–Р–Љ–Є–ї–Њ–Є–і–Њ–Ј: —Б–Њ–≤—А–µ–Љ–µ–љ–љ—Л–µ –Љ–µ—В–Њ–і—Л –і–Є–∞–≥–љ–Њ—Б—В–Є–Ї–Є –Є –ї–µ—З–µ–љ–Є—П
- –Р–љ–љ–Њ—В–∞—Ж–Є—П
- –°—В–∞—В—М—П
- –°—Б—Л–ї–Ї–Є
–Ґ–µ—А–Љ–Є–љ ¬Ђ–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј¬ї –Њ–±—К–µ–і–Є–љ—П–µ—В –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П, –Ї–Њ—В–Њ—А—Л–µ —Е–∞—А–∞–Ї—В–µ—А–Є–Ј—Г—О—В—Б—П –≤–љ–µ–Ї–ї–µ—В–Њ—З–љ—Л–Љ –Њ—В–ї–Њ–ґ–µ–љ–Є–µ–Љ —Б–њ–µ—Ж–Є—Д–Є—З–µ—Б–Ї–Њ–≥–Њ –љ–µ—А–∞—Б—В–≤–Њ—А–Є–Љ–Њ–≥–Њ —Д–Є–±—А–Є–ї–ї—П—А–љ–Њ–≥–Њ –±–µ–ї–Ї–∞ –∞–Љ–Є–ї–Њ–Є–і–∞. –Э–µ–Љ–µ—Ж–Ї–Є–є –њ–∞—В–Њ–ї–Њ–≥ –†—Г–і–Њ–ї—М—Д –Т–Є—А—Е–Њ–≤ –≤¬†1853¬†–≥. –њ—А–µ–і–ї–Њ–ґ–Є–ї —В–µ—А–Љ–Є–љ ¬Ђ–∞–Љ–Є–ї–Њ–Є–і¬ї –і–ї—П –Њ–±–Њ–Ј–љ–∞—З–µ–љ–Є—П –≤–µ—Й–µ—Б—В–≤–∞, –Њ—В–Ї–ї–∞–і—Л–≤–∞—О—Й–µ–≥–Њ—Б—П –≤¬†–Њ—А–≥–∞–љ–∞—Е –±–Њ–ї—М–љ—Л—Е ¬Ђ—Б–∞–ї—М–љ–Њ–є –±–Њ–ї–µ–Ј–љ—М—О¬ї (–Ї–∞–Ї –љ–∞–Ј—Л–≤–∞–ї–Є –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј –≤¬†–љ–∞—З–∞–ї–µ XIX¬†–≤–µ–Ї–∞) –њ—А–Є —В—Г–±–µ—А–Ї—Г–ї–µ–Ј–µ, —Б–Є—Д–Є–ї–Є—Б–µ, –ї–µ–њ—А–µ. –≠—В–Њ –≤–µ—Й–µ—Б—В–≤–Њ –Њ–љ –Њ—И–Є–±–Њ—З–љ–Њ –њ–Њ—Б—З–Є—В–∞–ї –Ї—А–∞—Е–Љ–∞–ї–Њ–њ–Њ–і–Њ–±–љ—Л–Љ –Є–Ј-–Ј–∞ —Е–∞—А–∞–Ї—В–µ—А–љ–Њ–є —А–µ–∞–Ї—Ж–Є–Є —Б¬†–Є–Њ–і–Њ–Љ. –Т¬†–љ–∞—Б—В–Њ—П—Й–µ–µ –≤—А–µ–Љ—П —Г—Б—В–∞–љ–Њ–≤–ї–µ–љ–Њ, —З—В–Њ –њ–Њ–ї–Є—Б–∞—Е–∞—А–Є–і—Л —Б–Њ—Б—В–∞–≤–ї—П—О—В –љ–µ –±–Њ–ї–µ–µ 4% –Њ—В –Љ–∞—Б—Б—Л –∞–Љ–Є–ї–Њ–Є–і–∞, –Њ–і–љ–∞–Ї–Њ —В–µ—А–Љ–Є–љ—Л ¬Ђ–∞–Љ–Є–ї–Њ–Є–і¬ї –Є¬†¬Ђ–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј¬ї –Ј–∞–Ї—А–µ–њ–Є–ї–Є—Б—М.
–≠–њ–Є–і–µ–Љ–Є–Њ–ї–Њ–≥–Є—П –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞
–†–∞—Б–њ—А–Њ—Б—В—А–∞–љ–µ–љ–љ–Њ—Б—В—М –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ –і–Њ –љ–∞—Б—В–Њ—П—Й–µ–≥–Њ –≤—А–µ–Љ–µ–љ–Є –Є–Ј—Г—З–µ–љ–∞ –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ. –Я–Њ –і–∞–љ–љ—Л–Љ S.Y. Tan –Є¬†—Б–Њ–∞–≤—В. (1995) [1], —А–µ–∞–Ї—В–Є–≤–љ—Л–є –Р–Р-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј, –Њ–і–Є–љ –Є–Ј –љ–∞–Є–±–Њ–ї–µ–µ —З–∞—Б—В—Л—Е –≤–∞—А–Є–∞–љ—В–Њ–≤ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞, —А–∞–Ј–≤–Є–≤–∞–µ—В—Б—П —Г¬†5% –±–Њ–ї—М–љ—Л—Е —Б¬†—Е—А–Њ–љ–Є—З–µ—Б–Ї–Є–Љ–Є –≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ—Л–Љ–Є –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П–Љ–Є –≤¬†–Х–≤—А–Њ–њ–µ; –њ–Њ –і–∞–љ–љ—Л–Љ –і—А—Г–≥–Є—Е –Є—Б—В–Њ—З–љ–Є–Ї–Њ–≤, –Р–Р-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј –Њ—Б–ї–Њ–ґ–љ—П–µ—В —В–µ—З–µ–љ–Є–µ —А–µ–≤–Љ–∞—В–Њ–Є–і–љ–Њ–≥–Њ –∞—А—В—А–Є—В–∞ –≤¬†6вАУ10% —Б–ї—Г—З–∞–µ–≤. –Т¬†—Б—А–µ–і–љ–µ–Љ –і–Њ–ї—П –Р–Р-–∞–Љ–Є–ї–Њ–Є–і–љ–Њ–є –љ–µ—Д—А–Њ–њ–∞—В–Є–Є –≤¬†—Б—В—А—Г–Ї—В—Г—А–µ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є –њ–Њ—З–µ–Ї –≤¬†–Х–≤—А–Њ–њ–µ —Б–Њ—Б—В–∞–≤–ї—П–µ—В 2,5вАУ2,8%, –∞¬†–≤ —Б—В—А—Г–Ї—В—Г—А–µ –±–Њ–ї–µ–Ј–љ–µ–є, –њ—А–Є–≤–µ–і—И–Є—Е –Ї¬†—Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–є –њ–Њ—З–µ—З–љ–Њ–є –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В–Є (–•–Я–Э),¬†вАУ 1% (–≤ —Б–Њ–Њ—В–≤–µ—В—Б—В–≤–Є–Є —Б¬†–і–∞–љ–љ—Л–Љ–Є –Х–≤—А–Њ–њ–µ–є—Б–Ї–Њ–є –∞—Б—Б–Њ—Ж–Є–∞—Ж–Є–Є –і–Є–∞–ї–Є–Ј–∞ –Є¬†—В—А–∞–љ—Б–њ–ї–∞–љ—В–∞—Ж–Є–Є).
–Т –°–®–Р —З–∞—Б—В–Њ—В–∞ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ –≤–∞—А—М–Є—А—Г–µ—В –Њ—В 5,1¬†–і–Њ 12,8¬†—Б–ї—Г—З–∞–µ–≤ –љ–∞ 100¬†—В—Л—Б. –љ–∞—Б–µ–ї–µ–љ–Є—П –≤¬†–≥–Њ–і [2]. –≠—В–Є –і–∞–љ–љ—Л–µ –Ї–∞—Б–∞—О—В—Б—П –њ—А–µ–Є–Љ—Г—Й–µ—Б—В–≤–µ–љ–љ–Њ —А–∞—Б–њ—А–Њ—Б—В—А–∞–љ–µ–љ–љ–Њ—Б—В–Є –њ–µ—А–≤–Є—З–љ–Њ–≥–Њ AL-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞, –Є–ї–Є –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ –≤¬†—А–∞–Љ–Ї–∞—Е –Љ–Є–µ–ї–Њ–Љ–љ–Њ–є –±–Њ–ї–µ–Ј–љ–Є –Є¬†–і—А—Г–≥–Є—Е –Т-–≥–µ–Љ–Њ–±–ї–∞—Б—В–Њ–Ј–Њ–≤. –Т¬†—Б—В—А–∞–љ–∞—Е —В—А–µ—В—М–µ–≥–Њ –Љ–Є—А–∞, –њ–Њ –Љ–љ–µ–љ–Є—О S.Y. Tan (1995), —Б–Љ–µ—А—В–љ–Њ—Б—В—М –Њ—В AL-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ —Б–Њ—Б—В–∞–≤–ї—П–µ—В 1 –љ–∞ 2000¬†–љ–∞—Б–µ–ї–µ–љ–Є—П (0,05%). –Я–Њ-–≤–Є–і–Є–Љ–Њ–Љ—Г, –њ—А–µ–і—Б—В–∞–≤–ї–µ–љ–љ—Л–µ –і–∞–љ–љ—Л–µ –Њ¬†—А–∞—Б–њ—А–Њ—Б—В—А–∞–љ–µ–љ–љ–Њ—Б—В–Є —А–∞–Ј–ї–Є—З–љ—Л—Е –≤–∞—А–Є–∞–љ—В–Њ–≤ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ –≤¬†—Ж–µ–ї–Њ–Љ –Љ–Њ–≥—Г—В –±—Л—В—М —Н–Ї—Б—В—А–∞–њ–Њ–ї–Є—А–Њ–≤–∞–љ—Л –Є¬†–љ–∞ –і—А—Г–≥–Є–µ —Б—В—А–∞–љ—Л.
–Ь–Њ—А—Д–Њ–ї–Њ–≥–Є—П –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞
–Ю—Б–љ–Њ–≤—Г —В–Ї–∞–љ–µ–≤—Л—Е –Њ—В–ї–Њ–ґ–µ–љ–Є–є –∞–Љ–Є–ї–Њ–Є–і–∞ —Б–Њ—Б—В–∞–≤–ї—П—О—В –∞–Љ–Є–ї–Њ–Є–і–љ—Л–µ —Д–Є–±—А–Є–ї–ї—Л¬†вАУ –Њ—Б–Њ–±—Л–µ –±–µ–ї–Ї–Њ–≤—Л–µ —Б—В—А—Г–Ї—В—Г—А—Л –і–Є–∞–Љ–µ—В—А–Њ–Љ 5вАУ10¬†–љ–Љ –Є¬†–і–ї–Є–љ–Њ–є –і–Њ 800¬†–љ–Љ, —Б–Њ—Б—В–Њ—П—Й–Є–µ –Є–Ј 2 –Є¬†–±–Њ–ї–µ–µ –њ–∞—А–∞–ї–ї–µ–ї—М–љ—Л—Е —А–∞–Ј–љ–Њ–љ–∞–њ—А–∞–≤–ї–µ–љ–љ—Л—Е (–∞–љ—В–Є–њ–∞—А–∞–ї–ї–µ–ї—М–љ—Л—Е) —Д–Є–ї–∞–Љ–µ–љ—В–Њ–≤, –Њ–±—А–∞–Ј—Г—О—Й–Є—Е –Ї—А–Њ—Б—Б-–±–µ—В–∞-—Б–Ї–ї–∞–і—З–∞—В—Г—О –Ї–Њ–љ—Д–Њ—А–Љ–∞—Ж–Є—О. –Ш–Љ–µ–љ–љ–Њ –Њ–љ–∞ –Њ–њ—А–µ–і–µ–ї—П–µ—В —Б–њ–µ—Ж–Є—Д–Є—З–µ—Б–Ї–Њ–µ –Њ–њ—В–Є—З–µ—Б–Ї–Њ–µ —Б–≤–Њ–є—Б—В–≤–Њ –∞–Љ–Є–ї–Њ–Є–і–∞¬†вАУ —Б–њ–Њ—Б–Њ–±–љ–Њ—Б—В—М –Ї¬†–і–≤–Њ–є–љ–Њ–Љ—Г –ї—Г—З–µ–њ—А–µ–ї–Њ–Љ–ї–µ–љ–Є—О. –Я—А–Є –Љ–Є–Ї—А–Њ—Б–Ї–Њ–њ–Є–Є –Њ–Ї—А–∞—И–µ–љ–љ—Л—Е –Ї–Њ–љ–≥–Њ –Ї—А–∞—Б–љ—Л–Љ –њ—А–µ–њ–∞—А–∞—В–Њ–≤ –≤¬†–њ–Њ–ї—П—А–Є–Ј–Њ–≤–∞–љ–љ–Њ–Љ —Б–≤–µ—В–µ –∞–Љ–Є–ї–Њ–Є–і –Є–Ј–Љ–µ–љ—П–µ—В –Ї—А–∞—Б–љ—Л–є —Ж–≤–µ—В –Њ–Ї—А–∞—Б–Ї–Є –љ–∞ —П–±–ї–Њ—З–љ–Њ-–Ј–µ–ї–µ–љ–Њ–µ —Б–≤–µ—З–µ–љ–Є–µ. –Ю–±–љ–∞—А—Г–ґ–µ–љ–Є–µ —Н—В–Њ–≥–Њ —Б–≤–Њ–є—Б—В–≤–∞ –њ–Њ–ї–Њ–ґ–µ–љ–Њ –≤¬†–Њ—Б–љ–Њ–≤—Г –і–Є–∞–≥–љ–Њ—Б—В–Є–Ї–Є –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞.
–°—В—А—Г–Ї—В—Г—А–љ—Л–µ –Є¬†—Е–Є–Љ–Є–Ї–Њ-—Д–Є–Ј–Є—З–µ—Б–Ї–Є–µ –Њ—Б–Њ–±–µ–љ–љ–Њ—Б—В–Є –∞–Љ–Є–ї–Њ–Є–і–∞ –Њ–њ—А–µ–і–µ–ї—П—О—В—Б—П –Њ—Б–љ–Њ–≤–љ—Л–Љ –±–µ–ї–Ї–Њ–Љ-–њ—А–µ–і—И–µ—Б—В–≤–µ–љ–љ–Є–Ї–Њ–Љ, —Б–Њ–і–µ—А–ґ–∞–љ–Є–µ –Ї–Њ—В–Њ—А–Њ–≥–Њ –≤¬†—Д–Є–±—А–Є–ї–ї–µ –і–Њ—Б—В–Є–≥–∞–µ—В 80% –Є¬†—П–≤–ї—П–µ—В—Б—П —Б–њ–µ—Ж–Є—Д–Є—З–љ—Л–Љ –њ—А–Є–Ј–љ–∞–Ї–Њ–Љ –і–ї—П –Ї–∞–ґ–і–Њ–≥–Њ —В–Є–њ–∞ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞.
–•–∞—А–∞–Ї—В–µ—А–Є—Б—В–Є–Ї–∞ –Њ—Б–љ–Њ–≤–љ—Л—Е —В–Є–њ–Њ–≤ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞
–Т –≥—А—Г–њ–њ—Г –Р–Р-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ –≤—Е–Њ–і—П—В —А–µ–∞–Ї—В–Є–≤–љ—Л–є (–≤—В–Њ—А–Є—З–љ—Л–є) –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј, –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј –њ—А–Є –њ–µ—А–Є–Њ–і–Є—З–µ—Б–Ї–Њ–є –±–Њ–ї–µ–Ј–љ–Є, –Ї—А–Є–Њ–њ–Є—А–Є–љ–Њ–њ–∞—В–Є—П—Е (–њ—А–µ–Є–Љ—Г—Й–µ—Б—В–≤–µ–љ–љ–Њ –њ—А–Є —Б–Є–љ–і—А–Њ–Љ–µ –Ь–∞–Ї–ї–∞¬†вАУ –£—Н–ї–ї—Б–∞¬†вАУ —Б–µ–Љ–µ–є–љ–Њ–є –њ–µ—А–Є–Њ–і–Є—З–µ—Б–Ї–Њ–є –ї–Є—Е–Њ—А–∞–і–Ї–µ –≤¬†—Б–Њ—З–µ—В–∞–љ–Є–Є —Б¬†–≥–ї—Г—Е–Њ—В–Њ–є –Є¬†–Ї—А–∞–њ–Є–≤–љ–Є—Ж–µ–є). –Э–∞–Є–±–Њ–ї–µ–µ —З–∞—Б—В—Л–Љ–Є –њ—А–Є—З–Є–љ–∞–Љ–Є –≤—В–Њ—А–Є—З–љ–Њ–≥–Њ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ –≤¬†–љ–∞—Б—В–Њ—П—Й–µ–µ –≤—А–µ–Љ—П —Б–ї—Г–ґ–∞—В —А–µ–≤–Љ–∞—В–Њ–Є–і–љ—Л–є –∞—А—В—А–Є—В (30вАУ50%), —Е—А–Њ–љ–Є—З–µ—Б–Ї–Є–µ –≥–љ–Њ–є–љ–Њ-–і–µ—Б—В—А—Г–Ї—В–Є–≤–љ—Л–µ –±–Њ–ї–µ–Ј–љ–Є (–Њ—Б—В–µ–Њ–Љ–Є–µ–ї–Є—В, –±—А–Њ–љ—Е–Њ—Н–Ї—В–∞—В–Є—З–µ—Б–Ї–∞—П –±–Њ–ї–µ–Ј–љ—М), –≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ—Л–µ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П –Ї–Є—И–µ—З–љ–Є–Ї–∞ (—П–Ј–≤–µ–љ–љ—Л–є –Ї–Њ–ї–Є—В, –±–Њ–ї–µ–Ј–љ—М –Ъ—А–Њ–љ–∞), —В—Г–±–µ—А–Ї—Г–ї–µ–Ј, –Њ–њ—Г—Е–Њ–ї–Є (—З–∞—Й–µ –ї–Є–Љ—Д–Њ–≥—А–∞–љ—Г–ї–µ–Љ–∞—В–Њ–Ј –Є¬†—А–∞–Ї –њ–Њ—З–Ї–Є) [3].
–Р–Р-–∞–Љ–Є–ї–Њ–Є–і –Њ–±—А–∞–Ј—Г–µ—В—Б—П –Є–Ј —Б—Л–≤–Њ—А–Њ—В–Њ—З–љ–Њ–≥–Њ –±–µ–ї–Ї–∞-–њ—А–µ–і—И–µ—Б—В–≤–µ–љ–љ–Є–Ї–∞ SAA¬†вАУ –Њ—Б—В—А–Њ—Д–∞–Ј–Њ–≤–Њ–≥–Њ –±–µ–ї–Ї–∞, –≤¬†–љ–Њ—А–Љ–µ —Б–Є–љ—В–µ–Ј–Є—А—Г–µ–Љ–Њ–≥–Њ –≥–µ–њ–∞—В–Њ—Ж–Є—В–∞–Љ–Є, –љ–µ–є—В—А–Њ—Д–Є–ї–∞–Љ–Є –Є¬†—Д–Є–±—А–Њ–±–ї–∞—Б—В–∞–Љ–Є –≤¬†—Б–ї–µ–і–Њ–≤—Л—Е –Ї–Њ–ї–Є—З–µ—Б—В–≤–∞—Е. –Х–≥–Њ –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є—П –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ –≤–Њ–Ј—А–∞—Б—В–∞–µ—В –њ–Њ–і –≤–Њ–Ј–і–µ–є—Б—В–≤–Є–µ–Љ –Є–љ—В–µ—А–ї–µ–є–Ї–Є–љ–Њ–≤ 1 –Є¬†6, —Д–∞–Ї—В–Њ—А–∞ –љ–µ–Ї—А–Њ–Ј–∞ –Њ–њ—Г—Е–Њ–ї–Є –≤¬†–Њ—В–≤–µ—В –љ–∞ –≤–Њ—Б–њ–∞–ї–µ–љ–Є–µ, –Њ–њ—Г—Е–Њ–ї–µ–≤—Л–є —А–Њ—Б—В. –Я–Њ–≤—Л—И–µ–љ–Є–µ —Б–Њ–і–µ—А–ґ–∞–љ–Є—П SAA –≤¬†–Ї—А–Њ–≤–Є –Є–≥—А–∞–µ—В –Њ—Б–љ–Њ–≤–љ—Г—О —А–Њ–ї—М –≤¬†–њ–∞—В–Њ–≥–µ–љ–µ–Ј–µ –Р–Р-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞.
–Ю–і–љ–∞–Ї–Њ –і–ї—П —А–∞–Ј–≤–Є—В–Є—П –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ —В–Њ–ї—М–Ї–Њ –≤—Л—Б–Њ–Ї–Њ–є –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є–Є SAA, –љ–µ–Њ–±—Е–Њ–і–Є–Љ–Њ —В–∞–Ї–ґ–µ –љ–∞–ї–Є—З–Є–µ —Г¬†–±–µ–ї–Ї–∞-–њ—А–µ–і—И–µ—Б—В–≤–µ–љ–љ–Є–Ї–∞ –∞–Љ–Є–ї–Њ–Є–і–Њ–≥–µ–љ–љ–Њ—Б—В–Є. –†–∞–Ј–≤–Є—В–Є–µ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ —Г¬†—З–µ–ї–Њ–≤–µ–Ї–∞ —Б–≤—П–Ј—Л–≤–∞—О—В —Б¬†–і–µ–њ–Њ–Ј–Є—Ж–Є–µ–є SAA1; –≤¬†–љ–∞—Б—В–Њ—П—Й–µ–µ –≤—А–µ–Љ—П –Є–Ј–≤–µ—Б—В–љ–Њ 5¬†–Є–Ј–Њ—В–Є–њ–Њ–≤ SAA1, –Є–Ј –Ї–Њ—В–Њ—А—Л—Е –љ–∞–Є–±–Њ–ї—М—И—Г—О –∞–Љ–Є–ї–Њ–Є–і–Њ–≥–µ–љ–љ–Њ—Б—В—М –њ—А–Є–њ–Є—Б—Л–≤–∞—О—В –Є–Ј–Њ—В–Є–њ–∞–Љ 1.1¬†–Є¬†1.5¬†[4]. –Ъ–Њ–љ–µ—З–љ—Л–є —Н—В–∞–њ –∞–Љ–Є–ї–Њ–Є–і–Њ–≥–µ–љ–µ–Ј–∞¬†вАУ –Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є–µ —Д–Є–±—А–Є–ї–ї –∞–Љ–Є–ї–Њ–Є–і–∞ –Є–Ј –±–µ–ї–Ї–∞-–њ—А–µ–і—И–µ—Б—В–≤–µ–љ–љ–Є–Ї–∞¬†вАУ –Њ—Б—Г—Й–µ—Б—В–≤–ї—П–µ—В—Б—П –њ—А–Є –љ–µ–њ–Њ–ї–љ–Њ–Љ —А–∞—Б—Й–µ–њ–ї–µ–љ–Є–Є –њ—А–Њ—В–µ–∞–Ј–∞–Љ–Є –Љ–Њ–љ–Њ—Ж–Є—В–Њ–≤-–Љ–∞–Ї—А–Њ—Д–∞–≥–Њ–≤. –°—В–∞–±–Є–ї–Є–Ј–∞—Ж–Є—П –∞–Љ–Є–ї–Њ–Є–і–љ–Њ–є —Д–Є–±—А–Є–ї–ї—Л –Є¬†—А–µ–Ј–Ї–Њ–µ —Б–љ–Є–ґ–µ–љ–Є–µ —А–∞—Б—В–≤–Њ—А–Є–Љ–Њ—Б—В–Є —Н—В–Њ–≥–Њ –Љ–∞–Ї—А–Њ–Љ–Њ–ї–µ–Ї—Г–ї—П—А–љ–Њ–≥–Њ –Ї–Њ–Љ–њ–ї–µ–Ї—Б–∞ –≤–Њ –Љ–љ–Њ–≥–Њ–Љ –Њ–±—Г—Б–ї–Њ–≤–ї–µ–љ—Л –≤–Ј–∞–Є–Љ–Њ–і–µ–є—Б—В–≤–Є–µ–Љ —Б¬†–њ–Њ–ї–Є—Б–∞—Е–∞—А–Є–і–∞–Љ–Є –Є–љ—В–µ—А—Б—В–Є—Ж–Є—П. –Я—А–Є –Р–Р-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–µ –∞–Љ–Є–ї–Њ–Є–і –Њ–±–љ–∞—А—Г–ґ–Є–≤–∞—О—В –≤¬†—А–∞–Ј–ї–Є—З–љ—Л—Е –Њ—А–≥–∞–љ–∞—Е, –Њ–і–љ–∞–Ї–Њ –Ї–ї–Є–љ–Є—З–µ—Б–Ї–∞—П –Ї–∞—А—В–Є–љ–∞ –Є¬†–њ—А–Њ–≥–љ–Њ–Ј –Њ–±—Г—Б–ї–Њ–≤–ї–µ–љ—Л –њ–Њ—А–∞–ґ–µ–љ–Є–µ–Љ –њ–Њ—З–µ–Ї.
AL-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј –≤–Ї–ї—О—З–∞–µ—В –≤¬†—Б–µ–±—П –њ–µ—А–≤–Є—З–љ—Л–є (–Є–і–Є–Њ–њ–∞—В–Є—З–µ—Б–Ї–Є–є) –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј –Є¬†–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј, –∞—Б—Б–Њ—Ж–Є–Є—А–Њ–≤–∞–љ–љ—Л–є —Б¬†–Љ–Є–µ–ї–Њ–Љ–љ–Њ–є –±–Њ–ї–µ–Ј–љ—М—О, —А–∞–Ј–≤–Є–≤–∞—О—Й–Є–є—Б—П —Г¬†7вАУ10% –±–Њ–ї—М–љ—Л—Е —Б¬†–Љ–љ–Њ–ґ–µ—Б—В–≤–µ–љ–љ–Њ–є –Љ–Є–µ–ї–Њ–Љ–Њ–є. –Я–Њ —Б–Њ–≤—А–µ–Љ–µ–љ–љ—Л–Љ –њ—А–µ–і—Б—В–∞–≤–ї–µ–љ–Є—П–Љ, –њ–µ—А–≤–Є—З–љ—Л–є AL-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј –Є¬†–Љ–Є–µ–ї–Њ–Љ–љ—Г—О –±–Њ–ї–µ–Ј–љ—М (–Ї–∞–Ї –∞—Б—Б–Њ—Ж–Є–Є—А–Њ–≤–∞–љ–љ—Г—О, —В–∞–Ї –Є¬†–љ–µ –∞—Б—Б–Њ—Ж–Є–Є—А–Њ–≤–∞–љ–љ—Г—О —Б¬†–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–Њ–Љ) —А–∞—Б—Б–Љ–∞—В—А–Є–≤–∞—О—В –≤¬†—А–∞–Љ–Ї–∞—Е –µ–і–Є–љ–Њ–є –Т-–ї–Є–Љ—Д–Њ—Ж–Є—В–∞—А–љ–Њ–є –і–Є—Б–Ї—А–∞–Ј–Є–Є, –Ї–Њ—В–Њ—А–∞—П —Е–∞—А–∞–Ї—В–µ—А–Є–Ј—Г–µ—В—Б—П –њ—А–Њ–ї–Є—Д–µ—А–∞—Ж–Є–µ–є –∞–љ–Њ–Љ–∞–ї—М–љ–Њ–≥–Њ –Ї–ї–Њ–љ–∞ –њ–ї–∞–Ј–Љ–∞—В–Є—З–µ—Б–Ї–Є—Е –Ї–ї–µ—В–Њ–Ї –Є–ї–Є –Т-–Ї–ї–µ—В–Њ–Ї –≤¬†–Ї–Њ—Б—В–љ–Њ–Љ –Љ–Њ–Ј–≥–µ —Б¬†–Є–Ј–±—Л—В–Њ—З–љ–Њ–є –њ—А–Њ–і—Г–Ї—Ж–Є–µ–є –Љ–Њ–љ–Њ–Ї–ї–Њ–љ–∞–ї—М–љ–Њ–≥–Њ –Є–Љ–Љ—Г–љ–Њ–≥–ї–Њ–±—Г–ї–Є–љ–∞, –Њ–±–ї–∞–і–∞—О—Й–µ–≥–Њ –∞–Љ–Є–ї–Њ–Є–і–Њ–≥–µ–љ–љ–Њ—Б—В—М—О. –С–µ–ї–Ї–Њ–Љ-–њ—А–µ–і—И–µ—Б—В–≤–µ–љ–љ–Є–Ї–Њ–Љ –њ—А–Є AL-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–µ —Б—З–Є—В–∞—О—В –Љ–Њ–љ–Њ–Ї–ї–Њ–љ–∞–ї—М–љ—Л–µ –ї–µ–≥–Ї–Є–µ —Ж–µ–њ–Є –Є–Љ–Љ—Г–љ–Њ–≥–ї–Њ–±—Г–ї–Є–љ–Њ–≤ (–Ы–¶–Ш), –Є–Ј –љ–∞–Ј–≤–∞–љ–Є—П –Ї–Њ—В–Њ—А—Л—Е –њ—А–Њ–Є—Б—Е–Њ–і–Є—В –∞–±–±—А–µ–≤–Є–∞—В—Г—А–∞ L [3]. AL-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј¬†вАУ —Н—В–Њ –≥–µ–љ–µ—А–∞–ї–Є–Ј–Њ–≤–∞–љ–љ—Л–є –њ—А–Њ—Ж–µ—Б—Б —Б¬†–њ—А–µ–Є–Љ—Г—Й–µ—Б—В–≤–µ–љ–љ—Л–Љ –њ–Њ—А–∞–ґ–µ–љ–Є–µ–Љ —Б–µ—А–і—Ж–∞, –њ–Њ—З–µ–Ї, –ґ–µ–ї—Г–і–Њ—З–љ–Њ-–Ї–Є—И–µ—З–љ–Њ–≥–Њ —В—А–∞–Ї—В–∞ (–Ц–Ъ–Ґ), –љ–µ—А–≤–љ–Њ–є —Б–Є—Б—В–µ–Љ—Л –Є¬†–Ї–Њ–ґ–Є.
