Дифференциальный диагноз патологии печени при синдроме цитолиза: разбор клинического случая
- Аннотация
- Статья
- Ссылки
- English

![Рис. 2. Алгоритм обследования пациента с синдромом цитолиза (адаптировано из [6])](/upload/resize_cache/iblock/d6a/800_800_1/Livzan3.jpg)
![Таблица 2. Клинические признаки и диагностика заболеваний печени (адаптировано из [1])](/upload/resize_cache/iblock/ed4/800_800_1/Livzan4.jpg)
![Рис. 3. Корреляция уровня аминотрансфераз при различных патологиях печени (адаптировано из [6])](/upload/iblock/355/Livzan5.jpg)
Синдром цитолиза (СЦ) – лабораторный синдром, который характеризуется повышением активности аланинаминотрансферазы (АЛТ), аспартатаминотрансферазы (АСТ), лактатдегидрогеназы (ЛДГ 4 и ЛДГ 5), специфических печеночных ферментов (сорбитдегидрогеназы, альдолазы, орнитин-карбамилтрансферазы), митохондриальных ферментов (глутаматдегидрогеназы, сукцинатдегидрогеназы), а также билирубина (главным образом вследствие повышения прямой фракции) [1].
Впервые повышение аминотрансфераз (АТ) в сыворотке крови и его значение для клинической диагностики были описаны в 1955 г. [2]. С тех пор определение данных биохимических показателей стало рутинным тестом, который проводится как с целью диагностики заболеваний внутренних органов, в том числе печени, так и в качестве скринингового теста, включенного в плановый профилактический осмотр.
Повышение уровня АТ в сыворотке крови встречается при заболеваниях печени, миокарда, скелетных мышц, почек и других органов. Это обусловлено тем, что АЛТ и АСТ являются ферментами, которые регулируют углеводно-белковый обмен во многих органах и тканях (табл. 1).
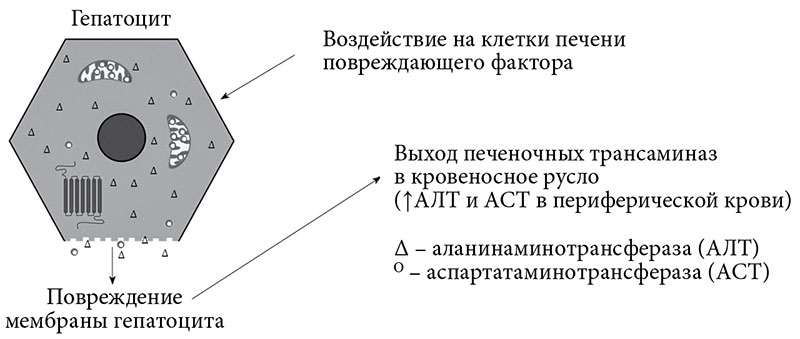
Сущность СЦ заключается в разрушении мембраны клеточной стенки, что приводит к выходу внутриклеточных ферментов в межклеточную среду с последующим проникновением в кровеносное русло, при этом становится возможным определение повышенного уровня АТ в периферической крови (рис. 1) [3, 4]. Для косвенной оценки тяжести повреждения клетки и внутриклеточных структур важно ориентироваться во внутриклеточной локализации ферментов. АЛТ локализуется исключительно в цитоплазме клеток, и ее появление в сыворотке крови свидетельствует о повреждении клеточной стенки, в то время как большая часть АСТ находится в митохондриях и в меньшей степени в цитоплазме, появление ее в сывороточной крови говорит о разрушении клеточных органелл, в первую очередь митохондрий [4].
Уровень АТ в периферической крови измеряют количественно, интерпретируя результаты в изменениях кратности полученных результатов к референсным значениям. Умеренным считается цитолиз до пятикратного увеличения показателей, выраженным – увеличение в 5–10 раз, а превышение показателей более чем в 10 раз следует трактовать как высокий цитолиз [1, 3, 5]. Данная классификация СЦ по уровню АТ имеет клиническую значимость для динамической оценки состояния пациента и эффективности терапии.
