Патофизиология трофологических нарушений при первичном билиарном холангите
- Аннотация
- Статья
- Ссылки
- English

1. Введение
Состояние недостаточного питания часто наблюдается у пациентов с первичным билиарным холангитом (ПБХ). Недостаточность питания (НП), сопутствующая этому заболеванию, ухудшает его течение, прогноз и качество жизни больного, отрицательно влияет на исход заболевания и чаще всего распознается только на поздних стадиях [1]. Этиология и патофизиология НП при ПБХ многофакторна [2]. Нарушение процессов желчевыделения при ПБХ, сопровождающееся холестазом и снижением функции гепатоцитов, влияет на метаболизм как макронутриентов, так и микронутриентов и зависит от стадии заболевания. Для своевременной диагностики и коррекции нарушений трофологического статуса важно понимание, когда, на какой стадии заболевания и с помощью каких механизмов развивается энергетическая, белковая, витаминная и минеральная НП у пациентов с ПБХ. Для улучшения результатов лечения пациентов с ПБХ необходимо уделять внимание проблеме развития у них НП и вопросам ее профилактики, лечения уже на ранних стадиях заболевания. Поздние, терминальные стадии ПБХ сопровождаются дисбалансом между процессами катаболизма и анаболизма с преобладанием первых над вторыми. Целями нутриционной терапии пациентов с холестатическими заболеваниями печени являются поддержка регенерации печени, профилактика или коррекция НП, а также профилактика и/или лечение возникающих при этом осложнений заболевания печени. Очень важно уже при первичном осмотре пациентов акцентировать внимание не только на специфических признаках заболевания, но и на оценке статуса питания. При этом должны учитываться особенности и механизмы нарушения метаболических процессов на разных стадиях ПБХ, чтобы своевременно распознать НП и учесть известные научные данные при проведении базисного лечения у этих пациентов.
2. Определение НП
В отечественной литературе нет общепринятого термина для определения статуса питания [3]. НП (синонимы: белково-энергетическая, нутритивная, трофологическая недостаточность, гипотрофия) – патологическое состояние, обусловленное несоответствием (дисбалансом) поступления и расхода органических питательных веществ, энергии, макро- и микронутриентов, приводящим к снижению массы тела, измеримому отрицательному изменению компонентного состава организма, сопровождающемуся нарушением его функционирования и ухудшением клинического исхода [4, 5]. Согласно определению Всемирной организации здравоохранения, недостаточность питания (malnutrition) является результатом недостаточного потребления или усвоения питательных веществ, необходимых для стимулирования роста и предотвращения хронических или острых заболеваний, и часто характеризуется задержкой роста, истощением, недостаточным весом и дефицитом питательных микроэлементов [6]. НП сопровождается потерей массы тела, снижением физической работоспособности, ухудшением самочувствия, а также вызывает серьезные нарушения в обмене веществ, ослабление иммунной защиты и эндокринные дисфункции [7, 8].
3. Распространенность недостаточности питания при заболеваниях печени
Истинную распространенность НП оценить очень сложно по следующим причинам [5]:
- крайне низкое внимание врачей к состоянию трофологического статуса;
- трудности в оценке НП, маскирование потерь мышечной ткани на фоне избытка жировой массы и задержки жидкости.
Около двух миллиардов человек в мире страдают от различных форм НП [9]. У пациентов с различными заболеваниями отмечается вторичная (эндогенная) НП. Исследования показывают, что НП наблюдается у 20–80% пациентов с заболеваниями печени в зависимости от клинической стадии заболевания [10]. Практически любое хроническое заболевание может привести к прогрессирующей потере массы тела. Исследование, проведенное Carvalho и Parise, показало, что до 75% пациентов с хроническими заболеваниями печени имеют различную степень НП [11]. Недостаточное питание и саркопения часто встречаются у пациентов с хроническими заболеваниями печени и связаны с повышенным риском декомпенсации, инфекций, а также зачастую являются независимым фактором риска смертности этих пациентов и ухудшения результатов лечения после трансплантации печени [2, 12]. Важно отметить, что частота трофологической недостаточности у этих пациентов увеличивается по мере прогрессирования заболевания. В исследованиях 90-х годов прошлого столетия оценка статуса питания пациентов с циррозом печени различной этиологии и различной степенью печеночной недостаточности [13, 14] позволила прийти к консенсусу о том, что НП распознается при всех формах цирроза [15] и, по оценкам различных авторов, составляет от 40 до 100% [16–19]. Отмечается высокая распространенность недостаточного питания у лиц с декомпенсированным циррозом печени. У пациентов с циррозом печени и тяжестью состояния, оцениваемой по шкале Чайлд-Пью A, уровень НП составляет 46% по сравнению с 84 и 95% пациентов с Чайлд-Пью B и Чайлд-Пью C соответственно [11]. У пациентов, включенных в список на трансплантацию печени, НП может достигать 100% случаев [20, 21].
4. Первичный билиарный холангит и недостаточность питания
НП развивается у пациентов с ПБХ как с установленным циррозом, так и без него [22, 23]. По данным Wicks и соавт., НП обнаруживается у 33% пациентов с различными стадиями ПБХ [22]. Первичный билиарный холангит (Primary Biliary Cholangitis – PBC; МКБ-10: K.74.3; МКБ-11 (бета-версия): DB37.2) – заболевание, которое ранее (до 2015 г.) было известно как первичный билиарный цирроз (ПБЦ, Primary Biliary Cirrhosis – PBC) [24], − это хроническое холестатическое прогрессирующее заболевание печени аутоиммунного генеза, протекающее с деструкцией, некрозом и апоптозом эпителия преимущественно внутридольковых и септальных желчных протоков, в терминальной стадии которого развивается цирроз печени [25, 26]. ПБХ характеризуется Т-клеточным опосредованным разрушением эпителиальных клеток, выстилающих мелкие внутрипеченочные желчные протоки [27]. Это ведет к дуктулопении и персистирующему холестазу с развитием в терминальной стадии цирроза с печеночно-клеточной недостаточностью [26].
Заболевание на ранней стадии может протекать бессимптомно или с неспецифическими симптомами, такими как слабость, утомляемость, снижение работоспособности, анорексия/гипорексия и недомогание, которые легко спутать с другими заболеваниями. И в этот период НП, обусловленная самим заболеванием, как правило, практически незаметна, так как нет значительных повреждений печеночных клеток, участвующих в обменных процессах. На ранних стадиях ПБХ происходит незначительное снижение потребления энергии, не приводящее к клинически выраженной белково-энергетической недостаточности, но потенциально поддающаяся изменению НП в этот период уже имеется [28–30].
По мере прогрессирования холестаза избыточные желчные кислоты оказывают хроническое (непрерывное) агрессивное воздействие на паренхиму печени, что проявляется развитием постепенно нарастающей печеночно-клеточной недостаточности. Также снижается и трофологический статус пациентов с ПБХ по мере прогрессирования заболевания, что отчасти связано со значительным уменьшением потребления энергии [22]. У пациентов с ПБХ на поздних стадиях развивается цирроз печени, который может сопровождаться асцитом, портальной гипертензией, кровоизлиянием из варикозно расширенных вен пищевода/желудка и печеночной энцефалопатией (ПЭ) [31]. Портальная гипертензия может развиться у пациентов с холестатическими заболеваниями печени до установления цирроза [32, 33]. Существует почти прямая взаимосвязь между тяжестью заболевания печени и степенью НП [30]. Состояние питания при этом нарушено вторично по отношению к симптомам заболевания [33]. Тяжелая белково-энергетическая недостаточность чаще развивается и наблюдается при далеко зашедших и терминальных стадиях ПБХ, как правило, у пациентов, страдающих этим заболеванием более одного десятилетия [29], и при снижении общего количества функционирующих гепатоцитов до 25% и менее [34]. Трофологическая недостаточность становится более легко выявляемой при развитии у пациентов с ПБХ тяжелого цирроза с асцитом.
