Диабетическая ретинопатия: можно ли предотвратить слепоту?
- Аннотация
- Статья
- Ссылки
- English
Введение
Сахарный диабет (СД) является проблемой здравоохранения во всем мире. По прогнозам эпидемиологов, к 2030 г. численность больных СД превысит 550 млн. В Российской Федерации, по данным государственного регистра, на сегодняшний день насчитывается около 4 млн пациентов с СД. За 15 лет численность больных увеличилась практически в два раза.
Медико-социальная значимость СД обусловлена хроническими осложнениями (преимущественно сосудистыми: нефропатией, ретинопатией, поражением магистральных сосудов сердца, головного мозга, периферических сосудов нижних конечностей), длительной потерей трудоспособности и смертностью пациентов, которая в два – шесть раз превышает таковую в общей популяции [1]. Очевидно, что вопрос о возможности предотвращения хронических осложнений СД является актуальным не только для современной диабетологии, но и медицинской науки в целом.
Патогенез сосудистых осложнений при сахарном диабете и способы их профилактики
Патогенез сосудистых осложнений изучен, но не до конца. Главная роль отводится гипергликемии. Фундаментальные экспериментальные работы показали, что глюкоза (как химически активное соединение) в высоких концентрациях оказывает патологическое воздействие на внутриклеточную среду органов-мишеней, нарушая их нормальное функционирование [2].
В течение последних 10 лет изучается роль инсулинорезистентности в патогенезе как макрососудистых, так и микрососудистых осложнений СД 2 типа. Так, в ходе популяционного исследования инсулинорезистентность была выявлена у 58% пациентов с артериальной гипертензией, у 84% – с гипертриглицеридемией, у 42% – с гиперхолестеринемией, у 66% – с нарушением толерантности к глюкозе и у 95% – с метаболическим синдромом [3]. Важное значение также придают окислительному стрессу [4], иммунному генезу, генетической предрасположенности [5], эндотелиальной дисфункции. Однако определить, что стало первопричиной инсулинорезистентности, зачастую бывает трудно.
Необходимо помнить, что гипергликемия способна влиять на все звенья патогенеза, в том числе и на развитие окислительного стресса, эндотелиальной дисфункции, активацию полиолового пути утилизации глюкозы.
В исследовании UKPDS в 1998 г. была показана взаимосвязь между риском развития макро- и микрососудистых осложнений у больных СД и степенью нарушения углеводного обмена. Так, повышение уровня НbА1с на 1% сопровождается увеличением риска развития не только микрососудистых осложнений (ретинопатии, нефропатии, нейропатии) на 35%, но и смертельных и несмертельных инфарктов миокарда на 18%, смертельных исходов, связанных с СД, на 25%, общей смертности на 7% [6].
В ряде исследований было доказано, что коррекция гипергликемии и достижение целевых значений гликолизированного гемоглобина (НbА1с) приводят к значительному снижению риска развития микрососудистых осложнений [7]. Например, результаты исследования UKPDS продемонстрировали, что интенсивный контроль гликемии у пациентов СД 2 типа с помощью препаратов сульфонилмочевины или инсулина снижает риск развития диабетической нефропатии и других микрососудистых осложнений, но не макрососудистых [8]. В рандомизированном шестилетнем исследовании было показано, что благодаря интенсивной инсулинотерапии частота развития или прогрессирования диабетической микроангиопатии снизилась по сравнению с таковой на фоне инсулинотерапии в виде одной-двух инъекций в сутки (7,7 против 28%) [9]. Исследование ADVANCE также подтвердило роль интенсивного контроля гликемии в предотвращении микрососудистых осложнений СД 2 типа [10].
Надо отметить, что не все органы и ткани одинаково чувствительны к высокой концентрации глюкозы – только те, которые не имеют механизма регуляции транспорта глюкозы внутрь клетки. К «незащищенным» этим механизмом клеткам относятся клетки эндотелия сосудов, мезангиальные клетки в почечных клубочках, нейроны и шванновские клетки, окружающие периферические нервы. При гипергликемии внутри этих клеток создается высокая концентрация глюкозы, которая вступает в соединение с внутриклеточными структурами, вызывая их повреждение [11].
