Деменция при болезни Паркинсона
- Аннотация
- Статья
- Ссылки
- English

Определение. Эпидемиология. Особенности течения
Болезнь Паркинсона (БП) – мультисистемное заболевание, проявляющееся моторными и многочисленными немоторными симптомами, важное место среди которых занимают когнитивные расстройства.
Согласно Диагностическому и статистическому руководству по психическим расстройствам (Diagnostic and Statistical Manual of Mental Disorders IV – DSM-IV), деменция – приобретенное снижение когнитивных функций, которое вызвано органическим повреждением головного мозга, определяется на фоне ясного сознания и приводит к ограничению повседневной жизнедеятельности.
По результатам проведенных исследований, когнитивные нарушения разной степени выраженности (легкие, умеренные, тяжелые) обнаруживаются при нейропсихологическом обследовании у 90–95% пациентов с БП [1]. Метаанализ 13 популяционных исследований показал, что средняя распространенность деменции у пациентов с БП равна 31,5% [2]. Согласно данным лонгитудинальных исследований, кумулятивная распространенность деменции у пациентов с БП составляет 90% [3], что делает связь деменции с БП почти неизбежной.
Как установили T.A. Hughes (2000) и D. Aarsland (2003), от момента развития моторных проявлений БП до развития деменции в среднем проходит десять лет [4], но этот временной интервал широко варьируется. У некоторых пациентов деменция развивается вскоре после дебюта паркинсонизма, у других, напротив, когнитивные нарушения, достигающие степени деменции, возникают через 20 и более лет [5]. А. Schrag и соавт. (1998) считают, что возраст – ключевой фактор развития деменции. У пациентов с ранним началом БП деменция редко развивается в течение первых десяти лет болезни [6].
Развившись, деменция при БП обычно неуклонно прогрессирует. D. Aarsland (2004) установил, что ежегодное ухудшение у пациентов с деменцией при БП составляет в среднем 2,3 балла по Краткой шкале оценки психического статуса (динамика состояния пациентов оценивалась в течение четырех лет). В целом скорость прогрессии сопоставима с развитием деменции при деменции с тельцами Леви [7] и значительно превосходит таковую у пациентов с БП без деменции (около 1 балла по Краткой шкале оценки психического статуса в год) [5].
Факторы риска
К факторам риска развития деменции при БП относят:
- пожилой возраст [8];
- тяжелый паркинсонизм, особенно ригидность, постуральную неустойчивость и нарушения походки [8–10], трансформацию дрожательной формы БП в акинетико-ригидную [7];
- легкое когнитивное снижение в дебюте БП [10] с дефицитом кортикальных функций в когнитивном профиле, локализующихся в височно-теменно-затылочных отделах коры головного мозга: семантическая афазия, зрительно-пространственные нарушения. При этом дефицит исполнительных функций не ассоциируется с высоким риском развития деменции. Описанные клинические данные подкреплены специфическими генетическими изменениями, обусловленными клиническим паттерном. Так, гаплотип H1 в гене тау-протеина связан с задним корковым дефицитом и высоким риском развития деменции, в то время как генотип катехол-О-метилтрансферазы ассоциировался с нарушением исполнительных функций и не сопровождался риском развития деменции [10];
- раннее развитие зрительных галлюцинаций и иллюзий, что, вероятно, связано с отложением телец Леви в височной коре, миндалине и развитием холинергического дефицита [11, 12];
- апатию [8, 13];
- нарушение поведения в фазу быстрого сна (отсутствие атонии в фазу быстрого сна) [14];
- длительное использование холинолитиков (циклодола, акинетона), в том числе антидепрессантов с холинолитической активностью (амитриптилина) [15], в связи с ускорением амилоидогенеза на фоне их приема [12];
- семейный анамнез (страдающие деменцией близкие родственники) [4].