5. Патогенез недостаточности питания при первичном билиарном холангите
Причины и механизмы, приводящие к НП и потере массы тела у пациентов с ПБХ, многофакторны и могут быть разделены на три группы:
- недостаточное поступление нутриентов;
- нарушения переваривания и всасывания (синдромы мальдигестии и мальабсорбции);
- повышенная скорость метаболизма (ускоренный катаболизм).
5.1. Недостаточное поступление нутриентов у пациентов с первичным билиарным холангитом
На ранних стадиях ПБХ изменения трофологического статуса связаны с повышенным содержанием желчных кислот в плазме крови этих пациентов, что приводит к появлению раннего и чаще всего единственного в течение нескольких месяцев и даже лет патогномоничного симптома болезни – локального или диффузного (распространенность), умеренного или резко выраженного (выраженность), постоянного или волнообразного (продолжительность) кожного зуда [26, 35]. Причиной возникновения кожного зуда считается отложение в эпидермисе желчных кислот, содержащихся в повышенном количестве в крови пациентов с ПБХ уже в асимптоматической и начальной стадии заболевания, задолго до развития у них желтухи. При этом в крови увеличиваются все фракции конъюгированных желчных кислот.
В ответ на избыточное содержание желчных кислот в плазме крови пациентов с ПБХ организм больного пытается вывести из общего кровотока токсичные, обладающие детергентными свойствами желчные кислоты через почки и кожу. В коже при этом обнаруживают 50–85% желчных кислот, неконъюгированных с глицином или таурином, и менее 20% желчных кислот находят в виде сульфоэфиров [25, 26, 36]. Кожный зуд более выражен в ночное время и часто усиливается при соприкосновении с тканями, а также в тепле. Зуд не снимается симптоматическими (антигистаминными, седативными) препаратами, часто вызывает мучительную бессонницу, изменение эмоционального состояния, тревогу, депрессию [24]. Все это приводит к снижению аппетита и недостаточному поступлению пищевых ингредиентов, сопровождающимся повышением гликогенолиза и снижением гликогеногенеза.
Гликогенолиз – биохимический процесс расщепления гликогена до глюкозы, осуществляется главным образом в печени и мышцах. Основная задача гликогенолиза – поддержание постоянного уровня глюкозы в крови для обеспечения организма энергией. Запасы гликогена в процессе гликогенолиза в печени обеспечивают до 75% потребности организма в глюкозе, основном быстро используемом для пополнения энергии субстрате. Гликогеногенез – это метаболический путь синтеза гликогена из глюкозы. Этот процесс происходит во всех тканях, однако в основном он имеет место в печени и мышцах. «Стартовой точкой» гликогеногенеза служит глюкозо-6-фосфат, образованный из глюкозы в ходе реакции, катализируемой глюкокиназой в печени и гексокиназой в мышцах. Как известно, гликоген печени используется в качестве энергетического материала всеми тканями и органами. При этом гликоген мышц используется ими в качестве энергетического материала исключительно для своих нужд.
Представленные Green и соавт. данные указывают на то, что уже на начальных стадиях ПБХ в печени постепенно происходит снижение запасов гликогена, связанное с повышением гликогенолиза и снижением гликогеногенеза [37]. Авторы убедительно показали, что у пациентов с ПБХ значительно (вплоть до нуля) падает активность глюкокиназы, что свидетельствует о снижении образования гликогена в печени [37]. При этом гексокиназа (производит фосфорилирование гексоз), отвечающая за синтез гликогена преимущественно в мышцах, у пациентов с ПБХ в этот период достоверно увеличивается по сравнению со здоровыми лицами [37].
Мучительная бессонница, изменение эмоционального состояния, тревога, депрессия, развивающиеся уже на ранних стадиях заболевания, способствуют снижению запасов гликогена и развитию постепенно нарастающей энергетической недостаточности (снижение количества глюкозы, используемой в качестве энергетического субстрата) с клиническими проявлениями выраженной слабости, быстрой утомляемости, снижения работоспособности, функционального статуса и качества жизни пациентов с ПБХ [24, 33, 35, 38–40]. При этом астенический синдром у пациентов с ПБХ более выражен, чем при других хронических заболеваниях печени [24]. Имеются данные, что в механизме развития усталости важную роль играют ароматические аминокислоты тирозин, фенилаланин и триптофан, которые у пациентов с ПБХ содержатся в крови в повышенном количестве [39, 41, 42].
Итак, на ранних стадиях ПБХ незаметно развивается изменение трофологического статуса в виде энергетической НП, которая длительное время проявляется лишь общей слабостью и/или снижением работоспособности [40, 43, 44].
Развивающееся нарушение процессов желчевыделения (накопление желчных кислот в крови) у пациентов с ПБХ уже в асимптоматической и ранней стадиях заболевания способствует развитию энергетической НП, что требует включения в рацион этих пациентов продуктов с повышенной калорийностью.
Даже незначительный дефицит нутриентов, сопровождающийся постепенно нарастающим гликогенолизом и снижением гликогеногенеза, приводит к включению механизмов компенсации, которые призваны защитить жизненно важные органы, нуждающиеся в повышенном потреблении энергии (мозг, сердечная мышца, эритроциты и др.), от дефицита энергии путем перераспределения пластических и энергетических ресурсов [5]. Это приводит к мобилизации энергоресурсов жировой ткани и потреблению в качестве энергетического материала жирных кислот. Последние становятся важными субстратами для выработки энергии. Вследствие ускорения процессов β-окисления жирных кислот наблюдается прогрессирующее уменьшение запасов жира в организме пациентов с ПБХ [45, 46]. Активация этих процессов происходит по мере нарастания холестаза.
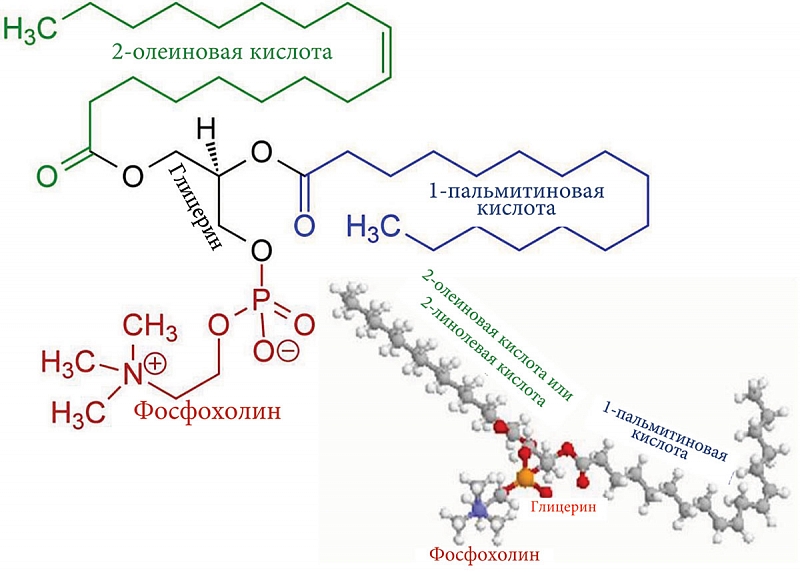
Наряду с этим у пациентов с ПБХ наблюдается повышенный уровень пальмитиновой и олеиновой жирных кислот [37]. Последние являются основными компонентами фосфолипидов желчи (рис. 1), участвующих в образовании мицеллярно-ламеллярных структур, состоящих из фосфолипидов, холестерола и желчных кислот. Содержание в плазме крови пациентов с ПБХ пальмитиновой и олеиновой кислот, а также фосфолипидов и холестерола повышается уже на ранних стадиях заболевания и направлено на нейтрализацию детергентного действия избыточных желчных кислот, попадающих в общий кровоток по мере нарастания холестаза [26, 32]. Холестерол, фосфолипиды, пальмитиновая и олеиновая кислоты могут попадать в общий кровоток пациентов с ПБХ за счет увеличения их синтеза в печени и попадания компонентов желчи в кровь в результате развивающегося холестаза.
