Микроваскулярное поражение нервной системы при сахарном диабете
- Аннотация
- Статья
- Ссылки
- English
В статье рассмотрены механизмы микроваскулярных поражений и их роль в патогенезе неврологических осложнений сахарного диабета, а также возможные методы коррекции данных осложнений.
В статье рассмотрены механизмы микроваскулярных поражений и их роль в патогенезе неврологических осложнений сахарного диабета, а также возможные методы коррекции данных осложнений.

Нервная система – одна из основных мишеней сахарного диабета (СД) [1, 2]. Поражение периферической нервной системы (диабетические невропатии) при СД многократно и подробно описано, однако к изучению патологии центральной нервной системы, в первую очередь головного мозга, исследователи приступили сравнительно недавно. Наиболее частым проявлением дисфункции головного мозга при СД являются когнитивные нарушения.
Гипергликемия, являющаяся прямым следствием СД, несомненно, играет определенную роль в развитии церебральной и периферической дисфункции. Однако более важное значение в развитии как полиневропатии, так и когнитивном снижении может иметь цереброваскулярная патология, ускоряемая метаболическими расстройствами, характерными для СД [1–3].
Один из основных механизмов развития диабетической невропатии может быть связан с микроангиопатией, поражающей vasa nervorum и вызывающей ишемическое повреждение и дегенерацию нервных волокон. Электронная микроскопия позволила выявить в эндотелиоцитах дегенерацию перицитов – клеток, формирующих наружный слой по отношению к эндотелию микрососудов и, возможно, участвующих в поддержании гематоневрального барьера, а также удвоение базальной мембраны. Утолщение базальной мембраны приводит к сужению просвета капилляров. Как следствие, повышается внутрисосудистое сопротивление и нарушается микроциркуляция в пораженных областях. При биопсии нервов у больных СД определяются утолщение стенок и изменение калибра капилляров, микротромбозы, частичная или полная закупорка капилляров [4, 5].
Немаловажное значение в развитии диабетической невропатии имеют также повышенная продукция свободных радикалов и истощение антиоксидантных механизмов, обусловливающих развитие окислительного стресса. В настоящее время именно окислительный стресс считается конечным путем, опосредующим различные варианты осложнений при СД. К факторам, повышающим чувствительность нервной ткани к окислительному стрессу, относятся расстройство аксоплазматического транспорта, нарушение продукции энергии в нейронах, сопровождаемое вторичным повреждением мембранного и ионного транспорта.
Ишемическое поражение может запускать нейровоспаление, которое участвует в развитии разных форм диабетической невропатии, особенно радикулоплексопатии и множественной мононевропатии. Так, у пациентов с диабетической радикулоплексопатией отмечались повышенная продукция провоспалительных цитокинов, а при исследовании биоптата нервов – признаки васкулита. У больных СД 2 типа и полиневропатией в сыворотке крови определяли аутоиммунный иммуноглобулин, индуцирующий кальцийзависимый апоптоз нервных клеток, причем его содержание коррелировало с тяжестью полиневропатии, особенно ее сенсорных и вегетативных проявлений.
Варианты диабетической невропатии различаются по клинике, патогенезу, гистологическим изменениям, течению, реакции на лечение, прогнозу (табл. 1). Нередко они сочетаются друг с другом.
В клинической практике используется упрощенная классификация диабетической невропатии, предусматривающая три основные стадии:
- первая стадия соответствует асимптомной (субклинической) полиневропатии. Ее выявляют с помощью электронейромиографии, количественного исследования чувствительности и/или вегетативных тестов;
- вторая стадия – легко или умеренно выраженной полиневропатии, которую можно выявить при обычном неврологическом осмотре по характерным субъективным проявлениям, выпадению рефлексов, снижению чувствительности и т.д.;
- третья стадия – тяжелой полиневропатии, сопровождающейся развитием выраженного сенсорного или сенсомоторного дефекта (существенно ограничивающего функциональные возможности, инвалидизирующего), вегетативной недостаточности, тяжелого болевого синдрома и таких осложнений, как трофические язвы, нейроартропатия, синдром диабетической стопы.
Причины дисфункции головного мозга при сахарном диабете
Дисфункция головного мозга при СД обусловлена прежде всего поражением мелких сосудов. Оно вызывает диффузную патологию белого вещества больших полушарий, образование множественных лакунарных очагов и микроинфаркт [6, 7].
