Наследственный транстиретиновый амилоидоз: новая выявленная амилоидогенная мутация
- Аннотация
- Статья
- Ссылки
- English
Введение
Наследственный транстиретиновый амилоидоз (ATTR-амилоидоз) – редкое заболевание, обусловленное диффузным отложением патологического фибриллярного белка (амилоида) и приводящее к поражению различных органов и систем [1]. Описаны как семейные случаи ATTR-амилоидоза с аутосомно-доминантным типом передачи, так и спорадические. Распространенность ATTR-амилоидоза в Европе и США составляет 1 на 100 тыс. человек [2].
Заболевание вызывает мутация в гене, отвечающем за синтез транспортного белка транстиретина. Транстиретин синтезируется печенью (90–95%), а также сосудистыми сплетениями желудочков головного мозга, пигментным слоем сетчатки глаза и обеспечивает перенос тироксина и ретинола в организме. В кровеносном русле белок циркулирует в виде тетрамера. Однако в результате мутации его структура становится нестабильной, что ведет к распаду транстиретина на мономеры, агрегация которых в свою очередь приводит к образованию нерастворимых фибрилл амилоида [1, 3].
В настоящее время известно более 120 мутаций, но не все они относятся к амилоидогенным, то есть вызывающим заболевание [4]. В зависимости от обнаруженной мутации клиническая картина ATTR-амилоидоза может значительно варьироваться, даже при одной и той же мутации у родственников первой линии. Наиболее изученной и распространенной амилоидогенной мутацией в мире является Val30Met, для которой характерно развитие неуклонно прогрессирующей сенсорно-моторной полиневропатии в сочетании с вегетативными нарушениями (ортостатической гипотензией, нарушением моторики желудочно-кишечного тракта, потоотделением, тазовыми нарушениями, эректильной дисфункцией) [4–6].
Далее в статье рассмотрен клинический случай спорадической мутации гена транстиретина, которую ранее не относили к амилоидогенным, но результаты генетического и морфологического исследований подтвердили ATTR-амилоидоз.
Клинический случай
Жалобы
В неврологическое отделение поступила пациентка Ф., 62 года, с жалобами:
- на частые эпизоды предобморочных и обморочных состояний на фоне выраженного снижения артериального давления;
- колебания артериального давления при перемене положения тела;
- ощущение жжения в области лопаток, распространяющееся вниз по спине, задне-наружной поверхности бедер и голеней до уровня стоп, усиливающееся в ночное время;
- нарушение мочеиспускания в виде упускания порции мочи без предшествующего позыва;
- плохой аппетит, чувство тяжести в желудке после приема небольшой порции пищи;
- общую слабость, утомляемость;
- трудности при засыпании, сноговорение.
Анамнез жизни
Сопутствующие заболевания – нарушение толерантности к глюкозе, язвенная болезнь желудка, хронический пиелонефрит. В течение 15 лет отмечалось повышение артериального давления до 180–190/100 мм рт. ст., пациентка получала гипотензивную терапию. С 30-летнего возраста – сноговорение. Наследственный анамнез по неврологическим заболеваниям не отягощен.
Анамнез заболевания
В 2013 г. в возрасте 57 лет пациентка впервые отметила эпизоды упускания мочи при отсутствии позыва к мочеиспусканию. Наблюдалась гинекологом, урологом, но доказательств патологии органов мочеполовой системы не выявлено.
Через год появились эпизоды снижения артериального давления при переходе в положение стоя, которые со временем участились, а также преходящее ощущение жжения между лопаток.
Еще через год на фоне выраженного снижения артериального давления до 70/50 мм рт. ст. впервые развилось обморочное состояние. Ощущение жжения между лопаток стало усиливаться и распространяться вниз по спине на нижние конечности. В связи с подозрением на патологию сердечно-сосудистой системы было проведено кардиологическое обследование – холтеровское мониторирование, эхокардиография, коронарография. Данных, подтверждающих кардиальную патологию, не получено.