Хроническая гипергликемия активирует ряд биохимических процессов, в ходе которых образуются конечные необратимые продукты гликирования (КПГ), протеинкиназа С и высокореактивные соединения кислорода, повреждающие сосудистую стенку [12]. Именно поэтому можно предположить, что гипергликемия является первичным фактором поражения эндотелия сосудистой стенки.
КПГ – результат неферментативного гликозилирования внутриклеточных белков, регулирующих транскрипцию генов. КПГ могут диффундировать из клеток в межклеточное пространство, нарушая структуру внеклеточного пространства, внеклеточного матрикса и передачу сигнала от клетки. Они также могут поступать в кровь, связываться с циркулирующими белками (например, альбуминами), присоединяться к клеточным рецепторам КПГ на макрофагах, стимулируя образование цитокинов, факторов роста и других факторов, повреждающих ткани, или к рецепторам на поверхности эндотелиальных клеток, провоцируя выброс вазоактивных факторов [13].
Наибольшее повреждающее воздействие оказывают реактивные КПГ, изменяющие структуру и метаболизм основных белков, липидов и ДНК. Последствиями необратимого гликозилирования являются изменение структуры и функции базальной мембраны сосудов, активация пролиферативных, воспалительных и склеротических процессов, изменение экспрессии ряда генов и повышение частоты мутаций.
Кроме того, гипергликемия усиливает выработку ферментов, участвующих в синтезе коллагена, а также задерживает репликацию эндотелиоцитов и способствует гибели клеток отчасти через активизацию окислительных процессов и гликозилирования.
Окислительный стресс при СД может быть следствием повышенного образования реактивных окислителей, снижения активности антиоксидантной системы, нарушения функционирования ферментов, изменения концентрации или процессов при участии глутатиона и ионов металлов переменной валентности.
Повышение гликирования и образования КПГ в совокупности с избытком свободных радикалов вызывает дисфункцию эндотелия, инициируя развитие ангиопатий. КПГ реагируют со специфическими рецепторами на поверхности эндотелиальных клеток, макрофагов, нейронов, гладкомышечных клеток и др., что способствует усилению локального окислительного стресса и активации ядерного фактора каппа B (NF-κB), являющегося первичным регулятором ответа на окислительный стресс. Это сопровождается экспрессией генов, контролирующих синтез разных белков, и опосредует высвобождение фактора некроза опухоли и интерлейкина 1 (ИЛ-1), которые участвуют в процессах, приводящих не только к изменениям в сосудистой стенке, но и к дефициту секреции инсулина и развитию инсулинорезистентности.
В последние годы все больше внимания уделяется роли эндотелиальных клеток, выстилающих сосудистое русло, в развитии органной патологии. Клетки эндотелия являются первым барьером между циркулирующей кровью и тканями, с помощью которого регулируется проникновение жидкости и растворенных в ней веществ, макромолекул, клеточных элементов и лекарств из крови в прилежащую ткань [14].
Однако более вероятным представляется мнение, что патогенез микрососудистых осложнений един и связан с воздействием гипергликемии на основную клетку-мишень – эндотелий сосудов. Высокая восприимчивость сосудов к воздействию гипергликемии при СД обусловлена тем, что эндотелиальные клетки как крупных, так и мелких сосудов являются инсулиннезависимыми. Именно поэтому глюкоза беспрепятственно проникает в эндотелий сосудов, вызывая их дисфункцию.
Нарушение функционального состояния эндотелия сосудов можно диагностировать по биохимическим и функциональным маркерам. К биохимическим маркерам поврежденного эндотелия относится повышение концентрации в крови биологически активных веществ, синтезируемых эндотелием или экспрессируемых на его поверхности. Наиболее значимыми считаются фактор Виллебранда, эндотелин 1, молекулы адгезии, тканевой активатор плазминогена, тромбомодулин, фибронектин.
