–Ґ—А–Є—Е–Њ–≥–µ–њ–∞—В–Њ—Н–љ—В–µ—А–∞–ї—М–љ—Л–ЄћЖ —Б–Є–љ–і—А–Њ–Љ
- –Р–љ–љ–Њ—В–∞—Ж–Є—П
- –°—В–∞—В—М—П
- –°—Б—Л–ї–Ї–Є
- English
–†–∞—Б–њ—А–Њ—Б—В—А–∞–љ–µ–љ–љ–Њ—Б—В—М
—В—А–Є—Е–Њ–≥–µ–њ–∞—В–Њ—Н–љ—В–µ—А–∞–ї—М–љ–Њ–≥–Њ —Б–Є–љ–і—А–Њ–Љ–∞
–Ґ—А–Є—Е–Њ–≥–µ–њ–∞—В–Њ—Н–љ—В–µ—А–∞–ї—М–љ—Л–ЄћЖ —Б–Є–љ–і—А–Њ–Љ (—Б–Є–љ–і—А–Њ–Љ–љ–∞—П, –Є–ї–Є —Д–µ–љ–Њ—В–Є–њ–Є—З–µ—Б–Ї–∞—П, –і–Є–∞—А–µ—П) –±—Л–ї –≤–њ–µ—А–≤—Л–µ –Њ–њ–Є—Б–∞–љ –≤¬†1982¬†–≥.¬†L. Stankler –Є¬†—Б–Њ–∞–≤—В. —Г¬†–і–≤—Г—Е —Б–Є–±–ї–Є–љ–≥–Њ–≤ —Б¬†–љ–Є–Ј–Ї–Є–Љ –≤–µ—Б–Њ–Љ –њ—А–Є —А–Њ–ґ–і–µ–љ–Є–Є, –і–Є—Б–Љ–Њ—А—Д–Є—З–љ—Л–Љ–Є —З–µ—А—В–∞–Љ–Є –ї–Є—Ж–∞, –њ–ї–Њ—Б–Ї–Є–Љ —И–Є—А–Њ–Ї–Є–Љ –љ–Њ—Б–Њ–Љ, –≤—Л—Б—В—Г–њ–∞—О—Й–Є–Љ –ї–±–Њ–Љ, —В—П–ґ–µ–ї–Њ–є –і–Є–∞—А–µ–µ–є [1]. –Ч–љ–∞—З–Є—В–µ–ї—М–љ–∞—П —З–∞—Б—В—М –Њ–њ—Г–±–ї–Є–Ї–Њ–≤–∞–љ–љ—Л—Е –≤¬†–Љ–Є—А–Њ–≤–Њ–є –ї–Є—В–µ—А–∞—В—Г—А–µ –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є—Е —Б–ї—Г—З–∞–µ–≤ —Б–Њ–±—А–∞–љ–∞ –Є¬†–Њ–њ–Є—Б–∞–љ–∞ —Д—А–∞–љ—Ж—Г–Ј—Б–Ї–Є–Љ–Є –≤—А–∞—З–∞–Љ–Є –≤¬†2007 –Є¬†2013¬†–≥–≥. [2, 3]. –Т¬†–љ–∞—Б—В–Њ—П—Й–µ–µ –≤—А–µ–Љ—П –њ—А–µ–і—Б—В–∞–≤–ї–µ–љ–Њ –≤—Б–µ–≥–Њ –Њ–Ї–Њ–ї–Њ 80 —Б–ї—Г—З–∞–µ–≤ –і–∞–љ–љ–Њ–≥–Њ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П. –Х–≥–Њ —А–∞—Б–њ—А–Њ—Б—В—А–∞–љ–µ–љ–љ–Њ—Б—В—М —Б–Њ—Б—В–∞–≤–ї—П–µ—В –Њ–Ї–Њ–ї–Њ 1:1 000 000 –љ–Њ–≤–Њ—А–Њ–ґ–і–µ–љ–љ—Л—Е [4, 5]. –Т¬†2015¬†–≥.¬†–±—Л–ї–Є –њ—А–Є–≤–µ–і–µ–љ—Л –њ–µ—А–≤—Л–µ –і–≤–∞ –Њ–њ–Є—Б–∞–љ–Є—П —В—А–Є—Е–Њ–≥–µ–њ–∞—В–Њ—Н–љ—В–µ—А–∞–ї—М–љ–Њ–≥–Њ —Б–Є–љ–і—А–Њ–Љ–∞ –≤¬†–†–Њ—Б—Б–Є–Є [6].
–У–µ–љ–µ—В–Є—З–µ—Б–Ї–Њ–µ –Ї–Њ–љ—Б—Г–ї—М—В–Є—А–Њ–≤–∞–љ–Є–µ
–Ґ—А–Є—Е–Њ–≥–µ–њ–∞—В–Њ—Н–љ—В–µ—А–∞–ї—М–љ—Л–є —Б–Є–љ–і—А–Њ–Љ¬†вАУ –Њ—А—Д–∞–љ–љ–Њ–µ, –љ–∞—Б–ї–µ–і—Г–µ–Љ–Њ–µ –њ–Њ¬†–∞—Г—В–Њ—Б–Њ–Љ–љ–Њ-—А–µ—Ж–µ—Б—Б–Є–≤–љ–Њ–Љ—Г —В–Є–њ—Г¬†–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Њ–µ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–µ, —Б–≤—П–Ј–∞–љ–љ–Њ–µ —Б¬†–Љ—Г—В–∞—Ж–Є—П–Љ–Є –≤¬†–і–≤—Г—Е¬†–≥–µ–љ–∞—Е: TTC37 (5q15) –Є–ї–Є SKIV2L (6p21.3)¬†вАУ –Ї–Њ–Љ–њ–Њ–љ–µ–љ—В–∞—Е¬†–≥–Є–њ–Њ—В–µ—В–Є—З–µ—Б–Ї–Њ–≥–Њ –Њ—В–≤–µ—В—Б—В–≤–µ–љ–љ–Њ–≥–Њ –Ј–∞ —Н–Ї–Ј–Њ—Б–Њ–Љ–Њ–њ–Њ—Б—А–µ–і–Њ–≤–∞–љ–љ—Л–є –Ї–Њ–љ—В—А–Њ–ї—М –†–Э–Ъ Ski-–Ї–Њ–Љ–њ–ї–µ–Ї—Б–∞ —З–µ–ї–Њ–≤–µ–Ї–∞ [7вАУ10]. –°–Є–љ–і—А–Њ–Љ –Љ–Њ–ґ–µ—В –њ—А–Њ—П–≤–ї—П—В—М—Б—П —И–Є—А–Њ–Ї–Є–Љ —Б–њ–µ–Ї—В—А–Њ–Љ –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є—Е —Б–Є–Љ–њ—В–Њ–Љ–Њ–≤ –≤¬†–љ–µ–Њ–љ–∞—В–∞–ї—М–љ–Њ–Љ –њ–µ—А–Є–Њ–і–µ. –°¬†—Г—З–µ—В–Њ–Љ —Н—В–Њ–≥–Њ —Б–µ–Ї–≤–µ–љ–Є—А–Њ–≤–∞–љ–Є–µ –њ–Њ–ї–љ–Њ–≥–Њ —Н–Ї–Ј–Њ–Љ–∞ —Б—З–Є—В–∞–µ—В—Б—П –µ–і–Є–љ—Б—В–≤–µ–љ–љ—Л–Љ –і–Њ—Б—В–Њ–≤–µ—А–љ—Л–Љ –і–Є–∞–≥–љ–Њ—Б—В–Є—З–µ—Б–Ї–Є–Љ –Є–љ—Б—В—А—Г–Љ–µ–љ—В–Њ–Љ –≤¬†–µ–≥–Њ –Њ–њ—А–µ–і–µ–ї–µ–љ–Є–Є [8, 10].
