–Ь–µ–і–Є—Ж–Є–љ—Б–Ї–Є–є –њ–Њ—А—В–∞–ї –і–ї—П –≤—А–∞—З–µ–є
–Ї–Њ–ї–Є—З–µ—Б—В–≤–Њ —Б—В–∞—В–µ–є
7525
–Ч–∞–≥—А—Г–Ј–Ї–∞...
–Я–Њ–ґ–∞–ї—Г–є—Б—В–∞, –∞–≤—В–Њ—А–Є–Ј—Г–є—В–µ—Б—М:
–Ш—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П
–Э–∞—Б–ї–µ–і—Б—В–≤–µ–љ–љ—Л–µ –Љ–µ—В–∞–±–Њ–ї–Є—З–µ—Б–Ї–Є–µ –±–Њ–ї–µ–Ј–љ–Є –њ–µ—З–µ–љ–Є
–≠—Д—Д–µ–Ї—В–Є–≤–љ–∞—П —Д–∞—А–Љ–∞–Ї–Њ—В–µ—А–∞–њ–Є—П. –Я–µ–і–Є–∞—В—А–Є—П. –°–Я–Х–¶–Т–Ђ–Я–£–°–Ъ.
- –Р–љ–љ–Њ—В–∞—Ж–Є—П
- –°—В–∞—В—М—П
- –°—Б—Л–ї–Ї–Є
–С–Њ–ї–µ–Ј–љ–Є –Њ–±–Љ–µ–љ–∞ вАУ –≥—А—Г–њ–њ–∞ –љ–∞—Б–ї–µ–і—Б—В–≤–µ–љ–љ—Л—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є, –≤–Њ–Ј–љ–Є–Ї–∞—О—Й–Є—Е –≤ —А–µ–Ј—Г–ї—М—В–∞—В–µ —Б—В—А—Г–Ї—В—Г—А–љ—Л—Е –љ–∞—А—Г—И–µ–љ–Є–є
–≥–µ–љ–Њ–≤, –Њ—В–≤–µ—В—Б—В–≤–µ–љ–љ—Л—Е –Ј–∞ —Б–Є–љ—В–µ–Ј —А–∞–Ј–ї–Є—З–љ—Л—Е —Д—Г–љ–Ї—Ж–Є–Њ–љ–∞–ї—М–љ—Л—Е –±–µ–ї–Ї–Њ–≤. –Т –Њ—Б–љ–Њ–≤–µ –њ–∞—В–Њ–≥–µ–љ–µ–Ј–∞ –і–∞–љ–љ—Л—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є –ї–µ–ґ–∞—В –љ–∞—А—Г—И–µ–љ–Є—П –Њ–њ—А–µ–і–µ–ї–µ–љ–љ—Л—Е –±–Є–Њ—Е–Є–Љ–Є—З–µ—Б–Ї–Є—Е –њ—А–Њ—Ж–µ—Б—Б–Њ–≤, –њ—А–Є–≤–Њ–і—П—Й–Є—Е –Ї —Д–µ—А–Љ–µ–љ—В–∞—В–Є–≤–љ–Њ–Љ—Г –±–ї–Њ–Ї—Г –Є–ї–Є –Ї –і–µ—Д–Є—Ж–Є—В—Г –Ї–Њ–љ–µ—З–љ—Л—Е –њ—А–Њ–і—Г–Ї—В–Њ–≤ —А–µ–∞–Ї—Ж–Є–Є. –Т —А–µ–Ј—Г–ї—М—В–∞—В–µ –≤ –±–Њ–ї—М—И–Є–љ—Б—В–≤–µ —Б–ї—Г—З–∞–µ–≤ –њ—А–Њ–Є—Б—Е–Њ–і–Є—В –љ–∞–Ї–Њ–њ–ї–µ–љ–Є–µ –≤–µ—Й–µ—Б—В–≤,
–љ–∞—А—Г—И–∞—О—Й–Є—Е —Д—Г–љ–Ї—Ж–Є–Є —Ж–µ–ї–Њ–≥–Њ —А—П–і–∞ –Њ—А–≥–∞–љ–Њ–≤ –Є —Б–Є—Б—В–µ–Љ.