Генетические факторы риска
Открытие локусов генов, ответственных за развитие наследственных форм БП, позволило по-новому взглянуть на проблемы этиопатогенеза и диагностики БП. Хотя, как правило, БП носит спорадический характер, у 15% пациентов диагностируется семейная форма БП, когда заболевание имеет место у нескольких родственников из одного или разных поколений. Доказано, что мутации по крайней мере в пяти генах определенно приводят к развитию менделирующих заболеваний [16].
Гены альфа-синуклеина (SNCA) и обогащенной лейциновыми повторами киназы 2 (Leucine-Rich Repeat Kinase 2 – LRRK2) ассоциированы с развитием аутосомно-доминантных форм БП [16].
Миссенс-мутации гена SNCA связаны с быстрым прогрессированием БП, а также с высокой распространенностью деменции и других психотических симптомов. Множественные повторы SNCA выявляются и при семейных формах БП, и при спорадических случаях БП. Клиническая картина БП зависит от количества повторов SNCA. Например, пациенты с трипликацией SNCA демонстрируют раннее начало болезни и высокую скорость развития деменции, в то время как у пациентов с дупликацией SNCA БП начинается позднее и развивается типично.
Пациенты с БП – носители мутации LRRK2 имеют клинический паттерн, похожий на спорадическую БП, но с несколько более доброкачественной прогрессией и медленным развитием деменции.
Гены паркина (PARK2), DJ-1, PINK1 ассоциированы c аутосомно-рецессивной формой заболевания с ранним началом [16, 17]. Для PINK1 и DJ-1 типичны раннее начало, медленное прогрессирование БП и медленное развитие деменции.
К генам высокого риска БП относят ген глюкоцереброзидазы [18], мутации которого в 2,4 раза увеличивают риск когнитивных нарушений [19].
В то же время полиморфизмы связанного с микротрубочками тау-протеина (Microtubule-Associated Protein Tau), возможно, имеют некоторое значение для развития деменции при БП [19].
Защитные факторы
Предполагается, что некоторые факторы могут защитить пациентов с болезнью Паркинсона от развития деменции. Особый интерес представляют данные о защитном действии нормального кишечного микробиома, ведущего к снижению проницаемости интерстициального эпителиального барьера и контролю периферических иммунных реакций, в противовес хроническому воспалению в желудочно-кишечном тракте, которое, по мнению T.R. Sampson (2016), является триггером нейродегенеративных заболеваний, в том числе БП [20].
Есть мнение, что снижение инфекционной нагрузки на организм за счет элиминации бактерий и подавления репликации вирусов, вызывающих хроническое воспаление, обеспечивает уменьшение риска дегенеративных заболеваний нервной системы [21].
В ходе лонгитудинальных исследований были получены противоречивые результаты о протективном действии курения в отношении развития когнитивных нарушений при БП [22]. Так, воздействие на никотиновые рецепторы, участвующие в обучении, может замедлять когнитивное снижение, однако курение усиливает окислительный стресс и провоцирует развитие сердечно-сосудистых заболеваний.
R. Inzelberg и соавт. (2003) установили снижение риска развития деменции при использовании амантадина для лечения БП [23]. Аналогичные данные получены для мемантина, имеющего похожий механизм действия и демонстрирующего позитивное действие на когнитивные функции у пациентов с деменцией на фоне БП и болезни диффузных телец Леви (деменции с тельцами Леви) [10, 24].
Предполагалось, что снижение уровня половых гормонов в постменопаузальном периоде может влиять на скорость развития когнитивных нарушений. Например, заместительная терапия эстрогенами способствовала снижению риска деменции.
Многократно оценивалось влияние приема статинов на частоту развития когнитивных расстройств. Большое исследование, посвященное изучению взаимосвязи когнитивных нарушений при БП и приема статинов, показало, что применение симвастатина может снизить риск развития деменции при БП более чем на 50% [25]. Благоприятный эффект статинов может быть обусловлен не только влиянием на сопутствующие сосудистые факторы риска, но и блокированием водород-потребляющей активности метаногенной микрофлоры кишечника (без формирования дисбиоза) и ингибирующим влиянием молекулярного водорода на экспрессию мРНК-индуцибельной NO-синтазы и провоспалительных цитокинов (интерлейкина 1b, 6, фактора некроза опухоли альфа) [21].
