Трихогепатоэнтеральный синдром
- Аннотация
- Статья
- Ссылки
- English
Распространенность
трихогепатоэнтерального синдрома
Трихогепатоэнтеральный синдром (синдромная, или фенотипическая, диарея) был впервые описан в 1982 г. L. Stankler и соавт. у двух сиблингов с низким весом при рождении, дисморфичными чертами лица, плоским широким носом, выступающим лбом, тяжелой диареей [1]. Значительная часть опубликованных в мировой литературе клинических случаев собрана и описана французскими врачами в 2007 и 2013 гг. [2, 3]. В настоящее время представлено всего около 80 случаев данного заболевания. Его распространенность составляет около 1:1 000 000 новорожденных [4, 5]. В 2015 г. были приведены первые два описания трихогепатоэнтерального синдрома в России [6].
Генетическое консультирование
Трихогепатоэнтеральный синдром – орфанное, наследуемое по аутосомно-рецессивному типу генетическое заболевание, связанное с мутациями в двух генах: TTC37 (5q15) или SKIV2L (6p21.3) – компонентах гипотетического ответственного за экзосомопосредованный контроль РНК Ski-комплекса человека [7–10]. Синдром может проявляться широким спектром клинических симптомов в неонатальном периоде. С учетом этого секвенирование полного экзома считается единственным достоверным диагностическим инструментом в его определении [8, 10].
При зачатии между гетерозиготными носителями данной мутации каждый сибс имеет 25%-ную вероятность родиться с данным синдромом, 50%-ную вероятность – быть бессимптомным носителем и 25%-ную – быть незатронутым и не быть носителем. После того как патогенные варианты идентифицированы у пострадавшего члена семьи, возможны тестирование родственников, находящихся в группе риска, пренатальное тестирование на беременность с повышенным риском и генетическое тестирование на предымплантацию [5].
Клинические характеристики
Трихогепатоэнтеральный синдром характеризуется следующими основными симптомами [6]:
- задержка внутриутробного развития плода;
- хроническая диарея, приводящая к значительным кишечным потерям и тяжелой гипотрофии;
- аномалия роста волос (сухие и ломкие, склонны к быстрому выпадению);
- лицевой дисморфизм (выступающие лоб и щеки, широкое основание носа, широкая переносица);
- вторичный иммунодефицит (снижение продукции иммуноглобулинов всех классов);
- задержка психомоторного развития как следствие тяжелой гипотрофии;
- кожные аномалии (гипо- и гиперпигментация) [7];
- тромбоцитоз (редко) [6, 8].
При эндоскопическом исследовании верхних отделов желудочно-кишечного тракта обнаруживают изменения, характерные для синдрома мальабсорбции [3, 6]. Редки случаи, когда заболевание имеет кроноподобное течение [6].
Дифференциальная диагностика
Дифференциальную диагностику следует проводить с другими синдромами врожденной хронической диареи, прежде всего врожденной тафтинговой энтеропатией, IPEX-синдромом (I – иммунная дисрегуляция, P – полиэндокринопатия, E – энтеропатия, X – сцепленная с X-хромосомой), первичными иммунодефицитами с кишечным синдромом [5].
Особое внимание уделяют синдрому мальабсорбции, вызванному непереносимостью глютена и лактозы [8].
Лечение
Лечение включает в себя парентеральное питание и в ряде случаев внутривенное введение человеческого иммуноглобулина [4].
Дети, не получающие парентерального питания, должны находиться под постоянным наблюдением педиатра-диетолога и гастроэнтеролога для обеспечения парентерального питания в случае, когда дефицит питательных веществ станет выраженным [4]. Кроме того, необходимо ежегодно проводить [5]:
- ультразвуковое исследование брюшной полости и почек;
- измерение ферментов печени (аспартатаминотрансфераза, аланинаминотрансфераза, гамма-глутамилтранспептидаза), общего билирубина;
- определение уровней иммуноглобулинов классов G, M, A в сыворотке крови;
- оценку когнитивного развития, речи, а также психосоциальных навыков в возрасте 2, 4, 8, 12 и 15 лет.
Поскольку эволюция дерматологических особенностей (в основном гипо- и гиперпигментных пятен и аномалий роста волос) неизвестна, целесообразна также ежегодная консультация дерматолога [5].
