Клинический случай пациента школьного возраста с заболеванием спектра синдрома Ретта и делецией в гене MECP2
- Аннотация
- Статья
- Ссылки
- English
![Таблица 1. Диагностические критерии синдрома Ретта, 2010 г. [5]](/upload/resize_cache/iblock/640/800_800_1/Toropina1.jpg)


Введение
Синдром Ретта (СР) – тяжело протекающее генетическое заболевание, для которого характерно поражение центральной нервной системы. Встречается почти исключительно у девочек с частотой 1 случай на 10 000–20 000 лиц женского пола [1]. Заболевание названо в честь венского педиатра Андреаса Ретта, в 1960 г. описавшего двух девочек, ожидавших приема в клинике. Внимание врача привлекли одинаковое выражение их лиц и идентичные стереотипные движения в руках по типу моющих. При обследовании у обеих пациенток оказался схожий анамнез: нормальное раннее развитие с последующим регрессом и появлением бесцельных движений в руках. В дальнейшем Ретт выявил еще несколько похожих клинических случаев и предположил, что они могут быть связаны с гиперамониемией [2]. Удивительно, но опубликованное описание клинических случаев не вызвало широкого интереса, и лишь спустя 17 лет синдром Ретта был признан медицинским сообществом благодаря английской публикации шведского врача Бенгта Харберга, который проанализировал 35 случаев заболевания и предложил эпоним для его названия [3]. В 1999 г. была установлена связь СР с мутацией гена MECP2 (methyl-CpG-bilding protein 2) – регулятора транскрипции methyl-CpG-связывающего протеина 2 на Х-хромосоме [4]. Затем были идентифицированы другие мутации, ассоциированные с СР – гена CDKL5 (Х-linked cyclin-dependent kinase-like 5) и гена FOXG1 (forkhead box G1) [5].
В классическом варианте заболевание дебютирует после периода нормального развития ребенка. С 2–4 месяцев жизни может отмечаться замедление прироста окружности головы, что служит важным критерием диагностики (задержка внутриутробного развития и микроцефалия при рождении относятся к критериям, исключающим типичный СР). На втором полугодии жизни ребенка происходит регресс в психомоторном развитии, утрачивается способность целенаправленных движений в руках. В период от года до трех лет присоединяются симптомы аутистического поведения. Речь, если начинала формироваться, полностью утрачивается, возникает умственная отсталость, появляются стереотипные движения рук, нарушается походка, обнаруживается расстройство движений по типу дистонии, хореоатетоза, атаксии и миоклонуса, а также развиваются нарушения дыхания: чередование эпизодов гипервентиляции и апноэ. Имеют место характерные стереотипии рук: моющие движения, потирание, заламывание, вращение, хлопанье в ладоши, держание рук во рту. Пациентам свойственна общая заторможенность. Выражение лица безучастное, взгляд пристальный или в одну точку, возможны насильственный смех и приступы импульсивного поведения. Затем следует период стабилизации состояния или частичного восстановления утраченных функций: аутистические черты и общее беспокойство пациентов уменьшаются, улучшается сон.
Эпилептические припадки развиваются у 60–80% больных, чаще в возрасте 5–15 лет, с пиком развития в 7–12 лет [6, 7]. Относительно типа припадков данные разнятся. Зарубежные авторы указывают на преимущественно фокальные и билатеральные тонико-клонические приступы, в том числе фебрильные в раннем возрасте [7]. По данным отечественных авторов, чаще наблюдаются эпилептический миоклонус и абсансы [3]. Припадки часто фармакорезистентны.
На поздней стадии заболевания, обычно в пубертатном периоде, ухудшается двигательный статус пациентов, развиваются и нарастают спастические парезы, дистония, не исключен паркинсонизм. Нарастание выраженности двигательных расстройств приводит к формированию ортопедических деформаций по типу сколиоза или кифосколиоза, стопы «балерины». В конечном итоге пациенты перестают самостоятельно передвигаться и имеют склонность к кахексии [3, 7].
Такая картина заболевания характерна для типичного СР, однако описаны и атипичные случаи с более ранним или поздним дебютом заболевания, более мягким фенотипом, легким интеллектуальным дефицитом, относительной сохранностью речевой функции, минимальными двигательными нарушениями или, напротив, с более выраженной клинической симптоматикой и дебютом эпилепсии до одного года [5, 8].
