–ü–į—ā–ĺ—Ą–ł–∑–ł–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ł–Ķ –ĺ—Ā–Ĺ–ĺ–≤—č –Ĺ–ĺ–≤–ĺ–≥–ĺ –Ĺ–į–Ņ—Ä–į–≤–Ľ–Ķ–Ĺ–ł—Ź —ā–Ķ—Ä–į–Ņ–ł–ł –≥–Ķ–ĺ–≥—Ä–į—Ą–ł—á–Ķ—Ā–ļ–ĺ–Ļ –į—ā—Ä–ĺ—Ą–ł–ł ‚Äď –ł–Ĺ–≥–ł–Ī–ł—ā–ĺ—Ä–ĺ–≤ –ļ–ĺ–ľ–Ņ–Ľ–Ķ–ľ–Ķ–Ĺ—ā–į
- –ź–Ĺ–Ĺ–ĺ—ā–į—Ü–ł—Ź
- –°—ā–į—ā—Ć—Ź
- –°—Ā—č–Ľ–ļ–ł
- English
–í–≤–Ķ–ī–Ķ–Ĺ–ł–Ķ
–í–ĺ–∑—Ä–į—Ā—ā–Ĺ–į—Ź –ľ–į–ļ—É–Ľ—Ź—Ä–Ĺ–į—Ź –ī–Ķ–≥–Ķ–Ĺ–Ķ—Ä–į—Ü–ł—Ź (–í–ú–Ē), –≤¬†—á–į—Ā—ā–Ĺ–ĺ—Ā—ā–ł —Ą–ł–Ĺ–į–Ľ—Ć–Ĺ–į—Ź —Ā—ā–į–ī–ł—Ź –Ķ–Ķ —Ā—É—Ö–ĺ–Ļ —Ą–ĺ—Ä–ľ—謆‚Äst–≥–Ķ–ĺ–≥—Ä–į—Ą–ł—á–Ķ—Ā–ļ–į—Ź –į—ā—Ä–ĺ—Ą–ł—Ź (–ď–ź), –ĺ—Ā—ā–į–Ķ—ā—Ā—Ź –≤–Ķ–ī—É—Č–Ķ–Ļ –Ņ—Ä–ł—á–ł–Ĺ–ĺ–Ļ –ł–Ĺ–≤–į–Ľ–ł–ī–Ĺ–ĺ—Ā—ā–ł –Ņ–嬆–∑—Ä–Ķ–Ĺ–ł—é –≤¬†—Ä–į–∑–≤–ł—ā—č—Ö —Ā—ā—Ä–į–Ĺ–į—Ö —Ā—Ä–Ķ–ī–ł –Ľ–ł—Ü —Ā—ā–į—Ä—ą–Ķ 60 –Ľ–Ķ—ā. –°–ĺ–≥–Ľ–į—Ā–Ĺ–ĺ —ć–ļ—Ā–Ņ–Ķ—Ä—ā–Ĺ–ĺ–Ļ –ĺ—Ü–Ķ–Ĺ–ļ–Ķ, –≤¬†–Ĺ–į—Ā—ā–ĺ—Ź—Č–Ķ–Ķ –≤—Ä–Ķ–ľ—Ź –≤¬†–†–ĺ—Ā—Ā–ł–Ļ—Ā–ļ–ĺ–Ļ –§–Ķ–ī–Ķ—Ä–į—Ü–ł–ł –Ĺ–į—Ā—á–ł—ā—č–≤–į–Ķ—ā—Ā—Ź –ĺ–ļ–ĺ–Ľ–ĺ 370 —ā—č—Ā. –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ —Ā¬†–ď–ź [1]. –ú–Ĺ–ĺ–≥–ĺ—Ą–į–ļ—ā–ĺ—Ä–Ĺ–ĺ—Ā—ā—Ć –Ņ–į—ā–ĺ–≥–Ķ–Ĺ–Ķ–∑–į, –ĺ–Ī—É—Ā–Ľ–ĺ–≤–Ľ–Ķ–Ĺ–Ĺ–į—Ź —Ā–Ľ–ĺ–∂–Ĺ—č–ľ –≤–∑–į–ł–ľ–ĺ–ī–Ķ–Ļ—Ā—ā–≤–ł–Ķ–ľ –ľ–Ķ—ā–į–Ī–ĺ–Ľ–ł—á–Ķ—Ā–ļ–ł—Ö,¬†–≥–Ķ–Ĺ–Ķ—ā–ł—á–Ķ—Ā–ļ–ł—Ö –ł¬†–ł–ľ–ľ—É–Ĺ–Ĺ—č—Ö —Ą–į–ļ—ā–ĺ—Ä–ĺ–≤, –∑–į—ā—Ä—É–ī–Ĺ—Ź–Ķ—ā —Ä–į–∑—Ä–į–Ī–ĺ—ā–ļ—É —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ–≥–ĺ –Ľ–Ķ—á–Ķ–Ĺ–ł—Ź –ī–į–Ĺ–Ĺ–ĺ–≥–ĺ –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł—Ź. –ē—Ā–Ľ–ł –≤¬†—ā–Ķ—Ä–į–Ņ–ł–ł —ć–ļ—Ā—Ā—É–ī–į—ā–ł–≤–Ĺ–ĺ–Ļ (–≤–Ľ–į–∂–Ĺ–ĺ–Ļ) —Ą–ĺ—Ä–ľ—č –í–ú–Ē –ī–ĺ—Ā—ā–ł–≥–Ĺ—É—ā—č –∑–Ĺ–į—á–ł—ā–Ķ–Ľ—Ć–Ĺ—č–Ķ —É—Ā–Ņ–Ķ—Ö–ł, —ā–ĺ –Ľ–Ķ—á–Ķ–Ĺ–ł–Ķ —Ā—É—Ö–ĺ–Ļ —Ą–ĺ—Ä–ľ—č, —Ą–ł–Ĺ–į–Ľ—Ć–Ĺ–ĺ–Ļ —Ā—ā–į–ī–ł–Ķ–Ļ –ļ–ĺ—ā–ĺ—Ä–ĺ–Ļ —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź –ď–ź, –≤–Ņ–Ľ–ĺ—ā—Ć –ī–ĺ –Ĺ–Ķ–ī–į–≤–Ĺ–Ķ–≥–ĺ –≤—Ä–Ķ–ľ–Ķ–Ĺ–ł –ĺ—Ā—ā–į–≤–į–Ľ–ĺ—Ā—Ć —Ā–ł–ľ–Ņ—ā–ĺ–ľ–į—ā–ł—á–Ķ—Ā–ļ–ł–ľ.
–í–ú–Ē¬†‚Äď —Ö—Ä–ĺ–Ĺ–ł—á–Ķ—Ā–ļ–ĺ–Ķ –Ņ—Ä–ĺ–≥—Ä–Ķ—Ā—Ā–ł—Ä—É—é—Č–Ķ–Ķ –Ĺ–Ķ–Ļ—Ä–ĺ–ī–Ķ–≥–Ķ–Ĺ–Ķ—Ä–į—ā–ł–≤–Ĺ–ĺ–Ķ –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł–Ķ —Ā–Ķ—ā—á–į—ā–ļ–ł, —Ö–į—Ä–į–ļ—ā–Ķ—Ä–ł–∑—É—é—Č–Ķ–Ķ—Ā—Ź –Ĺ–Ķ–ĺ–Ī—Ä–į—ā–ł–ľ–ĺ–Ļ –Ņ–ĺ—ā–Ķ—Ä–Ķ–Ļ —Ą–ĺ—ā–ĺ—Ä–Ķ—Ü–Ķ–Ņ—ā–ĺ—Ä–Ĺ—č—Ö –ļ–Ľ–Ķ—ā–ĺ–ļ –ł¬†–ļ–Ľ–Ķ—ā–ĺ–ļ –Ņ–ł–≥–ľ–Ķ–Ĺ—ā–Ĺ–ĺ–≥–ĺ —ć–Ņ–ł—ā–Ķ–Ľ–ł—Ź —Ā–Ķ—ā—á–į—ā–ļ–ł (–ü–≠–°). –ü–≠–° –≤—č–Ņ–ĺ–Ľ–Ĺ—Ź–Ķ—ā —Ä—Ź–ī –≤–į–∂–Ĺ—č—Ö —Ą—É–Ĺ–ļ—Ü–ł–Ļ –ī–Ľ—Ź –Ņ–ĺ–ī–ī–Ķ—Ä–∂–į–Ĺ–ł—Ź —Ā—ā—Ä—É–ļ—ā—É—Ä—č —Ā–Ķ—ā—á–į—ā–ļ–ł: —Ā–Ľ—É–∂–ł—ā¬†–≥–Ķ–ľ–į—ā–ĺ—Ä–Ķ—ā–ł–Ĺ–į–Ľ—Ć–Ĺ—č–ľ –Ī–į—Ä—Ć–Ķ—Ä–ĺ–ľ, –ĺ–Ī–Ķ—Ā–Ņ–Ķ—á–ł–≤–į–Ķ—ā —ā—Ä–ĺ—Ą–ł–ļ—É –ł¬†—É—á–į—Ā—ā–≤—É–Ķ—ā –≤¬†—Ą–į–≥–ĺ—Ü–ł—ā–ĺ–∑–Ķ [2]. –í¬†–ĺ—Ā–Ĺ–ĺ–≤–Ķ –Ņ–į—ā–ĺ—Ą–ł–∑–ł–ĺ–Ľ–ĺ–≥–ł–ł –í–ú–Ē –Ľ–Ķ–∂–ł—ā —Ā–Ľ–ĺ–∂–Ĺ—č–Ļ –ļ–į—Ā–ļ–į–ī –ľ–Ķ—Ö–į–Ĺ–ł–∑–ľ–ĺ–≤, –≤–ļ–Ľ—é—á–į—é—Č–ł–Ļ –Ľ–ĺ–ļ–į–Ľ—Ć–Ĺ–ĺ–Ķ –≤–ĺ—Ā–Ņ–į–Ľ–Ķ–Ĺ–ł–Ķ –ł¬†–Ĺ–Ķ–Ļ—Ä–ĺ–ī–Ķ–≥–Ķ–Ĺ–Ķ—Ä–į—Ü–ł—é –ľ–į–ļ—É–Ľ—č. –†–į–∑–≤–ł—ā–ł–Ķ –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł—Ź –į—Ā—Ā–ĺ—Ü–ł–ł—Ä—É–Ķ—ā—Ā—Ź —Ā¬†–Ņ–ĺ–∂–ł–Ľ—č–ľ –≤–ĺ–∑—Ä–į—Ā—ā–ĺ–ľ, —ć–Ĺ–ī–ĺ–≥–Ķ–Ĺ–Ĺ—č–ľ–ł (–ľ–Ķ—ā–į–Ī–ĺ–Ľ–ł—á–Ķ—Ā–ļ–ł–Ļ –ī–ł—Ā–Ī–į–Ľ–į–Ĺ—Ā, –ĺ–ļ–ł—Ā–Ľ–ł—ā–Ķ–Ľ—Ć–Ĺ—č–Ļ —Ā—ā—Ä–Ķ—Ā—Ā) –ł¬†—ć–ļ–∑–ĺ–≥–Ķ–Ĺ–Ĺ—č–ľ–ł (–Ņ—Ä–Ķ–∂–ī–Ķ –≤—Ā–Ķ–≥–ĺ —ā–į–Ī–į–ļ–ĺ–ļ—É—Ä–Ķ–Ĺ–ł–Ķ) —Ą–į–ļ—ā–ĺ—Ä–į–ľ–ł [3].
