Семилетний опыт терапии нитизиноном наследственной тирозинемии 1-го типа в России
- Аннотация
- Статья
- Ссылки
- English


Актуальность
Наследственная тирозинемия 1-го типа (НТ1), или гепаторенальная тирозинемия, относится к редким генетическим заболеваниям с аутосомно-рецессивным типом наследования, обусловлена мутациями в гене фермента фумарилацетоацетазы (фумарилацетогидролазы (FAH)), OMIM 276700, код по Международной классификации болезней 10-го пересмотра – Е70.2. Частота НТ1 в разных популяциях колеблется от 1:100–200 тыс. живых новорожденных. Частота носительства мутаций НТ1 в популяциях – 1:150–100 человек. Половых различий по частоте встречаемости и тяжести течения НТ1 не выявлено [1, 2].
Особенностью российских больных НТ1 является поздняя диагностика. На долю хронических больных приходится более половины всех больных. В некоторых штатах США и стран Европы, где тирозинемия включена в перечень болезней, подлежащих неонатальному скринингу, хронических форм НТ1 не встречается из-за раннего начала лечения [3]. Но ретроспективно отмечалось, что 3/4 пациентов страдали острой формой заболевания (НТ1А) и только 1/4 – хронической (НТ1Б). Большинство (до 90%) пациентов без специфического лечения и трансплантации печени умирали в возрасте до десяти лет. В исследовании F.J. van Spronsen и соавт. (1994), охватившем 125 центров Европы, США, Канады и Японии, участвовали 108 детей с НТ1. Как показали результаты, выживаемость зависела от сроков появления симптомов. При дебюте заболевания до двух месяцев к году умерли практически все дети (96%). При дебюте от двух до шести месяцев выживаемость была чуть выше (до шести лет летальный исход имел место в 74% случаев). При появлении симптомов в возрасте старше шести месяцев в течение десяти лет умерли 38% пациентов [4].
Ген FAH локализован на длинном плече 15-й хромосомы (15q23–q25), состоит из 14 экзонов. Известно 20 различных мутаций, четыре – частые. Четких взаимосвязей между генотипом и фенотипом не установлено. Разные клинические варианты могут присутствовать у членов одной семьи с одинаковыми мутациями [2, 5, 6].
Патогенез
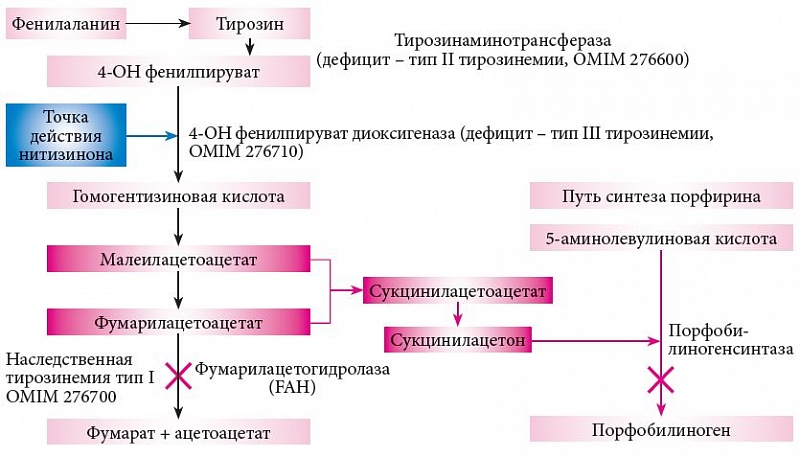
Патогенез НТ1 заключается в интоксикации продуктами аномального распада тирозина – фумарилацетоацетатом и малеилацетоацетатом и их конечными метаболитами – сукцинилацетоном и сукцинилацетоацетатом (рис. 1). Накопление этих метаболитов приводит к развитию печеночной недостаточности, циррозу, тубулопатии с формированием синдрома Фанкони, полинейропатии, кардиомиопатии, а на более поздней стадии гепатоцеллюлярной карциномы (ГЦК) [7]. Кроме того, сукцинилацетон ингибирует дельта-аминолевулинат-дегидратазу, промежуточный медиатор порфобилиногена, что приводит к нарушению биосинтеза гема и клинически может проявляться симптомами острой перемежающейся порфирии – болью в животе, артериальной гипертензией, нейропатией.