С конца 2017 г. колебания артериального давления (от 70/50 до 180/90 мм рт. ст.) и ощущение жжения стали беспокоить ежедневно, ортостатические обмороки участились до нескольких раз в месяц. Пациентка стала отмечать чувство тяжести в желудке после приема пищи.
В связи с отсутствием органических нарушений со стороны сердечно-сосудистой системы пациентка получила направление в неврологическое отделение для дообследования. В неврологическом статусе обращает внимание двусторонний полуптоз. Косоглазия, диплопии нет. Движения глазных яблок в полном объеме. Сила мышц конечностей и туловища сохранена. Глубокие рефлексы на руках и ногах живые, симметричные. Положительный патологический кистевой симптом Тремнера с двух сторон. Патологические стопные рефлексы не вызываются. При исследовании болевой чувствительности выявляется гиперестезия в стопах. Глубокая чувствительность сохранена. Динамические координаторные пробы выполняет удовлетворительно. В позе Ромберга отмечается неустойчивость из-за ортостатической гипотензии. Ортостатическая проба положительная: на четвертой минуте исследование было прервано в связи с развитием липотимического состояния. Тазовые функции: упускание мочи, отсутствие позывов к мочеиспусканию.
Лабораторные и инструментальные исследования
Общий анализ крови и мочи, биохимический анализ крови – без патологии.
Для исключения эпилептической природы пароксизмальных расстройств сознания проводилось электроэнцефалографическое исследование. Эпилептическая активность не выявлена.
Магнитно-резонансная томография головы: в перивентрикулярных зонах обнаружен лейкоареоз у передних рогов боковых желудочков, в остальном – в пределах возрастной нормы.
С учетом того, что эндокринные нарушения могли стать причиной колебаний артериального давления, проведена компьютерная томография надпочечников – без патологии.
При электронейромиографическом исследовании нервов конечностей установлено поражение моторных и сенсорных волокон правого локтевого нерва в области локтевого сустава (синдром кубитального канала).
Количественное сенсорное тестирование показало повышение порогов тепловой чувствительности в дистальных отделах ног, свидетельствующее о вовлечении в патологический процесс тонких волокон нервов нижних конечностей. Во время количественного сенсорного тестирования отмечались парестезии (жжение, покалывание) и дизестезия (восприятие холода как тепло).
С целью выяснения причины прогрессирующей сенсорно-вегетативной невропатии проведено иммунохимическое исследование белков сыворотки и мочи для исключения системного AL-амилоидоза. Концентрация поликлональных иммуноглобулинов была в пределах нормы, моноклональная секреция отсутствовала.
Молекулярно-генетическое исследование: выявлен вариант нуклеотидной последовательности в первом экзоне гена TTR в гетерозиготном состоянии Arg5His, которая, согласно базе данных ClinVar, в настоящее время трактуется как вариант неопределенной значимости и не может рассматриваться в качестве причины заболевания.
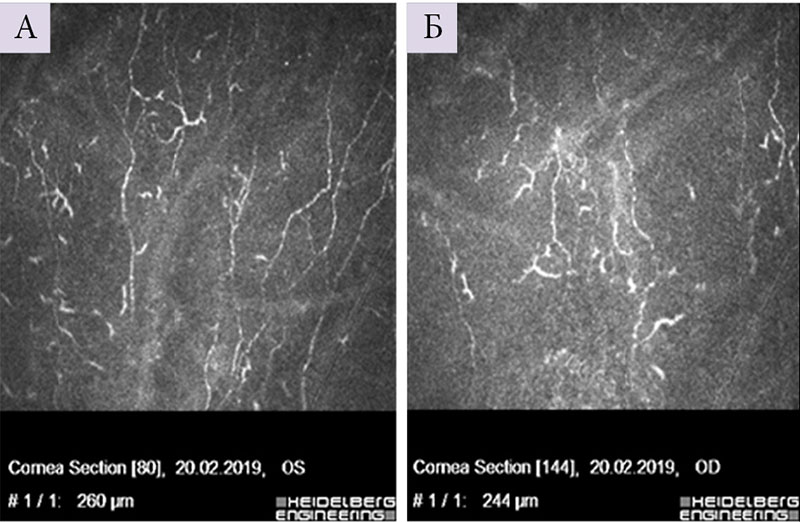
Офтальмолог: признаки псевдоэксфолиативного синдрома в виде легкой атрофии радужки, неравномерной структуры зрачковой каймы, на хрусталике наслоения амилоида не визуализируются. Нервные волокна роговицы сильно извиты, имеют четкообразную структуру, что свидетельствует об их поражении (рис. 1).