–ú–į—ā–Ķ—Ä–ł–į–Ľ –ł¬†–ľ–Ķ—ā–ĺ–ī—č
–ü–ĺ–ł—Ā–ļ –Ĺ–į—É—á–Ĺ—č—Ö —Ä–į–Ī–ĺ—ā, —Ä–į–∑–ľ–Ķ—Č–Ķ–Ĺ–Ĺ—č—Ö –≤¬†–ĺ—ā–Ķ—á–Ķ—Ā—ā–≤–Ķ–Ĺ–Ĺ—č—Ö –ł¬†–∑–į—Ä—É–Ī–Ķ–∂–Ĺ—č—Ö –Ī–į–∑–į—Ö –ī–į–Ĺ–Ĺ—č—Ö, –≤¬†—á–į—Ā—ā–Ĺ–ĺ—Ā—ā–ł PubMed –ł¬†Scopus, –ĺ—Ā—É—Č–Ķ—Ā—ā–≤–Ľ—Ź–Ľ—Ā—Ź –Ņ–嬆–ļ–Ľ—é—á–Ķ–≤—č–ľ —Ā–Ľ–ĺ–≤–į–ľ, —Ā–ĺ–ĺ—ā–≤–Ķ—ā—Ā—ā–≤—É—é—Č–ł–ľ —ā–Ķ–ľ–į—ā–ł–ļ–Ķ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź. –ü–ĺ–Ľ—É—á–Ķ–Ĺ–Ĺ–į—Ź –ł–Ĺ—Ą–ĺ—Ä–ľ–į—Ü–ł—Ź —Ā–ł—Ā—ā–Ķ–ľ–į—ā–ł–∑–ł—Ä–ĺ–≤–į–Ĺ–į –ł¬†–ĺ–Ī–ĺ–Ī—Č–Ķ–Ĺ–į –ī–Ľ—Ź —Ą–ĺ—Ä–ľ–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź —Ü–Ķ–Ľ–ĺ—Ā—ā–Ĺ–ĺ–≥–ĺ –Ņ—Ä–Ķ–ī—Ā—ā–į–≤–Ľ–Ķ–Ĺ–ł—Ź –嬆–ļ–Ľ—é—á–Ķ–≤—č—Ö –∑–≤–Ķ–Ĺ—Ć—Ź—Ö –Ņ–į—ā–ĺ–≥–Ķ–Ĺ–Ķ–∑–į –ď–ź –≤¬†—Ā–ĺ–≤—Ä–Ķ–ľ–Ķ–Ĺ–Ĺ–ĺ–Ļ –ĺ—Ą—ā–į–Ľ—Ć–ľ–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ĺ–Ļ –Ņ—Ä–į–ļ—ā–ł–ļ–Ķ. –í¬†–ĺ—ā–ĺ–Ī—Ä–į–Ĺ–Ĺ—č—Ö –ł—Ā—ā–ĺ—á–Ĺ–ł–ļ–į—Ö –Ņ—Ä–ĺ–į–Ĺ–į–Ľ–ł–∑–ł—Ä–ĺ–≤–į–Ĺ—č –ī–į–Ĺ–Ĺ—č–Ķ –嬆–Ī–ł–ĺ—Ö–ł–ľ–ł—á–Ķ—Ā–ļ–ł—Ö –ļ–į—Ā–ļ–į–ī–į—Ö,¬†–≥–Ķ–Ĺ–Ķ—ā–ł—á–Ķ—Ā–ļ–ł—Ö –Ņ–ĺ–Ľ–ł–ľ–ĺ—Ä—Ą–ł–∑–ľ–į—Ö –ł¬†–ļ–Ľ–Ķ—ā–ĺ—á–Ĺ—č—Ö –≤–∑–į–ł–ľ–ĺ–ī–Ķ–Ļ—Ā—ā–≤–ł—Ź—Ö —Ā¬†—Ü–Ķ–Ľ—Ć—é —É—Ā—ā–į–Ĺ–ĺ–≤–Ľ–Ķ–Ĺ–ł—Ź –≤–∑–į–ł–ľ–ĺ—Ā–≤—Ź–∑–ł –ľ–Ķ–∂–ī—É –ľ–Ķ—ā–į–Ī–ĺ–Ľ–ł—á–Ķ—Ā–ļ–ł–ľ–ł –Ĺ–į—Ä—É—ą–Ķ–Ĺ–ł—Ź–ľ–ł, –ł–ľ–ľ—É–Ĺ–Ĺ—č–ľ –ĺ—ā–≤–Ķ—ā–ĺ–ľ –ł¬†–Ņ–ĺ—Ā–Ľ–Ķ–ī—É—é—Č–Ķ–Ļ¬†–≥–ł–Ī–Ķ–Ľ—Ć—é –ļ–Ľ–Ķ—ā–ĺ–ļ.
–¶–Ķ–Ľ—Ƭ†‚Äď —Ā–ł—Ā—ā–Ķ–ľ–į—ā–ł–∑–ł—Ä–ĺ–≤–į—ā—Ć, –ĺ–Ī–ĺ–Ī—Č–ł—ā—Ć –ł¬†–Ņ—Ä–ĺ–į–Ĺ–į–Ľ–ł–∑–ł—Ä–ĺ–≤–į—ā—Ć —Ā–ĺ–≤—Ä–Ķ–ľ–Ķ–Ĺ–Ĺ—č–Ķ –ī–į–Ĺ–Ĺ—č–Ķ –嬆–Ņ–į—ā–ĺ–≥–Ķ–Ĺ–Ķ–∑–Ķ –ď–ź –Ĺ–į¬†–ľ–ĺ–Ľ–Ķ–ļ—É–Ľ—Ź—Ä–Ĺ–ĺ–ľ, –ļ–Ľ–Ķ—ā–ĺ—á–Ĺ–ĺ–ľ –ł¬†—Ā–ĺ—Ā—É–ī–ł—Ā—ā–ĺ–ľ —É—Ä–ĺ–≤–Ĺ—Ź—Ö –ī–Ľ—Ź –Ī–ĺ–Ľ–Ķ–Ķ¬†–≥–Ľ—É–Ī–ĺ–ļ–ĺ–≥–ĺ –Ņ–ĺ–Ĺ–ł–ľ–į–Ĺ–ł—Ź –Ņ—Ä–ł—á–ł–Ĺ –Ķ–Ķ —Ä–į–∑–≤–ł—ā–ł—Ź –ł¬†–ĺ–Ī–ĺ–∑–Ĺ–į—á–Ķ–Ĺ–ł—Ź –Ĺ–į–ł–Ī–ĺ–Ľ–Ķ–Ķ –Ņ–Ķ—Ä—Ā–Ņ–Ķ–ļ—ā–ł–≤–Ĺ—č—Ö –Ĺ–į–Ņ—Ä–į–≤–Ľ–Ķ–Ĺ–ł–Ļ —ā–Ķ—Ä–į–Ņ–ł–ł.
–ö–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ł–Ļ —Ā–Ľ—É—á–į–Ļ
–ü–į—Ü–ł–Ķ–Ĺ—ā –ö., 65 –Ľ–Ķ—ā, –Ņ—Ä–Ķ–ī—ä—Ź–≤–Ľ—Ź–Ķ—ā –∂–į–Ľ–ĺ–Ī—č –Ĺ–į¬†–≤—č—Ä–į–∂–Ķ–Ĺ–Ĺ–ĺ–Ķ –Ī–Ķ–∑–Ī–ĺ–Ľ–Ķ–∑–Ĺ–Ķ–Ĺ–Ĺ–ĺ–Ķ —Ā–Ĺ–ł–∂–Ķ–Ĺ–ł–Ķ –ĺ—Ā—ā—Ä–ĺ—ā—č —Ü–Ķ–Ĺ—ā—Ä–į–Ľ—Ć–Ĺ–ĺ–≥–ĺ –∑—Ä–Ķ–Ĺ–ł—Ź –≤¬†—ā–Ķ—á–Ķ–Ĺ–ł–Ķ –Ņ–ĺ—Ā–Ľ–Ķ–ī–Ĺ–ł—Ö –Ņ—Ź—ā–ł –Ľ–Ķ—ā, –Ĺ–į–Ľ–ł—á–ł–Ķ –Ņ–ĺ—Ā—ā–ĺ—Ź–Ĺ–Ĺ–ĺ–≥–ĺ —Ā–Ķ—Ä–ĺ–≥–ĺ –Ņ—Ź—ā–Ĺ–į –Ņ–Ķ—Ä–Ķ–ī –Ľ–Ķ–≤—č–ľ¬†–≥–Ľ–į–∑–ĺ–ľ (–Ņ–ĺ–Ľ–ĺ–∂–ł—ā–Ķ–Ľ—Ć–Ĺ–į—Ź —Ü–Ķ–Ĺ—ā—Ä–į–Ľ—Ć–Ĺ–į—Ź —Ā–ļ–ĺ—ā–ĺ–ľ–į). –ė—Ā–Ņ—č—ā—č–≤–į–Ķ—ā –∑–į—ā—Ä—É–ī–Ĺ–Ķ–Ĺ–ł—Ź –Ņ—Ä–ł —á—ā–Ķ–Ĺ–ł–ł, –Ņ–ł—Ā—Ć–ľ–Ķ, —Ä–į–Ī–ĺ—ā–Ķ —Ā¬†–ľ–Ķ–Ľ–ļ–ł–ľ–ł –Ņ—Ä–Ķ–ī–ľ–Ķ—ā–į–ľ–ł (–∑–į–Ĺ–ł–ľ–į–Ķ—ā—Ā—Ź —ą–ł—ā—Ć–Ķ–ľ), –ĺ—ā–ľ–Ķ—á–į–Ķ—ā –ł—Ā–ļ–į–∂–Ķ–Ĺ–ł–Ķ –ļ–ĺ–Ĺ—ā—É—Ä–ĺ–≤ –Ņ—Ä–Ķ–ī–ľ–Ķ—ā–ĺ–≤ (–ľ–Ķ—ā–į–ľ–ĺ—Ä—Ą–ĺ–Ņ—Ā–ł–ł).