–Я—А–Є –Ј–∞—З–∞—В–Є–Є –Љ–µ–ґ–і—Г¬†–≥–µ—В–µ—А–Њ–Ј–Є–≥–Њ—В–љ—Л–Љ–Є –љ–Њ—Б–Є—В–µ–ї—П–Љ–Є –і–∞–љ–љ–Њ–є –Љ—Г—В–∞—Ж–Є–Є –Ї–∞–ґ–і—Л–є —Б–Є–±—Б –Є–Љ–µ–µ—В 25%-–љ—Г—О –≤–µ—А–Њ—П—В–љ–Њ—Б—В—М —А–Њ–і–Є—В—М—Б—П —Б¬†–і–∞–љ–љ—Л–Љ —Б–Є–љ–і—А–Њ–Љ–Њ–Љ, 50%-–љ—Г—О –≤–µ—А–Њ—П—В–љ–Њ—Б—В—М¬†вАУ –±—Л—В—М –±–µ—Б—Б–Є–Љ–њ—В–Њ–Љ–љ—Л–Љ –љ–Њ—Б–Є—В–µ–ї–µ–Љ –Є¬†25%-–љ—Г—О¬†вАУ –±—Л—В—М –љ–µ–Ј–∞—В—А–Њ–љ—Г—В—Л–Љ –Є¬†–љ–µ –±—Л—В—М –љ–Њ—Б–Є—В–µ–ї–µ–Љ. –Я–Њ—Б–ї–µ —В–Њ–≥–Њ –Ї–∞–Ї –њ–∞—В–Њ–≥–µ–љ–љ—Л–µ –≤–∞—А–Є–∞–љ—В—Л –Є–і–µ–љ—В–Є—Д–Є—Ж–Є—А–Њ–≤–∞–љ—Л —Г¬†–њ–Њ—Б—В—А–∞–і–∞–≤—И–µ–≥–Њ —З–ї–µ–љ–∞ —Б–µ–Љ—М–Є, –≤–Њ–Ј–Љ–Њ–ґ–љ—Л —В–µ—Б—В–Є—А–Њ–≤–∞–љ–Є–µ —А–Њ–і—Б—В–≤–µ–љ–љ–Є–Ї–Њ–≤, –љ–∞—Е–Њ–і—П—Й–Є—Е—Б—П –≤¬†–≥—А—Г–њ–њ–µ —А–Є—Б–Ї–∞, –њ—А–µ–љ–∞—В–∞–ї—М–љ–Њ–µ —В–µ—Б—В–Є—А–Њ–≤–∞–љ–Є–µ –љ–∞¬†–±–µ—А–µ–Љ–µ–љ–љ–Њ—Б—В—М —Б¬†–њ–Њ–≤—Л—И–µ–љ–љ—Л–Љ —А–Є—Б¬≠–Ї–Њ–Љ –Є¬†–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Њ–µ —В–µ—Б—В–Є—А–Њ–≤–∞–љ–Є–µ –љ–∞¬†–њ—А–µ–і—Л–Љ–њ–ї–∞–љ—В–∞—Ж–Є—О [5].
–Ъ–ї–Є–љ–Є—З–µ—Б–Ї–Є–µ —Е–∞—А–∞–Ї—В–µ—А–Є—Б—В–Є–Ї–Є
–Ґ—А–Є—Е–Њ–≥–µ–њ–∞—В–Њ—Н–љ—В–µ—А–∞–ї—М–љ—Л–ЄћЖ —Б–Є–љ–і—А–Њ–Љ —Е–∞—А–∞–Ї—В–µ—А–Є–Ј—Г–µ—В—Б—П —Б–ї–µ–і—Г—О—Й–Є–Љ–Є –Њ—Б–љ–Њ–≤–љ—Л–Љ–Є —Б–Є–Љ–њ—В–Њ–Љ–∞–Љ–Є [6]:
- –Ј–∞–і–µ—А–ґ–Ї–∞ –≤–љ—Г—В—А–Є—Г—В—А–Њ–±–љ–Њ–≥–Њ —А–∞–Ј–≤–Є—В–Є—П –њ–ї–Њ–і–∞;
- —Е—А–Њ–љ–Є—З–µ—Б–Ї–∞—П –і–Є–∞—А–µ—П, –њ—А–Є–≤–Њ–і—П—Й–∞—П –Ї¬†–Ј–љ–∞—З–Є—В–µ–ї—М–љ—Л–Љ –Ї–Є—И–µ—З–љ—Л–Љ –њ–Њ—В–µ—А—П–Љ –Є¬†—В—П–ґ–µ–ї–Њ–є¬†–≥–Є–њ–Њ—В—А–Њ—Д–Є–Є;
- –∞–љ–Њ–Љ–∞–ї–Є—П —А–Њ—Б—В–∞ –≤–Њ–ї–Њ—Б (—Б—Г—Е–Є–µ –Є¬†–ї–Њ–Љ–Ї–Є–µ, —Б–Ї–ї–Њ–љ–љ—Л –Ї¬†–±—Л—Б—В—А–Њ–Љ—Г –≤—Л–њ–∞–і–µ–љ–Є—О);
- –ї–Є—Ж–µ–≤–Њ–є –і–Є—Б–Љ–Њ—А—Д–Є–Ј–Љ (–≤—Л—Б—В—Г–њ–∞—О—Й–Є–µ –ї–Њ–± –Є¬†—Й–µ–Ї–Є, —И–Є—А–Њ–Ї–Њ–µ –Њ—Б–љ–Њ–≤–∞–љ–Є–µ –љ–Њ—Б–∞, —И–Є—А–Њ–Ї–∞—П –њ–µ—А–µ–љ–Њ—Б–Є—Ж–∞);
- –≤—В–Њ—А–Є—З–љ—Л–є –Є–Љ–Љ—Г–љ–Њ–і–µ—Д–Є—Ж–Є—В (—Б–љ–Є–ґ–µ–љ–Є–µ –њ—А–Њ–і—Г–Ї—Ж–Є–Є –Є–Љ–Љ—Г–љ–Њ–≥–ї–Њ–±—Г–ї–Є–љ–Њ–≤ –≤—Б–µ—Е –Ї–ї–∞—Б—Б–Њ–≤);
- –Ј–∞–і–µ—А–ґ–Ї–∞ –њ—Б–Є—Е–Њ–Љ–Њ—В–Њ—А–љ–Њ–≥–Њ —А–∞–Ј–≤–Є—В–Є—П –Ї–∞–Ї —Б–ї–µ–і—Б—В–≤–Є–µ —В—П–ґ–µ–ї–Њ–є¬†–≥–Є–њ–Њ—В—А–Њ—Д–Є–Є;
- –Ї–Њ–ґ–љ—Л–µ –∞–љ–Њ–Љ–∞–ї–Є–Є (–≥–Є–њ–Њ- –Є¬†–≥–Є–њ–µ—А–њ–Є–≥–Љ–µ–љ—В–∞—Ж–Є—П) [7];
- —В—А–Њ–Љ–±–Њ—Ж–Є—В–Њ–Ј (—А–µ–і–Ї–Њ) [6, 8].
–Я—А–Є —Н–љ–і–Њ—Б–Ї–Њ–њ–Є—З–µ—Б–Ї–Њ–Љ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–Є –≤–µ—А—Е–љ–Є—Е –Њ—В–і–µ–ї–Њ–≤ –ґ–µ–ї—Г–і–Њ—З–љ–Њ-–Ї–Є—И–µ—З–љ–Њ–≥–Њ —В—А–∞–Ї—В–∞ –Њ–±–љ–∞—А—Г–ґ–Є–≤–∞—О—В –Є–Ј–Љ–µ–љ–µ–љ–Є—П, —Е–∞—А–∞–Ї—В–µ—А–љ—Л–µ –і–ї—П —Б–Є–љ–і—А–Њ–Љ–∞ –Љ–∞–ї—М–∞–±—Б–Њ—А–±—Ж–Є–Є [3, 6]. –†–µ–і–Ї–Є —Б–ї—Г—З–∞–Є, –Ї–Њ–≥–і–∞ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–µ –Є–Љ–µ–µ—В –Ї—А–Њ–љ–Њ–њ–Њ–і–Њ–±–љ–Њ–µ —В–µ—З–µ–љ–Є–µ [6].