Эндокринолог: нарушение толерантности к глюкозе. Ортостатическая гипотензия не связана с нарушением углеводного обмена.
Сомнолог: хроническая инсомния, нарушение восприятия сна, парасомнии.
Проведена сцинтиграфия миокарда с пирофосфатом: сцинтиграфическая картина не характерна для транстиретинового амилоидоза.
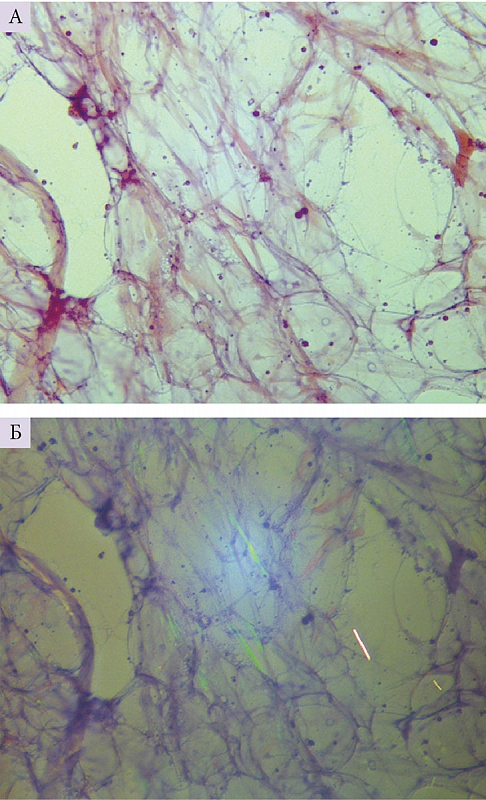
С целью морфологической верификации амилоидоза изучены биоптаты слизистой оболочки прямой кишки, кожи и подкожной жировой клетчатки живота. При исследовании биоптатов слизистой прямой кишки и кожного лоскута тыльной поверхности стопы депозитов амилоида не обнаружено. В подкожной жировой клетчатке живота при окраске конго красным и исследовании в поляризованном свете определялись множественные мелкие депозиты амилоида между мембранами липоцитов по Визуальной шкале оценки от CR2+ до СR3+ (рис. 2). Типирование выявленного амилоида не проводилось из-за скудности субстрата.
Диагноз
На основании клинической картины, генетического исследования и обнаружения депозитов амилоида в подкожной жировой клетчатке установлен диагноз «транстиретиновый амилоидоз с преимущественным поражением периферической вегетативной нервной системы».
Лечение
Назначено симптоматическое лечение:
- флудрокортизон 100 мкг/сут;
- габапентин 900 мг/сут, флуоксетин 20 мг/сут (с целью купирования невропатического болевого синдрома);
- метформин 1500 мг/сут.
На фоне терапии уменьшились суточные колебания артериального давления, не отмечалось синкопальных состояний, менее выраженным стало ощущение жжения в области спины и нижних конечностей.
В качестве патогенетической терапии с целью замедления прогрессирования заболевания пациентке рекомендован пожизненный прием тафамидиса 20 мг/сут.