Морфология
В настоящее время болезнь Паркинсона относят к классу конформационных болезней мозга. Эти болезни вызваны нарушением конформации и внутриклеточного процессинга определенного белка, которое приводит к формированию белковых агрегатов, инициирующих процесс нейродегенерации. В основе молекулярных механизмов БП – нарушение фолдинга белка альфа-синуклеина в сочетании с его полимеризацией [16].
Альфа-синуклеин – пресинаптический белок, участвующий в везикулярном нейрональном транспорте. В клетке альфа-синуклеин существует в мембрансвязанной и нативной формах. Связывание альфа-синуклеина с мембранами сопровождается его переходом в альфа-спираль. В нативной форме он представляет собой растворимый белок со слабо упорядоченной структурой. При повышенной концентрации альфа-синуклеина в растворе образуются фибриллы и агрегаты, в частности процесс агрегации фибрилл тау-белка типичен для образования телец Леви [26]. Агрегаты альфа-синуклеина нарушают работу протеосом и лизосом, стимулируя дальнейшее накопление измененного белка [24].
Есть данные о прионоподобных свойствах альфа-синуклеина. Показана способность агрегатов альфа-синуклеина к секреции с последующим захватом соседними клетками и постепенным распространением патологии по нейронным путям [27].
Н. Braak (2002) предположил, что при БП имеет место восходящий тип патологического нейродегенеративного процесса, который проходит шесть стадий:
- отложение альфа-синуклеина в периферических вегетативных ганглиях кишечника и кожи;
- вовлечение стволовых ядерных структур (магноцеллюлярные части ретикулярной формации, голубое пятно);
- поражение компактной части черной субстанции, где происходит потеря 80–85% пигментированных дофаминергических нейронов, а также нейродегенеративные изменения в педункулопонтинном ядре, оральном ядре шва, холинергических магноцеллюлярных ядрах базальных отделов переднего мозга (в том числе в базальном ядре Мейнерта), туберомаммилярном ядре гипоталамуса;
- поражение височного мезокортекса (предположительно, включая лимбическую систему) и гиппокампа;
- вовлечение ассоциативных зон префронтальной, височной, теменной, затылочной коры;
- поражение первичных моторных и сенсорных зон коры больших полушарий головного мозга [28].
Согласно H. Hall и соавт. (2014), у пациентов с БП и деменцией достоверно чаще обнаруживались накопления альфа-синуклеина в базальном переднем мозге и гиппокампе по сравнению с пациентами с БП без деменции. Авторы предполагают, что накопление альфа-синуклеина связано с нейрональной дисфункцией и развитием значительных когнитивных расстройств [29]. Другие исследователи считают, что к деменции при БП приводят отложение телец Леви и нейродегенерация лимбических и кортикальных зон [30], в частности коры лобных долей, поясной извилины [31], а также коры височных долей головного мозга [32].
Морфологический субстрат деменции при БП – предмет оживленных дискуссий. Накоплено много данных, свидетельствующих об обнаружении при аутопсии у части пациентов (от 17,6% [33] до 40%) с БП и деменцией помимо телец Леви значительного количества амилоидных бляшек и нейрофибриллярных сплетений в стриатуме, коре головного мозга, в том числе в поясной извилине, – типичных для болезни Альцгеймера морфологических изменений [30, 34]. Н. Hall и соавт. (2014), М. Hely и соавт. (2008) считают, что сочетание патологии амилоидного белка и синуклеина может приводить к ускорению гибели клеток, вследствие чего деменция развивается на ранней стадии заболевания [30, 33, 35].
В литературе есть единичные сообщения об обнаружении при БП с деменцией морфологических изменений, характерных для болезни Пика: микровакуолизация нейронов и баллонообразные клетки во фронтолатеральных и орбитофронтальных кортикальных областях и поясной извилине, тау-позитивные цитоплазматические нейрональные включения. Следует отметить, что возможный вклад нейродегенеративных изменений по типу лобно-височной дегенерации при БП с деменцией явно изучен недостаточно [36].