–ě–Ī—ä–Ķ–ļ—ā–ł–≤–Ĺ—č–Ļ —Ā—ā–į—ā—É—Ā: Visus OS¬†‚Äď 0,07, –Ĺ–Ķ¬†–ļ–ĺ—Ä—Ä–ł–≥–ł—Ä—É–Ķ—ā—Ā—Ź. –í–Ĺ—É—ā—Ä–ł–≥–Ľ–į–∑–Ĺ–ĺ–Ķ –ī–į–≤–Ľ–Ķ–Ĺ–ł–Ķ (–í–ď–Ē) OS¬†‚Äď 16 –ľ–ľ —Ä—ā. —Ā—ā. (–Ņ–嬆–Ņ–Ĺ–Ķ–≤–ľ–ĺ–ľ–Ķ—ā—Ä–ł–ł). –Ď–ł–ĺ–ľ–ł–ļ—Ä–ĺ—Ā–ļ–ĺ–Ņ–ł—Ź –Ņ–Ķ—Ä–Ķ–ī–Ĺ–Ķ–≥–ĺ –ĺ—ā—Ä–Ķ–∑–ļ–į¬†–≥–Ľ–į–∑–į –Ī–Ķ–∑ –ĺ—Ā–ĺ–Ī–Ķ–Ĺ–Ĺ–ĺ—Ā—ā–Ķ–Ļ. –ü–ĺ–ľ—É—ā–Ĺ–Ķ–Ĺ–ł–Ķ —Ö—Ä—É—Ā—ā–į–Ľ–ł–ļ–į –≤¬†–ļ–ĺ—Ä—ā–ł–ļ–į–Ľ—Ć–Ĺ—č—Ö —Ā–Ľ–ĺ—Ź—Ö. –Ē–Ķ—Ā—ā—Ä—É–ļ—Ü–ł—Ź —Ā—ā–Ķ–ļ–Ľ–ĺ–≤–ł–ī–Ĺ–ĺ–≥–ĺ —ā–Ķ–Ľ–į. –ü–ĺ–Ľ–Ĺ–į—Ź –∑–į–ī–Ĺ—Ź—Ź –ĺ—ā—Ā–Ľ–ĺ–Ļ–ļ–į —Ā—ā–Ķ–ļ–Ľ–ĺ–≤–ł–ī–Ĺ–ĺ–≥–ĺ —ā–Ķ–Ľ–į.
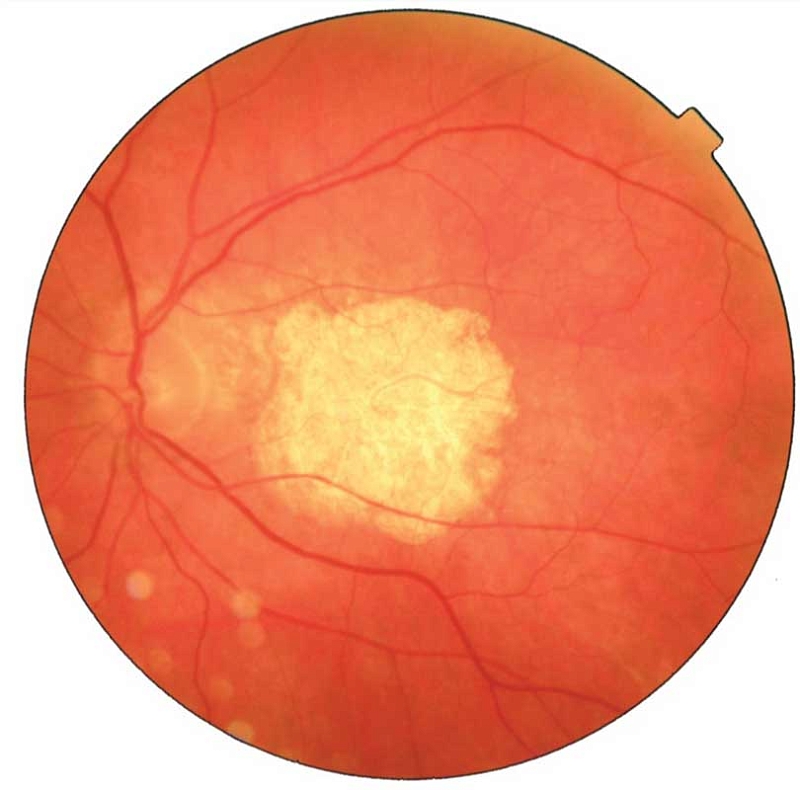
–ě—Ą—ā–į–Ľ—Ć–ľ–ĺ—Ā–ļ–ĺ–Ņ–ł—Ź¬†–≥–Ľ–į–∑–Ĺ–ĺ–≥–ĺ –ī–Ĺ–į: –≤¬†–ľ–į–ļ—É–Ľ—Ź—Ä–Ĺ–ĺ–Ļ –ĺ–Ī–Ľ–į—Ā—ā–ł –≤–ł–∑—É–į–Ľ–ł–∑–ł—Ä—É–Ķ—ā—Ā—Ź –ĺ–ļ—Ä—É–≥–Ľ—č–Ļ –ĺ—á–į–≥ –ī–Ķ–Ņ–ł–≥–ľ–Ķ–Ĺ—ā–į—Ü–ł–ł (–į—ā—Ä–ĺ—Ą–ł–ł) —Ā¬†—á–Ķ—ā–ļ–ł–ľ–ł –ļ—Ä–į—Ź–ľ–ł¬†‚Äď 3,5 –ī–ł–į–ľ–Ķ—ā—Ä–į –ī–ł—Ā–ļ–į –∑—Ä–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ–≥–ĺ –Ĺ–Ķ—Ä–≤–į. –í—Ā–Ľ–Ķ–ī—Ā—ā–≤–ł–Ķ –Ņ–ĺ–Ľ–Ĺ–ĺ–Ļ –į—ā—Ä–ĺ—Ą–ł–ł –ü–≠–° –ł¬†—Ö–ĺ—Ä–ł–ĺ–ļ–į–Ņ–ł–Ľ–Ľ—Ź—Ä–ĺ–≤ –ĺ–Ņ—Ä–Ķ–ī–Ķ–Ľ—Ź—é—ā—Ā—Ź —Ā–ļ–Ľ–Ķ—Ä–ĺ–∑–ł—Ä–ĺ–≤–į–Ĺ–Ĺ—č–Ķ —Ö–ĺ—Ä–ł–ĺ–ł–ī–į–Ľ—Ć–Ĺ—č–Ķ —Ā–ĺ—Ā—É–ī—č –ł¬†–ĺ–Ī–Ĺ–į–∂–Ķ–Ĺ–Ĺ–į—Ź —Ā–ļ–Ľ–Ķ—Ä–į (—Ä–ł—Ā.¬†1).
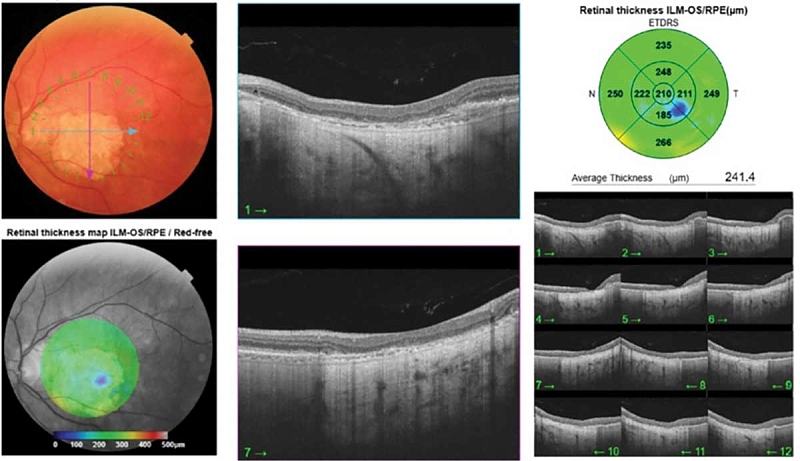
–Ē–į–Ĺ–Ĺ—č–Ķ –ĺ–Ņ—ā–ł—á–Ķ—Ā–ļ–ĺ–Ļ –ļ–ĺ–≥–Ķ—Ä–Ķ–Ĺ—ā–Ĺ–ĺ–Ļ —ā–ĺ–ľ–ĺ–≥—Ä–į—Ą–ł–ł (–ě–ö–Ę): –≤—č—Ä–į–∂–Ķ–Ĺ–Ĺ–ĺ–Ķ –ł—Ā—ā–ĺ–Ĺ—á–Ķ–Ĺ–ł–Ķ —Ā–Ķ—ā—á–į—ā–ļ–ł –≤¬†—Ü–Ķ–Ĺ—ā—Ä–į–Ľ—Ć–Ĺ–ĺ–Ļ –∑–ĺ–Ĺ–Ķ –ī–ĺ 185 –ľ–ļ–ľ. –Ē–Ķ—Ā—ā—Ä—É–ļ—Ü–ł—Ź –Ĺ–į—Ä—É–∂–Ĺ—č—Ö —Ā–Ľ–ĺ–Ķ–≤ —Ā–Ķ—ā—á–į—ā–ļ–ł: –Ĺ–į—Ä—É–∂–Ĺ–ĺ–Ļ –Ņ–ĺ–≥—Ä–į–Ĺ–ł—á–Ĺ–ĺ–Ļ –ľ–Ķ–ľ–Ī—Ä–į–Ĺ—č, –ľ–ł–ĺ–ł–ī–Ĺ–ĺ–Ļ –ł¬†—ć–Ľ–Ľ–ł–Ņ—Ā–ĺ–ł–ī–Ĺ–ĺ–Ļ –∑–ĺ–Ĺ, –į¬†—ā–į–ļ–∂–Ķ –ü–≠–° –ł¬†–Ņ–ĺ–ī–Ľ–Ķ–∂–į—Č–ł—Ö —Ö–ĺ—Ä–ł–ĺ–ļ–į–Ņ–ł–Ľ–Ľ—Ź—Ä–ĺ–≤. –≠—Ą—Ą–Ķ–ļ—ā¬†–≥–ł–Ņ–Ķ—Ä—ā—Ä–į–Ĺ—Ā–ľ–ł—Ā—Ā–ł–ł (–Ņ–ĺ–≤—č—ą–Ķ–Ĺ–Ĺ–ĺ–Ķ –Ņ—Ä–ĺ—Ö–ĺ–∂–ī–Ķ–Ĺ–ł–Ķ –ě–ö–Ę-—Ā–ł–≥–Ĺ–į–Ľ–į) –∑–į —Ā—á–Ķ—ā –ĺ—ā—Ā—É—ā—Ā—ā–≤–ł—Ź —ć–ļ—Ä–į–Ĺ–ł—Ä—É—é—Č–Ķ–≥–ĺ –Ņ–ł–≥–ľ–Ķ–Ĺ—ā–Ĺ–ĺ–≥–ĺ —ć–Ņ–ł—ā–Ķ–Ľ–ł—Ź (—Ä–ł—Ā.¬†2).