С классической формой СР ассоциируются преимущественно патогенные варианты гена MECP2, возникающие de novo. Эту мутацию выявляют более чем в 95% случаев типичного СР. При атипичном СР она также встречается, но в меньшей пропорции – у 75% пациентов [9]. MECP2 экспрессируется во всех тканях, но особенно обильно в нейронах головного мозга. Уровень MECP2 повышается во время созревания нейронов и синаптогенеза, достигая пика в постмиграционной фазе. Предполагается, что MECP2 оказывает модулирующее влияние на активность и пластичность нейронов во время их созревания; изменение строения MECP2 приводит к повреждению этого процесса, нарушению функции прежде всего лобных отделов мозга и, как следствие, деафферентации иерархически ниже стоящих отделов коры и подкорковых структур [3]. Мутация MECP2 описана не только при СР. Так, аналогичный дефект выявляется у пациентов с расстройством аутистического спектра, при изолированной умственной отсталости, детской шизофрении, системной красной волчанке [3, 10].
Спектр мутаций MECP2 включает точечные вставки, дупликации, делеции разного размера. Из всего разнообразия выделяют восемь основных точечных мутаций (типа миссенс- и нонсенс-мутаций), которые составляют примерно 70% всех встречающихся при СР (следует отметить, что крупные делеции наблюдаются только в 5% случаев). Статус мутации MECP2 определяет тяжесть заболевания. Наиболее тяжелые клинические проявления описаны у пациентов с крупными делециями. Тем не менее у пациентов с одинаковой мутацией фенотип, как правило, сильно варьирует, что обусловлено различиями в инактивации Х-хромосомы и возможной мутацией второго гена, модифицирующего проявления MECP2 [10]. Х-сцепленная мутация MECP2 предполагает высокую эмбриональную смертность среди мальчиков. Тем не менее единичные младенцы мужского пола рождаются жизнеспособными и могут иметь широкий диапазон расстройств – от тяжелой неонатальной энцефалопатии с ранней смертью в младенчестве до менее тяжелого нейропсихиатрического фенотипа СР [11].
Мутации CDKL5 и FOXG1 в большей степени связаны с атипичным СР или фенотипом, похожим на СР. Общность фенотипических проявлений при мутациях разных генов образует так называемый спектр СР [9]. Ген CDKL5 принимает непосредственное участие в создании белка, необходимого для развития нейрональных связей. Он широко экспрессируется в тканях организма, но преимущественно в головном мозге в перинатальном и раннем постнатальном периодах. Мутация CDKL5 ассоциируется с ранней энцефалопатией и ранним дебютом эпилепсии рефрактерного течения [12]. Ген FOXG1 относится к семейству транскрипционных факторов, также играющих роль в развитии мозга. При мутациях FOXG1 описаны такие нарушения, как постнатальная микроцефалия, нарушения формирования корковых извилин, микро- и пахигирия, задержка миелинизации, гиперкинезы, эпилептические припадки [13].
Таким образом, мутации MECP2, CDKL5 и FOXG1 могут наблюдаться не только при СР и не могут служить необходимым или достаточным критерием его диагностики. Диагноз СР устанавливают на основании фенотипа пациента с учетом критериев, представленных в табл. 1. Для постановки диагноза типичной формы СР требуется выполнение всех основных критериев и всех критериев исключения, для атипичной – выполнение двух из четырех основных критериев и пяти из 11 поддерживающих [5]. Термин «заболевание спектра синдрома Ретта» или «Ретт-подобный синдром» используется для описания заболевания, фенотипически сходного с СР, но не удовлетворяющего критериям его типичной или атипичной формы [7].
В соответствии с клиническими проявлениями, в разные фазы заболевания меняется картина электроэнцефалограммы (ЭЭГ). Последовательность изменений ЭЭГ при типичной форме, ассоциированной с мутацией MECP2, представлена в табл. 2. На ранних этапах ЭЭГ может быть нормальной. Во второй фазе появляются центротемпоральные спайки, указывающие на вовлечение моторной коры и коррелирующие с началом двигательных расстройств. В третьей фазе нарушается ЭЭГ-картина сна, регистрируются билатерально-синхронные вспышки псевдопериодических дельта-волн и генерализованные ритмические разряды спайков. В четвертой фазе наблюдаются выраженное замедление ЭЭГ, мультифокальные эпилептические разряды и генерализованная медленная спайк-волновая активность во сне. При атипичном СР эпилептические разряды могут регистрироваться уже в первой фазе, во второй фазе формируется паттерн модифицированной гипсаритмии с очень медленными дельта-волнами и мультифокальными или фокальными спайками. На поздних стадиях картина схожа с типичным СР [7, 14].