–Ъ ATTR-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј—Г –Њ—В–љ–Њ—Б—П—В —Б–µ–Љ–µ–є–љ—Г—О –∞–Љ–Є–ї–Њ–Є–і–љ—Г—О –њ–Њ–ї–Є–љ–µ–≤—А–Њ–њ–∞—В–Є—О, –љ–∞—Б–ї–µ–і—Г–µ–Љ—Г—О –њ–Њ –∞—Г—В–Њ—Б–Њ–Љ–љ–Њ-–і–Њ–Љ–Є–љ–∞–љ—В–љ–Њ–Љ—Г —В–Є–њ—Г, –Є¬†—Б–Є—Б—В–µ–Љ–љ—Л–є —Б—В–∞—А—З–µ—Б–Ї–Є–є –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј. –С–µ–ї–Ї–Њ–Љ-–њ—А–µ–і—И–µ—Б—В–≤–µ–љ–љ–Є–Ї–Њ–Љ –њ—А–Є —Н—В–Њ–є —Д–Њ—А–Љ–µ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ —Б–ї—Г–ґ–Є—В —В—А–∞–љ—Б—В–Є—А–µ—В–Є–љ¬†вАУ –Ї–Њ–Љ–њ–Њ–љ–µ–љ—В —Д—А–∞–Ї—Ж–Є–Є –њ—А–µ–∞–ї—М–±—Г–Љ–Є–љ–∞, —Б–Є–љ—В–µ–Ј–Є—А—Г–µ–Љ—Л–є –њ–µ—З–µ–љ—М—О –Є¬†–≤—Л–њ–Њ–ї–љ—П—О—Й–Є–є —Д—Г–љ–Ї—Ж–Є–Є —В—А–∞–љ—Б–њ–Њ—А—В–љ–Њ–≥–Њ –±–µ–ї–Ї–∞ —В–Є—А–Њ–Ї—Б–Є–љ–∞ –Є¬†—А–µ—В–Є–љ–Њ–ї–∞. –£—Б—В–∞–љ–Њ–≤–ї–µ–љ–Њ, —З—В–Њ –РTTR-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј –±—Л–≤–∞–µ—В —А–µ–Ј—Г–ї—М—В–∞—В–Њ–Љ –Љ—Г—В–∞—Ж–Є–Є –≤¬†–≥–µ–љ–µ, –Ї–Њ–і–Є—А—Г—О—Й–µ–Љ —Б–Є–љ—В–µ–Ј —В—А–∞–љ—Б—В–Є—А–µ—В–Є–љ–∞, —З—В–Њ –њ—А–Є–≤–Њ–і–Є—В –Ї¬†–Ј–∞–Љ–µ–љ–µ –∞–Љ–Є–љ–Њ–Ї–Є—Б–ї–Њ—В –≤¬†–Љ–Њ–ї–µ–Ї—Г–ї–µ TTR. –Т¬†—А–µ–Ј—Г–ї—М—В–∞—В–µ –њ–Њ–≤—Л—И–∞–µ—В—Б—П –∞–Љ–Є–ї–Њ–Є–і–Њ–≥–µ–љ–љ–Њ—Б—В—М –±–µ–ї–Ї–∞-–њ—А–µ–і—И–µ—Б—В–≤–µ–љ–љ–Є–Ї–∞ –Є¬†–Њ–±–ї–µ–≥—З–∞–µ—В—Б—П –µ–≥–Њ –њ–Њ–ї–Є–Љ–µ—А–Є–Ј–∞—Ж–Є—П –≤¬†–∞–Љ–Є–ї–Њ–Є–і–љ—Л–µ —Д–Є–±—А–Є–ї–ї—Л. –Т¬†–љ–∞—Б—В–Њ—П—Й–µ–µ –≤—А–µ–Љ—П –Є–Ј–≤–µ—Б—В–љ–Њ –Љ–љ–Њ–ґ–µ—Б—В–≤–Њ –≤–∞—А–Є–∞–љ—В–љ—Л—Е —В—А–∞–љ—Б—В–Є—А–µ—В–Є–љ–Њ–≤, —З–µ–Љ –Є¬†–Њ–±—Г—Б–ї–Њ–≤–ї–µ–љ–Њ —А–∞–Ј–љ–Њ–Њ–±—А–∞–Ј–Є–µ –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є—Е —Д–Њ—А–Љ –љ–∞—Б–ї–µ–і—Б—В–≤–µ–љ–љ–Њ–є –љ–µ–≤—А–Њ–њ–∞—В–Є–Є [5]. –≠—В–∞ –≥—А—Г–њ–њ–∞ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є —Е–∞—А–∞–Ї—В–µ—А–Є–Ј—Г–µ—В—Б—П –њ—А–Њ–≥—А–µ—Б—Б–Є—А—Г—О—Й–µ–є –њ–µ—А–Є—Д–µ—А–Є—З–µ—Б–Ї–Њ–є –Є¬†–≤–µ–≥–µ—В–∞—В–Є–≤–љ–Њ–є –љ–µ–≤—А–Њ–њ–∞—В–Є–µ–є, –Ї–Њ—В–Њ—А–∞—П —Б–Њ—З–µ—В–∞–µ—В—Б—П —Б¬†–њ–Њ—А–∞–ґ–µ–љ–Є–µ–Љ —Б–µ—А–і—Ж–∞, –њ–Њ—З–µ–Ї –Є¬†–і—А—Г–≥–Є—Е –Њ—А–≥–∞–љ–Њ–≤ —А–∞–Ј–ї–Є—З–љ–Њ–є —Б—В–µ–њ–µ–љ–Є.
–°–Є—Б—В–µ–Љ–љ—Л–є —Б—В–∞—А—З–µ—Б–Ї–Є–є –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј —А–∞–Ј–≤–Є–≤–∞–µ—В—Б—П –њ–Њ—Б–ї–µ 70¬†–ї–µ—В –≤¬†—А–µ–Ј—Г–ї—М—В–∞—В–µ –≤–Њ–Ј—А–∞—Б—В–љ—Л—Е –Є–Ј–Љ–µ–љ–µ–љ–Є–є —Б—В—А—Г–Ї—В—Г—А—Л –љ–Њ—А–Љ–∞–ї—М–љ–Њ–≥–Њ —В—А–∞–љ—Б—В–Є—А–µ—В–Є–љ–∞, —Г—Б–Є–ї–Є–≤–∞—О—Й–Є—Е –µ–≥–Њ –∞–Љ–Є–ї–Њ–Є–і–Њ–≥–µ–љ–љ–Њ—Б—В—М. –Ъ¬†–Њ—А–≥–∞–љ–∞–Љ-–Љ–Є—И–µ–љ—П–Љ —Б—В–∞—А—З–µ—Б–Ї–Њ–≥–Њ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ –Њ—В–љ–Њ—Б—П—В —Б–µ—А–і—Ж–µ, —Б–Њ—Б—Г–і—Л –≥–Њ–ї–Њ–≤–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞ –Є¬†–∞–Њ—А—В—Г.
–Ъ —Б–µ–Љ–µ–є–љ—Л–Љ —Д–Њ—А–Љ–∞–Љ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ –Њ—В–љ–Њ—Б—П—В—Б—П —В–∞–Ї–ґ–µ –±–Њ–ї–µ–µ —А–µ–і–Ї–Є–µ AGel-, AFib-, ALys-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј—Л, –њ—А–Є –Ї–Њ—В–Њ—А—Л—Е –∞–Љ–Є–ї–Њ–Є–і–Њ–≥–µ–љ–љ–Њ—Б—В—М—О –Њ–±–ї–∞–і–∞—О—В –Љ—Г—В–∞–љ—В–љ—Л–µ —Д–Њ—А–Љ—Л —Б–Њ–Њ—В–≤–µ—В—Б—В–≤–µ–љ–љ–Њ –≥–µ–ї—Б–Њ–ї–Є–љ–∞, —Д–Є–±—А–Є–љ–Њ–≥–µ–љ–∞, –ї–Є–Ј–Њ—Ж–Є–Љ–∞. –Я—А–Є —Н—В–Є—Е –≤–∞—А–Є–∞–љ—В–∞—Е –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ —В–∞–Ї–ґ–µ –Њ—В–Љ–µ—З–∞–µ—В—Б—П –њ—А–µ–Є–Љ—Г—Й–µ—Б—В–≤–µ–љ–љ–Њ–µ –њ–Њ—А–∞–ґ–µ–љ–Є–µ –њ–Њ—З–µ–Ї, –Њ–і–љ–∞–Ї–Њ –і–ї—П –≥–µ–ї—Б–Њ–ї–Є–љ–Њ–≤–Њ–≥–Њ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ —Е–∞—А–∞–Ї—В–µ—А–љ–Њ —Б–Њ—З–µ—В–∞–љ–Є–µ –љ–µ—Д—А–Њ–њ–∞—В–Є–Є —Б¬†—Б–µ—В—З–∞—В–Њ–є –і–Є—Б—В—А–Њ—Д–Є–µ–є —А–Њ–≥–Њ–≤–Є—Ж—Л –Є¬†–њ–µ—А–Є—Д–µ—А–Є—З–µ—Б–Ї–Њ–є –љ–µ–≤—А–Њ–њ–∞—В–Є–µ–є (–њ—А–µ–Є–Љ—Г—Й–µ—Б—В–≤–µ–љ–љ–Њ –њ–Њ—А–∞–ґ–∞—О—В—Б—П —З–µ—А–µ–њ–љ–Њ-–Љ–Њ–Ј–≥–Њ–≤—Л–µ –љ–µ—А–≤—Л).
–Т –љ–∞—Б—В–Њ—П—Й–µ–µ –≤—А–µ–Љ—П –Є–Ј–≤–µ—Б—В–љ–Њ –±–Њ–ї–µ–µ 20¬†–∞–Љ–Є–ї–Њ–Є–і–Њ–≥–µ–љ–љ—Л—Е –±–µ–ї–Ї–Њ–≤-–њ—А–µ–і—И–µ—Б—В–≤–µ–љ–љ–Є–Ї–Њ–≤ –Є, —Б–Њ–Њ—В–≤–µ—В—Б—В–≤–µ–љ–љ–Њ, –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є—Е –≤–∞—А–Є–∞–љ—В–Њ–≤ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞. –Ґ–∞–Ї, –Р-–±–µ—В–∞-–∞–Љ–Є–ї–Њ–Є–і —П–≤–ї—П–µ—В—Б—П –Љ–Њ—А—Д–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–є –Њ—Б–љ–Њ–≤–Њ–є –±–Њ–ї–µ–Ј–љ–Є –Р–ї—М—Ж–≥–µ–є–Љ–µ—А–∞, AIAPP-–∞–Љ–Є–ї–Њ–Є–і¬†вАУ —Б–∞—Е–∞—А–љ–Њ–≥–Њ –і–Є–∞–±–µ—В–∞ 2¬†—В–Є–њ–∞ –Є¬†–і—А. [3].
–Т –љ–µ—Д—А–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–є –њ—А–∞–Ї—В–Є–Ї–µ –±–Њ–ї—М—И–Њ–µ –Ј–љ–∞—З–µ–љ–Є–µ –Є–Љ–µ–µ—В –Р-–±–µ—В–∞-2-–Ь-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј (–∞—Б—Б–Њ—Ж–Є–Є—А–Њ–≤–∞–љ–љ—Л–є —Б¬†—Е—А–Њ–љ–Є—З–µ—Б–Ї–Є–Љ –≥–µ–Љ–Њ–і–Є–∞–ї–Є–Ј–Њ–Љ). –С–µ–ї–Ї–Њ–Љ-–њ—А–µ–і—И–µ—Б—В–≤–µ–љ–љ–Є–Ї–Њ–Љ –њ—А–Є —Н—В–Њ–є —Д–Њ—А–Љ–µ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ —Б–ї—Г–ґ–Є—В –±–µ—В–∞-2-–Љ–Є–Ї—А–Њ–≥–ї–Њ–±—Г–ї–Є–љ, –Ї–Њ—В–Њ—А—Л–є –≤¬†–љ–Њ—А–Љ–µ –њ—А–Є—Б—Г—В—Б—В–≤—Г–µ—В –≤¬†–Ї—А–Њ–≤–Є, –Љ–Њ—З–µ, —Б–њ–Є–љ–љ–Њ–Љ–Њ–Ј–≥–Њ–≤–Њ–є –Є¬†—Б–Є–љ–Њ–≤–Є–∞–ї—М–љ–Њ–є –ґ–Є–і–Ї–Њ—Б—В—П—Е. –Я—А–Є –љ–Њ—А–Љ–∞–ї—М–љ–Њ–є —Д—Г–љ–Ї—Ж–Є–Є –њ–Њ—З–µ–Ї –µ–≥–Њ –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є—П –≤¬†–Ї—А–Њ–≤–Є —Б–Њ—Б—В–∞–≤–ї—П–µ—В 1вАУ2¬†–Љ–≥/–ї. –≠—В–Њ—В –±–µ–ї–Њ–Ї —Д–Є–ї—М—В—А—Г–µ—В—Б—П –≤¬†–Ї–ї—Г–±–Њ—З–Ї–∞—Е –њ–Њ—З–µ–Ї –Є¬†–Љ–µ—В–∞–±–Њ–ї–Є–Ј–Є—А—Г–µ—В—Б—П –њ–Њ—Б–ї–µ —А–µ–∞–±—Б–Њ—А–±—Ж–Є–Є –≤¬†–њ—А–Њ–Ї—Б–Є–Љ–∞–ї—М–љ—Л—Е –Ї–∞–љ–∞–ї—М—Ж–∞—Е. –£¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–•–Я–Э –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є—П —Н—В–Њ–≥–Њ –±–µ–ї–Ї–∞ –≤¬†–Ї—А–Њ–≤–Є –≤–Њ–Ј—А–∞—Б—В–∞–µ—В, –Ї–Њ—А—А–µ–ї–Є—А—Г—П —Б¬†—Б–Њ–і–µ—А–ґ–∞–љ–Є–µ–Љ –Ї—А–µ–∞—В–Є–љ–Є–љ–∞, –Њ–і–љ–∞–Ї–Њ –Љ–∞–Ї—Б–Є–Љ–∞–ї—М–љ—Л—Е –Ј–љ–∞—З–µ–љ–Є–є (–≤¬†20вАУ70¬†—А–∞–Ј –њ—А–µ–≤—Л—И–∞—О—Й–Є—Е –љ–Њ—А–Љ—Г) –Њ–љ–∞ –і–Њ—Б—В–Є–≥–∞–µ—В —З–µ—А–µ–Ј –љ–µ—Б–Ї–Њ–ї—М–Ї–Њ –ї–µ—В –њ—А–Њ–≤–µ–і–µ–љ–Є—П —А–µ–≥—Г–ї—П—А–љ–Њ–≥–Њ –≥–µ–Љ–Њ–і–Є–∞–ї–Є–Ј–∞. –С–µ—В–∞-2-–Љ–Є–Ї—А–Њ–≥–ї–Њ–±—Г–ї–Є–љ –њ–ї–Њ—Е–Њ —Г–і–∞–ї—П–µ—В—Б—П –њ—А–Є –њ—А–Њ–≤–µ–і–µ–љ–Є–Є —Б—В–∞–љ–і–∞—А—В–љ–Њ–≥–Њ –≥–µ–Љ–Њ–і–Є–∞–ї–Є–Ј–∞, —З—В–Њ –љ–µ–Є–Ј–±–µ–ґ–љ–Њ –њ—А–Є–≤–Њ–і–Є—В –Ї¬†—А–∞–Ј–≤–Є—В–Є—О –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ [3]. –Я–µ—А–≤—Л–µ –і–µ–њ–Њ–Ј–Є—В—Л –±–µ—В–∞-2-–Ь-–∞–Љ–Є–ї–Њ–Є–і–∞ –Њ–±–љ–∞—А—Г–ґ–Є–≤–∞—О—В –≤¬†—Б–Є–љ–Њ–≤–Є–∞–ї—М–љ–Њ–є –Њ–±–Њ–ї–Њ—З–Ї–µ —Б—Г—Б—В–∞–≤–Њ–≤ —Г–ґ–µ —З–µ—А–µ–Ј 1вАУ2¬†–≥–Њ–і–∞ –Ј–∞–Љ–µ—Б—В–Є—В–µ–ї—М–љ–Њ–є –њ–Њ—З–µ—З–љ–Њ–є —В–µ—А–∞–њ–Є–Є, –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є–µ —Б–Є–Љ–њ—В–Њ–Љ—Л –њ–Њ—П–≤–ї—П—О—В—Б—П —З–µ—А–µ–Ј 7¬†–Є¬†–±–Њ–ї–µ–µ –ї–µ—В —В–µ—А–∞–њ–Є–Є [3]. –£¬†–±–Њ–ї—М–љ—Л—Е —Б—В–∞—А—И–µ 60¬†–ї–µ—В –і–Є–∞–ї–Є–Ј–љ—Л–є –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј —А–∞–Ј–≤–Є–≤–∞–µ—В—Б—П –±—Л—Б—В—А–µ–µ. –Ъ—А–Њ–Љ–µ –≤—Л—Б–Њ–Ї–Њ–є –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є–Є –±–µ–ї–Ї–∞-–њ—А–µ–і—И–µ—Б—В–≤–µ–љ–љ–Є–Ї–∞, –≤¬†–њ–∞—В–Њ–≥–µ–љ–µ–Ј–µ –і–Є–∞–ї–Є–Ј–љ–Њ–≥–Њ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ —Б—Г—Й–µ—Б—В–≤–µ–љ–љ—Г—О —А–Њ–ї—М –Є–≥—А–∞—О—В –Є¬†–і—А—Г–≥–Є–µ —Д–∞–Ї—В–Њ—А—Л. –Р–Љ–Є–ї–Њ–Є–і–Њ–≥–µ–љ–љ–Њ—Б—В—М –±–µ—В–∞-2-–Љ–Є–Ї—А–Њ–≥–ї–Њ–±—Г–ї–Є–љ–∞ –≤–Њ–Ј—А–∞—Б—В–∞–µ—В –њ—А–Є –Љ–Њ–і–Є—Д–Є–Ї–∞—Ж–Є—П—Е, –Њ–±—Г—Б–ї–Њ–≤–ї–µ–љ–љ—Л—Е –≤–Ј–∞–Є–Љ–Њ–і–µ–є—Б—В–≤–Є–µ–Љ —Б¬†–њ—А–Њ–і—Г–Ї—В–∞–Љ–Є –љ–µ–њ–Њ–ї–љ–Њ–≥–Њ –≥–ї–Є–Ї–Є—А–Њ–≤–∞–љ–Є—П, –∞¬†—В–∞–Ї–ґ–µ –њ—А–Є –Њ–Ї–Є—Б–ї–µ–љ–Є–Є –±–µ—В–∞-2-–Љ–Є–Ї—А–Њ–≥–ї–Њ–±—Г–ї–Є–љ–∞ –Є¬†–∞—Ж–Є–і–Є—Д–Є–Ї–∞—Ж–Є–Є —Б—А–µ–і—Л [3]. –Ю–і–љ–Є–Љ –Є–Ј –Њ—Б–љ–Њ–≤–љ—Л—Е –Є—Б—В–Њ—З–љ–Є–Ї–Њ–≤ —Н—В–Є—Е –Љ–Њ–і–Є—Д–Є–Ї–∞—Ж–Є–є —П–≤–ї—П—О—В—Б—П –Ї–Њ–Љ–њ–Њ–љ–µ–љ—В—Л –і–Є–∞–ї–Є–Ј–∞—В–∞, –≤¬†—В–Њ–Љ —З–Є—Б–ї–µ –±–∞–Ї—В–µ—А–Є–∞–ї—М–љ—Л–є –ї–Є–њ–Њ–њ–Њ–ї–Є—Б–∞—Е–∞—А–Є–і; –≥–ї–Є–Ї–Є—А–Њ–≤–∞–љ–Є–µ –±–µ—В–∞-2-–Љ–Є–Ї—А–Њ–≥–ї–Њ–±—Г–ї–Є–љ–∞ –≤–Њ–Ј–Љ–Њ–ґ–љ–Њ —В–∞–Ї–ґ–µ –њ—А–Є –Ї–Њ–љ—В–∞–Ї—В–µ —Б¬†—Ж–µ–ї–ї—О–ї–Њ–Ј–љ—Л–Љ–Є –і–Є–∞–ї–Є–Ј–љ—Л–Љ–Є –Љ–µ–Љ–±—А–∞–љ–∞–Љ–Є. –Ґ–∞–Ї–Є–Љ –Њ–±—А–∞–Ј–Њ–Љ, –њ—А–Њ—Д–Є–ї–∞–Ї—В–Є–Ї–∞ –і–Є–∞–ї–Є–Ј–љ–Њ–≥–Њ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ –≤–Њ –Љ–љ–Њ–≥–Њ–Љ —Б–≤—П–Ј–∞–љ–∞ —Б¬†–Њ—В–Ї–∞–Ј–Њ–Љ –Њ—В —Ж–µ–ї–ї—О–ї–Њ–Ј–љ—Л—Е –і–Є–∞–ї–Є–Ј–љ—Л—Е –Љ–µ–Љ–±—А–∞–љ –Є¬†—В—Й–∞—В–µ–ї—М–љ–Њ—Б—В—М—О –Ї–Њ–љ—В—А–Њ–ї—П –Ї–∞—З–µ—Б—В–≤–∞ –њ—А–Є–Љ–µ–љ—П–µ–Љ–Њ–≥–Њ –і–Є–∞–ї–Є–Ј–∞—В–∞. –Ь–Њ–і–Є—Д–Є—Ж–Є—А–Њ–≤–∞–љ–љ—Л–є –±–µ—В–∞-2-–Љ–Є–Ї—А–Њ–≥–ї–Њ–±—Г–ї–Є–љ —П–≤–ї—П–µ—В—Б—П –Љ–Њ—Й–љ—Л–Љ –Є–љ–і—Г–Ї—В–Њ—А–Њ–Љ –Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є—П —Ж–Є—В–Њ–Ї–Є–љ–Њ–≤ (–Є–љ—В–µ—А–ї–µ–є–Ї–Є–љ—Л 1¬†–Є¬†6, —Д–∞–Ї—В–Њ—А –љ–µ–Ї—А–Њ–Ј–∞ –Њ–њ—Г—Е–Њ–ї–Є –∞–ї—М—Д–∞). –†–µ–Ј—Г–ї—М—В–∞—В–Њ–Љ –∞–Ї—В–Є–≤–∞—Ж–Є–Є —Ж–Є—В–Њ–Ї–Є–љ–Њ–≤–Њ–≥–Њ –Ї–∞—Б–Ї–∞–і–∞ —П–≤–ї—П–µ—В—Б—П —В—П–ґ–µ–ї–Њ–µ —Б–Є–љ–Њ–≤–Є–∞–ї—М–љ–Њ–µ –≤–Њ—Б–њ–∞–ї–µ–љ–Є–µ —Б¬†–њ—А–Є–≤–ї–µ—З–µ–љ–Є–µ–Љ –Љ–∞–Ї—А–Њ—Д–∞–≥–Њ–≤ [3]. –Ь–∞–Ї—А–Њ—Д–∞–≥–∞–ї—М–љ–Њ–µ –≤–Њ—Б–њ–∞–ї–µ–љ–Є–µ –Њ–њ—А–µ–і–µ–ї—П–µ—В –≤—Л—Б–Њ–Ї–Є–є –і–µ—Б—В—А—Г–Ї—В–Є–≤–љ—Л–є –њ–Њ—В–µ–љ—Ж–Є–∞–ї –≤–Њ—Б–њ–∞–ї–µ–љ–Є—П –≤¬†—Б–Є–љ–Њ–≤–Є–Є —Б¬†—А–∞–Ј–≤–Є—В–Є–µ–Љ –Ї–Њ—Б—В–љ—Л—Е —Н—А–Њ–Ј–Є–є –Є¬†–Ї–Є—Б—В, —А–Њ—Б—В –Ї–Њ—В–Њ—А—Л—Е –Њ–±—Г—Б–ї–Њ–≤–ї–Є–≤–∞–µ—В —А–∞–Ј–≤–Є—В–Є–µ –њ–µ—А–µ–ї–Њ–Љ–Њ–≤ –Ї–Њ—Б—В–µ–є. –С—Л–ї–Њ —Г—Б—В–∞–љ–Њ–≤–ї–µ–љ–Њ, —З—В–Њ –±–µ—В–∞-2-–Љ–Є–Ї—А–Њ–≥–ї–Њ–±—Г–ї–Є–љ –Њ–±–ї–∞–і–∞–µ—В –≤—Л—Б–Њ–Ї–Њ–є –Ї–Њ–ї–ї–∞–≥–µ–љ—Б–≤—П–Ј—Л–≤–∞—О—Й–µ–є –∞–Ї—В–Є–≤–љ–Њ—Б—В—М—О, –≤–Њ–Ј—А–∞—Б—В–∞—О—Й–µ–є –њ–Њ –Љ–µ—А–µ —Г–≤–µ–ї–Є—З–µ–љ–Є—П –µ–≥–Њ –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є–Є –≤¬†–Ї—А–Њ–≤–Є. –Ъ—А–Њ–Љ–µ —В–Њ–≥–Њ, –≤—Л—П–≤–ї–µ–љ–∞ —Б–≤—П–Ј—М –±–µ—В–∞-2-–Љ–Є–Ї—А–Њ–≥–ї–Њ–±—Г–ї–Є–љ–∞ —Б¬†–≥–ї–Є–Ї–Њ–Ј–Њ–∞–Љ–Є–љ–Њ–≥–ї–Є–Ї–∞–љ–∞–Љ–Є —Е—А—П—Й–∞. –≠—В–Є–Љ–Є —Д–∞–Ї—В–∞–Љ–Є –Љ–Њ–ґ–љ–Њ –Њ–±—К—П—Б–љ–Є—В—М –њ—А–µ–Є–Љ—Г—Й–µ—Б—В–≤–µ–љ–љ–Њ–µ –Њ—В–ї–Њ–ґ–µ–љ–Є–µ —Д–Є–±—А–Є–ї–ї –∞–Љ–Є–ї–Њ–Є–і–∞ –≤¬†—Б—Г—Б—В–∞–≤–љ—Л—Е —В–Ї–∞–љ—П—Е. –Ґ–∞–Ї–Є–Љ –Њ–±—А–∞–Ј–Њ–Љ, –њ—А–Є –Р-–±–µ—В–∞-2-–Ь-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–µ –Њ—В–Љ–µ—З–∞—О—В –њ–Њ—А–∞–ґ–µ–љ–Є–µ –Ї–Њ—Б—В–µ–є –Є¬†–њ–µ—А–Є–∞—А—В–Є–Ї—Г–ї—П—А–љ—Л—Е —В–Ї–∞–љ–µ–є, —А–µ–ґ–µ¬†вАУ —Б–Њ—Б—Г–і–Њ–≤.
–Я–∞—В–Њ–≥–µ–љ–µ–Ј –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞
–Э–µ—Б–Љ–Њ—В—А—П –љ–∞ —А–∞–Ј–ї–Є—З–Є–µ –≤¬†—В–Є–њ–∞—Е –∞–Љ–Є–ї–Њ–Є–і–љ–Њ–≥–Њ –±–µ–ї–Ї–∞, —Б—Г—Й–µ—Б—В–≤—Г–µ—В –Њ–±—Й–љ–Њ—Б—В—М –њ–∞—В–Њ–≥–µ–љ–µ–Ј–∞ —А–∞–Ј–ї–Є—З–љ—Л—Е –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є—Е —Д–Њ—А–Љ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞. –Ю—Б–љ–Њ–≤–љ–Њ–є –њ—А–Є—З–Є–љ–Њ–є —А–∞–Ј–≤–Є—В–Є—П –±–Њ–ї–µ–Ј–љ–Є —Б–ї—Г–ґ–Є—В –љ–∞–ї–Є—З–Є–µ –Њ–њ—А–µ–і–µ–ї–µ–љ–љ–Њ–≥–Њ, –љ–µ—А–µ–і–Ї–Њ –њ–Њ–≤—Л—И–µ–љ–љ–Њ–≥–Њ –Ї–Њ–ї–Є—З–µ—Б—В–≤–∞ –∞–Љ–Є–ї–Њ–Є–і–Њ–≥–µ–љ–љ–Њ–≥–Њ –њ—А–µ–і—И–µ—Б—В–≤–µ–љ–љ–Є–Ї–∞. –Ъ–∞–Ї —Г–ґ–µ —Г–Ї–∞–Ј—Л–≤–∞–ї–Њ—Б—М, –њ–Њ—П–≤–ї–µ–љ–Є–µ –Є–ї–Є —Г—Б–Є–ї–µ–љ–Є–µ –∞–Љ–Є–ї–Њ–Є–і–Њ–≥–µ–љ–љ–Њ—Б—В–Є –Љ–Њ–ґ–µ—В –±—Л—В—М –Њ–±—Г—Б–ї–Њ–≤–ї–µ–љ–Њ —Ж–Є—А–Ї—Г–ї—П—Ж–Є–µ–є –≤–∞—А–Є–∞–љ—В–Њ–≤ –±–µ–ї–Ї–Њ–≤ —Б¬†–њ–Њ–≤—Л—И–µ–љ–љ–Њ–є –Њ–±—Й–µ–є –≥–Є–і—А–Њ—Д–Њ–±–љ–Њ—Б—В—М—О –Љ–Њ–ї–µ–Ї—Г–ї—Л, –љ–∞—А—Г—И–µ–љ–љ—Л–Љ —Б–Њ–Њ—В–љ–Њ—И–µ–љ–Є–µ–Љ –њ–Њ–≤–µ—А—Е–љ–Њ—Б—В–љ—Л—Е –Љ–Њ–ї–µ–Ї—Г–ї—П—А–љ—Л—Е –Ј–∞—А—П–і–Њ–≤, —З—В–Њ –њ—А–Є–≤–Њ–і–Є—В –Ї¬†–љ–µ—Б—В–∞–±–Є–ї—М–љ–Њ—Б—В–Є –±–µ–ї–Ї–Њ–≤–Њ–є –Љ–Њ–ї–µ–Ї—Г–ї—Л –Є¬†—Б–њ–Њ—Б–Њ–±—Б—В–≤—Г–µ—В –µ–µ –∞–≥—А–µ–≥–∞—Ж–Є–Є –≤¬†–∞–Љ–Є–ї–Њ–Є–і–љ—Г—О —Д–Є–±—А–Є–ї–ї—Г. –≠—В–Є –Љ–µ—Е–∞–љ–Є–Ј–Љ—Л –Њ—Б–Њ–±–µ–љ–љ–Њ —П—А–Ї–Њ –њ—А–Њ—Б–ї–µ–ґ–Є–≤–∞—О—В—Б—П –љ–∞ –њ—А–Є–Љ–µ—А–µ –±–µ–ї–Ї–Њ–≤, –≤¬†—Д—Г–љ–Ї—Ж–Є—О –Ї–Њ—В–Њ—А—Л—Е –Ј–∞–ї–Њ–ґ–µ–љ–∞ –љ–µ–Њ–±—Е–Њ–і–Є–Љ–Њ—Б—В—М —Д–Є–Ј–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–≥–Њ –Є–Ј–Љ–µ–љ–µ–љ–Є—П –Ї–Њ–љ—Д–Њ—А–Љ–∞—Ж–Є–Є. –Ґ–∞–Ї, –њ—А–∞–Ї—В–Є—З–µ—Б–Ї–Є –≤—Б–µ –∞–њ–Њ–ї–Є–њ–Њ–њ—А–Њ—В–µ–Є–љ—Л, –≤—Л–љ—Г–ґ–і–µ–љ–љ—Л–µ —А–∞–Ј–≤–Њ—А–∞—З–Є–≤–∞—В—М —Б–≤–Њ—О –≤—В–Њ—А–Є—З–љ—Г—О —Б—В—А—Г–Ї—В—Г—А—Г –≤¬†–њ—А–Њ—Ж–µ—Б—Б–µ —В—А–∞–љ—Б–ї–Њ–Ї–∞—Ж–Є–Є —Е–Њ–ї–µ—Б—В–µ—А–Є–љ–∞ —З–µ—А–µ–Ј —Б—В–µ–љ–Ї—Г —Б–Њ—Б—Г–і–∞, —Г—З–∞—Б—В–≤—Г—О—В –≤¬†–њ–∞—В–Њ–≥–µ–љ–µ–Ј–µ —А–∞–Ј–ї–Є—З–љ—Л—Е —Д–Њ—А–Љ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞. –Э–∞ –њ–Њ—Б–ї–µ–і–љ–µ–Љ —Н—В–∞–њ–µ –∞–Љ–Є–ї–Њ–Є–і–Њ–≥–µ–љ–µ–Ј–∞ –њ—А–Њ–Є—Б—Е–Њ–і–Є—В –≤–Ј–∞–Є–Љ–Њ–і–µ–є—Б—В–≤–Є–µ –∞–Љ–Є–ї–Њ–Є–і–љ–Њ–≥–Њ –±–µ–ї–Ї–∞ —Б¬†–±–µ–ї–Ї–∞–Љ–Є –њ–ї–∞–Ј–Љ—Л –Ї—А–Њ–≤–Є –Є¬†–≥–ї–Є–Ї–Њ–Ј–Њ–∞–Љ–Є–љ–Њ–≥–ї–Є–Ї–∞–љ–∞–Љ–Є —В–Ї–∞–љ–µ–є. –Ъ—А–Њ–Љ–µ —Б—В—А—Г–Ї—В—Г—А–љ—Л—Е –Њ—Б–Њ–±–µ–љ–љ–Њ—Б—В–µ–є, –Є–Љ–µ—О—В –Ј–љ–∞—З–µ–љ–Є–µ —В–∞–Ї–ґ–µ —Д–Є–Ј–Є–Ї–Њ-—Е–Є–Љ–Є—З–µ—Б–Ї–Є–µ —Б–≤–Њ–є—Б—В–≤–∞ –Љ–µ–ґ–Ї–ї–µ—В–Њ—З–љ–Њ–≥–Њ –Љ–∞—В—А–Є–Ї—Б–∞, –≥–і–µ –њ—А–Њ–Є—Б—Е–Њ–і–Є—В —Б–±–Њ—А–Ї–∞ –∞–Љ–Є–ї–Њ–Є–і–љ–Њ–є —Д–Є–±—А–Є–ї–ї—Л. –Т¬†–њ—А–∞–Ї—В–Є–Ї–µ —Н–Ї—Б–њ–µ—А–Є–Љ–µ–љ—В–∞–ї—М–љ–Њ–≥–Њ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ —Е–Њ—А–Њ—И–Њ –Є–Ј–≤–µ—Б—В–љ–∞ —Б–њ–Њ—Б–Њ–±–љ–Њ—Б—В—М —Б—Г—Б–њ–µ–љ–Ј–Є–Є –∞–Љ–Є–ї–Њ–Є–і–љ—Л—Е –Љ–∞—Б—Б (–∞–Љ–Є–ї–Њ–Є–і—Г—Б–Ї–Њ—А—П—О—Й–∞—П —Б—Г–±—Б—В–∞–љ—Ж–Є—П), –њ–Њ–ї—Г—З–µ–љ–љ–Њ–є –Є–Ј —В–Ї–∞–љ–µ–є –ґ–Є–≤–Њ—В–љ—Л—Е, –њ–Њ—А–∞–ґ–µ–љ–љ—Л—Е –∞–Љ–Є–ї–Њ–Є–і–Њ–Љ, –њ—А–Њ–≤–Њ—Ж–Є—А–Њ–≤–∞—В—М –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј –њ—А–Є –≤–≤–µ–і–µ–љ–Є–Є –Ј–і–Њ—А–Њ–≤—Л–Љ –ґ–Є–≤–Њ—В–љ—Л–Љ. –Т¬†–Ї–ї–Є–љ–Є—З–µ—Б–Ї–Њ–є –њ—А–∞–Ї—В–Є–Ї–µ —Г¬†–±–Њ–ї—М–љ—Л—Е ATTR-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–Њ–Љ, –љ–µ—Б–Љ–Њ—В—А—П –љ–∞ –њ—А–µ–Ї—А–∞—Й–µ–љ–Є–µ —Ж–Є—А–Ї—Г–ї—П—Ж–Є–Є –њ–∞—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–≥–Њ —В—А–∞–љ—Б—В–Є—А–µ—В–Є–љ–∞ –њ–Њ—Б–ї–µ —В—А–∞–љ—Б–њ–ї–∞–љ—В–∞—Ж–Є–Є –Ј–і–Њ—А–Њ–≤–Њ–є –њ–µ—З–µ–љ–Є, –њ—А–Њ–і–Њ–ї–ґ–∞–µ—В—Б—П –љ–∞—А–∞—Б—В–∞–љ–Є–µ –Љ–∞—Б—Б—Л –∞–Љ–Є–ї–Њ–Є–і–љ—Л—Е –і–µ–њ–Њ–Ј–Є—В–Њ–≤ –≤¬†—Б–µ—А–і—Ж–µ –Ј–∞ —Б—З–µ—В –Ј–∞—Е–≤–∞—В–∞ –љ–Њ—А–Љ–∞–ї—М–љ–Њ–≥–Њ –љ–µ–Є–Ј–Љ–µ–љ–µ–љ–љ–Њ–≥–Њ —В—А–∞–љ—Б—В–Є—А–µ—В–Є–љ–∞ [6]. –Ь–љ–Њ–≥–Є–µ —Д–Њ—А–Љ—Л –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ –Љ–Њ–ґ–љ–Њ –Њ–±—К–µ–і–Є–љ–Є—В—М —В–∞–Ї–ґ–µ –њ–Њ –њ—А–Є–Ј–љ–∞–Ї—Г –≤–Њ–Ј–љ–Є–Ї–љ–Њ–≤–µ–љ–Є—П –≤¬†–њ–Њ–ґ–Є–ї–Њ–Љ –Є¬†—Б—В–∞—А—З–µ—Б–Ї–Њ–Љ –≤–Њ–Ј—А–∞—Б—В–µ (AL, ATTR, AIAPP, AApoA1, AFib, ALys, AANF, A-–±–µ—В–∞), —З—В–Њ —Г–Ї–∞–Ј—Л–≤–∞–µ—В –љ–∞ –љ–∞–ї–Є—З–Є–µ –Љ–µ—Е–∞–љ–Є–Ј–Љ–Њ–≤ –≤–Њ–Ј—А–∞—Б—В–љ–Њ–є —Н–≤–Њ–ї—О—Ж–Є–Є —Б—В—А—Г–Ї—В—Г—А—Л –Њ–њ—А–µ–і–µ–ї–µ–љ–љ—Л—Е –±–µ–ї–Ї–Њ–≤ –≤¬†—Б—В–Њ—А–Њ–љ—Г –њ–Њ–≤—Л—И–µ–љ–Є—П –∞–Љ–Є–ї–Њ–Є–і–Њ–≥–µ–љ–љ–Њ—Б—В–Є –Є¬†–њ–Њ–Ј–≤–Њ–ї—П–µ—В —А–∞—Б—Б–Љ–∞—В—А–Є–≤–∞—В—М –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј –Ї–∞–Ї –Њ–і–љ—Г –Є–Ј –Љ–Њ–і–µ–ї–µ–є —Б—В–∞—А–µ–љ–Є—П –Њ—А–≥–∞–љ–Є–Ј–Љ–∞.