–Ф–Є—Д—Д–µ—А–µ–љ—Ж–Є–∞–ї—М–љ–∞—П –і–Є–∞–≥–љ–Њ—Б—В–Є–Ї–∞
–Ф–Є—Д—Д–µ—А–µ–љ—Ж–Є–∞–ї—М–љ—Г—О –і–Є–∞–≥–љ–Њ—Б—В–Є–Ї—Г —Б–ї–µ–і—Г–µ—В –њ—А–Њ–≤–Њ–і–Є—В—М —Б¬†–і—А—Г–≥–Є–Љ–Є —Б–Є–љ–і—А–Њ–Љ–∞–Љ–Є –≤—А–Њ–ґ–і–µ–љ–љ–Њ–є —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–є –і–Є–∞—А–µ–Є, –њ—А–µ–ґ–і–µ –≤—Б–µ–≥–Њ –≤—А–Њ–ґ–і–µ–љ–љ–Њ–є —В–∞—Д—В–Є–љ–≥–Њ–≤–Њ–є —Н–љ—В–µ—А–Њ–њ–∞—В–Є–µ–є, IPEX-—Б–Є–љ–і—А–Њ–Љ–Њ–Љ (I¬†вАУ –Є–Љ–Љ—Г–љ–љ–∞—П –і–Є—Б—А–µ–≥—Г–ї—П—Ж–Є—П, P¬†вАУ –њ–Њ–ї–Є—Н–љ–і–Њ–Ї—А–Є–љ–Њ–њ–∞—В–Є—П, E¬†вАУ —Н–љ—В–µ—А–Њ–њ–∞—В–Є—П, X¬†вАУ —Б—Ж–µ–њ–ї–µ–љ–љ–∞—П —Б¬†X-—Е—А–Њ–Љ–Њ—Б–Њ–Љ–Њ–є), –њ–µ—А–≤–Є—З–љ—Л–Љ–Є –Є–Љ–Љ—Г–љ–Њ–і–µ—Д–Є—Ж–Є—В–∞–Љ–Є —Б¬†–Ї–Є—И–µ—З–љ—Л–Љ —Б–Є–љ–і—А–Њ–Љ–Њ–Љ [5].
–Ю—Б–Њ–±–Њ–µ –≤–љ–Є–Љ–∞–љ–Є–µ —Г–і–µ–ї—П—О—В —Б–Є–љ–і—А–Њ–Љ—Г –Љ–∞–ї—М–∞–±—Б–Њ—А–±—Ж–Є–Є, –≤—Л–Ј–≤–∞–љ–љ–Њ–Љ—Г –љ–µ–њ–µ—А–µ–љ–Њ—Б–Є–Љ–Њ—Б—В—М—О¬†–≥–ї—О—В–µ–љ–∞ –Є¬†–ї–∞–Ї—В–Њ–Ј—Л [8].
–Ы–µ—З–µ–љ–Є–µ
–Ы–µ—З–µ–љ–Є–µ –≤–Ї–ї—О—З–∞–µ—В –≤¬†—Б–µ–±—П –њ–∞—А–µ–љ—В–µ—А–∞–ї—М–љ–Њ–µ –њ–Є—В–∞–љ–Є–µ –Є¬†–≤ —А—П–і–µ —Б–ї—Г—З–∞–µ–≤ –≤–љ—Г—В—А–Є–≤–µ–љ–љ–Њ–µ –≤–≤–µ–і–µ–љ–Є–µ —З–µ–ї–Њ–≤–µ—З–µ—Б–Ї–Њ–≥–Њ –Є–Љ–Љ—Г–љ–Њ–≥–ї–Њ–±—Г–ї–Є–љ–∞ [4].
–Ф–µ—В–Є, –љ–µ¬†–њ–Њ–ї—Г—З–∞—О—Й–Є–µ –њ–∞—А–µ–љ—В–µ—А–∞–ї—М–љ–Њ–≥–Њ –њ–Є—В–∞–љ–Є—П, –і–Њ–ї–ґ–љ—Л –љ–∞—Е–Њ–і–Є—В—М—Б—П –њ–Њ–і –њ–Њ—Б—В–Њ—П–љ–љ—Л–Љ –љ–∞–±–ї—О–і–µ–љ–Є–µ–Љ –њ–µ–і–Є–∞—В—А–∞-–і–Є–µ—В–Њ–ї–Њ–≥–∞ –Є¬†–≥–∞—Б—В—А–Њ—Н–љ—В–µ—А–Њ–ї–Њ–≥–∞ –і–ї—П –Њ–±–µ—Б–њ–µ—З–µ–љ–Є—П –њ–∞—А–µ–љ—В–µ—А–∞–ї—М–љ–Њ–≥–Њ –њ–Є—В–∞–љ–Є—П –≤¬†—Б–ї—Г—З–∞–µ, –Ї–Њ–≥–і–∞ –і–µ—Д–Є—Ж–Є—В –њ–Є—В–∞—В–µ–ї—М–љ—Л—Е –≤–µ—Й–µ—Б—В–≤ —Б—В–∞–љ–µ—В –≤—Л—А–∞–ґ–µ–љ–љ—Л–Љ [4]. –Ъ—А–Њ–Љ–µ —В–Њ–≥–Њ, –љ–µ–Њ–±—Е–Њ–і–Є–Љ–Њ –µ–ґ–µ–≥–Њ–і–љ–Њ –њ—А–Њ–≤–Њ–і–Є—В—М [5]:
- —Г–ї—М—В—А–∞–Ј–≤—Г–Ї–Њ–≤–Њ–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ –±—А—О—И–љ–Њ–є –њ–Њ–ї–Њ—Б¬≠—В–Є –Є¬†–њ–Њ—З–µ–Ї;
- –Є–Ј–Љ–µ—А–µ–љ–Є–µ —Д–µ—А–Љ–µ–љ—В–Њ–≤ –њ–µ—З–µ–љ–Є (–∞—Б–њ–∞—А—В–∞—В–∞–Љ–Є–љ–Њ—В—А–∞–љ—Б—Д–µ—А–∞–Ј–∞, –∞–ї–∞–љ–Є–љ–∞–Љ–Є–љ–Њ—В—А–∞–љ—Б—Д–µ—А–∞–Ј–∞,¬†–≥–∞–Љ–Љ–∞-–≥–ї—Г—В–∞–Љ–Є–ї—В—А–∞–љ—Б–њ–µ–њ—В–Є–і–∞–Ј–∞), –Њ–±—Й–µ–≥–Њ –±–Є–ї–Є—А—Г–±–Є–љ–∞;
- –Њ–њ—А–µ–і–µ–ї–µ–љ–Є–µ —Г—А–Њ–≤–љ–µ–є –Є–Љ–Љ—Г–љ–Њ–≥–ї–Њ–±—Г–ї–Є–љ–Њ–≤ –Ї–ї–∞—Б—Б–Њ–≤ G, M,¬†A –≤¬†—Б—Л–≤–Њ—А–Њ—В–Ї–µ –Ї—А–Њ–≤–Є;
- –Њ—Ж–µ–љ–Ї—Г –Ї–Њ–≥–љ–Є—В–Є–≤–љ–Њ–≥–Њ —А–∞–Ј–≤–Є—В–Є—П, —А–µ—З–Є, –∞¬†—В–∞–Ї–ґ–µ –њ—Б–Є—Е–Њ—Б–Њ—Ж–Є–∞–ї—М–љ—Л—Е –љ–∞–≤—Л–Ї–Њ–≤ –≤¬†–≤–Њ–Ј—А–∞—Б—В–µ 2, 4, 8, 12 –Є¬†15¬†–ї–µ—В.
–Я–Њ—Б–Ї–Њ–ї—М–Ї—Г —Н–≤–Њ–ї—О—Ж–Є—П –і–µ—А–Љ–∞—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є—Е –Њ—Б–Њ–±–µ–љ–љ–Њ—Б—В–µ–є (–≤¬†–Њ—Б–љ–Њ–≤–љ–Њ–Љ¬†–≥–Є–њ–Њ- –Є¬†–≥–Є–њ–µ—А–њ–Є–≥–Љ–µ–љ—В–љ—Л—Е –њ—П—В–µ–љ –Є¬†–∞–љ–Њ–Љ–∞–ї–Є–є —А–Њ—Б—В–∞ –≤–Њ–ї–Њ—Б) –љ–µ–Є–Ј–≤–µ—Б—В–љ–∞, —Ж–µ–ї–µ—Б–Њ–Њ–±—А–∞–Ј–љ–∞ —В–∞–Ї–ґ–µ –µ–ґ–µ–≥–Њ–і–љ–∞—П –Ї–Њ–љ—Б—Г–ї—М—В–∞—Ж–Є—П –і–µ—А–Љ–∞—В–Њ–ї–Њ–≥–∞ [5].