–ü—Ä–ł –ď–ź –Ņ–ĺ—Ä–į–∂–į—é—ā—Ā—Ź –Ĺ–į—Ä—É–∂–Ĺ—č–Ķ —Ā–Ľ–ĺ–ł —Ā–Ķ—ā—á–į—ā–ļ–ł, —á—ā–ĺ –Ņ—Ä–ł–≤–ĺ–ī–ł—ā –ļ¬†–Ņ—Ä–ĺ–≥—Ä–Ķ—Ā—Ā–ł—Ä—É—é—Č–Ķ–ľ—É —Ā–Ĺ–ł–∂–Ķ–Ĺ–ł—é —á–ł—Ā–Ľ–į –ļ–Ľ–Ķ—ā–ĺ–ļ –Ņ–ł–≥–ľ–Ķ–Ĺ—ā–Ĺ–ĺ–≥–ĺ —ć–Ņ–ł—ā–Ķ–Ľ–ł—Ź, —Ą–ĺ—ā–ĺ—Ä–Ķ—Ü–Ķ–Ņ—ā–ĺ—Ä–ĺ–≤ –ł¬†–Ņ–Ľ–ĺ—ā–Ĺ–ĺ—Ā—ā–ł —Ā–ĺ—Ā—É–ī–ĺ–≤ —Ö–ĺ—Ä–ł–ĺ–ļ–į–Ņ–ł–Ľ–Ľ—Ź—Ä–ĺ–≤ [4, 5]. –¶–Ķ–Ĺ—ā—Ä–į–Ľ—Ć–Ĺ–į—Ź —Ź–ľ–ļ–į, —Ä–į—Ā–Ņ–ĺ–Ľ–ĺ–∂–Ķ–Ĺ–Ĺ–į—Ź –≤¬†—Ü–Ķ–Ĺ—ā—Ä–Ķ –ľ–į–ļ—É–Ľ—č, —Ā–ĺ–ī–Ķ—Ä–∂–ł—ā –Ī–ĺ–Ľ—Ć—ą–ĺ–Ķ –ļ–ĺ–Ľ–ł—á–Ķ—Ā—ā–≤–ĺ –ļ–ĺ–Ľ–Ī–ĺ—á–ļ–ĺ–≤—č—Ö —Ą–ĺ—ā–ĺ—Ä–Ķ—Ü–Ķ–Ņ—ā–ĺ—Ä–ĺ–≤ –ł¬†–ĺ—ā–≤–Ķ—á–į–Ķ—ā –∑–į —Ü–Ķ–Ĺ—ā—Ä–į–Ľ—Ć–Ĺ–ĺ–Ķ –ł¬†—Ü–≤–Ķ—ā–ĺ–≤–ĺ–Ķ –∑—Ä–Ķ–Ĺ–ł–Ķ. –°–ļ–ĺ—Ä–ĺ—Ā—ā—Ć –Ņ—Ä–ĺ–≥—Ä–Ķ—Ā—Ā–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź –í–ú–Ē –∑–į–≤–ł—Ā–ł—ā –ĺ—ā¬†—Ä–į–∑–ľ–Ķ—Ä–į –ł¬†–Ľ–ĺ–ļ–į–Ľ–ł–∑–į—Ü–ł–ł –į—ā—Ä–ĺ—Ą–ł–ł, –į¬†—ā–į–ļ–∂–Ķ –ĺ—ā¬†—Ā—ā–Ķ–Ņ–Ķ–Ĺ–ł –≤–ĺ–≤–Ľ–Ķ—á–Ķ–Ĺ–ł—Ź —Ü–Ķ–Ĺ—ā—Ä–į–Ľ—Ć–Ĺ–ĺ–Ļ —Ź–ľ–ļ–ł [3, 6]. –ü–Ķ—Ä–≤–ĺ–Ĺ–į—á–į–Ľ—Ć–Ĺ–ĺ –ł–∑–ľ–Ķ–Ĺ–Ķ–Ĺ–ł—Ź –Ņ—Ä–ł –í–ú–Ē –Ņ–ĺ—Ź–≤–Ľ—Ź—é—ā—Ā—Ź –≤¬†–Ņ–Ķ—Ä–ł—Ą–ĺ–≤–Ķ–į–Ľ—Ć–Ĺ–ĺ–Ļ –ĺ–Ī–Ľ–į—Ā—ā–ł –ľ–į–ļ—É–Ľ—č, –į¬†–∑–į—ā–Ķ–ľ, –ĺ–Ī—č—á–Ĺ–ĺ –≤¬†—ā–Ķ—á–Ķ–Ĺ–ł–Ķ 1,5‚Äď2,4¬†–≥–ĺ–ī–į, —Ä–į—Ā–Ņ—Ä–ĺ—Ā—ā—Ä–į–Ĺ—Ź—é—ā—Ā—Ź –Ĺ–į¬†—Ü–Ķ–Ĺ—ā—Ä–į–Ľ—Ć–Ĺ—É—é —Ź–ľ–ļ—É [6, 7]. –ü–Ķ—Ä–ł—Ą–ĺ–≤–Ķ–į–Ľ—Ć–Ĺ–į—Ź –į—ā—Ä–ĺ—Ą–ł—Ź –Ņ—Ä–ł–≤–ĺ–ī–ł—ā –ļ¬†–∑–Ĺ–į—á–ł—ā–Ķ–Ľ—Ć–Ĺ—č–ľ —ā—Ä—É–ī–Ĺ–ĺ—Ā—ā—Ź–ľ –Ņ—Ä–ł –≤—č–Ņ–ĺ–Ľ–Ĺ–Ķ–Ĺ–ł–ł –Ņ–ĺ–≤—Ā–Ķ–ī–Ĺ–Ķ–≤–Ĺ—č—Ö –∑–į–ī–į—á (—á—ā–Ķ–Ĺ–ł–Ķ, –≤–ĺ–∂–ī–Ķ–Ĺ–ł–Ķ –į–≤—ā–ĺ–ľ–ĺ–Ī–ł–Ľ—Ź, –ĺ—Ä–ł–Ķ–Ĺ—ā–į—Ü–ł—Ź –≤¬†—É—Ā–Ľ–ĺ–≤–ł—Ź—Ö –Ĺ–Ķ–ī–ĺ—Ā—ā–į—ā–ĺ—á–Ĺ–ĺ–Ļ –ĺ—Ā–≤–Ķ—Č–Ķ–Ĺ–Ĺ–ĺ—Ā—ā–ł). –ź—ā—Ä–ĺ—Ą–ł—á–Ķ—Ā–ļ–ł–Ķ –ł–∑–ľ–Ķ–Ĺ–Ķ–Ĺ–ł—Ź, –∑–į—ā—Ä–į–≥–ł–≤–į—é—Č–ł–Ķ —Ü–Ķ–Ĺ—ā—Ä–į–Ľ—Ć–Ĺ—É—é —Ź–ľ–ļ—É, –ľ–ĺ–≥—É—ā —Ā—ā–į—ā—Ć –Ņ—Ä–ł—á–ł–Ĺ–ĺ–Ļ —Ā—É—Č–Ķ—Ā—ā–≤–Ķ–Ĺ–Ĺ–ĺ–≥–ĺ —É—Ö—É–ī—ą–Ķ–Ĺ–ł—Ź —Ü–Ķ–Ĺ—ā—Ä–į–Ľ—Ć–Ĺ–ĺ–≥–ĺ –∑—Ä–Ķ–Ĺ–ł—Ź [8].
–ü–į—ā–ĺ–≥–Ķ–Ĺ–Ķ–∑ –ď–ź –Ņ—Ä–Ķ–ī—Ā—ā–į–≤–Ľ—Ź–Ķ—ā —Ā–ĺ–Ī–ĺ–Ļ —Ā–Ľ–ĺ–∂–Ĺ—č–Ļ –ľ–Ĺ–ĺ–≥–ĺ—Ą–į–ļ—ā–ĺ—Ä–Ĺ—č–Ļ –Ņ—Ä–ĺ—Ü–Ķ—Ā—Ā. –†–į–Ĺ–Ĺ—Ź—Ź —Ā—ā–į–ī–ł—Ź –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł—Ź —Ö–į—Ä–į–ļ—ā–Ķ—Ä–ł–∑—É–Ķ—ā—Ā—Ź –Ņ–ĺ—Ź–≤–Ľ–Ķ–Ĺ–ł–Ķ–ľ –≤–Ĺ–Ķ–ļ–Ľ–Ķ—ā–ĺ—á–Ĺ—č—Ö –Ľ–ł–Ņ–ł–ī-—Ā–ĺ–ī–Ķ—Ä–∂–į—Č–ł—Ö —Ā–ļ–ĺ–Ņ–Ľ–Ķ–Ĺ–ł–Ļ¬†‚Äď –ī—Ä—É–∑. –ě–Ĺ–ł –Ľ–ĺ–ļ–į–Ľ–ł–∑—É—é—ā—Ā—Ź –Ņ—Ä–Ķ–ł–ľ—É—Č–Ķ—Ā—ā–≤–Ķ–Ĺ–Ĺ–ĺ –≤¬†–ľ–į–ļ—É–Ľ—Ź—Ä–Ĺ–ĺ–Ļ –∑–ĺ–Ĺ–Ķ –ľ–Ķ–∂–ī—É –Ī–į–∑–į–Ľ—Ć–Ĺ–ĺ–Ļ –Ņ–Ľ–į—Ā—ā–ł–Ĺ–ļ–ĺ–Ļ –ü–≠–° –ł¬†–≤–Ĺ—É—ā—Ä–Ķ–Ĺ–Ĺ–ł–ľ –ļ–ĺ–Ľ–Ľ–į–≥–Ķ–Ĺ–ĺ–≤—č–ľ —Ā–Ľ–ĺ–Ķ–ľ –ľ–Ķ–ľ–Ī—Ä–į–Ĺ—č –Ď—Ä—É—Ö–į [9]. –Ē—Ä—É–∑—č —Ā–ĺ—Ā—ā–ĺ—Ź—ā –ł–∑¬†–Ľ–ł–Ņ–ł–ī–ĺ–≤, –Ī–Ķ–Ľ–ļ–ĺ–≤, –ļ–Ľ–Ķ—ā–ĺ—á–Ĺ–ĺ–≥–ĺ –ī–Ķ—ā—Ä–ł—ā–į, –ĺ—ā–Ľ–ĺ–∂–Ķ–Ĺ–ł–Ļ –Ī–Ķ—ā–į-–į–ľ–ł–Ľ–ĺ–ł–ī–į (–Ņ—Ä–ĺ–ī—É–ļ—ā–ĺ–≤ –ļ–Ľ–Ķ—ā–ĺ—á–Ĺ–ĺ–≥–ĺ —Ä–į—Ā–Ņ–į–ī–į), –į–Ņ–ĺ–Ľ–ł–Ņ–ĺ–Ņ—Ä–ĺ—ā–Ķ–ł–Ĺ–ĺ–≤, –ł–ĺ–Ĺ–ĺ–≤ –∂–Ķ–Ľ–Ķ–∑–į –ł¬†—Ü–ł–Ĺ–ļ–į [10].–Ě–į–Ľ–ł—á–ł–Ķ –ļ—Ä—É–Ņ–Ĺ—č—Ö –ł–Ľ–ł –ľ—Ź–≥–ļ–ł—Ö –ī—Ä—É–∑ —Ā¬†–Ĺ–Ķ—á–Ķ—ā–ļ–ł–ľ–ł¬†–≥—Ä–į–Ĺ–ł—Ü–į–ľ–ł —É–ļ–į–∑—č–≤–į–Ķ—ā –Ĺ–į¬†–≤—č—Ā–ĺ–ļ–ł–Ļ —Ä–ł—Ā–ļ¬†–Ņ—Ä–ĺ–≥—Ä–Ķ—Ā—Ā–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł—Ź.