О роли микроваскулярной патологии в генезе деменции свидетельствует корреляция между когнитивным снижением и некоторыми изменениями головного мозга, выявляемыми при проведении магнитно-резонансной томографии (лейкоареоз, лакунарные инфаркты, микрогеморрагии, церебральная атрофия и т.д.), а также патологиями сосудов сетчатки и почек [8]. Причина поражения мелких церебральных сосудов при СД не до конца ясна. Предполагают, что формирование конечных продуктов гликирования, как и активация альтернативных путей углеводного метаболизма, способствует развитию окислительного стресса, что приводит к повреждению эндотелия сосудов и развитию ишемии мозга. Однако, согласно результатам исследования J.A. Sonnen и соавт. (2009), молекулярный маркер окислительного стресса F2-изопростан у пациентов с СД был ниже, чем у лиц с деменцией, но без СД [9].
При СД для микроангиопатии характерно изменение стенок сосудов и реологических свойств крови. Главным фактором, вызывающим повреждение сосудистых стенок, по-видимому, является гипергликемия, на фоне которой происходит увеличение поглощения глюкозы эндотелиоцитами и повреждение их из-за накопления гликогенсодержащих структур. Эндотелий наряду с нервной тканью, хрусталиком, сетчаткой, почечными гломерулярными клетками и, возможно, кардиомиоцитами относится к тканям, которые поглощают глюкозу независимо от наличия инсулина. Такие ткани особенно чувствительны к гипергликемии. Внутриклеточный уровень глюкозы в них напрямую зависит от уровня глюкозы в крови [10].
Накопление полиолов, конечных продуктов гликирования, продуктов воспаления, интенсификация перекисного окисления липидов выступают в качестве основных факторов дисфункции эндотелия. О дисфункции эндотелиальных клеток свидетельствует повышение уровня эндотелина в плазме. Эндотелин, будучи вазоконстриктором, усугубляет поражение сосудистой стенки.
Дисфункция эндотелия приводит к снижению продукции оксида азота (NO), простациклина PGI2 и увеличению содержания в плазме фактора фон Виллебранда. В результате могут повышаться свертывающая активность крови и уровень активации тромбоцитов. Дисфункция тромбоцитов при СД выражается в повышении метаболизма арахидоновой кислоты и продукции тромбоксана, а также в снижении продукции NO. Таким образом, снижение продукции NO может быть результатом дисфункции как эндотелиальных клеток, так и тромбоцитов.
Установлено также, что изменение продукции ангионевринов (например, фактора роста сосудистого эндотелия) может опосредовать повреждение мелких сосудов и гибель нейронов.
Микроваскулярное поражение способно инициировать нейродегенеративный процесс. Общим звеном может выступать воспалительный процесс, связанный с повышенной выработкой провоспалительных цитокинов и активацией микроглии в головном мозге [11]. В упомянутом ранее исследовании J.A. Sonnen и соавт. (2009) у пациентов с деменцией, страдавших СД, зафиксирован повышенный уровень интерлейкина 6 [9]. Вклад в когнитивное снижение может также вносить активация гипоталамо-гипофизарно-надпочечниковой оси, что приводит к увеличению уровня кортизола в крови.
Повышению свертываемости крови способствуют усиление синтеза фибриногена и тромбина, снижение уровня белков С и S в плазме. Несмотря на повышение содержания в плазме антитромбина III, его функциональная активность снижается, возможно, вследствие гликирования. Фибринолитическая активность плазмы уменьшается, вероятно, в результате недостаточной выработки тканевых активаторов плазминогена или повышенной продукции ингибитора тканевых активаторов плазминогена.
Повышенное содержание в плазме фибриногена и альфа-2-глобулина при сниженном уровне альбумина приводит к повышению вязкости крови, которое становится еще более выраженным вследствие гемоконцентрации, сопровождающей стойкую гипергликемию. Тяжесть этих нарушений коррелирует со степенью гипергликемии, поэтому антитромбоцитарные агенты оказываются недостаточно эффективными без одновременного проведения коррекции метаболических нарушений.
По-видимому, нарушение метаболизма в эндотелии сосудов и нервной ткани происходит параллельно, более того – во взаимосвязи друг с другом. Одно из наиболее важных связующих звеньев – снижение продукции NO. Последний обладает сосудорасширяющим действием и опосредованно воздействует на разные метаболические процессы, в частности может влиять на активность альдозоредуктазы.
