В случае обнаружения атрофии 2‑й или 3‑й степени (укорочение ворсин, углубление крипт, густая равномерная лимфоплазмоцитарная инфильтрация собственной пластинки, увеличение межэпителиальных лимфоцитов – МЭЛ) (рис. 3) у ребенка, который уже получал глютен, необходимо провести исследование уровня антиглиадиновых антител (IgA, IgG), антител к тканевой трансглютаминазе (IgA, IgG) и эндомизию. При повышенном уровне этих иммуноглобулинов устанавливается диагноз целиакии, ребенку назначается пожизненная строгая безглютеновая диета. Классическими признаками целиакии в раннем возрасте являются недостаточная прибавка веса с развитием гипотрофии и увеличение живота.
Сходный с целиакией энтеральный синдром нередко наблюдается у детей с пищевой аллергией. Симптомы аллергической энтеропатии могут проявляться уже в первые месяцы жизни в виде беспокойства, кишечной колики, срыгиваний, позднее может присоединяться железодефицитная анемия. Однако в ряде случаев у детей до 2 лет могут доминировать симптомы аллергического колита с появлением крови и слизи в стуле. Кишечные симптомы у большинства детей сочетаются с картиной атопического дерматита. Морфологическая картина аллергической энтеропатии характеризуется признаками хронического воспаления СОТК с нормальной высотой (или незначительным укорочением) и глубиной ворсин и наличием эозинофилов в инфильтрате (рис. 4). Диагноз может подтверждаться повышением в крови общего и специфических IgE, но поскольку гастроинтестинальная аллергия чаще реализуется через клеточные механизмы сенсибилизации, нормальный уровень IgE у ребенка вовсе не исключает вероятность аллергического поражения кишечника. Сходная морфологическая картина свойственна также лямблиозу. Алгоритм диагностики на основании морфологических признаков представлен на рис. 5.
Сочетание хронической диареи с поражениями кожи свойственно также энтеропатическому акродерматиту, вызванному дефицитом цинка. Поскольку цинк входит в состав более 40 металлосодержащих ферментов и участвует в обеспечении иммунологической защиты, диарея при этом заболевании может поддерживаться эндогенной микрофлорой, в частности нередко развивается вторичный кандидоз. Морфологические изменения СОТК характеризуются сочетанием воспаления и умеренной атрофии на фоне сниженного количества МЭЛ, плазмоцитов в собственной пластинке и сниженной регенераторной способностью эпителия. Кожные проявления заболевания характеризуются четко отграниченными от нормальной кожи зонами гиперемии, инфильтрации и экскориации, локализующимися обычно на ягодицах, вокруг рта, на подбородке и щеках. Энтеропатический акродерматит проявляется после отмены грудного вскармливания, поскольку женское молоко содержит цинк-всасывающий лиганд, препятствующий развитию дефицита этого микроэлемента. Иногда болезнь начинается на фоне острой кишечной инфекции, что связано с значительной энтеральной потерей цинка. Таким образом, формируется порочный круг: острая диарея вызывает дефицит цинка, который в свою очередь поддерживает и усугубляет диарею, способствуя ее переходу в хроническую. Уровень цинка в крови обычно опускается ниже 8 ммоль/л. Пероральное назначение препаратов цинка в высоких дозах (сульфат цинка 50–100 мг/сут) быстро купирует диарею и кожные симптомы.
Диарея с кровью и слизью в стуле
Появление крови и слизи в стуле свойственно колитическому синдрому. После исключения инфекций (дизентерии, сальмонеллеза, иерсиниоза, кампилобактериоза) установление причин диареи предполагает прежде всего морфологическую и иммунологическую верификацию. В любом случае диарея со слизью и кровью в стуле указывает на высокую активность воспаления в кишечнике и наличие дефектов слизистой оболочки. Косвенным доказательством активности воспалительного процесса может служить оценка уровня кальпротектина в кале, уровень которого в норме не превышает 50, а при воспалении в кишечнике возрастает в несколько раз.
Для морфологической верификации заболевания пациенту с хроническим колитическим синдромом показано проведение колоноскопии с илеоскопией. Эндоскопическое исследование может выявить специфические изменения, свойственные болезни Крона или язвенному колиту.