–Ъ–ї–∞—Б—Б–Є—Д–Є–Ї–∞—Ж–Є—П –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞
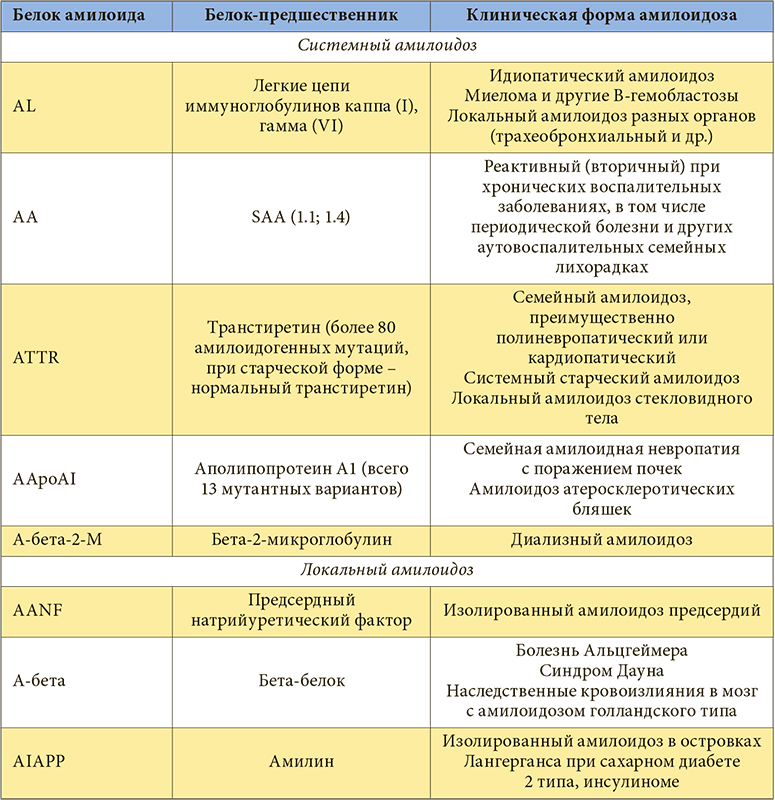
–Ю–±—К—П—Б–љ–µ–љ–Є–µ –Љ–љ–Њ–≥–Њ–Њ–±—А–∞–Ј–Є—П –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є—Е —Д–Њ—А–Љ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ –љ–∞–ї–Є—З–Є–µ–Љ –Љ–љ–Њ–≥–Њ—З–Є—Б–ї–µ–љ–љ—Л—Е —Б—Л–≤–Њ—А–Њ—В–Њ—З–љ—Л—Е –±–µ–ї–Ї–Њ–≤-–њ—А–µ–і—И–µ—Б—В–≤–µ–љ–љ–Є–Ї–Њ–≤ –њ—А–Є–≤–µ–ї–Њ –Ї¬†—Б–Њ–Ј–і–∞–љ–Є—О —Б–Њ–≤—А–µ–Љ–µ–љ–љ–Њ–є –Ї–ї–∞—Б—Б–Є—Д–Є–Ї–∞—Ж–Є–Є –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞, –≤¬†–Њ—Б–љ–Њ–≤—Г –Ї–Њ—В–Њ—А–Њ–є –њ–Њ–ї–Њ–ґ–µ–љ –±–Є–Њ—Е–Є–Љ–Є—З–µ—Б–Ї–Є–є —В–Є–њ –±–µ–ї–Ї–∞-–њ—А–µ–і—И–µ—Б—В–≤–µ–љ–љ–Є–Ї–∞ (—В–∞–±–ї.). –Т—Б–µ —В–Є–њ—Л –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ –Њ–±–Њ–Ј–љ–∞—З–∞—О—В –∞–±–±—А–µ–≤–Є–∞—В—Г—А–Њ–є, –≤¬†–Ї–Њ—В–Њ—А–Њ–є –њ–µ—А–≤–∞—П –±—Г–Ї–≤–∞ –Р¬†–Њ–Ј–љ–∞—З–∞–µ—В ¬Ђ–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј¬ї, –∞¬†–њ–Њ—Б–ї–µ–і—Г—О—Й–Є–µ¬†вАУ —Б–Њ–Ї—А–∞—Й–µ–љ–љ–Њ–µ –љ–∞–Ј–≤–∞–љ–Є–µ –Њ—Б–љ–Њ–≤–љ—Л—Е —Д–Є–±—А–Є–ї–ї—П—А–љ—Л—Е –±–µ–ї–Ї–Њ–≤ –∞–Љ–Є–ї–Њ–Є–і–∞: –Р¬†вАУ –∞–Љ–Є–ї–Њ–Є–і–љ—Л–є –±–µ–ї–Њ–Ї –Р, L¬†вАУ –ї–µ–≥–Ї–Є–µ —Ж–µ–њ–Є –Є–Љ–Љ—Г–љ–Њ–≥–ї–Њ–±—Г–ї–Є–љ–Њ–≤, TTR¬†вАУ —В—А–∞–љ—Б—В–Є—А–µ—В–Є–љ, –±–µ—В–∞-2-–Ь¬†вАУ –±–µ—В–∞2-–Љ–Є–Ї—А–Њ–≥–ї–Њ–±—Г–ї–Є–љ –Є¬†–і—А. –°¬†–Ї–ї–Є–љ–Є—З–µ—Б–Ї–Њ–є —В–Њ—З–Ї–Є –Ј—А–µ–љ–Є—П —Ж–µ–ї–µ—Б–Њ–Њ–±—А–∞–Ј–љ–Њ –≤—Л–і–µ–ї—П—В—М –ї–Њ–Ї–∞–ї—М–љ—Л–µ –Є¬†—Б–Є—Б—В–µ–Љ–љ—Л–µ, –Є–ї–Є –≥–µ–љ–µ—А–∞–ї–Є–Ј–Њ–≤–∞–љ–љ—Л–µ, —Д–Њ—А–Љ—Л –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞. –°—А–µ–і–Є –њ–Њ—Б–ї–µ–і–љ–Є—Е –Њ—Б–љ–Њ–≤–љ—Л–Љ–Є —Б—З–Є—В–∞—О—В –Р–Р-, AL-, –РTTR- –Є¬†–Р-–±–µ—В–∞-2-–Ь-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј.
–°–Є–Љ–њ—В–Њ–Љ—Л –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ –њ–Њ—З–µ–Ї
–Т –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Њ–є –њ—А–∞–Ї—В–Є–Ї–µ –љ–∞–Є–±–Њ–ї—М—И–µ–µ –Ј–љ–∞—З–µ–љ–Є–µ –Є–Љ–µ—О—В –Р–Р- –Є¬†AL-—В–Є–њ—Л —Б–Є—Б—В–µ–Љ–љ–Њ–≥–Њ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞, –Ї–Њ—В–Њ—А—Л–µ –њ—А–Њ—В–µ–Ї–∞—О—В —Б¬†–≤–Њ–≤–ї–µ—З–µ–љ–Є–µ–Љ –≤¬†–њ–∞—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–є –њ—А–Њ—Ж–µ—Б—Б –Љ–љ–Њ–≥–Є—Е –Њ—А–≥–∞–љ–Њ–≤, –Њ–і–љ–∞–Ї–Њ —З–∞—Й–µ –Љ–∞–љ–Є—Д–µ—Б—В–Є—А—Г—О—В —Б–Є–Љ–њ—В–Њ–Љ–∞–Љ–Є –Љ–Њ–љ–Њ–Њ—А–≥–∞–љ–љ–Њ–≥–Њ –њ–Њ—А–∞–ґ–µ–љ–Є—П. –Т¬†–і–∞–ї—М–љ–µ–є—И–µ–Љ —А–∞–Ј–≤–Є–≤–∞–µ—В—Б—П, –Ї–∞–Ї –њ—А–∞–≤–Є–ї–Њ, —Б—В–Њ–ї—М —Е–∞—А–∞–Ї—В–µ—А–љ—Л–є –і–ї—П —Н—В–Є—Е —В–Є–њ–Њ–≤ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ –њ–Њ–ї–Є–Љ–Њ—А—Д–Є–Ј–Љ –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є—Е –њ—А–Њ—П–≤–ї–µ–љ–Є–є. –Р–Р- –Є¬†AL-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј —Г¬†–Љ—Г–ґ—З–Є–љ –Њ—В–Љ–µ—З–∞—О—В –≤¬†1,8¬†—А–∞–Ј–∞ —З–∞—Й–µ, —З–µ–Љ —Г¬†–ґ–µ–љ—Й–Є–љ. –Ф–ї—П –≤—В–Њ—А–Є—З–љ–Њ–≥–Њ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ —Е–∞—А–∞–Ї—В–µ—А–љ–Њ –±–Њ–ї–µ–µ —А–∞–љ–љ–µ–µ –љ–∞—З–∞–ї–Њ, —З–µ–Љ –і–ї—П –њ–µ—А–≤–Є—З–љ–Њ–≥–Њ (—Б—А–µ–і–љ–Є–є –≤–Њ–Ј—А–∞—Б—В –Ј–∞–±–Њ–ї–µ–≤—И–Є—Е¬†вАУ –Њ–Ї–Њ–ї–Њ 40¬†–Є¬†65¬†–ї–µ—В —Б–Њ–Њ—В–≤–µ—В—Б—В–≤–µ–љ–љ–Њ). –°—А–µ–і–Є –Љ–љ–Њ–≥–Њ—З–Є—Б–ї–µ–љ–љ—Л—Е –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є—Е –њ—А–Њ—П–≤–ї–µ–љ–Є–є –Р–Р- –Є¬†AL-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ –љ–∞—А—П–і—Г —Б¬†–Њ–±—Й–Є–Љ–Є –і–ї—П –Њ–±–Њ–Є—Е —В–Є–њ–Њ–≤ —Б–Є–Љ–њ—В–Њ–Љ–∞–Љ–Є —Б—Г—Й–µ—Б—В–≤—Г—О—В —Е–∞—А–∞–Ї—В–µ—А–љ—Л–µ –ї–Є—И—М –і–ї—П AL-—В–Є–њ–∞ –њ—А–Є–Ј–љ–∞–Ї–Є (–њ–µ—А–Є–Њ—А–±–Є—В–∞–ї—М–љ—Л–µ –≥–µ–Љ–Њ—А—А–∞–≥–Є–Є, –Љ–∞–Ї—А–Њ–≥–ї–Њ—Б—Б–Є—П, –њ–Њ—А–∞–ґ–µ–љ–Є–µ –Ї–Њ–ґ–Є –Є¬†–і—А.) [7вАУ9]. –Ъ—А–Њ–Љ–µ —В–Њ–≥–Њ, –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є–µ –њ—А–Њ—П–≤–ї–µ–љ–Є—П, –љ–∞–њ–Њ–Љ–Є–љ–∞—О—Й–Є–µ –њ—А–Є–Ј–љ–∞–Ї–Є –њ–µ—А–≤–Є—З–љ–Њ–≥–Њ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞, –≤–Њ–Ј–Љ–Њ–ґ–љ—Л –њ—А–Є –РTTR- –Є¬†–Р-–±–µ—В–∞-2-–Ь-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–µ (—Б–Є–љ–і—А–Њ–Љ –Ј–∞–њ—П—Б—В–љ–Њ–≥–Њ –Ї–∞–љ–∞–ї–∞ –Є¬†–і—А.).
–Я–Њ—А–∞–ґ–µ–љ–Є–µ –њ–Њ—З–µ–Ї¬†вАУ –≤–µ–і—Г—Й–Є–є –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є–є –њ—А–Є–Ј–љ–∞–Ї –Р–Р- –Є¬†AL-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞. –Я—А–Є –Р–Р-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–µ –њ–Њ—З–Ї–Є –≤–Њ–≤–ї–µ—З–µ–љ—Л –≤¬†–њ–∞—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–є –њ—А–Њ—Ж–µ—Б—Б –њ—А–∞–Ї—В–Є—З–µ—Б–Ї–Є —Г¬†–≤—Б–µ—Е –±–Њ–ї—М–љ—Л—Е, –њ—А–Є AL-—В–Є–њ–µ —З–∞—Б—В–Њ—В–∞ –љ–µ—Д—А–Њ–њ–∞—В–Є–Є —В–∞–Ї–ґ–µ –≤—Л—Б–Њ–Ї–∞ –Є¬†–њ—А–Є–±–ї–Є–ґ–∞–µ—В—Б—П –Ї¬†80%. –Я–Њ—А–∞–ґ–µ–љ–Є–µ –њ–Њ—З–µ–Ї –љ–∞–±–ї—О–і–∞—О—В –Є¬†–њ—А–Є –РTTR-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–µ, –Њ–і–љ–∞–Ї–Њ —Г¬†–±–Њ–ї—М—И–Є–љ—Б—В–≤–∞ –±–Њ–ї—М–љ—Л—Е —Б–µ–Љ–µ–є–љ–Њ–є –∞–Љ–Є–ї–Њ–Є–і–љ–Њ–є –љ–µ–≤—А–Њ–њ–∞—В–Є–µ–є –Њ—В–Љ–µ—З–∞—О—В –љ–µ—Б–Њ–Њ—В–≤–µ—В—Б—В–≤–Є–µ –Љ–µ–ґ–і—Г –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є–Љ–Є –Є¬†–Љ–Њ—А—Д–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–Љ–Є –њ—А–Є–Ј–љ–∞–Ї–∞–Љ–Є –љ–∞—А—Г—И–µ–љ–Є–є –њ–Њ—З–µ–Ї.
–Р–Љ–Є–ї–Њ–Є–і –њ—А–Є –Р–Р- –Є¬†AL-—В–Є–њ–∞—Е –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ –ї–Њ–Ї–∞–ї–Є–Ј—Г–µ—В—Б—П –њ—А–µ–Є–Љ—Г—Й–µ—Б—В–≤–µ–љ–љ–Њ –≤¬†–Ї–ї—Г–±–Њ—З–Ї–∞—Е, –Њ–і–љ–∞–Ї–Њ —Г¬†10% –±–Њ–ї—М–љ—Л—Е –њ–µ—А–≤–Є—З–љ—Л–Љ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–Њ–Љ –Є¬†—Г –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ–є —З–∞—Б—В–Є –±–Њ–ї—М–љ—Л—Е –љ–∞—Б–ї–µ–і—Б—В–≤–µ–љ–љ–Њ–є –љ–µ–≤—А–Њ–њ–∞—В–Є–µ–є –Њ—В–Љ–µ—З–∞—О—В —В–Њ–ї—М–Ї–Њ –Њ—В–ї–Њ–ґ–µ–љ–Є—П –≤–љ–µ –Ї–ї—Г–±–Њ—З–Ї–Њ–≤. –Р–Љ–Є–ї–Њ–Є–і –Њ—В–Ї–ї–∞–і—Л–≤–∞–µ—В—Б—П —В–∞–Ї–ґ–µ –≤¬†–і—А—Г–≥–Є—Е –њ–Њ—З–µ—З–љ—Л—Е —Б—В—А—Г–Ї—В—Г—А–∞—Е: –≤¬†–±–∞–Ј–∞–ї—М–љ–Њ–є –Љ–µ–Љ–±—А–∞–љ–µ –Ї–∞–љ–∞–ї—М—Ж–µ–≤ (–њ—А–µ–Є–Љ—Г—Й–µ—Б—В–≤–µ–љ–љ–Њ –і–Є—Б—В–∞–ї—М–љ—Л—Е –Є¬†–њ–µ—В–ї–Є –У–µ–љ–ї–µ), –Є–љ—В–µ—А—Б—В–Є—Ж–Є–Є, —Б—В–µ–љ–Ї–∞—Е —Б–Њ—Б—Г–і–Њ–≤.
–Ъ–ї–Є–љ–Є—З–µ—Б–Ї–Є –∞–Љ–Є–ї–Њ–Є–і–љ–∞—П –љ–µ—Д—А–Њ–њ–∞—В–Є—П –Љ–∞–љ–Є—Д–µ—Б—В–Є—А—Г–µ—В, –Ї–∞–Ї –њ—А–∞–≤–Є–ї–Њ, –Є–Ј–Њ–ї–Є—А–Њ–≤–∞–љ–љ–Њ –њ—А–Њ—В–µ–Є–љ—Г—А–Є–µ–є –Є¬†—Е–∞—А–∞–Ї—В–µ—А–Є–Ј—Г–µ—В—Б—П –љ–µ—Г–Ї–ї–Њ–љ–љ–Њ –њ—А–Њ–≥—А–µ—Б—Б–Є—А—Г—О—Й–Є–Љ —В–µ—З–µ–љ–Є–µ–Љ —Б¬†–њ–Њ—Б–ї–µ–і–Њ–≤–∞—В–µ–ї—М–љ–Њ–є —Б–Љ–µ–љ–Њ–є —Б—В–∞–і–Є–є: –њ—А–Њ—В–µ–Є–љ—Г—А–Є—З–µ—Б–Ї–∞—П, –љ–µ—Д—А–Њ—В–Є—З–µ—Б–Ї–∞—П, –•–Я–Э. –Ґ–Њ–ї—М–Ї–Њ —Г¬†20% –±–Њ–ї—М–љ—Л—Е –Р–Р-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–Њ–Љ –•–Я–Э —А–∞–Ј–≤–Є–≤–∞–µ—В—Б—П –±–µ–Ј –њ—А–µ–і—И–µ—Б—В–≤—Г—О—Й–µ–≥–Њ –љ–µ—Д—А–Њ—В–Є—З–µ—Б–Ї–Њ–≥–Њ —Б–Є–љ–і—А–Њ–Љ–∞. –Я—А–Є AL-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–µ —Б—В–∞–і–Є–є–љ–Њ—Б—В—М —В–µ—З–µ–љ–Є—П –∞–Љ–Є–ї–Њ–Є–і–љ–Њ–є –љ–µ—Д—А–Њ–њ–∞—В–Є–Є –њ—А–Њ—П–≤–ї—П–µ—В—Б—П –Љ–µ–љ–µ–µ –Њ—В—З–µ—В–ї–Є–≤–Њ [9].
–Ъ –Њ—Б–Њ–±–µ–љ–љ–Њ—Б—В—П–Љ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ –њ–Њ—З–µ–Ї –Њ—В–љ–Њ—Б—П—В —А–µ–і–Ї–Њ—Б—В—М –≥–µ–Љ–∞—В—Г—А–Є–Є –Є¬†–ї–µ–є–Ї–Њ—Ж–Є—В—Г—А–Є–Є (¬Ђ—Б–Ї—Г–і–љ—Л–є¬ї –Љ–Њ—З–µ–≤–Њ–є –Њ—Б–∞–і–Њ–Ї), –∞¬†—В–∞–Ї–ґ–µ –∞—А—В–µ—А–Є–∞–ї—М–љ–Њ–є –≥–Є–њ–µ—А—В–Њ–љ–Є–Є, –Ї–Њ—В–Њ—А—Г—О –і–∞–ґ–µ –њ—А–Є –•–Я–Э –Њ—В–Љ–µ—З–∞—О—В –ї–Є—И—М —Г¬†20% –±–Њ–ї—М–љ—Л—Е –Р–Р-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–Њ–Љ –Є¬†–µ—Й–µ —А–µ–ґ–µ –њ—А–Є AL-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–µ. –Э–µ—Д—А–Њ—В–Є—З–µ—Б–Ї–Є–є —Б–Є–љ–і—А–Њ–Љ –Є¬†–±–Њ–ї—М—И–Є–µ —А–∞–Ј–Љ–µ—А—Л –њ–Њ—З–µ–Ї —Б–Њ—Е—А–∞–љ—П—О—В—Б—П –і–∞–ґ–µ –њ—А–Є —А–∞–Ј–≤–Є—В–Є–Є –Є¬†–њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є–Є –•–Я–Э.
–Т–µ–ї–Є—З–Є–љ–∞ –њ—А–Њ—В–µ–Є–љ—Г—А–Є–Є –љ–µ –Ї–Њ—А—А–µ–ї–Є—А—Г–µ—В —Б¬†–≤—Л—А–∞–ґ–µ–љ–љ–Њ—Б—В—М—О –∞–Љ–Є–ї–Њ–Є–і–љ—Л—Е –Њ—В–ї–Њ–ґ–µ–љ–Є–є –≤¬†–њ–Њ—З–Ї–∞—Е (–њ—А–Є –њ—А–µ–Є–Љ—Г—Й–µ—Б—В–≤–µ–љ–љ–Њ —Б–Њ—Б—Г–і–Є—Б—В–Њ–Љ –њ–Њ—А–∞–ґ–µ–љ–Є–Є –њ—А–Њ—В–µ–Є–љ—Г—А–Є—П –Љ–Њ–ґ–µ—В –±—Л—В—М –Љ–Є–љ–Є–Љ–∞–ї—М–љ–Њ–є) –Є¬†–Ј–∞–≤–Є—Б–Є—В –Њ—В —Б—В–µ–њ–µ–љ–Є –і–µ—Б—В—А—Г–Ї—Ж–Є–Є –њ–Њ–і–Њ—Ж–Є—В–Њ–≤. –Ь–∞–Ї—Б–Є–Љ–∞–ї—М–љ—Г—О –њ–Њ—В–µ—А—О –±–µ–ї–Ї–∞ –Њ–±–љ–∞—А—Г–ґ–Є–≤–∞—О—В —З–µ—А–µ–Ј —Г—З–∞—Б—В–Ї–Є –±–∞–Ј–∞–ї—М–љ–Њ–є –Љ–µ–Љ–±—А–∞–љ—Л, –Ї–Њ—В–Њ—А—Л–µ –њ—А–Њ–њ–Є—В–∞–љ—Л –∞–Љ–Є–ї–Њ–Є–і–Њ–Љ –Є¬†–ї–Є—И–µ–љ—Л —Н–њ–Є—В–µ–ї–Є–∞–ї—М–љ–Њ–≥–Њ –њ–Њ–Ї—А—Л—В–Є—П.
–Р–Љ–Є–ї–Њ–Є–і–Њ–Ј –њ–Њ—З–µ–Ї —Г¬†–±–Њ–ї—М—И–Є–љ—Б—В–≤–∞ –±–Њ–ї—М–љ—Л—Е –і–Є–∞–≥–љ–Њ—Б—В–Є—А—Г—О—В –љ–∞ —Б—В–∞–і–Є–Є –љ–µ—Д—А–Њ—В–Є—З–µ—Б–Ї–Њ–≥–Њ —Б–Є–љ–і—А–Њ–Љ–∞, —Г¬†33%¬†вАУ –љ–∞ —Б—В–∞–і–Є–Є –•–Я–Э. –Т¬†—А–µ–і–Ї–Є—Е —Б–ї—Г—З–∞—П—Е –∞–Љ–Є–ї–Њ–Є–і–љ–∞—П –љ–µ—Д—А–Њ–њ–∞—В–Є—П –Љ–Њ–ґ–µ—В –њ—А–Њ—П–≤–ї—П—В—М—Б—П –Њ—Б—В—А–Њ–љ–µ—Д—А–Њ—В–Є—З–µ—Б–Ї–Є–Љ —Б–Є–љ–і—А–Њ–Љ–Њ–Љ –Є¬†–Љ–∞–Ї—А–Њ–≥–µ–Љ–∞—В—Г—А–Є–µ–є. –Ю–њ–Є—Б–∞–љ—Л —В–∞–Ї–ґ–µ —Б–Є–љ–і—А–Њ–Љ –§–∞–љ–Ї–Њ–љ–Є –Є¬†—В—А–Њ–Љ–±–Њ–Ј –њ–Њ—З–µ—З–љ—Л—Е –≤–µ–љ.
–Я–Њ—А–∞–ґ–µ–љ–Є–µ —Б–µ—А–і—Ж–∞ –Њ—В–Љ–µ—З–∞—О—В —Г¬†–њ–Њ–і–∞–≤–ї—П—О—Й–µ–≥–Њ –±–Њ–ї—М—И–Є–љ—Б—В–≤–∞ –±–Њ–ї—М–љ—Л—Е AL-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–Њ–Љ –Є¬†—Г —З–∞—Б—В–Є –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–РTTR-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–Њ–Љ, –≤¬†—В–Њ –≤—А–µ–Љ—П –Ї–∞–Ї –і–ї—П –Р–Р-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ —Н—В–Њ—В —Б–Є–Љ–њ—В–Њ–Љ –љ–µ —Е–∞—А–∞–Ї—В–µ—А–µ–љ. –Т¬†—А–µ–Ј—Г–ї—М—В–∞—В–µ –Ј–∞–Љ–µ—Й–µ–љ–Є—П –Љ–Є–Њ–Ї–∞—А–і–∞ –∞–Љ–Є–ї–Њ–Є–і–љ—Л–Љ–Є –Љ–∞—Б—Б–∞–Љ–Є —А–∞–Ј–≤–Є–≤–∞–µ—В—Б—П —А–µ—Б—В—А–Є–Ї—В–Є–≤–љ–∞—П –Ї–∞—А–і–Є–Њ–Љ–Є–Њ–њ–∞—В–Є—П [10]. –Ъ–ї–Є–љ–Є—З–µ—Б–Ї–Є –Њ–њ—А–µ–і–µ–ї—П—О—В –Ї–∞—А–і–Є–Њ–Љ–µ–≥–∞–ї–Є—О, —А–∞–љ–Њ —А–∞–Ј–≤–Є–≤–∞–µ—В—Б—П —Б–µ—А–і–µ—З–љ–∞—П –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В—М (—Г 22% –±–Њ–ї—М–љ—Л—Е —Г–ґ–µ –≤¬†–і–µ–±—О—В–µ –±–Њ–ї–µ–Ј–љ–Є), –Ї–Њ—В–Њ—А–∞—П –±—Л—Б—В—А–Њ –њ—А–Њ–≥—А–µ—Б—Б–Є—А—Г–µ—В –Є¬†–њ–Њ—З—В–Є —Г¬†50% –њ–∞—Ж–Є–µ–љ—В–Њ–≤, –љ–∞—А—П–і—Г —Б¬†–∞—А–Є—В–Љ–Є—П–Љ–Є, –±—Л–≤–∞–µ—В –њ—А–Є—З–Є–љ–Њ–є —Б–Љ–µ—А—В–Є. –Ю—Б–Њ–±–µ–љ–љ–Њ—Б—В—М—О —Б–µ—А–і–µ—З–љ–Њ–є –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В–Є –њ—А–Є –њ–µ—А–≤–Є—З–љ–Њ–Љ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–µ —Б–ї—Г–ґ–Є—В –µ–µ —А–µ—Д—А–∞–Ї—В–µ—А–љ–Њ—Б—В—М –Ї¬†—В–µ—А–∞–њ–Є–Є, –≤¬†–њ–µ—А–≤—Г—О –Њ—З–µ—А–µ–і—М —Б–µ—А–і–µ—З–љ—Л–Љ–Є –≥–ї–Є–Ї–Њ–Ј–Є–і–∞–Љ–Є [11].