Функциональным маркером поврежденного эндотелия является нарушение эндотелийзависимой вазодилатации сосудов, сохранность которой обеспечивается секрецией сосудорасширяющей молекулы NO. Именно этой молекуле принадлежит роль модератора основных функций эндотелия: регуляция активности и последовательности запуска всех остальных биологически активных веществ, продуцируемых эндотелием. NO не только вызывает расширение сосудов, но и блокирует пролиферацию гладкомышечных клеток, препятствует адгезии клеток крови. Соединение обладает также антиагрегантными свойствами. Таким образом, NO – базовый фактор антиатерогенеза. NO-продуцирующая функция эндотелия наиболее ранима из-за нестабильности молекулы NO, являющейся по своей природе свободным радикалом. В результате благоприятное антиатерогенное действие NO нивелируется и уступает токсическому атерогенному действию других факторов поврежденного эндотелия.
Беспрепятственное проникновение глюкозы в эндотелиальные клетки нарушает сбалансированную выработку эндотелием вазоактивных веществ, факторов свертывающей системы крови, факторов роста и пролиферацию. В результате нарушается внутриорганная и тканевая гемодинамика, активируются тромбообразование, пролиферация сосудов и гладкомышечных клеток. Восстановление функции эндотелиальных клеток возможно только при устранении патологического фактора, вызвавшего дисфункцию эндотелия.
Развитие диабетической микроангиопатии обусловлено в первую очередь воздействием разных факторов роста и цитокинов. Наиболее изученными из них являются трансформирующий фактор роста бета (TGF-бета), фактор роста соединительной ткани (CTGF), сосудистый эндотелиальный фактор роста (VEGF), инсулиноподобный фактор роста (IGF), тромбоцитарный фактор роста (PDGF), фактор роста пигментного эпителия (PEGF) [15]. VEGF – главный фактор ангиогенеза. Он синтезируется преимущественно подоцитами; гипергликемия увеличивает его продукцию. Этот фактор роста способен повышать проницаемость капилляров, его гиперпродукция может вносить вклад в развитие неоваскуляризации [16, 17]. VEGF повышает синтез коллагена типа IV в подоцитах и усиливает пролиферацию мезангиальных клеток.
PEGF – мощный ингибитор ангиогенеза. Снижение уровня этого фактора ассоциируется с развитием ретинопатии. В условиях in vitrо показано, что секреция PEGF значительно снижается при гипергликемии. PEGF блокирует повышенную экспрессию трансформирующего фактора роста и фибронектина, индуцируемую гипергликемией.
По данным Национального исследования здоровья и питания в США, 69% больных диабетом имеют нарушения липидного обмена. У таких пациентов определенные нарушения липидного обмена сохраняются и после коррекции показателей глюкозы крови. Эти нарушения получили название «диабетическая дислипидемия». К диабетической дислипидемии относят гипертриглицеридемию, повышение уровня липопротеинов низкой плотности (ЛПНП) и снижение уровня липопротеинов высокой плотности (ЛПВП). Эти метаболические изменения сопровождаются развитием системного воспаления, окислительного стресса, что в совокупности приводит к нарушению функции эндотелия сосудов и, как следствие, развитию воспаления непосредственно в стенке сосудов [18].
Установлено, что атерогенные ЛПНП являются классом атерогенных липопротеинов плазмы крови, наиболее подверженных свободнорадикальному окислению. В результате окисления изменяется структура частицы ЛПНП. Измененные ЛПНП стимулируют продукцию моноцитарного хемоаттрактантного белка (МСР-1) в мезангиоцитах. Это способствует притоку в мезангий моноцитов и макрофагов, которые захватывают большое количество измененных липопротеинов и превращаются в пенистые клетки. Моноциты и макрофаги также способны вырабатывать молекулы МСР-1. Данный белок стимулирует продукцию молекулы межклеточной адгезии (ICAM-1), ИЛ-6 и цитокинов [19].
Образующиеся в результате хронической гипергликемии свободные радикалы, которые являются высокореактивными соединениями, связываются с молекулами липидов и приводят к раннему развитию атеросклероза.