–Ґ—А–∞–і–Є—Ж–Є–Њ–љ–љ–Њ –љ–∞—Б–ї–µ–і—Б—В–≤–µ–љ–љ—Л–µ –±–Њ–ї–µ–Ј–љ–Є –Њ–±–Љ–µ–љ–∞ –≤–µ—Й–µ—Б—В–≤ —А–∞–Ј–і–µ–ї—П—О—В—Б—П –љ–∞ –љ–∞—А—Г—И–µ–љ–Є—П –Њ–±–Љ–µ–љ–∞ —Г–≥–ї–µ–≤–Њ–і–Њ–≤, –∞–Љ–Є–љ–Њ–Ї–Є—Б–ї–Њ—В, –Њ—А–≥–∞–љ–Є—З–µ—Б–Ї–Є—Е –Ї–Є—Б–ї–Њ—В, –ї–Є–њ–Є–і–Њ–≤, –Љ–µ—В–∞–ї–ї–Њ–≤. –Т –њ–Њ—Б–ї–µ–і–љ–Є–µ –і–µ—Б—П—В–Є–ї–µ—В–Є—П –±—Л–ї–Є –Њ—В–Ї—А—Л—В—Л —Б–Њ—В–љ–Є –љ–Њ–≤—Л—Е –љ–∞—Б–ї–µ–і—Б—В–≤–µ–љ–љ—Л—Е –љ–∞—А—Г—И–µ–љ–Є–є –Њ–±–Љ–µ–љ–∞. –Ф–ї—П –љ–∞—Б–ї–µ–і—Б—В–≤–µ–љ–љ—Л—Е –±–Њ–ї–µ–Ј–љ–µ–є –Њ–±–Љ–µ–љ–∞ —Е–∞—А–∞–Ї—В–µ—А–µ–љ –њ–Њ–ї–Є–Љ–Њ—А—Д–Є–Ј–Љ –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є—Е –њ—А–Њ—П–≤–ї–µ–љ–Є–є, –Њ–±—Г—Б–ї–Њ–≤–ї–µ–љ–љ—Л–є –≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Њ–є –≥–µ—В–µ—А–Њ–≥–µ–љ–љ–Њ—Б—В—М—О, –≤–Њ–Ј–Љ–Њ–ґ–љ–Њ—Б—В—М—О –≤–Њ–Ј–љ–Є–Ї–љ–Њ–≤–µ–љ–Є—П –Љ—Г—В–∞—Ж–Є–є –≤ —А–∞–Ј–ї–Є—З–љ—Л—Е –≥–µ–љ–∞—Е. –°—Г—Й–µ—Б—В–≤—Г–µ—В –Љ–љ–Њ–ґ–µ—Б—В–≤–Њ –љ–Њ–Ј–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є—Е —Д–Њ—А–Љ –љ–∞—Б–ї–µ–і—Б—В–≤–µ–љ–љ—Л—Е –±–Њ–ї–µ–Ј–љ–µ–є –Њ–±–Љ–µ–љ–∞, —Б—Е–Њ–і–љ—Л—Е –њ–Њ –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є–Љ –њ—А–Њ—П–≤–ї–µ–љ–Є—П–Љ, –љ–Њ —А–∞–Ј–ї–Є—З–∞—О—Й–Є—Е—Б—П –њ–Њ —Д–µ—А–Љ–µ–љ—В–∞—В–Є–≤–љ–Њ–Љ—Г –і–µ—Д–µ–Ї—В—Г, —З—В–Њ –Њ–±—К—П—Б–љ—П–µ—В—Б—П –Ї–∞–Ї —Д—Г–љ–Ї—Ж–Є–Њ–љ–Є—А–Њ–≤–∞–љ–Є–µ–Љ —А–∞–Ј–ї–Є—З–љ—Л—Е —Д–µ—А–Љ–µ–љ—В–Њ–≤
–≤ –Њ–і–љ–Њ–є –Є —В–Њ–є –ґ–µ —Ж–µ–њ–Є –Њ–±–Љ–µ–љ–љ—Л—Е –њ—А–Њ—Ж–µ—Б—Б–Њ–≤, —В–∞–Ї –Є —Г—З–∞—Б—В–Є–µ–Љ –Њ–і–љ–Њ–≥–Њ –Є —В–Њ–≥–Њ –ґ–µ —Д–µ—А–Љ–µ–љ—В–∞ –≤ –љ–µ—Б–Ї–Њ–ї—М–Ї–Є—Е –±–Є–Њ—Е–Є–Љ–Є—З–µ—Б–Ї–Є—Е —А–µ–∞–Ї—Ж–Є—П—Е. –Э–µ—Б–Љ–Њ—В—А—П –љ–∞ —Н—В–Њ, –Љ–Њ–ґ–љ–Њ –≤—Л–і–µ–ї–Є—В—М –љ–µ–Ї–Њ—В–Њ—А—Л–µ –Њ–±—Й–Є–µ –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є–µ –њ—А–Є–Ј–љ–∞–Ї–Є, –њ–Њ–Ј–≤–Њ–ї—П—О—Й–Є–µ –Ј–∞–њ–Њ–і–Њ–Ј—А–Є—В—М