Повышенный синтез фосфолипидов требует повышенного количества не только пальмитиновой и олеиновой жирных кислот, но и ортофосфата. В связи с этим уже на начальных стадиях развития заболевания в плазме крови пациентов с ПБХ выявляется умеренное повышение активности печеночной фракции щелочной фосфатазы (ЩФ) и 5’-нуклеотидазы (5’-НК), что указывает на изменения в обмене фосфора [26]. Эти ферменты участвуют в гидролизе фосфомоноэфиров с образованием ортофосфата, необходимого в качестве основного компонента для биосинтеза фосфолипидов, которые в свою очередь необходимы для нейтрализации повышенного содержания желчных кислот в плазме крови пациентов с ПБХ.
Длительное повышенное содержание холестерола в плазме крови пациентов с ПБХ в результате увеличения его синтеза в печени может приводить к появлению ксантелазм – одиночных или множественных слегка приподнятых над кожей бледно-желтого цвета образований. Повышение уровня холестерола у пациентов с ПБХ, так же как увеличение содержания фосфолипидов, направлено на нейтрализацию детергентного действия желчных кислот, попавших в общий кровоток. При этом у пациентов с ПБХ, несмотря на увеличение у них общего холестерола в плазме крови, выявляются низкая степень стеатоза печени, низкий риск развития атеросклероза и сердечно-сосудистых событий [48].
Развивающееся нарушение процессов желчевыделения (накопление желчных кислот в плазме крови) у пациентов с ПБХ уже на ранних стадиях заболевания приводит к изменениям в метаболизме жиров, которые направлены на компенсацию развивающейся энергетической недостаточности (ускоренное β-окисление жирных кислот) и на нейтрализацию детергентного действия избыточных желчных кислот (повышенный синтез фосфолипидов и холестерола). Поэтому на ранних стадиях заболевания стандартные пищевые продукты, как правило, нормально переносятся пациентами с ПБХ, и им не требуется назначение диеты с низким содержанием холестерола. Возможно включение в рацион продуктов с повышенным содержанием фосфора для поддержания достаточного синтеза фосфолипидов. Диеты с низким содержанием жира для уменьшения ксантелазм признаны неудачными и даже вредными [49].
5.2. Нарушения переваривания и/или всасывания (синдромы мальдигестии и мальабсорбции)
Внутрипеченочный холестаз при ПБХ – это многофакторный процесс, который связан с повреждением субклеточных структур в эпителиоцитах внутрипеченочных желчных протоков и изменением метаболизма желчных кислот вследствие нарушения процессов желчевыделения. Недостаточное поступление желчных кислот в просвет кишечника у пациентов с ПБХ приводит к снижению скорости процессов гидролиза жиров и недостаточному всасыванию в тонком кишечнике жиров и жирорастворимых витаминов (А, D, Е, K). Это способствует прогрессированию НП за счет стеатореи (потеря жира с фекалиями более 7 г/сут) и постепенному развитию витаминно-минеральной недостаточности [25, 26]. Тяжесть стеатореи коррелирует со снижением продукции и концентрации желчных кислот (r = 0,82; р < 0,0001), уровнем повышения сывороточного билирубина (r = 0,88; р < 0,001) и поздними гистологическими стадиями ПБХ (р < 0,005) [52]. Все пациенты с уровнем общего билирубина в сыворотке крови более чем 4,5 мг/дл имеют тяжелую стеаторею (потеря жира с фекалиями составляет более 25 г/сут) [26, 50].
Механизм развития стеатореи [51] связан с недостаточным эмульгированием жиров в результате сниженного поступления желчных кислот в просвет кишечника. При этом процессы гидролиза жиров панкреатическими липазами не нарушены. Результаты, полученные E. Ros и соавт., указывают на то, что функция поджелудочной железы, как правило, сохранена и не является причиной развития стеатореи при ПБХ [52]. Активность ЩФ в сыворотке крови пациентов с ПБХ не коррелирует с тяжестью стеатореи, а активность амилазы поджелудочной железы находится в пределах нормы [52]. Эмульгирование жиров необходимо для увеличения площади соприкосновения субстрата с ферментами липазами.
Уменьшение процессов эмульгирования жиров способствует снижению скорости гидролиза жиров, что приводит к неполному их перевариванию в процессе продвижения по кишечнику и постепенному развитию стеатореи.
Кроме эмульгирования жиров желчные кислоты участвуют в абсорбции гидролизованных жиров и жирорастворимых витаминов. Жирные кислоты и моноглицериды, образующиеся из нейтральных жиров и фосфолипидов с участием желчных кислот и под воздействием липаз, в верхних отделах тонкой кишки подвергаются всасыванию энтероцитами в виде эмульсии липоидно-желчных комплексов (рис. 2).
Желчные кислоты, являясь сильными детергентами, образуют с жирными кислотами и моноглицеридами мицеллярные или ламеллярные структуры для абсорбции энтероцитами [25, 26]. Внутри энтероцитов комплексы распадаются, и жирные кислоты с моноглицеридами остаются в энтероцитах (используются клеткой в качестве строительного, энергетического материала или включаются в хиломикроны), а желчные кислоты снова выходят в просвет кишечника и принимают участие в эмульгировании жиров и в образовании новых липоидно-желчных комплексов для доставки жирных кислот, моноглицеридов и жирорастворимых витаминов в энтероциты. За время продвижения по тонкой кишке желчные кислоты способны 4–6 раз участвовать в процессах доставки жирных кислот и моноглицеридов внутрь энтероцитов [53]. Таким образом, недостаточное количество желчных кислот при ПБХ нарушает процессы абсорбции жиров и жирорастворимых витаминов.
Дефицит желчных кислот в кишечнике приводит не только к снижению эмульгирования жиров и всасывания гидролизованных продуктов – жирных кислот и моноглицеридов у пациентов с ПБХ [52], но и к дисбиозу микробиома кишечника [54]. J.K. DiBaise и соавт. предполагают, что дисбиоз также играет значимую роль в развитии стеатореи у пациентов с ПБХ и что у этих больных следует обязательно проводить оценку избыточного роста бактерий [55].
Так как недостаточное поступление желчных кислот в кишечник является одним из первых признаков патологии, то уже на ранних стадиях заболевания в кале пациентов с ПБХ можно обнаружить примеси не полностью переваренных жиров – один из признаков стеатореи. По мере прогрессирования заболевания и развития стеатореи у большинства больных стул приобретает кашицеобразную консистенцию, вплоть до диареи различной степени выраженности. Наряду с этим у отдельных пациентов с ПБХ наблюдаются запоры. Последние можно объяснить невыраженной (легкой) степенью стеатореи, определенным изменением микробиома кишечника и недостаточным влиянием малого количества желчных кислот на моторику кишечника.
Стеаторея на фоне постепенно и незаметно развивающейся энергетической НП приводит к развитию медленно прогрессирующего похудания пациентов с ПБХ. Окружность средней трети плеча, толщина кожно-жировой складки над трицепсом и процентное содержание жира, измеренные с помощью двухэнергетической рентгеновской абсорбциометрии (DEXA), были значительно ниже у пациентов с прогрессированием заболевания (p < 0,001) и особенно с установленным циррозом печени и асцитом [22].
Развитию медленно прогрессирующего похудания может способствовать применение некоторых лекарственных препаратов. Так, холестирамин, назначаемый для снятия кожного зуда, может вызвать вздутие живота, запор или диарею, что в свою очередь приводит к недостаточному поступлению пищевых ингредиентов и усилению энергетической недостаточности [32].
Развивающееся нарушение процессов желчевыделения (недостаточное поступление желчных кислот в двенадцатиперстную кишку) у пациентов с ПБХ уже на ранних стадиях заболевания способствует развитию медленно прогрессирующего похудания, что требует назначения препаратов урсодезоксихолевой кислоты и включения в рацион продуктов с повышенной калорийностью. Поскольку у таких пациентов происходит более раннее насыщение, им следует рекомендовать более частое дробное питание небольшими порциями с повышенной энергией блюд [28, 29]. При этом ограничение жиров в рационе следует вводить только в случае, если при их потреблении у пациентов наблюдается выраженная стеаторея с симптомами расстройства пищеварения. Однако необходимо учитывать, что пища без жира, с низким содержанием жира и/или триглицеридов, содержащих жирные кислоты со средней длиной углеродной цепи, как правило, переносится лучше. Поэтому необходим индивидуальный подход к оценке толерантности пациентов к различным жирам.