Клинический случай
Девочка с делецией в 4-м экзоне гена MECP2 находилась под наблюдением в Сеченовском центре материнства и детства с семи до десяти лет.
Ребенок от здоровых родителей, неродственного брака, второй беременности (сибс здоров), протекавшей на фоне постоянной угрозы прерывания, железодефицитной анемии с первого триместра, уреаплазмоза в третьем триместре беременности. Родилась самостоятельно в срок. Вес при рождении – 3850 г, рост – 55 см, оценка по шкале Апгар – 7/8 баллов. Выписана домой на четвертые сутки. Моторное развитие с темповой задержкой: голову удерживала с 3,5 месяца, переворачивалась самостоятельно со спины на живот с 8,5 месяца, начала сидеть с восьми месяцев, ходить в 1 год 7 месяцев. С инициацией самостоятельной ходьбы диагностирована атаксия. Речевое развитие также с задержкой – появление первых слов в 2,5 года. Тогда же ребенку установлен диагноз «детский церебральный паралич». Проходила курсы реабилитации по неврологическому профилю с умеренной положительной динамикой. Однако сохранялись шаткость походки, нарушения мелкой моторики, отставала в психическом и речевом развитии.
Впервые в клинику поступила в возрасте семи лет по поводу пароксизмальных состояний, которые появились через неделю после перенесенного острого инфекционного заболевания. Приступы возникали в основном в утренние часы, после пробуждения, протекали в виде заведения глазных яблок вверх, миоклоний век, затрудненного дыхания и сглатывающих движений длительностью до 20 секунд, иногда сериями по два-три.
В статусе на момент поступления: окружность головы – 53 см (норма – 50–52,5 см), диффузная мышечная гипотония, высокие сухожильные рефлексы с нижних конечностей, патологический рефлекс Бабинского с двух сторон, мозжечковая атаксия при ходьбе, интенция и дисметрия в руках.
Нейропсихологический статус в возрасте семи лет: понимает обращенную речь на бытовом уровне, экспрессивная речь заключается в десяти словах, знает основных животных, части тела, цвета. Навыки опрятности сформированы, самостоятельно ест, одевается и раздевается. Любит рассматривать цветы и раскрашивать картинки. Выделяет близких и врача, улыбается им, проявляет бесцельное любопытство, часто заглядывает в ординаторскую, любит гулять, с детьми не общается. На просьбы врача выполнить задание закрывает лицо, складывает руки вместе, перебирает пальцами, инструкции выполняет по подражанию. По тесту Векслера уровень интеллектуального развития (IQ) общий – 44, вербальный – 51, невербальный – 48 баллов, что соответствует умственному дефекту умеренной степени выраженности.
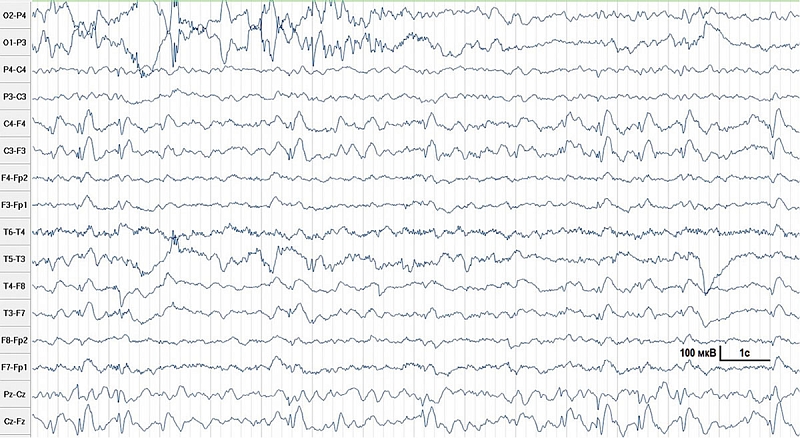
В лабораторных анализах и при ультразвуковом обследовании отклонений от нормы не обнаружено. Магнитно-резонансная томография (МРТ) головного мозга показала уменьшение объема мозжечка (рис. 1).