–Э–∞—А—Г—И–µ–љ–Є—П —А–Є—В–Љ–∞ –Є¬†–њ—А–Њ–≤–Њ–і–Є–Љ–Њ—Б—В–Є –њ—А–Є AL-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–µ –Љ–љ–Њ–≥–Њ–Њ–±—А–∞–Ј–љ—Л: –Љ–µ—А—Ж–∞—В–µ–ї—М–љ–∞—П –∞—А–Є—В–Љ–Є—П, –љ–∞–і–ґ–µ–ї—Г–і–Њ—З–Ї–Њ–≤–∞—П —В–∞—Е–Є–Ї–∞—А–і–Є—П, —Б–Є–љ–і—А–Њ–Љ –њ—А–µ–ґ–і–µ–≤—А–µ–Љ–µ–љ–љ–Њ–≥–Њ –≤–Њ–Ј–±—Г–ґ–і–µ–љ–Є—П –ґ–µ–ї—Г–і–Њ—З–Ї–Њ–≤, —А–∞–Ј–ї–Є—З–љ—Л–µ –±–ї–Њ–Ї–∞–і—Л –Є¬†—Б–Є–љ–і—А–Њ–Љ —Б–ї–∞–±–Њ—Б—В–Є —Б–Є–љ—Г—Б–Њ–≤–Њ–≥–Њ —Г–Ј–ї–∞. –Т—Б–ї–µ–і—Б—В–≤–Є–µ –Њ—В–ї–Њ–ґ–µ–љ–Є—П –∞–Љ–Є–ї–Њ–Є–і–∞ –≤¬†–Ї–Њ—А–Њ–љ–∞—А–љ—Л—Е –∞—А—В–µ—А–Є—П—Е –≤–Њ–Ј–Љ–Њ–ґ–љ–Њ —А–∞–Ј–≤–Є—В–Є–µ –Є–љ—Д–∞—А–Ї—В–∞ –Љ–Є–Њ–Ї–∞—А–і–∞, –Њ–±–љ–∞—А—Г–ґ–Є–≤–∞–µ–Љ–Њ–≥–Њ –љ–∞ –∞—Г—В–Њ–њ—Б–Є–Є —Г¬†6% –±–Њ–ї—М–љ—Л—Е. –Р–Љ–Є–ї–Њ–Є–і–љ—Л–µ –Њ—В–ї–Њ–ґ–µ–љ–Є—П –≤¬†–Ї–ї–∞–њ–∞–љ–љ—Л—Е —Б—В—А—Г–Ї—В—Г—А–∞—Е —Б–Є–Љ—Г–ї–Є—А—Г—О—В –Ї–∞—А—В–Є–љ—Г –Ї–ї–∞–њ–∞–љ–љ–Њ–≥–Њ –њ–Њ—А–Њ–Ї–∞. –Ю—Б–љ–Њ–≤–љ—Л–Љ –њ—А–Є–Ј–љ–∞–Ї–Њ–Љ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ —Б–µ—А–і—Ж–∞ –љ–∞ —Н–ї–µ–Ї—В—А–Њ–Ї–∞—А–і–Є–Њ–≥—А–∞–Љ–Љ–µ –±—Л–≤–∞–µ—В —Б–љ–Є–ґ–µ–љ–Є–µ –≤–Њ–ї—М—В–∞–ґ–∞ –Ј—Г–±—Ж–Њ–≤ –Ї–Њ–Љ–њ–ї–µ–Ї—Б–∞ QRS. –Ю–њ–Є—Б–∞–љ –Є–љ—Д–∞—А–Ї—В–Њ–њ–Њ–і–Њ–±–љ—Л–є —В–Є–њ —Н–ї–µ–Ї—В—А–Њ–Ї–∞—А–і–Є–Њ–≥—А–∞–Љ–Љ—Л. –Э–∞–Є–±–Њ–ї–µ–µ –∞–і–µ–Ї–≤–∞—В–љ—Л–Љ –Љ–µ—В–Њ–і–Њ–Љ –і–Є–∞–≥–љ–Њ—Б—В–Є–Ї–Є –∞–Љ–Є–ї–Њ–Є–і–љ–Њ–є –Ї–∞—А–і–Є–Њ–Љ–Є–Њ–њ–∞—В–Є–Є —Б—З–Є—В–∞—О—В —Н—Е–Њ–Ї–∞—А–і–Є–Њ–≥—А–∞—Д–Є—О, —Б¬†–њ–Њ–Љ–Њ—Й—М—О –Ї–Њ—В–Њ—А–Њ–є –Љ–Њ–ґ–љ–Њ –і–Є–∞–≥–љ–Њ—Б—В–Є—А–Њ–≤–∞—В—М —Б–Є–Љ–Љ–µ—В—А–Є—З–љ–Њ–µ —Г—В–Њ–ї—Й–µ–љ–Є–µ —Б—В–µ–љ–Њ–Ї –ґ–µ–ї—Г–і–Њ—З–Ї–Њ–≤, –і–Є–ї–∞—В–∞—Ж–Є—О –њ—А–µ–і—Б–µ—А–і–Є–є, –љ–∞—А—Г—И–µ–љ–Є—П –≥–µ–Љ–Њ–і–Є–љ–∞–Љ–Є–Ї–Є.
–°–µ—А—М–µ–Ј–љ—Л–Љ –њ–∞—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–Љ –њ—А–Є–Ј–љ–∞–Ї–Њ–Љ –њ—А–Є AL-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–µ —Б–ї—Г–ґ–Є—В –Њ—А—В–Њ—Б—В–∞—В–Є—З–µ—Б–Ї–∞—П –∞—А—В–µ—А–Є–∞–ї—М–љ–∞—П –≥–Є–њ–Њ—В–Њ–љ–Є—П, –Ї–Њ—В–Њ—А—Г—О –љ–∞–±–ї—О–і–∞—О—В —Г¬†11% –±–Њ–ї—М–љ—Л—Е –≤¬†–Љ–Њ–Љ–µ–љ—В –њ–Њ—Б—В–∞–љ–Њ–≤–Ї–Є –і–Є–∞–≥–љ–Њ–Ј–∞. –Ю–±—Л—З–љ–Њ —Н—В–Њ—В —Б–Є–Љ–њ—В–Њ–Љ —Б–≤—П–Ј–∞–љ —Б¬†–∞–≤—В–Њ–љ–Њ–Љ–љ–Њ–є –і–Є—Б—Д—Г–љ–Ї—Ж–Є–µ–є (–њ–Њ—А–∞–ґ–µ–љ–Є–µ –≤–µ–≥–µ—В–∞—В–Є–≤–љ–Њ–є –љ–µ—А–≤–љ–Њ–є —Б–Є—Б—В–µ–Љ—Л) –Є¬†–≤ —В—П–ґ–µ–ї—Л—Е —Б–ї—Г—З–∞—П—Е —Б–Њ–њ—А–Њ–≤–Њ–ґ–і–∞–µ—В—Б—П —Б–Є–љ–Ї–Њ–њ–∞–ї—М–љ—Л–Љ–Є —Б–Њ—Б—В–Њ—П–љ–Є—П–Љ–Є. –Р—А—В–µ—А–Є–∞–ї—М–љ–∞—П –≥–Є–њ–Њ—В–Њ–љ–Є—П –±—Л–≤–∞–µ—В —В–∞–Ї–ґ–µ —Г¬†–±–Њ–ї—М–љ—Л—Е –Р–Р-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–Њ–Љ, –љ–Њ –≤¬†—Н—В–Њ–Љ —Б–ї—Г—З–∞–µ –µ–µ –њ—А–Є—З–Є–љ–Њ–є —Б–ї—Г–ґ–Є—В –љ–∞–і–њ–Њ—З–µ—З–љ–Є–Ї–Њ–≤–∞—П –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В—М –≤—Б–ї–µ–і—Б—В–≤–Є–µ –Њ—В–ї–Њ–ґ–µ–љ–Є—П –∞–Љ–Є–ї–Њ–Є–і–∞ –≤¬†–љ–∞–і–њ–Њ—З–µ—З–љ–Є–Ї–∞—Е [3].
–Я–Њ—А–∞–ґ–µ–љ–Є–µ –і—Л—Е–∞—В–µ–ї—М–љ–Њ–є —Б–Є—Б—В–µ–Љ—Л –≤–Њ–Ј–љ–Є–Ї–∞–µ—В –њ—А–Є –њ–µ—А–≤–Є—З–љ–Њ–Љ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–µ –њ—А–Є–Љ–µ—А–љ–Њ —Г¬†50% –±–Њ–ї—М–љ—Л—Е, –∞¬†–њ—А–Є –≤—В–Њ—А–Є—З–љ–Њ–Љ¬†вАУ —Г¬†10вАУ14%. –Т¬†–±–Њ–ї—М—И–Є–љ—Б—В–≤–µ —Б–ї—Г—З–∞–µ–≤ –Њ–љ–Њ –њ—А–Њ—В–µ–Ї–∞–µ—В –±–µ—Б—Б–Є–Љ–њ—В–Њ–Љ–љ–Њ –Є–ї–Є —Б–Њ —Б–Ї—Г–і–љ–Њ–є –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Њ–є —Б–Є–Љ–њ—В–Њ–Љ–∞—В–Є–Ї–Њ–є [12]. –Я—А–Є AL-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–µ –Њ–і–љ–Є–Љ –Є–Ј —А–∞–љ–љ–Є—Е –њ—А–Є–Ј–љ–∞–Ї–Њ–≤ –±–Њ–ї–µ–Ј–љ–Є –Љ–Њ–ґ–µ—В –±—Л—В—М –Њ—Е—А–Є–њ–ї–Њ—Б—В—М –Є–ї–Є –Є–Ј–Љ–µ–љ–µ–љ–Є–µ —В–µ–Љ–±—А–∞ –≥–Њ–ї–Њ—Б–∞ –≤—Б–ї–µ–і—Б—В–≤–Є–µ –Њ—В–ї–Њ–ґ–µ–љ–Є—П –∞–Љ–Є–ї–Њ–Є–і–∞ –≤¬†–≥–Њ–ї–Њ—Б–Њ–≤—Л—Е —Б–≤—П–Ј–Ї–∞—Е, –Њ–њ–µ—А–µ–ґ–∞—О—Й–µ–≥–Њ –µ–≥–Њ –њ–Њ—П–≤–ї–µ–љ–Є–µ –≤¬†–і–Є—Б—В–∞–ї—М–љ—Л—Е –Њ—В–і–µ–ї–∞—Е –і—Л—Е–∞—В–µ–ї—М–љ—Л—Е –њ—Г—В–µ–є. –Т¬†–ї–µ–≥–Ї–Є—Е –∞–Љ–Є–ї–Њ–Є–і –Њ—В–Ї–ї–∞–і—Л–≤–∞–µ—В—Б—П –њ—А–µ–Є–Љ—Г—Й–µ—Б—В–≤–µ–љ–љ–Њ –≤¬†–∞–ї—М–≤–µ–Њ–ї—П—А–љ—Л—Е –њ–µ—А–µ–≥–Њ—А–Њ–і–Ї–∞—Е (—З—В–Њ –њ—А–Є–≤–Њ–і–Є—В –Ї¬†—А–∞–Ј–≤–Є—В–Є—О –Ї–∞—И–ї—П –Є¬†–Њ–і—Л—И–Ї–Є) –Є¬†—Б—В–µ–љ–Ї–∞—Е —Б–Њ—Б—Г–і–Њ–≤. –Ю–њ–Є—Б–∞–љ—Л —В–∞–Ї–ґ–µ –∞—В–µ–ї–µ–Ї—В–∞–Ј—Л –Є¬†–Є–љ—Д–Є–ї—М—В—А–∞—В—Л –≤¬†–ї–µ–≥–Ї–Є—Е. –†–µ–љ—В–≥–µ–љ–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–∞—П –Ї–∞—А—В–Є–љ–∞ –љ–µ —Б–њ–µ—Ж–Є—Д–Є—З–љ–∞, —Б–Љ–µ—А—В—М –Њ—В –њ—А–Њ–≥—А–µ—Б—Б–Є—А—Г—О—Й–µ–є –і—Л—Е–∞—В–µ–ї—М–љ–Њ–є –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В–Є –љ–∞—Б—В—Г–њ–∞–µ—В —А–µ–і–Ї–Њ [13].
–Я–Њ—А–∞–ґ–µ–љ–Є–µ –Њ—А–≥–∞–љ–Њ–≤ –њ–Є—Й–µ–≤–∞—А–µ–љ–Є—П –љ–∞–±–ї—О–і–∞—О—В –њ—А–Є –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–µ –≤¬†70% —Б–ї—Г—З–∞–µ–≤, –њ—А–Є—З–µ–Љ —Г¬†–±–Њ–ї—М–љ—Л—Е —Б¬†AL- –Є¬†–Р–Р-—В–Є–њ–∞–Љ–Є –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ —З–∞—Б—В–Њ—В–∞ –њ–Њ—А–∞–ґ–µ–љ–Є—П —В–µ—Е –Є–ї–Є –Є–љ—Л—Е –Њ—В–і–µ–ї–Њ–≤ –Ц–Ъ–Ґ —А–∞–Ј–ї–Є—З–љ–∞ [3]. –£¬†25% –±–Њ–ї—М–љ—Л—Е –њ–µ—А–≤–Є—З–љ—Л–Љ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–Њ–Љ –Њ—В–Љ–µ—З–∞—О—В –∞–Љ–Є–ї–Њ–Є–і–љ–Њ–µ –њ–Њ—А–∞–ґ–µ–љ–Є–µ –њ–Є—Й–µ–≤–Њ–і–∞, –њ—А–Њ—П–≤–ї—П—О—Й–µ–µ—Б—П –њ—А–µ–Є–Љ—Г—Й–µ—Б—В–≤–µ–љ–љ–Њ –і–Є—Б—Д–∞–≥–Є–µ–є, –Ї–Њ—В–Њ—А–∞—П –Љ–Њ–ґ–µ—В –±—Л—В—М –Њ–і–љ–Є–Љ –Є–Ј —А–∞–љ–љ–Є—Е —Б–Є–Љ–њ—В–Њ–Љ–Њ–≤ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П. –Ю¬†–њ–Њ—А–∞–ґ–µ–љ–Є–Є –ґ–µ–ї—Г–і–Ї–∞ –Є¬†–Ї–Є—И–µ—З–љ–Є–Ї–∞ –Љ–Њ–≥—Г—В —Б–≤–Є–і–µ—В–µ–ї—М—Б—В–≤–Њ–≤–∞—В—М –Є–Ј—К—П–Ј–≤–ї–µ–љ–Є—П –Є¬†–њ–µ—А—Д–Њ—А–∞—Ж–Є–Є –Є—Е —Б—В–µ–љ–Њ–Ї —Б¬†–≤–Њ–Ј–Љ–Њ–ґ–љ—Л–Љ –Ї—А–Њ–≤–Њ—В–µ—З–µ–љ–Є–µ–Љ, –∞¬†—В–∞–Ї–ґ–µ –њ—А–µ–њ–Є–ї–Њ—А–Є—З–µ—Б–Ї–∞—П –љ–µ–њ—А–Њ—Е–Њ–і–Є–Љ–Њ—Б—В—М –ґ–µ–ї—Г–і–Ї–∞ –Є–ї–Є –Љ–µ—Е–∞–љ–Є—З–µ—Б–Ї–∞—П –Ї–Є—И–µ—З–љ–∞—П –љ–µ–њ—А–Њ—Е–Њ–і–Є–Љ–Њ—Б—В—М –Є–Ј-–Ј–∞ –Њ—В–ї–Њ–ґ–µ–љ–Є—П –∞–Љ–Є–ї–Њ–Є–і–љ—Л—Е –Љ–∞—Б—Б. –£¬†–±–Њ–ї—М–љ—Л—Е —Б¬†–њ—А–µ–Є–Љ—Г—Й–µ—Б—В–≤–µ–љ–љ—Л–Љ –њ–Њ—А–∞–ґ–µ–љ–Є–µ–Љ —В–Њ–ї—Б—В–Њ–є –Ї–Є—И–Ї–Є –≤–Њ–Ј–Љ–Њ–ґ–љ–Њ –њ–Њ—П–≤–ї–µ–љ–Є–µ –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є—Е —Б–Є–Љ–њ—В–Њ–Љ–Њ–≤, –Є–Љ–Є—В–Є—А—Г—О—Й–Є—Е —П–Ј–≤–µ–љ–љ—Л–є –Ї–Њ–ї–Є—В. –Э–∞–Є–±–Њ–ї–µ–µ —З–∞—Б—В—Л–Љ –ґ–µ–ї—Г–і–Њ—З–љ–Њ-–Ї–Є—И–µ—З–љ—Л–Љ –њ—А–Њ—П–≤–ї–µ–љ–Є–µ–Љ AL-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞, –Њ—В–Љ–µ—З–∞–µ–Љ—Л–Љ –њ–Њ—З—В–Є —Г¬†25% –њ–∞—Ж–Є–µ–љ—В–Њ–≤, –±—Л–≤–∞–µ—В —В—П–ґ–µ–ї–∞—П –Љ–Њ—В–Њ—А–љ–∞—П –і–Є–∞—А–µ—П —Б¬†–≤—В–Њ—А–Є—З–љ—Л–Љ –љ–∞—А—Г—И–µ–љ–Є–µ–Љ –≤—Б–∞—Б—Л–≤–∞–љ–Є—П. –Т–Њ–Ј–Љ–Њ–ґ–љ–Њ–є –њ—А–Є—З–Є–љ–Њ–є —В—П–ґ–µ–ї–Њ–є –і–Є–∞—А–µ–Є –њ—А–Є —Н—В–Њ–Љ, –љ–∞—А—П–і—Г —Б¬†–Є–љ—Д–Є–ї—М—В—А–∞—Ж–Є–µ–є –Ї–Є—И–µ—З–љ–Њ–є —Б—В–µ–љ–Ї–Є, –Є¬†–≤ —В–Њ–Љ —З–Є—Б–ї–µ –≤–Њ—А—Б–Є–љ, –∞–Љ–Є–ї–Њ–Є–і–Њ–Љ, —Г¬†–±–Њ–ї—М–љ—Л—Е AL-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–Њ–Љ —Б–ї—Г–ґ–Є—В –∞–≤—В–Њ–љ–Њ–Љ–љ–∞—П (–≤–µ–≥–µ—В–∞—В–Є–≤–љ–∞—П) –і–Є—Б—Д—Г–љ–Ї—Ж–Є—П. –Ш—Б—В–Є–љ–љ—Л–є —Б–Є–љ–і—А–Њ–Љ –љ–∞—А—Г—И–µ–љ–љ–Њ–≥–Њ –≤—Б–∞—Б—Л–≤–∞–љ–Є—П —А–∞–Ј–≤–Є–≤–∞–µ—В—Б—П –њ—А–Є–±–ї–Є–Ј–Є—В–µ–ї—М–љ–Њ —Г¬†4вАУ5% –±–Њ–ї—М–љ—Л—Е. –Я—А–Є –Р–Р-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–µ —Н—В–Є —Б–Є–Љ–њ—В–Њ–Љ—Л –Є–љ–Њ–≥–і–∞ —В–∞–Ї–ґ–µ –≤–Њ–Ј–Љ–Њ–ґ–љ—Л, –≤¬†—В–Њ–Љ —З–Є—Б–ї–µ –Ї–∞–Ї –µ–і–Є–љ—Б—В–≤–µ–љ–љ–Њ–µ –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Њ–µ –њ—А–Њ—П–≤–ї–µ–љ–Є–µ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞.
–Я–Њ—А–∞–ґ–µ–љ–Є–µ –њ–µ—З–µ–љ–Є –њ—А–Є –Р–Р- –Є¬†AL-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–µ –љ–∞–±–ї—О–і–∞—О—В –њ—А–∞–Ї—В–Є—З–µ—Б–Ї–Є –≤¬†100% —Б–ї—Г—З–∞–µ–≤, –њ—А–Є —Н—В–Њ–Љ –Њ–±—Л—З–љ–Њ –Њ—В–Љ–µ—З–∞—О—В –љ–µ–±–Њ–ї—М—И–Њ–µ —Г–≤–µ–ї–Є—З–µ–љ–Є–µ –њ–µ—З–µ–љ–Є –Є¬†3вАУ4-–Ї—А–∞—В–љ–Њ–µ –њ–Њ–≤—Л—И–µ–љ–Є–µ –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є –≥–∞–Љ–Љ–∞-–≥–ї—Г—В–∞–Љ–Є–ї—В—А–∞–љ—Б–њ–µ–њ—В–Є–і–∞–Ј—Л –Є¬†—Й–µ–ї–Њ—З–љ–Њ–є —Д–Њ—Б—Д–∞—В–∞–Ј—Л [14]. –£–≤–µ–ї–Є—З–µ–љ–Є—О –њ–µ—З–µ–љ–Є –Њ–±—Л—З–љ–Њ —Б–Њ–њ—Г—В—Б—В–≤—Г–µ—В –љ–µ–±–Њ–ї—М—И–Њ–µ —Г–≤–µ–ї–Є—З–µ–љ–Є–µ —Б–µ–ї–µ–Ј–µ–љ–Ї–Є. –Ґ—П–ґ–µ–ї–Њ–µ –њ–Њ—А–∞–ґ–µ–љ–Є–µ –њ–µ—З–µ–љ–Є —Б¬†–≤—Л—А–∞–ґ–µ–љ–љ–Њ–є –≥–µ–њ–∞—В–Њ–Љ–µ–≥–∞–ї–Є–µ–є –Є¬†—А–∞–Ј–≤–µ—А–љ—Г—В—Л–Љ–Є –њ—А–Є–Ј–љ–∞–Ї–∞–Љ–Є —В—П–ґ–µ–ї–Њ–≥–Њ —Е–Њ–ї–µ—Б—В–∞–Ј–∞ –Њ—В–Љ–µ—З–∞–µ—В—Б—П –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ —А–µ–ґ–µ (—Г 15вАУ25% –±–Њ–ї—М–љ—Л—Е) –Є¬†–±–Њ–ї–µ–µ —Е–∞—А–∞–Ї—В–µ—А–љ–Њ –і–ї—П AL-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞. –Э–µ—Б–Љ–Њ—В—А—П –љ–∞ –≤—Л—А–∞–ґ–µ–љ–љ—Г—О –≥–µ–њ–∞—В–Њ–Љ–µ–≥–∞–ї–Є—О, —Д—Г–љ–Ї—Ж–Є—П –њ–µ—З–µ–љ–Є –Њ–±—Л—З–љ–Њ –Њ—Б—В–∞–µ—В—Б—П —Б–Њ—Е—А–∞–љ–љ–Њ–є. –†–µ–і–Ї–Є–Љ –њ—А–Є–Ј–љ–∞–Ї–Њ–Љ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ –њ–µ—З–µ–љ–Є –±—Л–≤–∞–µ—В –≤–љ—Г—В—А–Є–њ–µ—З–µ–љ–Њ—З–љ–∞—П –њ–Њ—А—В–∞–ї—М–љ–∞—П –≥–Є–њ–µ—А—В–µ–љ–Ј–Є—П, –Ї–Њ—В–Њ—А–∞—П —З–∞—Й–µ —Б–Њ—З–µ—В–∞–µ—В—Б—П —Б¬†–≤—Л—А–∞–ґ–µ–љ–љ–Њ–є –ґ–µ–ї—В—Г—Е–Њ–є, —Е–Њ–ї–µ—Б—В–∞–Ј–Њ–Љ, –њ–µ—З–µ–љ–Њ—З–љ–Њ–є –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В—М—О –Є¬†—Б–≤–Є–і–µ—В–µ–ї—М—Б—В–≤—Г–µ—В –Њ¬†–і–∞–ї–µ–Ї–Њ –Ј–∞—И–µ–і—И–µ–Љ –њ–Њ—А–∞–ґ–µ–љ–Є–Є –њ–µ—З–µ–љ–Є —Б¬†—А–Є—Б–Ї–Њ–Љ –њ–Є—Й–µ–≤–Њ–і–љ–Њ–≥–Њ –Ї—А–Њ–≤–Њ—В–µ—З–µ–љ–Є—П, –њ–µ—З–µ–љ–Њ—З–љ–Њ–є –Ї–Њ–Љ—Л. –Я—А–Є –љ–µ–Ї–Њ—В–Њ—А—Л—Е –≤–∞—А–Є–∞–љ—В–∞—Е —Б–µ–Љ–µ–є–љ–Њ–≥–Њ ALys-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ –Њ–њ–Є—Б–∞–љ—Л —В—П–ґ–µ–ї—Л–µ —Б–њ–Њ–љ—В–∞–љ–љ—Л–µ –≤–љ—Г—В—А–Є–њ–µ—З–µ–љ–Њ—З–љ—Л–µ –Ї—А–Њ–≤–Њ—В–µ—З–µ–љ–Є—П [15]. –†–µ–і–Ї–Є–Љ –њ—А–Њ—П–≤–ї–µ–љ–Є–µ–Љ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ —Б–µ–ї–µ–Ј–µ–љ–Ї–Є –±—Л–≤–∞–µ—В –µ–µ —Б–њ–Њ–љ—В–∞–љ–љ—Л–є —А–∞–Ј—А—Л–≤.
–Я–Њ—А–∞–ґ–µ–љ–Є–µ –љ–µ—А–≤–љ–Њ–є —Б–Є—Б—В–µ–Љ—Л, –њ—А–µ–і—Б—В–∞–≤–ї–µ–љ–љ–Њ–µ —Б–Є–Љ–њ—В–Њ–Љ–∞–Љ–Є –њ–µ—А–Є—Д–µ—А–Є—З–µ—Б–Ї–Њ–є –љ–µ–≤—А–Њ–њ–∞—В–Є–Є –Є¬†–≤–µ–≥–µ—В–∞—В–Є–≤–љ–Њ–є –і–Є—Б—Д—Г–љ–Ї—Ж–Є–Є, –Њ—В–Љ–µ—З–∞—О—В —Г¬†17% –±–Њ–ї—М–љ—Л—Е AL-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–Њ–Љ –Є¬†—Г –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†—Б–µ–Љ–µ–є–љ–Њ–є –∞–Љ–Є–ї–Њ–Є–і–љ–Њ–є –љ–µ–≤—А–Њ–њ–∞—В–Є–µ–є —А–∞–Ј–љ—Л—Е —В–Є–њ–Њ–≤ (ATTR, AApoA1¬†–Є¬†–і—А.). –Ъ–ї–Є–љ–Є—З–µ—Б–Ї–∞—П –Ї–∞—А—В–Є–љ–∞ –љ–µ–≤—А–Њ–њ–∞—В–Є–Є –њ—А–Є –≤—Б–µ—Е —В–Є–њ–∞—Е –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ –њ—А–∞–Ї—В–Є—З–µ—Б–Ї–Є –Њ–і–Є–љ–∞–Ї–Њ–≤–∞, –њ–Њ—Б–Ї–Њ–ї—М–Ї—Г –Њ–±—Г—Б–ї–Њ–≤–ї–µ–љ–∞ —Б—Е–Њ–і–љ—Л–Љ–Є –њ—А–Њ—Ж–µ—Б—Б–∞–Љ–Є: –≤¬†–њ–µ—А–≤—Г—О –Њ—З–µ—А–µ–і—М, –і–µ–≥–µ–љ–µ—А–∞—Ж–Є–µ–є –Љ–Є–µ–ї–Є–љ–Њ–≤–Њ–є –Њ–±–Њ–ї–Њ—З–Ї–Є –љ–µ—А–≤–Њ–≤, –∞¬†—В–∞–Ї–ґ–µ –Ї–Њ–Љ–њ—А–µ—Б—Б–Є–µ–є –љ–µ—А–≤–љ—Л—Е —Б—В–≤–Њ–ї–Њ–≤ –Њ—В–ї–Њ–ґ–µ–љ–Є—П–Љ–Є –∞–Љ–Є–ї–Њ–Є–і–∞ –Є¬†–Є—И–µ–Љ–Є–µ–є –≤¬†—А–µ–Ј—Г–ї—М—В–∞—В–µ –∞–Љ–Є–ї–Њ–Є–і–љ—Л—Е –і–µ–њ–Њ–Ј–Є—В–Њ–≤ –≤¬†—Б—В–µ–љ–Ї–∞—Е —Б–Њ—Б—Г–і–Њ–≤ [3].
–Т –±–Њ–ї—М—И–Є–љ—Б—В–≤–µ —Б–ї—Г—З–∞–µ–≤ –≤–Њ–Ј–љ–Є–Ї–∞–µ—В —Б–Є–Љ–Љ–µ—В—А–Є—З–љ–∞—П –і–Є—Б—В–∞–ї—М–љ–∞—П –љ–µ–≤—А–Њ–њ–∞—В–Є—П —Б¬†–љ–µ—Г–Ї–ї–Њ–љ–љ—Л–Љ –њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є–µ–Љ. –Т¬†–і–µ–±—О—В–µ –њ–Њ—А–∞–ґ–µ–љ–Є—П –љ–µ—А–≤–љ–Њ–є —Б–Є—Б—В–µ–Љ—Л –љ–∞–±–ї—О–і–∞—О—В, –≥–ї–∞–≤–љ—Л–Љ –Њ–±—А–∞–Ј–Њ–Љ, —Б–µ–љ—Б–Њ—А–љ—Л–µ –љ–∞—А—Г—И–µ–љ–Є—П, –≤¬†–њ–µ—А–≤—Г—О –Њ—З–µ—А–µ–і—М –±–Њ–ї–µ–≤–Њ–є –Є¬†—В–µ–Љ–њ–µ—А–∞—В—Г—А–љ–Њ–є —З—Г–≤—Б—В–≤–Є—В–µ–ї—М–љ–Њ—Б—В–Є, –њ–Њ–Ј–ґ–µ –њ—А–Є—Б–Њ–µ–і–Є–љ—П—О—В—Б—П –љ–∞—А—Г—И–µ–љ–Є—П –≤–Є–±—А–∞—Ж–Є–Њ–љ–љ–Њ–є –Є¬†–њ–Њ–Ј–Є—Ж–Є–Њ–љ–љ–Њ–є —З—Г–≤—Б—В–≤–Є—В–µ–ї—М–љ–Њ—Б—В–Є, –і–≤–Є–≥–∞—В–µ–ї—М–љ—Л–µ –љ–∞—А—Г—И–µ–љ–Є—П. –†–∞–љ–љ–Є–Љ–Є —Б–Є–Љ–њ—В–Њ–Љ–∞–Љ–Є –љ–µ–≤—А–Њ–њ–∞—В–Є–Є –±—Л–≤–∞—О—В –њ–∞—А–µ—Б—В–µ–Ј–Є–Є –Є–ї–Є –Љ—Г—З–Є—В–µ–ї—М–љ—Л–µ –і–Є–Ј–µ—Б—В–µ–Ј–Є–Є (—З—Г–≤—Б—В–≤–Њ –Њ–љ–µ–Љ–µ–љ–Є—П). –Э–Є–ґ–љ–Є–µ –Ї–Њ–љ–µ—З–љ–Њ—Б—В–Є –≤–Њ–≤–ї–µ–Ї–∞—О—В—Б—П –≤¬†–њ–∞—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–є –њ—А–Њ—Ж–µ—Б—Б —З–∞—Й–µ, —З–µ–Љ –≤–µ—А—Е–љ–Є–µ.