–Ъ–ї–Є–љ–Є—З–µ—Б–Ї–Є–є —Б–ї—Г—З–∞–є
–Ь–∞–ї—М—З–Є–Ї –Њ—В¬†—В—А–µ—В—М–µ–є –±–µ—А–µ–Љ–µ–љ–љ–Њ—Б—В–Є, –њ—А–Њ—В–µ–Ї–∞–≤—И–µ–є –љ–∞¬†—Д–Њ–љ–µ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є –Љ–∞—В–µ—А–Є (—Е—А–Њ–љ–Є—З–µ—Б–Ї–∞—П –∞—А—В–µ—А–Є–∞–ї—М–љ–∞—П¬†–≥–Є–њ–µ—А—В–µ–љ–Ј–Є—П, –Љ–Є–Њ–Љ–∞ –Љ–∞—В–Ї–Є). –Э–∞¬†30-–є –љ–µ–і–µ–ї–µ¬†–≥–µ—Б—В–∞—Ж–Є–Є –≤—Л—П–≤–ї–µ–љ—Л –≤—А–Њ–ґ–і–µ–љ–љ—Л–є –њ–Њ—А–Њ–Ї —Б–µ—А–і—Ж–∞ (–Т–Я–°) (–Ї–Њ–∞—А–Ї—В–∞—Ж–Є—П –∞–Њ—А—В—Л) –Є¬†–≤—А–Њ–ґ–і–µ–љ–љ—Л–є –њ–Њ—А–Њ–Ї —А–∞–Ј–≤–Є—В–Є—П –њ–Њ—З–µ–Ї (–≥–Є–і—А–Њ–љ–µ—Д—А–Њ–Ј). –£¬†—А–µ–±–µ–љ–Ї–∞ –Њ—В—П–≥–Њ—Й–µ–љ–љ—Л–є –∞–љ–∞–Љ–љ–µ–Ј: —Б—В–∞—А—И–Є–є —Б–Є–±—Б –≤¬†—Б–µ–Љ—М–µ¬†вАУ –і–µ–≤–Њ—З–Ї–∞ —Б—В—А–∞–і–∞–µ—В —В—А–Є—Е–Њ–≥–µ–њ–∞—В–Њ—Н–љ—В–µ—А–∞–ї—М–љ—Л–Љ —Б–Є–љ–і¬≠—А–Њ–Љ–Њ–Љ, –Є–Љ–µ–µ—В –љ—Г–Ї–ї–µ–Њ—В–Є–і–љ—Л–є –≤–∞—А–Є–∞–љ—В —Б.2164–°>–Ґ –≤¬†–≥–Њ–Љ–Њ–Ј–Є–≥–Њ—В–љ–Њ–Љ —Б–Њ—Б—В–Њ—П–љ–Є–Є –≤¬†–≥–µ–љ–µ SKIV2L, —А–Њ–і–Є—В–µ–ї–Є –њ—А–Њ–±–∞–љ–і–∞¬†вАУ¬†–≥–µ—В–µ—А–Њ–Ј–Є–≥–Њ—В–љ—Л–µ –љ–Њ—Б–Є—В–µ–ї–Є –≤–∞—А–Є–∞–љ—В–∞ —Б.2164–°>–Ґ.
–†–Њ–і—Л –љ–∞¬†36-–є –љ–µ–і–µ–ї–µ —Б¬†–њ–Њ–Љ–Њ—Й—М—О –Њ–њ–µ—А–∞—Ж–Є–Є —Н–Ї—Б—В—А–µ–љ–љ–Њ–≥–Њ –Ї–µ—Б–∞—А–µ–≤–∞ —Б–µ—З–µ–љ–Є—П (–њ—А–µ—Н–Ї–ї–∞–Љ–њ—Б–Є—П —Г¬†–Љ–∞—В–µ—А–Є). –Ь–∞—Б—Б–∞ —В–µ–ї–∞ –њ—А–Є —А–Њ–ґ–і–µ–љ–Є–Є¬†вАУ 2140¬†–≥,¬†–і–ї–Є–љ–∞ —В–µ–ї–∞¬†вАУ 47¬†—Б–Љ, –Њ–Ї—А—Г–ґ–љ–Њ—Б—В—М¬†–≥–Њ–ї–Њ–≤—Л¬†вАУ 32 —Б–Љ, –Њ–Ї—А—Г–ґ–љ–Њ—Б—В—М¬†–≥—А—Г–і–љ–Њ–є –Ї–ї–µ—В–Ї–Є¬†вАУ 26 —Б–Љ. –Ю—Ж–µ–љ–Ї–∞ –њ–Њ¬†—И–Ї–∞–ї–µ –Р–њ–≥–∞—А¬†вАУ 7/8 –±–∞–ї–ї–Њ–≤. –°–Њ—Б—В–Њ—П–љ–Є–µ —А–µ–±–µ–љ–Ї–∞ —Б¬†—А–Њ–ґ–і–µ–љ–Є—П –Њ—Ж–µ–љ–Є–≤–∞–ї–Њ—Б—М –Ї–∞–Ї —В—П–ґ–µ–ї–Њ–µ, –Њ–±—Г—Б–ї–Њ–≤–ї–µ–љ–љ–Њ–µ –Њ—Б–љ–Њ–≤–љ—Л–Љ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–µ–Љ, –Љ–Њ—А—Д–Њ—Д—Г–љ–Ї—Ж–Є–Њ–љ–∞–ї—М–љ–Њ–є –љ–µ–Ј—А–µ–ї–Њ—Б—В—М—О, –Т–Я–°. –І–µ—А–µ–Ј –њ—П—В—М –і–љ–µ–є –њ–Њ—Б–ї–µ —Б—В–∞–±–Є–ї–Є–Ј–∞—Ж–Є–Є —Б–Њ—Б—В–Њ—П–љ–Є—П —А–µ–±–µ–љ–Ї–∞ –њ–µ—А–µ–≤–µ–ї–Є –Є–Ј¬†–Њ–±–ї–∞—Б—В–љ–Њ–є –±–Њ–ї—М–љ–Є—Ж—Л –≤¬†–§–µ–і–µ—А–∞–ї—М–љ—Л–є —Ж–µ–љ—В—А —Б–µ—А–і–µ—З–љ–Њ-—Б–Њ—Б—Г–і–Є—Б—В–Њ–є —Е–Є—А—Г—А–≥–Є–Є –Є–Љ. –°.–У. –°—Г—Е–∞–љ–Њ–≤–∞ (–Я–µ—А–Љ—М) –і–ї—П –Њ–њ–µ—А–∞—В–Є–≤–љ–Њ–≥–Њ –ї–µ—З–µ–љ–Є—П¬†вАУ —А–µ–Ј–µ–Ї—Ж–Є–Є –Ї–Њ–∞—А–Ї—В–∞—Ж–Є–Є –∞–Њ—А—В—Л —Б¬†—А–∞—Б—И–Є—А–µ–љ–љ—Л–Љ –∞–љ–∞—Б—В–Њ–Љ–Њ–Ј–Њ–Љ ¬Ђ–Ї–Њ–љ–µ—Ж –≤¬†–Ї–Њ–љ–µ—Ж¬ї.
–І–µ—А–µ–Ј —З–µ—В—Л—А–µ –і–љ—П –њ–Њ—Б–ї–µ –Њ–њ–µ—А–∞—Ж–Є–Є –Љ–∞–ї—М—З–Є–Ї —В—А–∞–љ—Б–њ–Њ—А—В–Є—А–Њ–≤–∞–љ –≤¬†–±–Њ–ї—М–љ–Є—Ж—Г –њ–Њ¬†–Љ–µ—Б—В—Г –ґ–Є—В–µ–ї—М—Б—В–≤–∞ –і–ї—П –і–∞–ї—М–љ–µ–є—И–µ–≥–Њ –љ–∞–±–ї—О–і–µ–љ–Є—П.