Во многих странах разработаны практические руководства при обнаружении у пациента СЦ, позволяющие минимизировать риски ошибочного диагноза. Важно соблюдение пошагового алгоритма диагностики (рис. 2). На первом этапе важно исключить внепеченочные причины, при необходимости применить уточняющий лабораторный скрининг и консультации узких специалистов. Очень важно умение врача выстроить доверительные взаимоотношения с пациентом для подробного уточнения жалоб (при их наличии) и давности их возникновения. Необходимо собрать анамнез о перенесенных заболеваниях в прошлом, наличии сопутствующих патологий на данный момент. С целью исключения токсического повреждения печени следует уточнить информацию о приеме лекарственных препаратов, биологически активных добавок и сведения об употреблении алкоголя, его количестве и кратности. При общем осмотре пациента в пользу патологии печени могут свидетельствовать телеангиэктазии, изменение окраски кожных покровов, экскориации, расширение венозной сети на передней брюшной стенке, асцит, увеличение печени в размерах при перкуссии печени и пальпации ее нижнего края.
Следующим шагом при СЦ проводится дифференциальная диагностика среди патологии печени (табл. 2) для назначения эффективной фармакотерапии. С этой целью всем пациентам с СЦ показан диагностический минимум: скрининг на вирусные гепатиты, ультразвуковое исследование органов брюшной полости, а также оценка других этиологических факторов.
Для исключения вирусного поражения печени производится определение маркеров вирусных гепатитов В и С (HBsAg, Anti-HCV), при этом следует помнить о наличии ложноотрицательных результатов, что определяет необходимость определения маркеров репликации вирусов (HBеAg, HCVcAg) и проведения полимеразной цепной реакции для определения ДНК и РНК вирусов в крови.
Женщинам молодого возраста при наличии аутоиммунных патологий в анамнезе следует провести скрининг на аутоиммунный гепатит, для которого характерно наличие антинуклеарных антител (ANA), антител к гладкой мускулатуре (ASMA), антител к микросомам печени и почек (anti-LKM), антимитохондриальных антител (AMA) в диагностическом титре [6]. Данные маркеры аутоиммунного поражения также позволяют исключить первичный билиарный холангит. Дополнительным критерием аутоиммунной патологии печени является повышение уровня γ-глобулинов.
Алкогольная болезнь печени включает в себя несколько вариантов повреждения паренхимы печени вследствие злоупотребления алкоголем от стеатоза до алкогольного гепатита (стеатогепатита), приводящего к развитию последовательных стадий – фиброза, цирроза печени и гепатоцеллюлярной карциномы [7, 8]. Гепатотоксичной дозой этанола является 40–80 г/сут для мужчин, 20 г/сут для женщин в пересчете на чистый этанол. Являясь растворителем, этанол разрушает фосфолипиды мембран митохондрий и гепатоцитов. Повреждение мембран митохондрий лежит в основе жировой дистрофии печени, так как теряется их способность метаболизировать триглицериды [8–10]. У большинства пациентов алкогольный стеатоз протекает бессимптомно и обнаруживается случайно при обследовании. Среди форм острого алкогольного гепатита выделяют латентную, желтушную (желтуха без кожного зуда, выраженная слабость), холестатическую (желтуха, кожный зуд, ахолия кала, потемнение мочи) и фульминантную (желтуха, геморрагический синдром, синдром печеночной энцефалопатии и печеночной недостаточности) формы [8]. Стадия цирроза печени характеризуется присоединением синдрома печеночно-клеточной недостаточности и портальной гипертензии. Заподозрить алкогольное поражение печени можно по преобладанию повышенного уровня АСТ [11, 12], изолированному повышению гамма-глутамилтранспептидазы (ГГТП), повышенному уровню в сыворотке крови углевод-дефицитного трансферрина, среднего корпускулярного объема эритроцитов (MCV), мочевой кислоты, а также определение этилглюкуронида в моче [8].