5.2.1. Витаминно-минеральная недостаточность
Желчные кислоты играют важную роль в абсорбции жирорастворимых витаминов A, D, E, K из кишечника. Желчные кислоты могут включать в состав липоидно-желчных комплексов жирорастворимые витамины и таким образом осуществлять транспортировку их в энтероцит. Недостаточное поступление желчных кислот в кишечник при ПБХ приводит к уменьшению процессов всасывания жирорастворимых витаминов и развитию витаминной недостаточности [56]. При ПБХ у 33,5; 13,2; 1,9 и 7,8% пациентов был обнаружен дефицит витаминов A, D, E и K соответственно [56].
Несмотря на то что недостаточное поступление желчных кислот при ПБХ имеет место уже на ранних стадиях заболевания, дефицит жирорастворимых витаминов чаще проявляется на стадии развернутого холестаза с выраженными признаками заболевания или на стадии развития цирроза. Способность жирорастворимых витаминов накапливаться в значительных количествах и храниться в печени и адипоцитах, а также возможность отдельных из них синтезироваться в организме приводит к тому, что на ранних стадиях ПБХ дефицит жирорастворимых витаминов, как правило, не развивается. Так, витамин D синтезируется в коже из холестерола под воздействием ультрафиолетовых лучей, а витамин К – микрофлорой кишечника. Но по мере прогрессирования заболевания и развития печеночно-клеточной недостаточности отмечается дефицит именно этих жирорастворимых витаминов, так как они подвергаются метаболизму в печени.
5.2.1.1. Витамин D
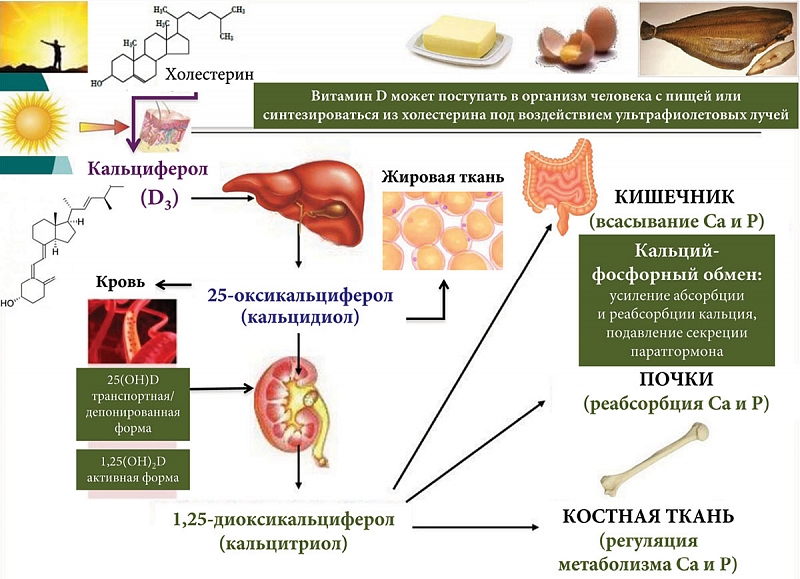
Витамин D принимает активное участие в фосфорно-кальциевом обмене. Пищевой (диетический) и вновь синтезированный из холестерола под воздействием ультрафиолетового излучения витамин D3 (кальциферол) является неактивной формой витамина. В печени происходит его гидроксилирование до 25-гидроксикальциферола (кальцидиол), который может накапливаться и храниться в печени и жировой ткани. Концентрация 25-гидроксикальциферола в сыворотке крови считается самым надежным показателем общего обмена витамина D, поэтому этот показатель может быть использован для определения обеспеченности организма витамином D [25, 26]. При снижении кальция в крови увеличивается синтез паратиреоидного гормона, стимулирующего в почках гидроксилирование кальцидиола до 1,25-дигидроксикальциферола (кальцитриол, активная форма витамина D, которая и участвует в регуляции обмена кальция и фосфора): активирует всасывание кальция и фосфора в кишечнике, реабсорбцию кальция и фосфора в почечных канальцах, регулирует обмен кальция и фосфора в костях (рис. 3).
При ПБХ по мере развития печеночно-клеточной недостаточности развивается постепенно прогрессирующий дефицит предшественника активной формы витамина D – кальцидиола, что приводит к остеодистрофии, сопровождающейся остеопенией. Остеопения является признанным осложнением холестатических заболеваний печени с распространенностью от 10 до 56%, в зависимости от стадии развития заболевания [57]. ПБХ является состоянием, вызывающим остеопению чаще, чем другие хронические заболевания печени [58–61], что клинически проявляется развитием признаков остеопороза [62].
5.2.1.1.1. Остеопороз
Остеопороз является системным заболеванием скелета, характеризующимся снижением массы и нарушением микроархитектоники костной ткани, что приводит к повышенной хрупкости костей и увеличению риска немотивированных переломов [63–65]. Распространенность остеопороза среди пациентов с ПБХ колеблется от 20 до 37% и больше, что выше, чем в общей популяции (10–11%) [59–61, 66]. По данным K.D. Lindor и соавт., частота остеопороза при ПБХ составляет 30% [32]. Остеопороз усиливается с прогрессированием заболевания печени [67].
Молекулярные механизмы остеопороза у пациентов с ПБХ связаны с нарушением энтерогепатической циркуляции желчных кислот, сопровождающейся снижением концентрации желчных кислот в тонком кишечнике и мальабсорбцией жирорастворимого витамина D [68]. У пациентов с ПБХ на стадии выраженного холестаза наряду с развитием дефицита абсорбции диетического витамина D в кишечнике происходит замедление превращения кальциферола в кальцидиол в печени по мере прогрессирования печеночно-клеточной недостаточности [69] и вследствие конкурентного ингибирования монооксигеназ [70]. Уменьшение количества кальцидиола у пациентов с ПБХ приводит к снижению образования активной формы витамина D (кальцитриола) в почках. В результате нарушается фосфорно-кальциевый обмен, что способствует развитию остеодистрофии [58, 71, 72], которая может проявляться болями в костях уже на ранних стадиях ПБХ. Развитие денситометрии костей позволило оценить костную массу и риск развития переломов [58], который коррелирует с минеральной плотностью костной ткани [62]. При этом лабораторные данные дают важную информацию о метаболическом статусе кости. Уровень кальция и фосфора в сыворотке крови пациентов ПБХ, как правило, несколько снижен [58]. По мнению S. Sherlock, нарушению фосфорно-кальциевого обмена у пациентов с ПБХ способствует развивающаяся стеаторея – из-за повышенного содержания жира в кишечнике могут образовываться нерастворимые мыла с кальцием, препятствуя дальнейшему его всасыванию энтероцитами [51]. Снижение всасывания кальция хорошо коррелирует с увеличением экскреции фекального жира и в меньшей степени с интенсивностью желтухи [73]. В развитии остеопороза играют роль и генетические факторы [74–76]. При R-графии костей, денситометрии и морфологическом исследовании биоптата костной ткани при ПБХ чаще всего выявляются признаки остеопороза [58]. На поздних стадиях заболевания встречаются патологические переломы позвонков и ребер, реже – костей таза и длинных трубчатых костей [66].
К клинически значимой потере костной массы с увеличением риска переломов более чем в два раза у пациентов с ПБХ может привести длительная стероидная терапия, которая ускоряет и усугубляет развитие остеопороза [77]. Атравматические переломы особенно опасны у пациентов с ПБХ, перенесших ортотопическую трансплантацию печени и получающих лечение высокими дозами кортикостероидов [62].