–Р–≤—В–Њ–љ–Њ–Љ–љ—Л–µ –і–Є—Б—Д—Г–љ–Ї—Ж–Є–Є —З–∞—Б—В–Њ –Љ–∞–љ–Є—Д–µ—Б—В–Є—А—Г—О—В –Њ—А—В–Њ—Б—В–∞—В–Є—З–µ—Б–Ї–Њ–є –∞—А—В–µ—А–Є–∞–ї—М–љ–Њ–є –≥–Є–њ–Њ—В–Њ–љ–Є–µ–є (—Б–Љ. –≤—Л—И–µ), –Є–љ–Њ–≥–і–∞ —Б¬†–Њ–±–Љ–Њ—А–Њ—З–љ—Л–Љ–Є —Б–Њ—Б—В–Њ—П–љ–Є—П–Љ–Є, –і–Є–∞—А–µ–µ–є, –љ–∞—А—Г—И–µ–љ–Є–µ–Љ —Д—Г–љ–Ї—Ж–Є–Є –Љ–Њ—З–µ–≤–Њ–≥–Њ –њ—Г–Ј—Л—А—П, –Є–Љ–њ–Њ—В–µ–љ—Ж–Є–µ–є —Г¬†–Љ—Г–ґ—З–Є–љ.
–£ 20% –±–Њ–ї—М–љ—Л—Е AL-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–Њ–Љ –Є¬†—Г –±–Њ–ї—М—И–Є–љ—Б—В–≤–∞ –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–Њ–Љ –љ–∞ —Д–Њ–љ–µ –њ—А–Њ–≤–µ–і–µ–љ–Є—П –≥–µ–Љ–Њ–і–Є–∞–ї–Є–Ј–∞ –≤—Л—П–≤–ї—П—О—В —Б–Є–љ–і—А–Њ–Љ –Ј–∞–њ—П—Б—В–љ–Њ–≥–Њ –Ї–∞–љ–∞–ї–∞, –Њ–±—Г—Б–ї–Њ–≤–ї–µ–љ–љ—Л–є —Б–і–∞–≤–ї–µ–љ–Є–µ–Љ —Б—А–µ–і–Є–љ–љ–Њ–≥–Њ –љ–µ—А–≤–∞ –≤¬†–Ј–∞–њ—П—Б—В–љ–Њ–Љ –Ї–∞–љ–∞–ї–µ –∞–Љ–Є–ї–Њ–Є–і–Њ–Љ, –Њ—В–Ї–ї–∞–і—Л–≤–∞—О—Й–Є–Љ—Б—П –≤¬†—Б–≤—П–Ј–Ї–∞—Е –Ј–∞–њ—П—Б—В—М—П [2, 8, 9]. –Ъ–ї–Є–љ–Є—З–µ—Б–Ї–Є —Н—В–Њ—В —Б–Є–љ–і—А–Њ–Љ –њ—А–Њ—П–≤–ї—П–µ—В—Б—П –Є–љ—В–µ–љ—Б–Є–≤–љ—Л–Љ–Є –±–Њ–ї—П–Љ–Є –Є¬†–њ–∞—А–µ—Б—В–µ–Ј–Є—П–Љ–Є –≤¬†IвАУIII –њ–∞–ї—М—Ж–∞—Е –Ї–Є—Б—В–Є —Б¬†–њ–Њ—Б—В–µ–њ–µ–љ–љ–Њ–є –∞—В—А–Њ—Д–Є–µ–є –Љ—Л—И—Ж —В–µ–љ–∞—А–∞. –Ъ¬†–Њ—Б–Њ–±–µ–љ–љ–Њ—Б—В—П–Љ —Б–Є–љ–і—А–Њ–Љ–∞ –Ј–∞–њ—П—Б—В–љ–Њ–≥–Њ –Ї–∞–љ–∞–ї–∞ –њ—А–Є –і–Є–∞–ї–Є–Ј–љ–Њ–Љ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–µ –Њ—В–љ–Њ—Б—П—В –µ–≥–Њ –њ—А–µ–Є–Љ—Г—Й–µ—Б—В–≤–µ–љ–љ–Њ–µ —А–∞–Ј–≤–Є—В–Є–µ –љ–∞ —В–Њ–є —А—Г–Ї–µ, –≥–і–µ —Б—Д–Њ—А–Љ–Є—А–Њ–≤–∞–љ–∞ —Д–Є—Б—В—Г–ї–∞, –∞¬†—В–∞–Ї–ґ–µ —Г—Б–Є–ї–µ–љ–Є–µ –±–Њ–ї–µ–є –≤–Њ –≤—А–µ–Љ—П –њ—А–Њ—Ж–µ–і—Г—А—Л –≥–µ–Љ–Њ–і–Є–∞–ї–Є–Ј–∞, –≤–Њ–Ј–Љ–Њ–ґ–љ–Њ, –≤¬†—А–µ–Ј—Г–ї—М—В–∞—В–µ —А–∞–Ј–≤–Є—В–Є—П —Д–µ–љ–Њ–Љ–µ–љ–∞ –Њ–±–Ї—А–∞–і—Л–≤–∞–љ–Є—П, –Є–љ–і—Г—Ж–Є—А–Њ–≤–∞–љ–љ–Њ–≥–Њ —Д–Є—Б—В—Г–ї–Њ–є, —З—В–Њ –њ—А–Є–≤–Њ–і–Є—В –Ї¬†–Є—И–µ–Љ–Є–Є —Б—А–µ–і–Є–љ–љ–Њ–≥–Њ –љ–µ—А–≤–∞.
–Я–Њ—А–∞–ґ–µ–љ–Є–µ –Ї–Њ–ґ–Є –љ–∞–±–ї—О–і–∞—О—В –њ–Њ—З—В–Є —Г¬†40% –±–Њ–ї—М–љ—Л—Е –њ–µ—А–≤–Є—З–љ—Л–Љ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–Њ–Љ. –Ф–ї—П –љ–µ–≥–Њ —Е–∞—А–∞–Ї—В–µ—А–љ–Њ —А–∞–Ј–љ–Њ–Њ–±—А–∞–Ј–Є–µ –њ—А–Њ—П–≤–ї–µ–љ–Є–є, –љ–∞–Є–±–Њ–ї–µ–µ —З–∞—Б—В—Л–Љ–Є –Є–Ј –Ї–Њ—В–Њ—А—Л—Е –±—Л–≤–∞—О—В –њ–∞—А–∞–Њ—А–±–Є—В–∞–ї—М–љ—Л–µ –≥–µ–Љ–Њ—А—А–∞–≥–Є–Є (–њ–∞—В–Њ–≥–љ–Њ–Љ–Њ–љ–Є—З–љ—Л –і–ї—П AL-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞), –≤–Њ–Ј–љ–Є–Ї–∞—О—Й–Є–µ –њ—А–Є –Љ–∞–ї–µ–є—И–µ–Љ –љ–∞–њ—А—П–ґ–µ–љ–Є–Є. –Ю–њ–Є—Б–∞–љ—Л —В–∞–Ї–ґ–µ –њ–∞–њ—Г–ї—Л, –±–ї—П—И–Ї–Є, —Г–Ј–µ–ї–Ї–Є, –њ—Г–Ј—Л—А—М–Ї–Њ–≤—Л–µ –≤—Л—Б—Л–њ–∞–љ–Є—П. –Э–µ—А–µ–і–Ї–Њ –љ–∞–±–ї—О–і–∞—О—В –Є–љ–і—Г—А–∞—Ж–Є—О –Ї–Њ–ґ–Є, –∞–љ–∞–ї–Њ–≥–Є—З–љ—Г—О —Б–Ї–ї–µ—А–Њ–і–µ—А–Љ–Є—З–µ—Б–Ї–Њ–є. –†–µ–і–Ї–Є–Љ –≤–∞—А–Є–∞–љ—В–Њ–Љ –њ–Њ—А–∞–ґ–µ–љ–Є—П –Ї–Њ–ґ–Є –њ—А–Є AL-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–µ —Б–ї—Г–ґ–∞—В –љ–∞—А—Г—И–µ–љ–Є—П –њ–Є–≥–Љ–µ–љ—В–∞—Ж–Є–Є (–Њ—В –≤—Л—А–∞–ґ–µ–љ–љ–Њ–≥–Њ —Г—Б–Є–ї–µ–љ–Є—П –і–Њ —В–Њ—В–∞–ї—М–љ–Њ–≥–Њ –∞–ї—М–±–Є–љ–Є–Ј–Љ–∞), –∞–ї–Њ–њ–µ—Ж–Є—П, —В—А–Њ—Д–Є—З–µ—Б–Ї–Є–µ –љ–∞—А—Г—И–µ–љ–Є—П.
–Я–Њ—А–∞–ґ–µ–љ–Є–µ –Њ–њ–Њ—А–љ–Њ-–і–≤–Є–≥–∞—В–µ–ї—М–љ–Њ–≥–Њ –∞–њ–њ–∞—А–∞—В–∞ —Е–∞—А–∞–Ї—В–µ—А–љ–Њ –і–ї—П –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–і–Є–∞–ї–Є–Ј–љ—Л–Љ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–Њ–Љ –Є¬†—А–µ–і–Ї–Њ (–≤ 5вАУ10% —Б–ї—Г—З–∞–µ–≤) –≤–Њ–Ј–љ–Є–Ї–∞–µ—В —Г¬†–±–Њ–ї—М–љ—Л—Е AL-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–Њ–Љ (–Є—Б–Ї–ї—О—З–∞—П –Ї–Њ—Б—В–љ—Л–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П –њ—А–Є –Љ–Є–µ–ї–Њ–Љ–љ–Њ–є –±–Њ–ї–µ–Ј–љ–Є). –Я—А–Є —Н—В–Њ–Љ —Е–∞—А–∞–Ї—В–µ—А —В–Ї–∞–љ–µ–≤–Њ–≥–Њ –Њ—В–ї–Њ–ґ–µ–љ–Є—П –∞–Љ–Є–ї–Њ–Є–і–∞ —Б—Е–Њ–і–µ–љ –њ—А–Є –Њ–±–Њ–Є—Е —Н—В–Є—Е —В–Є–њ–∞—Е –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞: –∞–Љ–Є–ї–Њ–Є–і –Њ—В–Ї–ї–∞–і—Л–≤–∞–µ—В—Б—П –≤¬†–Ї–Њ—Б—В—П—Е, —Б—Г—Б—В–∞–≤–љ–Њ–Љ —Е—А—П—Й–µ, —Б–Є–љ–Њ–≤–Є–Є, —Б–≤—П–Ј–Ї–∞—Е –Є¬†–Љ—Л—И—Ж–∞—Е [16, 17].
–Я—А–Є –і–Є–∞–ї–Є–Ј–љ–Њ–Љ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–µ –љ–∞–Є–±–Њ–ї–µ–µ —З–∞—Б—В–Њ –Њ—В–Љ–µ—З–∞—О—В —В—А–Є–∞–і—Г –њ—А–Є–Ј–љ–∞–Ї–Њ–≤¬†вАУ –њ–ї–µ—З–µ-–ї–Њ–њ–∞—В–Њ—З–љ—Л–є –њ–µ—А–Є–∞—А—В—А–Є—В, —Б–Є–љ–і—А–Њ–Љ –Ј–∞–њ—П—Б—В–љ–Њ–≥–Њ –Ї–∞–љ–∞–ї–∞ –Є¬†–њ–Њ—А–∞–ґ–µ–љ–Є–µ —Б—Г—Е–Њ–ґ–Є–ї—М–љ—Л—Е –≤–ї–∞–≥–∞–ї–Є—Й —Б–≥–Є–±–∞—В–µ–ї–µ–є –Ї–Є—Б—В–Є, –њ—А–Є–≤–Њ–і—П—Й–µ–µ –Ї¬†—А–∞–Ј–≤–Є—В–Є—О —Б–≥–Є–±–∞—В–µ–ї—М–љ—Л—Е –Ї–Њ–љ—В—А–∞–Ї—В—Г—А –њ–∞–ї—М—Ж–µ–≤. –Ъ—А–Њ–Љ–µ –љ–Є—Е, —Е–∞—А–∞–Ї—В–µ—А–љ–Њ —А–∞–Ј–≤–Є—В–Є–µ –Ї–Є—Б—В–Њ–Ј–љ–Њ–≥–Њ –њ–Њ—А–∞–ґ–µ–љ–Є—П –Ї–Њ—Б—В–µ–є –Є–Ј-–Ј–∞ –Њ—В–ї–Њ–ґ–µ–љ–Є—П –∞–Љ–Є–ї–Њ–Є–і–∞. –Ґ–Є–њ–Є—З–љ—Л –∞–Љ–Є–ї–Њ–Є–і–љ—Л–µ –Ї–Є—Б—В—Л –≤¬†–Ї–Њ—Б—В—П—Е –Ј–∞–њ—П—Б—В—М—П –Є¬†–≥–Њ–ї–Њ–≤–Ї–∞—Е —В—А—Г–±—З–∞—В—Л—Е –Ї–Њ—Б—В–µ–є. –°–Њ –≤—А–µ–Љ–µ–љ–µ–Љ —Н—В–Є –Њ—В–ї–Њ–ґ–µ–љ–Є—П —Г–≤–µ–ї–Є—З–Є–≤–∞—О—В—Б—П –≤¬†—А–∞–Ј–Љ–µ—А–∞—Е, —Б—В–∞–љ–Њ–≤—П—Б—М –њ—А–Є—З–Є–љ–Њ–є –њ–∞—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є—Е –њ–µ—А–µ–ї–Њ–Љ–Њ–≤.
–І–∞—Б—В—Л–Љ –њ—А–Є–Ј–љ–∞–Ї–Њ–Љ –і–Є–∞–ї–Є–Ј–љ–Њ–≥–Њ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ –±—Л–≤–∞–µ—В —В–∞–Ї–ґ–µ –і–µ—Б—В—А—Г–Ї—В–Є–≤–љ–∞—П —Б–њ–Њ–љ–і–Є–ї–Њ–∞—А—В—А–Њ–њ–∞—В–Є—П –≤¬†—А–µ–Ј—Г–ї—М—В–∞—В–µ –∞–Љ–Є–ї–Њ–Є–і–љ–Њ–≥–Њ –њ–Њ—А–∞–ґ–µ–љ–Є—П –Љ–µ–ґ–њ–Њ–Ј–≤–Њ–љ–Ї–Њ–≤—Л—Е –і–Є—Б–Ї–Њ–≤, –њ—А–µ–Є–Љ—Г—Й–µ—Б—В–≤–µ–љ–љ–Њ –≤¬†—И–µ–є–љ–Њ–Љ –Њ—В–і–µ–ї–µ –њ–Њ–Ј–≤–Њ–љ–Њ—З–љ–Є–Ї–∞.
–Р–Љ–Є–ї–Њ–Є–і–љ—Л–µ –Њ—В–ї–Њ–ґ–µ–љ–Є—П –≤¬†–Љ—Л—И—Ж–∞—Е —З–∞—Й–µ –љ–∞–±–ї—О–і–∞—О—В –њ—А–Є –њ–µ—А–≤–Є—З–љ–Њ–Љ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–µ. –Ю–љ–Є –њ—А–Њ—П–≤–ї—П—О—В—Б—П –њ—Б–µ–≤–і–Њ–≥–Є–њ–µ—А—В—А–Њ—Д–Є–µ–є –Є–ї–Є –∞—В—А–Њ—Д–Є–µ–є –Љ—Л—И—Ж, –Ј–∞—В—А—Г–і–љ—П—О—Й–Є–Љ–Є –і–≤–Є–ґ–µ–љ–Є—П, –Љ—Л—И–µ—З–љ—Л–Љ–Є –±–Њ–ї—П–Љ–Є. –Ь–∞–Ї—А–Њ–≥–ї–Њ—Б—Б–Є—П, –Њ–±—Г—Б–ї–Њ–≤–ї–µ–љ–љ–∞—П –≤—Л—А–∞–ґ–µ–љ–љ–Њ–є –Є–љ—Д–Є–ї—М—В—А–∞—Ж–Є–µ–є –Љ—Л—И—Ж –∞–Љ–Є–ї–Њ–Є–і–Њ–Љ,¬†вАУ —Н—В–Њ –њ–∞—В–Њ–≥–љ–Њ–Љ–Њ–љ–Є—З–љ—Л–є —Б–Є–Љ–њ—В–Њ–Љ AL-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞, –Ї–Њ—В–Њ—А—Л–є –≤—Б—В—А–µ—З–∞–µ—В—Б—П –њ—А–Є–Љ–µ—А–љ–Њ —Г¬†20% –њ–∞—Ж–Є–µ–љ—В–Њ–≤ –Є¬†–љ–µ—А–µ–і–Ї–Њ —Б–Њ—З–µ—В–∞–µ—В—Б—П —Б¬†–њ—Б–µ–≤–і–Њ–≥–Є–њ–µ—А—В—А–Њ—Д–Є–µ–є –і—А—Г–≥–Є—Е –≥—А—Г–њ–њ –њ–Њ–њ–µ—А–µ—З–љ–Њ-–њ–Њ–ї–Њ—Б–∞—В–Њ–є –Љ—Г—Б–Ї—Г–ї–∞—В—Г—А—Л. –Т¬†—В—П–ґ–µ–ї—Л—Е —Б–ї—Г—З–∞—П—Е –Љ–∞–Ї—А–Њ–≥–ї–Њ—Б—Б–Є—П –љ–µ —В–Њ–ї—М–Ї–Њ –Ј–∞—В—А—Г–і–љ—П–µ—В –њ—А–Є–µ–Љ –њ–Є—Й–Є –Є¬†—А–µ—З—М, –љ–Њ –Є¬†–њ—А–Є–≤–Њ–і–Є—В –Ї¬†–Њ–±—Б—В—А—Г–Ї—Ж–Є–Є –і—Л—Е–∞—В–µ–ї—М–љ—Л—Е –њ—Г—В–µ–є [3]. –Я—А–Є –Р–Р-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–µ –Њ–љ–∞ –љ–µ —А–∞–Ј–≤–Є–≤–∞–µ—В—Б—П.
–°—А–µ–і–Є –і—А—Г–≥–Є—Е –Њ—А–≥–∞–љ–љ—Л—Е –њ–Њ—А–∞–ґ–µ–љ–Є–є –њ—А–Є –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–µ –Є–Ј–≤–µ—Б—В–љ—Л –њ–Њ—А–∞–ґ–µ–љ–Є–µ —Й–Є—В–Њ–≤–Є–і–љ–Њ–є –ґ–µ–ї–µ–Ј—Л —Б¬†—А–∞–Ј–≤–Є—В–Є–µ–Љ –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Њ–є –Ї–∞—А—В–Є–љ—Л –≥–Є–њ–Њ—В–Є—А–µ–Њ–Ј–∞ (AL-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј), –љ–∞–і–њ–Њ—З–µ—З–љ–Є–Ї–Њ–≤ —Б¬†–њ–Њ—П–≤–ї–µ–љ–Є–µ–Љ —Б–Є–Љ–њ—В–Њ–Љ–Њ–≤ –Є—Е –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В–Є (—З–∞—Й–µ –њ—А–Є –Р–Р-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–µ), —Н–Ї–Ј–Њ–Ї—А–Є–љ–љ—Л—Е –ґ–µ–ї–µ–Ј, –њ—А–Є–≤–Њ–і—П—Й–µ–µ –Ї¬†–≤–Њ–Ј–љ–Є–Ї–љ–Њ–≤–µ–љ–Є—О —Б—Г—Е–Њ–≥–Њ —Б–Є–љ–і—А–Њ–Љ–∞, –ї–Є–Љ—Д–∞–і–µ–љ–Њ–њ–∞—В–Є—П. –†–µ–і–Ї–Є–Љ –њ—А–Њ—П–≤–ї–µ–љ–Є–µ–Љ, –Њ–њ–Є—Б–∞–љ–љ—Л–Љ –њ—А–Є AL- –Є¬†–РTTR- –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–µ, –±—Л–≤–∞–µ—В –њ–Њ—А–∞–ґ–µ–љ–Є–µ –≥–ї–∞–Ј.
–Ф–Є–∞–≥–љ–Њ—Б—В–Є–Ї–∞ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞
–Я—А–µ–і–њ–Њ–ї–∞–≥–∞–µ–Љ—Л–є –љ–∞ –Њ—Б–љ–Њ–≤–∞–љ–Є–Є –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є—Е –Є¬†–ї–∞–±–Њ—А–∞—В–Њ—А–љ—Л—Е –і–∞–љ–љ—Л—Е –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј –љ–µ–Њ–±—Е–Њ–і–Є–Љ–Њ –њ–Њ–і—В–≤–µ—А–і–Є—В—М –Љ–Њ—А—Д–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є –Њ–±–љ–∞—А—Г–ґ–µ–љ–Є–µ–Љ –∞–Љ–Є–ї–Њ–Є–і–∞ –≤¬†–±–Є–Њ–њ—В–∞—В–∞—Е —В–Ї–∞–љ–µ–є.
–Я—А–Є –њ–Њ–і–Њ–Ј—А–µ–љ–Є–Є –љ–∞ AL-—В–Є–њ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ —А–µ–Ї–Њ–Љ–µ–љ–і—Г—О—В –њ—А–Њ–Є–Ј–≤–Њ–і–Є—В—М –њ—Г–љ–Ї—Ж–Є—О –Ї–Њ—Б—В–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞. –Я–Њ–і—Б—З–µ—В –њ–ї–∞–Ј–Љ–∞—В–Є—З–µ—Б–Ї–Є—Е –Ї–ї–µ—В–Њ–Ї –Є¬†–Њ–Ї—А–∞—Б–Ї–∞ –њ—Г–љ–Ї—В–∞—В–∞ –љ–∞ –∞–Љ–Є–ї–Њ–Є–і –њ–Њ–Ј–≤–Њ–ї—П—О—В –љ–µ —В–Њ–ї—М–Ї–Њ –і–Є–∞–≥–љ–Њ—Б—В–Є—А–Њ–≤–∞—В—М –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј, –љ–Њ –Є¬†–і–Є—Д—Д–µ—А–µ–љ—Ж–Є—А–Њ–≤–∞—В—М –њ–µ—А–≤–Є—З–љ—Л–є –Є¬†–∞—Б—Б–Њ—Ж–Є–Є—А–Њ–≤–∞–љ–љ—Л–є —Б¬†–Љ–Є–µ–ї–Њ–Љ–Њ–є –≤–∞—А–Є–∞–љ—В—Л AL-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞. –Я–Њ–ї–Њ–ґ–Є—В–µ–ї—М–љ—Л–є —А–µ–Ј—Г–ї—М—В–∞—В –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П –Ї–Њ—Б—В–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞ –љ–∞ –∞–Љ–Є–ї–Њ–Є–і –Њ—В–Љ–µ—З–∞—О—В —Г¬†60% –±–Њ–ї—М–љ—Л—Е AL-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–Њ–Љ.
–Я—А–Њ—Б—В–Њ–є –Є¬†–±–µ–Ј–Њ–њ–∞—Б–љ–Њ–є –і–Є–∞–≥–љ–Њ—Б—В–Є—З–µ—Б–Ї–Њ–є –њ—А–Њ—Ж–µ–і—Г—А–Њ–є —Б—З–Є—В–∞—О—В –∞—Б–њ–Є—А–∞—Ж–Є–Њ–љ–љ—Г—О –±–Є–Њ–њ—Б–Є—О –њ–Њ–і–Ї–Њ–ґ–љ–Њ–є –ґ–Є—А–Њ–≤–Њ–є –Ї–ї–µ—В—З–∞—В–Ї–Є, –њ—А–Є –Ї–Њ—В–Њ—А–Њ–є –≤—Л—П–≤–ї—П—О—В –∞–Љ–Є–ї–Њ–Є–і –≤¬†80% —Б–ї—Г—З–∞–µ–≤ AL-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞. –Ъ¬†–њ—А–µ–Є–Љ—Г—Й–µ—Б—В–≤–∞–Љ —Н—В–Њ–є –њ—А–Њ—Ж–µ–і—Г—А—Л, –Ї—А–Њ–Љ–µ –Є–љ—Д–Њ—А–Љ–∞—В–Є–≤–љ–Њ—Б—В–Є, –Њ—В–љ–Њ—Б—П—В —В–∞–Ї–ґ–µ —А–µ–і–Ї–Њ—Б—В—М —А–∞–Ј–≤–Є—В–Є—П –Ї—А–Њ–≤–Њ—В–µ—З–µ–љ–Є–є, —З—В–Њ –њ–Њ–Ј–≤–Њ–ї—П–µ—В –Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞—В—М —Н—В–Њ—В –Љ–µ—В–Њ–і —Г¬†–±–Њ–ї—М–љ—Л—Е —Б¬†–љ–∞—А—Г—И–µ–љ–Є—П–Љ–Є —Б–≤–µ—А—В—Л–≤–∞–љ–Є—П –Ї—А–Њ–≤–Є (–±–Њ–ї—М–љ—Л–µ –њ–µ—А–≤–Є—З–љ—Л–Љ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–Њ–Љ –љ–µ—А–µ–і–Ї–Њ –Є–Љ–µ—О—В –і–µ—Д–Є—Ж–Є—В –• —Д–∞–Ї—В–Њ—А–∞ —Б–≤–µ—А—В—Л–≤–∞–љ–Є—П, –њ—А–Є –Ї–Њ—В–Њ—А–Њ–Љ –Љ–Њ–≥—Г—В —А–∞–Ј–≤–Є—В—М—Б—П –≥–µ–Љ–Њ—А—А–∞–≥–Є–Є [8]).
–Э–∞–Є–±–Њ–ї–µ–µ —З–∞—Б—В–Њ –і–ї—П –і–Є–∞–≥–љ–Њ—Б—В–Є–Ї–Є —А–∞–Ј–љ—Л—Е —В–Є–њ–Њ–≤ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ –њ—А–Њ–≤–Њ–і—П—В –±–Є–Њ–њ—Б–Є—О —Б–ї–Є–Ј–Є—Б—В–Њ–є –Њ–±–Њ–ї–Њ—З–Ї–Є –њ—А—П–Љ–Њ–є –Ї–Є—И–Ї–Є, –њ–Њ—З–Ї–Є, –њ–µ—З–µ–љ–Є. –С–Є–Њ–њ—Б–Є—П —Б–ї–Є–Ј–Є—Б—В–Њ–≥–Њ –Є¬†–њ–Њ–і—Б–ї–Є–Ј–Є—Б—В–Њ–≥–Њ —Б–ї–Њ–µ–≤ –њ—А—П–Љ–Њ–є –Ї–Є—И–Ї–Є –њ–Њ–Ј–≤–Њ–ї—П–µ—В –≤—Л—П–≤–Є—В—М –∞–Љ–Є–ї–Њ–Є–і —Г¬†70% –±–Њ–ї—М–љ—Л—Е, –∞¬†–±–Є–Њ–њ—Б–Є—П –њ–Њ—З–Ї–Є¬†вАУ –њ—А–∞–Ї—В–Є—З–µ—Б–Ї–Є –≤¬†100% —Б–ї—Г—З–∞–µ–≤. –£¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†—Б–Є–љ–і—А–Њ–Љ–Њ–Љ –Ј–∞–њ—П—Б—В–љ–Њ–≥–Њ –Ї–∞–љ–∞–ї–∞ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—О –љ–∞ –∞–Љ–Є–ї–Њ–Є–і –љ–µ–Њ–±—Е–Њ–і–Є–Љ–Њ –њ–Њ–і–≤–µ—А–≥–∞—В—М —В–Ї–∞–љ—М, —Г–і–∞–ї–µ–љ–љ—Г—О –њ—А–Є –Њ–њ–µ—А–∞—Ж–Є–Є –і–µ–Ї–Њ–Љ–њ—А–µ—Б—Б–Є–Є –Ј–∞–њ—П—Б—В–љ–Њ–≥–Њ –Ї–∞–љ–∞–ї–∞.
–С–Є–Њ–њ—Б–Є–є–љ—Л–є –Љ–∞—В–µ—А–Є–∞–ї –і–ї—П –≤—Л—П–≤–ї–µ–љ–Є—П –∞–Љ–Є–ї–Њ–Є–і–∞ –љ–µ–Њ–±—Е–Њ–і–Є–Љ–Њ –Њ–Ї—А–∞—И–Є–≤–∞—В—М –Ї–Њ–љ–≥–Њ –Ї—А–∞—Б–љ—Л–Љ —Б¬†–њ–Њ—Б–ї–µ–і—Г—О—Й–µ–є –Љ–Є–Ї—А–Њ—Б–Ї–Њ–њ–Є–µ–є –≤¬†–њ–Њ–ї—П—А–Є–Ј–Њ–≤–∞–љ–љ–Њ–Љ —Б–≤–µ—В–µ –і–ї—П –≤—Л—П–≤–ї–µ–љ–Є—П —Б–њ–Њ—Б–Њ–±–љ–Њ—Б—В–Є –Ї¬†–і–≤–Њ–є–љ–Њ–Љ—Г –ї—Г—З–µ–њ—А–µ–ї–Њ–Љ–ї–µ–љ–Є—О. –°–Њ–≤—А–µ–Љ–µ–љ–љ–∞—П –Љ–Њ—А—Д–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–∞—П –і–Є–∞–≥–љ–Њ—Б—В–Є–Ї–∞ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ –≤–Ї–ї—О—З–∞–µ—В –љ–µ —В–Њ–ї—М–Ї–Њ –Њ–±–љ–∞—А—Г–ґ–µ–љ–Є–µ, –љ–Њ –Є¬†—В–Є–њ–Є—А–Њ–≤–∞–љ–Є–µ –∞–Љ–Є–ї–Њ–Є–і–∞, –њ–Њ—Б–Ї–Њ–ї—М–Ї—Г —В–Є–њ –∞–Љ–Є–ї–Њ–Є–і–∞ –Њ–њ—А–µ–і–µ–ї—П–µ—В —В–µ—А–∞–њ–µ–≤—В–Є—З–µ—Б–Ї—Г—О —В–∞–Ї—В–Є–Ї—Г. –Ф–ї—П —В–Є–њ–Є—А–Њ–≤–∞–љ–Є—П —З–∞—Б—В–Њ –њ—А–Є–Љ–µ–љ—П—О—В –њ—А–Њ–±—Г —Б¬†–њ–µ—А–Љ–∞–љ–≥–∞–љ–∞—В–Њ–Љ –Ї–∞–ї–Є—П. –Я—А–Є –Њ–±—А–∞–±–Њ—В–Ї–µ –Њ–Ї—А–∞—И–µ–љ–љ—Л—Е –Ї–Њ–љ–≥–Њ –Ї—А–∞—Б–љ—Л–Љ –њ—А–µ–њ–∞—А–∞—В–Њ–≤ 5%-–љ—Л–Љ —А–∞—Б—В–≤–Њ—А–Њ–Љ –њ–µ—А–Љ–∞–љ–≥–∞–љ–∞—В–∞ –Ї–∞–ї–Є—П –Р–Р-—В–Є–њ –∞–Љ–Є–ї–Њ–Є–і–∞ —В–µ—А—П–µ—В –Њ–Ї—А–∞—Б–Ї—Г –Є¬†—Г—В—А–∞—З–Є–≤–∞–µ—В —Б–≤–Њ–є—Б—В–≤–Њ –і–≤–Њ–є–љ–Њ–≥–Њ –ї—Г—З–µ–њ—А–µ–ї–Њ–Љ–ї–µ–љ–Є—П, —В–Њ–≥–і–∞ –Ї–∞–Ї AL-—В–Є–њ –∞–Љ–Є–ї–Њ–Є–і–∞ —Б–Њ—Е—А–∞–љ—П–µ—В –Є—Е. –Ш—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–љ–Є–µ —Й–µ–ї–Њ—З–љ–Њ–≥–Њ –≥—Г–∞–љ–Є–і–Є–љ–∞ –њ–Њ–Ј–≤–Њ–ї—П–µ—В –±–Њ–ї–µ–µ —В–Њ—З–љ–Њ –і–Є—Д—Д–µ—А–µ–љ—Ж–Є—А–Њ–≤–∞—В—М –Р–Р- –Є¬†AL-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј.