Для лекарственного поражения печени характерен прием лекарственных препаратов или биологически активных добавок в анамнезе. Наиболее часто проявляется на фоне приема амиодарона, синтетических эстрогенов, аспирина, антагонистов кальция [13]. Среди факторов риска выделяют женский пол, пожилой возраст и повышенный индекс массы тела (ИМТ), беременность, сахарный диабет, заболевание печени и почек, курение, употребление алкоголя, длительный прием препаратов и полипрагмазию [14–17].
К основным клиническим проявлениям перегрузки организма железом (гемохроматоз) относят немотивированную слабость, утомляемость и сонливость. При общем осмотре обращают на себя внимание участки пигментации кожных покровов бурого цвета, обусловленные отложением гемосидерина в коже. При лабораторном исследовании характерно повышение концентрации сывороточного железа и ферритина. Диагноз должен обязательно дополняться генетическим тестированием [18].
Болезнь Вильсона – Коновалова (гепатолентикулярная дегенерация) является наследственным заболеванием с аутосомно-рецессивным типом наследования, обусловленным нарушением обмена меди, в результате чего происходят сбой выведения печенью избытка меди в желчь и накопление ее в гепатоцитах. Избыток меди вызывает повреждение митохондрий и активацию перекисного окисления липидов, разрушение клеточной мембраны гепатоцитов, что приводит к формированию острого или хронического гепатита с риском развития цирроза печени. Повреждение мембраны гепатоцитов сопровождается утечкой и отложением меди в других органах и тканях [16, 19], основной мишенью при этом является головной мозг, что обусловливает появление неврологической симптоматики, дебют которой происходит в 20–30 лет [16]. В качестве подтверждения диагноза используются диагностические критерии: низкий уровень церулоплазмина (< 20 мг/дл), увеличение 24-часовой экскреции меди с мочой (> 80 мкг/сут), концентрация меди в ткани печени > 200 мкг/г сухой массы, наличие роговичного «медного» кольца Кайзера – Флейшера (зеленовато-коричневые пигментные кольца на периферии радужной оболочки глаз) [20–22].
При недостаточности α1-антитрипсина характерно снижение фермента в сыворотке крови (< 11 мкмоль/л), в результате чего происходит задержка его полимеризованных молекул в эндоплазматическом ретикулуме печеночных клеток с развитием гепатотоксического эффекта [23, 24].
В качестве диагноза исключения при СЦ следует рассмотреть неалкогольную жировую болезнь печени (НАЖБП). Это приобретенное заболевание, имеющее сходную с алкогольным поражением гистологическую картину, возникающее при отсутствии злоупотребления алкоголем или других возможных причин развития патологии печени при наличии стеатоза [25]. При исследовании АТ выявляется нормальный уровень или повышение активности АЛТ и АСТ, а повышение показателей более пяти верхних границ нормы может указывать на сопутствующее острое поражение печени [11, 12].
При наличии стеатоза в сочетании с метаболическими состояниями (избыточный вес или ожирение, сахарный диабет второго типа, наличие двух и более критериев метаболического синдрома) необходимо акцентировать внимание на новом адаптивном понятии – метаболически ассоциированная жировая болезнь печени (МАЖБП) [26, 27] с целью персонализации объемов и направления лечебно-диагностической помощи при различных клинических вариантах МАЖБП-ассоциированной коморбидности [28, 29].
Кроме того, существует относительная корреляция уровня АТ в зависимости от заболевания (рис. 3), которая может быть полезной для прицельного диагностического поиска.
Далее на примере клинического случая показано проведение диагностического поиска.
Клинический случай
Пациент А., 50 лет, обратился вместе с супругой к гастроэнтерологу на кафедру факультетской терапии и гастроэнтерологии Омского государственного медицинского университета.
Основные жалобы: общая слабость, быстрая утомляемость, со слов супруги, за последние четыре года цвет кожных покровов приобрел загорелый оттенок при отсутствии на это причин.