Обсуждение
Рассматриваемый клинический случай представляет большой интерес, поскольку выявленная у пациентки мутация гена транстиретина ранее считалась клинически незначимой. Симптомы заболевания развивались постепенно в течение нескольких лет, отмечалось прогредиентное течение болезни без ремиссий и обострений. Доминировали симптомы вегетативной недостаточности, начавшиеся с тазовых нарушений с дальнейшим присоединением ортостатической гипотензии и нарушений моторики желудочно-кишечного тракта. При поступлении в клинику основным дезадаптирующим фактором было выраженное колебание артериального давления, сопровождающееся потерей сознания. В качестве основных неврологических синдромов у пациентки следует рассматривать:
- синдром вегетативных нарушений – ортостатическую гипотензию, нарушение моторики желудочно-кишечного тракта, нейрогенную дисфункцию мочевого пузыря;
- синдром чувствительных нарушений – полиневропатический тип расстройства поверхностной (болевой, температурной) чувствительности в дистальных отделах нижних конечностей;
- нарушения сна – хроническую инсомнию, парасомнии (расстройство поведения в быструю фазу сна).
Таким образом, у пациентки отмечалась прогредиентно текущая сенсорно-вегетативная невропатия, обусловленная поражением чувствительных и вегетативных волокон нервов конечностей. Кроме того, не исключалось вовлечение в патологический процесс вегетативных ганглиев и сплетений. Двусторонний полуптоз, наблюдаемый в течение длительного времени, возможно, обусловлен нарушением симпатической иннервации мюллеровой мышцы, участвующей в подъеме века. Выявленная при генетическом исследовании мутация гена транстиретина, а также отложения амилоида в подкожно-жировой клетчатке живота позволили диагностировать ATTR-амилоидоз.
Дифференциальный диагноз проводился с наиболее частыми причинами сенсорно-вегетативных нарушений метаболического генеза, поскольку у пациентки имело место нарушение толерантности к глюкозе. Однако выраженная ортостатическая гипотензия в совокупности с другими симптомами не позволила рассматривать нарушение углеводного обмена в качестве этиологического фактора развития и прогрессирования неврологических расстройств. Результаты иммуногистохимического исследования сыворотки крови и мочи исключили диагноз AL-амилоидоза как причину невропатии тонких волокон [7]. Из других более редких причин вегетативных нарушений стоит отметить первичную прогрессирующую вегетативную недостаточность (идиопатическую изолированную вегетативную недостаточность, синдром Шая – Дрейджера, различные наследственные сенсорно-вегетативные полиневропатии) [8].
Генетическое исследование, проведенное детям пациентки (сын 34 года и дочь 40 лет), не подтвердило носительство мутации, что говорит о спорадическом случае с поздним (после 50 лет) дебютом.
Критерии диагностики транстиретинового амилоидоза – наличие прогрессирующей сенсорно-моторной полиневропатии, и/или вегетативной дисфункции, и/или туннельной невропатии в сочетании с более чем одним из следующих признаков: семейным анамнезом, необъяснимым снижением веса, патологией висцеральных органов и органа зрения [9]. Характерные для ATTR-амилоидоза клинические симптомы требуют подтверждения диагноза генетическими и морфологическими методами, в частности выявления амилоидогенной мутации гена транстиретина и отложения амилоида в тканях. Необходимо подчеркнуть, что для случаев ATTR-амилоидоза характерен выраженный клинический полиморфизм, затрудняющий диагностику, особенно на ранних стадиях болезни. В частности, в приведенном клиническом случае у пациентки на протяжении длительного времени превалировали расстройства вегетативной нервной системы и отсутствовали характерные симптомы сенсорно-моторной полиневропатии.