Поскольку гипергликемия является главным патогенетическим фактором развития и прогрессирования сосудистых осложнений у больных СД 2 типа, жесткий контроль уровня глюкозы в крови рассматривается как закономерный способ профилактики развития и прогрессирования сосудистых осложнений. Помимо этого доказанным методом профилактики прогрессирования сосудистых осложнений является оптимальная коррекция метаболических нарушений.
Диабетическая ретинопатия: диагностика, профилактика и лечение
Диабетическая ретинопатия – специфичное позднее сосудистое осложнение СД, развивающееся, как правило, последовательно: от изменений, связанных с повышенной проницаемостью и окклюзией ретинальных сосудов, до появления новообразованных сосудов и соединительной ткани [20].
Данное заболевание является основной причиной слепоты среди лиц трудоспособного возраста. У больных СД ретинопатия развивается в 25 раз чаще, чем в общей популяции.
Снижение зрения происходит в основном из-за поражения центральной зоны глазного дна (макулярной области): отека, резкого нарушения кровотока (вплоть до полного отсутствия перфузии капилляров) и тракционной деформации сетчатки в результате разрастания и сокращения фиброзной ткани. Полная потеря зрения связана, как правило, с развитием пролиферативного процесса: кровоизлиянием в стекловидное тело, отслойкой сетчатки и развитием неоваскулярной глаукомы, возникающей вследствие блокирования новообразованными сосудами оттока внутриглазной жидкости и приводящей к неконтролируемому повышению внутриглазного давления. Следует учитывать, что даже далеко зашедший процесс, в подавляющем большинстве случаев при отсутствии лечения приводящий к слепоте, может протекать бессимптомно – пациенты не догадываются об осложнении вплоть до значительного снижения зрения, которое на этих стадиях часто необратимо.
Проблема предотвращения слепоты при СД во многом носит организационный характер. Ее решение требует:
- четкого взаимодействия врачей разных специальностей при ведении больных;
- своевременного направления пациента к офтальмологу;
- адекватного офтальмологического обследования;
- оценки степени риска прогрессирования и снижения остроты зрения (при наличии диабетической ретинопатии и макулярного отека);
- своевременного начала лечения.
В основе патогенеза диабетической ретинопатии лежит ишемия, развивающаяся из-за окклюзии сосудов сетчатки. Причина нарушения перфузии сетчатки – поражение эндотелия сосудов (эндотелиальный стресс), возникающее вследствие резкого усиления ретинального кровотока в условиях гипергликемии и приводящее к образованию тромбов в капиллярном русле. Открытие шунтов в ответ на значительное снижение кровоснабжения лишь усугубляет ситуацию, поскольку перераспределяет кровоток в обход неперфузируемой сетчатки. Повреждение эндотелия, усиление агрегации тромбоцитов и активация факторов коагуляции способствуют окклюзии капилляров. Важная роль в этом процессе принадлежит свободным радикалам, которые оказывают негативное воздействие на эндотелиальные клетки. Свободные радикалы – активные окислители, продукты нормального метаболизма. При СД их выработка резко повышается. Свободные радикалы денатурируют белки, вызывая их агрегацию, и окисляют липиды с формированием липидных пероксидов, участвующих в различных процессах, в том числе метаболизме арахидоновой кислоты, усиливая агрегацию тромбоцитов.
Увеличение площади ишемии повышает экспрессию VEGF. Выработка VEGF выше критического уровня приводит к развитию основных клинических проявлений диабетического поражения сетчатки – макулярного отека и неоваскуляризации [15, 21, 22].
Надо отметить, что нормальное функционирование сетчатки обеспечивается в значительной степени полноценностью гематоретинальных барьеров. Стенка капилляров сетчатки представляет собой внутренний гематоретинальный барьер, который регулирует метаболический обмен между кровью и сетчаткой и поддерживает сетчатку в дегидратированном и прозрачном состоянии. Воздействуя на эндотелиальные белки плотных межклеточных контактов, VEGF увеличивает сосудистую проницаемость. Это в свою очередь усиливает экссудацию и накопление внеклеточной жидкости и белков в ткани сетчатки. Жидкость, которая проходит через стенку капилляров, в норме должна реабсорбироваться пигментным эпителием (наружный гематоретинальный барьер) и соседними капиллярами сетчатки. Когда диффузия превышает потенциальные возможности пигментного эпителия и капилляров к реабсорбции жидкости, возникают клинические признаки макулярного отека.