Анамнез заболевания: считает себя больным в течение пяти лет, когда впервые стал отмечать появление немотивированной слабости, быстрой утомляемости. В 2017 г. обращался к участковому терапевту, назначены обследования: антропометрия, общий анализ крови, биохимический анализ крови, общий анализ мочи, кал на яйца гельминтов, цифровая флюорография (ввиду отсутствия прохождения за последний год), рекомендовано посещение смотрового кабинета. По результатам обследований: рост – 175 см, вес – 102 кг, в биохимическом анализе крови выявлен СЦ (АЛТ – 205 Ед/л (↑), АСТ – 187 Ед/л (↑), прямой билирубин – 6,2 мкмоль/л (↑), глюкоза – 5,7 ммоль/л, общий холестерин – 4,8 ммоль/л), по результатам остальных методов исследования – без патологических изменений. При повторном сборе анамнеза жизни пациент отрицает вирусные гепатиты, прием алкоголя и наркотических веществ, в течение десяти лет получает антигипертензивную терапию. Участковым терапевтом исключено лекарственное поражение печени ввиду отсутствия СЦ по результатам прохождения диспансеризации годом ранее на фоне приема антигипертензивного препарата. Пациенту назначено дополнительное обследование: скрининг на вирусные гепатиты В и С, ультразвуковое исследование органов брюшной полости. По данным ультразвуковой диагностики, обнаружены диффузные изменения печени по типу жирового гепатоза, признаки билиарного сладжа. Маркеры вирусных гепатитов (HBsAg, anti-HCV) отрицательные. Пациенту выставлен диагноз: «Неалкогольная жировая болезнь печени. Синдром билиарного сладжа. Артериальная гипертензия (стадия II), контролируемое течение, риск 2». Назначено лечение: урсодезоксихолевая кислота (УДХК) 500 мг на прием вечером на три месяца, индапамид + периндоприл 2,5/10 мг на прием утром длительно. На фоне лечения пациент положительной динамики не отмечал. При повторной сдаче крови через три месяца на фоне продолжающегося приема УДХК пациенту было проведено контрольное биохимическое исследование крови (АЛТ – 193 Ед/л (↑), АСТ – 155 Ед/л (↑), глюкоза – 6,2 ммоль/л (↑)). По результатам анализов терапевтом по месту жительства рекомендованы консультация гастроэнтеролога, проведение перорального глюкозотолерантного теста (глюкоза через два часа после глюкозотолерантного теста – 8,2 ммоль/л).
Анамнез жизни: вирусные гепатиты, ВИЧ-инфекцию, туберкулез отрицает. Травм, операций не было, чужую кровь не переливали. Наблюдается у кардиолога по поводу гипертонической болезни на протяжении десяти лет. На регулярной основе принимает индапамид + периндоприл 2,5/10 мг на прием один раз в день. Наследственность отягощена по сахарному диабету (у матери). Аллергоанамнез без особенностей.
Объективно: рост – 175 см, вес – 105 кг, ИМТ – 34,3 кг/м2. Состояние пациента удовлетворительное, в сознании. Питание повышенное. Кожные покровы сухие, на открытых участках отмечается пигментация бурого цвета, остальные участки кожных покровов серого цвета с коричневым оттенком. Подкожно-жировая клетчатка увеличена в области живота. Пальпируемые лимфатические узлы не увеличены, безболезненные. Щитовидная железа –степень 0 (по ВОЗ). Дыхание везикулярное, хрипов нет, частота дыхательных движений – 16 в минуту. Тоны сердца ритмичные без патологических шумов, акцент тона II на аорте. Отеков нет. Артериальное давление – 130/80 мм рт. ст.; пульс – 66 ударов в минуту. Tемпература тела – 36,6 °С. SpO2 – 99%. Живот мягкий, безболезненный, увеличен вследствие увеличения объема подкожной жировой клетчатки. Печень выступает на 1 см из-под края реберной дуги. Симптомы Ортнера и Мерфи отрицательные, симптом Кера слабоположительный. При пальпации по методу Гротта поджелудочная железа болезненна в зоне Шоффара. Симптом поколачивания по поясничной области отрицательный с обеих сторон. Стул, диурез в норме.
Пациенту установлен предварительный диагноз: «Стеатогепатит смешанного генеза (ожирение, нарушение толерантности к глюкозе), клинико-биохимическая активность 1, фиброз (?). Дисфункция сфинктера Одди по билиарному типу. Синдром билиарного сладжа. Хронический панкреатит, метаболический без внешнесекреторной недостаточности, латентное течение».