—Г –±–Њ–ї—М–љ–Њ–≥–Њ —А–∞—Б—Б—В—А–Њ–є—Б—В–≤–Њ –Љ–µ—В–∞–±–Њ–ї–Є–Ј–Љ–∞. –Т—Л—П–≤–ї–µ–љ—Л —Б–ї–µ–і—Г—О—Й–Є–µ –Њ—Б–љ–Њ–≤–љ—Л–µ –њ—А–Є–Ј–љ–∞–Ї–Є –љ–∞—Б–ї–µ–і—Б—В–≤–µ–љ–љ—Л—Е –Љ–µ—В–∞–±–Њ–ї–Є—З–µ—Б–Ї–Є—Е –±–Њ–ї–µ–Ј–љ–µ–є –њ–µ—З–µ–љ–Є: –≥–µ–њ–∞—В–Њ- –Є —Б–њ–ї–µ–љ–Њ–Љ–µ–≥–∞–ї–Є—П, –≤–Њ–Ј–Љ–Њ–ґ–љ—Л –Ј–∞–і–µ—А–ґ–Ї–∞ –њ—Б–Є—Е–Њ–Љ–Њ—В–Њ—А–љ–Њ–≥–Њ —А–∞–Ј–≤–Є—В–Є—П, —Б—Г–і–Њ—А–Њ–ґ–љ—Л–є —Б–Є–љ–і—А–Њ–Љ; –Љ–Є–Њ–њ–∞—В–Є–Є; –Ї–µ—В–Њ–∞—Ж–Є–і–Њ–Ј; –Ї–∞—В–∞—А–∞–Ї—В–∞; —Б–њ–µ—Ж–Є—Д–Є—З–µ—Б–Ї–Є–є –Ј–∞–њ–∞—Е —В–µ–ї–∞ –Є –≤—Л–і–µ–ї–µ–љ–Є–є, –љ–µ—Д—А–Њ–Љ–µ–≥–∞–ї–Є—П, –њ–∞—В–Њ–ї–Њ–≥–Є—П —Б–Ї–µ–ї–µ—В–∞. –Ъ–ї–Є–љ–Є—З–µ—Б–Ї–Є–µ –њ—А–Њ—П–≤–ї–µ–љ–Є—П –љ–∞—Б–ї–µ–і—Б—В–≤–µ–љ–љ—Л—Е –±–Њ–ї–µ–Ј–љ–µ–є –Њ–±–Љ–µ–љ–∞ —З–∞—Б—В–Њ —Б–Њ–њ—А—П–ґ–µ–љ—Л —Б –љ–∞–ї–Є—З–Є–µ–Љ –њ–Њ—А–∞–ґ–µ–љ–Є—П –љ–µ—А–≤–љ–Њ–є —Б–Є—Б—В–µ–Љ—Л. –Ф–ї—П –Ї–∞–ґ–і–Њ–є –≥—А—Г–њ–њ—Л –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є —Е–∞—А–∞–Ї—В–µ—А–љ—Л —Б–≤–Њ–Є —Б–њ–µ—Ж–Є—Д–Є—З–µ—Б–Ї–Є–µ —Б–Є–Љ–њ—В–Њ–Љ—Л.
–Ю–і–љ–∞ –Є–Ј —В—А—Г–і–љ–Њ—Б—В–µ–є —А–∞–љ–љ–µ–є –і–Є–∞–≥–љ–Њ—Б—В–Є–Ї–Є –љ–∞—Б–ї–µ–і—Б—В–≤–µ–љ–љ—Л—Е –Љ–µ—В–∞–±–Њ–ї–Є—З–µ—Б–Ї–Є—Е –±–Њ–ї–µ–Ј–љ–µ–є –њ–µ—З–µ–љ–Є —Б–≤—П–Ј–∞–љ–∞ —Б —В–µ–Љ,
—З—В–Њ –≤ –њ–µ—А–Є–Њ–і –љ–Њ–≤–Њ—А–Њ–ґ–і–µ–љ–љ–Њ—Б—В–Є —Г –љ–µ–Ї–Њ—В–Њ—А—Л—Е –і–µ—В–µ–є –Њ—В—Б—Г—В—Б—В–≤—Г—О—В —Б–њ–µ—Ж–Є—Д–Є—З–µ—Б–Ї–Є–µ —А–∞—Б—Б—В—А–Њ–є—Б—В–≤–∞, –∞ –њ–Њ–Ј–і–љ–Є–µ –њ—А–Њ—П–≤–ї–µ–љ–Є—П —Б—Е–Њ–ґ–Є —Б –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П–Љ–Є –љ–µ–љ–∞—Б–ї–µ–і—Б—В–≤–µ–љ–љ–Њ–≥–Њ –≥–µ–љ–µ–Ј–∞ —Б –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є–Љ–Є –њ—А–Њ—П–≤–ї–µ–љ–Є—П–Љ–Є
–≥–µ–њ–∞—В–Є—В–∞ —А–∞–Ј–ї–Є—З–љ–Њ–є —Б—В–µ–њ–µ–љ–Є –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є.