Классификация дисфункции головного мозга
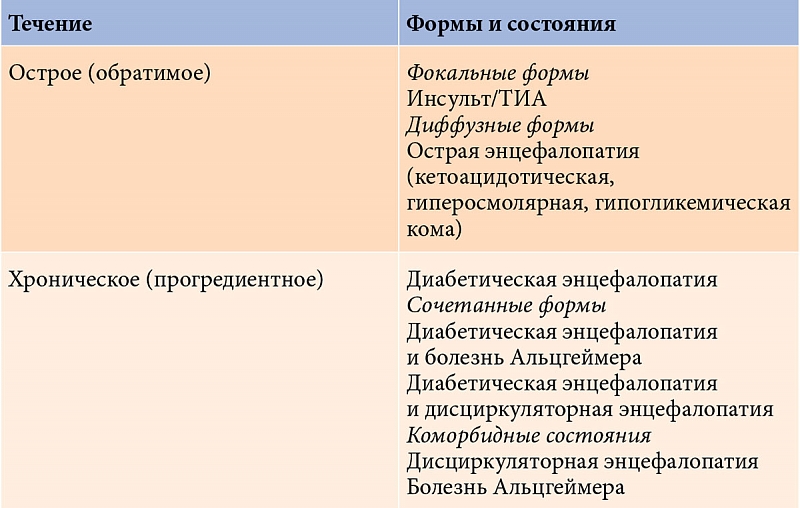
Поражение головного мозга при СД может быть острым или хроническим (табл. 2). Острые поражения могут быть вызваны непосредственно метаболическими нарушениями (острые метаболические энцефалопатии, или комы гипергликемические, кетоацидотические, гипогликемические и т.д.). Как правило, такие состояния являются обратимыми, однако могут оставлять резидуальный дефект. К острым поражениям головного мозга условно можно отнести инсульт и транзиторную ишемическую атаку (ТИА).
К хроническим, как правило прогредиентным формам поражения головного мозга относятся диабетическая энцефалопатия и условно такие коморбидные состояния, как дисциркуляторная энцефалопатия и болезнь Альцгеймера, которые патогенетически связаны с основным заболеванием. Кроме того, существуют переходные (сочетанные) формы. Они занимают положение между диабетической энцефалопатией и болезнью Альцгеймера или дисциркуляторной энцефалопатией [12].
Общие принципы лечения
Поскольку гипергликемия является одним из основных патогенетических факторов, оптимальный контроль уровня глюкозы в крови считается неотъемлемым условием стабилизации и регресса проявлений как диабетической невропатии, так и дисфункции головного мозга. Немаловажно снижать уровень липидов в крови. Для коррекции метаболических нарушений также необходимо поддерживать адекватную физическую активность и нормализовать массу тела. Кроме того, следует контролировать артериальную гипертензию.
Хороший контроль СД позволяет существенно снизить риск развития осложнений со стороны нервной системы. Согласно результатам исследования DCCT (1993), включавшего более тысячи больных СД 1 типа, через 6,5 года интенсивная инсулинотерапия в отличие от традиционной терапии инсулином сокращала частоту развития невропатии и микроангиопатий на 60%. Это позволило предположить, что целью антидиабетической терапии должна быть нормогликемия (эугликемия) или близкий к нормальному уровень глюкозы в крови [13].
Аналогичные выводы были сделаны по результатам исследования UKPDS, включавшего более 5000 больных СД 2 типа. Было также показано, что при проведении интенсивной терапии (уровень глюкозы < 6 ммоль/л) к концу девятого года наблюдения частота полиневропатий уменьшилась на 16%, а к концу 15-го года – на 40% по сравнению с группой, в которой концентрация глюкозы в крови поддерживалась на более высоком уровне (≤ 15 ммоль/л).
После улучшения контроля гликемии могут отмечаться замедление прогрессирования, стабилизация или частичный регресс симптомов уже развившейся полиневропатии. В среднем проявления полиневропатии начинают уменьшаться через шесть месяцев после достижения оптимального контроля глюкозы. При этом ослабляются не только субъективные сенсорные симптомы, но и проявления вегетативной дисфункции (снижается частота диареи, сглаживаются проявления ортостатической гипотензии), а также увеличивается скорость проведения импульсов по нервам.
Наилучшим показателем, на который следует ориентироваться при лечении СД, является уровень гликированного гемоглобина (HbA1c). Он должен быть менее 7%. На фоне интенсивной инсулинотерапии, приводившей к стойкому снижению HbA1c, частота выявления нарушения нервной проводимости снизилась на 44%, вегетативной дисфункции, выявляемой при проведении кардиоваскулярных тестов, – на 53%.