Рекомендовано к обследованию: общий анализ крови, биохимический анализ крови (АЛТ, АСТ, щелочная фосфатаза (ЩФ), ГГТП, глюкоза, билирубин прямой, общий белок, общий холестерин, глюкоза, панкреатическая амилаза, мочевая кислота, креатинин, сывороточное железо, ферритин, общая железосвязывающая способность сыворотки (ОЖСС), витамин D), общий анализ мочи, копрограмма, кал на паразитов и яйца гельминтов трехкратно, кал на цисты лямблий, кал на углеводы, определение фекального кальпротектина, иммуноферментный анализ (ИФА) на антитела описторхисов и лямблий (IgM, IgG), эластометрия печени.
Результаты дополнительных обследований:
- общий анализ крови: лейкоциты – 6,54 × 109/л, нейтрофилы – 59,1%, лимфоциты – 33,0%, моноциты – 4,6%, эозинофилы – 2,7%, базофилы – 0,6%, эритроциты – 4,94 × 1012/л, гемоглобин – 150 г/л, тромбоциты – 377 × 109/л, СОЭ – 10 мм/ч;
- биохимический анализ крови: АЛТ – 192 Ед/л (↑), АСТ – 156 Ед/л (↑), ЩФ – 110 МЕ/л, ГГТП – 40 ЕД/л, глюкоза – 7,2 ммоль/л (↑), мочевая кислота – 352 ммоль/л, креатинин – 86 ммоль/л, общий белок – 70,2 г/л, общий билирубин – 18,4 мкмоль/л, холестерин – 6,43 ммоль/л (↑), амилаза – 56 Ед/л, сывороточное железо – 41 мкмоль/л (↑), ОЖСС – 47,3 мкмоль/л, латентная железосвязывающая способность сыворотки – 6,3 мкмоль/л (↓), ферритин – 1200 мкг/л (↑), витамин D – 11,2 нг/мл (↓);
- общий анализ мочи: цвет – желтый, прозрачность – прозрачная, pH – 5,0, плотность – 1020, белок – нет, глюкоза – нет, лейкоциты – 0–1 в п/з, эритроциты – 0 в п/з, эпителий – нет в п/з;
- кал на паразитов и яйца гельминтов методом PARASEP: не обнаружено;
- кал на цисты лямблий (Giardia lamblia) иммуногистохимическим методом: не обнаружено;
- кал на определение уровня кальпротектина: 28,7 мкг/г;
- копрограмма: без патологических изменений;
- ИФА крови на выявление антител к описторхисам (IgM, IgG) и лямблиям (IgM, IgG): не обнаружено;
- эластометрия печени на аппарате FibroScan: степень ригидности печени составила 5,8 кПа (интерквартильный разброс – 0,9 кПа), что соответствует стадии фиброза F1 по шкале METAVIR.
С учетом высокого уровня сывороточного железа и ферритина, по данным биохимического анализа крови, пациенту рекомендовано генетическое тестирование – гемохроматоз, определение мутаций: снижение функциональной активности фермента HFE: 845 G > A (rs 1800562) А/А.
При проведении консилиума в составе гастроэнтеролога, кардиолога и эндокринолога выставлен заключительный диагноз. Основной диагноз: «Хронический гепатит смешанного генеза (МАЖБП, ассоциированная с ожирением и нарушением толерантности к глюкозе; гемохроматоз (HFE: 845 G > A (rs 1800562) А/А)), клинико-биохимическая активность I, фиброз I (по данным эластометрии). Дисфункция сфинктера Одди по билиарному типу. Синдром билиарного сладжа. Хронический панкреатит, метаболический, без внешнесекреторной недостаточности, латентное течение. Дефицит витамина D»; сопутствующий диагноз: «Гипертоническая болезнь (стадия II). Артериальная гипертензия – контролируемое течение, риск 3 (высокий). Дислипидемия, целевой уровень липопротеинов низкой плотности < 2,6 ммоль/л. Ожирение первой степени, ИМТ – 34,3 кг/м2. Хроническая сердечная недостаточность (стадия IIа, функциональный класс I). Целевой уровень артериального давления < 140/90 мм рт. ст. Нарушение толерантности к глюкозе».