Клиническая картина
Клиническая картина НТ1 чрезвычайно полиморфна. Как уже отмечалось, различают два клинических варианта – острую и хроническую формы. Острая форма – НТ1А обычно развивается в возрасте до шести месяцев. Наиболее тяжелая форма с дебютом до двух месяцев характеризуется витамин K-зависимой коагулопатией на фоне острой печеночной недостаточности [8]. Как правило, ставится ошибочный диагноз сепсиса, цитомегаловирусного гепатита, фетального гепатита. У детей с НТ1А выражена интоксикация, гипогликемия, лейкоцитоз, возможен асцит, динамическая непроходимость, парез кишечника. При этом отмечается повышенный уровень сукцинилацетона (≥ 10–100 раз), альфа-фетопротеина (АФП) (≥ 100–1000 раз). Цитолиз обычно не превышает трех-четырех норм, умеренная гипербилирубинемия в пределах 100–150 мкмоль/л, в равном соотношении прямой и непрямой билирубин.
Хроническая форма – НТ1Б характеризуется слабой интоксикацией, диагностируется обычно в возрасте старше одного года на стадии сформированного цирроза и/или рахитических деформаций скелета. Цитолиз минимальный (в пределах трех норм). Уровень АФП повышен в пределах 1000 нг/дл, сукцинилацетона – не более чем в 20 раз [9].
Исследование системы свертывания крови необходимо в связи с витамин K-зависимой коагулопатией – дефицитом всех печеночных факторов свертывания (II, V, VII, X, IX, XI, XII), коагулопатией с удлинением протромбинового и активированного частичного тромбопластинового времени, снижением протромбинового индекса, фибриногена, снижением антитромбина III. Коагулопатия чаще встречается при НТ1А и протекает значительно тяжелее, чем при других болезнях печени. Активность трансаминаз и содержание билирубина, напротив, выражены умеренно как за счет аспартатаминотрансферазы (АСТ), так и аланинаминотрансферазы (АЛТ). Высокая степень цитолиза встречается редко.
При НТ1А холестаз умеренный, внутрипеченочный, при НТ1Б может быть диссоциированным – с нормальным уровнем билирубина, но с высоким уровнем гаммаглутамилтранспептидазы (ГГТП), щелочной фосфатазы (ЩФ). Последняя может быть проявлением острого рахита.
У детей старше двух-трех лет показаниями к диагностике НТ1 является наличие синдрома Фанкони – фосфат-диабета, рахитоподобного заболевания, тяжелых деформаций скелета.
Вторичный гиперпаратиреоз сопряжен с гипокальциемией и гипофосфатемией и соответствующими им кальций- и фосфатурией. Признаки синдрома Фанкони включают глюкозурию, генерализованную аминоацидурию, почечный канальцевый ацидоз, фосфатурию. Синдром Фанкони может быть как полным, так и неполным.
Гипогликемия – один из частых и тяжелых симптомов острой тирозинемии, плохо поддается диетотерапии, по мнению ряда авторов, обусловлен гиперпродукцией инсулина, что нами не подтверждено ни в одном случае НТ1А.
Классификация
В основе классификации НТ1 лежат клинические варианты ее течения. НТ1А характеризуется острым течением, ранним дебютом от двух до пяти – семи месяцев, встречается в три раза чаще, чем тип Б с подострым или хроническим течением. НТ1А характеризуется задержкой развития, фебрильной лихорадкой, рвотой, диареей, гепатомегалией. Живот увеличен, напряжен как за счет гепатомегалии, в меньшей степени спленомегалии, так и за счет асцита и динамической непроходимости. На стадии острого гепатита не исключена желтуха. При остром желудочно-кишечном кровотечении возможна мелена, рвота кофейной гущей. При снижении белково-синтетической функции печени появляются безбелковые отеки, анасарка, кровотечения.
Причиной летального исхода при естественном течении НТ1А обычно становится острая печеночная недостаточность и катастрофическое кровотечение на ее фоне. Диагноз затрудняется такими сопутствующими состояниями, как внутриутробная инфекция, неонатальный гепатит, сепсис.