У пациентов с деменцией и БП в ходе магнитно-резонансной томографии (МРТ) головного мозга нередко обнаруживаются диффузные изменения белого вещества в виде перивентрикулярного и субкортикального лейкоареоза. Как правило, обширность и локализация данных изменений невелики и не могут быть субстратом деменции, но, предположительно, влияют на скорость развития когнитивных нарушений [1].
Таким образом, морфологические изменения при БП с деменцией неоднородны. Самый частый морфологический вариант – распространенная нейродегенерация церебральных структур с диффузным отложением телец Леви. Для развития деменции важно поражение гиппокампа, лимбической системы и коры больших полушарий головного мозга. У 40% пациентов, страдающих БП и деменцией, типичная для БП морфологическая картина сочетается с изменениями, характерными для болезни Альцгеймера. Реже при БП и деменции отмечается морфологическая картина лобно-височной дегенерации. Возможно также сочетание ишемического и нейродегенеративного процесса у одного пациента. В этом случае церебральная ишемия, вероятно, будет ускорять развитие нейродегенерации.
Нейромедиаторные нарушения
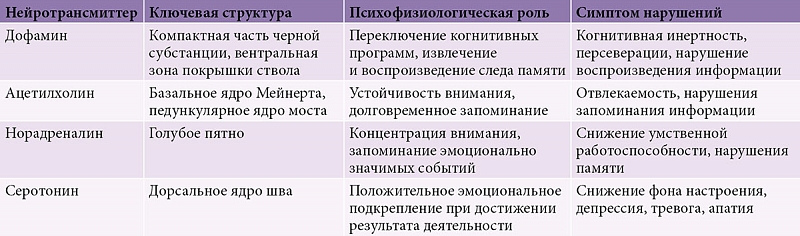
В настоящее время БП считается мультимедиаторным расстройством, связанным с дисфункцией различных отделов центральной и периферической нервной системы (табл. 1) [37].
Снижение численности дофаминергических нейронов в компактной части черной субстанции приводит к уменьшению содержания дофамина в полосатом теле, что вызывает дисфункцию нейронов других базальных ганглиев, прежде всего растормаживание нейронов внутреннего сегмента бледного шара и ретикулярной части черной субстанции [38], торможение таламокортикальных нейронов и дефицит активации нейронов дополнительной моторной коры. С указанными изменениями связывают развитие основных двигательных симптомов БП и когнитивных дизрегуляторных расстройств [23, 39]. Нейродегенеративный процесс при БП поражает также вентральные отделы среднего мозга и голубое пятно [40], что проявляется норадренергической и дофаминергической недостаточностью [29, 40].
Результат поражения ацетилхолинергического ядра Мейнерта – ацетилхолинергический дефицит нейронов коры теменных, височных и затылочных отделов больших полушарий головного мозга. Согласно проведенным исследованиям, атрофия коры медиальных отделов височных долей, гиппокампа и сопутствующий этим морфологическим изменениям ацетилхолинергический дефицит могут быть основанием для развития гиппокампальных нарушений памяти, обычно не характерных для БП [29]. В то же время ацетилхолинергическая недостаточность при БП все же выражена меньше, чем при болезни Альцгеймера, что, предположительно, объясняет лучший ответ на лечение ацетилхолинергическими препаратами у пациентов с БП и деменцией, а также у пациентов с деменцией с тельцами Леви.
Серотонинергические дорсальные ядра шва ствола головного мозга тоже подвержены нейродегенеративному процессу при БП с деменцией, в результате чего снижается уровень серотонина в стриатуме и коре. Серотонин участвует в формировании положительного эмоционального подкрепления при достижении результата деятельности. Снижение уровня серотонина в соответствующих отделах головного мозга может стать причиной развития эмоционально-поведенческих нарушений (депрессии и апатии) [39].
Поражение нейронов в голубом пятне вызывает недостаток норадреналина. В свою очередь снижение уровня церебрального норадреналина может приводить к нарушению процесса запоминания эмоционально значимых событий и колебанию либо недостаточности уровня внимания [41, 42], умственной работоспособности, памяти [39].