Даны следующие рекомендации по питанию: средиземноморская диета, обогащенная полиненасыщенными и мононенасыщенными жирами, исключение продуктов, содержащих лактозу. Ограничение поступления продуктов с высоким содержанием железа, продуктов, способствующих всасыванию железа (аскорбиновая кислота, фруктовые соки, красные сорта мяса, рыба).
Рекомендации по физической активности включали дозированные аэробные физические нагрузки по 30–60 минут средней интенсивности не менее пяти дней в неделю.
Назначено лечение:
- адеметионин 400 мг на прием два раза в день утром и в обед в течение восьми недель;
- УДХК 500 мг на прием утром, 1000 мг на прием вечером длительно (из расчета 10–15 мг/кг в сутки);
- панкреатин 25 000 ЕД на более обильный прием пищи в режиме «по требованию»;
- водорастворимый витамин D 7000 МЕ в день (14 капель) на прием один раз в день утром в течение восьми недель с последующим контролем уровня витамина D в сыворотке крови;
- индапамид + периндоприл 2,5/10 мг на прием один раз в день длительно;
- питавастатин 1 мг на прием один дин раз в день длительно под контролем липидного спектра, АСТ, АЛТ, креатинфосфокиназы с последующей индивидуальной интенсификацией дозировки;
- наблюдение у кардиолога, эндокринолога;
- эритроцитоферез.
На этапе диагностического поиска ввиду наличия у пациента СЦ участковый терапевт провел скрининг на вирусные гепатиты. При получении отрицательных результатов и с учетом наличия диффузных изменений печени по типу жирового гепатоза по данным ультразвуковой диагностики терапевт прекратил дальнейший поиск, расценив состояние пациента как НАЖБП. Приняв во внимание наличие у пациента немотивированной слабости, быстрой утомляемости, бурой окраски кожных покровов и стойкого повышения АТ, гастроэнтеролог Омского государственного медицинского университета рекомендовал дополнительные обследования согласно протоколам пошагового диагностического поиска. Были выявлены лабораторные признаки накопления железа в печени, которые были дополнены генетическим тестированием на мутации HFE (845 G > A (rs 1800562) А/А), что подтвердило наличие гемохроматоза. К сожалению, назначенная пациенту ранее УДХК в дозировке 500 мг на прием вечером не способствовала устранению СЦ, однако скорректированное лечение из расчета УДХК 10–15 мг/кг в сутки позволило достичь нормализации уровня АТ через четыре недели. Пациент взят под диспансерное наблюдение.
Литературная справка
Наследственный гемохроматоз (НГХ) – генетически детерминированное заболевание, характеризующееся повышенной абсорбцией железа в кишечнике и высвобождением железа из макрофагов, что приводит к увеличению циркулирующего пула железа в периферической крови, избыточному накоплению его в органах и тканях и, как следствие, нарушению их функций [18].
НГХ является аутосомно-рецессивным заболеванием, обусловленным мутацией C282Y – заменой цистеина на тирозин в 282-й аминокислоте гена HFE, который играет роль в регуляции гепсидина, в результате чего происходит избыточное накопление не-трансферин-связанного железа (NTBI – non-transferrin-bound iron) в печени, поджелудочной железе, сердце и других органах [30]. Частота встречаемости достигает одного случая на 200–250 человек населения [30, 31]. Также известны редкие мутации в других генах (в совокупности называемых гемохроматозом без HFE), включая HJV (кодирующий гемоджувелин), HAMP (кодирующий гепсидин), TFR2 (кодирующий рецепторный белок трансферрина 2) и SLC40A1 (также известный как FPN1, кодирующий синтез транспортного белка ферропортина) [32].