Глюкокортикостероиды (ГК) снижают абсорбцию кальция в кишечнике вследствие уменьшения продукции кальцитриола (1,25-дигидрокси витамин D3), подавления канальцевой реабсорбции кальция в почках и увеличения его экскреции с мочой. Снижение уровня кальция в крови приводит к компенсаторному увеличению выработки паратиреоидного гормона (ПТГ) и резорбции костной ткани. Кроме того, ГК напрямую увеличивают высвобождение ПТГ, непосредственно подавляют функцию остеобластов, повышая активность остеокластов, а также опосредованно ингибируют образование костной ткани, подавляя синтез тестостерона в гонадах и снижая выработку гормона роста, инсулиноподобного фактора роста 1 (IGF-I – синтезируется печенью и стимулирует синтез костного коллагена первого типа и функцию остеобластов) [58, 78, 79].
Таким образом, патогенез остеопороза у пациентов с ПБХ сложен и многофакторен [58, 62, 80] и включает: нарушения всасывания и метаболизма витаминов D и К [81]; дефицит ионов магния, снижение абсорбции ионов кальция в кишечнике и реабсорбции его в почечных канальцах, увеличение резорбции костной ткани [82, 83]; повышение уровня билирубина, ингибирующего функцию остеобластов [58, 66, 84]; генетическую предрасположенность [85] и побочные эффекты лекарств (например, кортикостероиды и холестирамин, которые используются для лечения пациентов с ПБХ) [86]. Развитие остеопороза связывают с тяжестью заболевания, а не с его длительностью. Остеопороз может повлиять на качество жизни пациента и течение заболевания [58]. Дефицит активной формы витамина D (кальцитриола) является фактором риска остеосаркопении [66, 87].
5.2.1.2. Витамин К
Витамин К необходим для синтеза в печени факторов свертывания VII, IX, X и протромбина [88–90]. На ранних стадиях ПБХ дефицит витамина K, как правило, отсутствует. По мере прогрессирования мальабсорбции и нарушения белково-синтетической функции печени на поздних стадиях ПБХ появляется угроза снижения синтеза факторов свертывания крови [91]. Снижение уровня витамина К в плазме крови пациентов с ПБХ наблюдается в 23% случаев, что обычно сопровождается увеличением протромбинового времени [92]. Дефицит витамина К у больных ПБХ в терминальной стадии заболевания с портальной гипертензией и варикозно расширенными венами пищевода/желудка повышает вероятность развития массивного трудноостанавливаемого кровотечения.
5.2.1.3. Витамин А
Абсорбция витамина А требует интактной энтерогепатической циркуляции желчных кислот и образования липоидно-желчных мицеллярно-ламеллярных структур в кишечнике. Значительное прогрессирование мальабсорбции у пациентов с ПБХ, особенно на стадии выраженного холестаза, может приводить к снижению всасывания в кишечнике витамина А, сопровождающемуся уменьшением уровня ретинола в сыворотке крови. На стадии печеночно-клеточной недостаточности происходит нарушение синтеза печеночного ретинолсвязывающего белка, что также способствует снижению уровня витамина А в сыворотке крови [29]. С клинической точки зрения, дефицит витамина А не является распространенным явлением у пациентов с ПБХ. Тем не менее недостаточная адаптация к темноте (ночная, куриная слепота – гемералопия) иногда развивается у пациентов с тяжелым течением ПБХ [29]. Можно отметить другие потенциальные проявления дефицита витамина А: сухость кожи, потерю ее эластичности, дерматологические расстройства, нарушение гуморального и клеточно-опосредованного иммунитета [29].
Развивающееся нарушение процессов желчевыделения (недостаточное поступление желчных кислот в двенадцатиперстную кишку) у пациентов с ПБХ в стадию выраженного холестаза и печеночно-клеточной недостаточности способствует постепенному развитию дефицита жирорастворимых витаминов, что требует назначения препаратов урсодезоксихолевой кислоты, контроля уровня жирорастворимых витаминов в плазме крови и включения в рацион продуктов, обогащенных соответствующими витаминами, при их низком содержании. При дефиците витамина D назначается активная форма этого витамина (кальцитриол) совместно с препаратами кальция и бифосфонатами.
5.2.2. Изменения в метаболизме меди
Как известно, печень играет важную роль в метаболизме меди за счет образования в гепатоцитах комплексов «церулоплазмин-медь» и экскреции ее с желчью. В норме около 80% поступающей в организм меди экскретируется в желчь и выделяется с фекалиями.
На поздних стадиях ПБХ при развитии печеночно-клеточной недостаточности происходит накопление меди в печени, иногда до уровня 25 мг на 100 г сухого веса ткани печени (при норме до 6 мг на 100 г) [93]. При этом вследствие связывания с церулоплазмином отсутствуют клинические признаки токсического воздействия меди на организм человека, а также не выявляется кольцо Кайзера – Флейшера.
Накопление меди в организме пациентов с ПБХ приводит к активации медьсодержащего фермента тирозиназы. В результате увеличивается выработка меланина, что приводит к развитию у этих пациентов гиперпигментации кожных покровов. Наряду с этим организм пытается вывести избыток меди не только через почки, но и через кожу. Это приводит к отложению меди в эпидермисе, что придает бронзовый оттенок гиперпигментированной коже пациентов с ПБХ.
Развивающееся нарушение процессов желчевыделения (накопление желчных кислот в гепатоцитах) у пациентов с ПБХ в стадию выраженной печеночно-клеточной недостаточности сопровождается нарушением обмена меди, что требует осторожного включения в рацион продуктов, содержащих больше 0,5 мг меди на 100 г продукта, и исключения использования медной посуды для приготовления и хранения пищи.
5.3. Повышенная скорость метаболизма (ускоренный катаболизм)
По мере развития энергетической НП и стеатореи у пациентов с ПБХ адаптивная реакция организма приводит к увеличению потребности внутренних органов в кислороде, что сопровождается активацией процессов катаболизма, мобилизацией энергоресурсов жировой ткани и потреблением в качестве энергетического материала мышечного белка наряду с активным использованием углеводов [54].
Потребность в глюкозе для глюкозозависимых тканей по мере истощения запасов гликогена у пациентов с ПБХ компенсаторно покрывается за счет активации процессов глюконеогенеза. Последний служит важным источником поддержания нормального уровня глюкозы в организме и представляет собой метаболический путь, приводящий к образованию глюкозы из неуглеводных соединений. Процесс протекает в основном в печени и менее интенсивно – в корковом веществе почек, а также в слизистой оболочке кишечника [94]. Важными предшественниками глюкозы при глюконеогенезе выступают трехуглеродные соединения, такие как лактат, пируват, глицерол, образующиеся в результате гидролиза жиров в адипоцитах, а также аминокислот соматических (мышечных) белков. Известно, что метаболизм ароматических аминокислот происходит преимущественно в печени, а метаболизм аминокислот с разветвленной цепью – преимущественно в мышцах [43]. В сыворотке крови пациентов с ПБХ выявлено снижение концентрации аминокислот с разветвленной цепью и повышение уровня ароматических аминокислот [95–97]. Увеличение ароматических аминокислот в плазме крови пациентов с ПБХ коррелирует с нарастанием печеночно-клеточной недостаточности и служит одним из маркеров степени развития печеночно-клеточной недостаточности.
В отличие от углеводов и жиров белки и аминокислоты имеют ограниченную способность запасаться в организме человека [94]. Аминокислоты, как правило, либо используются организмом в качестве пластического материала, либо подвергаются метаболической деградации [94]. Содержащийся в аминокислотах азот при их деградации превращается в мочевину и креатинин и выделяется почками, а углеродный скелет может использоваться для биосинтеза глюкозы (глюконеогенез) или жирных кислот либо окисляться до углекислого газа и воды с образованием энергии, в том числе и в виде АТФ.
Мышцы играют важную роль в метаболизме аминокислот, в том числе и через глюконеогенез. Аминокислоты мышечных белков являются важным источником образования глюкозы и метаболической энергии [94]. Дефицит гликогена и глюкозы в организме пациентов с ПБХ усиливает катаболизм мышечных (соматических) белков с освобождением свободных аминокислот, многие из которых (прежде всего аминокислоты с разветвленной цепью) сразу превращаются в пируват или сначала в оксалоацетат, а затем в пируват. Последний превращается в аланин, приобретая аминогруппу от других аминокислот. Аланин из мышц переносится кровью в печень, где снова преобразуется в пируват, который используется в качестве энергетического субстрата или включается в глюконеогенез [94]. Повышенный глюконеогенез у пациентов с ПБХ постепенно приводит к массивному распаду и дефициту мышечного белка.