Клинический случай
Мальчик от третьей беременности, протекавшей на фоне заболеваний матери (хроническая артериальная гипертензия, миома матки). На 30-й неделе гестации выявлены врожденный порок сердца (ВПС) (коарктация аорты) и врожденный порок развития почек (гидронефроз). У ребенка отягощенный анамнез: старший сибс в семье – девочка страдает трихогепатоэнтеральным синдромом, имеет нуклеотидный вариант с.2164С>Т в гомозиготном состоянии в гене SKIV2L, родители пробанда – гетерозиготные носители варианта с.2164С>Т.
Роды на 36-й неделе с помощью операции экстренного кесарева сечения (преэклампсия у матери). Масса тела при рождении – 2140 г, длина тела – 47 см, окружность головы – 32 см, окружность грудной клетки – 26 см. Оценка по шкале Апгар – 7/8 баллов. Состояние ребенка с рождения оценивалось как тяжелое, обусловленное основным заболеванием, морфофункциональной незрелостью, ВПС. Через пять дней после стабилизации состояния ребенка перевели из областной больницы в Федеральный центр сердечно-сосудистой хирургии им. С.Г. Суханова (Пермь) для оперативного лечения – резекции коарктации аорты с расширенным анастомозом «конец в конец».
Через четыре дня после операции мальчик транспортирован в больницу по месту жительства для дальнейшего наблюдения.
При госпитализации состояние тяжелое из-за сердечной недостаточности 2-й степени, дыхательной недостаточности 2-й степени и стридора, кислородной зависимости, энтеропатии, выраженной неврологической симптоматики на фоне возбудимости. Ребенок на зондовом кормлении, срыгивает.
В анализах крови – тромбоцитопения, анемия, выраженное повышение уровня щелочной фосфатазы, умеренный цитолиз.
Данные кардиографии: состояние после коррекции ВПС.
Проведены инфузионная терапия, постепенное расширение энтерального питания под контролем переносимости.
В Сеченовский центр материнства и детства ребенок поступил на втором месяце жизни для проведения обследования, постановки диагноза и расширения объема энтерального питания.
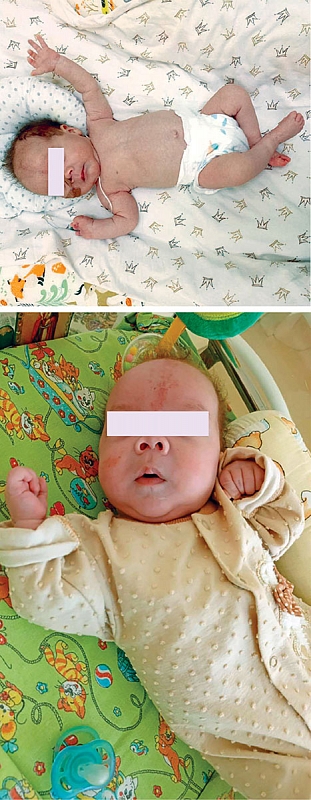
В объективном статусе – тяжесть состояния, низкая масса тела (2750 г), обильные срыгивания (до 15–20 мл) после каждого кормления, выраженный кишечный синдром (жидкий стул до 7–9 раз в сутки), живот при пальпации вздут, чувствителен по ходу кишечника. Фенотипически отмечались тонкие волосы на голове с участками гипертрихоза, гипоплазия надбровных дуг, длинный уплощенный фильтр.
При госпитализации состояние ребенка расценивалось как тяжелое: в анализе крови – умеренный метаболический ацидоз, гипопротеинемия, гипоальбуминемия, тромбоцитопения до 165 × 109/л, анемия средней степени.
С учетом тяжести основного заболевания, выраженности гипопротеинемии и гипоальбуминемии, раннего возраста, недоношенности установлен периферический венозный катетер, затем введен человеческий иммуноглобулин. С целью коррекции волемического объема выполнена инфузионная терапия. В силу отягощенного анамнеза, раннего возраста, умеренного нарастания маркеров воспаления, вероятности врожденной энтеропатии в целях деконтаминации показано назначение антибактериальной (ванкомицин, амикацин), а также противогрибковой терапии (флуконазол).