М. Ray и I. Bohr (2004) установили, что изменения в никотиновых рецепторах могут быть связаны с нарушениями внимания, памяти и обусловливать развитие зрительных галлюцинаций [42]. В исследованиях, посвященных активности глутаматергической системы при БП с деменцией, выявлено снижение функции нескольких глутаматных рецепторов [43].
Диагностика
Диагностические критерии
Подход к диагностике деменции при БП менялся, особенно значительно за последние 20 лет. Так, до 2007 г. для установления этого диагноза использовались критерии Международной классификации болезней 10-го пересмотра и/или DSM-IV и даже показатели нейропсихологических шкал (чаще всего Краткой шкалы оценки психического статуса, деменция устанавливалась при сумме баллов 26 или менее). Деменция должна была развиться не ранее чем через год от дебюта двигательных расстройств. В 2007 г. Комитет экспертов Международного общества расстройств движения (Movement Disorder Society Task Force) опубликовал Клинические диагностические критерии деменции, ассоциированной с болезнью Паркинсона (табл. 2).
Дополнительные методы исследования
Биологические маркеры. Количественно определяемые биологические параметры, индикаторы физиологических и патологических биологических процессов.
Найдено несколько потенциальных биомаркеров, позволяющих выявить нейродегенеративные заболевания с образованием телец Леви, в том числе БП с деменцией: альфа-синуклеин, бета-амилоид 42, фосфорилированный тау-протеин.
Альфа-синуклеин – основной биомаркер синуклеинопатий – может быть выявлен и в крови, и в спинномозговой жидкости. В исследованиях показано, что увеличение уровня олигомерного альфа-синуклеина в ликворе и плазме крови высоко специфично для БП (85% случаев). Дальнейший рост концентрации альфа-синуклеина в плазме крови прямо связан с развитием деменции. У пациентов с БП и деменцией уровень альфа-синуклеина выше, чем у пациентов с БП без деменции [44]. Еще специфичнее повышение концентрации фосфорилированного альфа-синуклеина. У здоровых людей фосфорилировано не более 4% всего альфа-синуклеина, у пациентов с БП этот показатель существенно возрастает [45].
При развитии нейродегенерации с накоплением амилоидного белка в веществе головного мозга его концентрация в спинномозговой жидкости снижается. Таким образом, у пациентов с БП и болезнью Альцгеймера концентрация амилоидного белка в ликворе будет ниже таковой у пациентов с БП без деменции и здоровых пожилых людей [46]. Часто снижение уровня амилоидного белка сопровождается двумя процессами. Первый – реципрокное повышение уровня тау-протеина, что свидетельствует о гибели нейронов головного мозга и может отмечаться при целом ряде заболеваний – инсульте, после генерализованного эпилептического припадка, при энцефалите, нейродегенерации и т.д. Второй – появление в спинномозговой жидкости гиперфосфорилированного тау-белка, что в сочетании со снижением уровня амилоидного белка высоко специфично для болезни Альцгеймера.
Установлено, что одновременное снижение уровня бета-амилоида, повышение уровня альфа-синуклеина и тау-протеина, в том числе гиперфосфорилированного тау-протеина, в спинномозговой жидкости ассоциируется с деменцией у пациентов с БП [34].
МРТ головного мозга. В диагностике БП до недавнего времени МРТ рассматривалась в основном для исключения или подтверждения заболеваний, вызывающих вторичный паркинсонизм: опухолей, субдуральных гематом, сосудистого поражения головного мозга, гидроцефалии и т.д. [39, 47].
В последние годы описаны некоторые нейровизуализационные признаки, характерные для БП. И.В. Литвиненко и М.М. Одинак (2011) показали, что у пациентов с БП и деменцией наблюдается атрофия коры височных и затылочных долей головного мозга. Указанные изменения достоверно отличают пациентов с БП и деменцией от пациентов с БП без деменции при равной выраженности двигательных расстройств [47].