Баланс между синтезом и деградацией соматического белка нарушается, что приводит к развитию атрофии мышц (саркопении). Саркопения характеризуется потерей массы и силы скелетных мышц и при ПБХ классифицируется как вторичная саркопения [66]. S. Fülster с соавт. показали, что атрофия скелетных мышц, развивающаяся при хронических заболеваниях, связана также с низкой толерантностью к физическим нагрузкам [98]. Точный механизм, способствующий развитию саркопении при ПБХ, четко не определен. Повышенная деградация аминокислот с разветвленной цепью, мышечная аутофагия, кортикостероиды, гипераммониемия, миостатин и низкая физическая активность считаются потенциальными факторами, способствующими саркопении [99, 100]. При недостатке аминокислот и энергии активируется аутофагия – процесс, при котором компоненты клетки подвергаются деградации ферментами лизосом. При ПБХ потеря печеночного гликогена с последующим ускоренным глюконеогенезом, повышенным катаболизмом аминокислот с разветвленной цепью, прием глюкокортикоидов могут приводить к мышечной аутофагии и являться механизмом истощения мышц у таких пациентов [101]. Вторичная саркопения, вызванная ПБХ, ухудшает качество жизни и прогноз этих пациентов [102–104].
Остеопороз и саркопения тесно взаимосвязаны друг с другом и часто сосуществуют у пациентов с хроническими заболеваниями печени [105, 106]. Появился новый термин «остеосаркопения», который подразумевает сосуществование саркопении и остеопороза [106]. В исследовании C. Saeki и соавт. распространенность остеосаркопении у пациентов с ПБХ составила 15,4% [66]. Остеосаркопения является «опасным дуэтом», поскольку вызывает как склонность к частым падениям (из-за саркопении), так и уязвимость костей (из-за остеопороза) [106]. Особенно проблематичными остеопороз и саркопения являются у пациенток с ПБХ в постменопаузе [66].
На фоне развившейся на ранних стадиях заболевания энергетической недостаточности у пациентов с явной клинической картиной холестаза при ПБХ постепенно возникают белково-энергетическая недостаточность (БЭН) и незаметно прогрессирующая саркопения (атрофия мышц) [19]. При развитии цирроза у пациентов с ПБХ скорость глюконеогенеза с использованием аминокислот существенно возрастает [94]. Несмотря на повышенную деградацию соматических белков, связанную с энергетической недостаточностью питания, висцеральный пул белка, синтезируемый в гепатоцитах, при ПБХ сохраняется в нормальных пределах (с незначительными отклонениями) до развития печеночно-клеточной недостаточности. Содержание альбуминов и глобулинов в крови больных ПБХ на ранних стадиях и в период выраженного холестаза находится в пределах нормы [25, 26]. Вместе с тем в сыворотке крови пациентов уже в асимптоматической стадии заболевания обнаруживают антимитохондриальные антитела в диагностическом титре 1:40 и выше. По мере прогрессирования холестаза наблюдается повышение уровня γ-глобулинов [25, 26].
Развивающееся нарушение процессов желчевыделения (накопление желчных кислот в плазме крови и гепатоцитах) у пациентов с ПБХ в стадию выраженного холестаза приводит к постепенному развитию БЭН по типу «маразм» и к саркопении. Это требует включения в их рацион продуктов с повышенным содержанием белка (с преимущественным содержанием аминокислот с разветвленной цепью).
По мере прогрессирования ПБХ за счет нарастания процессов катаболизма увеличиваются скорость метаболизма в состоянии покоя и общий термогенез [37, 54]. Создается метаболическая ситуация перераспределения ресурсов, которая усиливается по мере нарастания холестаза и развития печеночно-клеточной недостаточности. Развитие печеночно-клеточной недостаточности в терминальной стадии ПБХ сопровождается нарушением белково-синтетической функции гепатоцитов [54]. В дополнение к БЭН у пациентов с ПБХ в период декомпенсации печеночно-клеточной недостаточности уменьшается синтез мочевины, белков сыворотки крови в печени и усиливается распад висцерального пула белков, что приводит к резкому снижению уровня циркулирующих в плазме альбуминов и повышению экскреции азота с мочой [107]. Наряду с продолжающимся повышенным катаболизмом соматических белков происходит развитие недостаточности висцерального пула белков с последующим развитием отеков и асцита [29]. Клинические проявления нарушения трофологического статуса у пациентов с ПБХ в терминальной стадии заболевания приобретают промежуточную форму БЭН – маразм-квашиоркор. Развитию БЭН способствует снижение всасывания белков в кишечнике. Портальная гипертензия, приводящая к циркуляторной гипоксии слизистой оболочки кишечника и увеличению ее проницаемости, также приводит к повышенной потере белков.
Развивающееся нарушение процессов желчевыделения (накопление желчных кислот в гепатоцитах и плазме крови) у пациентов с ПБХ в стадии выраженной печеночно-клеточной недостаточности (потеря 75% и более функционирующих печеночных клеток) сопровождается нарушением белково-синтетической функции гепатоцитов, что приводит к дефициту висцерального белка и, как следствие, к развитию отеков и асцита. Происходит переход клинической формы БЭН по типу «маразм» в смешанную – «маразм-квашиоркор» – НП. Это требует сокращения приема соли, жидкости и, если отсутствуют признаки ПЭ, включения в рацион продуктов с повышенным содержанием белка. Нутриционная поддержка в этот период должна включать белковые модули с преимущественным содержанием аминокислот с разветвленной цепью, а также разным количеством и соотношением заменимых и незаменимых аминокислот [94]. Для предотвращения катаболизма белка и поддержания баланса азота рекомендуют прием пищи, содержащей 50 г углеводов, перед сном [108, 109].
Заметное улучшение статуса питания пациентов с ПБХ на стадии развития цирроза и резистентного асцита отмечено после успешного лечения последнего, что подчеркивает важность применения нутритивной поддержки у таких пациентов [110].
6. Печеночная энцефалопатия
При ПБХ в терминальной стадии заболевания прогрессирующая печеночно-клеточная недостаточность, портальная гипертензия и портосистемное шунтирование приводят к развитию ПЭ [111, 112]. Под ПЭ понимают потенциально обратимые нейропсихические расстройства, развивающиеся на фоне тяжелых поражений печени, в основе формирования которых лежат нарушение дезинтоксикационной функции и шунтирование портальной крови [111, 113]. ПЭ – классический признак далеко зашедшей печеночно-клеточной недостаточности [111, 112]. Энцефалопатия, связанная с нарастающей гибелью гепатоцитов, в прогностическом плане становится грозным и почти всегда фатальным осложнением ПБХ. Распространенность минимальной ПЭ среди пациентов с циррозом печени составляет от 30 до 84% [114].
Существует метаболическая теория развития ПЭ, которая основывается на обратимости ее основных симптомов при весьма обширных церебральных нарушениях [115]. При ПБХ можно выделить два фактора, определяющие взаимосвязь печени и нервной системы и играющие роль в патогенезе ПЭ [113].
- Способность печени осуществлять детоксикацию нейротоксичных ядов (аммиак, меркаптан, скатол, индол, фенолы и др.), образующихся в кишечнике в процессе переваривания пищевых ингредиентов и в результате жизнедеятельности микроорганизмов [116, 117].
- Церебральный метаболизм сильно зависит от поддержания нормального уровня гликемии, который в период между приемами пищи в значительной степени определяется запасом гликогена в печени и скоростью гликолиза. Как упоминалось выше, запасы гликогена при ПБХ истощаются. Снижение интенсивности метаболизма кислорода и глюкозы при ПБХ сопровождается уменьшением выработки энергии, снижением активности нейронов, что способствует развитию ПЭ [115]. Согласно результатам позитронной эмиссионной томографии при ПЭ и по результатам нейропсихологических тестов, имеется сильная корреляция между уменьшением церебрального кровотока (в лобной и теменной долях коры головного мозга), сопровождающаяся снижением метаболизма глюкозы и результатами нейропсихологических тестов [113].