При ЭЭГ-мониторинге основная активность по частотным характеристикам соответствовала возрасту. Сон модулирован по стадиям, физиологические паттерны сна интактны. Во сне зарегистрированы единичные комплексы «острая волна – медленная волна» в левой центрально-височной области с индексом менее 1%.
Рабочий диагноз: эпилепсия неуточненной этиологии, вероятно генетическая.
Через два-три месяца после дебюта приступы стали серийными – до 15 подряд, сопровождались снижением мышечного тонуса – подкашивались ноги, повисала голова, иногда случались падения. Применялась противосудорожная терапия: леветирацетам – без эффекта, вальпроевая кислота купировала приступы на шесть недель, затем они возобновились с прежней частотой. На фоне приема карбамазепина кратковременно сократились частота и длительность приступов. Характер приступов изменился – они стали протекать по типу остановки двигательной активности на несколько секунд.
В возрасте восьми лет приступы с резкой потерей мышечного тонуса до 15 секунд стали провоцироваться неожиданным звуковым или тактильным раздражителем с заведением глазных яблок вверх. Нейропсихологический статус и поведение существенно не изменились.
ЭЭГ-мониторинг в этом возрасте демонстрировал устойчивый альфа-ритм 8–9 Гц амплитудой 50 мкВ в сочетании с частыми затылочными медленными волнами юношества. В центральных отделах мю-ритм до 70 мкВ. В левой и правой гемисфере в задневисочно-теменно-затылочной областях одиночные и сгруппированные острые волны и комплексы «острая волна – медленная волна» до 100 мкВ с индексом 5–10% в бодрствовании и до 30% во сне. Физиологические паттерны сна оставались сохранными.
Повторная МРТ головного мозга с контрастным усилением не выявила участков накопления контрастного вещества, сохранялись ранее обнаруженные изменения.
Проведено исследование ликвора: цитоз – 1/3, лимфоциты – 1, белок – 0,4 г/л. При анализе олигоклональных антител в ликворе и крови обнаружен 1-й тип синтеза. Соотношение глюкозы ликвора и глюкозы крови – 0,68 (норма > 0,45).
После замены карбамазепина ламотриджином добавился новый тип приступов: при утреннем пробуждении наблюдались серийные замирания с тоническим сгибанием правой руки и движениями в ней по типу перебирания пальцами, сохранялись также приступы с потерей мышечного тонуса.
В схему терапии был введен клобазам, на фоне которого приступы утратили серийность. На ЭЭГ сохранялась региональная эпилептиформная активность.
В возрасте 9 лет 6 месяцев появилась сонливость, снизились двигательная активность и инициативность: предпочитала лежать в постели или безучастно сидеть, практически перестала интересоваться картинками и рисовать. В остальном существенного регресса не отмечалось.
На ЭЭГ наблюдалась отрицательная динамика: основная активность стала дезорганизованной, альфа-ритм не выражен, преобладали колебания тета-диапазона, участились эпилептические разряды в центральных областях билатерально (рис. 2). Во время исследования зарегистрирован привычный приступ из положения сидя: внезапно повисла голова, откинулась на постель, глаза закатила вверх, через 15 секунд тонус восстановился, повернула голову, несколько раз открывала рот, затем улыбалась. Проверить уровень сознания не представлялось возможным из-за отсутствия контакта с пациенткой. На ЭЭГ в момент приступа двигательные артефакты, центральные эпилептические разряды прекратились и возобновились через 20 секунд, что было расценено как иктальные проявления.
Проведена пульс-терапия метилпреднизолоном с положительным эффектом в виде восстановления двигательной активности ребенка, но без эффекта в отношении приступов.
В возрасте 9 лет 10 месяцев отмечался эпизод стремительного ухудшения состояния в течение шести суток: перестала самостоятельно ходить, затем садиться и сидеть, усилилась сонливость, резко снизился аппетит. Приступы продолжались с прежней частотой 5–10 в сутки, их характер не изменился. При повторном обследовании по месту жительства изменений в клиническом и биохимическом анализах крови не выявлено. Тандемная масс-спектрометрия (ТМС) не показала аминоацидопатий, органических ацидурий, нарушений бета-окисления жирных кислот. При выполнении газовой хроматографии мочи органических ацидурий не обнаружено. Однократно отмечалось повышение уровня аммиака в крови до 152,6 мкмоль/л (норма до 80 мкмоль/л) и лактата до 4,8 ммоль/л (норма до 2,0 ммоль/л). При повторных исследованиях кислотно-щелочной статус (КЩС) и уровень аммиака крови были в норме.
