Дефицит лизосомной кислой липазы: клиническая и патоморфологическая характеристика
- Аннотация
- Статья
- Ссылки
- English
История открытия болезней накопления липидов
В 1882 г. французский врач Филипп Гоше впервые описал неизвестное ранее заболевание у 32-летней женщины с выраженной спленомегалией и необычными большими клетками в селезенке. Болезнь Гоше стала первым из описанных заболеваний, относящихся к лизосомным болезням накопления, и дала толчок последующему изучению энзимных, генетических и молекулярных нарушений в данной группе. Открытие Филиппа Гоше позволило составить фенотипическую характеристику сходных заболеваний и обосновать заместительную ферментную терапию.
На сегодняшний день известно свыше 50 лизосомных болезней накопления, вошедших в перечень орфанных (редких). Термин «орфанные заболевания» появился в 1983 г. в США. Орфанные заболевания имеют аутосомно-рецессивный тип наследования, встречаются в среднем с частотой 1/75 000 (частота ряда орфанных заболеваний – 1/350 000–1/700 000) и отличаются неблагоприятным прогнозом. Наиболее изученными считаются лизосомные болезни накопления липидов: болезнь Гоше, болезнь Ниманна – Пика и дефицит лизосомной кислой липазы (ДЛКЛ).
Врожденный дефицит лизосомной кислой липазы
ДЛКЛ у младенцев впервые был описан в 1956 г. A. Abramov, S. Schorr и M. Wolman. В 1961 г. M. Wolman, V.V. Sterk, S. Gatt, M. Frenkel дали полное клиническое описание болезни у трех сибсов. Болезнь получила название болезни Вольмана в честь невролога Moshe Wolman (1914–2009). В 1963 г. был описан относительно благоприятный вариант течения ДЛКЛ – болезнь накопления эфиров холестерина. В 1969 г. A.D. Patrick и B.D Lake, а в 1972 г. J.A. Burkle, W.K. Schubert, H.R. Sloan и D.S. Fredrickson [1] установили причину болезни: дефицит фермента кислой липазы в лизосомах обусловлен мутацией гена LIPA, локализованного на 10-й хромосоме, локус 10q23.2–23.3 [2]. Врожденный ДЛКЛ в 1990 г. был подробно описан и у крыс [3]. В 2015 г. был зарегистрирован препарат для заместительной ферментной терапии себелипаза-альфа (Канума), позволивший значительно улучшить прогноз болезни [4–8].
ДЛКЛ до настоящего времени остается мало изученным не только в нашей стране, но и за рубежом. Не случайно каждый вновь описанный подтвержденный случай ДЛКЛ тщательно анализируется для уточнения клинических и морфологических особенностей течения у детей. Генетический анализ не является ведущим в диагностике ДЛКЛ, поскольку описано свыше 40 мутаций гена LIPA [9]. Основным подтверждением диагноза служит определение активности лизосомной кислой липазы в сухих пятнах крови.
Значение фермента лизосомной кислой липазы
Лизосомная кислая липаза является ключевым ферментом, участвующим в обмене липидов, катализирующим гидролиз эфиров холестерина и триглицеридов из липопротеинов низкой плотности в лизосомах, обеспечивая клетки свободным холестерином [9, 10]. В свою очередь свободный холестерин участвует в синтезе компонентов клеточных мембран, стероидных гормонов и желчных кислот. Следует отметить, что по химическим свойствам холестерин относится к природным полициклическим липофильным спиртам и в англоязычной литературе именуется холестеролом. В отечественной литературе сохраняется старое название (холестерин), предложенное в 1815 г. Мишелем Шеврелем.
Холестерин составляет до 30–40% общего количества липидов клетки, 60–80% распределены в плазматической мембране, 0,5–1,0% холестерина входят в структуру эндоплазматического ретикулума. При ДЛКЛ эфиры холестерина кумулируются в различных органах и тканях (печени, селезенке, эпителии кишечника, лимфатических узлах, надпочечниках, эндотелии капилляров, клетках макрофагально-моноцитарной системы), для которых высокая активность фермента считается нормой.
Клинические варианты ДЛКЛ
Дефицит лизосомной кислой липазы имеет два клинических проявления: болезнь Вольмана (другое название – первичный семейный ксантоматоз с вовлечением и кальцификацией надпочечников) и болезнь накопления эфиров холестерина.
Болезнь Вольмана, или инфантильная форма ДЛКЛ, характеризуется крайне неблагоприятным прогнозом: манифестация болезни отмечается уже в первый месяц жизни ребенка, средняя продолжительность жизни составляет 3,7 месяца [11]. Уровень липазы при болезни Вольмана снижен в 200 и более раз.