Заболевание чаще встречается у мужчин в возрасте 30–40 лет, женщины заболевают реже и позже, поскольку избыток железа у них удаляется естественным путем при менструальных кровотечениях [17]. На ранних стадиях заболевание, как правило, протекает бессимптомно. Его дебют сопровождается повышенной утомляемостью, немотивированной слабостью, в большинстве случаев серовато-коричневой окраской кожных покровов [18, 32]. Для заболевания характерны поражение печени с формированием фиброза и развитием гепатоцеллюлярной карциномы, поражение сердца с формированием токсической кардиомиопатии, которая может проявляться фибрилляцией предсердий, экстрасистолией и другими формами аритмий. В патологический процесс могут вовлекаться суставы с формированием моно- или олигоартрита, чаще всего голеностопные и второй-третий пястно-фаланговые. Возрастает риск формирования сахарного диабета и дисфункции половых желез [31, 33, 34].
Согласно действующим рекомендациям Европейской ассоциации по изучению печени (European Association for the Study of the Liver – EASL) [18], у пациентов с клиническими признаками НГХ необходимо исследовать уровень сывороточного железа, трансферрина и ферритина. При обнаружении биохимических признаков перегрузки железом (женщины с насыщением трансферрина > 45% и сывороточным ферритином > 200 мкг/л, мужчины с насыщением трансферрина > 50% и сывороточным ферритином > 300 мкг/л) необходимо проведение генетического тестирования (выявление мутации гена HFE). Генетическое тестирование на НГХ также показано пациентам с повышенным содержанием железа по данным биопсии печени и результатам магнитно-резонансной томографии, кровным родственникам первой линии (в случае генетически подтвержденной мутации гена HFE).
Стандартом медицинской помощи пациентам с НГХ является кровопускание (флеботомия) под контролем уровня гемоглобина и трансферрина крови, которая уменьшает накопление железа в организме вследствие мобилизации железа для эритропоэза. Флеботомия состоит из двух фаз: индукции – еженедельное кровопускание с целью истощения запасов железа (целевой уровень сывороточного ферритина – 50 мкг/л) и поддерживающей фазы – для поддержания сывороточного ферритина в диапазоне 50–100 мкг/л. На ранних стадиях лечебное кровопускание снижает утомляемость у большинства пациентов, предотвращает или останавливает прогрессирование фиброза печени и нормализует ожидаемую продолжительность жизни. При отсутствии противопоказаний в поддерживающую фазу пациенту может быть рекомендована сдача донорской крови. При наличии у пациента противопоказаний к кровопусканию назначается хелатная терапия препаратом деферазирокс [16, 18].
Таким образом, обнаружение повышенного уровня АТ является распространенным в клинической практике явлением. Нет строгой специфичности для АЛТ и АСТ в качестве маркеров повреждения гепатоцитов, что диктует необходимость исключения внепеченочных причин СЦ. На представленном клиническом примере показано, что диагностический поиск причины СЦ при заболеваниях печени должен складываться таким образом, чтобы рассмотреть все возможные причины его возникновения, в том числе и редкие, с соблюдением пошаговых алгоритмов диагностики для предотвращения ошибки диагностического поиска.
M.A. Livzan, PhD, Prof., Corresponding member of the RAS, O.V. Gaus, PhD, M.A. Lisovskiy
Omsk State Medical University
Contact person: Olga V. Gaus, gaus_olga@bk.ru
Assessment of the level of aminotransferases in the blood serum is a routine diagnostic method for diseases of the digestive system and preventive medical examinations. The purpose of this article is to systematize data on the doctor's tactics when an elevated level of aminotransferases in the blood serum is detected in order to minimize the erroneous error of the cytolysis syndrome, using a step-by-step diagnostic algorithm in clinical practice.
Уважаемый посетитель uMEDp!
Уведомляем Вас о том, что здесь содержится информация, предназначенная исключительно для специалистов здравоохранения.
Если Вы не являетесь специалистом здравоохранения, администрация не несет ответственности за возможные отрицательные последствия, возникшие в результате самостоятельного использования Вами информации с портала без предварительной консультации с врачом.
Нажимая на кнопку «Войти», Вы подтверждаете, что являетесь врачом или студентом медицинского вуза.