На ЭЭГ описано замедление базовой активности до тета-диапазона и частые разряды генерализованных комплексов «острая волна – медленная волна» высокой амплитуды. Повторная МРТ головного мозга без отрицательной динамики.
Получены результаты генетического обследования с полным секвенированием экзома: выявлена ранее не описанная мутация в 49-м экзоне гена TRIO (chr5:14449270А > G), приводящая к замене аминокислоты в 2596-й позиции белка (с.7786А > G/p.Thr2596Ala, NM_007118.3), которая может наблюдаться преимущественно в статусе de novo у пациентов с аутосомно-доминантным нарушением интеллектуального развития, тип 63, с макроцефалией (ОМІМ: 618825) и аутосомно-доминантным нарушением интеллектуального развития, тип 44, с микроцефалией (ОМІМ: 617061).
Проводились инфузии глюкозо-солевых растворов, левокарнитина, получала внутрь рибофлавин, преднизолон, клобазам был отменен. На этом фоне зарегистрировано учащение эпилептических приступов того же характера до 20–25 в сутки. При введении топирамата в приступах появился аксиальный тонический компонент – поднимала плечи и прижимала обе руки к туловищу. На фоне применения комбинации вальпроевой кислоты с леветирацетамом, топираматом и перампанелом приступы прекратились на две недели, уровень бодрствования полностью восстановился через 6–8 суток, однако девочка перестала вставать, ходить, самостоятельно принимать пищу, утратила навыки опрятности и элементарных целенаправленных движений. Атонические рефлекторные приступы возобновились через две недели с частотой до 15 в сутки.
Через месяц отмечалось повторное ухудшение состояния. Ребенок поступил в реанимационное отделение Сеченовского центра материнства и детства в состоянии сопора (9 баллов по педиатрической шкале комы Глазго). Наблюдалась диффузная мышечная атония, голову не удерживала, эпизодически вздрагивала, тазовые органы не контролировала. При нормотермии на компьютерной томограмме органов грудной клетки обнаружены признаки правосторонней сегментарной пневмонии. Клинический и биохимический анализы крови, КЩС, лактат, аммиак, ТМС крови и органические кислоты в моче в норме.
МРТ головного мозга: картина минимального расширения внутренних ликворных пространств, изолированная гипоплазия каудальных отделов червя, заднемедиальных отделов обеих гемисфер мозжечка с умеренным расширением субарахноидального пространства заднечерепной ямки и широким сообщением с полостью четвертого желудочка. Патологических изменений МР-сигнала от вещества головного мозга не выявлено (рис. 3).
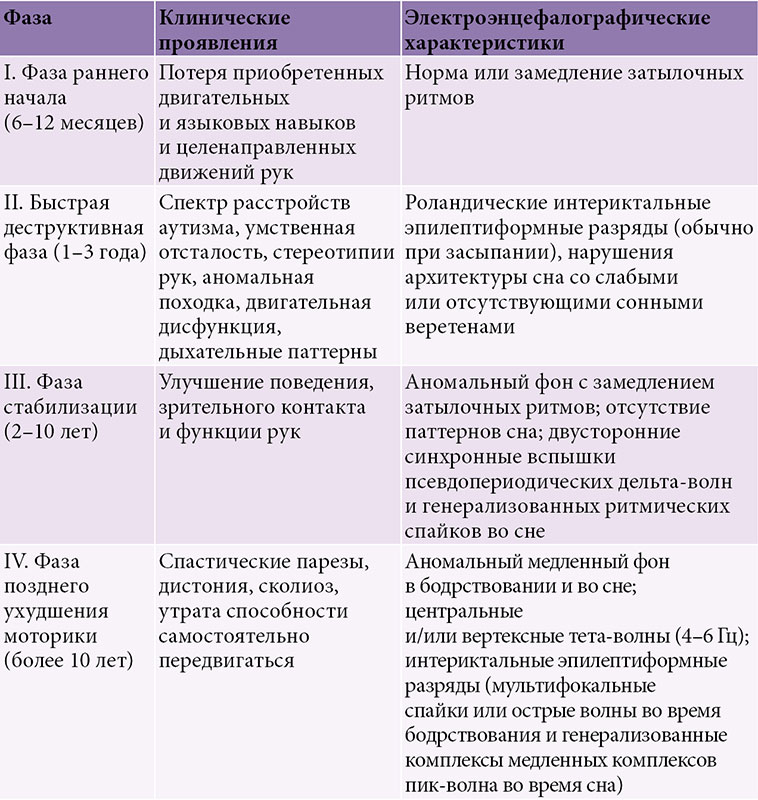
ЭЭГ-мониторинг проведен на второй день поступления в стационар в состоянии оглушения: большую часть времени регистрировались мультифокальные эпилептические разряды на фоне значительно замедленной базовой активности довольно высокой амплитуды с акцентом в передних отделах (рис. 4А). В состоянии, поведенчески похожем на глубокий сон, появлялись периоды неполного угнетения длительностью 1–3 секунды, далее следовало несколько эпизодов ритмичных разрядов высокой амплитуды с периодом 4 секунды, последний эпизод был самым длительным – около 6 минут (рис. 4Б). Затем ритмичные разряды стали смешиваться с неритмичными дельта-волнами, и восстановилась картина мультифокальных разрядов.