НТ1Б протекает легче, особенно когда дети отказываются от белковой пищи, а родители не настаивают на ее приеме. Гепатомегалия выявляется у всех детей. Поражение почек определяет различную степень рахита от минимальной гипофосфатемии и вальгусной или варусной деформации ног до тяжелой деформации скелета, позвоночника, грудной клетки, вплоть до полной потери опорной способности. Кардиомиопатия и артериальная гипертензия проявляются в результате метаболических процессов и вторично на фоне поражения почек.
Еще одно осложнение НТ1 – ГЦК. Частота ее развития в 40 раз превышает популяционную.
Диагностика
1. Патогномоничный признак НТ1 – высокий уровень сукцинилацетона в моче (норма 0–2,0 ммоль/моль креатинина) и плазме крови. Однако сукцинилацетон наблюдается в крови и моче при обратимой ингибиции фермента (например, белковом перекорме, приводящем к функциональной недостаточности FAH, кормлении неразведенным козьим молоком).
2. Повышенное содержание ароматических аминокислот (тирозина, метионина, пролина, фенилаланина и др.), определяемых методом тандемной масс-спектрометрии, появление «капустного» запаха, который скорее отражает печеночно-клеточную недостаточность.
3. АФП – маркер пролиферации желчных ходов, при НТ1Б повышен в десятки, а при НТ1А в тысячи раз. Повышение АФП – не специфичный, но чувствительный признак. При нормальном уровне АФП диагноз НТ1 сомнителен (АФП у детей от нуля до трех месяцев менее 1000 нг/мл, от трех месяцев до 18 лет – менее 12 нг/мл).
4. Косвенный признак НТ1 – повышение дельта-аминолевуленовой кислоты, поскольку сукцинилацетон ингибирует дегидрогеназу дельта-аминолевуленовой кислоты в печени и эритроцитах.
5. Генетическое исследование – подтверждающий метод. Выявление двух мутаций (гомозиготы или компаунд-гетерозиготы) верифицирует диагноз.
Лечение
До 1992 г. единственным радикальным методом лечения НТ1 считалась трансплантация печени (охватывала только 10% детей, в 10–20% случаев имела место послеоперационная смертность). Даже при удачной трансплантации поражение почек прогрессировало. Требовалась пожизненная иммуносупрессивная терапия. Редуцирующая безбелковая диета лишь тормозила прогрессирование заболевания и позволяла дожить до трансплантации.
На протяжении 22 лет в мире в качестве единственного патогенетического средства лечения НТ1 используется нитизинон (Орфадин, Swedish Orphan Biovitrum (Швеция)). Аналогов Орфадина в мире не существует. Союз педиатров России рекомендует использовать его в лечении НТ1 до развития ГЦК.
Доза подбирается индивидуально в зависимости от эффективности, которая оценивается по уровню сукцинилацетона в моче и крови и уровню тирозина в сыворотке крови. Обычная терапевтическая доза составляет 1 мг/кг/сут, начальная доза – 1–1,5 мг/кг/сут, если уровень тирозина менее 600–800 мкмоль/л, но не выше 2 мг/кг/сут. Суточная доза применяется внутрь однократно. Увеличение дозы до 2 мг/кг/сут возможно при отсутствии эффекта (не улучшаются показатели коагулограммы, уровень сукцинилацетона не снижается в течение двух недель, уровень тирозина превышает 800 мкмоль/л). Чем выше уровень тирозина в крови, тем жестче должны быть ограничения белка в диете (не более 1 г/кг). Потребность в других незаменимых аминокислотах восполняется назначением лечебного питания (до 2 г/кг) по специальным формулам, не содержащим тирозина и его предшественника фенилаланина. Это позволяет добиться нормального роста и развития. Доза нитизинона корректируется в зависимости от биохимических показателей и нарастания массы тела ребенка. Около 10% больных на терапию нитизиноном не отвечают, что обусловливает необходимость биохимического мониторинга (уровень сукцинилацетона в моче, функциональные пробы печени, АФП). Такие пациенты – потенциальные претенденты на трансплантацию печени [10, 11, 12].
Нитизинон блокирует аномальный распад тирозина, способствует переводу НТ1 в НТ2. При этом содержание тирозина увеличивается, но интоксикации продуктами его распада не происходит. Снижение концентрации тирозина в сыворотке достигается низкобелковой диетой и применением специализированного диетического продукта.