–Я—А–Є¬†–≥–Њ—Б–њ–Є—В–∞–ї–Є–Ј–∞—Ж–Є–Є —Б–Њ—Б—В–Њ—П–љ–Є–µ —В—П–ґ–µ–ї–Њ–µ –Є–Ј-–Ј–∞ —Б–µ—А–і–µ—З–љ–Њ–є –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В–Є 2-–є¬†—Б—В–µ–њ–µ–љ–Є, –і—Л—Е–∞—В–µ–ї—М–љ–Њ–є –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В–Є 2-–є¬†—Б—В–µ–њ–µ–љ–Є –Є¬†—Б—В—А–Є–і–Њ—А–∞, –Ї–Є—Б–ї–Њ—А–Њ–і–љ–Њ–є –Ј–∞–≤–Є—Б–Є–Љ–Њ—Б—В–Є, —Н–љ—В–µ—А–Њ–њ–∞—В–Є–Є, –≤—Л—А–∞–ґ–µ–љ–љ–Њ–є –љ–µ–≤—А–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–є —Б–Є–Љ–њ—В–Њ–Љ–∞—В–Є–Ї–Є –љ–∞¬†—Д–Њ–љ–µ –≤–Њ–Ј–±—Г–і–Є–Љ–Њ—Б—В–Є. –†–µ–±–µ–љ–Њ–Ї –љ–∞¬†–Ј–Њ–љ–і–Њ–≤–Њ–Љ –Ї–Њ—А–Љ–ї–µ–љ–Є–Є, —Б—А—Л–≥–Є–≤–∞–µ—В.
–Т¬†–∞–љ–∞–ї–Є–Ј–∞—Е –Ї—А–Њ–≤–Є¬†вАУ —В—А–Њ–Љ–±–Њ—Ж–Є—В–Њ–њ–µ–љ–Є—П, –∞–љ–µ–Љ–Є—П, –≤—Л—А–∞–ґ–µ–љ–љ–Њ–µ –њ–Њ–≤—Л—И–µ–љ–Є–µ —Г—А–Њ–≤–љ—П —Й–µ–ї–Њ—З–љ–Њ–є —Д–Њ—Б—Д–∞—В–∞–Ј—Л, —Г–Љ–µ—А–µ–љ–љ—Л–є —Ж–Є—В–Њ–ї–Є–Ј.
–Ф–∞–љ–љ—Л–µ –Ї–∞—А–і–Є–Њ–≥—А–∞—Д–Є–Є: —Б–Њ—Б—В–Њ—П–љ–Є–µ –њ–Њ—Б–ї–µ –Ї–Њ—А—А–µ–Ї—Ж–Є–Є –Т–Я–°.
–Я—А–Њ–≤–µ–і–µ–љ—Л –Є–љ—Д—Г–Ј–Є–Њ–љ–љ–∞—П —В–µ—А–∞–њ–Є—П, –њ–Њ—Б—В–µ–њ–µ–љ–љ–Њ–µ —А–∞—Б—И–Є—А–µ–љ–Є–µ —Н–љ—В–µ—А–∞–ї—М–љ–Њ–≥–Њ –њ–Є—В–∞–љ–Є—П –њ–Њ–і –Ї–Њ–љ—В¬≠—А–Њ–ї–µ–Љ –њ–µ—А–µ–љ–Њ—Б–Є–Љ–Њ—Б—В–Є.
–Т¬†–°–µ—З–µ–љ–Њ–≤—Б–Ї–Є–є —Ж–µ–љ—В—А –Љ–∞—В–µ—А–Є–љ—Б—В–≤–∞ –Є¬†–і–µ—В—Б—В–≤–∞ —А–µ–±–µ–љ–Њ–Ї –њ–Њ—Б—В—Г–њ–Є–ї –љ–∞¬†–≤—В–Њ—А–Њ–Љ –Љ–µ—Б—П—Ж–µ –ґ–Є–Ј–љ–Є –і–ї—П –њ—А–Њ–≤–µ–і–µ–љ–Є—П –Њ–±—Б–ї–µ–і–Њ–≤–∞–љ–Є—П, –њ–Њ—Б—В–∞–љ–Њ–≤–Ї–Є –і–Є–∞–≥–љ–Њ–Ј–∞ –Є¬†—А–∞—Б—И–Є—А–µ–љ–Є—П –Њ–±—К–µ–Љ–∞ —Н–љ—В–µ—А–∞–ї—М–љ–Њ–≥–Њ –њ–Є—В–∞–љ–Є—П.
–Т¬†–Њ–±—К–µ–Ї—В–Є–≤–љ–Њ–Љ —Б—В–∞—В—Г—Б–µ¬†вАУ —В—П–ґ–µ—Б—В—М —Б–Њ—Б—В–Њ—П–љ–Є—П, –љ–Є–Ј–Ї–∞—П –Љ–∞—Б—Б–∞ —В–µ–ї–∞ (2750¬†–≥), –Њ–±–Є–ї—М–љ—Л–µ —Б—А—Л–≥–Є–≤–∞–љ–Є—П (–і–Њ¬†15вАУ20 –Љ–ї) –њ–Њ—Б–ї–µ –Ї–∞–ґ–і–Њ–≥–Њ –Ї–Њ—А–Љ–ї–µ–љ–Є—П, –≤—Л—А–∞–ґ–µ–љ–љ—Л–є –Ї–Є—И–µ—З–љ—Л–є —Б–Є–љ–і—А–Њ–Љ (–ґ–Є–і–Ї–Є–є —Б—В—Г–ї –і–Њ 7вАУ9¬†—А–∞–Ј –≤¬†—Б—Г—В–Ї–Є), –ґ–Є–≤–Њ—В –њ—А–Є –њ–∞–ї—М–њ–∞—Ж–Є–Є –≤–Ј–і—Г—В, —З—Г–≤—Б—В¬≠–≤–Є—В–µ–ї–µ–љ –њ–Њ¬†—Е–Њ–і—Г –Ї–Є—И–µ—З–љ–Є–Ї–∞. –§–µ–љ–Њ—В–Є–њ–Є—З–µ—Б–Ї–Є –Њ—В–Љ–µ—З–∞–ї–Є—Б—М —В–Њ–љ–Ї–Є–µ –≤–Њ–ї–Њ—Б—Л –љ–∞¬†–≥–Њ–ї–Њ–≤–µ —Б¬†—Г—З–∞—Б—В–Ї–∞–Љ–Є¬†–≥–Є–њ–µ—А—В—А–Є—Е–Њ–Ј–∞,¬†–≥–Є–њ–Њ–њ–ї–∞–Ј–Є—П –љ–∞–і–±—А–Њ–≤–љ—Л—Е –і—Г–≥, –і–ї–Є–љ–љ—Л–є —Г–њ–ї–Њ—Й–µ–љ–љ—Л–є —Д–Є–ї—М—В—А.
–Я—А–Є¬†–≥–Њ—Б–њ–Є—В–∞–ї–Є–Ј–∞—Ж–Є–Є —Б–Њ—Б—В–Њ—П–љ–Є–µ —А–µ–±–µ–љ–Ї–∞ —А–∞—Б—Ж–µ–љ–Є–≤–∞–ї–Њ—Б—М –Ї–∞–Ї —В—П–ґ–µ–ї–Њ–µ: –≤¬†–∞–љ–∞–ї–Є–Ј–µ –Ї—А–Њ–≤–Є¬†вАУ —Г–Љ–µ—А–µ–љ–љ—Л–є –Љ–µ—В–∞–±–Њ–ї–Є—З–µ—Б–Ї–Є–є –∞—Ж–Є–і–Њ–Ј,¬†–≥–Є–њ–Њ–њ—А–Њ—В–µ–Є–љ–µ–Љ–Є—П,¬†–≥–Є–њ–Њ–∞–ї—М–±—Г–Љ–Є–љ–µ–Љ–Є—П, —В—А–Њ–Љ–±–Њ—Ж–Є—В–Њ–њ–µ–љ–Є—П –і–Њ 165 √Ч 109/–ї, –∞–љ–µ–Љ–Є—П —Б—А–µ–і–љ–µ–є —Б—В–µ–њ–µ–љ–Є.