Проводимая терапия: инфузии глюкозо-солевых растворов, дексаметазон, цефтриаксон, левокарнитин. Получала также парентеральное питание, внутрь убидекаренон, рибофлавин. Отменена вальпроевая кислота, увеличена доза перампанела и леветирацетама. На фоне терапии на седьмые сутки стала открывать глаза и активнее реагировать на внешние стимулы. В течение последующих двух недель смогла садиться и ходить с поддержкой вдоль кровати, однако полностью отсутствовал пищевой интерес, не жевала, питание получала через назогастральный зонд. В положении сидя отмечались выраженная туловищная атаксия и короткие атонические эпизоды по типу кивания головой. В неврологическом статусе к ранее выявляемой симптоматике добавился расторможенный хватательный рефлекс. В когнитивной и речевой сфере наблюдался более выраженный регресс: не узнавала и не дифференцировала родных, абсолютно не понимала обращенную к ней речь и жесты, собственная речь полностью отсутствовала, могла издавать только звуки, не выполняла никаких инструкций, в том числе по подражанию, с игрушкой производила стереотипные манипуляции (стучала, ковыряла, ломала). Кроме того, появились множественные двигательные стереотипии в руках – размахивала руками, трясла кистями, складывала и перебирала пальцами. Продолжались приступы потери мышечного тонуса с заведением глаз вверх и автоматизмами по типу жевания длительностью до одной минуты, которые провоцировались резким звуком или тактильным стимулом с частотой до шести в сутки. Кроме того, впервые зафиксированы билатеральные тонико-клонические приступы, всего пять, два из них спровоцированы установкой гастростомы и пропуском антиконвульсантов.
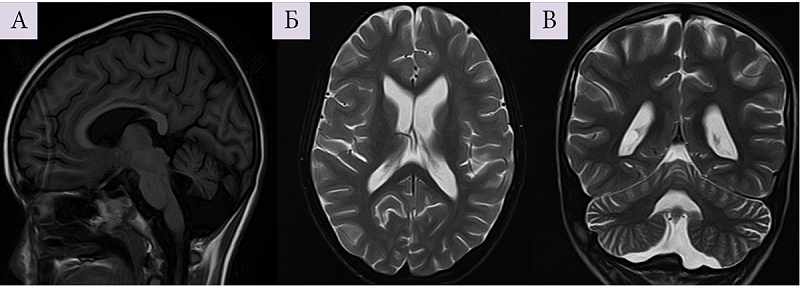
ЭЭГ на этом этапе: грубое замедление, основная активность представлена квазиритмичными дельта-волнами 1,2–1,6 Гц амплитудой до 450 мкВ, которые преобладали в передних отделах и комбинировались со спайками и острыми волнами, формируя комплексы «спайк – волна» и «острая – медленная волна» разной степени выраженности. Картина соответствовала паттерну псевдопериодических генерализованных разрядов (рис. 5). Сон отличался от бодрствования усилением разрядов эпилептических комплексов и появлением после части разрядов периодов угнетения длительностью 1–3 секунды. Физиологические паттерны сна отсутствовали. Во время записи отмечались единичные вздрагивания, которые не совпадали с эпилептическими разрядами.