На фоне сохранения кишечных потерь, нарастания анемии и интоксикации объем энтерального питания уменьшен до трофического, установлен центральный венозный катетер для налаживания частичного парентерального питания. Консультирован трансфузиологом (выполнена гемотрансфузия).
После проведенной терапии отмечалась стойкая положительная динамика: ребенок гемодинамически стабилен, купирована интоксикация, достигнута нормализация показателей анализа крови, уменьшились объемы кишечных потерь (стул до четырех-пяти раз в сутки, кашицеобразный). За шестинедельный период госпитализации вес увеличился на 1130 г.
Консультация генетика: с учетом отягощенного клинико-генеалогического анамнеза (старший сибс в семье – девочка страдает данным заболеванием, имеет нуклеотидный вариант с.2164С>Т в гомозиготном состоянии в гене SKIV2L), клинической картины (особенности лицевого фенотипа, поперечные ладонные борозды, ВПС, коарктация аорты, гидронефроз, кислородзависимость в анамнезе, энтеропатия) и результатов проведенного молекулярно-генетического исследования (выявлен нуклеотидный вариант chr63 1933752C>T (с.2164C>T, NM_000006.11) в гомозиготном состоянии в гене SKIV2L) у ребенка диагностирован трихогепатоэнтеральный синдром, тип 2 (OMIM 614602). Диагноз подтвержден результатами молекулярно-генетических исследований. Заболевание имеет аутосомно-рецессивный тип наследования. Риск повторного рождения у родителей ребенка с данным заболеванием – 25%. Рекомендовано проведение пренатального генетического тестирования.
В силу особенности течения трихогепатоэнтерального синдрома и потребности ребенка в частичном парентеральном питании в отделении стационара проводится обучение родителей по уходу и выхаживанию ребенка в домашних условиях. Состояние ребенка остается стабильным: мальчик гемодинамически стабилен, прибавляет в весе, объем энтерального питания увеличен до 40–45 мл каждые три часа, усваивает, находится на частичном парентеральном питании (фото).
Заключение
Прогноз при трихогепатоэнтеральном синдроме в целом неблагоприятный. Обычно смерть наступает в раннем возрасте в результате печеночной недостаточности, рецидивирующей инфекции или осложнений, связанных с длительным парентеральным питанием [2, 8]. Парентеральное питание и лечение рецидивирующей инфекции могут положительно повлиять на выживаемость детей с трихогепатоэнтеральным синдромом. Именно поэтому осведомленность врачей-педиатров, неонатологов, гастроэнтерологов об этом синдроме позволяет обеспечить его раннюю диагностику, выбор адекватной тактики ведения пациентов и надлежащее лечение на ранних стадиях.
Конфликт интересов. Авторы заявляют об отсутствии конфликта интересов.
Финансирование. Работа выполнена без спонсорской поддержки.
Соблюдение прав пациентов и правил биоэтики. Родители ребенка не возражают против публикации данных из истории болезни и фото ребенка.
K.A. Kazakova, PhD1, M.A. Varichkina, G.S. Golosnaya, PhD, Prof., G.V. Galstyan, L.G. Khachatryan, PhD, Prof., Ye.K. Kulikova
I.M. Sechenov First Moscow State Medical University
N.N. Burdenko Voronezh State Medical University
Contact person: Klavdiya A. Kazakova, kazakova_k_a@staff.sechenov.ru
Tricho-hepato-enteric syndrome is a rare genetic disease with a leading intestinal syndrome caused by impaired differentiation and polarization of intestinal enterocytes. A disease that develops due to mutations in the genes TTC37 (5q15) or SKIV2L (6p21.3), characterized by intrauterine fetal growth retardation, severe chronic diarrhea with onset in infancy, specific facial features, hair growth features and secondary immunodeficiency. A clinical case of trichohepatoenteral syndrome diagnosed in a child at an early age is presented. In the Russian medical literature, this syndrome is described in sibling.
Уважаемый посетитель uMEDp!
Уведомляем Вас о том, что здесь содержится информация, предназначенная исключительно для специалистов здравоохранения.
Если Вы не являетесь специалистом здравоохранения, администрация не несет ответственности за возможные отрицательные последствия, возникшие в результате самостоятельного использования Вами информации с портала без предварительной консультации с врачом.
Нажимая на кнопку «Войти», Вы подтверждаете, что являетесь врачом или студентом медицинского вуза.