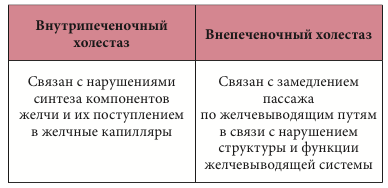
–Т–Є–і—Л —Е–Њ–ї–µ—Б—В–∞–Ј–∞
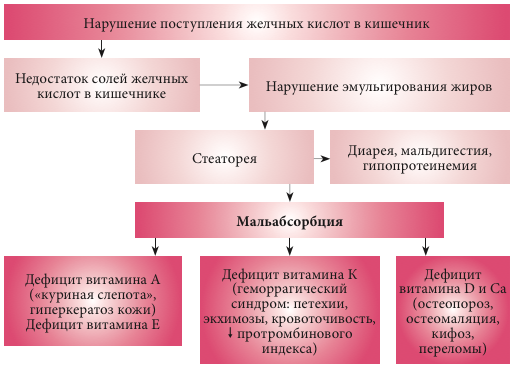
–Я–∞—В–Њ–≥–µ–љ–µ–Ј –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є—Е –њ—А–Њ—П–≤–ї–µ–љ–Є–є —Е–Њ–ї–µ—Б—В–∞–Ј–∞

–Ъ–ї–Є–љ–Є—З–µ—Б–Ї–Є–µ –њ—А–Њ—П–≤–ї–µ–љ–Є—П —Е–Њ–ї–µ—Б—В–∞–Ј–∞
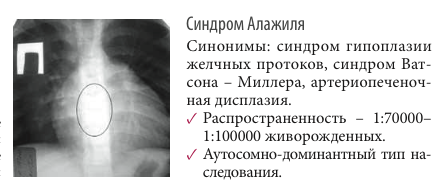
–У—А—Г–і–љ—Л–µ –њ–Њ–Ј–≤–Њ–љ–Ї–Є –≤ —Д–Њ—А–Љ–µ –±–∞–±–Њ—З–Ї–Є

–≠—В–Є–Њ–ї–Њ–≥–Є—П –Я–°–Т–•

–Ъ–ї–Є–љ–Є—З–µ—Б–Ї–Є–µ –њ—А–Њ—П–≤–ї–µ–љ–Є—П –Я–°–Т–•/–С–Є–Њ—Е–Є–Љ–Є—З–µ—Б–Ї–Є–µ –њ—А–Њ—П–≤–ї–µ–љ–Є—П –Я–°–Т–•

–Ъ–ї–Є–љ–Є—З–µ—Б–Ї–Є–µ –њ—А–Њ—П–≤–ї–µ–љ–Є—П –Я–°–Т–•

–≠—В–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–µ —Д–∞–Ї—В–Њ—А—Л –С–Э–У
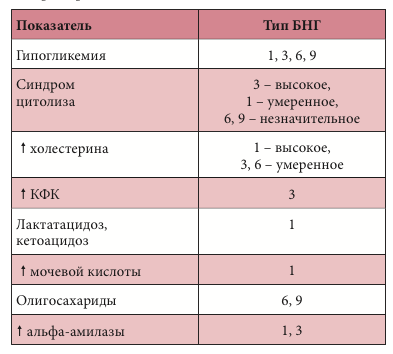
–Ы–∞–±–Њ—А–∞—В–Њ—А–љ—Л–µ –њ–Њ–Ї–∞–Ј–∞—В–µ–ї–Є –С–Э–У
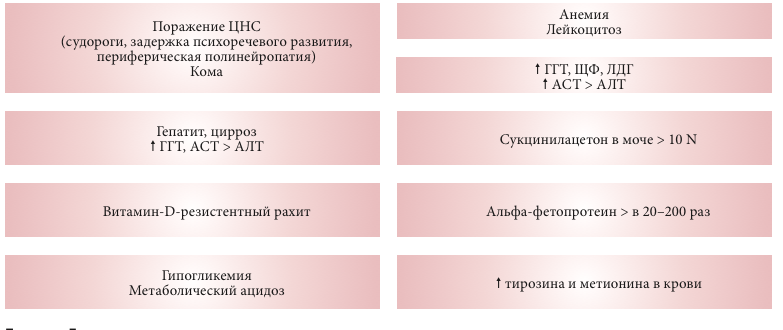
–Ъ–ї–Є–љ–Є–Ї–Њ-–ї–∞–±–Њ—А–∞—В–Њ—А–љ—Л–µ –њ–Њ–Ї–∞–Ј–∞—В–µ–ї–Є —В–Є—А–Њ–Ј–Є–љ–µ–Љ–Є–Є I —В–Є–њ–∞

–°—В—А–∞—В–µ–≥–Є—П –њ–Њ—Б—В–∞–љ–Њ–≤–Ї–Є –і–Є–∞–≥–љ–Њ–Ј–∞ ¬Ђ–±–Њ–ї–µ–Ј–љ—М –У–Њ—И–µ¬ї

–С–Њ–ї–µ–Ј–љ—М –Т–Є–ї—М—Б–Њ–љ–∞
–Э–∞—Б–ї–µ–і—Б—В–≤–µ–љ–љ—Л–µ –±–Њ–ї–µ–Ј–љ–Є –Њ–±–Љ–µ–љ–∞ –≤–µ—Й–µ—Б—В–≤ вАУ —Н—В–Њ –≥—А—Г–њ–њ–∞ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є, –≤–Њ–Ј–љ–Є–Ї–∞—О—Й–Є—Е –≤ —А–µ–Ј—Г–ї—М—В–∞—В–µ —Б—В—А—Г–Ї—В—Г—А–љ—Л—Е –љ–∞—А—Г—И–µ–љ–Є–є –≥–µ–љ–Њ–≤, –Њ—В–≤–µ—В—Б—В–≤–µ–љ–љ—Л—Е –Ј–∞ —Б–Є–љ—В–µ–Ј —А–∞–Ј–ї–Є—З–љ—Л—Е —Д—Г–љ–Ї—Ж–Є–Њ–љ–∞–ї—М–љ—Л—Е –±–µ–ї–Ї–Њ–≤.