Получены результаты полногеномного ДНК-секвенирования: обнаружена гетерозиготная делеция сегмента хромосомы Х с координатами 154030384–154030612, соответствующая делеции 228 нуклеотидов в 4-м экзоне гена МЕСР2, приводящая к сдвигу рамки считывания, начиная с 406-го кодона (с.1217_1445del / p.Gln406Profs*30, NM_004992.4). Данная мутация описана у пациенток женского пола с вариабельным течением СР (OMIM: 312750) и в ряде случаев ассоциирована с фармакорезистентной эпилепсией. Делеция подтверждена путем определения протяженных делеций в 4-м экзоне гена MECP2 протяженностью 206 п.н. (NCB136/hg18: chrX;152948980–152949186).
Таким образом, окончательный диагноз был сформулирован следующим образом: заболевание спектра синдрома Ретта, тяжелое течение, стадия позднего ухудшения, генетическая эпилепсия, фармакорезистентное течение.
Обсуждение
Диагностика атипичных форм СР может вызывать значительные затруднения, что демонстрирует представленный случай. До поступления в клинику девочка наблюдалась с диагнозом «детский церебральный паралич», установленным на основании отягощенного перинатального анамнеза, задержки темпов становления моторных навыков на первом году жизни, доминирования в клинической картине двигательных и когнитивных нарушений, а также отсутствия прогрессирующего течения заболевания в течение первых семи лет жизни. С учетом характера двигательного дефекта и данных МРТ был проведен дифференциальный диагноз с непрогрессирующими наследственными атаксиями хронического течения, которые были исключены после генетических консультаций и исследований крови и мочи.
Инфекционное заболевание, которое пациентка перенесла в семилетнем возрасте перед дебютом эпилептических приступов, дало основание провести обследование на предмет дизиммунной природы пароксизмов, что также не подтвердилось.
Сочетание когнитивного дефицита, атаксии и фармакорезистентного течения эпилепсии в отсутствие структурной эпилептогенной патологии на МРТ вызывало подозрение на синдром дефицита переносчика глюкозы 1-го типа GLUT1. Тем не менее отсутствие микрокрании, билатеральных тонико-клонических и классических миоклонических приступов, а также флюктуации симптоматики в течение дня вызывали сомнение в предполагаемом диагнозе. В конечном итоге он также был исключен путем анализа соотношения уровня глюкозы крови и ликвора.
Дальнейшее течение заболевания характеризовалось появлением эпизодов снижения уровня бодрствования и угнетения сознания до состояния сопора. Определенный эффект интенсивной метаболической терапии этих эпизодов позволил предположить наследственную болезнь обмена веществ. Однако неоднократные исследования крови методом ТМС на предмет аминоацидопатии, аминоацидурии и нарушения бета-окисления жирных кислот, а также мочи на содержание органических кислот не подтвердили и этот диагноз.
В ходе параллельных генетических обследований у ребенка были выявлены две мутации:
- мутация в 49-м экзоме гена TRIO, ассоциированная с аутосомно-доминантным нарушением интеллектуального развития;
- делеция 4-го экзона гена MECP2, ассоциированная с СР.
Последняя находка оказалась неожиданной, поскольку мутация не соответствовала клиническому фенотипу пациентки. У девочки отсутствовали обязательные критерии СР и имелись исключающие его признаки. Так, отсутствовал период первоначального нормального развития (с рождения развивалась с задержкой), темпы прироста окружности головы соответствовали нормативным значениям, а специфичные для СР стереотипные движения рук стали заметны лишь в возрасте десяти лет.
Первое ухудшение состояния у девочки было связано с развитием пароксизмальных состояний, классифицировать которые также оказалось непросто. Клинические проявления первых приступов могли соответствовать сложным абсансам, что не противоречит данным отечественных исследователей, отмечающих преимущественно миоклонический и абсансный типы припадков в серии наблюдаемых больных с СР [3]. Однако многочисленные исследования ЭЭГ ни разу не выявили паттерн абсанса, даже в периоды серийных ежедневных приступов. В дальнейшем главным иктальным проявлением приступов стал атонический компонент. Известно, что атонические приступы чаще наблюдаются у пациентов с диффузной или мультифокальной церебральной патологией. Аксиальная билатеральная атония обычно характерна для генерализованных однофазных припадков и генерализованных эпилептических синдромов. Такому предположению не противоречила иктальная ЭЭГ нашей пациентки, демонстрировавшая исчезновение двусторонних интериктальных разрядов в роландической области на период атонии, а также чувствительность приступов к неожиданным тактильным и звуковым стимулам по типу гиперэкплексических пароксизмов. Генерализованные эпилептические припадки с гиперэкплексией (стартл-припадки) типичны для пациентов с задержкой умственного и моторного развития, в рамках прогрессирующих энцефалопатий и генетической патологии [15]. С этой точки зрения характер эпилептического синдрома вполне соответствует поставленному диагнозу. Правда, есть одна особенность – позднее присоединение генерализованных тонико-клонических припадков. Тем не менее нельзя полностью исключить фокальный тип приступов, учитывая наличие в клинической картине алиментарных автоматизмов и пароксизмов с асимметричным тоническим компонентом. ЭЭГ при таких приступах может не демонстрировать четкого иктального паттерна. Кроме того, нет полной уверенности, что по крайней мере часть пароксизмальных состояний имела неэпилептический генез. Согласно результатам проведенных ранее исследований, у пациентов с СР нередко поведенческие и подкорковые нарушения ошибочно принимают за эпилептические приступы, в том числе стереотипии, нарушения дыхания с цианозом, окулогирные кризы, эпизоды моргания или оральной дискинезии, приступы смеха, дистонические или атонические атаки [14].