Стабилизация уровня глюкозы в крови также позволяет уменьшить выраженность болевого синдрома, тогда как резкие колебания гликемии способствуют усилению боли.
Неблагоприятное воздействие оказывает не только хроническая гипергликемия, но и резкое преходящее повышение концентрации глюкозы (например, после приема пищи). Вместе с тем следует избегать и гипогликемии, которая может усугублять повреждение периферических нервных волокон.
В тяжелых случаях для компенсации метаболических нарушений прибегают к трансплантации поджелудочной железы. Успешная трансплантация ослабляет проявления полиневропатии у 50% больных.
Связь между улучшением контроля гликемии и более благоприятным течением полиневропатии особенно четко прослеживается у пациентов с СД 1 типа, у пациентов с СД 2 типа ситуация менее определенная. В нескольких исследованиях не удалось замедлить прогрессирования полиневропатии на фоне интенсивной терапии. Возможно, достижению положительного результата противодействует инерция ранее запущенного патологического процесса, так называемая метаболическая память.
Нет убедительных данных и о том, что перевод больных СД 2 типа, страдающих диабетической полиневропатией, с пероральных противодиабетических препаратов на инсулин способствует стабилизации или улучшению состояния. Тем не менее при развитии диабетической радикулоплексопатии (диабетической амиотрофии) на фоне плохо контролируемого СД 2 типа часто рекомендуют временно перейти на использование инсулина.
В целом следует отметить, что оптимизация контроля гликемии является единственным известным методом предупреждения развития диабетической невропатии и главной составляющей ее лечения, но, к сожалению, она не решает всех проблем, связанных с этим осложнением СД. Поэтому сохраняется потребность в лекарственных средствах, воздействующих на различные звенья патогенеза диабетической полиневропатии.
Экспериментальные работы по поиску препаратов для лечения дисфункции головного мозга ведутся в направлении усиления активности инсулизина, увеличения чувствительности к инсулину, что может способствовать повышению клиренса бета-амилоида. Модуляторы инсулизина в виде небольших пептидов могут усилить гидролиз бета-амилоида, не повышая распад самого инсулина. Применение пиоглитазона – агониста гамма-рецепторов, активируемых пролифератором пероксисом, позволяет улучшить чувствительность к инсулину.
Системное применение препаратов для лечения заболеваний центральной нервной системы малоэффективно для большинства малых молекул и почти всех крупных молекул, на пути которых встает гематоэнцефалический барьер. Инсулин может проникать через гематоэнцефалический барьер с помощью специальной транспортной системы. Однако он должен вводиться в количествах, не вызывающих гипогликемию. Альтернативой системному введению может стать интраназальное введение пептидов, таких как инсулин, проникающих в мозг через систему обонятельного и тройничного нервов. Установлено, что интраназальное введение инсулина способно оказывать положительное влияние на когнитивное снижение, степень атрофии головного мозга и изменения белого вещества, при условии относительного дефицита инсулина, что может наблюдаться как при СД 1 типа, так и при СД 2 типа.
Пентоксифиллин
Пентоксифиллин является дериватом метилксантина. При приеме в терапевтических дозах (400 мг три раза в день) он улучшает реологические свойства крови: снижает вязкость плазмы и крови преимущественно за счет уменьшения уровня фибриногена, повышает растяжимость эритроцитов, уменьшает агрегацию тромбоцитов и эритроцитов за счет супрессии нейтрофильной активации. Вследствие активации нейтрофилов они становятся менее растяжимыми из-за увеличения внутриклеточного актинового каркаса. Таким образом, пентоксифиллин повышает способность эритроцитов проходить через капиллярное русло, что особенно важно при уменьшении градиента давления из-за вышестоящего стеноза. Этим может объясняться эффективность пентоксифиллина при перемежающейся хромоте [14–16].
Пентоксифиллин обладает также антиоксидантным и противовоспалительным эффектами. Его антиоксидантное действие обусловлено прежде всего снижением активации нейтрофилов. Пентоксифиллин уменьшает уровень воспалительных цитокинов в плазме крови, таких как фактор некроза опухоли альфа, интерлейкины 1 и 6 [17–19].