–Ē–Ľ—Ź –Ņ–ĺ–Ĺ–ł–ľ–į–Ĺ–ł—Ź –Ņ–į—ā–ĺ–≥–Ķ–Ĺ–Ķ–∑–į –ď–ź –Ĺ–Ķ–ĺ–Ī—Ö–ĺ–ī–ł–ľ–ĺ –ĺ–Ī—Ä–į—ā–ł—ā—Ć—Ā—Ź –ļ¬†–ľ–Ķ—Ö–į–Ĺ–ł–∑–ľ—É –∑—Ä–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ–≥–ĺ –≤–ĺ—Ā–Ņ—Ä–ł—Ź—ā–ł—Ź¬†‚Äď —Ą–ł–∑–ł–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ĺ–ľ—É –Ņ—Ä–ĺ—Ü–Ķ—Ā—Ā—É –Ņ—Ä–Ķ–ĺ–Ī—Ä–į–∑–ĺ–≤–į–Ĺ–ł—Ź —Ā–≤–Ķ—ā–ĺ–≤–ĺ–Ļ —ć–Ĺ–Ķ—Ä–≥–ł–ł –≤¬†–Ĺ–Ķ—Ä–≤–Ĺ—č–Ļ –ł–ľ–Ņ—É–Ľ—Ć—Ā. –§–ĺ—ā–ĺ–Ĺ —Ā–≤–Ķ—ā–į –Ņ–ĺ–Ņ–į–ī–į–Ķ—ā –Ĺ–į¬†—Ā–Ķ—ā—á–į—ā–ļ—É –ł¬†–Ņ–ĺ–≥–Ľ–ĺ—Č–į–Ķ—ā—Ā—Ź –ľ–ĺ–Ľ–Ķ–ļ—É–Ľ–ĺ–Ļ —Ä–ĺ–ī–ĺ–Ņ—Ā–ł–Ĺ–į. –í¬†—Ā–Ķ—ā—á–į—ā–ļ–Ķ –Ņ—Ä–ĺ–ł—Ā—Ö–ĺ–ī–ł—ā –Ĺ–Ķ–Ņ—Ä–Ķ—Ä—č–≤–Ĺ–į—Ź —Ü–Ķ–Ņ—Ć –Ī–ł–ĺ—Ö–ł–ľ–ł—á–Ķ—Ā–ļ–ł—Ö —Ä–Ķ–į–ļ—Ü–ł–Ļ, –ĺ–Ī–Ķ—Ā–Ņ–Ķ—á–ł–≤–į—é—Č–ł—Ö –≤–ĺ—Ā—Ā—ā–į–Ĺ–ĺ–≤–Ľ–Ķ–Ĺ–ł–Ķ –ł¬†—Ä–Ķ–≥–Ķ–Ĺ–Ķ—Ä–į—Ü–ł—é –∑—Ä–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ–≥–ĺ –Ņ–ł–≥–ľ–Ķ–Ĺ—ā–į, —á—ā–ĺ –Ņ—Ä–ł–≤–ĺ–ī–ł—ā –ļ¬†–Ķ–≥–ĺ –Ņ–ĺ—Ā—ā–ĺ—Ź–Ĺ–Ĺ–ĺ–ľ—É –ĺ–Ī–Ĺ–ĺ–≤–Ľ–Ķ–Ĺ–ł—é. –ö–Ľ—é—á–Ķ–≤—č–ľ –ļ–ĺ–ľ–Ņ–ĺ–Ĺ–Ķ–Ĺ—ā–ĺ–ľ —ć—ā–ĺ–≥–ĺ —Ü–ł–ļ–Ľ–į —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź –≤–ł—ā–į–ľ–ł–Ŭ†A (—Ä–Ķ—ā–ł–Ĺ–ĺ–Ľ), –ļ–ĺ—ā–ĺ—Ä—č–Ļ –≤¬†—Ą–ĺ—Ä–ľ–Ķ 11-—Ü–ł—Ā-—Ä–Ķ—ā–ł–Ĺ–į–Ľ—Ź —Ä–į—Ā—Ā–ľ–į—ā—Ä–ł–≤–į–Ķ—ā—Ā—Ź –ļ–į–ļ –Ĺ–Ķ–ĺ–Ī—Ö–ĺ–ī–ł–ľ—č–Ļ –ļ–ĺ—Ą–į–ļ—ā–ĺ—Ä –∑—Ä–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ–≥–ĺ —Ü–ł–ļ–Ľ–į [11]. –ü—Ä–ł —ć—ā–ĺ–ľ –ł–Ĺ–ĺ–≥–ī–į –ĺ–Ī—Ä–į–∑—É—é—ā—Ā—Ź –Ņ–ĺ–Ī–ĺ—á–Ĺ—č–Ķ –Ņ—Ä–ĺ–ī—É–ļ—ā—č –ī–ł–ľ–Ķ—Ä–ł–∑–į—Ü–ł–ł –≤–ł—ā–į–ľ–ł–Ĺ–į A, –≤¬†—á–į—Ā—ā–Ĺ–ĺ—Ā—ā–ł N-—Ä–Ķ—ā–ł–Ĺ–ł–Ľ–ł–ī–Ķ–Ĺ-N-—Ä–Ķ—ā–ł–Ĺ–ł–Ľ—ć—ā–į–Ĺ–ĺ–Ľ–į–ľ–ł–Ĺ (A2E)¬†‚Äď —Ā–ļ–Ľ–Ķ–Ķ–Ĺ–Ĺ–į—Ź –ľ–ĺ–Ľ–Ķ–ļ—É–Ľ–į, —ā–ĺ–ļ—Ā–ł—á–Ĺ–į—Ź –ł¬†–Ĺ–Ķ—Ā–Ņ–ĺ—Ā–ĺ–Ī–Ĺ–į—Ź —É—ā–ł–Ľ–ł–∑–ł—Ä–ĺ–≤–į—ā—Ć—Ā—Ź –ļ–Ľ–Ķ—ā–ļ–į–ľ–ł —Ā–Ķ—ā—á–į—ā–ļ–ł,¬†‚Äď –ł–∑¬†–ī–ł–ľ–Ķ—Ä–į —ā—Ä–į–Ĺ—Ā-—Ä–Ķ—ā–ł–Ĺ–į–Ľ—Ć–ī–Ķ–≥–ł–ī–į. –£—Ā–ļ–ĺ—Ä–Ķ–Ĺ–Ĺ–į—Ź –ī–ł–ľ–Ķ—Ä–ł–∑–į—Ü–ł—Ź –≤–ł—ā–į–ľ–ł–Ĺ–į A –Ņ—Ä–ł–≤–ĺ–ī–ł—ā –ļ¬†–Ī—č—Ā—ā—Ä–ĺ–ľ—É –Ĺ–į–ļ–ĺ–Ņ–Ľ–Ķ–Ĺ–ł—é –ī–ł–ľ–Ķ—Ä–ĺ–≤ –ł¬†–ĺ—ā–Ľ–ĺ–∂–Ķ–Ĺ–ł–Ļ –Ľ–ł–Ņ–ĺ—Ą—É—Ā—Ü–ł–Ĺ–į, —á—ā–ĺ –≤—č–∑—č–≤–į–Ķ—ā –į—ā—Ä–ĺ—Ą–ł—é –Ņ–ł–≥–ľ–Ķ–Ĺ—ā–Ĺ–ĺ–≥–ĺ —ć–Ņ–ł—ā–Ķ–Ľ–ł—Ź [12]. –ė–∑–Ī—č—ā–ĺ—á–Ĺ–ĺ–Ķ –Ĺ–į–ļ–ĺ–Ņ–Ľ–Ķ–Ĺ–ł–Ķ —ā–ĺ–ļ—Ā–ł—á–Ĺ—č—Ö –ī–ł–ľ–Ķ—Ä–ĺ–≤, –Ĺ–į–Ņ—Ä–ł–ľ–Ķ—Ä A2E, –Ņ—Ä–ĺ–≤–ĺ—Ü–ł—Ä—É–Ķ—ā –Ņ–ĺ—Ā—ā–ĺ—Ź–Ĺ–Ĺ—É—é –ī–Ķ–≥—Ä–į–ī–į—Ü–ł—é –ļ–Ľ–Ķ—ā–ĺ–ļ –ü–≠–°, –Ľ–Ķ–∂–į—Č—É—é –≤¬†–ĺ—Ā–Ĺ–ĺ–≤–Ķ —Ä–į–∑–≤–ł—ā–ł—Ź –í–ú–Ē [12, 13].
–Ě–į–ļ–ĺ–Ņ–Ľ–Ķ–Ĺ–ł–Ķ A2E –Ņ–ĺ–≤—č—ą–į–Ķ—ā pH –Ľ–ł–∑–ĺ—Ā–ĺ–ľ –≤¬†–ļ–Ľ–Ķ—ā–ļ–į—Ö –ü–≠–°, –Ĺ–į—Ä—É—ą–į—Ź –ł—Ö —Ą–Ķ—Ä–ľ–Ķ–Ĺ—ā–į—ā–ł–≤–Ĺ—É—é –į–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć –ł¬†–ī–Ķ–Ľ–į—Ź –ł—Ö –Ĺ–Ķ—Ā–Ņ–ĺ—Ā–ĺ–Ī–Ĺ—č–ľ–ł —É—ā–ł–Ľ–ł–∑–ł—Ä–ĺ–≤–į—ā—Ć –ĺ—ā—Ä–į–Ī–ĺ—ā–į–Ĺ–Ĺ—č–Ķ –ļ–Ľ–Ķ—ā–ĺ—á–Ĺ—č–Ķ —Ā—ā—Ä—É–ļ—ā—É—Ä—č [14, 15]. –ö—Ä–ĺ–ľ–Ķ —ā–ĺ–≥–ĺ, –Ņ–ĺ–ī –ī–Ķ–Ļ—Ā—ā–≤–ł–Ķ–ľ —Ā–≤–Ķ—ā–į –Ľ–ł–Ņ–ĺ—Ą—É—Ā—Ü–ł–Ŭ†–≥–Ķ–Ĺ–Ķ—Ä–ł—Ä—É–Ķ—ā –į–ļ—ā–ł–≤–Ĺ—č–Ķ —Ą–ĺ—Ä–ľ—č –ļ–ł—Ā–Ľ–ĺ—Ä–ĺ–ī–į [16], –≤—č–∑—č–≤–į—é—Č–ł–Ķ –ĺ–ļ–ł—Ā–Ľ–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ–Ķ –Ņ–ĺ–≤—Ä–Ķ–∂–ī–Ķ–Ĺ–ł–Ķ –ļ–Ľ–Ķ—ā–ĺ–ļ –Ņ–ł–≥–ľ–Ķ–Ĺ—ā–Ĺ–ĺ–≥–ĺ —ć–Ņ–ł—ā–Ķ–Ľ–ł—Ź¬†[15]. –°–ł—ā—É–į—Ü–ł—Ź —É—Ā—É–≥—É–Ī–Ľ—Ź–Ķ—ā—Ā—Ź —ā–Ķ–ľ, —á—ā–ĺ –ü–≠–° —ā–Ķ—Ä—Ź–Ķ—ā —Ā–Ņ–ĺ—Ā–ĺ–Ī–Ĺ–ĺ—Ā—ā—Ć —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ —É—ā–ł–Ľ–ł–∑–ł—Ä–ĺ–≤–į—ā—Ć —Ą—Ä–į–≥–ľ–Ķ–Ĺ—ā—č –ľ–Ķ–ľ–Ī—Ä–į–Ĺ–Ĺ—č—Ö –ī–ł—Ā–ļ–ĺ–≤, –ĺ—ā–ī–Ķ–Ľ—Ź—é—Č–ł–Ķ—Ā—Ź –ĺ—ā¬†—Ą–ĺ—ā–ĺ—Ä–Ķ—Ü–Ķ–Ņ—ā–ĺ—Ä–ĺ–≤, —á—ā–ĺ –≤–Ķ–ī–Ķ—ā –ļ¬†–Ĺ–į–ļ–ĺ–Ņ–Ľ–Ķ–Ĺ–ł—é –Ľ–ł–Ņ–ł–ī–Ĺ—č—Ö –≤–ļ–Ľ—é—á–Ķ–Ĺ–ł–Ļ. –Ē–ĺ–ļ–į–∑–į–Ĺ–ĺ, —á—ā–ĺ —á—Ä–Ķ–∑–ľ–Ķ—Ä–Ĺ–ĺ–Ķ –≤–ĺ–∑–ī–Ķ–Ļ—Ā—ā–≤–ł–Ķ —Ā–ł–Ĺ–Ķ–≥–ĺ —Ā–≤–Ķ—ā–į –≤¬†–Ī–ĺ–Ľ—Ć—ą–Ķ–Ļ —Ā—ā–Ķ–Ņ–Ķ–Ĺ–ł –Ņ–ĺ–≤—Ä–Ķ–∂–ī–į–Ķ—ā –Ņ–ł–≥–ľ–Ķ–Ĺ—ā–Ĺ—č–Ļ —ć–Ņ–ł—ā–Ķ–Ľ–ł–Ļ, –į¬†–Ī–Ķ–Ľ–ĺ–≥–ĺ —Ā–≤–Ķ—ā–į¬†‚Äď —Ą–ĺ—ā–ĺ—Ä–Ķ—Ü–Ķ–Ņ—ā–ĺ—Ä—č. –°–ĺ—á–Ķ—ā–į–Ĺ–ł–Ķ¬†–≥–Ķ–Ĺ–Ķ—ā–ł—á–Ķ—Ā–ļ–ĺ–Ļ –Ņ—Ä–Ķ–ī—Ä–į—Ā–Ņ–ĺ–Ľ–ĺ–∂–Ķ–Ĺ–Ĺ–ĺ—Ā—ā–ł, –Ĺ–Ķ–Ī–Ľ–į–≥–ĺ–Ņ—Ä–ł—Ź—ā–Ĺ—č—Ö –≤–Ĺ–Ķ—ą–Ĺ–ł—Ö —Ą–į–ļ—ā–ĺ—Ä–ĺ–≤ –ł¬†—Ö—Ä–ĺ–Ĺ–ł—á–Ķ—Ā–ļ–ĺ–≥–ĺ –≤–ĺ—Ā–Ņ–į–Ľ–Ķ–Ĺ–ł—Ź –∑–į–Ņ—É—Ā–ļ–į–Ķ—ā –Ĺ–Ķ–ĺ–Ī—Ä–į—ā–ł–ľ—č–Ķ –ī–Ķ–≥–Ķ–Ĺ–Ķ—Ä–į—ā–ł–≤–Ĺ—č–Ķ –ł–∑–ľ–Ķ–Ĺ–Ķ–Ĺ–ł—Ź, —Ö–į—Ä–į–ļ—ā–Ķ—Ä–Ĺ—č–Ķ –ī–Ľ—Ź –ľ–į–ļ—É–Ľ—Ź—Ä–Ĺ–ĺ–Ļ –ī–ł—Ā—ā—Ä–ĺ—Ą–ł–ł, –≤¬†—ā–ĺ–ľ —á–ł—Ā–Ľ–Ķ –ď–ź.