–°¬†—Г—З–µ—В–Њ–Љ —В—П–ґ–µ—Б—В–Є –Њ—Б–љ–Њ–≤–љ–Њ–≥–Њ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П, –≤—Л—А–∞–ґ–µ–љ–љ–Њ—Б—В–Є¬†–≥–Є–њ–Њ–њ—А–Њ—В–µ–Є–љ–µ–Љ–Є–Є –Є¬†–≥–Є–њ–Њ–∞–ї—М–±—Г–Љ–Є–љ–µ–Љ–Є–Є, —А–∞–љ–љ–µ–≥–Њ –≤–Њ–Ј—А–∞—Б—В–∞, –љ–µ–і–Њ–љ–Њ—И–µ–љ–љ–Њ—Б—В–Є —Г—Б—В–∞–љ–Њ–≤–ї–µ–љ –њ–µ—А–Є—Д–µ—А–Є—З–µ—Б–Ї–Є–є –≤–µ–љ–Њ–Ј–љ—Л–є –Ї–∞—В–µ—В–µ—А, –Ј–∞—В–µ–Љ –≤–≤–µ–і–µ–љ —З–µ–ї–Њ–≤–µ—З–µ—Б–Ї–Є–є –Є–Љ–Љ—Г–љ–Њ–≥–ї–Њ–±—Г–ї–Є–љ. –°¬†—Ж–µ–ї—М—О –Ї–Њ—А—А–µ–Ї—Ж–Є–Є –≤–Њ–ї–µ–Љ–Є—З–µ—Б–Ї–Њ–≥–Њ –Њ–±—К–µ–Љ–∞ –≤—Л–њ–Њ–ї–љ–µ–љ–∞ –Є–љ—Д—Г–Ј–Є–Њ–љ–љ–∞—П —В–µ—А–∞–њ–Є—П. –Т¬†—Б–Є–ї—Г –Њ—В—П–≥–Њ—Й–µ–љ–љ–Њ–≥–Њ –∞–љ–∞–Љ–љ–µ–Ј–∞, —А–∞–љ–љ–µ–≥–Њ –≤–Њ–Ј—А–∞—Б—В–∞, —Г–Љ–µ—А–µ–љ–љ–Њ–≥–Њ –љ–∞—А–∞—Б—В–∞–љ–Є—П –Љ–∞—А–Ї–µ—А–Њ–≤ –≤–Њ—Б–њ–∞–ї–µ–љ–Є—П, –≤–µ—А–Њ—П—В–љ–Њ—Б—В–Є –≤—А–Њ–ґ–і–µ–љ–љ–Њ–є —Н–љ—В–µ—А–Њ–њ–∞—В–Є–Є –≤¬†—Ж–µ–ї—П—Е –і–µ–Ї–Њ–љ—В–∞–Љ–Є–љ–∞—Ж–Є–Є –њ–Њ–Ї–∞–Ј–∞–љ–Њ –љ–∞–Ј–љ–∞—З–µ–љ–Є–µ –∞–љ—В–Є–±–∞–Ї—В–µ—А–Є–∞–ї—М–љ–Њ–є (–≤–∞–љ–Ї–Њ–Љ–Є—Ж–Є–љ, –∞–Љ–Є–Ї–∞—Ж–Є–љ), –∞¬†—В–∞–Ї–ґ–µ –њ—А–Њ—В–Є–≤–Њ–≥—А–Є–±–Ї–Њ–≤–Њ–є —В–µ—А–∞–њ–Є–Є (—Д–ї—Г–Ї–Њ–љ–∞–Ј–Њ–ї).
–Э–∞¬†—Д–Њ–љ–µ —Б–Њ—Е—А–∞–љ–µ–љ–Є—П –Ї–Є—И–µ—З–љ—Л—Е –њ–Њ—В–µ—А—М, –љ–∞—А–∞—Б—В–∞–љ–Є—П –∞–љ–µ–Љ–Є–Є –Є¬†–Є–љ—В–Њ–Ї—Б–Є–Ї–∞—Ж–Є–Є –Њ–±—К–µ–Љ —Н–љ—В–µ—А–∞–ї—М–љ–Њ–≥–Њ –њ–Є—В–∞–љ–Є—П —Г–Љ–µ–љ—М—И–µ–љ –і–Њ —В—А–Њ—Д–Є—З–µ—Б–Ї–Њ–≥–Њ, —Г—Б—В–∞–љ–Њ–≤–ї–µ–љ —Ж–µ–љ—В—А–∞–ї—М–љ—Л–є –≤–µ–љ–Њ–Ј–љ—Л–є –Ї–∞—В–µ—В–µ—А –і–ї—П –љ–∞–ї–∞–ґ–Є–≤–∞–љ–Є—П —З–∞—Б—В–Є—З–љ–Њ–≥–Њ –њ–∞—А–µ–љ—В–µ—А–∞–ї—М–љ–Њ–≥–Њ –њ–Є—В–∞–љ–Є—П. –Ъ–Њ–љ—Б—Г–ї—М—В–Є—А–Њ–≤–∞–љ —В—А–∞–љ—Б—Д—Г–Ј–Є–Њ–ї–Њ–≥–Њ–Љ (–≤—Л–њ–Њ–ї–љ–µ–љ–∞¬†–≥–µ–Љ–Њ—В—А–∞–љ—Б—Д—Г–Ј–Є—П).
–Я–Њ—Б–ї–µ –њ—А–Њ–≤–µ–і–µ–љ–љ–Њ–є —В–µ—А–∞–њ–Є–Є –Њ—В–Љ–µ—З–∞–ї–∞—Б—М —Б—В–Њ–є–Ї–∞—П –њ–Њ–ї–Њ–ґ–Є—В–µ–ї—М–љ–∞—П –і–Є–љ–∞–Љ–Є–Ї–∞: —А–µ–±–µ–љ–Њ–Ї¬†–≥–µ–Љ–Њ–і–Є–љ–∞–Љ–Є—З–µ—Б–Ї–Є —Б—В–∞–±–Є–ї–µ–љ, –Ї—Г–њ–Є—А–Њ–≤–∞–љ–∞ –Є–љ—В–Њ–Ї—Б–Є–Ї–∞—Ж–Є—П, –і–Њ—Б—В–Є–≥–љ—Г—В–∞ –љ–Њ—А–Љ–∞–ї–Є–Ј–∞—Ж–Є—П –њ–Њ–Ї–∞–Ј–∞—В–µ–ї–µ–є –∞–љ–∞–ї–Є–Ј–∞ –Ї—А–Њ–≤–Є, —Г–Љ–µ–љ—М—И–Є–ї–Є—Б—М –Њ–±—К–µ–Љ—Л –Ї–Є—И–µ—З–љ—Л—Е –њ–Њ—В–µ—А—М (—Б—В—Г–ї –і–Њ —З–µ—В—Л—А–µ—Е-–њ—П—В–Є —А–∞–Ј –≤¬†—Б—Г—В–Ї–Є, –Ї–∞—И–Є—Ж–µ–Њ–±—А–∞–Ј–љ—Л–є). –Ч–∞ —И–µ—Б—В–Є–љ–µ–і–µ–ї—М–љ—Л–є –њ–µ—А–Є–Њ–і¬†–≥–Њ—Б–њ–Є—В–∞–ї–Є–Ј–∞—Ж–Є–Є –≤–µ—Б —Г–≤–µ–ї–Є—З–Є–ї—Б—П –љ–∞¬†1130¬†–≥.