–С–Њ–ї—М—И–Є–љ—Б—В–≤–Њ –±–Њ–ї–µ–Ј–љ–µ–є –Њ–±–Љ–µ–љ–∞ —П–≤–ї—П—О—В—Б—П —А–µ—Ж–µ—Б—Б–Є–≤–љ—Л–Љ–Є –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П–Љ–Є, –Ї–∞–Ї –њ—А–∞–≤–Є–ї–Њ, –∞—Г—В–Њ—Б–Њ–Љ–љ—Л–Љ–Є, —А–µ–ґ–µ —Б—Ж–µ–њ–ї–µ–љ–љ—Л–Љ–Є —Б X-—Е—А–Њ–Љ–Њ—Б–Њ–Љ–Њ–є.
–Э–∞–Є–±–Њ–ї–µ–µ —З–∞—Б—В–Њ —Н—В–Є –±–Њ–ї–µ–Ј–љ–Є –Њ–±—Г—Б–ї–Њ–≤–ї–µ–љ—Л –Ї–∞—З–µ—Б—В–≤–µ–љ–љ—Л–Љ –Є–ї–Є –Ї–Њ–ї–Є—З–µ—Б—В–≤–µ–љ–љ—Л–Љ –і–µ—Д–µ–Ї—В–Њ–Љ —Д–µ—А–Љ–µ–љ—В–Њ–≤ (—Ж–Є—В–Њ–њ–ї–∞–Ј–Љ–∞—В–Є—З–µ—Б–Ї–Є—Е, –ї–Є–Ј–Њ—Б–Њ–Љ–љ—Л—Е, –њ–µ—А–Њ–Ї—Б–Є—Б–Њ–Љ–љ—Л—Е) –Є–ї–Є —В—А–∞–љ—Б–њ–Њ—А—В–љ—Л—Е –±–µ–ї–Ї–Њ–≤.
–Т —А–µ–Ј—Г–ї—М—В–∞—В–µ –њ—А–Њ–Є—Б—Е–Њ–і–Є—В –Є–Ј–±—Л—В–Њ—З–љ–Њ–µ –љ–∞–Ї–Њ–њ–ї–µ–љ–Є–µ –≤–µ—Й–µ—Б—В–≤–∞-–њ—А–µ–і—И–µ—Б—В–≤–µ–љ–љ–Є–Ї–∞, –µ–≥–Њ —В–Њ–Ї—Б–Є—З–µ—Б–Ї–Є—Е –Љ–µ—В–∞–±–Њ–ї–Є—В–Њ–≤ –Є–ї–Є –љ–∞–±–ї—О–і–∞–µ—В—Б—П –љ–µ—Е–≤–∞—В–Ї–∞ –Ї–Њ–љ–µ—З–љ–Њ–≥–Њ –њ—А–Њ–і—Г–Ї—В–∞ —А–µ–∞–Ї—Ж–Є–Є.
–С–Њ–ї–µ–Ј–љ–Є, –њ—А–Є –Ї–Њ—В–Њ—А—Л—Е –њ—А–Њ–і—Г–Ї—В—Л –љ–∞—А—Г—И–µ–љ–љ–Њ–≥–Њ –Њ–±–Љ–µ–љ–∞ –љ–∞–Ї–∞–њ–ї–Є–≤–∞—О—В—Б—П –≤ –Ї–ї–µ—В–Ї–∞—Е –Є —В–Ї–∞–љ—П—Е, –љ–∞–Ј—Л–≤–∞—О—В —В–∞–Ї–ґ–µ –±–Њ–ї–µ–Ј–љ—П–Љ–Є –љ–∞–Ї–Њ–њ–ї–µ–љ–Є—П (—В–µ–Ј–∞—Г—А–Є—Б–Љ–Њ–Ј—Л).