–ē—Č–Ķ –ĺ–ī–ł–Ĺ —Ą–į–ļ—ā–ĺ—Ä —Ä–ł—Ā–ļ–į —Ā–≤—Ź–∑–į–Ĺ —Ā¬†–Ņ–ĺ—ā–Ķ—Ä–Ķ–Ļ —Ą–Ķ—Ä–ľ–Ķ–Ĺ—ā–į DICER1 –≤—Ā–Ľ–Ķ–ī—Ā—ā–≤–ł–Ķ –≤–ĺ–∑—Ä–į—Ā—ā–Ĺ–ĺ–≥–ĺ –ĺ–ļ–ł—Ā–Ľ–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ–≥–ĺ —Ā—ā—Ä–Ķ—Ā—Ā–į [17]. DICER1¬†‚Äď —Ä–ł–Ī–ĺ–Ĺ—É–ļ–Ľ–Ķ–į–∑–į, —Ä–į–∑—Ä–Ķ–∑–į—é—Č–į—Ź –ī–Ľ–ł–Ĺ–Ĺ—č–Ķ –ī–≤—É—Ö—Ü–Ķ–Ņ–ĺ—á–Ķ—á–Ĺ—č–Ķ –†–Ě–ö –Ĺ–į¬†–ļ–ĺ—Ä–ĺ—ā–ļ–ł–Ķ —Ą—Ä–į–≥–ľ–Ķ–Ĺ—ā—č. –ü—Ä–ł –ī–ł—Ā—Ą—É–Ĺ–ļ—Ü–ł–ł DICER1 –≤¬†–ļ–Ľ–Ķ—ā–ļ–į—Ö –Ņ–ł–≥–ľ–Ķ–Ĺ—ā–Ĺ–ĺ–≥–ĺ —ć–Ņ–ł—ā–Ķ–Ľ–ł—Ź –Ĺ–į–ļ–į–Ņ–Ľ–ł–≤–į—é—ā—Ā—Ź –ĺ–Ņ–į—Ā–Ĺ—č–Ķ —Ą—Ä–į–≥–ľ–Ķ–Ĺ—ā—謆‚Äď Alu-–†–Ě–ö [18, 19]. –≠—ā–ł –ľ–ĺ–Ľ–Ķ–ļ—É–Ľ—č –Ī–Ľ–ĺ–ļ–ł—Ä—É—é—ā –į–Ĺ—ā–ł–ĺ–ļ—Ā–ł–ī–į–Ĺ—ā–Ĺ—É—é –∑–į—Č–ł—ā—É –ļ–Ľ–Ķ—ā–ļ–ł, –ī–Ķ–Ľ–į—Ź –Ķ–Ķ —É—Ź–∑–≤–ł–ľ–ĺ–Ļ –ļ¬†–Ņ–ĺ–≤—Ä–Ķ–∂–ī–Ķ–Ĺ–ł—Ź–ľ [20].
–ö—É—Ä–Ķ–Ĺ–ł–Ķ —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź –ĺ–ī–Ĺ–ł–ľ –ł–∑¬†–≤–Ķ–ī—É—Č–ł—Ö —Ą–į–ļ—ā–ĺ—Ä–ĺ–≤ —Ä–ł—Ā–ļ–į —Ä–į–∑–≤–ł—ā–ł—Ź –í–ú–Ē [21]. –ö–ĺ–ľ–Ņ–ĺ–Ĺ–Ķ–Ĺ—ā—č —ā–į–Ī–į—á–Ĺ–ĺ–≥–ĺ –ī—č–ľ–į, –Ĺ–į–Ņ—Ä–ł–ľ–Ķ—Ĭ†–≥–ł–ī—Ä–ĺ—Ö–ł–Ĺ–ĺ–Ĺ, –≤—č–∑—č–≤–į—é—ā –ĺ–ļ–ł—Ā–Ľ–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ–Ķ –Ņ–ĺ–≤—Ä–Ķ–∂–ī–Ķ–Ĺ–ł–Ķ –ļ–Ľ–Ķ—ā–ĺ–ļ –ł¬†–ĺ–Ī—Ä–į–∑–ĺ–≤–į–Ĺ–ł–Ķ –į–ļ—ā–ł–≤–Ĺ—č—Ö —Ą–ĺ—Ä–ľ –ļ–ł—Ā–Ľ–ĺ—Ä–ĺ–ī–į [22]. –ě–ļ–ł—Ā–Ľ–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ–ľ—É —Ā—ā—Ä–Ķ—Ā—Ā—É –≤¬†—Ā–Ķ—ā—á–į—ā–ļ–Ķ —ā–į–ļ–∂–Ķ —Ā–Ņ–ĺ—Ā–ĺ–Ī—Ā—ā–≤—É—é—ā –ĺ—ā—Ā–Ľ–ĺ–Ļ–ļ–į —Ą–ĺ—ā–ĺ—Ä–Ķ—Ü–Ķ–Ņ—ā–ĺ—Ä–ĺ–≤, –ł–∑–Ī—č—ā–ĺ—á–Ĺ–ĺ–Ķ –ĺ—Ā–≤–Ķ—Č–Ķ–Ĺ–ł–Ķ –ł¬†—Ā—ā–į—Ä–Ķ–Ĺ–ł–Ķ [23]. –í¬†–Ĺ–ĺ—Ä–ľ–Ķ —Ą–ĺ—ā–ĺ—Ä–Ķ—Ü–Ķ–Ņ—ā–ĺ—Ä—č –ł—Ā–Ņ–ĺ–Ľ—Ć–∑—É—é—ā¬†–≥–Ľ—é–ļ–ĺ–∑—É –ī–Ľ—Ź¬†–≥–Ľ–ł–ļ–ĺ–Ľ–ł–∑–į –ł¬†–≤—č—Ä–į–Ī–į—ā—č–≤–į—é—ā –Ī–ĺ–Ľ—Ć—ą–ĺ–Ķ –ļ–ĺ–Ľ–ł—á–Ķ—Ā—ā–≤–ĺ –Ľ–į–ļ—ā–į—ā–į, –ļ–ĺ—ā–ĺ—Ä—č–Ļ –Ņ–ĺ–≥–Ľ–ĺ—Č–į–Ķ—ā—Ā—Ź –ļ–Ľ–Ķ—ā–ļ–į–ľ–ł –Ņ–ł–≥–ľ–Ķ–Ĺ—ā–Ĺ–ĺ–≥–ĺ —ć–Ņ–ł—ā–Ķ–Ľ–ł—Ź –ī–Ľ—Ź –Ņ—Ä–ĺ–ī—É–ļ—Ü–ł–ł —ć–Ĺ–Ķ—Ä–≥–ł–ł —á–Ķ—Ä–Ķ–∑ –ĺ–ļ–ł—Ā–Ľ–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ–Ķ —Ą–ĺ—Ā—Ą–ĺ—Ä–ł–Ľ–ł—Ä–ĺ–≤–į–Ĺ–ł–Ķ –≤¬†–ľ–ł—ā–ĺ—Ö–ĺ–Ĺ–ī—Ä–ł—Ź—Ö [24]. –ě–ī–Ĺ–į–ļ–ĺ –Ņ–ĺ–ī –ī–Ķ–Ļ—Ā—ā–≤–ł–Ķ–ľ –ĺ–ļ–ł—Ā–Ľ–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ–≥–ĺ —Ā—ā—Ä–Ķ—Ā—Ā–į –≤¬†–ľ–ł—ā–ĺ—Ö–ĺ–Ĺ–ī—Ä–ł—Ź—Ö —É—Ā–ł–Ľ–ł–≤–į–Ķ—ā—Ā—Ź¬†–≥–Ľ–ł–ļ–ĺ–Ľ–ł—ā–ł—á–Ķ—Ā–ļ–ł–Ļ –ľ–Ķ—ā–į–Ī–ĺ–Ľ–ł–∑–ľ –≤—Ā–Ľ–Ķ–ī—Ā—ā–≤–ł–Ķ –ľ–Ķ—ā–į–Ī–ĺ–Ľ–ł—á–Ķ—Ā–ļ–ĺ–≥–ĺ –Ņ–Ķ—Ä–Ķ–Ņ—Ä–ĺ–≥—Ä–į–ľ–ľ–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź, —á—ā–ĺ –Ĺ–į—Ä—É—ą–į–Ķ—ā –ł—Ö —Ą—É–Ĺ–ļ—Ü–ł—é –ł¬†–Ņ—Ä–ł–≤–ĺ–ī–ł—ā –ļ¬†–≥–ł–Ī–Ķ–Ľ–ł —Ą–ĺ—ā–ĺ—Ä–Ķ—Ü–Ķ–Ņ—ā–ĺ—Ä–ĺ–≤ [25].