До начала терапии нитизиноном в период предполагаемого диагноза, когда уже требуется ограничение белка, но еще не верифицирована НТ1 у детей первых двух-трех месяцев жизни, целесообразно продолжать грудное вскармливание или изыскать возможность донорского вскармливания. У детей трех – пяти месяцев следует ограничить объем вскармливания на 1/3 с заменой частичным парентеральным питанием инфузиями 10%-ной глюкозы (150 мл/кг/сут). Детям старше пяти месяцев вводят прикормы на воде (1,4 г белка/100 мл смеси). Пересматривая питание, исходят из расчета 1,5 г белка на 1 кг веса ребенка. Калорическая недостаточность восполняется жирами.
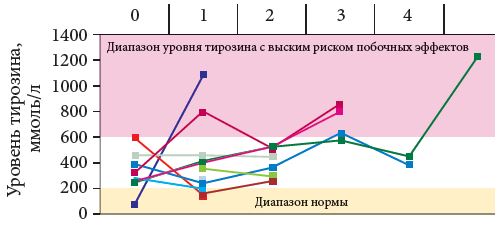
После назначения нитизинона в дозе 1–2 мг/1 кг веса коррекция белка проводится из расчета 2 г/1 кг веса. После начала терапии нитизиноном соотношение белка пищевого и белка тирозидона составляет 1:1. Средняя потребность в белке колеблется от 1,5 до 2,0 г/1 кг веса. Дефицит незаменимых аминокислот мониторируется как минимум один раз в полгода после изучения аминокислотного спектра сыворотки любым способом (тандемной масс-спектрометрией, АК-анализатором). Последующая коррекция белкового дефицита проводится при снижении фенилаланина менее 20 мкмоль/л (норма 20–125 мкмоль/л). При концентрации тирозина выше 600 мкмоль/л высока вероятность побочных явлений в виде фоточувствительности глаз вследствие отложения кристаллов тирозина в роговице. Это требует более четкого соблюдения рекомендаций по ограничению белков.
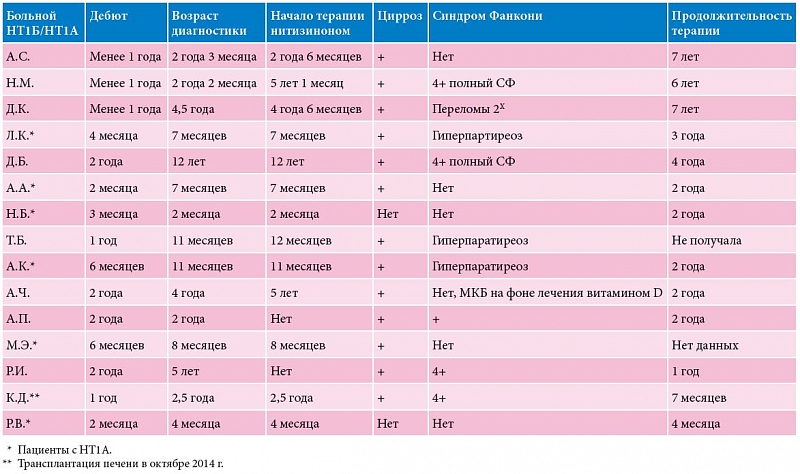
План мониторирования других биохимических параметров направлен главным образом на контроль уровня АФП, поскольку риск развития ГЦК сохраняется, несмотря на его многократное снижение. В тех же целях проводится систематически ультразвуковое исследование (УЗИ), компьютерная томография (КТ) и магнитно-резонансная томография (МРТ). Периодичность исследований отражена в табл. 1.
Важный момент: в доступной литературе нет сведений о скорости рецидива НТ1 при отмене терапии. На сегодняшний день трое из восьми детей прервали терапию из-за того, что органами местного здравоохранения не закупается жизненно необходимое лекарство детям-инвалидам.
Оценка и контроль эффективности терапии
Чем раньше установлен диагноз, тем лучше результат лечения. У 90% пациентов на фоне приема нитизинона печеночная недостаточность становится контролируемой, внепеченочные проявления, как правило, купируются. Нитизинон обладает хорошей переносимостью, ожидаемое повышение в плазме крови уровня тирозина клинически не проявляется у большинства детей. Опыт применения нитизинона у детей, начавших лечение им в возрасте до двух месяцев, показал, что риск ГЦК снижается с 40 до 5%.
