
Введение
Гастроинтестинальные стромальные опухоли (gastrointestinal stromal tumours, GIST) являются наиболее частыми мезенхимальными опухолями желудочно-кишечного тракта (ЖКТ). Считается, что GIST происходят из клеток Кахала (Cajal), которые отвечают за моторику ЖКТ. В пользу этого говорит не только схожее гистологическое строение на ультраструктурном уровне, но и экспрессия гена c-kit как клетками Кахала, так и стромальными опухолями. Экспрессия гена c-kit, определяемая с помощью моноклонального антитела CD117, считается суррогатным маркером GIST. В 95% случаев GIST являются антиген-позитивными по CD117, однако при отрицательной реакции и исключении лейомиосаркомы может потребоваться молекулярно-генетический анализ с целью выявления антиген-негативной опухоли.
В 2001 г. для лечения больной с диссеминированной формой GIST впервые был применен препарат Гливек (иматиниб), который оказался чрезвычайно эффективным и вскоре стал широко применяться в качестве монотерапии заболевания, которое считалось нечувствительным к химиолучевому лечению. Практически сразу начали проводиться исследования по изучению эффективности Гливека в адъювантном и неоадъювантном режимах. В 2011 г. на съезде Американского общества клинической онкологии (American Society of Clinical Oncology, ASCO) было сообщено об эффективности 3-летней адъювантной терапии Гливеком у больных с высоким риском прогрессирования. Многочисленные исследования также показали, что основные гистологические характеристики (размер опухоли, локализация, митотический индекс), а также молекулярно-генетический статус являются важными факторами прогноза.
Так, например, пациенты с метастатической формой болезни и мутацией в 9-м экзоне гена c-kit получают выгоду в плане безрецидивной выживаемости, если сразу получают двойную дозу лекарства. Пациенты с крайне редкой мутацией D842V полностью нечувствительны к Гливеку. Несмотря на высокую эффективность терапии Гливеком, минорная часть больных демонстрирует первичную резистентность, а у большинства пациентов даже после многолетней успешной терапии наступает вторичная резистентность. В качестве выбора при прогрессировании патологического процесса дозу Гливека повышают с 400 до 800 мг либо осуществляют переход на препарат второй линии Сутент. Эффективность Сутента после приема Гливека составляет около 3–4 мес. и, как оказалось, также зависит от мутационного статуса. В качестве ингибиторов тирозинкиназ также исследуются нилотиниб, дазатиниб, сорафениб, регорафениб, ваталаниб, мазитиниб, пазопаниб, креноланиб. Ряд препаратов с иным механизмом действия проходят исследования II фазы [1, 2].
Основной механизм патогенеза развития GIST – мутации в генах c-kit и тромбоцитарного фактора роста (PDGFRA)
В 1998 г. S. Hirota и соавт. опубликовали в журнале Science статью «Доминантно-негативные мутации c-kit при гастроинтестинальных стромальных опухолях у человека» [3]. Среди 58 опухолей мезенхимального происхождения с помощью иммуногистохимического метода было выявлено 49 GIST. Также было показано, что экспрессия CD117 наблюдается не только на клетках опухоли, но и на клетках Кахала, что позволило высказать предположение о происхождении GIST именно из этих клеток. В 5 из 6 образцов опухолей авторы выявили мутацию в гене c-kit. В последующем подтвердилось, что мутация гена c-kit – это основной механизм развития GIST. В сентябре 1998 г. в Хельсинки, Финляндия, прошел первый международный симпозиум по изучению GIST, на котором присутствовали 23 делегата. На симпозиуме было высказано предположение о потенциальной эффективности ингибиторов c-kit при GIST.