В основе патогенеза ПЭ лежат [115]:
- печеночно-клеточная недостаточность, сопровождающаяся снижением печеночного клиренса нейротоксичных ядов, образующихся в кишечнике в процессе переваривания пищевых ингредиентов и в результате жизнедеятельности микроорганизмов;
- развитие портосистемного шунтирования;
- нарушение метаболизма аминокислот, приводящее к образованию ложных нейротрансмиттеров.
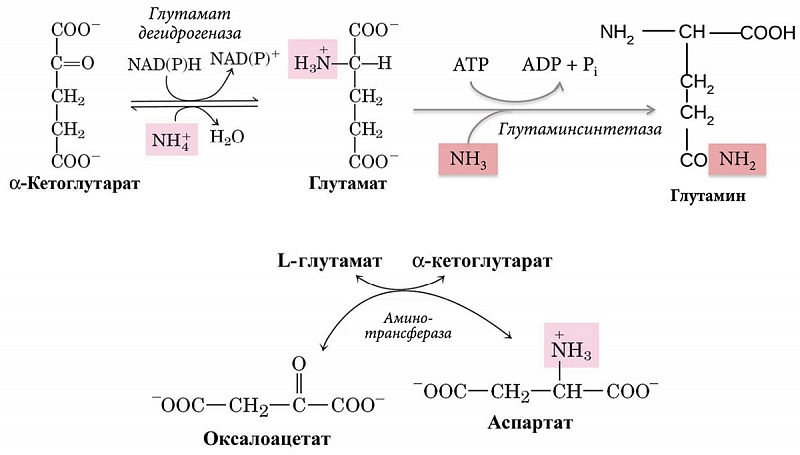
В ходе деградации белка и аминокислот образуется аминный азот, который, в отличие от углеводородной части аминокислот, непригоден для получения энергии [94]. Поэтому аминогруппы, которые не могут быть использованы повторно, например, в реакциях трансаминирования, превращаются в аммиак. Аммиак в клетках образуется при дезаминировании аминокислот, нуклеотидов, биогенных аминов. Аммиак является токсическим веществом, и его концентрация в крови в норме не превышает 50 мкмоль/л. В норме около 7% образующегося в организме аммиака проходит через ткань мозга, не вызывая изменений его функций [111]. Основной реакцией обезвреживания аммиака, протекающей во всех тканях, является связывание NH3 с глутаматом с образованием глутамина. Основными тканями-поставщиками глутамина являются мышцы, головной мозг, печень.
Помимо аммиака, образующегося в тканях, значительное количество NH3 вырабатывается в кишечнике бактериальной микрофлорой и в результате гидролиза белков пищи. Всасывание аммиака в кишечнике может служить причиной значительного его поступления в печень. Это происходит при повышенном потреблении белка с пищей, нарушении эвакуации содержимого из кишечника, защелачивании кишечного содержимого, избыточном росте условно-патогенной флоры, кровотечениях из варикозно расширенных вен пищевода/желудка при развитии портальной гипертензии [112]. Концентрация токсических продуктов, прежде всего аммиака, а также скатола, индола и фенолов в кишечнике может при этом увеличиваться. В норме эти вещества попадают в систему портальной вены и в печени включаются в орнитиновый цикл, чтобы через реакции дезаминирования, переаминирования и декарбоксилирования превратиться в относительно безвредный для организма продукт – мочевину [94]. Последняя является основным конечным продуктом азотистого обмена (85% всего азота выводится из организма с мочевиной). Мочевина в организме человека синтезируется только в печени [94].
Нарушение нейрональной функции происходит под влиянием повышенного содержания в крови нейротоксичного аммиака – при гипераммониемии [111]. Последняя наблюдается у пациентов с ПБХ на стадии развития цирроза и обусловлена повышенным всасыванием аммиака в кишечнике, нарушением детоксикации аммиака в гепатоцитах (снижение активности ферментов цикла синтеза мочевины) и уменьшением степени связывания аммиака в гипотрофичных скелетных мышцах (снижение активности глутаминсинтетазы) [112, 118]. Большое значение в развитии ПЭ играет нарушение печеночного кровотока. Развитие цирроза в терминальной стадии ПБХ приводит к тому, что кровь шунтируется как внутри самой печени (вокруг долек образуются портопеченочные венозные анастомозы, которые функционируют как внутрипеченочные шунты), так и за счет поступления крови из воротной вены в системный кровоток, минуя печень по естественным коллатералям [115]. В результате портосистемного шунтирования и коллатеральных путей кровотока оттекающая от кишечника кровь попадает в системный кровоток, минуя печень. Содержащиеся в крови портальной системы токсические вещества, и прежде всего аммиак, поступают необезвреженными в общий кровоток.
Гипераммониемия у пациентов с ПБХ запускает компенсаторные механизмы метаболизма и клиренса аммиака за счет активизации процессов его обезвреживания в скелетных мышцах и нейронах [119]. Повышенный уровень аммиака в крови приводит к увеличению его проникновения через гематоэнцефалический барьер в головной мозг, что оказывает неблагоприятное воздействие на астроциты. Детоксикация аммиака в астроцитах происходит под действием глутаматсинтетазы, что приводит к связыванию аммиака с глутаматом с образованием глутамина (рис. 4) [120].
Избыток аммиака в мышечной ткани также может инактивироваться за счет его взаимодействия как с глутаматом, так и с аспартатом с синтезом глутамина (рис. 4) [113, 121]. В условиях избытка аммиака запасы глутамата и аспартата истощаются (при одновременном накоплении глутамина). Повышенное количество образующегося глутамина высвобождается в кровоток в обмен на аминокислоты с разветвленной цепью [122].
Гипераммониемия при ПБХ требует, таким образом, повышенного образования глутамата и аспартата из α-кетоглутарата и оксалоацетата. Это приводит к тому, что часть α-кетоглутарата и оксалоацетата выключается из цикла трикарбоновых кислот, что сопровождается снижением синтеза АТФ. Так как нейроны особенно чувствительны к уменьшению выработки энергии, то это играет определенную роль в механизме развития клинических признаков ПЭ, а также приводит к усилению энергетической недостаточности у пациентов с ПБХ.
Мышцы также чувствительны к уменьшению выработки энергии АТФ. Поэтому для повышения содержания в мышцах α-кетоглутарата и оксалоацетата, необходимых для цикла Кребса, с одной стороны, и поддержания достаточного уровня глутамата – с другой, при гипераммониемии у пациентов с ПБХ происходит ускорение катаболизма аминокислот с разветвленной цепью. Это приводит к недостаточному синтезу мышечного белка и развитию истощения мышц [123, 124]. Гипераммониемия сопровождается ПЭ, повышенным катаболизмом аминокислот с разветвленной цепью и саркопенией [125]. Саркопения усугубляет ПЭ, что в свою очередь приводит к снижению потребления пищи и развитию НП. Возникает трудно разрываемый порочный круг.
Имеются сведения, что гипераммониемия сказывается на работе центра насыщения в гипоталамусе и способствует подавлению аппетита, что у пациентов с ПБХ может усиливать и БЭН [111, 113].