Контроль терапии Орфадином включает определение уровня фенилаланина и тирозина. При низком (менее 20 мкмоль/л) уровне фенилаланина необходимо увеличить применение белка. Уровень тирозина желательно удерживать в диапазоне 200–400 мкмоль/л во избежание развития побочных эффектов. Последние связаны с офтальмологическими симптомами в результате раздражения роговицы и склер высокой концентрацией тирозина (фотобоязнь, ощущение песка в глазах).
УЗИ брюшной полости надлежит проводить каждые шесть месяцев, а МРТ печени ежегодно. При выявлении подозрительных узловых образований показаны методы визуализации с контрастированием (КТ, МРТ, радиоизотопное исследование). Это позволит подтвердить ГЦК. Уровень АФП следует контролировать каждые три – шесть месяцев. Повторное повышение АФП служит поводом для углубленного обследования на предмет ГЦК.
Заболевание детей, имеющих печеночную недостаточность, часто протекает крайне тяжело с выраженной коагулопатией и асцитом. Применение нитизинона обычно приводит к ощутимому клиническому улучшению в течение нескольких дней. 90% пациентов отвечают на терапию. Если профиль коагуляции не улучшается в течение одной недели, дозу нитизинона повышают. У большинства детей с клиническими проявлениями цирроза и портальной гипертензии лечение приводит к компенсации и даже регрессу цирротических изменений.
Функция почечных канальцев нормализуется, прекращаются потери кальция и фосфора с мочой.
Лечение нитизиноном в течение первых трех месяцев жизни предотвращает развитие кардиомиопатии. При более позднем начале ее течение улучшается независимо от формы заболевания.
Применение нитизинона обеспечивает полное купирование неврологических кризов по типу порфирии.
Задержка развития не является следствием НТ1. Однако терапия нитизиноном вызывает развитие фенотипа НТ2 (рис. 1). Естественный ход развития НТ2 представляет собой генетический дефицит 4-гидроксифенилпируват-диоксигеназы, что встречается чрезвычайно редко. Но задержка интеллектуального развития является компонентом в структуре данного заболевания. В этой связи вполне обоснованно рекомендовать регулярный контроль уровня развития у детей с НТ1, получающих нитизинон.
Показания к трансплантации печени, в том числе при лечении нитизиноном
В состоянии острой печеночной недостаточности трансплантацию печени проводят, если профиль коагуляции не улучшается через одну неделю лечения.
При хронической печеночной недостаточности показаниями для трансплантации печени служат в основном подозрения на ГЦК, а именно рецидив повышения уровня АФП.
У детей старше двух лет, не принимающих нитизинон, риск развития ГЦК остается высоким (40%). В этом случае тактика спорна – проводить трансплантацию или отложить ее до появления признаков ГЦК. Рекомендации сугубо индивидуальны.
Рецидив нарастания АФП в сыворотке крови требует комплексного исследования, при котором особое значение придается различным методам визуализации (МРТ, КТ) подозрительного на ГЦК объемного образования печени и морфологическому исследованию ткани печени, подтверждающему ГЦК.
Характеристики детей, находящихся под нашим наблюдением
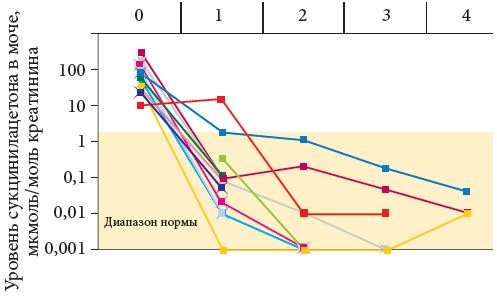
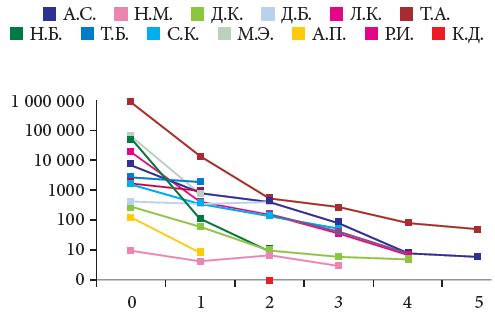
Диагноз НТ1 установлен у 18 детей с 2006 по 2014 г. (табл. 2). Трое из них умерли до того, как появилась возможность применения терапии Орфадином. У всех троих отмечалось подострое течение заболевания с дебютом до одного года. В то же время поздняя госпитализация через несколько месяцев от дебюта сочеталась с регрессом навыков, гипогликемией и острым течением рахита. На рис. 2, 3 и 4 представлены наиболее значимые параметры – данные до начала терапии, через месяц терапии и далее через шесть месяцев. Эти параметры наиболее значимы: с одной стороны, они подтверждают диагноз, с другой – отражают эффективность терапии. АФП – наиболее специфичный маркер вероятной ГЦК.