–Ъ–ї–Є–љ–Є—З–µ—Б–Ї–Є–µ –њ—А–Њ—П–≤–ї–µ–љ–Є—П –Њ–±—Г—Б–ї–Њ–≤–ї–µ–љ—Л –Њ—В—Б—Г—В—Б—В–≤–Є–µ–Љ –Є–ї–Є –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ—Л–Љ —Б–Є–љ—В–µ–Ј–Њ–Љ –Њ–і–љ–Њ–≥–Њ –Є–ї–Є –љ–µ—Б–Ї–Њ–ї—М–Ї–Є—Е —Д–µ—А–Љ–µ–љ—В–Њ–≤, —Г—З–∞—Б—В–≤—Г—О—Й–Є—Е –≤ –Њ–±–Љ–µ–љ–µ –∞–Љ–Є–љ–Њ–Ї–Є—Б–ї–Њ—В, —Г–≥–ї–µ–≤–Њ–і–Њ–≤, –ґ–Є—А–љ—Л—Е –Ї–Є—Б–ї–Њ—В –Є–ї–Є –±–Њ–ї–µ–µ —Б–ї–Њ–ґ–љ—Л—Е –њ—А–Њ–Љ–µ–ґ—Г—В–Њ—З–љ—Л—Е –њ—А–Њ–і—Г–Ї—В–Њ–≤.¬†–Ь–Њ–≥—Г—В –њ—А–Њ—П–≤–ї—П—В—М—Б—П –≤ –ї—О–±–Њ–Љ –≤–Њ–Ј—А–∞—Б—В–µ.
–Т–∞–ґ–µ–љ —В—Й–∞—В–µ–ї—М–љ—Л–є —Б–±–Њ—А —Б–µ–Љ–µ–є–љ–Њ–≥–Њ –∞–љ–∞–Љ–љ–µ–Ј–∞: –љ–∞–ї–Є—З–Є–µ –Ї—А–Њ–≤–љ–Њ—А–Њ–і—Б—В–≤–µ–љ–љ–Њ–≥–Њ –±—А–∞–Ї–∞ —А–Њ–і–Є—В–µ–ї–µ–є, –≤—Л–Ї–Є–і—Л—И–Є, –≥–Є–±–µ–ї—М –њ–ї–Њ–і–∞, —Б–Љ–µ—А—В—М –і–µ—В–µ–є –≤ —А–∞–љ–љ–µ–Љ –≤–Њ–Ј—А–∞—Б—В–µ, —Б–Є–љ–і—А–Њ–Љ –≤–љ–µ–Ј–∞–њ–љ–Њ–є –і–µ—В—Б–Ї–Њ–є —Б–Љ–µ—А—В–Є, –љ–µ–≤—А–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–µ —Б–Є–Љ–њ—В–Њ–Љ—Л –љ–µ—П—Б–љ–Њ–є —Н—В–Є–Њ–ї–Њ–≥–Є–Є, –Ј–∞–і–µ—А–ґ–Ї–∞ –њ—Б–Є—Е–Є—З–µ—Б–Ї–Њ–≥–Њ —А–∞–Ј–≤–Є—В–Є—П.
–Я—А–Є —А–∞–љ–љ–µ–Љ –і–µ–±—О—В–µ —Б–Є–Љ–њ—В–Њ–Љ—Л –њ–Њ—П–≤–ї—П—О—В—Б—П –њ–Њ—Б–ї–µ –љ–∞—З–∞–ї–∞ –≤—Б–Ї–∞—А–Љ–ї–Є–≤–∞–љ–Є—П.
–Ъ–ї–Є–љ–Є—З–µ—Б–Ї–∞—П –Ї–∞—А—В–Є–љ–∞: –≥–Є–њ–µ—А–≤–µ–љ—В–Є–ї—П—Ж–Є—П, —Н–њ–Є–ї–µ–њ—В–Є—З–µ—Б–Ї–Є–µ –њ—А–Є–њ–∞–і–Ї–Є, —Б–Њ–љ–ї–Є–≤–Њ—Б—В—М, –Ї–Њ–Љ–∞, –љ–∞—А—Г—И–µ–љ–Є—П –Љ—Л—И–µ—З–љ–Њ–≥–Њ —В–Њ–љ—Г—Б–∞, –≥–µ–њ–∞—В–Њ—Б–њ–ї–µ–љ–Њ–Љ–µ–≥–∞–ї–Є—П, –ґ–µ–ї—В—Г—Е–∞, —А–≤–Њ—В–∞, –≤—П–ї–Њ–µ —Б–Њ—Б–∞–љ–Є–µ, –љ–µ–Њ–±—Л—З–љ—Л–є –Ј–∞–њ–∞—Е –≤—Л–і–µ–ї–µ–љ–Є–є, —В–µ–ї–∞; –љ–µ–≤—А–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–µ –љ–∞—А—Г—И–µ–љ–Є—П.