Наиболее впечатляющей особенностью случая является нетипичное фазовое течение. После начального длительного периода достаточно стабильного состояния следовала быстро прогрессирующая деструктивная фаза, переходившая фактически в позднюю стадию заболевания. Резкое ухудшение состояния выражалось преимущественно в когнитивном регрессе и утрате жизненно важных навыков, тогда как двигательная сфера не демонстрировала характерного для СР нарастания моторного дефекта, спастических парезов и ортопедических изменений. В то же время ЭЭГ демонстрировала типичную последовательность изменений от роландических спайков до псевдопериодических разрядов, быстро нараставших в течение нескольких месяцев. Такое стремительное ухудшение, возможно, в определенной степени связано со статусом мутации MECP2: у девочки выявлена достаточно крупная делеция, что нечасто встречается при СР. Однако это не объясняет длительный начальный период относительно благополучного состояния.
Вызывает также интерес эффективность метаболической терапии, что можно объяснить описанной в литературе вторичной митохондриальной недостаточностью у пациентов с СР [16, 17]. Установлено, что MeCP2 прямо или косвенно регулирует экспрессию нескольких ядерных генов, кодирующих митохондриальные факторы [18]. В пользу этого предположения говорит связь эпизодов ухудшения с соматическим стрессом: развитие эпилептических припадков совпало с инфекционным заболеванием, а последнее ухудшение – с правосторонней сегментарной пневмонией. К тому же во время такого эпизода обнаружено повышение лактата крови, хотя и однократно.
Заключение
Представленный случай демонстрирует новые фенотипические особенности мутации MeCP2. Наличие в симптомокомплексе лишь некоторых черт типичного СР в отсутствие характерного течения дает основание классифицировать его как заболевание спектра СР. В таких ситуациях корректный диагноз требует настойчивого диагностического поиска и многочисленных исследований, включая генетические.
G.G. Toropina1, D.S. Razheva, I.Yu. Ozhegova, V.B. Idamzhapov, M.Yu. Kuzina, S.Ye. Kovaleva, I.S. Preobrazhenskaya
I.M. Sechenov First Moscow State Medical University
N.I. Pirogov Russian National Research Medical University
Contact person: Irina S. Preobrazhenskaya, preobrazhenskaya_i_s@staff.sechenov.ru
Rett syndrome is the most common genetic cause of developmental and intellectual disabilities in females. The main clinical manifestations of Rett syndrome are loss of cognitive and motor skills after a period of normal development, slow growth of head circumference, stereotypical hand movements, and gait abnormalities. Epileptic seizures develop in 60-80% of patients. There is significant variability in the phenotype of Rett syndrome, due to both the localization (MECP2, CDKL5, and FOXG1) and the nature of the genetic pathology (point mutation, duplication, deletion), as well as independent inactivation of the X chromosome. This publication presents a clinical case with an atypical course of the disease.
Уважаемый посетитель uMEDp!
Уведомляем Вас о том, что здесь содержится информация, предназначенная исключительно для специалистов здравоохранения.
Если Вы не являетесь специалистом здравоохранения, администрация не несет ответственности за возможные отрицательные последствия, возникшие в результате самостоятельного использования Вами информации с портала без предварительной консультации с врачом.
Нажимая на кнопку «Войти», Вы подтверждаете, что являетесь врачом или студентом медицинского вуза.