–Э–∞–Є–±–Њ–ї–µ–µ —Н—Д—Д–µ–Ї—В–Є–≤–љ—Л–Љ –Љ–µ—В–Њ–і–Њ–Љ —В–Є–њ–Є—А–Њ–≤–∞–љ–Є—П –∞–Љ–Є–ї–Њ–Є–і–∞ —Б–ї—Г–ґ–Є—В –Є–Љ–Љ—Г–љ–Њ–≥–Є—Б—В–Њ—Е–Є–Љ–Є—З–µ—Б–Ї–Њ–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ —Б¬†–њ—А–Є–Љ–µ–љ–µ–љ–Є–µ–Љ –∞–љ—В–Є—Б—Л–≤–Њ—А–Њ—В–Њ–Ї –Ї¬†–Њ—Б–љ–Њ–≤–љ—Л–Љ —В–Є–њ–∞–Љ –∞–Љ–Є–ї–Њ–Є–і–љ–Њ–≥–Њ –±–µ–ї–Ї–∞ (—Б–њ–µ—Ж–Є—Д–Є—З–µ—Б–Ї–Є–µ –∞–љ—В–Є—В–µ–ї–∞ –њ—А–Њ—В–Є–≤ –Р–Р-–±–µ–ї–Ї–∞, –ї–µ–≥–Ї–Є—Е —Ж–µ–њ–µ–є –Є–Љ–Љ—Г–љ–Њ–≥–ї–Њ–±—Г–ї–Є–љ–Њ–≤, —В—А–∞–љ—Б—В–Є—А–µ—В–Є–љ–∞ –Є¬†–±–µ—В–∞-2-–Љ–Є–Ї—А–Њ–≥–ї–Њ–±—Г–ї–Є–љ–∞) [3, 18].
–Ы–µ—З–µ–љ–Є–µ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞
–°–Њ–≥–ї–∞—Б–љ–Њ —Б–Њ–≤—А–µ–Љ–µ–љ–љ—Л–Љ –њ—А–µ–і—Б—В–∞–≤–ї–µ–љ–Є—П–Љ, —Ж–µ–ї—М—О —В–µ—А–∞–њ–Є–Є –ї—О–±–Њ–≥–Њ —В–Є–њ–∞ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ —П–≤–ї—П–µ—В—Б—П —Г–Љ–µ–љ—М—И–µ–љ–Є–µ –Ї–Њ–ї–Є—З–µ—Б—В–≤–∞ (–Є–ї–Є, –µ—Б–ї–Є –≤–Њ–Ј–Љ–Њ–ґ–љ–Њ, —Г–і–∞–ї–µ–љ–Є–µ) –±–µ–ї–Ї–Њ–≤-–њ—А–µ–і—И–µ—Б—В–≤–µ–љ–љ–Є–Ї–Њ–≤ –і–ї—П —В–Њ–≥–Њ, —З—В–Њ–±—Л –Ј–∞–Љ–µ–і–ї–Є—В—М –Є–ї–Є –њ—А–Є–Њ—Б—В–∞–љ–Њ–≤–Є—В—М –њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є–µ –±–Њ–ї–µ–Ј–љ–Є. –Э–µ–±–ї–∞–≥–Њ–њ—А–Є—П—В–љ—Л–є –њ—А–Њ–≥–љ–Њ–Ј –њ—А–Є –µ—Б—В–µ—Б—В–≤–µ–љ–љ–Њ–Љ —В–µ—З–µ–љ–Є–Є –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ –Њ–њ—А–∞–≤–і—Л–≤–∞–µ—В –њ—А–Є–Љ–µ–љ–µ–љ–Є–µ –љ–µ–Ї–Њ—В–Њ—А—Л—Е –∞–≥—А–µ—Б—Б–Є–≤–љ—Л—Е –ї–µ–Ї–∞—А—Б—В–≤–µ–љ–љ—Л—Е —А–µ–ґ–Є–Љ–Њ–≤ –Є–ї–Є –і—А—Г–≥–Є—Е —А–∞–і–Є–Ї–∞–ї—М–љ—Л—Е –Љ–µ—А. –Ъ–ї–Є–љ–Є—З–µ—Б–Ї–Њ–µ —Г–ї—Г—З—И–µ–љ–Є–µ, –і–Њ—Б—В–Є–≥–∞–µ–Љ–Њ–µ —Б¬†–њ–Њ–Љ–Њ—Й—М—О —Н—В–Є—Е –≤–Є–і–Њ–≤ –ї–µ—З–µ–љ–Є—П, –≤–Ї–ї—О—З–∞–µ—В —Б—В–∞–±–Є–ї–Є–Ј–∞—Ж–Є—О –Є–ї–Є –≤–Њ—Б—Б—В–∞–љ–Њ–≤–ї–µ–љ–Є–µ —Д—Г–љ–Ї—Ж–Є–Є –ґ–Є–Ј–љ–µ–љ–љ–Њ –≤–∞–ґ–љ—Л—Е –Њ—А–≥–∞–љ–Њ–≤, –∞¬†—В–∞–Ї–ґ–µ –њ—А–µ–і–Њ—В–≤—А–∞—Й–µ–љ–Є–µ —Д—Г–љ–Ї—Ж–Є–Њ–љ–∞–ї—М–љ—Л—Е –љ–∞—А—Г—И–µ–љ–Є–є, —З—В–Њ —Г–≤–µ–ї–Є—З–Є–≤–∞–µ—В –њ—А–Њ–і–Њ–ї–ґ–Є—В–µ–ї—М–љ–Њ—Б—В—М –ґ–Є–Ј–љ–Є –±–Њ–ї—М–љ—Л—Е. –Ь–Њ—А—Д–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–Љ –Ї—А–Є—В–µ—А–Є–µ–Љ —Н—Д—Д–µ–Ї—В–Є–≤–љ–Њ—Б—В–Є –ї–µ—З–µ–љ–Є—П —Б—З–Є—В–∞—О—В —Г–Љ–µ–љ—М—И–µ–љ–Є–µ –Њ—В–ї–Њ–ґ–µ–љ–Є–є –∞–Љ–Є–ї–Њ–Є–і–∞ –≤¬†—В–Ї–∞–љ—П—Е, —З—В–Њ –≤¬†–љ–∞—Б—В–Њ—П—Й–µ–µ –≤—А–µ–Љ—П –Љ–Њ–ґ–љ–Њ –Њ—Ж–µ–љ–Є—В—М, –њ—А–Є–Љ–µ–љ—П—П —А–∞–і–Є–Њ–Є–Ј–Њ—В–Њ–њ–љ—Г—О —Б—Ж–Є–љ—В–Є–≥—А–∞—Д–Є—О —Б¬†—Б—Л–≤–Њ—А–Њ—В–Њ—З–љ—Л–Љ –†-–Ї–Њ–Љ–њ–Њ–љ–µ–љ—В–Њ–Љ –∞–Љ–Є–ї–Њ–Є–і–∞. –Ъ—А–Њ–Љ–µ –Њ—Б–љ–Њ–≤–љ—Л—Е —В–µ—А–∞–њ–µ–≤—В–Є—З–µ—Б–Ї–Є—Е —А–µ–ґ–Є–Љ–Њ–≤, –ї–µ—З–µ–љ–Є–µ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ –і–Њ–ї–ґ–љ–Њ –≤–Ї–ї—О—З–∞—В—М —Б–Є–Љ–њ—В–Њ–Љ–∞—В–Є—З–µ—Б–Ї–Є–µ –Љ–µ—В–Њ–і—Л, –љ–∞–њ—А–∞–≤–ї–µ–љ–љ—Л–µ –љ–∞ —Г–Љ–µ–љ—М—И–µ–љ–Є–µ –≤—Л—А–∞–ґ–µ–љ–љ–Њ—Б—В–Є –Ј–∞—Б—В–Њ–є–љ–Њ–є –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В–Є –Ї—А–Њ–≤–Њ–Њ–±—А–∞—Й–µ–љ–Є—П, –∞—А–Є—В–Љ–Є–Є, –Њ—В–µ—З–љ–Њ–≥–Њ —Б–Є–љ–і—А–Њ–Љ–∞, –Ї–Њ—А—А–µ–Ї—Ж–Є—О –∞—А—В–µ—А–Є–∞–ї—М–љ–Њ–є –≥–Є–њ–µ—А- –Є¬†–≥–Є–њ–Њ—В–Њ–љ–Є–Є.
–¶–µ–ї—М—О —В–µ—А–∞–њ–Є–Є –≤—В–Њ—А–Є—З–љ–Њ–≥–Њ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ —Б–ї—Г–ґ–Є—В –њ–Њ–і–∞–≤–ї–µ–љ–Є–µ –њ—А–Њ–і—Г–Ї—Ж–Є–Є –±–µ–ї–Ї–∞-–њ—А–µ–і—И–µ—Б—В–≤–µ–љ–љ–Є–Ї–∞ SAA [19, 20], —З—В–Њ –і–Њ—Б—В–Є–≥–∞–µ—В—Б—П –ї–µ—З–µ–љ–Є–µ–Љ —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–≥–Њ –≤–Њ—Б–њ–∞–ї–µ–љ–Є—П, –≤¬†—В–Њ–Љ —З–Є—Б–ї–µ –Є¬†—Е–Є—А—Г—А–≥–Є—З–µ—Б–Ї–Є–Љ (—Б–µ–Ї–≤–µ—Б—В—А—Н–Ї—В–Њ–Љ–Є—П –њ—А–Є –Њ—Б—В–µ–Њ–Љ–Є–µ–ї–Є—В–µ, —Г–і–∞–ї–µ–љ–Є–µ –і–Њ–ї–Є –ї–µ–≥–Ї–Њ–≥–Њ –њ—А–Є –±—А–Њ–љ—Е–Њ—Н–Ї—В–∞—В–Є—З–µ—Б–Ї–Њ–є –±–Њ–ї–µ–Ј–љ–Є, –Њ–њ—Г—Е–Њ–ї–Є, —В—Г–±–µ—А–Ї—Г–ї–µ–Ј–µ).
–Ю—Б–Њ–±–Њ–µ –Ј–љ–∞—З–µ–љ–Є–µ –≤¬†–љ–∞—Б—В–Њ—П—Й–µ–µ –≤—А–µ–Љ—П –њ—А–Є–і–∞—О—В —В–µ—А–∞–њ–Є–Є —А–µ–≤–Љ–∞—В–Њ–Є–і–љ–Њ–≥–Њ –∞—А—В—А–Є—В–∞, —Г—З–Є—В—Л–≤–∞—П –µ–≥–Њ –ї–Є–і–Є—А—Г—О—Й–µ–µ –њ–Њ–ї–Њ–ґ–µ–љ–Є–µ —Б—А–µ–і–Є –њ—А–Є—З–Є–љ –≤—В–Њ—А–Є—З–љ–Њ–≥–Њ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞. –С–∞–Ј–Є—Б–љ–∞—П —В–µ—А–∞–њ–Є—П —А–µ–≤–Љ–∞—В–Њ–Є–і–љ–Њ–≥–Њ –∞—А—В—А–Є—В–∞ —Ж–Є—В–Њ—Б—В–∞—В–Є—З–µ—Б–Ї–Є–Љ–Є –ї–µ–Ї–∞—А—Б—В–≤–µ–љ–љ—Л–Љ–Є —Б—А–µ–і—Б—В–≤–∞–Љ–Є (–Љ–µ—В–Њ—В—А–µ–Ї—Б–∞—В–Њ–Љ, —Ж–Є–Ї–ї–Њ—Д–Њ—Б—Д–∞–Љ–Є–і–Њ–Љ, —Е–ї–Њ—А–∞–Љ–±—Г—Ж–Є–ї–Њ–Љ) –Є–ї–Є —Б–Њ–≤—А–µ–Љ–µ–љ–љ—Л–Љ–Є –∞–љ—В–Є—Ж–Є—В–Њ–Ї–Є–љ–Њ–≤—Л–Љ–Є —Б—А–µ–і—Б—В–≤–∞–Љ–Є (–±–ї–Њ–Ї–∞—В–Њ—А—Л —Н—Д—Д–µ–Ї—В–Њ–≤ —Д–∞–Ї—В–Њ—А–∞ –љ–µ–Ї—А–Њ–Ј–∞ –Њ–њ—Г—Е–Њ–ї–Є –∞–ї—М—Д–∞, –Є–љ—В–µ—А–ї–µ–є–Ї–Є–љ—Л 1¬†–Є¬†6, –±–ї–Њ–Ї–∞—В–Њ—А—Л –°D20¬†–Т-–ї–Є–Љ—Д–Њ—Ж–Є—В–Њ–≤ –Є¬†–і—А.), –љ–∞–Ј–љ–∞—З–∞–µ–Љ–∞—П –љ–∞ –і–ї–Є—В–µ–ї—М–љ—Л–є —Б—А–Њ–Ї (–±–Њ–ї–µ–µ 12¬†–Љ–µ—Б.), —Г–Љ–µ–љ—М—И–∞–µ—В —А–Є—Б–Ї —А–∞–Ј–≤–Є—В–Є—П –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞. –£¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†—Г–ґ–µ —А–∞–Ј–≤–Є–≤—И–Є–Љ—Б—П –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–Њ–Љ —Н—В–Њ –ї–µ—З–µ–љ–Є–µ –њ–Њ–Ј–≤–Њ–ї—П–µ—В –≤¬†–±–Њ–ї—М—И–Є–љ—Б—В–≤–µ —Б–ї—Г—З–∞–µ–≤ —Г–Љ–µ–љ—М—И–Є—В—М –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є–µ –њ—А–Њ—П–≤–ї–µ–љ–Є—П –∞–Љ–Є–ї–Њ–Є–і–љ–Њ–є –љ–µ—Д—А–Њ–њ–∞—В–Є–Є. –Т¬†—А–µ–Ј—Г–ї—М—В–∞—В–µ –њ–Њ–і–Њ–±–љ–Њ–є —В–µ—А–∞–њ–Є–Є –Њ—В–Љ–µ—З–∞—О—В —Б–љ–Є–ґ–µ–љ–Є–µ –≤—Л—А–∞–ґ–µ–љ–љ–Њ—Б—В–Є –њ—А–Њ—В–µ–Є–љ—Г—А–Є–Є, –Ї—Г–њ–Є—А–Њ–≤–∞–љ–Є–µ –љ–µ—Д—А–Њ—В–Є—З–µ—Б–Ї–Њ–≥–Њ —Б–Є–љ–і—А–Њ–Љ–∞, —Б—В–∞–±–Є–ї–Є–Ј–∞—Ж–Є—О —Д—Г–љ–Ї—Ж–Є–Є –њ–Њ—З–µ–Ї. –£¬†—З–∞—Б—В–Є –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Г–і–∞–µ—В—Б—П –њ—А–µ–і–Њ—В–≤—А–∞—В–Є—В—М —А–∞–Ј–≤–Є—В–Є–µ –•–Я–Э –Є–ї–Є –Ј–∞–Љ–µ–і–ї–Є—В—М –µ–µ –њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є–µ, —З—В–Њ —Б—Г—Й–µ—Б—В–≤–µ–љ–љ–Њ —Г–ї—Г—З—И–∞–µ—В –њ—А–Њ–≥–љ–Њ–Ј. –Ю–± —Н—Д—Д–µ–Ї—В–Є–≤–љ–Њ—Б—В–Є –ї–µ—З–µ–љ–Є—П —Б–≤–Є–і–µ—В–µ–ї—М—Б—В–≤—Г–µ—В —В–∞–Ї–ґ–µ –љ–Њ—А–Љ–∞–ї–Є–Ј–∞—Ж–Є—П –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є–Є –°-—А–µ–∞–Ї—В–Є–≤–љ–Њ–≥–Њ –±–µ–ї–Ї–∞ –≤¬†–Ї—А–Њ–≤–Є.
–°—А–µ–і—Б—В–≤–Њ–Љ –≤—Л–±–Њ—А–∞ –і–ї—П –ї–µ—З–µ–љ–Є—П –Р–Р-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ –њ—А–Є –њ–µ—А–Є–Њ–і–Є—З–µ—Б–Ї–Њ–є –±–Њ–ї–µ–Ј–љ–Є —Б–ї—Г–ґ–Є—В –Ї–Њ–ї—Е–Є—Ж–Є–љ. –Я—А–Є –µ–≥–Њ –њ–Њ—Б—В–Њ—П–љ–љ–Њ–Љ –њ—А–Є–µ–Љ–µ –Љ–Њ–ґ–љ–Њ –њ–Њ–ї–љ–Њ—Б—В—М—О –Є–Ј–±–µ–ґ–∞—В—М —А–µ—Ж–Є–і–Є–≤–Є—А–Њ–≤–∞–љ–Є—П –њ—А–Є—Б—В—Г–њ–Њ–≤ —Г¬†–±–Њ–ї—М—И–Є–љ—Б—В–≤–∞ –±–Њ–ї—М–љ—Л—Е, –њ—А–µ–і–Њ—В–≤—А–∞—Й–∞—П —Г¬†–љ–Є—Е —В–∞–Ї–ґ–µ —А–∞–Ј–≤–Є—В–Є–µ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞. –Я—А–Є —А–∞–Ј–≤–Є–≤—И–µ–Љ—Б—П –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–µ –і–ї–Є—В–µ–ї—М–љ—Л–є, –≤–Њ–Ј–Љ–Њ–ґ–љ–Њ –њ–Њ–ґ–Є–Ј–љ–µ–љ–љ—Л–є, –њ—А–Є–µ–Љ –Ї–Њ–ї—Е–Є—Ж–Є–љ–∞ –≤¬†–і–Њ–Ј–µ 1,8вАУ2¬†–Љ–≥/—Б—Г—В –њ—А–Є–≤–Њ–і–Є—В –Ї¬†—А–µ–Љ–Є—Б—Б–Є–Є, –≤—Л—А–∞–ґ–∞—О—Й–µ–є—Б—П –≤¬†–ї–Є–Ї–≤–Є–і–∞—Ж–Є–Є –љ–µ—Д—А–Њ—В–Є—З–µ—Б–Ї–Њ–≥–Њ —Б–Є–љ–і—А–Њ–Љ–∞, —Г–Љ–µ–љ—М—И–µ–љ–Є–Є –Є–ї–Є –Є—Б—З–µ–Ј–љ–Њ–≤–µ–љ–Є–Є –њ—А–Њ—В–µ–Є–љ—Г—А–Є–Є —Г¬†–±–Њ–ї—М–љ—Л—Е —Б¬†–љ–Њ—А–Љ–∞–ї—М–љ–Њ–є —Д—Г–љ–Ї—Ж–Є–µ–є –њ–Њ—З–µ–Ї. –Я—А–Є –љ–∞–ї–Є—З–Є–Є –•–Я–Э –љ–∞—З–∞–ї—М–љ—Г—О –і–Њ–Ј—Г –ї–µ–Ї–∞—А—Б—В–≤–µ–љ–љ—Л—Е —Б—А–µ–і—Б—В–≤ —Г–Љ–µ–љ—М—И–∞—О—В –≤¬†–Ј–∞–≤–Є—Б–Є–Љ–Њ—Б—В–Є –Њ—В –≤–µ–ї–Є—З–Є–љ—Л –Ї–ї—Г–±–Њ—З–Ї–Њ–≤–Њ–є —Д–Є–ї—М—В—А–∞—Ж–Є–Є, —Е–Њ—В—П –≤¬†—Б–ї—Г—З–∞–µ —Б–љ–Є–ґ–µ–љ–Є—П –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є–Є –Ї—А–µ–∞—В–Є–љ–Є–љ–∞ –≤¬†–Ї—А–Њ–≤–Є –≤–Њ–Ј–Љ–Њ–ґ–љ–Њ –њ–Њ–≤—Л—И–µ–љ–Є–µ –і–Њ–Ј—Л –і–Њ —Б—В–∞–љ–і–∞—А—В–љ–Њ–є. –Ъ–Њ–ї—Е–Є—Ж–Є–љ —В–∞–Ї–ґ–µ –њ—А–µ–і–Њ—В–≤—А–∞—Й–∞–µ—В —А–µ—Ж–Є–і–Є–≤ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ –≤¬†—В—А–∞–љ—Б–њ–ї–∞–љ—В–Є—А–Њ–≤–∞–љ–љ–Њ–є –њ–Њ—З–Ї–µ. –С–Њ–ї—М–љ—Л–µ —Е–Њ—А–Њ—И–Њ –њ–µ—А–µ–љ–Њ—Б—П—В –і–∞–љ–љ–Њ–µ –ї–µ–Ї–∞—А—Б—В–≤–µ–љ–љ–Њ–µ —Б—А–µ–і—Б—В–≤–Њ. –Х—Б–ї–Є —Н—Д—Д–µ–Ї—В–Є–≤–љ–Њ—Б—В—М –Ї–Њ–ї—Е–Є—Ж–Є–љ–∞ –њ—А–Є –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–µ –≤¬†—А–∞–Љ–Ї–∞—Е –њ–µ—А–Є–Њ–і–Є—З–µ—Б–Ї–Њ–є –±–Њ–ї–µ–Ј–љ–Є –љ–µ –≤—Л–Ј—Л–≤–∞–µ—В —Б–Њ–Љ–љ–µ–љ–Є–є, —В–Њ –Є–Љ–µ—О—В—Б—П –ї–Є—И—М –µ–і–Є–љ–Є—З–љ—Л–µ —А–∞–±–Њ—В—Л, —Б–≤–Є–і–µ—В–µ–ї—М—Б—В–≤—Г—О—Й–Є–µ –Њ¬†–µ–≥–Њ —Г—Б–њ–µ—И–љ–Њ–Љ –њ—А–Є–Љ–µ–љ–µ–љ–Є–Є —Г¬†–±–Њ–ї—М–љ—Л—Е –≤—В–Њ—А–Є—З–љ—Л–Љ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–Њ–Љ. –Ъ—А–Њ–Љ–µ –Ї–Њ–ї—Е–Є—Ж–Є–љ–∞, –њ—А–Є –Р–Р-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–µ –њ—А–Є–Љ–µ–љ—П—О—В –і–Є–Љ–µ—В–Є–ї—Б—Г–ї—М—Д–Њ–Ї—Б–Є–і, –≤—Л–Ј—Л–≤–∞—О—Й–Є–є —А–µ–Ј–Њ—А–±—Ж–Є—О –∞–Љ–Є–ї–Њ–Є–і–љ—Л—Е –Њ—В–ї–Њ–ґ–µ–љ–Є–є. –Ю–і–љ–∞–Ї–Њ –Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–љ–Є–µ –µ–≥–Њ –≤¬†–≤—Л—Б–Њ–Ї–Є—Е –і–Њ–Ј–∞—Е (–љ–µ –Љ–µ–љ–µ–µ 10¬†–≥/—Б—Г—В), –љ–µ–Њ–±—Е–Њ–і–Є–Љ—Л—Е –і–ї—П —Г—Б–њ–µ—И–љ–Њ–≥–Њ –ї–µ—З–µ–љ–Є—П, –Њ–≥—А–∞–љ–Є—З–µ–љ–Њ –Є–Ј-–Ј–∞ –Ї—А–∞–є–љ–µ –љ–µ–њ—А–Є—П—В–љ–Њ–≥–Њ –Ј–∞–њ–∞—Е–∞.
–Я—А–Є AL-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–µ, –Ї–∞–Ї –Є¬†–њ—А–Є –Љ–Є–µ–ї–Њ–Љ–љ–Њ–є –±–Њ–ї–µ–Ј–љ–Є, –ї–µ—З–µ–љ–Є–µ –і–Њ–ї–ґ–љ–Њ –±—Л—В—М –љ–∞–њ—А–∞–≤–ї–µ–љ–Њ –љ–∞ –њ–Њ–і–∞–≤–ї–µ–љ–Є–µ –њ—А–Њ–ї–Є—Д–µ—А–∞—Ж–Є–Є –Ї–ї–Њ–љ–∞ –њ–ї–∞–Ј–Љ–∞—В–Є—З–µ—Б–Ї–Є—Е –Ї–ї–µ—В–Њ–Ї –і–ї—П —Г–Љ–µ–љ—М—И–µ–љ–Є—П –њ—А–Њ–і—Г–Ї—Ж–Є–Є –Ы–¶–Ш. –Э–∞–Є–±–Њ–ї—М—И–Є–є –Њ–њ—Л—В –ї–µ—З–µ–љ–Є—П –љ–∞–Ї–Њ–њ–ї–µ–љ –і–ї—П –Љ–µ–ї—Д–∞–ї–∞–љ–∞ –≤¬†—Б–Њ—З–µ—В–∞–љ–Є–Є —Б¬†–њ—А–µ–і–љ–Є–Ј–Њ–ї–Њ–љ–Њ–Љ [21]. –Ы–µ—З–µ–љ–Є–µ –њ—А–Њ–і–Њ–ї–ґ–∞—О—В 7-–і–љ–µ–≤–љ—Л–Љ–Є –Ї—Г—А—Б–∞–Љ–Є —Б¬†–Є–љ—В–µ—А–≤–∞–ї–Њ–Љ 4вАУ6¬†–љ–µ–і–µ–ї—М –љ–∞ –њ—А–Њ—В—П–ґ–µ–љ–Є–Є 12вАУ24¬†–Љ–µ—Б—П—Ж–µ–≤. –Ф–Њ–Ј–∞ –Љ–µ–ї—Д–∞–ї–∞–љ–∞ —Б–Њ—Б—В–∞–≤–ї—П–µ—В 0,15¬†–Љ–≥/–Ї–≥ –Љ–∞—Б—Б—Л —В–µ–ї–∞ –≤¬†—Б—Г—В–Ї–Є, –њ—А–µ–і–љ–Є–Ј–Њ–ї–Њ–љ–∞¬†вАУ 0,8¬†–Љ–≥/–Ї–≥ –Љ–∞—Б—Б—Л —В–µ–ї–∞ –≤¬†—Б—Г—В–Ї–Є. –£¬†–±–Њ–ї—М–љ—Л—Е —Б¬†–•–Я–Э (—Б–Ї–Њ—А–Њ—Б—В—М –Ї–ї—Г–±–Њ—З–Ї–Њ–≤–Њ–є —Д–Є–ї—М—В—А–∞—Ж–Є–Є –Љ–µ–љ–µ–µ 40¬†–Љ–ї/–Љ–Є–љ) –і–Њ–Ј—Г –Љ–µ–ї—Д–∞–ї–∞–љ–∞ —Г–Љ–µ–љ—М—И–∞—О—В –љ–∞ 50%. –≠—В–∞ —Б—Е–µ–Љ–∞ –ї–µ—З–µ–љ–Є—П —Е–∞—А–∞–Ї—В–µ—А–Є–Ј—Г–µ—В—Б—П –љ–Є–Ј–Ї–Є–Љ —А–Є—Б–Ї–Њ–Љ –Њ—Б–ї–Њ–ґ–љ–µ–љ–Є–є —Е–Є–Љ–Є–Њ—В–µ—А–∞–њ–Є–Є –Є¬†–Љ–Њ–ґ–µ—В –њ—А–Є–Љ–µ–љ—П—В—М—Б—П —Г¬†–∞–±—Б–Њ–ї—О—В–љ–Њ–≥–Њ –±–Њ–ї—М—И–Є–љ—Б—В–≤–∞ –±–Њ–ї—М–љ—Л—Е, –і–∞–ґ–µ –њ—А–Є –љ–∞–ї–Є—З–Є–Є —В—П–ґ–µ–ї—Л—Е –≤–Є—Б—Ж–µ—А–∞–ї—М–љ—Л—Е –њ—А–Њ—П–≤–ї–µ–љ–Є–є –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞. –Ю–і–љ–∞–Ї–Њ —Н—Д—Д–µ–Ї—В —Н—В–Њ–є —В–µ—А–∞–њ–Є–Є –і–Њ—Б—В–Є–≥–∞–µ—В—Б—П —В–Њ–ї—М–Ї–Њ —Г¬†18вАУ30% –±–Њ–ї—М–љ—Л—Е.
–Т –љ–∞—Б—В–Њ—П—Й–µ–µ –≤—А–µ–Љ—П —А–∞–Ј—А–∞–±–Њ—В–∞–љ —Н—Д—Д–µ–Ї—В–Є–≤–љ—Л–є –Є–Љ–Љ—Г–љ–Њ—Е–Є–Љ–Є—З–µ—Б–Ї–Є–є –Љ–µ—В–Њ–і –Њ—Ж–µ–љ–Ї–Є —А–µ–Љ–Є—Б—Б–Є–Є –њ–ї–∞–Ј–Љ–Њ–Ї–ї–µ—В–Њ—З–љ–Њ–є –і–Є—Б–Ї—А–∞–Ј–Є–Є —Г¬†–±–Њ–ї—М–љ—Л—Е –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–Њ–Љ¬†вАУ –Њ–њ—А–µ–і–µ–ї–µ–љ–Є–µ —Б–≤–Њ–±–Њ–і–љ—Л—Е –Ы–¶–Ш –≤¬†—Б—Л–≤–Њ—А–Њ—В–Ї–µ (Freelite). –†–∞–љ–µ–µ –њ—А–Є–Љ–µ–љ—П–≤—И–Є–µ—Б—П –Љ–µ—В–Њ–і—Л –љ–µ –њ–Њ–Ј–≤–Њ–ї—П–ї–Є –Њ—В–ї–Є—З–Є—В—М –њ—Г–ї —Б–≤–Њ–±–Њ–і–љ—Л—Е –Ы–¶–Ш –Њ—В –њ—Г–ї–∞ —Б–≤—П–Ј–∞–љ–љ—Л—Е —Б¬†–Є–Љ–Љ—Г–љ–Њ–≥–ї–Њ–±—Г–ї–Є–љ–∞–Љ–Є, —З—В–Њ —А–µ–Ј–Ї–Њ —Б–љ–Є–ґ–∞–ї–Њ —З—Г–≤—Б—В–≤–Є—В–µ–ї—М–љ–Њ—Б—В—М –Љ–µ—В–Њ–і–Њ–≤. –С–ї–∞–≥–Њ–і–∞—А—П –Љ–µ—В–Њ–і—Г Freelite, –Ї–Њ—В–Њ—А—Л–є –±–∞–Ј–Є—А—Г–µ—В—Б—П –љ–∞ –њ—А–Є–Љ–µ–љ–µ–љ–Є–Є –∞–љ—В–Є—В–µ–ї –Ї¬†—Б–Ї—А—Л—В—Л–Љ —Н–њ–Є—В–Њ–њ–∞–Љ –Ы–¶–Ш, –њ–Њ—П–≤–Є–ї–∞—Б—М –≤–Њ–Ј–Љ–Њ–ґ–љ–Њ—Б—В—М –Ї–Њ–ї–Є—З–µ—Б—В–≤–µ–љ–љ–Њ–є –Њ—Ж–µ–љ–Ї–Є –њ—Г–ї–∞ —Б–≤–Њ–±–Њ–і–љ—Л—Е –Ы–¶–Ш, –≤–њ–µ—А–≤—Л–µ –±—Л–ї–Є —Б—Д–Њ—А–Љ—Г–ї–Є—А–Њ–≤–∞–љ—Л –Ї—А–Є—В–µ—А–Є–Є –љ–Њ—А–Љ–∞–ї—М–љ–Њ–є –њ—А–Њ–і—Г–Ї—Ж–Є–Є –Ы–¶–Ш. –Т¬†–љ–∞—Б—В–Њ—П—Й–µ–µ –≤—А–µ–Љ—П –≤–Њ –≤—Б–µ–Љ –Љ–Є—А–µ –Љ–µ—В–Њ–і Freelite —Б—В–∞–ї —Б—В–∞–љ–і–∞—А—В–Њ–Љ –Њ—Ж–µ–љ–Ї–Є —Н—Д—Д–µ–Ї—В–Є–≤–љ–Њ—Б—В–Є —В–µ—А–∞–њ–Є–Є –Є¬†–Љ–Њ–љ–Є—В–Њ—А–Є—А–Њ–≤–∞–љ–Є—П —В–µ—З–µ–љ–Є—П –±–Њ–ї–µ–Ј–љ–Є —Г¬†–±–Њ–ї—М–љ—Л—Е –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–Њ–Љ [22, 23].