–Ю—Б—В—А—Л–µ –±–Є–Њ—Е–Є–Љ–Є—З–µ—Б–Ї–Є–µ –љ–∞—А—Г—И–µ–љ–Є—П –≤–Ї–ї—О—З–∞—О—В –Љ–µ—В–∞–±–Њ–ї–Є—З–µ—Б–Ї–Є–є –∞—Ж–Є–і–Њ–Ј, –≥–Є–њ–Њ–≥–ї–Є–Ї–µ–Љ–Є—О, –≥–Є–њ–µ—А–∞–Љ–Љ–Њ–љ–Є–µ–Љ–Є—О, —Б–Є–љ–і—А–Њ–Љ —Ж–Є—В–Њ–ї–Є–Ј–∞, –≥–Є–њ–µ—А–±–Є–ї–Є—А—Г–±–Є–љ–µ–Љ–Є—О, –і–Є—Б–њ—А–Њ—В–µ–Є–љ–µ–Љ–Є—О.
–Я—А–Є –і–µ–±—О—В–µ –≤ —А–∞–љ–љ–µ–Љ –і–µ—В—Б–Ї–Њ–Љ –≤–Њ–Ј—А–∞—Б—В–µ —Е–∞—А–∞–Ї—В–µ—А–Є–Ј—Г—О—В—Б—П –њ—А–Њ–≥—А–µ—Б—Б–Є—А—Г—О—Й–µ–є –≥–µ–њ–∞—В–Њ—Б–њ–ї–µ–љ–Њ–Љ–µ–≥–∞–ї–Є–µ–є, —А–∞—Е–Є—В–Њ–Љ, –њ–Њ–Љ—Г—В–љ–µ–љ–Є–µ–Љ —А–Њ–≥–Њ–≤–Є—Ж—Л, –Є–Ј–Љ–µ–љ–µ–љ–Є—П–Љ–Є —Б–Њ —Б—В–Њ—А–Њ–љ—Л –≥–ї–∞–Ј: —Б–Є–Љ–њ—В–Њ–Љ ¬Ђ–≤–Є—И–љ–µ–≤–Њ–є –Ї–Њ—Б—В–Њ—З–Ї–Є¬ї, –љ–∞–ї–Є—З–Є–µ –Ї–Њ–ї—М—Ж–∞ –Ъ–∞–є–Ј–µ—А–∞¬†вАУ –§–ї—П–є—И–µ—А–∞.
–С–Њ–ї–µ–Ј–љ–Є –љ–∞–Ї–Њ–њ–ї–µ–љ–Є—П —Б –љ–∞—З–∞–ї–Њ–Љ –≤ –±–Њ–ї–µ–µ –њ–Њ–Ј–і–љ–µ–Љ –≤–Њ–Ј—А–∞—Б—В–µ –Љ–Њ–≥—Г—В –њ—А–Њ—П–≤–ї—П—В—М—Б—П —Б–љ–Є–ґ–µ–љ–Є–µ–Љ —Б–њ–Њ—Б–Њ–±–љ–Њ—Б—В–Є –Ї –Њ–±—Г—З–µ–љ–Є—О, –Љ—Л—И–µ—З–љ–Њ–є —Б–ї–∞–±–Њ—Б—В—М—О, –±–Њ–ї–µ–Ј–љ–µ–љ–љ—Л–Љ–Є —Б–њ–∞–Ј–Љ–∞–Љ–Є –Љ—Л—И—Ж, –Ј–∞—В—А—Г–і–љ–µ–љ–Є—П–Љ–Є –њ—А–Є —Е–Њ–і—М–±–µ, –љ–∞—А—Г—И–µ–љ–Є—П–Љ–Є –њ—Б–Є—Е–Є–Ї–Є –Є –њ–Њ–≤–µ–і–µ–љ–Є—П, –Ї–ї–Є–љ–Є–Ї–Њ-–ї–∞–±–Њ—А–∞—В–Њ—А–љ—Л–Љ–Є –њ—А–Њ—П–≤–ї–µ–љ–Є—П–Љ–Є —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–≥–Њ –≥–µ–њ–∞—В–Є—В–∞, —Ж–Є—А—А–Њ–Ј–∞ –њ–µ—З–µ–љ–Є.