Для диагностики амилоидной невропатии широко используются нейрофизиологические методы исследования: электронейромиография, количественное сенсорное тестирование, позволяющее оценить состояние толстых и тонких нервных волокон. Для установления вегетативной дисфункции и определения ее выраженности проводят ортостатическую пробу, оценку вариабельности сердечного ритма [10].
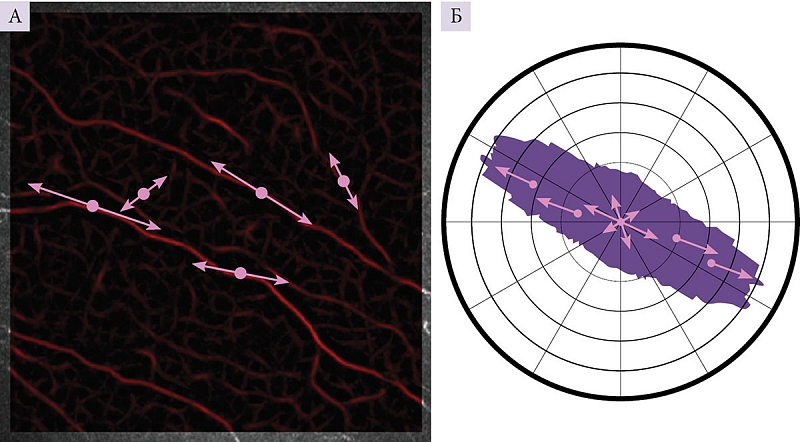
Все шире для диагностики невропатий с поражением тонких волокон периферических нервов, в том числе амилоидной невропатии, используется конфокальная микроскопия роговицы с возможностью визуализации и последующей оценки хода и структуры нервных волокон роговицы при системном амилоидозе [11]. В представленном наблюдении применялся авторский алгоритм при помощи разработанной компьютерной программы Liner 1.2 S, позволяющей учитывать состояние структуры нервных волокон роговицы на основе оценки направленности и извитости нервных волокон. Для оценки степени извитости нервных волокон роговицы были разработаны коэффициенты анизотропии (KΔL) и симметричности (Ksym) направленности нервных волокон роговицы. Формализация изображения состоит в том, что нервное волокно на конфокальном изображении представляет собой относительно более светлую полосу определенной ширины на темном фоне. После математического обсчета с применением модельной функции строится роза – диаграмма направленности. Чем более однонаправленны нервные волокна роговицы (и следовательно, более вытянута форма розы), тем меньше вероятность полиневропатии, и наоборот (рис. 3) [12–14]. При развитии полиневропатии структура безмиелиновых тонких нервных волокон роговицы страдает в первую очередь, что делает метод конфокальной микроскопии роговицы особенно информативным для выявления начальных изменений нервных волокон роговицы.
Среди основных направлений лечения транстиретинового амилоидоза необходимо выделить этиотропную терапию, направленную на подавление синтеза мутантных форм транстиретина, патогенетическую терапию, позволяющую предотвратить последующее отложение патологического белка в тканях, и симптоматическую поддержку.
До недавнего времени единственным методом лечения ATTR-амилоидоза была пересадка печени с целью устранения основного источника патологического белка – транстиретина. Трансплантация печени позволяет существенно улучшить прогноз, увеличить выживаемость больных и существенно замедлить темп развития заболевания, но не остановить его прогрессирование. Известно, что уже имеющиеся депозиты амилоида способны продолжать самостоятельный рост за счет неамилоидогенных вариантов белков-предшественников подобно кристаллу-затравке в насыщенном растворе соли. Было показано, что через пять лет после трансплантации печени в составе старых амилоидных депозитов начинает преобладать нормальный немутантный транстиретин [15].