–Ъ–Њ–љ—Б—Г–ї—М—В–∞—Ж–Є—П¬†–≥–µ–љ–µ—В–Є–Ї–∞: —Б¬†—Г—З–µ—В–Њ–Љ –Њ—В—П–≥–Њ—Й–µ–љ–љ–Њ–≥–Њ –Ї–ї–Є–љ–Є–Ї–Њ-–≥–µ–љ–µ–∞–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–≥–Њ –∞–љ–∞–Љ–љ–µ–Ј–∞ (—Б—В–∞—А—И–Є–є —Б–Є–±—Б –≤¬†—Б–µ–Љ—М–µ¬†вАУ –і–µ–≤–Њ—З–Ї–∞ —Б—В—А–∞–і–∞–µ—В –і–∞–љ–љ—Л–Љ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–µ–Љ, –Є–Љ–µ–µ—В –љ—Г–Ї–ї–µ–Њ—В–Є–і–љ—Л–є –≤–∞—А–Є–∞–љ—В —Б.2164–°>–Ґ –≤¬†–≥–Њ–Љ–Њ–Ј–Є–≥–Њ—В–љ–Њ–Љ —Б–Њ—Б—В–Њ—П–љ–Є–Є –≤¬†–≥–µ–љ–µ SKIV2L), –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Њ–є –Ї–∞—А—В–Є–љ—Л (–Њ—Б–Њ–±–µ–љ–љ–Њ—Б—В–Є –ї–Є—Ж–µ–≤–Њ–≥–Њ —Д–µ–љ–Њ—В–Є–њ–∞, –њ–Њ–њ–µ—А–µ—З–љ—Л–µ –ї–∞–і–Њ–љ–љ—Л–µ –±–Њ—А–Њ–Ј–і—Л, –Т–Я–°, –Ї–Њ–∞—А–Ї—В–∞—Ж–Є—П –∞–Њ—А—В—Л,¬†–≥–Є–і—А–Њ–љ–µ—Д—А–Њ–Ј, –Ї–Є—Б–ї–Њ—А–Њ–і–Ј–∞–≤–Є—Б–Є–Љ–Њ—Б—В—М –≤¬†–∞–љ–∞–Љ–љ–µ–Ј–µ, —Н–љ—В–µ—А–Њ–њ–∞—В–Є—П) –Є¬†—А–µ–Ј—Г–ї—М—В–∞—В–Њ–≤ –њ—А–Њ–≤–µ–і–µ–љ–љ–Њ–≥–Њ –Љ–Њ–ї–µ–Ї—Г–ї—П—А–љ–Њ-–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Њ–≥–Њ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П (–≤—Л—П–≤–ї–µ–љ –љ—Г–Ї–ї–µ–Њ—В–Є–і–љ—Л–є –≤–∞—А–Є–∞–љ—В chr63 1933752C>T (—Б.2164C>T, NM_000006.11) –≤¬†–≥–Њ–Љ–Њ–Ј–Є–≥–Њ—В–љ–Њ–Љ —Б–Њ—Б—В–Њ—П–љ–Є–Є –≤¬†–≥–µ–љ–µ SKIV2L) —Г¬†—А–µ–±–µ–љ–Ї–∞ –і–Є–∞–≥–љ–Њ—Б—В–Є—А–Њ–≤–∞–љ —В—А–Є—Е–Њ–≥–µ–њ–∞—В–Њ—Н–љ—В–µ—А–∞–ї—М–љ—Л–є —Б–Є–љ–і—А–Њ–Љ, —В–Є–њ 2 (OMIM 614602). –Ф–Є–∞–≥–љ–Њ–Ј –њ–Њ–і—В–≤–µ—А–ґ¬≠–і–µ–љ —А–µ–Ј—Г–ї—М—В–∞—В–∞–Љ–Є –Љ–Њ–ї–µ–Ї—Г–ї—П—А–љ–Њ-–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Є—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є. –Ч–∞–±–Њ–ї–µ–≤–∞–љ–Є–µ –Є–Љ–µ–µ—В –∞—Г—В–Њ—Б–Њ–Љ–љ–Њ-—А–µ—Ж–µ—Б—Б–Є–≤–љ—Л–є —В–Є–њ –љ–∞—Б–ї–µ–і–Њ–≤–∞–љ–Є—П. –†–Є—Б–Ї –њ–Њ–≤—В–Њ—А–љ–Њ–≥–Њ —А–Њ–ґ–і–µ–љ–Є—П —Г¬†—А–Њ–і–Є—В–µ–ї–µ–є —А–µ–±–µ–љ–Ї–∞ —Б¬†–і–∞–љ–љ—Л–Љ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–µ–Љ¬†вАУ 25%. –†–µ–Ї–Њ–Љ–µ–љ–і–Њ–≤–∞–љ–Њ –њ—А–Њ–≤–µ–і–µ–љ–Є–µ –њ—А–µ–љ–∞—В–∞–ї—М–љ–Њ–≥–Њ¬†–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Њ–≥–Њ —В–µ—Б—В–Є—А–Њ–≤–∞–љ–Є—П.
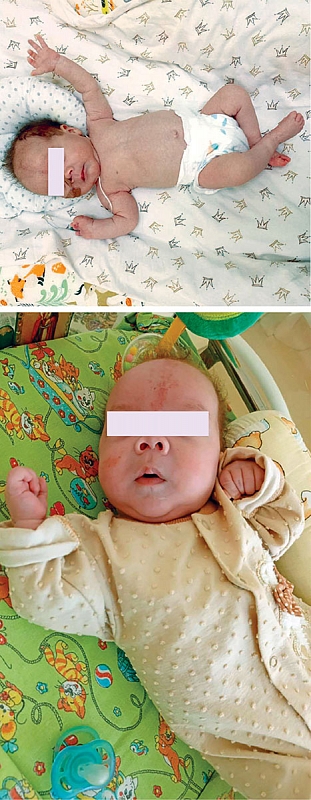
–Т¬†—Б–Є–ї—Г –Њ—Б–Њ–±–µ–љ–љ–Њ—Б—В–Є —В–µ—З–µ–љ–Є—П —В—А–Є—Е–Њ–≥–µ–њ–∞—В–Њ—Н–љ—В–µ—А–∞–ї—М–љ–Њ–≥–Њ —Б–Є–љ–і—А–Њ–Љ–∞ –Є¬†–њ–Њ—В—А–µ–±–љ–Њ—Б—В–Є —А–µ–±–µ–љ–Ї–∞ –≤¬†—З–∞—Б¬≠—В–Є—З–љ–Њ–Љ –њ–∞—А–µ–љ—В–µ—А–∞–ї—М–љ–Њ–Љ –њ–Є—В–∞–љ–Є–Є –≤¬†–Њ—В–і–µ–ї–µ–љ–Є–Є —Б—В–∞—Ж–Є–Њ–љ–∞—А–∞ –њ—А–Њ–≤–Њ–і–Є—В—Б—П –Њ–±—Г—З–µ–љ–Є–µ —А–Њ–і–Є—В–µ–ї–µ–є –њ–Њ¬†—Г—Е–Њ–і—Г –Є¬†–≤—Л—Е–∞–ґ–Є–≤–∞–љ–Є—О —А–µ–±–µ–љ–Ї–∞ –≤¬†–і–Њ–Љ–∞—И–љ–Є—Е —Г—Б–ї–Њ–≤–Є—П—Е. –°–Њ—Б—В–Њ—П–љ–Є–µ —А–µ–±–µ–љ–Ї–∞ –Њ—Б—В–∞–µ—В—Б—П —Б—В–∞–±–Є–ї—М–љ—Л–Љ: –Љ–∞–ї—М—З–Є–Ї¬†–≥–µ–Љ–Њ–і–Є–љ–∞–Љ–Є—З–µ—Б–Ї–Є —Б—В–∞–±–Є–ї–µ–љ, –њ—А–Є–±–∞–≤–ї—П–µ—В –≤¬†–≤–µ—Б–µ, –Њ–±—К–µ–Љ —Н–љ—В–µ—А–∞–ї—М–љ–Њ–≥–Њ –њ–Є—В–∞–љ–Є—П —Г–≤–µ–ї–Є—З–µ–љ –і–Њ 40вАУ45 –Љ–ї –Ї–∞–ґ–і—Л–µ —В—А–Є —З–∞—Б–∞, —Г—Б–≤–∞–Є–≤–∞–µ—В, –љ–∞—Е–Њ–і–Є—В—Б—П –љ–∞¬†—З–∞—Б—В–Є—З–љ–Њ–Љ –њ–∞—А–µ–љ—В–µ—А–∞–ї—М–љ–Њ–Љ –њ–Є—В–∞–љ–Є–Є (—Д–Њ—В–Њ).