Белок KIT, кодируемый геном c-kit, является членом семейства тирозинкиназ. В эту же группу входят альфа- и бета-рецепторы тромбоцитарного фактора роста (PDGFRA и PDRGFRB), рецептор колониестимулирующего фактора макрофагов (CSF1R) и рецептор цитокина Fl. Лигандом для KIT служит стволовый клеточный фактор (SCF). Результатами взаимодействия являются гомодимеризация рецептора и запуск киназной активации. При развитии GIST происходит независимая от наличия лиганда активация, что и приводит к развитию опухоли. Наиболее частая мутация происходит в 11-м экзоне гена, который кодирует внутриклеточный домен. При этом возможны различные типы мутаций: делеции, вставки, замены или комбинации. С делециями связан худший прогноз, для других типов мутаций характерно более благоприятное течение заболевания [4, 5].
Приблизительно в 10% случаев мутации происходят в 9-м экзоне гена c-kit, который кодирует внеклеточный домен белка. Киназный домен при мутации в 9-м экзоне похож на киназный домен при диком типе GIST, что предполагает чувствительность к терапии последних. Интересно, что мутации в 9-м экзоне встречаются практически исключительно в опухолях тонкой и толстой кишки, в опухолях желудка частота мутации – около 1% [6]. Мутации в экзонах 13 и 17 составляют 1–2% случаев. Предполагается, что данные экзоны отвечают за физиологическое подавление функции внутриклеточных доменов. Очевидно, что мутация гена является ключевой и начальной стадией заболевания. В дальнейшем запускается каскад самых различных сигнальных путей (MAPK, PI3K-AKT, MYC, mTOR). Исследования на клеточных линиях показали, что, в частности, подавление путей PI3K более эффективно по сравнению с ингибированием МАРК и mTOR [7].
Мутации в гене тромбоцитарного фактора роста встречаются в 5–10% случаев. Как и в случае с активацией рецептора KIT на мембране клеток, рецептор PDGFRA начинает активироваться без своего лиганда PDGFA, в результате чего запускается каскад реакций, схожих с реакциями, имеющими место при активации KIT [8]. Для подтверждения диагноза GIST панель иммуногистохимической реакции включает, кроме CD117, маркеры DOG1 и PKC-тета [9, 10].
В 10–15% случаев GIST не удается выявить мутации в генах c-kit и PDGFRA, таким образом, речь идет о диком типе этих генов. В то же время иммуногистохимически определяется экспрессия CD117, что говорит о запуске сигнальных путей, механизм которого не совсем ясен. В клинической практике часто проводится скрининг только на мутации в 11-м и 9-м экзонах, при отсутствии мутации формально предполагается дикий тип, а исследование на мутацию в 13-м или 17-м экзонах не проводится. Морфологически и макроскопически опухоли с диким типом ничем не отличаются от мутантных опухолей. В то же время тщательные исследования показали, что при диком типе выявляются другие механизмы активации и прогноз течения заболевания при этих опухолях также отличен от GIST с мутациями. Наиболее часто (в половине случаев) при диком типе выявляется повышенная экспрессия рецептора инсулинового фактора роста (IGF1R), который действует через сигнальные пути МАРК и PI3-AKT.
Интересно, что повышенная экспрессия IGF1R выявляется при забрюшинных саркомах и саркоме Юинга. Мутации в гене BRAF, которые более характерны для меланомы и рака щитовидной железы, выявляются при диком типе GIST в 13% случаев [11]. В ряде случаев при диком типе GIST удается обнаружить мутацию в гене сукцинатдегидрогеназы (SDH), который кодирует четыре субъединицы (SDHA, SDHB, SDHC, SDHD). Функция этого фермента – участие в цикле Кребса. Мутация указанного гена повышает риск развития не только GIST, но и синхронных параганглиом (синдром Карнея – Стратакиса, Carney – Stratakis syndrome) [12]. Механизм развития GIST при мутации гена сукцинатдегидрогеназы остается не совсем ясным, но предполагается, что фермент снижает активность пролилгидроксилазы, которая, в свою очередь, регулирует активность фактора, индуцируемого гипоксией (hypoxia-inducible factor, HIF1-альфа). В свою очередь HIF1-альфа является активатором для IGF2 и VEGF. Классификация GIST в зависимости от локализации мутации и чувствительность опухолей к препаратам представлены в таблице 1.