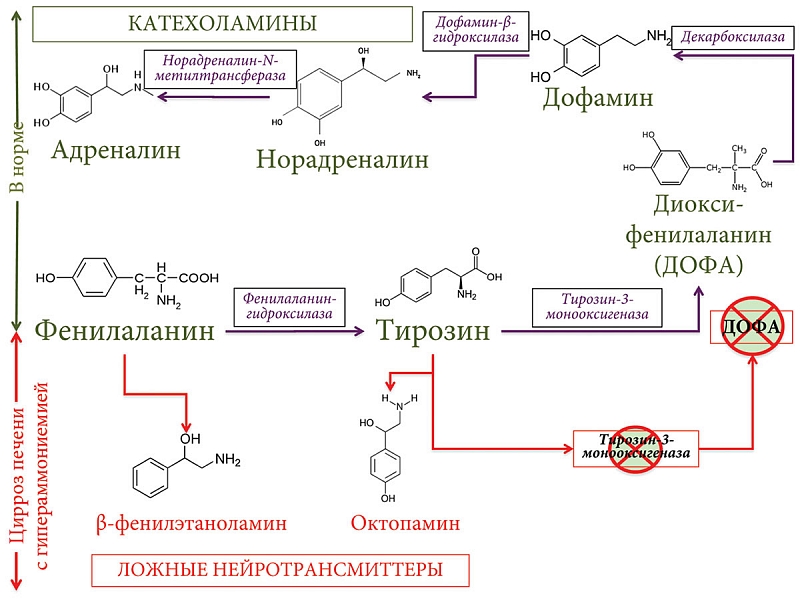
Наряду с гипераммониемией в патогенезе ПЭ у пациентов с ПБХ на стадии развития печеночно-клеточной недостаточности и портосистемного шунтирования играет важную роль нарушение синтеза и обмена основных нейромедиаторов, образующихся из ароматических аминокислот тирозина и фенилаланина [115]. P.C. ter Borg и соавт. обнаружили повышенные концентрации ароматических аминокислот тирозина и фенилаланина, а также пониженные концентрации аминокислот с разветвленной цепью валина, изолейцина и лейцина в крови у пациентов с ПБХ как в стадию развития цирроза, так и без цирроза [39]. Повышенное поступление в кровь ароматических аминокислот (в результате нарушения их катаболизма в печени) тормозит ферментные системы, участвующие в превращении ароматических аминокислот в катехоламины, что приводит к снижению биосинтеза дофамина, норадреналина и увеличению синтеза серотонина из триптофана. Проникая через гематоэнцефалический барьер в мозг, тирозин и фенилаланин участвуют в синтезе ложных нейротрансмиттеров – β-фенилэтаноламина и октопамина (рис. 5).
В кишечнике под действием бактериальных декарбоксилаз из аминокислоты тирозин образуется тирамин. Последний является физиологически активным и токсичным веществом. При развитии портосистемного шунтирования у пациентов с ПБХ он легко попадает в системный кровоток и, проникая через гематоэнцефалический барьер, влияет на процессы возбуждения и торможения в нервной системе. Ложные нейротрансмиттеры, тирамин наряду с гипераммониемией угнетают функцию нейронов и способствуют усилению развития ПЭ [39]. Нарушение когнитивных функций, забывчивость, сон во время приема пищи и перекусов, а также трудности с приготовлением пищи при развитии ПЭ на поздних стадиях ПБХ являются существенными препятствиями, с которыми сталкиваются пациенты этой группы [126]. Впоследствии недостаточность питания сама по себе становится независимым предиктором смертности у пациентов с ПБХ.
Развивающееся нарушение процессов желчевыделения (накопление желчных кислот в гепатоцитах) у пациентов с ПБХ приводит к выраженной печеночно-клеточной недостаточности, что сопровождается нарушением детоксикационной функции гепатоцитов, гипераммониемией, образованием ложных нейротрансмиттеров с развитием ПЭ. Это требует принятия мер, направленных на изменение соотношения нейромедиаторов, снижение образования и абсорбции аммиака и других токсинов, образующихся в кишечнике, увеличение элиминации аммиака, а именно: строгого ограничения белка в рационе; включения в рацион смесей, содержащих аминокислоты с разветвленной цепью (с минимальным количеством ароматических аминокислот); назначения антибиотиков (эффект которых основывается на их воздействии на микроорганизмы, продуцирующие азотистые соединения в желудочно-кишечном тракте). Строгая вегетарианская диета (содержание растительного белка до 120 г/сут) и белок молочного происхождения, как правило, хорошо переносятся (по-видимому, вследствие низкого содержания ароматических аминокислот). Пациенты с ПБХ в этот период, как правило, нуждаются в постановке в лист ожидания на проведение операции по трансплантации печени. Для удовлетворения потребности в энергии и белке пациентов с недостаточностью питания и потерей массы тела, находящихся в хирургических или реаниматологических отделениях, в клинической практике используются рекомендации ESPEN (European Society for Clinical Nutrition and Metabolism).
Плохой нутритивный статус влечет за собой серьезные последствия для послеоперационных осложнений среди кандидатов на трансплантацию печени, поскольку это важный прогностический критерий смертности и послеоперационных осложнений среди пациентов с ПБХ.
7. Заключение
ПБХ является хроническим медленно прогрессирующим заболеванием печени и желчевыводящих путей, которое приводит к изменению трофологического статуса этих пациентов. Причины НП при ПБХ сложны и многофакторны, поскольку печень участвует во многих метаболических процессах организма. Но ведущая роль в развитии НП у пациентов с ПБХ принадлежит нарушению процессов желчевыделения и, как следствие, изменению метаболизма макронутриентов и микронутриентов. Трофологическая недостаточность развивается постепенно и незаметно по мере развития холестаза с недостаточным поступлением желчных кислот в двенадцатиперстную кишку при одновременной их задержке в гепатоцитах и попадании в системный кровоток. Именно эти изменения уже в асимптоматической и ранней стадии заболевания запускают развитие энергетической НП. Со временем включаются компенсаторные механизмы получения энергии из жирных кислот и аминокислот соматического пула белков, что сопровождается БЭН (по типу «маразм») с медленно прогрессирующей потерей массы тела. Также развиваются нарушения в липидном обмене – происходит повышенный синтез холестерола и фосфолипидов для нейтрализации детергентного действия избыточных желчных кислот в плазме крови. Недостаточное поступление желчных кислот в кишечник способствует развитию стеатореи и дефицита жирорастворимых витаминов у этих пациентов, вследствие чего происходит нарастание БЭН и постепенно прогрессирует витаминно-минеральная недостаточность. Последняя приводит к развитию остеопороза, остеосаркопении. Длительное воздействие избыточных желчных кислот на гепатоциты приводит к развитию фиброза, цирроза печени, портальной гипертензии, портосистемного шунтирования, нарушаются белково-синтетическая и детоксикационная функции печени. Возникающий дефицит висцерального белка приводит к развитию отеков, асцита и усилению БЭН с переходом в смешанную форму «маразм-квашиоркор». Развивающаяся гипераммониемия и образующиеся ложные нейротрансмиттеры приводят к изменениям в центральной нервной системе – развивается ПЭ. Степень НП прогрессирует по мере увеличения тяжести заболевания. Все это делает коррекцию НП у пациентов с ПБХ особенно сложной. Таким образом, оценка состояния питания и борьба с НП имеют первостепенное значение для улучшения результатов лечения этих пациентов. Представленные в обзоре механизмы развития изменений трофологического статуса при ПБХ должны помочь своевременно распознать нарушения статуса питания и правильно подобрать схему нутриционной поддержки этим пациентам на разных стадиях развития заболевания наряду с симптоматической терапией.
Авторы выражают благодарность Татьяне Игоревне Карлович и Александру Игоревичу Бурмистрову за обсуждение и техническую помощь при подготовке обзора к публикации.
I.V. Maev, PhD, Prof., Academician of the RAS, V.I. Reshetnyak, PhD, Prof.
A.I. Yevdokimov Moscow State University of Medicine and Dentistry
Contact person: Vasily I. Reshetnyak, vasiliy.reshetnyak@yandex.ru
The review is devoted to the development of malnutrition in primary biliary cholangitis. Presented the factors contributing to the gradual progression of malnutrition signs in these patients at various stages of the disease. The pathogenesis of energy, protein-energy (form "marasmus") and protein (form "kwashiorkor") malnutrition as the disease progresses is considered. Taking into account the mechanisms of development of various signs and forms of malnutrition, presented the principles of diet therapy for primary biliary cholangitis.
Уважаемый посетитель uMEDp!
Уведомляем Вас о том, что здесь содержится информация, предназначенная исключительно для специалистов здравоохранения.
Если Вы не являетесь специалистом здравоохранения, администрация не несет ответственности за возможные отрицательные последствия, возникшие в результате самостоятельного использования Вами информации с портала без предварительной консультации с врачом.
Нажимая на кнопку «Войти», Вы подтверждаете, что являетесь врачом или студентом медицинского вуза.