МРТ-изменения, типичные для сосудистого поражения головного мозга (лакуны, перивентрикулярный и субкортикальный лейкоареоз), часто обнаруживаются у пожилых пациентов с сочетанием БП, артериальной гипертензии, сахарного диабета, гиперлипидемии, гиперкоагуляции, нарушений сердечного ритма. Согласно И.В. Литвиненко и соавт. (2011), у 36% пациентов с БП и деменцией наблюдался выраженный перивентрикулярный лейкоареоз в виде «шапочек» с неровными контурами у задних рогов боковых желудочков. Среди больных с БП без деменции подобная локализация лейкоареоза была отмечена лишь в 6,7% случаев. При сочетании БП с сосудистым поражением головного мозга у пациентов достоверно чаще обнаруживается нарушение ходьбы и равновесия. Кроме того, у них больше выражены когнитивные расстройства [47].
Воксел-ориентированная морфометрия головного мозга (Voxel-Based Morphometry). Заключается в выполнении высокопольной МРТ и анализе объема различных структур головного мозга.
М. Zarei (2011) выявил сильную отрицательную корреляцию объема хвостатого ядра с общим баллом в третьем разделе Унифицированной рейтинговой шкалы при БП (выраженность когнитивных и психических расстройств) [48].
Деменция при БП ассоциируется с распространенным истончением коры головного мозга в дорзальных и медиальных лобных областях, а также коры височных, заднетеменных, затылочных областей.
Оживленная дискуссия ведется по поводу типичных МРТ-проявлений при деменции на фоне БП. Высказываются различные точки зрения:
- деменция при БП ассоциируется главным образом с истончением коры височных долей и задней части поясной извилины;
- деменция при БП ассоциируется со снижением объема префронтальной, островковой, верхней височной извилины и прекунеуса, в то время как патологические изменения в височной доле и структурах гиппокампового круга больше связаны с болезнью Альцгеймера [49, 50].
Позитронно-эмиссионная томография. Исследование головного мозга, позволяющее измерить выброс радиоактивно меченных метаболически активных химических веществ, введенных в кровеносное русло.
В настоящее время наиболее часто используемым индикатором позитронно-эмиссионной томографии остается меченая форма глюкозы – 18F-дезоксиглюкоза (фтордезоксиглюкоза). Это маркер уровня метаболизма глюкозы в мозге, показывающий распределение активности клеток. Активно применяется и Питтсбургская субстанция B – маркер отложения бета-амилоида в головном мозге.
Согласно проведенным исследованиям, у больных БП на ранних стадиях был выявлен лишь незначительный гипометаболизм глюкозы в различных отделах коры головного мозга. На 2,5–3-й стадии БП без деменции отмечался гипометаболизм в хвостатых ядрах и дорсолатеральной префронтальной коре при сохранном метаболизме в других структурах мозга [49, 51]. Указанные изменения коррелировали с результатами нейропсихологических тестов, согласно которым у пациентов имели место снижение способности к обобщению, анализу и синтезу информации, негрубые нарушения памяти и внимания, зрительно-пространственных функций. В группе больных БП с деменцией наблюдалось значительное двустороннее снижение метаболизма коры больших полушарий головного мозга. У пациентов с БП, деменцией и зрительными галлюцинациями наибольшие изменения обнаружены в затылочной коре, поясных извилинах, орбитофронтальной коре и структурах гиппокампового круга [51].
Лечение
Нефармакологические методы
К общим мерам относятся информационные беседы с пациентом и лицами, участвующими в уходе за ним, рекомендации по сохранению достаточной психической и физической активности, исключение психоэмоциональных перегрузок, неблагоприятных воздействий окружающей среды.
Положительно влияют на когнитивные функции физическая активность и тренировки: аэробные нагрузки, упражнения на сопротивление, растяжение, силовой и баланс-тренинг. Предпочтение отдается аэробному тренингу, поскольку предполагается, что он способствует улучшению как функциональной церебральной активности, так и, возможно, формированию новых межсинаптических взаимодействий и снижает степень церебральной гипоперфузии и гипометаболизма [46].