Изменения во внеклеточном матриксе, обеспечивающие миграцию эндотелиальных клеток, повышенная экспрессия VEGF, разрушение контактов между эндотелиальными клетками способствуют появлению новообразованных сосудов, которые растут по задней поверхности стекловидного тела. Стенка новообразованного сосуда неполноценна, что приводит к выходу за его пределы как компонентов плазмы, так и цельной крови. Это стимулирует разрастание соединительной ткани в зонах неоваскуляризации. Поскольку соединительная ткань всегда стремится к сокращению, а адгезия фиброваскулярного конгломерата к задней поверхности стекловидного тела очень плотная, развивается отслойка стекловидного тела. Как правило, в этот момент происходит разрыв новообразованного сосуда с развитием преретинальных (перед поверхностью сетчатки) или витреальных (в полость стекловидного тела) кровоизлияний. Рецидивирующие кровоизлияния и происходящее вследствие этого рубцевание задних отделов стекловидного тела ведут к образованию патологических витреоретинальных сращений, которые могут вызвать тракционную отслойку сетчатки [23].
Распространенность диабетической ретинопатии и макулярного отека достаточно высока. В эпидемиологическом исследовании, проведенном в Санкт-Петербурге в 2003–2005 гг. (4989 пациентов с СД 1 типа и 2241 пациент с СД 2 типа, получавший инсулинотерапию), ретинопатия выявлялась в 65,4 и 83,9% случаев соответственно. Частота клинически значимого макулярного отека составила 7,4% среди больных СД 1 типа и 30,3% среди больных СД 2 типа. Более чем в половине случаев клинически значимый макулярный отек выявлялся на обоих глазах (у 57,3% пациентов с СД 1 типа и 66% пациентов с СД 2 типа на инсулинотерапии).
У больных СД 1 типа наиболее распространенными были следующие стадии ретинопатии: непролиферативная диабетическая ретинопатия – у 46,6% больных, пролиферативная диабетическая ретинопатия – у 14,2%. Причем в 5,5% случаев была диагностирована тяжелая форма пролиферативной диабетической ретинопатии (высокого риска значительного снижения зрения), а в 2,3% – далеко зашедшая. В группе СД 2 типа непролиферативная диабетическая ретинопатия была диагностирована у 53,6% пациентов, пролиферативная диабетическая ретинопатия – у 20,9%. При этом тяжелая ее форма отмечена у 9,7% больных, далеко зашедшая – у 4,1% [24].
В настоящее время выделяют два основных направления предупреждения слепоты: выявление (скрининг) ретинопатии и ее лечение с помощью лазерной коагуляции.
Скрининг – диагностическая процедура, проводимая у всех пациентов группы риска с целью выявления поражений, требующих дополнительного обследования и лечения [25]. К сожалению, даже в тех странах, где программы динамического наблюдения за больными СД существуют уже давно, менее половины пациентов, нуждающихся в офтальмологическом осмотре, обращаются за консультацией, а менее половины обратившихся получают адекватное офтальмологическое обследование [26]. Основной методикой скрининга диабетической ретинопатии в европейских странах является стандартная фотография сетчатки (в России – биомикроскопия глазного дна с использованием асферических линз).
Дополнительными методами обследования считаются оптическая когерентная томография и флюоресцентная ангиография. С их помощью можно выявить патологические изменения, неразличимые при биомикроскопии. Оптическая когерентная томография позволяет оценить толщину и топографию различных слоев сетчатки в центральной (макулярной) области. Она дает информацию о степени выраженности и характере макулярного отека. Процедура безопасна для пациента, поскольку в ее основе лежит принцип интерферометрии.
Флюоресцентная ангиография основана на явлении флюоресценции – свечении введенного в кровяное русло вещества (натриевой соли флюоресцеина) в ответ на световое воздействие. Для проведения флюоресцентной ангиографии используются аппараты, предназначенные для фотографирования глазного дна (ретинальные камеры или ангиографы).