Сочетание тяжелой энтеропатии с колитическим синдромом свойственно аутоиммунной энтеропатии. Заболевание чаще развивается у детей первых двух лет (иногда первых месяцев) жизни, кишечные симптомы сочетаются с признаками поражения других органов (почек, печени, легких, кожи, суставов и т.д.). Аутоиммунная энтеропатия у мальчиков может сочетаться с полиэндокринопатией (IPEX) или быть проявлением тяжелых иммунодефицитных заболеваний.
Морфологические изменения СОТК при аутоиммунной энтеропатии (рис. 6) характеризуются умеренной или выраженной атрофией ворсин, которая всегда ассоциирована с выраженной мононуклеарной инфильтрацией собственной пластинки. Гиперплазия крипт может быть вариабельна. В большинстве случаев тяжелая или тотальная атрофия ворсин сочетается с углублением крипт. Выраженная атрофия в сочетании с мононуклеарной инфильтрацией могут расцениваться первоначально как признаки целиакии (табл. 3), однако для нее характерно повышение митотической активности эпителия и увеличение количества МЭЛ, в то время как при аутоиммунной энтеропатии митотическая активность снижена, а инфильтрат локализуется в собственной пластинке без увеличения или с небольшим увеличением количества МЭЛ. МЭЛ имеют при этом в основном αβ-рецепторы, в то время как при целиакии возрастает число МЭЛ, несущих γδ-рецепторы. У некоторых больных тотальная атрофия ворсин ассоциирована с некрозами эпителиальных клеток и формированием крипт-абсцессов. Поверхностный эпителий уплощен, дистрофирован. Число бокаловидных клеток снижено. Мононуклеары представлены в основном CD4+ T-лимфоцитами и макрофагами. Морфологические изменения наиболее выражены в слизистой оболочке тонкой кишки, но могут обнаруживаться и в других отделах пищеварительного тракта – желудке или толстой кишке. Обширное поражение ЖКТ имеет наиболее тяжелые проявления и плохой прогноз.
В крови обнаруживают аутоантитела (преимущественно класса IgG) к кишечному эпителию, направленные против компонентов щеточной каймы или цитоплазмы энтероцитов нормальной интестинальной слизистой. Могут присутствовать антитела к бокаловидным клеткам. Неспецифические аутоантитела могут быть также направлены против ядер, ДНК, гладкой мускулатуры или митохондрий, что обычно сочетается с внекишечными проявлениями. В случаях ассоциации аутоиммунной энтеропатии с поражением почек описана циркуляция аутоантител против почечной ткани, в частности против 75 kDa-антигена. После установления диагноза аутоиммунной энтеропатии больному показана терапия глюкокортикостероидами в сочетании с цитостатиками.
Аутоиммунная диарея, наряду с врожденными аномалиями энтероцитов, относится к группе так называемых трудноизлечимых младенческих диарей. Следует еще раз подчеркнуть основные особенности заболеваний этой группы: генетическая предрасположенность;
- раннее начало;
- отсутствие связи с каким-либо инфекционным агентом или пищевым субстратом;
- упорный характер водянистой диареи секреторного типа, не купирующейся при переводе ребенка на парентеральное питание;
- выраженная атрофия СОТК.
Алгоритм клинической диагностики трудноизлечимых младенческих диарей представлен на рисунке 7, гистологические особенности СОТК – в таблице 4.
Таким образом, хроническая диарея у детей может быть обусловлена множеством различных причин. Успех в лечении этого крайне сложного в плане дифференциальной диагностики синдрома во многом зависит от знаний врача и его стремления найти истину.
Установление правильного диагноза, как известно, является ключом к выбору правильной терапии. Как указывалось выше, в лечении хронической диареи, используют прежде всего различные диетические подходы. Что касается медикаментозной терапии, то общей стратегией лечения всех диарейных заболеваний является коррекция вод-но-электролитных нарушений и усиление кишечного барьера. Последнее достигается назначением цитопротекторов и адсорбентов.
Уважаемый посетитель uMEDp!
Уведомляем Вас о том, что здесь содержится информация, предназначенная исключительно для специалистов здравоохранения.
Если Вы не являетесь специалистом здравоохранения, администрация не несет ответственности за возможные отрицательные последствия, возникшие в результате самостоятельного использования Вами информации с портала без предварительной консультации с врачом.
Нажимая на кнопку «Войти», Вы подтверждаете, что являетесь врачом или студентом медицинского вуза.