Когнитивный тренинг улучшает внимание, исполнительные функции, память и зрительно-пространственные функции у пациентов с БП и деменцией [52]. Проведено шесть рандомизированных контролируемых исследований по эффективности данного метода лечения, пять из которых показали положительный результат, в одном исследовании получены достоверные доказательства эффективности метода.
Когнитивный тренинг представляет собой специальные программы и методики для тренировки памяти, внимания и других когнитивных функций, направленные на поддержание оптимального интеллектуального уровня, развитие сниженных когнитивных функций, а также на обучение стратегиям компенсации. Выделяют два типа когнитивного тренинга: компенсаторный и восстановительный. В ходе компенсаторного когнитивного тренинга пациент обучается новым стратегиям решения поставленной задачи через сохранные когнитивные функции. При восстановительном когнитивном тренинге мероприятия нацелены на улучшение поврежденных когнитивных функций [46].
Медикаментозная терапия
Доказано, что при БП с деменцией имеет место холинергический дефицит, в связи с чем наиболее обоснованно применение ингибиторов ацетилхолинэстеразы. Клинические исследования показали, что таким пациентам могут назначаться все ингибиторы ацетилхолинэстеразы, зарегистрированные на настоящий момент: донепезил, ривастигмин, галантамин.
Согласно метаанализу исследований эффективности ингибиторов ацетилхолинэстеразы при БП с деменцией, на фоне лечения у пациентов уменьшается выраженность когнитивных нарушений, улучшаются поведение и общее качество жизни [53]. Назначение ингибиторов ацетилхолинэстеразы способствует уменьшению выраженности психических расстройств, таких как апатия, тревожность, галлюцинации и делирий [54]. На фоне лечения выраженность двигательных расстройств не нарастает.
Второй класс препаратов, использующийся при БП с деменцией, – антагонисты рецепторов к N-метил-D-аспартату. Представитель этого класса – мемантин. Проведенные клинические исследования показали, что препарат эффективен на стадии умеренных когнитивных расстройств и у пациентов с сочетанием БП и деменции, и у пациентов с БП и отсутствием деменции. На фоне приема мемантина достоверно снижается выраженность когнитивных нарушений, апатии, поведенческих расстройств. Выраженность зрительных галлюцинаций, однако, обычно остается неизменной или уменьшается незначительно [52].
У пациентов с БП без деменции можно ожидать положительный эффект от приема препаратов леводопы в том числе в отношении когнитивных функций. Так, согласно полученным данным, на фоне коррекции дозы леводопы у пациентов с БП без деменции ускорялись психические реакции, снижалась инертность мышления, облегчался переход с одного этапа задачи на другой. Однако результаты исследований эффективности препаратов леводопы у пациентов с сочетанием БП и деменции свидетельствуют, что никакого влияния на когнитивные функции у этих пациентов назначение или изменение дозы препаратов леводопы уже не оказывает [55].
В настоящее время проходит клинические испытания нилотиниб, способствующий деградации альфа-синуклеина с патологической конформацией (препарат зарегистрирован для лечения хронического миелолейкоза, обладает способностью ингибировать тирозинкиназу кластерного региона точечного разрыва Абельсона) [56].
Интенсивно разрабатывается иммунотерапия – создание антител против альфа-синуклеина. Несколько антител продемонстрировали активность in vitro и на животных моделях. Не так давно были обнародованы результаты исследования эффективности моноклональных антител PRX002 к альфа-синуклеину у 40 здоровых добровольцев [56]. Безусловно, сейчас исследования находятся на начальном этапе, требуется дальнейшее изучение как безопасности, так и эффективности этого метода лечения.