С учетом объема оперативного вмешательства, необходимости дальнейшего приема иммуносупрессивных препаратов, а также возможности рецидива заболевания вследствие других источников синтеза транстиретина разрабатывались консервативные методы лечения, позволяющие замедлить прогрессирование заболевания [10]. В частности, были созданы препараты тафамидис и дифлунизал – стабилизаторы транстиретина в физиологической тетрамерной форме циркуляции в крови, которые предотвращают ее распад на амилоидогенные мономеры транстиретина. Существенный недостаток дифлунизала – его побочные эффекты, характерные для всей группы нестероидных противовоспалительных средств, к которым он относится: ульцерогенное действие, лекарственный интерстициальный нефрит с развитием почечной недостаточности. Более благоприятный профиль безопасности имеет тафамидис. Он в отличие от дифлунизала зарегистрирован в России и рекомендован для приема в дозе 20 мг/сут внутрь.
За последний год были получены данные об эффективности новых лекарственных средств (например, патисирана), которые подавляют синтез белка транстиретина путем РНК-интерференции, что вызывает деградацию мРНК, приводит к уменьшению уровня сывороточного транстиретина и отложения его в тканях [16]. Предварительные результаты применения этих препаратов указывают на их высокую эффективность. Однако необходимо иметь в виду, что подобные лекарственные средства представляют собой неполный фармакологический аналог метода трансплантации печени и, значит, могут иметь схожие с трансплантацией недостатки – замедлять, но не останавливать прогрессирование амилоидоза.
Нужно также отметить, что физиологические функции транстиретина недостаточно изучены. Его функция как транспортного белка представляется маловостребованной организмом, транспорт гормонов щитовидной железы осуществляется преимущественно альбумином. Показано, что в процессе эволюции производство транстиретина печенью началось достаточно поздно, синтез этого белка гораздо раньше был освоен тканями головного мозга. Считается, что транстиретин необходим для элиминации бета-белка из головного мозга. Нельзя исключить, что блокада синтеза транстиретина может стать фактором накопления в ткани мозга бета-белка и, следовательно, причиной продукции бета-амилоида, стимулируя развитие или прогрессирование болезни Альцгеймера и амилоидной церебральной ангиопатии.
Заключение
Представленный случай иллюстрирует клинический полиморфизм ATTR-амилоидоза, затрудняющий постановку диагноза, особенно на ранних стадиях болезни. Неуклонно прогрессирующая вегетативная недостаточность в отсутствие наиболее частых метаболических причин ее развития позволила предположить амилоидогенную природу заболевания, что было подтверждено генетическим и морфологическим исследованием.
Авторы заявляют об отсутствии конфликта интересов.
O.Ye. Zinovyeva, MD, PhD, Prof., E.I. Safiulina, N.S. Shcheglova, PhD, V.V. Rameyev, PhD, Z.V. Surnina, PhD, Ye.A. Stepanova, Ye.M. Gordeyeva
I.M. Sechenov First Moscow State Medical University
Scientific Research Institute of Eye Diseases, Moscow
City Clinical Hospital named after V.M. Buyanov, Moscow
Russian Medical Academy of Continuous Professional Education, Moscow
Contact person: Olga Ye. Zinovyeva, zinovyevaolga@yandex.ru
The article describes a clinical case of transthyretin hereditary amyloidosis. Found gene mutation of Arg5His transthyretin previously assigned to mutations of uncertain significance. However, progressive neurological symptoms in the form of predominant damage of the autonomic nervous system and the detection of amyloid deposits in subcutaneous fat pointed to the amyloidogenic nature of the mutation. The genetic study showed the absence of the mutation in the patient's children, which speaks in favor of a sporadic case of the disease with a late debut.
Уважаемый посетитель uMEDp!
Уведомляем Вас о том, что здесь содержится информация, предназначенная исключительно для специалистов здравоохранения.
Если Вы не являетесь специалистом здравоохранения, администрация не несет ответственности за возможные отрицательные последствия, возникшие в результате самостоятельного использования Вами информации с портала без предварительной консультации с врачом.
Нажимая на кнопку «Войти», Вы подтверждаете, что являетесь врачом или студентом медицинского вуза.