Эпидемиология гастроинтестинальных стромальных опухолей
До открытия мутации гена c-kit и использования маркера CD117 GIST расценивались как лейомиосаркомы. Согласно классификации и стадированию злокачественных опухолей, лейомиосаркомы желудка и тонкой кишки были отнесены к злокачественным опухолям указанных органов и, соответственно, «растворились» среди диагнозов «рак желудка» и опухолей тонкой кишки. Точная заболеваемость была неизвестна [13]. За последние годы во многих странах, особенно в тех, где ведутся канцер-регистры и часто сохраняются парафиновые блоки и стекла удаленных образцов, удалось прояснить эпидемиологию GIST. Так, наиболее полноценные исследования проведены в скандинавских странах (Норвегия, Швеция, Финляндия, Дания) [14, 15]. В этих странах ретроспективно были пересмотрены блоки «лейомиом» и «лейомиосарком» и в подавляющем большинстве случаев диагноз был изменен на GIST. Оказалось, что в среднем заболеваемость составляет 10–15 человек на 1 млн населения. В других европейских странах этот показатель был ниже, что, скорее всего, было связано с меньшим охватом населения скринингом и более низким уровнем доступности медицинской помощи.
Кроме того, тщательное морфологическое исследование образцов желудка у больных, умерших от других заболеваний, позволило обнаружить так называемые микро-GIST, размерами до 1–2 мм. В 2006 г. в журнале Human Pathology была опубликована статья японских морфологов, в которой говорилось о высокой частоте встречаемости GIST в желудке [16]. Авторы исследовали 100 желудков, удаленных по поводу рака. В 35 желудках были обнаружены 50 микроскопических опухолей, которые оказались положительными на экспрессию KIT и/или CD34. Большинство опухолей (90%) локализовалось в проксимальных отделах желудка, в 2 случаях GIST выявлены мутации гена c-kit. В 28 желудках выявлена 51 лейомиома. В 12 желудках обнаружено сочетание микроскопических GIST и лейомиом. Средний размер опухоли составил 1,5 мм. Авторы предположили, что GIST, по крайней мере его микроскопические формы, распространены гораздо шире, чем принято полагать. Вероятно, что в большинстве случаев они не достигают клинически значимых размеров и в ряде случаев склонны к спонтанной регрессии.
Кроме того, стоит подчеркнуть, что такая высокая частота встречаемости ни в коей мере не означает истинного распространения GIST желудка, поскольку исследование проведено на выборке рака желудка, где механизмы канцерогенеза в определенной мере сами по себе могут способствовать появлению стромальных опухолей. В Северной Америке самое крупное исследование по эпидемиологии GIST проведено E.A. Perez и соавт. [17]. В работу были включены базы данных регистра SEER (Surveillance, Epidemiology and End Results database), который охватывает около 17% населения США, и база данных онкологического регистра Флориды (Florida Cancer Data System), охватывающая около 6% населения Америки. Данные SEER обработаны с 1992 по 2005 г., регистра Флориды – с 1984 г. Было установлено, что в 1994 г. 93% опухолей расценивались как гладкомышечные опухоли и только 6% – как GIST. В 2002 г. уже 82% составляли GIST и только 17% – гладкомышечные опухоли. Пропорция GIST желудка была еще выше – 96% для SEER и 93% для популяции Флориды. Заболеваемость GIST увеличилась с 0,028 на 100 тыс. населения в 1992 г. до 0,688 на 100 тыс. населения в 2005 г. Авторы подчеркнули, что повышение заболеваемости связано как с явным улучшением диагностики, так и с улучшением скрининга. После 2000 г. опухоли желудка составляли 47% случаев, опухоли тонкой кишки – 34%. Интересно, что заболеваемость GIST у афроамериканцев выше, чем у белого населения.