–Э–Њ–≤–Њ—Б—В–Є –љ–∞ —В–µ–Љ—Г
–Ш–Э–°–Ґ–†–£–Ь–Х–Э–Ґ–Ђ
–Ю—В–њ—А–∞–≤–Є—В—М —Б—В–∞—В—М—О –њ–Њ —Н–ї–µ–Ї—В—А–Њ–љ–љ–Њ–є –њ–Њ—З—В–µ
–Т–∞—И –∞–і—А–µ—Б —Н–ї–µ–Ї—В—А–Њ–љ–љ–Њ–є –њ–Њ—З—В—Л:
–Р–і—А–µ—Б —Н–ї–µ–Ї—В—А–Њ–љ–љ–Њ–є –њ–Њ—З—В—Л –њ–Њ–ї—Г—З–∞—В–µ–ї—П:
–†–∞–Ј–і–µ–ї–Є—В–µ –љ–µ—Б–Ї–Њ–ї—М–Ї–Њ –∞–і—А–µ—Б–Њ–≤ —Н–ї–µ–Ї—В—А–Њ–љ–љ–Њ–є –њ–Њ—З—В—Л –Ј–∞–њ—П—В–Њ–є
–°–Њ–Њ–±—Й–µ–љ–Є–µ(–љ–µ –Њ–±—П–Ј–∞—В–µ–ї—М–љ–Њ)
–Э–µ –±–Њ–ї–µ–µ 1500 —Б–Є–Љ–≤–Њ–ї–Њ–≤
–Р–љ—В–Є —Б–њ–∞–Љ:
–Ф–ї—П –њ—А–µ–і–Њ—В–≤—А–∞—Й–µ–љ–Є—П —Б–њ–∞–Љ–∞, –њ–Њ–ґ–∞–ї—Г–є—Б—В–∞, –≤–≤–µ–і–Є—В–µ –≤ –њ–Њ–ї–µ —Б–ї–Њ–≤–Њ, –Ї–Њ—В–Њ—А–Њ–µ –≤–Є–і–Є—В–µ –љ–Є–ґ–µ.
–Ю–±–љ–Њ–≤–Є—В—М –Ї–Њ–і
* –∞–і—А–µ—Б–∞ –њ—А–µ–і–Њ—Б—В–∞–≤–ї–µ–љ–љ—Л–µ –Т–∞–Љ–Є –±—Г–і—Г—В –Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞—В—М—Б—П —В–Њ–ї—М–Ї–Њ –і–ї—П –Њ—В–њ—А–∞–≤–Ї–Є —Н–ї–µ–Ї—В—А–Њ–љ–љ–Њ–є –њ–Њ—З—В—Л.
–£–≤–∞–ґ–∞–µ–Љ—Л–є –њ–Њ—Б–µ—В–Є—В–µ–ї—М uMEDp!
–£–≤–µ–і–Њ–Љ–ї—П–µ–Љ –Т–∞—Б –Њ —В–Њ–Љ, —З—В–Њ –Ј–і–µ—Б—М —Б–Њ–і–µ—А–ґ–Є—В—Б—П –Є–љ—Д–Њ—А–Љ–∞—Ж–Є—П, –њ—А–µ–і–љ–∞–Ј–љ–∞—З–µ–љ–љ–∞—П –Є—Б–Ї–ї—О—З–Є—В–µ–ї—М–љ–Њ –і–ї—П —Б–њ–µ—Ж–Є–∞–ї–Є—Б—В–Њ–≤ –Ј–і—А–∞–≤–Њ–Њ—Е—А–∞–љ–µ–љ–Є—П.
–Х—Б–ї–Є –Т—Л –љ–µ —П–≤–ї—П–µ—В–µ—Б—М —Б–њ–µ—Ж–Є–∞–ї–Є—Б—В–Њ–Љ –Ј–і—А–∞–≤–Њ–Њ—Е—А–∞–љ–µ–љ–Є—П, –∞–і–Љ–Є–љ–Є—Б—В—А–∞—Ж–Є—П –љ–µ –љ–µ—Б–µ—В –Њ—В–≤–µ—В—Б—В–≤–µ–љ–љ–Њ—Б—В–Є –Ј–∞ –≤–Њ–Ј–Љ–Њ–ґ–љ—Л–µ –Њ—В—А–Є—Ж–∞—В–µ–ї—М–љ—Л–µ –њ–Њ—Б–ї–µ–і—Б—В–≤–Є—П, –≤–Њ–Ј–љ–Є–Ї—И–Є–µ –≤ —А–µ–Ј—Г–ї—М—В–∞—В–µ —Б–∞–Љ–Њ—Б—В–Њ—П—В–µ–ї—М–љ–Њ–≥–Њ –Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–љ–Є—П –Т–∞–Љ–Є –Є–љ—Д–Њ—А–Љ–∞—Ж–Є–Є —Б –њ–Њ—А—В–∞–ї–∞ –±–µ–Ј –њ—А–µ–і–≤–∞—А–Є—В–µ–ї—М–љ–Њ–є –Ї–Њ–љ—Б—Г–ї—М—В–∞—Ж–Є–Є —Б –≤—А–∞—З–Њ–Љ.
–Э–∞–ґ–Є–Љ–∞—П –љ–∞ –Ї–љ–Њ–њ–Ї—Г ¬Ђ–Т–Њ–є—В–Є¬ї, –Т—Л –њ–Њ–і—В–≤–µ—А–ґ–і–∞–µ—В–µ, —З—В–Њ —П–≤–ї—П–µ—В–µ—Б—М –≤—А–∞—З–Њ–Љ –Є–ї–Є —Б—В—Г–і–µ–љ—В–Њ–Љ –Љ–µ–і–Є—Ж–Є–љ—Б–Ї–Њ–≥–Њ –≤—Г–Ј–∞.