Основной метод лечения клинически значимых проявлений диабетической ретинопатии в настоящее время – лазерная коагуляция сетчатки. Это подтверждается данными многочисленных исследований (отечественных и зарубежных). Именно информация о высокой эффективности лазеркоагуляции сетчатки как средства предупреждения потери зрения стала причиной разработки скрининговых программ для выявления диабетической ретинопатии [25, 27].
В 1968 г. L. Aiello и соавт. сообщили о первых результатах лечения пролиферативной диабетической ретинопатии с помощью коагуляции. При разработке этой методики авторы учитывали собственные наблюдения за больными СД, у которых кроме диабетических изменений были выявлены изменения, связанные с миопией высокой степени и хориоретинальной дистрофией. Было отмечено, что после процедуры пролиферация, как правило, не развивается, а диабетическая ретинопатия протекает в более легкой форме. Исследователи предположили, что при подобных изменениях потребность сетчатки в кислороде и активность ретинальных обменных процессов снижаются. В таких условиях продукция ростовых факторов недостаточна для запуска механизма пролиферации [28].
Примерно в это же время G. Meyer-Schwickerath и соавт. показали, что лазерная коагуляция микроаневризм, расположенных в центре колец «твердых» экссудатов, также дает положительный эффект [29]. Последующие работы подтвердили эффективность лазерного воздействия на кольца «твердых» экссудатов: исчезали экссудаты в зоне воздействия (несмотря на то, что методика лазеркоагуляции была различна). В 1976 г. Н. Schatz и соавт. сообщили о первых результатах перифовеолярной лазерной коагуляции сетчатки в виде решетки [30]. Высокая эффективность данного вида лазерной коагуляции отмечена и в работах G.W. Blankenship и соавт. [31, 32].
Однако только после проведения широкомасштабных исследований, выполненных группой по изучению диабетической ретинопатии (DRS – Diabetic Retinopathy Study) и группой по изучению раннего лечения диабетической ретинопатии (ETDRS – Early Treatment Diabetic Retinopathy Study), метод получил мировое признание [33–36]. В ходе этих работ была не только доказана эффективность данного метода лечения, но и сформулированы показания для лазерной коагуляции. Так, лазерное лечение на 50% уменьшило риск снижения остроты зрения при макулярном отеке и на 87% – риск развития пролиферативной диабетической ретинопатии.
Лазерная коагуляция при диабетическом поражении сетчатки направлена на выключение зон ретинальной ишемии, подавление неоваскуляризации и облитерацию сосудов с повышенной проницаемостью, а также на образование хориоретинальных сращений, которые снижают риск тракционной отслойки.
Принцип лазерной коагуляции сетчатки заключается в том, что при точно дозированном облучении энергия поглощается ретинальными структурами. Выделяющееся при этом тепло приводит к повышению температуры и образованию локальных участков ожога с последующим воспалением, которые через несколько дней превращаются в ограниченные участки рубцевания.
В последнее время широкое применение в мировой офтальмологической практике получили интравитреальные инъекции кортикостероидов и ингибиторов ангиогенеза. Интравитреальные инъекции значительно повышают шансы сохранить зрение у больных с диабетической ретинопатий и макулярным отеком, но, к сожалению, их нельзя рассматривать в качестве изолированной терапии [37, 38].
Витреальная хирургия показана при пролиферативной диабетической ретинопатии с массивными, длительно не рассасывающимися кровоизлияниями в стекловидное тело (более трех месяцев), фиброзными изменениями стекловидного тела, при витреоретинальных тракциях и отслойке сетчатки. Последнее время витрэктомия активно используется для лечения диабетического макулярного отека, резистентного к лазерной коагуляции, в сочетании с интравитреальными введениями ингибиторов VEGF и кортикостероидов. Однако, несмотря на эффективность этих методов лечения, все они сопровождаются различными (часто значительными) побочными эффектами.
Поскольку механизм возникновения и прогрессирования диабетической ретинопатии сложен и многообразен, профилактические мероприятия, направленные на сохранение зрения у больных диабетом, должны влиять на разные факторы патогенеза. Основным способом профилактики диабетических поражений сетчатки по-прежнему остается максимально стабильная компенсация СД: нормализация уровня гликемии (достижение целевых значений HbA1c) и артериального давления, коррекция дислипидемии.