Таргетная терапия метастатических форм GIST
Гливек начал применяться в 90-е годы прошлого столетия в качестве ингибитора тирозинкиназ для лечения хронического миелолейкоза благодаря способности ингибировать онкопротеин BCR-ABL. Впоследствии наблюдение, что белки BCR-ABL и KIT имеют структурную схожесть, инспирировало проведение исследований по использованию Гливека на клеточных линиях, в том числе и на клетках GIST [19]. Практически сразу же были инициированы протоколы по исследованию эффективности двойной дозы Гливека при метастатических формах заболевания. В целом Гливек был эффективен у 70–85% пациентов, медиана времени до прогрессирования составила 20–24 мес. [20].
Высокая эффективность Гливека породила убеждение, что возможно полное излечение больных с GIST: возникло желание прекратить прием препарата после успешной длительной ремиссии. В 2002 г. французскими исследователями инициировано рандомизированное исследование III фазы BFR14, которое поставило вопрос о продолжительности приема Гливека у больных с метастатической формой болезни. Больные были рандомизированы на группы постоянного приема препарата (C-group, continuous) и окончания приема через год успешного лечения Гливеком (S-group, stop). Дизайн подразумевал возобновление приема Гливека в S-группе в случае прогрессирования. В исследование было включено 58 пациентов. В последующем этот протокол трансформировался в рандомизацию С- и S-групп через 3 года (50 пациентов) и через 5 лет (25 пациентов). В stop-группе каждый раз после окончания приема Гливека достоверно быстрее наступало прогрессирование [25–27]. Так, через 5 лет после рандомизации ни у одного из 12 пациентов, вошедших в С-группу, не отмечено рецидивов, в S-группе – 7 рецидивов из 20 (р = 0,0317). Авторы сделали вывод, что пациенты с метастатической болезнью должны получать Гливек постоянно до прогрессирования либо до наступления непереносимости препарата. Достоверных различий в общей выживаемости между двумя группами не было. Таким образом, окончание приема лекарства даже после длительного успешного применения ассоциируется с быстрым прогрессированием болезни.
Нео- и адъювантная терапия GIST
Первый опыт применения Гливека при GIST во всех странах показал высокую эффективность препарата. Первые протоколы изучали метастатические формы болезни, затем почти одновременно были инициированы исследования по адъювантной и неоадъювантной терапии. К 2011 г. были накоплены данные, показавшие 25%-ную выживаемость за 9 лет при метастатической форме болезни [1] и эффективность трехлетней адъювантной терапии после радикальных операций [28, 29].
Основной целью предоперационной терапии большинство авторов считали уменьшение размеров опухоли. В частности, теоретически оправдана неоадъювантная терапия при локализации GIST в двенадцатиперстной и прямой кишке. Ретроспективный анализ 46 пациентов с первичными и рецидивными опухолями показал, что при применении Гливека в предоперационном режиме увеличивался шанс на полное удаление опухоли [30]. В рамках исследования пациентам выполняли компьютерную томографию каждые 2–3 мес. для оценки степени уменьшения опухоли. Одиннадцать из 46 больных получали терапию до операции с медианой 11,9 мес. При медиане наблюдения 19,5 мес. у 11 больных не наблюдалось признаков заболевания. В другом проспективном исследовании из 36 пациентов у 33 при медиане 11 мес. в предоперационном режиме размер опухоли в среднем уменьшился с 10,5 до 5,5 см. При этом 83% изначально неоперабельных пациентов стали операбельными [31]. Очевидно, что целесообразность применения Гливека до операции должна оцениваться вместе с хирургом и должна учитываться возможность уменьшить размеры опухоли, особенно при ее локализации в непосредственной близости к кардии, в двенадцатиперстной и прямой кишке.
Эффективность применения таргетной терапии при метастатических формах GIST не могла не инициировать исследования по адъювантной терапии GIST. Одно из самых крупных рандомизированных (плацебоконтролируемое, двойное слепое) исследований по адъювантной терапии GIST (ACOSOG Z9001) было проведено в США. С 2002 по 2007 г. в исследовании участвовали 230 клиник США и Канады. Критериями включения были следующие показатели: опухоль более 3 см в диаметре, радикальная (либо R1) операция, возраст пациента более 18 лет, CD117-положительная опухоль. При возникновении рецидива болезни «ослепление» прекращалось и больным из плацебо-группы предлагалось лечение Гливеком. Рандомизация была проведена у 713 пациентов.