Молекулярные механизмы эффектов пентоксифиллина до сих пор остаются плохо изученными. Известно, что препарат является неспецифическим ингибитором циклического аденозинмонофосфата фосфодиэстеразы и регулирует его эффекты. Снижение плазматических концентраций воспалительных цитокинов, скорее всего, свидетельствует о воздействии пентоксифиллина и его метаболитов на активность макрофагов/моноцитов. При оценке лечения пентоксифиллином установлено, что он значительно снижал уровень фактора некроза опухоли альфа. Вероятно, пентоксифиллин сам по себе может быть медиатором этого эффекта [19, 20].
Одно из последних рандомизированных контролируемых исследований эффективности пентоксифиллина у пациентов с СД 2 типа продемонстрировало, что его применение в течение шести месяцев способствовало значительному снижению концентрации протеинурии в группе пентоксифиллина по сравнению с группой плацебо – 23 и 4% соответственно. Кроме того, отмечалось небольшое снижение уровня HbA1c и инсулинорезистентности. Хотя воспалительные реакции в жировой ткани могут снизить системную чувствительность к инсулину, положительный эффект пентоксифиллина в отношении контроля гликемии может быть интерпретирован как его противовоспалительное действие [21].
В шестимесячном контролируемом исследовании, включавшем подростков с СД 1 типа, оценивался эффект препарата на толщину интимы внутренней сонной артерии (индекс атеромы). Индекс атеромы снижался в группе пациентов, принимавших пентоксифиллин, и незначительно повышался в группе плацебо. Препарат не влиял на уровень сывороточных липидов, но уровень малональдегида (маркера окислительного стресса) был на 32 и 37% ниже у принимавших его [22].
По меньшей мере в четырех исследованиях оценивалось влияние пентоксифиллина на когнитивные функции при сосудистой деменции. У получавших препарат наблюдалась тенденция к снижению прогрессирования когнитивных нарушений, что, возможно, связано с увеличением перфузии головного мозга на фоне улучшения реологических свойств крови.
В настоящее время получены данные о долгосрочном эффекте препарата у пациентов с сердечно-сосудистыми заболеваниями. Так, в шестимесячном рандомизированном плацебоконтролируемом исследовании пациентам с острым коронарным синдромом наряду с другими препаратами назначали пентоксифиллин в стандартной дозе (400 мг/сут) или плацебо. Критериями оценки были смерть, инфаркт или срочная повторная госпитализация. У пациентов, получавших пентоксифиллин, значительно снизился уровень С-реактивного белка и фактора некроза опухоли альфа [23].
Хотя нейтрофилы не являются значимым компонентом стабильной атеросклеротической бляшки и не играют решающей роли в формировании атеросклероза, они обнаруживаются в нестабильных бляшках у пациентов с острым коронарным синдромом. Более того, повышение уровня миелопероксидазы (характерно для активации нейтрофилов) является маркером повышенного риска инфаркта миокарда у пациентов со стенокардией. В ряде исследований было выявлено снижение риска смерти при застойной сердечной недостаточности у пациентов, принимавших пентоксифиллин в течение шести месяцев, по сравнению с получавшими плацебо.
В шестимесячном рандомизированном исследовании, включавшем 73 пациента, перенесших ТИА и принимавших аспирин с дипиридамолом, и 65 больных, использовавших пентоксифиллин, у первых зарегистрировано 80 случаев ТИА, у вторых – 19.
В четырех контролируемых исследованиях, включавших 763 пациента, оценивалось влияние в/в введения пентоксифиллина на прогноз при остром ишемическом инсульте. Ежедневная доза препарата составляла 600–1200 мг. Пентоксифиллин вводили в первые три – пять дней после начала инсульта. В трех исследованиях после трехдневного в/в введения препарата больных переводили на пероральный его прием. У трети пациентов наблюдалась тенденция к снижению риска ранней смерти.
Доказана защитная роль препарата при проведении тромболитической терапии.
Согласно результатам исследований, в случае повреждения ткани головного мозга после реперфузии ключевую роль может играть приток активированных нейтрофилов. На фоне введения пентоксифиллина наблюдалось снижение последствий реперфузии.
O.S. Levin, A.Sh. Tchimagomedova, I.I. Koloman
Russian Medical Academy of Continuing Professional Education
Contact person: Oleg Semyonovich Levin, oslevin@mail.ru
The nervous system is one of the main targets of diabetes. Hyperglycemia, which is a direct consequence of this pathology, is only one of the causes of cerebral and peripheral dysfunction. An important role in this belongs to microvascular lesions.
The article discusses the mechanisms of microvascular changes and their role in the pathogenesis of neurological complications of diabetes, as well as possible methods of these complications correction.