Так, по данным исследования DCCT, снижение уровня HbA1c на 2% у пациентов с СД 1 типа позволяет снизить риск прогрессирования ретинопатии на 63%, развития пролиферативной ретинопатии на 47%, макулярного отека на 26%, потребность в лазерном лечении на 51% [7, 39].
В исследовании UKPDS снижение уровня HbA1c на 1% у пациентов с СД 2 типа позволило снизить потребность в лазерном лечении на 29%, в удалении катаракты на 24%, риск прогрессирования ретинопатии на 17%, кровоизлияний в стекловидное тело на 23%, слепоты на один глаз на 16% [8, 40].
Однако быстрое снижение уровня глюкозы крови может привести к развитию транзиторной ретинопатии. Этот необычный клинический феномен встречается у пациентов с ранее выраженной декомпенсацией СД после интенсификации инсулинотерапии у больных СД 1 типа, перевода больных СД 2 типа на инсулинотерапию, постановки инсулиновой помпы, трансплантации поджелудочной железы. К развитию транзиторной диабетической ретинопатии может привести значительное ухудшение состояния сетчатки во время беременности (когда пациентки чаще (интенсивнее) стремятся улучшить компенсацию диабета). Скорее всего, это связано с тем, что в условиях гипергликемии происходит значительное усиление ретинального кровотока. Резкое снижение уровня глюкозы в крови приводит к замедлению кровотока, что при наличии грубых структурных изменений ретинальных сосудов усиливает ишемизацию сетчатки (транзиторная ретинопатия характеризуется появлением большого количества «мягких» экссудатов и ретинальных геморрагий). Причина этого, возможно, кроется в связанном с гипергликемией повышенном синтезе сосудорасширяющих простагландинов, поддерживающих перфузию сетчатки даже тогда, когда имеются значительные изменения капилляров. Можно предположить, что при хорошей компенсации диабета синтез сосудорасширяющих простагландинов снижается, а следовательно, уменьшается стимул для усиленного кровотока через сохранившиеся капилляры, что приводит к ишемии, за которой (крайне редко) могут последовать пролиферативные изменения.
В течение ряда лет проводились исследования эффективности препаратов разных фармакологических групп в отношении прогноза диабетической ретинопатии. Поскольку убедительных результатов получено не было, считалось, что лекарственных препаратов, способных предупредить развитие и прогрессирование диабетической ретинопатии, не существует, и при разработке скрининговых стратегий не должны рассматриваться никакие другие лечебные воздействия, кроме лазерной коагуляции сетчатки [26].
Ситуация кардинально изменилась после проведения двух крупномасштабных программ – FIELD и ACCORD-EYE [41, 42] с применением фенофибрата.
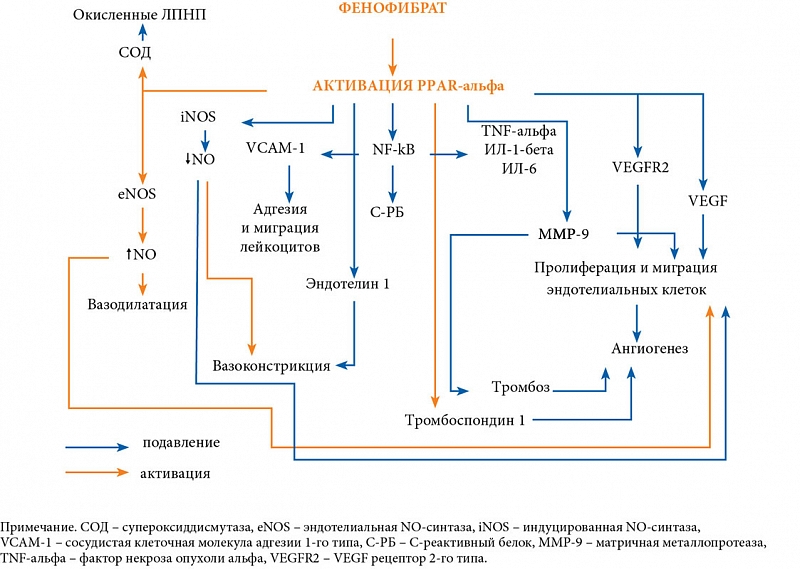
Фенофибрат является агонистом альфа-рецепторов, активируемых пероксисомными пролифераторами (PPAR-альфа). Он используется в качестве гиполипидемического препарата, оказывающего в разной степени положительное влияние на все составляющие липидного профиля: снижает уровень триглицеридов, общего холестерина, липопротеинов низкой и очень низкой плотности и повышает содержание ЛПВП. Помимо гиполипидемического эффекта фенофиброевая кислота оказывает благоприятное влияние на микрососудистое русло, в том числе на ретинальные сосуды. Наличие других положительных терапевтических эффектов, которые нельзя объяснить только гиполипидемическим действием, обусловливает повышенный интерес к этой группе препаратов и их механизму действия (рисунок).