В группе лечения Гливеком наблюдалось 30 событий и 5 летальных исходов от разных причин без рецидива. В группе плацебо зафиксировано 70 событий, при этом 69 возвратов болезни, 1 летальный исход без рецидива, 7 летальных исходов после развития рецидива болезни, 5 – от прогрессирования GIST, 2 – от других причин. Надо отметить, что первые промежуточные результаты этого исследования были опубликованы в 2007 г. При этом было установлено, что адъювантная терапия Гливеком в течение года способствовала улучшению показателей безрецидивной выживаемости с 83% в контрольной группе до 97% в группе лечения. Отсутствие различий в общей выживаемости объяснялось коротким сроком наблюдения и назначением терапии Гливеком в случае развития рецидива болезни. В 2009 г. опубликованы результаты трехлетнего наблюдения за больными [32]. Интересно, что даже в группе низкого риска рецидива были зафиксированы случаи рецидивов заболевания.
В группе высокого риска рецидива (опухоли более 10 см в диаметре) адъювантная терапия показала наиболее высокую эффективность, тем не менее после окончания приема Гливека у больных с высоким риском рецидива вновь стали отмечаться рецидивы заболевания. При сравнении с группой исторического контроля адъювантная терапия достоверно улучшала не только безрецидивную, но и общую выживаемость у больных с высоким риском рецидива. Исследователи предположили, что пациентам с высоким риском показана более продолжительная терапия Гливеком в адъювантном режиме. Учитывая высокую эффективность Гливека в улучшении безрецидивной выживаемости, в декабре 2008 г. в США и в 2009 г. в Европе и России препарат одобрен в качестве адъювантной терапии при CD117-положительных гастроинтестинальных стромальных опухолях. Субпопуляционный анализ, представленный на ASCO в 2010 г., показал, что наибольшую выгоду от адъювантной терапии в плане улучшения безрецидивной выживаемости получают больные с мутацией в 11-м экзоне [33].
В европейском исследовании (EORTC 62024) около 900 пациентов после радикальной операции по поводу GIST с высоким и промежуточным риском рецидива заболевания рандомизировали в 2 группы: группа больных, получавших адъювантную терапию 400 мг Гливека в сутки в течение 2 лет, сравнивалась с группой наблюдения, пациенты которой получали плацебо. Кроме того, планировалось изучить общую и безрецидивную выживаемость. Набор больных начался в 2004 г. и закончился в 2008 г. Результаты исследования ожидаются в ближайшее время.
Первичная и вторичная резистентность к Гливеку
В терапии GIST различают первичную и вторичную резистентность. Отсутствие чувствительности к Гливеку в течение первых 6 мес. считается первичной резистентностью. Она отмечается приблизительно у 10% больных. Первые же протоколы анализировали эффективность терапии в зависимости от мутационного статуса. Было отмечено, что тип мутации влияет на первичную резистентность: при мутации в 11-м экзоне – 5%, в 9-м экзоне – 16%, при диком типе – 2–3% [34]. В то же время относительная резистентность опухолей с мутацией в 9-м экзоне может быть связана с недостаточной дозой Гливека [20]. Исследования in vitro и клинические результаты показали, что больные с мутацией D842V абсолютно не чувствительны к Гливеку и в случае метастатической болезни имеют плохой прогноз. Пациенты с другими мутациями в гене PDGFRA в большинстве случаев чувствительны к терапии и демонстрируют продолжительный эффект на Гливеке. Поскольку при диком типе найдены иные механизмы прогрессии, то предполагается, что эффект может наступить от других таргетных препаратов: ингибиторов VEGFR – для педиатрических GIST, сукцинатдегидрогеназы – для SDH-мутантных, BRAF – для BRAF-мутантных GIST [35].