Купирование нейропсихиатрических симптомов
Психиатрические и поведенческие симптомы часто дезадаптируют пациентов и их родственников больше, чем двигательные проявления. Терапия ингибиторами ацетилхолинэстеразы может иметь положительный эффект в отношении ряда симптомов: зрительных галлюцинаций, нарушения цикла «сон – бодрствование», апатии, бредовых расстройств [54]. В меньшей степени она влияет на выраженность агрессии и психомоторного возбуждения, в таких случаях препаратами выбора являются антагонисты D2-рецепторов. Необходимо отметить, что применение типичных нейролептиков при БП с деменцией абсолютно недопустимо из-за высокой вероятности нарастания выраженности двигательных расстройств и развития нейролептического синдрома [57]. Возможен прием только атипичных нейролептиков и обязательно под контролем врача. Назначать препараты и наращивать дозу целесообразно в условиях стационара.
Наибольшие доказательства эффективности, согласно результатам проведенных клинических исследований, достигнуты для клозапина. Препарат не оказывает значимого влияния на моторные функции. Прием клозапина требует еженедельного мониторинга общего анализа крови в течение первых трех месяцев, а затем ежемесячно в связи с высоким риском развития агранулоцитоза [58].
Кветиапин также может использоваться в качестве препарата первой линии, хотя его эффективность, по оценкам, ниже, чем у клозапина [58]. Однако вследствие лучшей переносимости и более высокой безопасности его принимают даже чаще.
Необходимо отметить, что при БП с деменцией применение даже атипичных нейролептиков сопряжено с риском развития нейролептического синдрома, поэтому лечение препаратами этой группы должно проводиться обдуманно. Нейролептический синдром у пациентов с БП и деменцией был описан на фоне лечения рисперидоном, оланзепином, арипразолом. Препараты этой фармакотерапевтической группы должны с осторожностью назначаться пожилым пациентам с сердечно-сосудистой патологией, поскольку повышают риск сосудистых событий (ишемический инсульт, инфаркт миокарда) и смерти [57].
В 2016 г. в США был зарегистрирован новый таблетированный лекарственный препарат для лечения галлюцинаций и иллюзий при болезни Паркинсона – пимавансерин, обратный агонист 5-HT2A-рецепторов (подтип серотониновых рецепторов). Препарат не проявляет активности в отношении дофаминовых, гистаминовых, мускариновых и адренорецепторов. Изучению этого препарата было посвящено 25 клинических исследований (общее число испытуемых около 1200). В результате было доказано, что пимавансерин уменьшает частоту и/или тяжесть галлюцинаций и иллюзий, но не влияет на выраженность двигательных расстройств при БП [56].
Аффективные расстройства и депрессия – частые симптомы при БП с деменцией и без нее. Симптомы депрессии не всегда легко диагностируются у пациентов с БП – часто пациент не жалуется на снижение настроения. Одновременно с депрессией нарастает выраженность и двигательных, и когнитивных нарушений. В этой связи симптомы депрессии нуждаются в прицельной диагностике методами расспроса и анкетирования не только пациентов, но и их родственников.
Клинические исследования и метаанализ различных препаратов, применявшихся в лечении депрессии у пациентов с БП и деменцией, свидетельствуют о наибольшей активности дезипрамина и циталопрама, а также пароксетина и венлафаксина по сравнению с другими антидепрессантами и плацебо [59]. Таким образом, для лечения депрессии у пациентов с БП и деменцией предпочтительнее использовать селективные ингибиторы обратного захвата серотонина и селективные ингибиторы обратного захвата серотонина и норадреналина.
N.V. Trofimova, I.S. Preobrazhenskaya, M.A. Bykanova
I.M. Sechenov First Moscow State Medical University (Sechenovskiy University)
Contact person: Irina Sergeyevna Preobrazhenskaya, irinasp2@yandex.ru
The article provides the current data on the epidemiology, morphology, clinical manifestations of Parkinson's disease combined with dementia. On discussion different points of view about the nature of dementia in Parkinson's disease as well as the possible contribution to the development of vascular dementia and neurodegenerative diseases. The modern diagnostic criteria for dementia in Parkinson's disease are provided. The recommendation data and clinical studies of the efficacy of drugs used in the treatment of motor, cognitive, psychiatric disorders in patients with combination of dementia and Parkinson's disease are summarized.