–Ч–∞–Ї–ї—О—З–µ–љ–Є–µ
–Я—А–Њ–≥–љ–Њ–Ј –њ—А–Є —В—А–Є—Е–Њ–≥–µ–њ–∞—В–Њ—Н–љ—В–µ—А–∞–ї—М–љ–Њ–Љ —Б–Є–љ–і—А–Њ–Љ–µ –≤¬†—Ж–µ–ї–Њ–Љ –љ–µ–±–ї–∞–≥–Њ–њ—А–Є—П—В–љ—Л–є. –Ю–±—Л—З–љ–Њ —Б–Љ–µ—А—В—М –љ–∞—Б—В—Г–њ–∞–µ—В –≤¬†—А–∞–љ–љ–µ–Љ –≤–Њ–Ј—А–∞—Б—В–µ –≤¬†—А–µ–Ј—Г–ї—М—В–∞—В–µ –њ–µ—З–µ–љ–Њ—З–љ–Њ–є –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В–Є, —А–µ—Ж–Є–і–Є–≤–Є—А—Г—О—Й–µ–є –Є–љ—Д–µ–Ї—Ж–Є–Є –Є–ї–Є –Њ—Б–ї–Њ–ґ–љ–µ–љ–Є–є, —Б–≤—П–Ј–∞–љ–љ—Л—Е —Б¬†–і–ї–Є—В–µ–ї—М–љ—Л–Љ –њ–∞—А–µ–љ—В–µ—А–∞–ї—М–љ—Л–Љ –њ–Є—В–∞–љ–Є–µ–Љ [2, 8]. –Я–∞—А–µ–љ—В–µ—А–∞–ї—М–љ–Њ–µ –њ–Є—В–∞–љ–Є–µ –Є¬†–ї–µ—З–µ–љ–Є–µ —А–µ—Ж–Є–і–Є–≤–Є—А—Г—О—Й–µ–є –Є–љ—Д–µ–Ї—Ж–Є–Є –Љ–Њ–≥—Г—В –њ–Њ–ї–Њ–ґ–Є—В–µ–ї—М–љ–Њ –њ–Њ–≤–ї–Є—П—В—М –љ–∞¬†–≤—Л–ґ–Є–≤–∞–µ–Љ–Њ—Б—В—М –і–µ—В–µ–є —Б¬†—В—А–Є—Е–Њ–≥–µ–њ–∞—В–Њ—Н–љ—В–µ—А–∞–ї—М–љ—Л–Љ —Б–Є–љ–і—А–Њ–Љ–Њ–Љ. –Ш–Љ–µ–љ–љ–Њ –њ–Њ—Н—В–Њ–Љ—Г –Њ—Б–≤–µ–і–Њ–Љ–ї–µ–љ–љ–Њ—Б—В—М –≤—А–∞—З–µ–є-–њ–µ¬≠–і–Є–∞—В—А–Њ–≤, –љ–µ–Њ–љ–∞—В–Њ–ї–Њ–≥–Њ–≤,¬†–≥–∞—Б—В—А–Њ—Н–љ—В–µ—А–Њ–ї–Њ–≥–Њ–≤ –Њ–±¬†—Н—В–Њ–Љ —Б–Є–љ–і—А–Њ–Љ–µ –њ–Њ–Ј–≤–Њ–ї—П–µ—В –Њ–±–µ—Б–њ–µ—З–Є—В—М –µ–≥–Њ —А–∞–љ–љ—О—О –і–Є–∞¬≠–≥–љ–Њ—Б—В–Є–Ї—Г, –≤—Л–±–Њ—А –∞–і–µ–Ї–≤–∞—В–љ–Њ–є —В–∞–Ї—В–Є–Ї–Є –≤–µ–і–µ–љ–Є—П –њ–∞—Ж–Є–µ–љ—В–Њ–≤ –Є¬†–љ–∞–і–ї–µ–ґ–∞—Й–µ–µ –ї–µ—З–µ–љ–Є–µ –љ–∞¬†—А–∞–љ–љ–Є—Е —Б—В–∞–і–Є—П—Е.¬†
–Ъ–Њ–љ—Д–ї–Є–Ї—В –Є–љ—В–µ—А–µ—Б–Њ–≤. –Р–≤—В–Њ—А—Л –Ј–∞—П–≤–ї—П—О—В –Њ–±¬†–Њ—В—Б—Г—В—Б—В–≤–Є–Є –Ї–Њ–љ—Д–ї–Є–Ї—В–∞ –Є–љ—В–µ—А–µ—Б–Њ–≤.
–§–Є–љ–∞–љ—Б–Є—А–Њ–≤–∞–љ–Є–µ. –†–∞–±–Њ—В–∞ –≤—Л–њ–Њ–ї–љ–µ–љ–∞ –±–µ–Ј¬†—Б–њ–Њ–љ—Б–Њ—А—Б–Ї–Њ–є –њ–Њ–і–і–µ—А–ґ–Ї–Є.
–°–Њ–±–ї—О–і–µ–љ–Є–µ –њ—А–∞–≤ –њ–∞—Ж–Є–µ–љ—В–Њ–≤ –Є¬†–њ—А–∞–≤–Є–ї –±–Є–Њ—Н—В–Є–Ї–Є. –†–Њ–і–Є—В–µ–ї–Є —А–µ–±–µ–љ–Ї–∞ –љ–µ¬†–≤–Њ–Ј—А–∞–ґ–∞—О—В –њ—А–Њ—В–Є–≤ –њ—Г–±–ї–Є–Ї–∞—Ж–Є–Є –і–∞–љ–љ—Л—Е –Є–Ј¬†–Є—Б—В–Њ—А–Є–Є –±–Њ–ї–µ–Ј–љ–Є –Є¬†—Д–Њ—В–Њ —А–µ–±–µ–љ–Ї–∞.
K.A. Kazakova, PhD1, M.A. Varichkina, G.S. Golosnaya, PhD, Prof., G.V. Galstyan, L.G. Khachatryan, PhD, Prof., Ye.K. Kulikova
I.M. Sechenov First Moscow State Medical University
N.N. Burdenko Voronezh State Medical University
Contact person: Klavdiya A. Kazakova, kazakova_k_a@staff.sechenov.ru
Tricho-hepato-enteric syndrome is a rare genetic disease with a leading intestinal syndrome caused by impaired differentiation and polarization of intestinal enterocytes. A disease that develops due to mutations in the genes TTC37 (5q15) or SKIV2L (6p21.3), characterized by intrauterine fetal growth retardation, severe chronic diarrhea with onset in infancy, specific facial features, hair growth features and secondary immunodeficiency. A clinical case of trichohepatoenteral syndrome diagnosed in a child at an early age is presented. In the Russian medical literature, this syndrome is described in sibling.
–£–≤–∞–ґ–∞–µ–Љ—Л–є –њ–Њ—Б–µ—В–Є—В–µ–ї—М uMEDp!
–£–≤–µ–і–Њ–Љ–ї—П–µ–Љ –Т–∞—Б –Њ —В–Њ–Љ, —З—В–Њ –Ј–і–µ—Б—М —Б–Њ–і–µ—А–ґ–Є—В—Б—П –Є–љ—Д–Њ—А–Љ–∞—Ж–Є—П, –њ—А–µ–і–љ–∞–Ј–љ–∞—З–µ–љ–љ–∞—П –Є—Б–Ї–ї—О—З–Є—В–µ–ї—М–љ–Њ –і–ї—П —Б–њ–µ—Ж–Є–∞–ї–Є—Б—В–Њ–≤ –Ј–і—А–∞–≤–Њ–Њ—Е—А–∞–љ–µ–љ–Є—П.
–Х—Б–ї–Є –Т—Л –љ–µ —П–≤–ї—П–µ—В–µ—Б—М —Б–њ–µ—Ж–Є–∞–ї–Є—Б—В–Њ–Љ –Ј–і—А–∞–≤–Њ–Њ—Е—А–∞–љ–µ–љ–Є—П, –∞–і–Љ–Є–љ–Є—Б—В—А–∞—Ж–Є—П –љ–µ –љ–µ—Б–µ—В –Њ—В–≤–µ—В—Б—В–≤–µ–љ–љ–Њ—Б—В–Є –Ј–∞ –≤–Њ–Ј–Љ–Њ–ґ–љ—Л–µ –Њ—В—А–Є—Ж–∞—В–µ–ї—М–љ—Л–µ –њ–Њ—Б–ї–µ–і—Б—В–≤–Є—П, –≤–Њ–Ј–љ–Є–Ї—И–Є–µ –≤ —А–µ–Ј—Г–ї—М—В–∞—В–µ —Б–∞–Љ–Њ—Б—В–Њ—П—В–µ–ї—М–љ–Њ–≥–Њ –Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–љ–Є—П –Т–∞–Љ–Є –Є–љ—Д–Њ—А–Љ–∞—Ж–Є–Є —Б –њ–Њ—А—В–∞–ї–∞ –±–µ–Ј –њ—А–µ–і–≤–∞—А–Є—В–µ–ї—М–љ–Њ–є –Ї–Њ–љ—Б—Г–ї—М—В–∞—Ж–Є–Є —Б –≤—А–∞—З–Њ–Љ.
–Э–∞–ґ–Є–Љ–∞—П –љ–∞ –Ї–љ–Њ–њ–Ї—Г ¬Ђ–Т–Њ–є—В–Є¬ї, –Т—Л –њ–Њ–і—В–≤–µ—А–ґ–і–∞–µ—В–µ, —З—В–Њ —П–≤–ї—П–µ—В–µ—Б—М –≤—А–∞—З–Њ–Љ –Є–ї–Є —Б—В—Г–і–µ–љ—В–Њ–Љ –Љ–µ–і–Є—Ж–Є–љ—Б–Ї–Њ–≥–Њ –≤—Г–Ј–∞.