Несмотря на высокую эффективность терапии Гливеком при отсутствии первичной резистентности, у подавляющего большинства больных развивается вторичная резистентность к препарату. Вторичные мутации развиваются в том же самом гене, что и первичные мутации. При диком типе вторичные мутации не выявляются. Большинство вторичных мутаций KIT представлены точечными заменами одного нуклеотида, которые локализуются в тирозинкиназных доменах KIT и PDGFRA и в киназ-связывающих доменах KIT (15-й и 16-й экзоны). Как правило, вторичные мутации отличаются от первичных. Эти мутации могут определяться как единичные или как несколько клонов со специфической мутацией в одном опухолевом узле. С клинической точки зрения важно отметить, что количество вторичных мутаций может достигать 7 у одного пациента в различных метастазах [36]. Выраженная гетерогенность опухолевых клонов заставила отказаться от пункции метастазов при прогрессировании опухоли на фоне Гливека с целью определения «мишени» при прогрессировании. При диком типе привлекательной мишенью может быть IGF1R, гиперэкспрессия которого наблюдается при данном виде GIST и не зависит от KIT-мутации [37].
Препараты второй линии терапии и новые подходы
Для пациентов с первичной или вторичной резистентностью к Гливеку единственным препаратом второй линии лечения является сунитиниб (Сутент). Сунитиниб – пероральный ингибитор тирозинкиназ с эффективностью в отношении KIT, рецептора сосудистого фактора роста (VEGFR), тромбоцитарного фактора роста (PDGFR), рецептора макрофагального колониестимулирующего фактора (CSF-1R). Исследования ранних фаз по применению Сутента показали эффективность в отношении GIST с большинством видов мутаций [38]. Последние данные показали, что медианы до прогрессирования и общей выживаемости были дольше у пациентов с мутацией в 9-м экзоне и диким типом по сравнению с пациентами с мутацией в 11-м экзоне [39]. В крупном рандомизированном исследовании III фазы изучалась эффективность Сутента в сравнении с плацебо среди 312 пациентов с первичной резистентностью или непереносимостью Гливека.
Медиана времени до прогрессирования (TTP) составила 27,3 недели для пациентов, получающих Сутент, и 6,4 недели для группы плацебо (p < 0,0001). В лечебной группе наблюдалось 7% частичных ответов, 58% стабилизаций и достоверно чаще объективный ответ по сравнению с группой плацебо. Результаты исследования послужили основой для одобрения Сутента в качестве препарата второй линии при прогрессировании после Гливека или первичной непереносимости иматиниба [40]. Одним из существенных негативных отличий Сутента от Гливека явилась более выраженная токсичность, проявляющаяся лейкопенией и тромбоцитопенией у пациентов. Эффективность Сутента для больных с мутацией в 13-м и 14-м экзонах была выше, чем с мутацией в 17-м экзоне [39].
P. Rutkowski и соавт. в недавнем сообщении представили данные одноцентрового исследования, включавшего 137 пациентов с метастатической формой GIST Сутентом после прогрессирования на фоне терапии Гливеком [41]. Одногодичная выживаемость больных составила 42%. Общая выживаемость, рассчитанная от начала приема Гливека и Сутента, составила соответственно 74 недели и 51 месяц. Лучший ответ на терапию Сутентом отмечен у больных с мутацией в 9-м экзоне и при диком типе (медиана выживаемости 65,5 и 50,5 недели) по сравнению с мутацией в 11-м экзоне или PDGFRA (36,8 и 9 недель соответственно). Кроме того, вторым независимым фактором прогноза, достоверно влияющим на безрецидивную и общую выживаемость в одно- и многофакторном анализе, явилась артериальная гипертензия: пациенты, у которых фиксировался данный побочный эффект терапии Сутентом, жили достоверно дольше.