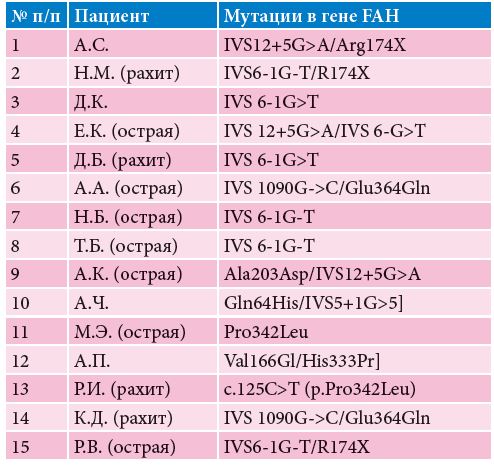
В таблице 3 показаны мутации, обнаруженные у наших пациентов. В лаборатории НБО МГНЦ РАМН установлена ранее не описанная мутация в гене FAH у одного больного якутского происхождения. Затем та же мутация была выявлена у детей из Бурятии и Дагестана. У остальных пациентов обнаружены ранее описанные мутации.
Собственные данные о результатах эффективности лечения НТ1 нитизиноном
Первой лечение нитизиноном в нашей стране начала (4 февраля 2008 г.) пациентка А.С. 2005 года рождения. Это произошло до момента регистрации препарата. Было получено разрешение локального этического комитета и информированное согласие матери. Терапия была начата на основании приказа Минздравсоцразвития России от 09.08.2005 № 494 «О порядке применения лекарственных средств у больных по жизненным показаниям». В результате проведенного лечения отмечались стабилизация состояния, частичный регресс цирроза и портальной гипертензии. Восстановился минеральный обмен. Развитие девочки стало соответствовать возрасту.
Продолжительность терапии составляет от четырех месяцев до 6,5 года. У троих детей отмечается перерыв в лечении, что до настоящего времени чревато развитием осложнений, поскольку высокие концентрации тирозина способны привести к тирозинемическому кризу. Наблюдение продолжается. Надеемся, что постановление Правительства РФ от 26.04.2012 № 403 «О порядке ведения федерального регистра лиц, страдающих жизнеугрожающими и хроническими прогрессирующими редкими (орфанными) заболеваниями, приводящими к сокращению продолжительности жизни граждан или их инвалидности, и его регионального сегмента» будет реализовано, и детям будет гарантировано бесперебойное лечение.
Эффективность лечения российских детей с поздно установленным диагнозом
Кроме основного метаболита – сукцинилацетона (рис. 2), собственно тирозина (рис. 3), онкомаркера – АФП (рис. 4), изучали уровни ГГТП, активность трансаминаз, ЩФ и других клинических и биохимических параметров крови, отражающих эффективность лечения НТ1. Нами систематически определяются биохимические параметры мочи – кальций- и фосфор-креатиновые коэффициенты, глюкозурия, проводится функциональная проба по Зимницкому, а также кислотно-основное состояние крови, показатели коагулограммы, в частности витамин К-зависимых факторов, гормоны костного метаболизма и уровень общего и ионизированного кальция и фосфора в крови.