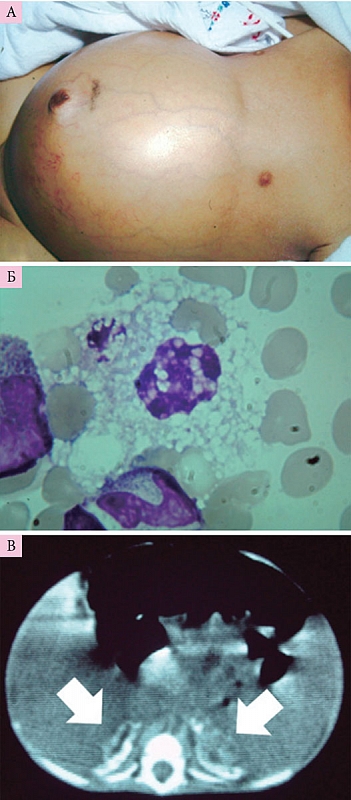
Общие клинические характеристики болезни Вольмана (рис. 1):
- увеличение размеров живота;
- тяжелая интестинальная мальабсорбция;
- анемия; гепатомегалия; гепатоспленомегалия (79% детей);
- диффузные кальцификаты надпочечников (патогномоничный признак);
- повышение уровня сывороточных трансаминаз;
- прогрессирующий печеночный фиброз и цирроз;
- задержка физического развития;
- быстро прогрессирующее течение болезни;
- наличие рвоты, диареи, стеатореи, изменений в биохимическом анализе крови (у 43% детей первые симптомы имеют место уже на первом месяце жизни);
- неблагоприятный исход в среднем в первые шесть месяцев жизни. В отсутствие патогенетического лечения к году умирают все дети. В исследовании 67% детей, получавших ферментозаместительную терапию, выжили, перешагнув 12-месячный рубеж [12].
В биохимическом анализе крови детей с болезнью Вольмана отмечаются повышенные уровни аланинаминотрансферазы (среднее значение – 56,5 Ед/л) (53,3% случаев), аспартатаминотрансферазы (среднее значение – 151 Ед/л) (94,7%), гамма-глутамилтранспептидазы (84,6%), билирубина (52%). В подавляющем большинстве случаев уровень холестерина в норме (1,54–4,58 ммоль/л). Повышенный уровень холестерина (> 5,17 ммоль/л) наблюдается в 5% случаев. Дети с повышенным уровнем холестерина не имеют задержки физического развития, продолжительность жизни может достигать 12 месяцев (при условии дальнейшей трансплантации печени).
Болезнь накопления эфиров холестерина
Болезнь имеет более медленный прогрессирующий характер и развивается не так стремительно, как инфантильная форма. Манифестация болезни возможна в любом возрасте, но у большинства пациентов начинается в детском возрасте. Вероятно, многие пациенты не доживают до взрослого возраста из-за развития жизнеугрожающих состояний (цирроза печени, печеночной недостаточности, атеросклероза, сердечно-сосудистых осложнений). Уровень фермента лизосомной кислой липазы может быть снижен в 50–100 раз. Как правило, уровень липазы в лейкоцитах периферической крови не превышает 5% нормы [13].
Болезнь накопления эфиров холестерина относится к прогрессирующим, метаболическим заболеваниям печени с аутосомно-рецессивным типом наследования. ДЛКЛ приводит к первичному накоплению эфиров холестерина и триглицеридов в гепатоцитах и макрофагах. Как следствие – развитие гепатомегалии, микровезикулярного стеатоза, цирроза, дислипидемии, раннего атеросклероза. Клиническая манифестация болезни возможна как на первом году жизни, так и в более старшем возрасте. Терапия статинами не приводит к обратному развитию болезни. Единственным методом лечения с доказанной эффективностью является заместительная ферментная терапия.
При болезни накопления эфиров холестерина в печени и селезенке накапливаются липиды. Уровень холестерина в ткани печени превышает норму в 70 раз и более. Отмечаются дислипидемия (повышение уровней общего холестерина, липопротеинов низкой плотности, иногда триглицеридов, снижение липопротеинов высокой плотности). Накопление липидов в кишечнике может вызывать нарушения функций ЖКТ. Накопление нейтрального жира и эфиров холестерина в артериях вызывает ранний атеросклероз. Гепатомегалия, печеночный фиброз, цирроз – результат накопления эфиров холестерина и триглицеридов в макрофагах и гепатоцитах.
Морфологические признаки ДЛКЛ
Болезнь Вольмана. В случае если удается провести прижизненную биопсию печени, у всех детей выявляются признаки фиброза и/или стеатоза. При аутопсии характерными признаками являются большие печень и селезенка, желто-оранжевая окраска печени, некроз гепатоцитов, фиброз и цирроз, большие надпочечники с микрокальцификатами.