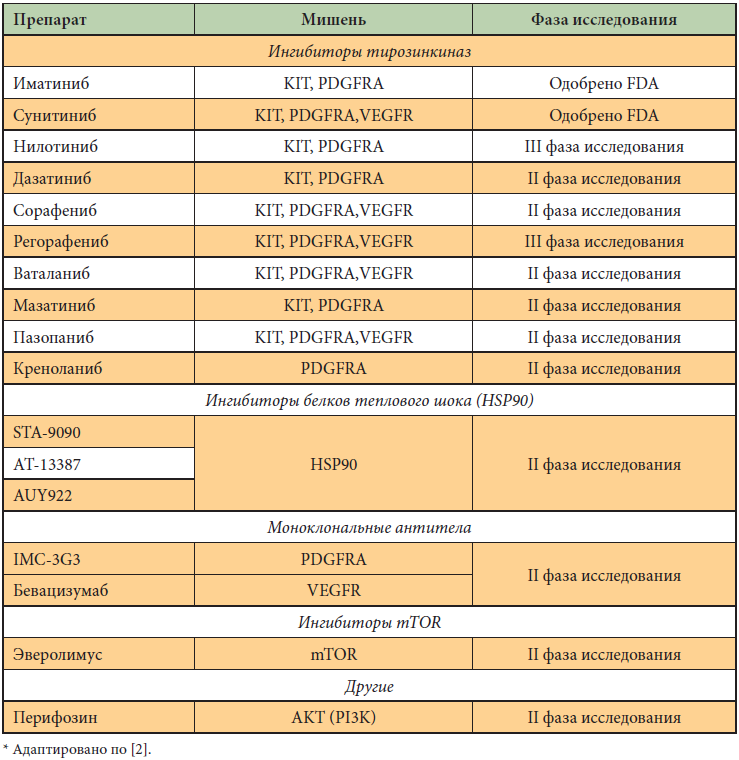
Другие таргетные препараты, ингибирующие тирозинкиназы (но не VEGFR), – мазатиниб и дазатиниб. Ингибиторы тирозинкиназ, активные не только в отношении KIT и PDGFR, но и подавляющие VEGFR: сорафениб, мотезаниб, ваталаниб – проходят исследования второй фазы. Бевацизумаб – моноклональное антитело, блокирующее VEGFR, – изучается в качестве дополнения к Гливеку в III фазе исследования. Изучаются и лекарства с непрямым механизмом действия – ингибиторы белка теплового шока (IPI-504) и механизма PI3K/AKT/mTOR (эверолимус). Многообразие вышеуказанных таргетных препаратов говорит о следующем. Несмотря на эффективность одного таргетного препарата (Гливек), рано или поздно опухоль находит новые пути развития. Возникает необходимость блокирования этих путей другими таргетными агентами (табл. 2). Поскольку потенциальных путей «ускользания» заболевания достаточно много, список возможных лекарств также расширяется, либо нужно отдавать предпочтение препарату с множественным механизмом действия, но тогда понятие «таргетная терапия» в известной мере размывается. Кроме того, множественный механизм действия неизбежно ведет за собой более тяжелые побочные эффекты препарата.
Заключение
Десятилетний опыт применения таргетной терапии гастроинтестинальных стромальных опухолей продемонстрировал небывалый успех в онкологии. Впервые в диагностике и лечении солидных опухолей удалось не только выявить ключевой механизм патогенеза, но и подобрать высокоточное «оружие» к «мишени» (англ. target – мишень). Применение Гливека улучшило выживаемость больных с метастатической формой болезни в три раза. Предоперационная терапия позволила улучшить результаты лечения при местнораспространенных формах. Адъювантная терапия снизила риск рецидива и увеличила общую выживаемость пациентов. Стромальные опухоли обладают выраженной гетерогенностью по мутационному статусу, который является фактором прогноза и, по возможности, должен выполняться всем больным. Развитие вторичных мутаций и прогрессирование на фоне лечения препаратом первой линии диктуют необходимость изменения тактики лечения и поиска новых путей блокады опухолевой прогрессии.