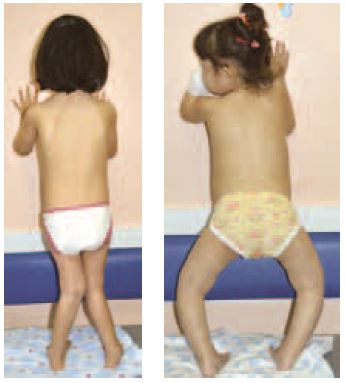
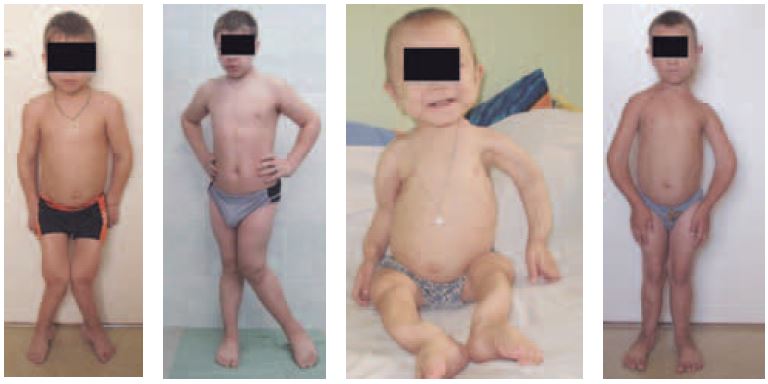
Из 15 детей у четверых (двух мальчиков и двух девочек) отмечались тяжелые деформации скелета с потерей опоры на ноги, саблевидная деформация трубчатых костей, сколиоз позвоночника и грубая варусная деформация стоп, что явилось результатом вторичного синдрома Фанкони, витамин D-резистентного рахита. Кроме того, у одного ребенка имели место сложная деформация коленных суставов, вывих коленных чашечек и тазобедренных суставов. За год терапии нитизиноном на фоне коррекции кислотно-щелочного состояния, назначения активных метаболитов витамина D (кальцитриола), препаратов кальция и двуосновных фосфатов купирован почечный канальцевый ацидоз, вторичный гиперпаратиреоз, восстановился минеральный обмен, нормализовались минеральная плотность костей (по данным денситометрии) и костный возраст. У одного ребенка отмечалась грубая деформация грудной клетки, приведшая к дыхательной недостаточности IIБ, которая после года терапии и эффективной дыхательной гимнастики с повышенным сопротивлением на выдохе привела к восстановлению вентиляционных показателей. Через год лечения нитизиноном нам совместно с сотрудниками ортопедического отделения удалось выполнить успешные операции корригирующей остеотомии у двух мальчиков и через полгода у одной девочки (рис. 5, 6 и 7).
У всех детей по данным фиброэластометрии печени отмечено снижение плотности печеночной паренхимы, что не свойственно циррозам печени другой этиологии. От контрольной биопсии печени по этическим соображениям, получив положительную динамику по другим параметрам, мы воздерживаемся.
Заключение
При анализе мониторирования биохимических показателей у детей, не прерывавших терапию, можно сделать следующие выводы.
Во-первых, ни у одного ребенка не выявлено побочных эффектов.
Во-вторых, лечение эффективно как на стадии цирроза (НТ1Б), так и на стадии острой печеночной недостаточности (НТ1А).
Во-вторых, хороший эффект получен даже при позднем старте терапии (НТ1Б). Но, конечно, чем раньше начата терапия, тем лучше результат.
В-третьих, в течение года регрессирует тубулопатия и восстанавливается минеральная плотность костей вследствие снижения потерь кальция и фосфора с мочой.
A 7-Year Experience of Therapy with Nitisinone of Hereditary Type 1 Tyrosinemia in Russia
L.S. Namazova-Baranova, S.I. Polyakova, T.E. Borovik, T.V. Bushuyeva, M.A. Varichkina, A.K. Gevorkyan
Scientific Center of Children’ s Health under the Russian Academy of Medical Sciences
Contact person: Svetlana Igorevna Polyakova, polyakova1963@list.ru
The main purpose of the current publication was to attract attention of medical audience to the problems related to hereditary tirosinemia. We refer to a timely and uninterrupted medical supply of such patients, the earliest possible diagnostics, by integrating efforts from physicians of different medical specialties (hepatologists, resuscitators, gastroenterologists, orthopedic surgeons, nephrologists, and neurologists) for development of appropriate individualized curative tactics.
Уважаемый посетитель uMEDp!
Уведомляем Вас о том, что здесь содержится информация, предназначенная исключительно для специалистов здравоохранения.
Если Вы не являетесь специалистом здравоохранения, администрация не несет ответственности за возможные отрицательные последствия, возникшие в результате самостоятельного использования Вами информации с портала без предварительной консультации с врачом.
Нажимая на кнопку «Войти», Вы подтверждаете, что являетесь врачом или студентом медицинского вуза.