–° —Ж–µ–ї—М—О –њ–Њ–≤—Л—И–µ–љ–Є—П —Н—Д—Д–µ–Ї—В–Є–≤–љ–Њ—Б—В–Є –ї–µ—З–µ–љ–Є—П —И–Є—А–Њ–Ї–Њ –њ—А–Є–Љ–µ–љ—П–ї–Є—Б—М –њ–Њ–њ—Л—В–Ї–Є –ї–µ—З–µ–љ–Є—П –±–Њ–ї–µ–µ –∞–≥—А–µ—Б—Б–Є–≤–љ—Л–Љ–Є —Б—Е–µ–Љ–∞–Љ–Є —Е–Є–Љ–Є–Њ—В–µ—А–∞–њ–Є–Є —Б¬†–≤–Ї–ї—О—З–µ–љ–Є–µ–Љ –≤–Є–љ–Ї—А–Є—Б—В–Є–љ–∞, –і–Њ–Ї—Б–Њ—А—Г–±–Є—Ж–Є–љ–∞, —Ж–Є–Ї–ї–Њ—Д–Њ—Б—Д–∞–љ–∞, –Љ–µ–ї—Д–∞–ї–∞–љ–∞, –і–µ–Ї—Б–∞–Љ–µ—В–∞–Ј–Њ–љ–∞ –≤¬†—А–∞–Ј–љ—Л—Е –Ї–Њ–Љ–±–Є–љ–∞—Ж–Є—П—Е. –Э–∞–Є–±–Њ–ї—М—И–∞—П —Н—Д—Д–µ–Ї—В–Є–≤–љ–Њ—Б—В—М –і–Њ—Б—В–Є–≥–љ—Г—В–∞ –њ—А–Є —Е–Є–Љ–Є–Њ—В–µ—А–∞–њ–Є–Є —Б¬†–Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–љ–Є–µ–Љ –≤—Л—Б–Њ–Ї–Є—Е –і–Њ–Ј. –Ґ–∞–Ї, –≤¬†1996¬†–≥. –±—Л–ї–Є –Њ–њ—Г–±–ї–Є–Ї–Њ–≤–∞–љ—Л –њ—А–µ–і–≤–∞—А–Є—В–µ–ї—М–љ—Л–µ —А–µ–Ј—Г–ї—М—В–∞—В—Л (R.L. Comenzo –Є¬†—Б–Њ–∞–≤—В.) –ї–µ—З–µ–љ–Є—П 5¬†–±–Њ–ї—М–љ—Л—Е AL-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–Њ–Љ –≤–љ—Г—В—А–Є–≤–µ–љ–љ—Л–Љ–Є –≤–ї–Є–≤–∞–љ–Є—П–Љ–Є –Љ–µ–ї—Д–∞–ї–∞–љ–∞ –≤¬†–і–Њ–Ј–µ 200¬†–Љ–≥/–Љ¬≤¬†–њ–Њ–≤–µ—А—Е–љ–Њ—Б—В–Є —В–µ–ї–∞ —Б¬†–њ–Њ—Б–ї–µ–і—Г—О—Й–Є–Љ –≤–≤–µ–і–µ–љ–Є–µ–Љ –∞—Г—В–Њ–ї–Њ–≥–Є—З–љ—Л—Е —Б—В–≤–Њ–ї–Њ–≤—Л—Е –Ї–ї–µ—В–Њ–Ї (CD34+) –Ї—А–Њ–≤–Є [24]. –Р—Г—В–Њ–ї–Њ–≥–Є—З–љ—Л–µ —Б—В–≤–Њ–ї–Њ–≤—Л–µ –Ї–ї–µ—В–Ї–Є –њ–Њ–ї—Г—З–∞—О—В –Љ–µ—В–Њ–і–Њ–Љ –ї–µ–є–Ї–Њ—Д–µ—А–µ–Ј–∞ –Ї—А–Њ–≤–Є –±–Њ–ї—М–љ–Њ–≥–Њ –њ–Њ—Б–ї–µ –њ—А–µ–і–≤–∞—А–Є—В–µ–ї—М–љ–Њ–є –Є—Е –Љ–Њ–±–Є–ї–Є–Ј–∞—Ж–Є–Є –Є–Ј –Ї–Њ—Б—В–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞ –њ–Њ–і –≤–ї–Є—П–љ–Є–µ–Љ –≤–≤–µ–і–µ–љ–љ–Њ–≥–Њ –Є–Ј–≤–љ–µ –≥—А–∞–љ—Г–ї–Њ—Ж–Є—В–∞—А–љ–Њ–≥–Њ –Ї–Њ–ї–Њ–љ–Є–µ—Б—В–Є–Љ—Г–ї–Є—А—Г—О—Й–µ–≥–Њ —Д–∞–Ї—В–Њ—А–∞. –Ю–і–љ–∞–Ї–Њ —В—П–ґ–µ–ї—Л–є –∞–≥—А–∞–љ—Г–ї–Њ—Ж–Є—В–Њ–Ј –Є¬†–і—А—Г–≥–Є–µ –Њ—Б–ї–Њ–ґ–љ–µ–љ–Є—П —Н—В–Њ–є —В–µ—А–∞–њ–Є–Є —Б—Г—Й–µ—Б—В–≤–µ–љ–љ–Њ –Њ–≥—А–∞–љ–Є—З–Є–≤–∞—О—В –њ—А–Є–Љ–µ–љ–µ–љ–Є–µ —В–µ—А–∞–њ–Є–Є —Б–≤–µ—А—Е–≤—Л—Б–Њ–Ї–Є–Љ–Є –і–Њ–Ј–∞–Љ–Є –Љ–µ–ї—Д–∞–ї–∞–љ–∞, –≤¬†—З–∞—Б—В–љ–Њ—Б—В–Є, —Г¬†–±–Њ–ї—М–љ—Л—Е —Б¬†–љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В—М—О –Ї—А–Њ–≤–Њ–Њ–±—А–∞—Й–µ–љ–Є—П. –Т¬†–њ–Њ—Б–ї–µ–і–љ–Є–µ –≥–Њ–і—Л –њ–Њ–Ї–∞–Ј–∞–љ–∞ –≤—Л—Б–Њ–Ї–∞—П —Н—Д—Д–µ–Ї—В–Є–≤–љ–Њ—Б—В—М –њ—А–Є –Њ—В–љ–Њ—Б–Є—В–µ–ї—М–љ–Њ —Г–і–Њ–≤–ї–µ—В–≤–Њ—А–Є—В–µ–ї—М–љ–Њ–є –њ–µ—А–µ–љ–Њ—Б–Є–Љ–Њ—Б—В–Є —Б—Е–µ–Љ—Л ¬Ђ–Љ–µ–ї—Д–∞–ї–∞–љ (0,15¬†–Љ–≥/–Ї–≥ 4¬†–і–љ—П) –≤¬†—Б–Њ—З–µ—В–∞–љ–Є–Є —Б¬†–і–µ–Ї—Б–∞–Љ–µ—В–∞–Ј–Њ–љ–Њ–Љ (20¬†–Љ–≥/—Б—Г—В –≤¬†1вАУ4-–µ –Є¬†9вАУ12-–µ –і–љ–Є)¬ї –µ–ґ–µ–Љ–µ—Б—П—З–љ—Л–Љ–Є –Ї—Г—А—Б–∞–Љ–Є [25]. –Т¬†–љ–∞—Б—В–Њ—П—Й–µ–µ –≤—А–µ–Љ—П —Н—В–∞ —Б—Е–µ–Љ–∞ —Б—В–∞–ї–∞ –њ–µ—А–≤–Њ–є –ї–Є–љ–Є–µ–є —В–µ—А–∞–њ–Є–Є AL-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞. –Т¬†–Ї–∞—З–µ—Б—В–≤–µ –≤—В–Њ—А–Њ–є –ї–Є–љ–Є–Є —В–µ—А–∞–њ–Є–Є –Њ–±—Б—Г–ґ–і–∞—О—В—Б—П —Б—Е–µ–Љ—Л —Б¬†–≤–Ї–ї—О—З–µ–љ–Є–µ–Љ –±–Њ—А—В–µ–Ј–Њ–Љ–Є–±–∞ –Є¬†—В–∞–ї–Є–і–Њ–Љ–Є–і–∞ [26, 27]. –Ъ–∞—А–і–Є–Њ—В–Њ–Ї—Б–Є—З–љ–Њ—Б—В—М –Є¬†–љ–µ–є—А–Њ—В–Њ–Ї—Б–Є—З–љ–Њ—Б—В—М —Н—В–Є—Е –њ—А–µ–њ–∞—А–∞—В–Њ–≤ –љ–µ –њ–Њ–Ј–≤–Њ–ї—П—О—В —А–µ–Ї–Њ–Љ–µ–љ–і–Њ–≤–∞—В—М –Є—Е –≤¬†–Ї–∞—З–µ—Б—В–≤–µ –њ—А–µ–њ–∞—А–∞—В–Њ–≤ –њ–µ—А–≤–Њ–є –ї–Є–љ–Є–Є –≤—Л–±–Њ—А–∞, –љ–µ—Б–Љ–Њ—В—А—П –љ–∞ –≤—Л—Б–Њ–Ї—Г—О —Н—Д—Д–µ–Ї—В–Є–≤–љ–Њ—Б—В—М –≤¬†–Њ—В–љ–Њ—И–µ–љ–Є–Є –њ–ї–∞–Ј–Љ–Њ–Ї–ї–µ—В–Њ—З–љ–Њ–є –і–Є—Б–Ї—А–∞–Ј–Є–Є.
–Ю—Б–љ–Њ–≤–љ–Њ–є —Ж–µ–ї—М—О –ї–µ—З–µ–љ–Є—П –Р-–±–µ—В–∞-2-–Ь-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ —П–≤–ї—П–µ—В—Б—П —Г–Љ–µ–љ—М—И–µ–љ–Є–µ –њ–Њ—Б—В—Г–њ–ї–µ–љ–Є—П –≤¬†—В–Ї–∞–љ–Є –±–µ—В–∞-2-–Љ–Є–Ї—А–Њ–≥–ї–Њ–±—Г–ї–Є–љ–∞. –Я–Њ—Б–Ї–Њ–ї—М–Ї—Г —Б–њ–Њ—Б–Њ–±—Л –њ–Њ–і–∞–≤–ї–µ–љ–Є—П –µ–≥–Њ –њ—А–Њ–і—Г–Ї—Ж–Є–Є –≤¬†–љ–∞—Б—В–Њ—П—Й–µ–µ –≤—А–µ–Љ—П –Њ—В—Б—Г—В—Б—В–≤—Г—О—В, –љ–µ–Њ–±—Е–Њ–і–Є–Љ–Њ —Г–≤–µ–ї–Є—З–Є—В—М –Ї–ї–Є—А–µ–љ—Б –±–µ—В–∞-2-–Љ–Є–Ї—А–Њ–≥–ї–Њ–±—Г–ї–Є–љ–∞ –њ—Г—В–µ–Љ –њ—А–Њ–≤–µ–і–µ–љ–Є—П —Б–Њ–≤—А–µ–Љ–µ–љ–љ—Л—Е –Љ–µ—В–Њ–і–Њ–≤ –Њ—З–Є—Й–µ–љ–Є—П –Ї—А–Њ–≤–Є.
–®–Є—А–Њ–Ї–Њ —А–∞—Б–њ—А–Њ—Б—В—А–∞–љ–µ–љ–Њ –Љ–љ–µ–љ–Є–µ –Њ¬†—В–Њ–Љ, —З—В–Њ –љ–∞–Є–±–Њ–ї–µ–µ —Н—Д—Д–µ–Ї—В–Є–≤–љ—Л–Љ –Љ–µ—В–Њ–і–Њ–Љ –≤—Л–≤–µ–і–µ–љ–Є—П –±–µ—В–∞-2-–Љ–Є–Ї—А–Њ–≥–ї–Њ–±—Г–ї–Є–љ–∞ —П–≤–ї—П–µ—В—Б—П —В—А–∞–љ—Б–њ–ї–∞–љ—В–∞—Ж–Є—П –њ–Њ—З–Ї–Є. –≠—В–∞ —В–Њ—З–Ї–∞ –Ј—А–µ–љ–Є—П –њ–Њ–і–Ї—А–µ–њ–ї–µ–љ–∞ –≤—Л—Б–Њ–Ї–Њ–є —Н—Д—Д–µ–Ї—В–Є–≤–љ–Њ—Б—В—М—О —В—А–∞–љ—Б–њ–ї–∞–љ—В–∞—Ж–Є–Є –≤¬†–Њ—В–љ–Њ—И–µ–љ–Є–Є –Ї–Њ—Б—В–љ—Л—Е –±–Њ–ї–µ–є, —Е–∞—А–∞–Ї—В–µ—А–љ—Л—Е –і–ї—П –і–Є–∞–ї–Є–Ј–љ–Њ–≥–Њ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞. –Т¬†–љ–∞—Б—В–Њ—П—Й–µ–µ –≤—А–µ–Љ—П —Г—Б—В–∞–љ–Њ–≤–ї–µ–љ–Њ, —З—В–Њ –њ—А–Є—З–Є–љ–Њ–є –Ї–Њ—Б—В–љ—Л—Е –±–Њ–ї–µ–є, —В–∞–Ї –ґ–µ –Ї–∞–Ї –Є¬†–Ї–Њ—Б—В–љ—Л—Е –Ї–Є—Б—В, —П–≤–ї—П–µ—В—Б—П –Љ–∞–Ї—А–Њ—Д–∞–≥–∞–ї—М–љ–Њ–µ –≤–Њ—Б–њ–∞–ї–µ–љ–Є–µ, —Е–∞—А–∞–Ї—В–µ—А–љ–Њ–µ –і–ї—П —Н—В–Њ–≥–Њ —В–Є–њ–∞ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞. –С—Л—Б—В—А–Њ–µ –Є—Б—З–µ–Ј–љ–Њ–≤–µ–љ–Є–µ –±–Њ–ї–µ–є –њ–Њ—Б–ї–µ —В—А–∞–љ—Б–њ–ї–∞–љ—В–∞—Ж–Є–Є —П–≤–ї—П–µ—В—Б—П —Б–ї–µ–і—Б—В–≤–Є–µ–Љ –љ–∞–Ј–љ–∞—З–µ–љ–Є—П —Б—В–µ—А–Њ–Є–і–Њ–≤ –Є¬†—Ж–Є—В–Њ—Б—В–∞—В–Є–Ї–Њ–≤, –љ–µ–Њ–±—Е–Њ–і–Є–Љ—Л—Е –і–ї—П –њ—А–µ–і–Њ—В–≤—А–∞—Й–µ–љ–Є—П –Њ—В—В–Њ—А–ґ–µ–љ–Є—П —В—А–∞–љ—Б–њ–ї–∞–љ—В–∞—В–∞ –Є¬†–Њ–і–љ–Њ–≤—А–µ–Љ–µ–љ–љ–Њ –њ–Њ–і–∞–≤–ї—П—О—Й–Є—Е –Љ–∞–Ї—А–Њ—Д–∞–≥–∞–ї—М–љ–Њ–µ –≤–Њ—Б–њ–∞–ї–µ–љ–Є–µ –≤¬†–Ј–Њ–љ–µ –і–µ–њ–Њ–Ј–Є—Ж–Є–Є –∞–Љ–Є–ї–Њ–Є–і–∞. –Э–∞ –њ–Њ–Ј–і–љ–Є—Е —Н—В–∞–њ–∞—Е –∞–Љ–Є–ї–Њ–Є–і–Њ–≥–µ–љ–µ–Ј–∞ –Ј–∞–≤–Є—Б–Є–Љ–Њ—Б—В—М –Љ–∞–Ї—А–Њ—Д–∞–≥–∞–ї—М–љ–Њ–≥–Њ –≤–Њ—Б–њ–∞–ї–µ–љ–Є—П –Њ—В –њ–Њ—Б—В–∞–≤–Ї–Є –±–µ—В–∞-2-–Љ–Є–Ї—А–Њ–≥–ї–Њ–±—Г–ї–Є–љ–∞ –≤¬†—В–Ї–∞–љ–Є —Б–љ–Є–ґ–∞–µ—В—Б—П, –Є¬†—Г–ї—Г—З—И–µ–љ–Є–µ –Ї–ї–Є—А–µ–љ—Б–∞ —Н—В–Њ–≥–Њ –±–µ–ї–Ї–∞ –њ–Њ—Б–ї–µ —В—А–∞–љ—Б–њ–ї–∞–љ—В–∞—Ж–Є–Є –љ–µ –њ—А–µ–і—Г–њ—А–µ–ґ–і–∞–µ—В –і–∞–ї—М–љ–µ–є—И–µ–µ –њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є–µ –Ї–Њ—Б—В–љ—Л—Е –Ї–Є—Б—В –Є¬†—Н—А–Њ–Ј–Є–є. –Ґ–∞–Ї–Є–Љ –Њ–±—А–∞–Ј–Њ–Љ, —В—А–∞–љ—Б–њ–ї–∞–љ—В–∞—Ж–Є—П —Б–њ–Њ—Б–Њ–±–љ–∞ –њ—А–µ–і–Њ—В–≤—А–∞—В–Є—В—М –њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є–µ –і–Є–∞–ї–Є–Ј–љ–Њ–≥–Њ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ —В–Њ–ї—М–Ї–Њ –љ–∞ —А–∞–љ–љ–Є—Е, –≤¬†–Њ—Б–љ–Њ–≤–љ–Њ–Љ –і–Њ–Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є—Е, —Б—В–∞–і–Є—П—Е –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ [28].
–Т —Н—В–Њ–є —Б–≤—П–Ј–Є –≤¬†–ї–µ—З–µ–љ–Є–Є –і–Є–∞–ї–Є–Ј–љ–Њ–≥–Њ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ –±–Њ–ї—М—И–Њ–µ –Ј–љ–∞—З–µ–љ–Є–µ –њ—А–Є–і–∞—О—В —Б–Њ–≤—А–µ–Љ–µ–љ–љ—Л–Љ –і–Є–∞–ї–Є–Ј–љ—Л–Љ —В–µ—Е–љ–Њ–ї–Њ–≥–Є—П–Љ —Г–≤–µ–ї–Є—З–µ–љ–Є—П –Ї–ї–Є—А–µ–љ—Б–∞ –±–µ—В–∞-2-–Љ–Є–Ї—А–Њ–≥–ї–Њ–±—Г–ї–Є–љ–∞. –Я—А–Є –њ—А–Њ–≤–µ–і–µ–љ–Є–Є —Б—В–∞–љ–і–∞—А—В–љ–Њ–є –њ—А–Њ—Ж–µ–і—Г—А—Л –≥–µ–Љ–Њ–і–Є–∞–ї–Є–Ј–∞ —Б¬†–Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–љ–Є–µ–Љ –љ–Є–Ј–Ї–Њ–њ–Њ—В–Њ—З–љ—Л—Е –Љ–µ–Љ–±—А–∞–љ (–і–Є—Д—Д—Г–Ј–Є–Њ–љ–љ–∞—П –Љ–Њ–і–µ–ї—М) –Ї–ї–Є—А–µ–љ—Б –±–µ—В–∞-2-–Љ–Є–Ї—А–Њ–≥–ї–Њ–±—Г–ї–Є–љ–∞ —Б—Г—Й–µ—Б—В–≤–µ–љ–љ–Њ –Њ–≥—А–∞–љ–Є—З–µ–љ, —В–∞–Ї –Ї–∞–Ї —Н—В–Њ—В –±–µ–ї–Њ–Ї –њ–Њ —Б–≤–Њ–µ–є –Љ–∞—Б—Б–µ (11 800¬†–Ф–∞) –њ—А–Є–±–ї–Є–ґ–∞–µ—В—Б—П –Ї¬†–Ї–ї–∞—Б—Б—Г —В–∞–Ї –љ–∞–Ј—Л–≤–∞–µ–Љ—Л—Е —Б—А–µ–і–љ–Є—Е –Љ–Њ–ї–µ–Ї—Г–ї. –Ю–і–љ–∞–Ї–Њ –њ—А–Є–Љ–µ–љ–µ–љ–Є–µ –≤—Л—Б–Њ–Ї–Њ–њ–Њ—В–Њ—З–љ—Л—Е —Б–Є–љ—В–µ—В–Є—З–µ—Б–Ї–Є—Е –Љ–µ–Љ–±—А–∞–љ –Є¬†—Б–Њ—З–µ—В–∞–љ–Є–µ –і–Є—Д—Д—Г–Ј–Є–Њ–љ–љ–Њ–≥–Њ –Љ–µ—Е–∞–љ–Є–Ј–Љ–∞ –Њ—З–Є—Й–µ–љ–Є—П –Ї—А–Њ–≤–Є —Б¬†–Ї–Њ–љ–≤–µ–Ї—В–Є–≤–љ—Л–Љ –њ—А–Є–≤–Њ–і—П—В –Ї¬†—Б—Г—Й–µ—Б—В–≤–µ–љ–љ–Њ–Љ—Г –њ–Њ–≤—Л—И–µ–љ–Є—О —Н—Д—Д–µ–Ї—В–Є–≤–љ–Њ—Б—В–Є –≤—Л–≤–µ–і–µ–љ–Є—П –±–µ—В–∞-2-–Љ–Є–Ї—А–Њ–≥–ї–Њ–±—Г–ї–Є–љ–∞. –Ґ–∞–Ї, –±—Л–ї–Њ –њ–Њ–Ї–∞–Ј–∞–љ–Њ, —З—В–Њ –ї–µ—З–µ–љ–Є–µ –≥–µ–Љ–Њ–і–Є–∞—Д–Є–ї—М—В—А–∞—Ж–Є–µ–є –≤¬†—А–µ–ґ–Є–Љ–µ online (—А–µ–ґ–Є–Љ –У–Ф–§ ONLINE –њ—А–µ–і—Г—Б–Љ–∞—В—А–Є–≤–∞–µ—В –Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–љ–Є–µ –≤¬†–Ї–∞—З–µ—Б—В–≤–µ –Ј–∞–Љ–µ—Й–∞—О—Й–µ–≥–Њ —А–∞—Б—В–≤–Њ—А–∞ —Б–≤–µ—А—Е—З–Є—Б—В–Њ–≥–Њ –і–Є–∞–ї–Є–Ј–∞—В–∞, –Ї–Њ—В–Њ—А—Л–є –≥–Њ—В–Њ–≤–Є—В—Б—П –љ–µ–њ–Њ—Б—А–µ–і—Б—В–≤–µ–љ–љ–Њ –≤¬†–∞–њ–њ–∞—А–∞—В–µ –і–ї—П –≥–µ–Љ–Њ–і–Є–∞–ї–Є–Ј–∞) –њ–Њ–Ј–≤–Њ–ї—П–µ—В –і–Њ–±–Є—В—М—Б—П –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ–≥–Њ —Б–љ–Є–ґ–µ–љ–Є—П –њ–Њ—Б—В–і–Є–∞–ї–Є–Ј–љ–Њ–≥–Њ —Г—А–Њ–≤–љ—П –±–µ—В–∞-2-–Љ–Є–Ї—А–Њ–≥–ї–Њ–±—Г–ї–Є–љ–∞ (–љ–∞ 70% –Є¬†–±–Њ–ї–µ–µ) [29]. –Т¬†—Н—В–Њ–є —Б–≤—П–Ј–Є –љ–∞–Є–±–Њ–ї–µ–µ –њ—А–µ–і–њ–Њ—З—В–Є—В–µ–ї—М–љ—Л–Љ –Љ–µ—В–Њ–і–Њ–Љ –≥–µ–Љ–Њ–і–Є–∞–ї–Є–Ј–∞ —Б¬†—В–Њ—З–Ї–Є –Ј—А–µ–љ–Є—П –њ—А–Њ—Д–Є–ї–∞–Ї—В–Є–Ї–Є –Є¬†–ї–µ—З–µ–љ–Є—П –і–Є–∞–ї–Є–Ј–љ–Њ–≥–Њ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ —П–≤–ї—П–µ—В—Б—П –≤—Л—Б–Њ–Ї–Њ—Н—Д—Д–µ–Ї—В–Є–≤–љ–∞—П –У–Ф–§ ONLINE —Б¬†–Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–љ–Є–µ–Љ —Б–Є–љ—В–µ—В–Є—З–µ—Б–Ї–Є—Е –Љ–µ–Љ–±—А–∞–љ –Є¬†—Б–Ї–Њ—А–Њ—Б—В—М—О –њ–Њ–і–∞—З–Є –Ј–∞–Љ–µ—Й–∞—О—Й–µ–≥–Њ —А–∞—Б—В–≤–Њ—А–∞ –љ–µ –Љ–µ–љ–µ–µ 100¬†–Љ–ї/–Љ–Є–љ. –Я–∞—Ж–Є–µ–љ—В—Л, –Ї–Њ—В–Њ—А—Л–µ –њ–Њ–ї—Г—З–∞–ї–Є –У–Ф–§ ONLINE —Б¬†–Њ–±—К–µ–Љ–Њ–Љ –Ј–∞–Љ–µ—Й–µ–љ–Є—П –љ–µ –Љ–µ–љ–µ–µ 24¬†–ї –Ј–∞ 4¬†—З–∞—Б–∞ –њ—А–Њ—Ж–µ–і—Г—А—Л, –Є–Љ–µ–ї–Є –і–Њ—Б—В–Њ–≤–µ—А–љ–Њ –±–Њ–ї–µ–µ —Б—Г—Й–µ—Б—В–≤–µ–љ–љ–Њ–µ (–љ–∞ 72,7%) —Б–љ–Є–ґ–µ–љ–Є–µ —Б—Л–≤–Њ—А–Њ—В–Њ—З–љ–Њ–≥–Њ —Г—А–Њ–≤–љ—П –±–µ—В–∞-2-–Љ–Є–Ї—А–Њ–≥–ї–Њ–±—Г–ї–Є–љ–∞ –њ–Њ —Б—А–∞–≤–љ–µ–љ–Є—О —Б¬†–њ–∞—Ж–Є–µ–љ—В–∞–Љ–Є, –њ–Њ–ї—Г—З–∞–≤—И–Є–Љ–Є —Б—В–∞–љ–і–∞—А—В–љ—Л–є –≥–µ–Љ–Њ–і–Є–∞–ї–Є–Ј –љ–∞ –љ–Є–Ј–Ї–Њ–њ–Њ—В–Њ—З–љ—Л—Е –Љ–µ–Љ–±—А–∞–љ–∞—Е (–љ–∞ 49,7%) [30].
–†–µ–Ј—Г–ї—М—В–∞—В—Л –љ–µ–і–∞–≤–љ–Њ –Ј–∞–≤–µ—А—И–µ–љ–љ–Њ–≥–Њ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П CONTRAST (CONvective TRAnsport STudy), –њ–Њ—Б–≤—П—Й–µ–љ–љ–Њ–≥–Њ –≤–ї–Є—П–љ–Є—О –Ї–Њ–љ–≤–µ–Ї—В–Є–≤–љ–Њ–≥–Њ —В—А–∞–љ—Б–њ–Њ—А—В–∞ –љ–∞ —Б—Л–≤–Њ—А–Њ—В–Њ—З–љ—Л–є —Г—А–Њ–≤–µ–љ—М –±–µ—В–∞-2-–Љ–Є–Ї—А–Њ–≥–ї–Њ–±—Г–ї–Є–љ–∞, —Б–≤–Є–і–µ—В–µ–ї—М—Б—В–≤—Г—О—В –Њ¬†—В–Њ–Љ, —З—В–Њ —З–µ—А–µ–Ј 6¬†–Љ–µ—Б—П—Ж–µ–≤ —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤, –Ї–Њ—В–Њ—А—Л–Љ –њ—А–Њ–≤–Њ–і–Є—В—Б—П –≥–µ–Љ–Њ–і–Є–∞—Д–Є–ї—М—В—А–∞—Ж–Є—П, —Г—А–Њ–≤–µ–љ—М –±–µ—В–∞-2-–Љ–Є–Ї—А–Њ–≥–ї–Њ–±—Г–ї–Є–љ–∞ –≤¬†—Б—Л–≤–Њ—А–Њ—В–Ї–µ –і–Њ—Б—В–Њ–≤–µ—А–љ–Њ –љ–Є–ґ–µ, —З–µ–Љ —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ –љ–∞ –љ–Є–Ј–Ї–Њ–њ–Њ—В–Њ—З–љ–Њ–Љ –≥–µ–Љ–Њ–і–Є–∞–ї–Є–Ј–µ [31].
–Ф–∞–ї—М–љ–µ–є—И–Є–µ –њ–µ—А—Б–њ–µ–Ї—В–Є–≤—Л –≤¬†—А–∞–Ј–≤–Є—В–Є–Є –≥–µ–Љ–Њ–і–Є–∞–ї–Є–Ј–∞ –≤–Њ –Љ–љ–Њ–≥–Њ–Љ —Б–≤—П–Ј–∞–љ—Л —Б¬†—В–µ—Е–љ–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–Љ —Г—Б–Њ–≤–µ—А—И–µ–љ—Б—В–≤–Њ–≤–∞–љ–Є–µ–Љ –Є¬†—И–Є—А–Њ–Ї–Є–Љ —А–∞—Б–њ—А–Њ—Б—В—А–∞–љ–µ–љ–Є–µ–Љ –Љ–µ—В–Њ–і–Є–Ї–Є –У–Ф–§ ONLINE –≤¬†–Ї–ї–Є–љ–Є—З–µ—Б–Ї–Њ–є –њ—А–∞–Ї—В–Є–Ї–µ. –Т¬†—З–∞—Б—В–љ–Њ—Б—В–Є, —Г—Б–Њ–≤–µ—А—И–µ–љ—Б—В–≤–Њ–≤–∞–љ–љ–∞—П —В–µ—Е–љ–Є–Ї–∞ –У–Ф–§ ONLINE¬†вАУ MIXED –У–Ф–§¬†вАУ –њ–Њ–Ј–≤–Њ–ї—П–µ—В —Б—Г—Й–µ—Б—В–≤–µ–љ–љ–Њ —Г–≤–µ–ї–Є—З–Є—В—М —Н—Д—Д–µ–Ї—В–Є–≤–љ–Њ—Б—В—М –Ї–Њ–љ–≤–µ–Ї—В–Є–≤–љ–Њ–≥–Њ —В—А–∞–љ—Б–њ–Њ—А—В–∞, –≤¬†—В–Њ–Љ —З–Є—Б–ї–µ –њ–µ—А–µ–љ–Њ—Б —Б—А–µ–і–љ–Є—Е –Љ–Њ–ї–µ–Ї—Г–ї, –≤¬†–±–µ–Ј–Њ–њ–∞—Б–љ–Њ–Љ –і–ї—П –њ–∞—Ж–Є–µ–љ—В–∞ —А–µ–ґ–Є–Љ–µ [32вАУ34].