В исследовании FIELD (9795 пациентов с СД 2 типа) оценивалась возможность применения фенофибрата для профилактики прогрессирования поражений глазного дна диабетического генеза и снижения потребности в лазерном лечении.
Лечение фенофибратом позволило снизить на 31% потребность в лазерной коагуляции сетчатки и на 79% частоту прогрессирования диабетической ретинопатии. При этом положительные эффекты фенофиброевой кислоты наблюдались независимо от степени гликемического контроля, уровня липидов крови и показателей артериального давления.
Исследование ACCORD-EYE (1593 пациента с СД 2 типа) оценивало риск развития и прогрессирования диабетических изменений сетчатки в зависимости от эффективности гликемического контроля, контроля артериального давления и комбинированной терапии фенофибратом и симвастатином против терапии симвастатином в комбинации с плацебо. Применение фенофиброевой кислоты снизило частоту прогрессирования диабетической ретинопатии на 40%. Необходимо подчеркнуть, что в исследовании ACCORD-EYE прогрессирование определялось по более жестким критериям: увеличение уровня ретинопатии на три шага и более по шкале ETDRS (Early Treatment Diabetic Retinopathy Study – исследование по раннему лечению диабетической ретинопатии), в то время как в исследовании FIELD – на два шага и более. Пациенты, принимавшие участие в ACCORD-EYE, имели худшие показатели в отношении длительности СД (10 лет против 5 лет) и уровня HbA1c (8,2 против 6,9%). Жесткий контроль гликемии (HbA1c менее 6,0% против HbA1c от 7,0 до 7,9%) привел к снижению прогрессирования ретинопатии только на 33%. При этом увеличилось количество случаев смертельных гипогликемий и сердечно-сосудистых смертей.
Таким образом, фенофибрат в настоящее время является единственным терапевтическим средством, доказавшим высокую эффективность в предотвращении прогрессирования диабетической ретинопатии и макулярного отека.
Фенофибрат показан больным СД 2 типа (как с дислипидемией, так и без нарушений липидного обмена) с любой стадией ретинопатии (от фоновой до тяжелой непролиферативной).
Заключение
Медико-социальная значимость СД обусловлена хроническими осложнениями, преимущественно сосудистыми. Одним из самых серьезных проявлений диабетической микроангиопатии является поражение сетчатки – ретинопатия.
В арсенале современной офтальмологии имеются достаточно эффективные методы лечения диабетической ретинопатии и макулярного отека – лазерная коагуляция сетчатки и витрэктомия, позволяющие (при своевременном выполнении) на длительный срок сохранить зрение пациенту. Однако эти методы воздействия обладают серьезными побочными эффектами.
Интравитреальное введение ингибиторов ангиогенеза и кортикостероидов в подавляющем большинстве случаев должно сочетаться с лазерной коагуляцией сетчатки или витрэктомией.
Основными способами профилактики развития и прогрессирования диабетической ретинопатии являются компенсация СД и сопутствующих состояний (то есть нормализация уровня гликемии и артериального давления, коррекция дислипидемии) и прием фенофибрата.