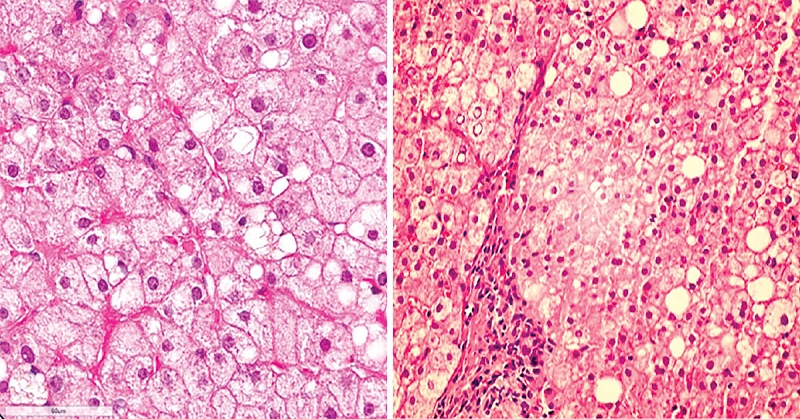
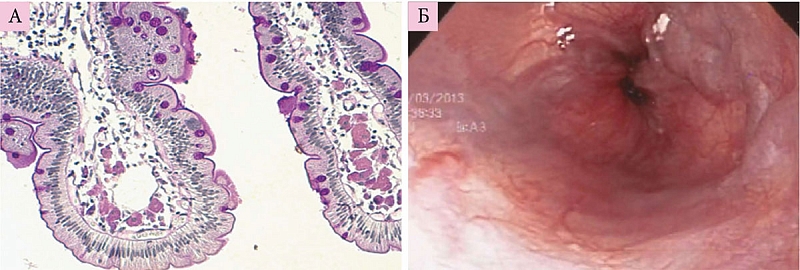
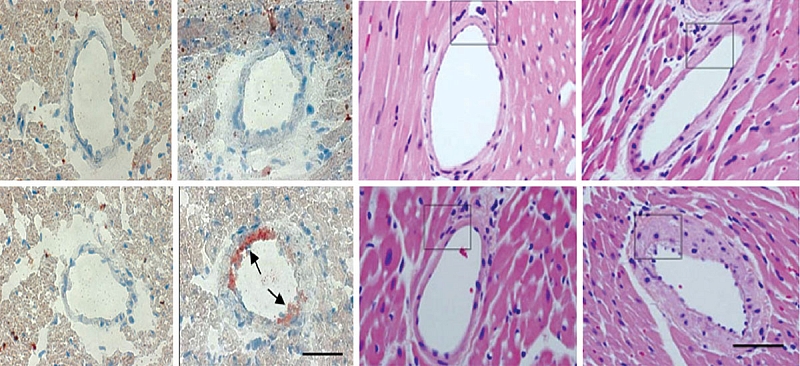
Болезнь накопления эфиров холестерина. В биоптате печени всех пациентов отмечаются характерные патологические прогрессирующие признаки: микровезикулярный стеатоз, способствующий развитию фиброза, микронодулярного цирроза и в конечном итоге печеночной недостаточности (рис. 2), наличие гистиоцитов с цероидным материалом в lamina propria ворсинок дуоденального биоптата (рис. 3) [2], формирование атеросклеротических бляшек в артериях [14], изменение стенок артерий (рис. 4) [14].
Диагностика ДЛКЛ
С учетом тесной взаимосвязи клинической и патоморфологической картины ДЛКЛ необходимы как можно более ранняя диагностика этого редкого заболевания и своевременное назначение заместительной ферментной терапии. Ключом к ранней идентификации ДЛКЛ и ранней постановке диагноза являются высокий уровень липопротеинов низкой плотности, низкий уровень липопротеинов высокой плотности в течение длительного периода времени и повышенные уровни трансаминаз. Необходимо помнить, что ДЛКЛ характеризуется прогрессирующим нарушением функций печени, часто приводящим к трансплантации. Для любого течения болезни характерно наличие стеатоза, нередко в сочетании с фиброзом и циррозом [4, 14]. ДЛКЛ необходимо исключать у пациентов с неалкогольной жировой болезнью печени и неалкогольным стеатогепатитом.
Основным лабораторным методом диагностики ДЛКЛ является определение активности кислой липазы в сухих пятнах крови. В России такое исследование проводят в нескольких научно-медицинских центрах, в том числе в Москве в НЦЗД и МГНЦ.
Ye.L. Tumanova, A.N. Goryaynova, A.N. Grishina
N.I. Pirogov Russian National Research Medical University
Russian Academy of Continuing Professional Education
Contact person: Aleksandra Nikitichna Goryaynova, alex.goriaynowa@yandex.ru
Deficiency of lysosomal acidic lipase (LAL-D) refers to a group of rare diseases of lipids accumulation, inherited in an autosomal recessive type. The disease has a progressive course, based on the violation of metabolism of cholesterol esters and triglycerides of low-density lipoproteins as a result of deficiency or complete absence of the enzyme lysosomal acid lipase. LAL-D has two clinical variants: Wolman disease (infantile form) and the disease is the accumulation of cholesterol esters. In Russia, LAL-D is being diagnosed rarely. The article presents the current literature data, and as well for the first time presents the results of domestic studies of morphological features of LAL-D in children, performed under the guidance of Ye.L. Tumanova, a professor.
Уважаемый посетитель uMEDp!
Уведомляем Вас о том, что здесь содержится информация, предназначенная исключительно для специалистов здравоохранения.
Если Вы не являетесь специалистом здравоохранения, администрация не несет ответственности за возможные отрицательные последствия, возникшие в результате самостоятельного использования Вами информации с портала без предварительной консультации с врачом.
Нажимая на кнопку «Войти», Вы подтверждаете, что являетесь врачом или студентом медицинского вуза.