–С–Њ–ї—М—И–Њ–µ –Ј–љ–∞—З–µ–љ–Є–µ –Є–Љ–µ–µ—В –њ—А–Є–Љ–µ–љ–µ–љ–Є–µ —Б–Њ–≤—А–µ–Љ–µ–љ–љ—Л—Е —Б–Є–љ—В–µ—В–Є—З–µ—Б–Ї–Є—Е –і–Є–∞–ї–Є–Ј–љ—Л—Е –Љ–µ–Љ–±—А–∞–љ, –Ї–Њ—В–Њ—А—Л–µ —Б–њ–Њ—Б–Њ–±–љ—Л –∞–і—Б–Њ—А–±–Є—А–Њ–≤–∞—В—М –Ј–љ–∞—З–Є—В–µ–ї—М–љ—Л–µ –Ї–Њ–ї–Є—З–µ—Б—В–≤–∞ –±–µ—В–∞-2-–Љ–Є–Ї—А–Њ–≥–ї–Њ–±—Г–ї–Є–љ–∞, –≤¬†—А–µ–Ј—Г–ї—М—В–∞—В–µ —З–µ–≥–Њ –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є—П —Н—В–Њ–≥–Њ –±–µ–ї–Ї–∞ –≤¬†–Ї—А–Њ–≤–Є –≤¬†—В–µ—З–µ–љ–Є–µ –њ—А–Њ—Ж–µ–і—Г—А—Л –≥–µ–Љ–Њ–і–Є–∞–ї–Є–Ј–∞ –Љ–Њ–ґ–µ—В —Б–љ–Є–ґ–∞—В—М—Б—П –љ–∞ 30вАУ40% [29]. –†–∞–Ј—А–∞–±–Њ—В–∞–љ—Л —В–∞–Ї–ґ–µ —Б–њ–µ—Ж–Є–∞–ї—М–љ—Л–µ —Б–Њ—А–±—Ж–Є–Њ–љ–љ—Л–µ –Ї–Њ–ї–Њ–љ–Ї–Є —Б¬†–≤—Л—Б–Њ–Ї–Є–Љ–Є –њ–Њ–Ї–∞–Ј–∞—В–µ–ї—П–Љ–Є –Њ—З–Є—Й–µ–љ–Є—П –Ї—А–Њ–≤–Є –Њ—В –±–µ—В–∞-2-–Љ–Є–Ї—А–Њ–≥–ї–Њ–±—Г–ї–Є–љ–∞, –Њ–і–љ–∞–Ї–Њ –Є—Е —И–Є—А–Њ–Ї–Њ–µ –њ—А–Є–Љ–µ–љ–µ–љ–Є–µ —Б–і–µ—А–ґ–Є–≤–∞–µ—В—Б—П –≤—Л—Б–Њ–Ї–Њ–є —Б–µ–±–µ—Б—В–Њ–Є–Љ–Њ—Б—В—М—О [35вАУ37].
–Я–Њ—Б–Ї–Њ–ї—М–Ї—Г –≤–∞–ґ–љ—Л–Љ –Ї–Њ–Љ–њ–Њ–љ–µ–љ—В–Њ–Љ –і–Є–∞–ї–Є–Ј–љ–Њ–≥–Њ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ —П–≤–ї—П–µ—В—Б—П –Љ–∞–Ї—А–Њ—Д–∞–≥–∞–ї—М–љ–Њ–µ –≤–Њ—Б–њ–∞–ї–µ–љ–Є–µ, –±–Њ–ї—М—И–Њ–µ –Ј–љ–∞—З–µ–љ–Є–µ –Є–Љ–µ–µ—В –Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–љ–Є–µ –≤—Л—Б–Њ–Ї–Њ–Њ—З–Є—Й–µ–љ–љ—Л—Е —А–∞—Б—В–≤–Њ—А–Њ–≤ –і–ї—П –њ—А–Є–≥–Њ—В–Њ–≤–ї–µ–љ–Є—П –і–Є–∞–ї–Є–Ј–∞—В–∞. G. Lonnemann –Є¬†–і—А—Г–≥–Є–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞—В–µ–ї–Є —Б–Њ–Њ–±—Й–∞—О—В –Њ¬†—В–Њ–Љ, —З—В–Њ –њ—А–Є–Љ–µ–љ–µ–љ–Є–µ —Г–ї—М—В—А–∞—З–Є—Б—В—Л—Е –і–Є–∞–ї–Є–Ј–љ—Л—Е –ґ–Є–і–Ї–Њ—Б—В–µ–є –≤–Њ –≤—А–µ–Љ—П –ї–µ—З–µ–љ–Є—П –Ї–Њ–љ–≤–µ–Ї—В–Є–≤–љ—Л–Љ–Є –Љ–µ—В–Њ–і–∞–Љ–Є –њ–Њ–Ј–≤–Њ–ї—П–µ—В —Б–љ–Є–Ј–Є—В—М —З–∞—Б—В–Њ—В—Г —Б–Є–љ–і—А–Њ–Љ–∞ –Ј–∞–њ—П—Б—В–љ–Њ–≥–Њ –Ї–∞–љ–∞–ї–∞ –љ–∞ 50% [37, 38].
–Т –Ї–∞—З–µ—Б—В–≤–µ —Б–Є–Љ–њ—В–Њ–Љ–∞—В–Є—З–µ—Б–Ї–Њ–є —В–µ—А–∞–њ–Є–Є –±–Њ–ї—М–љ—Л–Љ –і–Є–∞–ї–Є–Ј–љ—Л–Љ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–Њ–Љ –Љ–Њ–ґ–µ—В –±—Л—В—М —А–µ–Ї–Њ–Љ–µ–љ–і–Њ–≤–∞–љ–Њ –Љ–µ—Б—В–љ–Њ–µ –њ—А–Є–Љ–µ–љ–µ–љ–Є–µ —Б—В–µ—А–Њ–Є–і–љ—Л—Е –Љ–∞–Ј–µ–є –њ—А–Є –±–Њ–ї—П—Е, –≤¬†–ї–Є—В–µ—А–∞—В—Г—А–µ –Њ–±—Б—Г–ґ–і–∞—О—В—Б—П —В–∞–Ї–ґ–µ –њ–Њ–Ї–∞–Ј–∞–љ–Є—П –Ї¬†–њ—А–Є–Љ–µ–љ–µ–љ–Є—О —Б—В–µ—А–Њ–Є–і–Њ–≤ –≤–љ—Г—В—А—М –Є¬†–≤ –Є–љ—К–µ–Ї—Ж–Є—П—Е, –Њ–і–љ–∞–Ї–Њ –Є—Е –Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–љ–Є–µ –Њ–≥—А–∞–љ–Є—З–µ–љ–Њ –Є–Ј-–Ј–∞ —В—П–ґ–µ–ї–Њ–є —А–µ–љ–∞–ї—М–љ–Њ–є –Њ—Б—В–µ–Њ–і–Є—Б—В—А–Њ—Д–Є–Є. –Т¬†—Н—В–Њ–є —Б–≤—П–Ј–Є –њ—А–µ–і—Б—В–∞–≤–ї—П–µ—В—Б—П –њ–µ—А—Б–њ–µ–Ї—В–Є–≤–љ—Л–Љ –њ—А–Є–Љ–µ–љ–µ–љ–Є–µ —Г¬†—В–∞–Ї–Є—Е –±–Њ–ї—М–љ—Л—Е —Б–Њ–≤—А–µ–Љ–µ–љ–љ—Л—Е –њ—А–µ–њ–∞—А–∞—В–Њ–≤, –њ–Њ–і–∞–≤–ї—П—О—Й–Є—Е —Н—Д—Д–µ–Ї—В—Л —Д–∞–Ї—В–Њ—А–∞ –љ–µ–Ї—А–Њ–Ј–∞ –Њ–њ—Г—Е–Њ–ї–Є, –Є–љ—В–µ—А–ї–µ–є–Ї–Є–љ–∞-1 –Є¬†–Є–љ—В–µ—А–ї–µ–є–Ї–Є–љ–∞-6.
–°—А–µ–і—Б—В–≤–Њ–Љ –≤—Л–±–Њ—А–∞ –і–ї—П –ї–µ—З–µ–љ–Є—П –РTTR-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ —Б–ї—Г–ґ–Є—В —В—А–∞–љ—Б–њ–ї–∞–љ—В–∞—Ж–Є—П –њ–µ—З–µ–љ–Є, –њ—А–Є –Ї–Њ—В–Њ—А–Њ–є —Г–і–∞–µ—В—Б—П —Г–і–∞–ї–Є—В—М –Є—Б—В–Њ—З–љ–Є–Ї —Б–Є–љ—В–µ–Ј–∞ –∞–Љ–Є–ї–Њ–Є–і–Њ–≥–µ–љ–љ–Њ–≥–Њ –њ—А–µ–і—И–µ—Б—В–≤–µ–љ–љ–Є–Ї–∞. –Ґ—А–∞–љ—Б–њ–ї–∞–љ—В–∞—Ж–Є—П –њ–µ—З–µ–љ–Є —Б—Г—Й–µ—Б—В–≤–µ–љ–љ–Њ –Ј–∞–і–µ—А–ґ–Є–≤–∞–µ—В –њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є–µ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞, –≤¬†–њ–µ—А–≤—Г—О –Њ—З–µ—А–µ–і—М –љ–µ—А–≤–љ–Њ–є —Б–Є—Б—В–µ–Љ—Л [39]. –Т¬†—В–Њ –ґ–µ –≤—А–µ–Љ—П –љ–∞–Ї–Њ–њ–ї–µ–љ–љ—Л–µ –і–µ–њ–Њ–Ј–Є—В—Л –∞–Љ–Є–ї–Њ–Є–і–∞ –≤¬†—В–Ї–∞–љ—П—Е, –≤¬†–Њ—Б–Њ–±–µ–љ–љ–Њ—Б—В–Є –≤¬†—Б–µ—А–і—Ж–µ, —Б–њ–Њ—Б–Њ–±–љ—Л –∞–і—Б–Њ—А–±–Є—А–Њ–≤–∞—В—М —В–∞–Ї–ґ–µ –Є¬†–љ–Њ—А–Љ–∞–ї—М–љ—Л–є —В—А–∞–љ—Б—В–Є—А–µ—В–Є–љ –Є¬†—В–µ–Љ —Б–∞–Љ—Л–Љ —Б–њ–Њ—Б–Њ–±—Б—В–≤–Њ–≤–∞—В—М –і–∞–ї—М–љ–µ–є—И–µ–Љ—Г –њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є—О –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ [6]. –Т¬†—Н—В–Њ–є —Б–≤—П–Ј–Є –≤¬†–љ–∞—Б—В–Њ—П—Й–µ–µ –≤—А–µ–Љ—П —И–Є—А–Њ–Ї–Њ –Њ–±—Б—Г–ґ–і–∞—О—В—Б—П –≤–Њ–Ј–Љ–Њ–ґ–љ–Њ—Б—В–Є —Б—В–∞–±–Є–ї–Є–Ј–∞—Ж–Є–Є –љ–Њ—А–Љ–∞–ї—М–љ–Њ–≥–Њ —В—А–∞–љ—Б—В–Є—А–µ—В–Є–љ–∞ –Є¬†–њ—А–µ–і–Њ—В–≤—А–∞—Й–µ–љ–Є—П –µ–≥–Њ –∞–Љ–Є–ї–Њ–Є–і–Њ–≥–µ–љ–љ–Њ—Б—В–Є —Б¬†–њ–Њ–Љ–Њ—Й—М—О —Д–∞—А–Љ–∞–Ї–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є—Е –њ—А–µ–њ–∞—А–∞—В–Њ–≤ (diflunisal) [40].
–Я–Њ—Б–Ї–Њ–ї—М–Ї—Г –•–Я–Э —Б–ї—Г–ґ–Є—В –Њ–і–љ–Њ–є –Є–Ј –Њ—Б–љ–Њ–≤–љ—Л—Е –њ—А–Є—З–Є–љ —Б–Љ–µ—А—В–Є –±–Њ–ї—М–љ—Л—Е —Б–Є—Б—В–µ–Љ–љ—Л–Љ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–Њ–Љ, –њ—А–Њ–≤–µ–і–µ–љ–Є–µ –≥–µ–Љ–Њ–і–Є–∞–ї–Є–Ј–∞ –Є–ї–Є –њ–Њ—Б—В–Њ—П–љ–љ–Њ–≥–Њ –∞–Љ–±—Г–ї–∞—В–Њ—А–љ–Њ–≥–Њ –њ–µ—А–Є—В–Њ–љ–µ–∞–ї—М–љ–Њ–≥–Њ –і–Є–∞–ї–Є–Ј–∞ –њ–Њ–Ј–≤–Њ–ї—П–µ—В —Г–ї—Г—З—И–Є—В—М –њ—А–Њ–≥–љ–Њ–Ј —Н—В–Є—Е –њ–∞—Ж–Є–µ–љ—В–Њ–≤, –≤¬†—З–∞—Б—В–љ–Њ—Б—В–Є, –≤—Л–ґ–Є–≤–∞–µ–Љ–Њ—Б—В—М —Н—В–Є—Е –±–Њ–ї—М–љ—Л—Е —Б–≤—П–Ј—Л–≤–∞—О—В —Б¬†—Г—А–Њ–≤–љ–µ–Љ –±–µ—В–∞-2-–Љ–Є–Ї—А–Њ–≥–ї–Њ–±—Г–ї–Є–љ–∞ [41вАУ43].
–Т—Л–ґ–Є–≤–∞–µ–Љ–Њ—Б—В—М –±–Њ–ї—М–љ—Л—Е –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–Њ–Љ –њ—А–Є –њ—А–Њ–≤–µ–і–µ–љ–Є–Є –≥–µ–Љ–Њ–і–Є–∞–ї–Є–Ј–∞, –љ–µ–Ј–∞–≤–Є—Б–Є–Љ–Њ –Њ—В —В–Є–њ–∞ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞, —Б–Њ–њ–Њ—Б—В–∞–≤–Є–Љ–∞ —Б¬†–≤—Л–ґ–Є–≤–∞–µ–Љ–Њ—Б—В—М—О –±–Њ–ї—М–љ—Л—Е –і—А—Г–≥–Є–Љ–Є —Б–Є—Б—В–µ–Љ–љ—Л–Љ–Є –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П–Љ–Є –Є¬†—Б–∞—Е–∞—А–љ—Л–Љ –і–Є–∞–±–µ—В–Њ–Љ. –Я–Њ—Б—В–Њ—П–љ–љ—Л–є –∞–Љ–±—Г–ї–∞—В–Њ—А–љ—Л–є –њ–µ—А–Є—В–Њ–љ–µ–∞–ї—М–љ—Л–є –і–Є–∞–ї–Є–Ј –Є–Љ–µ–µ—В –љ–µ–Ї–Њ—В–Њ—А—Л–µ –њ—А–µ–Є–Љ—Г—Й–µ—Б—В–≤–∞ –њ–µ—А–µ–і –≥–µ–Љ–Њ–і–Є–∞–ї–Є–Ј–Њ–Љ, –њ–Њ—Б–Ї–Њ–ї—М–Ї—Г –љ–µ—В –љ–µ–Њ–±—Е–Њ–і–Є–Љ–Њ—Б—В–Є –≤¬†–њ–Њ—Б—В–Њ—П–љ–љ–Њ–Љ —Б–Њ—Б—Г–і–Є—Б—В–Њ–Љ –і–Њ—Б—В—Г–њ–µ, –Њ—В—Б—Г—В—Б—В–≤—Г–µ—В –∞—А—В–µ—А–Є–∞–ї—М–љ–∞—П –≥–Є–њ–Њ—В–Њ–љ–Є—П –≤–Њ –≤—А–µ–Љ—П –њ—А–Њ—Ж–µ–і—Г—А—Л –і–Є–∞–ї–Є–Ј–∞, –∞¬†—Г –±–Њ–ї—М–љ—Л—Е —Б¬†AL-—В–Є–њ–Њ–Љ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ –≤–Њ –≤—А–µ–Љ—П –њ—А–Њ—Ж–µ–і—Г—А—Л –≤–Њ–Ј–Љ–Њ–ґ–љ–Њ —Г–і–∞–ї–µ–љ–Є–µ –ї–µ–≥–Ї–Є—Е —Ж–µ–њ–µ–є –Є–Љ–Љ—Г–љ–Њ–≥–ї–Њ–±—Г–ї–Є–љ–Њ–≤.
–Ґ—А–∞–љ—Б–њ–ї–∞–љ—В–∞—Ж–Є—П –њ–Њ—З–Ї–Є –Њ–і–Є–љ–∞–Ї–Њ–≤–Њ —Н—Д—Д–µ–Ї—В–Є–≤–љ–∞ –њ—А–Є –Њ–±–Њ–Є—Е —В–Є–њ–∞—Е —Б–Є—Б—В–µ–Љ–љ–Њ–≥–Њ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞. –Я—П—В–Є–ї–µ—В–љ—П—П –≤—Л–ґ–Є–≤–∞–µ–Љ–Њ—Б—В—М –±–Њ–ї—М–љ—Л—Е –Є¬†—В—А–∞–љ—Б–њ–ї–∞–љ—В–∞—В–∞ —Б–Њ—Б—В–∞–≤–ї—П–µ—В 65 –Є¬†62% —Б–Њ–Њ—В–≤–µ—В—Б—В–≤–µ–љ–љ–Њ –Є¬†—Б–Њ–њ–Њ—Б—В–∞–≤–Є–Љ–∞ —Б¬†—В–∞–Ї–Њ–≤—Л–Љ–Є –њ–Њ–Ї–∞–Ј–∞—В–µ–ї—П–Љ–Є –і—А—Г–≥–Є—Е –≥—А—Г–њ–њ –±–Њ–ї—М–љ—Л—Е —Б¬†–•–Я–Э. –Ґ—А–∞–љ—Б–њ–ї–∞–љ—В–∞—Ж–Є—П –њ–Њ—З–Ї–Є –њ–Њ–Ї–∞–Ј–∞–љ–∞ –±–Њ–ї—М–љ—Л–Љ —Б¬†–Љ–µ–і–ї–µ–љ–љ—Л–Љ –њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є–µ–Љ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ –±–µ–Ј –њ–Њ—А–∞–ґ–µ–љ–Є—П —Б–µ—А–і—Ж–∞ –Є¬†–Ц–Ъ–Ґ. –Р–Љ–Є–ї–Њ–Є–і–Њ–Ј –≤¬†—В—А–∞–љ—Б–њ–ї–∞–љ—В–Є—А–Њ–≤–∞–љ–љ–Њ–є –њ–Њ—З–Ї–µ –≤–Њ–Ј–љ–Є–Ї–∞–µ—В, –њ–Њ —А–∞–Ј–љ—Л–Љ –і–∞–љ–љ—Л–Љ, –њ—А–Є–Љ–µ—А–љ–Њ —Г¬†30% –±–Њ–ї—М–љ—Л—Е, –Њ–і–љ–∞–Ї–Њ –Њ–љ —Б–ї—Г–ґ–Є—В –њ—А–Є—З–Є–љ–Њ–є –њ–Њ—В–µ—А–Є —В—А–∞–љ—Б–њ–ї–∞–љ—В–∞—В–∞ –≤—Б–µ–≥–Њ —Г¬†2вАУ3% –њ–∞—Ж–Є–µ–љ—В–Њ–≤.
–Я—А–Њ–≥–љ–Њ–Ј –њ—А–Є –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–µ
–Р–Љ–Є–ї–Њ–Є–і–Њ–Ј —Е–∞—А–∞–Ї—В–µ—А–Є–Ј—Г–µ—В—Б—П –љ–µ—Г–Ї–ї–Њ–љ–љ–Њ –њ—А–Њ–≥—А–µ—Б—Б–Є—А—Г—О—Й–Є–Љ —В–µ—З–µ–љ–Є–µ–Љ. –Я—А–Њ–≥–љ–Њ–Ј –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П –Ј–∞–≤–Є—Б–Є—В –Њ—В —В–Є–њ–∞ –∞–Љ–Є–ї–Њ–Є–і–∞, —Б—В–µ–њ–µ–љ–Є –≤–Њ–≤–ї–µ—З–µ–љ–Є—П —А–∞–Ј–ї–Є—З–љ—Л—Е –Њ—А–≥–∞–љ–Њ–≤, –≥–ї–∞–≤–љ—Л–Љ –Њ–±—А–∞–Ј–Њ–Љ —Б–µ—А–і—Ж–∞ –Є¬†–њ–Њ—З–µ–Ї, –љ–∞–ї–Є—З–Є—П –Є¬†—Е–∞—А–∞–Ї—В–µ—А–∞ –њ—А–µ–і—А–∞—Б–њ–Њ–ї–∞–≥–∞—О—Й–µ–≥–Њ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П.
–Э–∞–Є–±–Њ–ї–µ–µ —Б–µ—А—М–µ–Ј–µ–љ –њ—А–Њ–≥–љ–Њ–Ј –њ—А–Є AL-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–µ. –Я–Њ –і–∞–љ–љ—Л–Љ –Ї–ї–Є–љ–Є–Ї–Є –Ь–µ–є–Њ, —Б—А–µ–і–љ—П—П –њ—А–Њ–і–Њ–ї–ґ–Є—В–µ–ї—М–љ–Њ—Б—В—М –ґ–Є–Ј–љ–Є –±–Њ–ї—М–љ—Л—Е —Н—В–Є–Љ —В–Є–њ–Њ–Љ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ —Б–Њ—Б—В–∞–≤–ї—П–µ—В –ї–Є—И—М 13,2¬†–Љ–µ—Б., 5-–ї–µ—В–љ—П—П –≤—Л–ґ–Є–≤–∞–µ–Љ–Њ—Б—В—М¬†вАУ 7%, 10-–ї–µ—В–љ—П—П¬†вАУ –≤—Б–µ–≥–Њ 1%. –Я—А–Є —Н—В–Њ–Љ —Б–∞–Љ–∞—П –љ–Є–Ј–Ї–∞—П –њ—А–Њ–і–Њ–ї–ґ–Є—В–µ–ї—М–љ–Њ—Б—В—М –ґ–Є–Ј–љ–Є –Њ—В–Љ–µ—З–µ–љ–∞ —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–Ј–∞—Б—В–Њ–є–љ–Њ–є –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В—М—О –Ї—А–Њ–≤–Њ–Њ–±—А–∞—Й–µ–љ–Є—П (6¬†–Љ–µ—Б.) –Є¬†–Њ—А—В–Њ—Б—В–∞—В–Є—З–µ—Б–Ї–Њ–є –∞—А—В–µ—А–Є–∞–ї—М–љ–Њ–є –≥–Є–њ–Њ—В–Њ–љ–Є–µ–є (8¬†–Љ–µ—Б.) [2, 44, 45]. –Э–∞–Є–±–Њ–ї–µ–µ —З–∞—Б—В—Л–Љ–Є –њ—А–Є—З–Є–љ–∞–Љ–Є —Б–Љ–µ—А—В–Є –±–Њ–ї—М–љ—Л—Е —Б¬†AL-—В–Є–њ–Њ–Љ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ –±—Л–≤–∞—О—В —Б–µ—А–і–µ—З–љ–∞—П –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В—М –Є¬†–љ–∞—А—Г—И–µ–љ–Є—П —А–Є—В–Љ–∞ —Б–µ—А–і—Ж–∞ (48%), —Г—А–µ–Љ–Є—П (15%), —Б–µ–њ—Б–Є—Б –Є¬†–Є–љ—Д–µ–Ї—Ж–Є–Є (8%). –Э–µ—Б–Љ–Њ—В—А—П –љ–∞ —В–Њ —З—В–Њ —Б–Љ–µ—А—В—М –Њ—В —Г—А–µ–Љ–Є–Є –Њ—В–Љ–µ—З–∞—О—В –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ —А–µ–ґ–µ, —З–µ–Љ –Њ—В –Ї–∞—А–і–Є–∞–ї—М–љ—Л—Е –њ—А–Є—З–Є–љ, –•–Я–Э —А–∞–Ј–љ–Њ–є —Б—В–µ–њ–µ–љ–Є –≤—Л—А–∞–ґ–µ–љ–љ–Њ—Б—В–Є —А–µ–≥–Є—Б—В—А–Є—А—Г—О—В –±–Њ–ї–µ–µ —З–µ–Љ —Г¬†60% —Г–Љ–µ—А—И–Є—Е.
–Я—А–Њ–≥–љ–Њ–Ј –њ—А–Є –Р–Р-–∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–µ –±–Њ–ї–µ–µ –±–ї–∞–≥–Њ–њ—А–Є—П—В–µ–љ –Є¬†–Ј–∞–≤–Є—Б–Є—В, –≥–ї–∞–≤–љ—Л–Љ –Њ–±—А–∞–Ј–Њ–Љ, –Њ—В —Е–∞—А–∞–Ї—В–µ—А–∞ –њ—А–µ–і—А–∞—Б–њ–Њ–ї–∞–≥–∞—О—Й–µ–≥–Њ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П –Є¬†–≤–Њ–Ј–Љ–Њ–ґ–љ–Њ—Б—В–Є –µ–≥–Њ –Ї–Њ–љ—В—А–Њ–ї—П. –°—А–µ–і–љ—П—П –њ—А–Њ–і–Њ–ї–ґ–Є—В–µ–ї—М–љ–Њ—Б—В—М –ґ–Є–Ј–љ–Є –±–Њ–ї—М–љ—Л—Е —Б¬†—Н—В–Є–Љ —В–Є–њ–Њ–Љ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ –Њ—В –Љ–Њ–Љ–µ–љ—В–∞ –≤–µ—А–Є—Д–Є–Ї–∞—Ж–Є–Є –і–Є–∞–≥–љ–Њ–Ј–∞ —Б–Њ—Б—В–∞–≤–ї—П–µ—В 30вАУ60¬†–Љ–µ—Б. (–±–Њ–ї—М—И–∞—П –њ—А–Є –≤—В–Њ—А–Є—З–љ–Њ–Љ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–µ, –Љ–µ–љ—М—И–∞—П –њ—А–Є –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–µ –≤¬†—А–∞–Љ–Ї–∞—Е –њ–µ—А–Є–Њ–і–Є—З–µ—Б–Ї–Њ–є –±–Њ–ї–µ–Ј–љ–Є). –≠—Д—Д–µ–Ї—В–Є–≤–љ–Њ–µ –ї–µ—З–µ–љ–Є–µ –њ—А–µ–і—А–∞—Б–њ–Њ–ї–∞–≥–∞—О—Й–Є—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є, –≤¬†—В–Њ–Љ —З–Є—Б–ї–µ –њ–Њ–ї–љ–Њ–µ –Є–Ј–ї–µ—З–µ–љ–Є–µ —В—Г–±–µ—А–Ї—Г–ї–µ–Ј–∞ –Є–ї–Є —Е—А–Њ–љ–Є—З–µ—Б–Ї–Є—Е –љ–∞–≥–љ–Њ–µ–љ–Є–є, –љ–µ –Є—Б–Ї–ї—О—З–∞–µ—В –≤–Њ–Ј–Љ–Њ–ґ–љ–Њ—Б—В–Є —А–∞–Ј–≤–Є—В–Є—П –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ –≤¬†–і–∞–ї—М–љ–µ–є—И–µ–Љ, –Њ–і–љ–∞–Ї–Њ –Ј–∞–Љ–µ–і–ї—П–µ—В –µ–≥–Њ –њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є–µ, —Г–ї—Г—З—И–∞—П –њ—А–Њ–≥–љ–Њ–Ј. –≠—Д—Д–µ–Ї—В–Є–≤–љ–∞—П —В–µ—А–∞–њ–Є—П —А–µ–≤–Љ–∞—В–Њ–Є–і–љ–Њ–≥–Њ –∞—А—В—А–Є—В–∞ –њ–Њ–Ј–≤–Њ–ї—П–µ—В –њ—А–Њ–і–ї–Є—В—М —В–µ—З–µ–љ–Є–µ –∞–Љ–Є–ї–Њ–Є–і–љ–Њ–є –љ–µ—Д—А–Њ–њ–∞—В–Є–Є, –Ј–∞–Љ–µ–і–ї—П—П –љ–∞—Б—В—Г–њ–ї–µ–љ–Є–µ –•–Я–Э. –Ю—Б–љ–Њ–≤–љ–Њ–є –њ—А–Є—З–Є–љ–Њ–є —Б–Љ–µ—А—В–Є –±–Њ–ї—М–љ—Л—Е –Р–Р-—В–Є–њ–Њ–Љ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ —Б–ї—Г–ґ–Є—В –њ–Њ—З–µ—З–љ–∞—П –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В—М.
–Ф–Њ—Б—В–Є–ґ–µ–љ–Є—П –њ–Њ—Б–ї–µ–і–љ–Є—Е –ї–µ—В –≤¬†–Є–Ј—Г—З–µ–љ–Є–Є –њ—А–Њ–±–ї–µ–Љ—Л –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞, –њ–Њ–Ј–≤–Њ–ї–Є–≤—И–Є–µ —Б—Д–Њ—А–Љ—Г–ї–Є—А–Њ–≤–∞—В—М —З–µ—В–Ї–Є–µ –Ї—А–Є—В–µ—А–Є–Є –Ї–ї–∞—Б—Б–Є—Д–Є–Ї–∞—Ж–Є–Є –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є—Е —Д–Њ—А–Љ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ –Є¬†–њ–Њ–і—Е–Њ–і—Л –Ї¬†–ї–µ—З–µ–љ–Є—О, –і–∞–ї–Є –≤–Њ–Ј–Љ–Њ–ґ–љ–Њ—Б—В—М —Б—Г—Й–µ—Б—В–≤–µ–љ–љ–Њ —Г–ї—Г—З—И–Є—В—М –њ—А–Њ–≥–љ–Њ–Ј –±–Њ–ї—М–љ—Л—Е —А–∞–Ј–љ—Л–Љ–Є —В–Є–њ–∞–Љ–Є –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞.
–£–≤–∞–ґ–∞–µ–Љ—Л–є –њ–Њ—Б–µ—В–Є—В–µ–ї—М uMEDp!
–£–≤–µ–і–Њ–Љ–ї—П–µ–Љ –Т–∞—Б –Њ —В–Њ–Љ, —З—В–Њ –Ј–і–µ—Б—М —Б–Њ–і–µ—А–ґ–Є—В—Б—П –Є–љ—Д–Њ—А–Љ–∞—Ж–Є—П, –њ—А–µ–і–љ–∞–Ј–љ–∞—З–µ–љ–љ–∞—П –Є—Б–Ї–ї—О—З–Є—В–µ–ї—М–љ–Њ –і–ї—П —Б–њ–µ—Ж–Є–∞–ї–Є—Б—В–Њ–≤ –Ј–і—А–∞–≤–Њ–Њ—Е—А–∞–љ–µ–љ–Є—П.
–Х—Б–ї–Є –Т—Л –љ–µ —П–≤–ї—П–µ—В–µ—Б—М —Б–њ–µ—Ж–Є–∞–ї–Є—Б—В–Њ–Љ –Ј–і—А–∞–≤–Њ–Њ—Е—А–∞–љ–µ–љ–Є—П, –∞–і–Љ–Є–љ–Є—Б—В—А–∞—Ж–Є—П –љ–µ –љ–µ—Б–µ—В –Њ—В–≤–µ—В—Б—В–≤–µ–љ–љ–Њ—Б—В–Є –Ј–∞ –≤–Њ–Ј–Љ–Њ–ґ–љ—Л–µ –Њ—В—А–Є—Ж–∞—В–µ–ї—М–љ—Л–µ –њ–Њ—Б–ї–µ–і—Б—В–≤–Є—П, –≤–Њ–Ј–љ–Є–Ї—И–Є–µ –≤ —А–µ–Ј—Г–ї—М—В–∞—В–µ —Б–∞–Љ–Њ—Б—В–Њ—П—В–µ–ї—М–љ–Њ–≥–Њ –Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–љ–Є—П –Т–∞–Љ–Є –Є–љ—Д–Њ—А–Љ–∞—Ж–Є–Є —Б –њ–Њ—А—В–∞–ї–∞ –±–µ–Ј –њ—А–µ–і–≤–∞—А–Є—В–µ–ї—М–љ–Њ–є –Ї–Њ–љ—Б—Г–ї—М—В–∞—Ж–Є–Є —Б –≤—А–∞—З–Њ–Љ.
–Э–∞–ґ–Є–Љ–∞—П –љ–∞ –Ї–љ–Њ–њ–Ї—Г ¬Ђ–Т–Њ–є—В–Є¬ї, –Т—Л –њ–Њ–і—В–≤–µ—А–ґ–і–∞–µ—В–µ, —З—В–Њ —П–≤–ї—П–µ—В–µ—Б—М –≤—А–∞—З–Њ–Љ –Є–ї–Є —Б—В—Г–і–µ–љ—В–Њ–Љ –Љ–µ–і–Є—Ж–Є–љ—Б–Ї–Њ–≥–Њ –≤—Г–Ј–∞.