–†–ĺ–Ľ—Ć –ľ–ł–ļ—Ä–ĺ–≥–Ľ–ł–ł –ł¬†–ľ–ĺ–Ĺ–ĺ—Ü–ł—ā–ĺ–≤ –≤¬†–Ņ–į—ā–ĺ–Ī–ł–ĺ–Ľ–ĺ–≥–ł–ł –í–ú–Ē
–ú–ł–ļ—Ä–ĺ–≥–Ľ–ł—Ź —Ā–Ķ—ā—á–į—ā–ļ–ł –Ņ–ĺ–ī–ī–Ķ—Ä–∂–ł–≤–į–Ķ—ā¬†–≥–ĺ–ľ–Ķ–ĺ—Ā—ā–į–∑, —É–ī–į–Ľ—Ź—Ź —ā–ĺ–ļ—Ā–ł—á–Ĺ—č–Ķ –Ņ—Ä–ĺ–ī—É–ļ—ā—č, –≤—č—Ź–≤–Ľ—Ź—Ź –Ņ–ĺ–≤—Ä–Ķ–∂–ī–Ķ–Ĺ–ł—Ź –ł¬†–ĺ–Ī–Ķ—Ā–Ņ–Ķ—á–ł–≤–į—Ź –≤—Ä–ĺ–∂–ī–Ķ–Ĺ–Ĺ—č–Ļ –ł–ľ–ľ—É–Ĺ–Ĺ—č–Ļ –ĺ—ā–≤–Ķ—ā [26]. –ě–ī–Ĺ–į–ļ–ĺ —Ā–ĺ—á–Ķ—ā–į–Ĺ–Ĺ–ĺ–Ķ –≤–ĺ–∑–ī–Ķ–Ļ—Ā—ā–≤–ł–Ķ¬†–≥–Ķ–Ĺ–Ķ—ā–ł—á–Ķ—Ā–ļ–ł—Ö –ł¬†—Ā—Ä–Ķ–ī–ĺ–≤—č—Ö —Ą–į–ļ—ā–ĺ—Ä–ĺ–≤ –Ņ–Ķ—Ä–Ķ–≤–ĺ–ī–ł—ā –ľ–ł–ļ—Ä–ĺ–≥–Ľ–ł—é –≤¬†—Ä–Ķ–į–ļ—ā–ł–≤–Ĺ–ĺ–Ķ —Ā–ĺ—Ā—ā–ĺ—Ź–Ĺ–ł–Ķ: –ĺ–Ĺ–į –Ĺ–į—á–ł–Ĺ–į–Ķ—ā —Ā–Ķ–ļ—Ä–Ķ—ā–ł—Ä–ĺ–≤–į—ā—Ć –Ņ–į—ā–ĺ–≥–Ķ–Ĺ–Ĺ—č–Ķ —Ü–ł—ā–ĺ–ļ–ł–Ĺ—č, —Ā—ā–ł–ľ—É–Ľ–ł—Ä—É–Ķ—ā –ľ–ł–≥—Ä–į—Ü–ł—é –ľ–ĺ–Ĺ–ĺ—Ü–ł—ā–ĺ–≤ –≤¬†—Ā—É–Ī—Ä–Ķ—ā–ł–Ĺ–į–Ľ—Ć–Ĺ–ĺ–Ķ –Ņ—Ä–ĺ—Ā—ā—Ä–į–Ĺ—Ā—ā–≤–ĺ –ł¬†–Ĺ–į—Ä—É—ą–į–Ķ—ā —Ü–Ķ–Ľ–ĺ—Ā—ā–Ĺ–ĺ—Ā—ā—Ć —Ą–ĺ—ā–ĺ—Ä–Ķ—Ü–Ķ–Ņ—ā–ĺ—Ä–ĺ–≤ –ł¬†–Ņ–ł–≥–ľ–Ķ–Ĺ—ā–Ĺ–ĺ–≥–ĺ —ć–Ņ–ł—ā–Ķ–Ľ–ł—Ź [27]. –í¬†—É—Ā–Ľ–ĺ–≤–ł—Ź—Ö —Ö—Ä–ĺ–Ĺ–ł—á–Ķ—Ā–ļ–ĺ–≥–ĺ —Ā—ā—Ä–Ķ—Ā—Ā–į¬†–≥–ł–Ņ–Ķ—Ä–į–ļ—ā–ł–≤–Ĺ—č–Ļ –ł–ľ–ľ—É–Ĺ–Ĺ—č–Ļ –ĺ—ā–≤–Ķ—ā, –ĺ–Ī—É—Ā–Ľ–ĺ–≤–Ľ–Ķ–Ĺ–Ĺ—č–Ļ –ł–Ĺ—Ą–ł–Ľ—Ć—ā—Ä–į—Ü–ł–Ķ–Ļ –ľ–ĺ–Ĺ–ĺ–Ĺ—É–ļ–Ľ–Ķ–į—Ä–Ĺ—č—Ö —Ą–į–≥–ĺ—Ü–ł—ā–ĺ–≤ (–ľ–ł–ļ—Ä–ĺ–≥–Ľ–ł–ł, –ľ–į–ļ—Ä–ĺ—Ą–į–≥–ĺ–≤, –ľ–ĺ–Ĺ–ĺ—Ü–ł—ā–ĺ–≤), –į–ļ—ā–ł–≤–ł—Ä—É–Ķ—ā –Ņ—Ä–ĺ–≤–ĺ—Ā–Ņ–į–Ľ–ł—ā–Ķ–Ľ—Ć–Ĺ—č–Ļ –Ņ—É—ā—Ć NF-kB, —á—ā–ĺ –Ņ—Ä–ł–≤–ĺ–ī–ł—ā –ļ¬†–ľ–į—Ā—Ā–ł—Ä–ĺ–≤–į–Ĺ–Ĺ–ĺ–Ļ —Ā–Ķ–ļ—Ä–Ķ—Ü–ł–ł –≤–ĺ—Ā–Ņ–į–Ľ–ł—ā–Ķ–Ľ—Ć–Ĺ—č—Ö —Ü–ł—ā–ĺ–ļ–ł–Ĺ–ĺ–≤ –ł¬†—É—Ā–ļ–ĺ—Ä–Ķ–Ĺ–Ĺ–ĺ–Ļ¬†–≥–ł–Ī–Ķ–Ľ–ł –Ĺ–Ķ–Ļ—Ä–ĺ–Ĺ–ĺ–≤ —Ā–Ķ—ā—á–į—ā–ļ–ł [28].
–†–ĺ–Ľ—Ć —Ā–ł—Ā—ā–Ķ–ľ—č –ļ–ĺ–ľ–Ņ–Ľ–Ķ–ľ–Ķ–Ĺ—ā–į
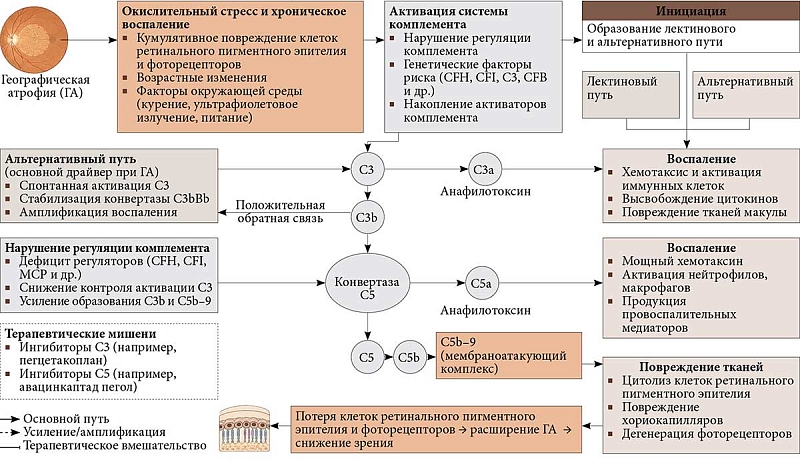
–ź–ļ—ā–ł–≤–Ĺ—É—é —Ä–ĺ–Ľ—Ć –≤¬†–Ņ–į—ā–ĺ–≥–Ķ–Ĺ–Ķ–∑–Ķ –ď–ź –ł–≥—Ä–į–Ķ—ā —Ā–ł—Ā—ā–Ķ–ľ–į –ļ–ĺ–ľ–Ņ–Ľ–Ķ–ľ–Ķ–Ĺ—ā–į¬†‚Äst–≥—Ä—É–Ņ–Ņ–į –Ī–ĺ–Ľ–Ķ–Ķ —á–Ķ–ľ –ł–∑¬†30 —Ü–ł—Ä–ļ—É–Ľ–ł—Ä—É—é—Č–ł—Ö —Ā–ł—Ā—ā–Ķ–ľ–Ĺ—č—Ö –Ī–Ķ–Ľ–ļ–ĺ–≤, —É—á–į—Ā—ā–≤—É—é—Č–ł—Ö –≤¬†–≤–ĺ—Ā–Ņ–į–Ľ–Ķ–Ĺ–ł–ł, —Ą–į–≥–ĺ—Ü–ł—ā–ĺ–∑–Ķ,¬†–≥–ł–Ī–Ķ–Ľ–ł –ļ–Ľ–Ķ—ā–ĺ–ļ, –į¬†—ā–į–ļ–∂–Ķ –≤¬†–ĺ–Ī–Ĺ–į—Ä—É–∂–Ķ–Ĺ–ł–ł –ł¬†—ć–Ľ–ł–ľ–ł–Ĺ–į—Ü–ł–ł –Ņ–į—ā–ĺ–≥–Ķ–Ĺ–ĺ–≤ [29]. –°–ł—Ā—ā–Ķ–ľ–į –ļ–ĺ–ľ–Ņ–Ľ–Ķ–ľ–Ķ–Ĺ—ā–į –ĺ–Ī–Ķ—Ā–Ņ–Ķ—á–ł–≤–į–Ķ—ā –ĺ–Ņ—Ā–ĺ–Ĺ–ł–∑–į—Ü–ł—鬆‚Äď –Ņ–ĺ–ļ—Ä—č—ā–ł–Ķ —á—É–∂–Ķ—Ä–ĺ–ī–Ĺ—č—Ö –į–≥–Ķ–Ĺ—ā–ĺ–≤ –ľ–ĺ–Ľ–Ķ–ļ—É–Ľ–į–ľ–ł-–ĺ–Ņ—Ā–ĺ–Ĺ–ł–Ĺ–į–ľ–ł, –ĺ–Ī–Ľ–Ķ–≥—á–į—é—Č–ł–ľ–ł –ł—Ö —É–Ĺ–ł—á—ā–ĺ–∂–Ķ–Ĺ–ł–Ķ. –ź–ļ—ā–ł–≤–į—Ü–ł—Ź –ļ–ĺ–ľ–Ņ–Ľ–Ķ–ľ–Ķ–Ĺ—ā–į –Ņ—Ä–ĺ–ł—Ā—Ö–ĺ–ī–ł—ā –Ņ–嬆—ā—Ä–Ķ–ľ –Ņ—É—ā—Ź–ľ: –ļ–Ľ–į—Ā—Ā–ł—á–Ķ—Ā–ļ–ĺ–ľ—É (–į–ļ—ā–ł–≤–ł—Ä—É–Ķ—ā—Ā—Ź –ļ–ĺ–ľ–Ņ–Ľ–Ķ–ļ—Ā–į–ľ–ł ¬ę–į–Ĺ—ā–ł–≥–Ķ–Ŭ†‚Äď –į–Ĺ—ā–ł—ā–Ķ–Ľ–嬼), –Ľ–Ķ–ļ—ā–ł–Ĺ–ĺ–≤–ĺ–ľ—É (–į–ļ—ā–ł–≤–ł—Ä—É–Ķ—ā—Ā—Ź –Ņ–ĺ–Ľ–ł—Ā–į—Ö–į—Ä–ł–ī–į–ľ–ł –Ĺ–į¬†–Ņ–ĺ–≤–Ķ—Ä—Ö–Ĺ–ĺ—Ā—ā–ł –ľ–ł–ļ—Ä–ĺ–Ī–ĺ–≤) –ł¬†–į–Ľ—Ć—ā–Ķ—Ä–Ĺ–į—ā–ł–≤–Ĺ–ĺ–ľ—É (–į–ļ—ā–ł–≤–ł—Ä—É–Ķ—ā—Ā—Ź –Ņ–ĺ–≤–Ķ—Ä—Ö–Ĺ–ĺ—Ā—ā–Ĺ—č–ľ–ł –Ī–Ķ–Ľ–ļ–į–ľ–ł –Ņ–į—ā–ĺ–≥–Ķ–Ĺ–ĺ–≤) [30]. C1q, –ļ–Ľ—é—á–Ķ–≤–ĺ–Ļ –Ī–Ķ–Ľ–ļ–ĺ–≤—č–Ļ –ļ–ĺ–ľ–Ņ–Ľ–Ķ–ļ—Ā, –∑–į–Ņ—É—Ā–ļ–į—é—Č–ł–Ļ –ļ–Ľ–į—Ā—Ā–ł—á–Ķ—Ā–ļ–ł–Ļ –Ņ—É—ā—Ć, –į–ļ—ā–ł–≤–ł—Ä—É–Ķ—ā—Ā—Ź –ļ–ĺ–ľ–Ņ–ĺ–Ĺ–Ķ–Ĺ—ā–į–ľ–ł –ī—Ä—É–∑¬†‚Äď –Ľ–ł–Ņ–ł–ī–į–ľ–ł (–Ľ–ł–∑–ĺ—Ą–ĺ—Ā—Ą–ĺ–Ľ–ł–Ņ–ł–ī–į–ľ–ł) –ł¬†–Ī–Ķ—ā–į-–į–ľ–ł–Ľ–ĺ–ł–ī–ĺ–ľ [31, 32]. –°¬†–≤–ĺ–∑—Ä–į—Ā—ā–ĺ–ľ –ł¬†–Ņ—Ä–ł –≤–ĺ—Ā–Ņ–į–Ľ–Ķ–Ĺ–ł–ł –Ņ—Ä–ĺ–ł—Ā—Ö–ĺ–ī–ł—ā –Ĺ–į–ļ–ĺ–Ņ–Ľ–Ķ–Ĺ–ł–Ķ C1q, —á—ā–ĺ –Ņ—Ä–ł–≤–ĺ–ī–ł—ā –ļ¬†–Ņ–ĺ–≤—Ä–Ķ–∂–ī–Ķ–Ĺ–ł—é —Ā–Ķ—ā—á–į—ā–ļ–ł –ł¬†—Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź –∑–Ĺ–į—á–ł–ľ—č–ľ —Ą–į–ļ—ā–ĺ—Ä–ĺ–ľ —Ä–ł—Ā–ļ–į —Ä–į–∑–≤–ł—ā–ł—Ź –í–ú–Ē [33].
–ź–ļ—ā–ł–≤–į—Ü–ł—Ź –ļ–ĺ–ľ–Ņ–Ľ–Ķ–ľ–Ķ–Ĺ—ā–į —Ā–Ņ–ĺ—Ā–ĺ–Ī—Ā—ā–≤—É–Ķ—ā –ĺ–Ī—Ä–į–∑–ĺ–≤–į–Ĺ–ł—é –ļ–ĺ–Ĺ–≤–Ķ—Ä—ā–į–∑ C3 –ł¬†C5, –ļ–ĺ—ā–ĺ—Ä—č–Ķ –≤–ĺ–∑–ī–Ķ–Ļ—Ā—ā–≤—É—é—ā –Ĺ–į¬†—Ü–Ķ–Ĺ—ā—Ä–į–Ľ—Ć–Ĺ—č–Ķ –Ī–Ķ–Ľ–ļ–ł C3 –ł¬†C5 [29]. –†–į—Ā—Č–Ķ–Ņ–Ľ–Ķ–Ĺ–ł–Ķ C3 –Ĺ–į¬†–Ņ—Ä–ĺ–≤–ĺ—Ā–Ņ–į–Ľ–ł—ā–Ķ–Ľ—Ć–Ĺ—č–Ļ –į–Ĺ–į—Ą–ł–Ľ–į—ā–ĺ–ļ—Ā–ł–Ĺ C3a –ł¬†–ĺ–Ņ—Ā–ĺ–Ĺ–ł–Ĺ C3b –≤—č–∑—č–≤–į–Ķ—ā –≤–ĺ—Ā–Ņ–į–Ľ–Ķ–Ĺ–ł–Ķ –ł¬†–ĺ–Ņ—Ā–ĺ–Ĺ–ł–∑–į—Ü–ł—é –ļ–Ľ–Ķ—ā–ĺ–ļ –ī–Ľ—Ź –Ņ–ĺ—Ā–Ľ–Ķ–ī—É—é—Č–Ķ–≥–ĺ —Ą–į–≥–ĺ—Ü–ł—ā–ĺ–∑–į. –£¬†–Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ —Ā¬†–í–ú–Ē –≤¬†—Ā—É–Ī—Ä–Ķ—ā–ł–Ĺ–į–Ľ—Ć–Ĺ–ĺ–ľ –Ņ—Ä–ĺ—Ā—ā—Ä–į–Ĺ—Ā—ā–≤–Ķ –ł¬†–ī—Ä—É–∑–į—Ö –ĺ–Ī–Ĺ–į—Ä—É–∂–ł–≤–į—é—ā—Ā—Ź –ļ–ĺ–ľ–Ņ–ĺ–Ĺ–Ķ–Ĺ—ā—č C5, C3 –ł¬†–ł—Ö —Ą—Ä–į–≥–ľ–Ķ–Ĺ—ā—č [33, 34]. –ü–ĺ–ļ–į–∑–į–Ĺ–ĺ, —á—ā–ĺ C3 –ł¬†–Ķ–≥–ĺ –Ņ—Ä–ĺ–ī—É–ļ—ā—č –į–ļ—ā–ł–≤–į—Ü–ł–ł –∑–į–Ņ—É—Ā–ļ–į—é—ā –ľ–Ĺ–ĺ–∂–Ķ—Ā—ā–≤–ĺ –Ņ—Ä–ĺ—Ü–Ķ—Ā—Ā–ĺ–≤, –≤–ļ–Ľ—é—á–į—Ź –Ņ—Ä–ł–≤–Ľ–Ķ—á–Ķ–Ĺ–ł–Ķ –ľ–į–ļ—Ä–ĺ—Ą–į–≥–ĺ–≤ –ł¬†–ľ–ł–ļ—Ä–ĺ–≥–Ľ–ł–ł –≤¬†—Ā—É–Ī—Ä–Ķ—ā–ł–Ĺ–į–Ľ—Ć–Ĺ–ĺ–Ķ –Ņ—Ä–ĺ—Ā—ā—Ä–į–Ĺ—Ā—ā–≤–ĺ, –Ĺ–į—Ä—É—ą–Ķ–Ĺ–ł–Ķ —Ą—É–Ĺ–ļ—Ü–ł–ł –Ľ–ł–∑–ĺ—Ā–ĺ–ľ –ł¬†–į–ļ—ā–ł–≤–į—Ü–ł—é –≤–Ĺ—É—ā—Ä–ł–ļ–Ľ–Ķ—ā–ĺ—á–Ĺ—č—Ö —Ā–ł–≥–Ĺ–į–Ľ–ĺ–≤ –≤¬†–Ņ–ł–≥–ľ–Ķ–Ĺ—ā–Ĺ–ĺ–ľ —ć–Ņ–ł—ā–Ķ–Ľ–ł–ł [35‚Äď37]. –°3¬†—ā–į–ļ–∂–Ķ —É—á–į—Ā—ā–≤—É–Ķ—ā –≤¬†–į–ļ—ā–ł–≤–į—Ü–ł–ł –ł–Ĺ—Ą–Ľ–į–ľ–ľ–į—Ā–ĺ–ľ¬†‚Äď –≤–Ĺ—É—ā—Ä–ł–ļ–Ľ–Ķ—ā–ĺ—á–Ĺ—č—Ö –Ī–Ķ–Ľ–ļ–ĺ–≤—č—Ö –ļ–ĺ–ľ–Ņ–Ľ–Ķ–ļ—Ā–ĺ–≤, –ł–Ĺ–ł—Ü–ł–ł—Ä—É—é—Č–ł—Ö –≤–ĺ—Ā–Ņ–į–Ľ–ł—ā–Ķ–Ľ—Ć–Ĺ—č–Ļ –ĺ—ā–≤–Ķ—ā –ł¬†–Ņ—Ä–ĺ–ī—É–ļ—Ü–ł—é –∑–į—Č–ł—ā–Ĺ—č—Ö —Ü–ł—ā–ĺ–ļ–ł–Ĺ–ĺ–≤. –ź–ļ—ā–ł–≤–į—Ü–ł—Ź –ł–Ĺ—Ą–Ľ–į–ľ–ľ–į—Ā–ĺ–ľ –ļ—Ä–ł—Ā—ā–į–Ľ–Ľ–į–ľ–ł —Ö–ĺ–Ľ–Ķ—Ā—ā–Ķ—Ä–ł–Ĺ–į, –Ľ–ł–Ņ–ĺ—Ą—É—Ā—Ü–ł–Ĺ–ĺ–ľ –ł–Ľ–ł –ļ–ĺ–ľ–Ņ–ĺ–Ĺ–Ķ–Ĺ—ā–į–ľ–ł –ī—Ä—É–∑ –Ņ–ĺ–≤—č—ą–į–Ķ—ā —É—Ä–ĺ–≤–Ķ–Ĺ—Ć –Ņ—Ä–ĺ–≤–ĺ—Ā–Ņ–į–Ľ–ł—ā–Ķ–Ľ—Ć–Ĺ—č—Ö –ł–Ĺ—ā–Ķ—Ä–Ľ–Ķ–Ļ–ļ–ł–Ĺ–ĺ–≤ 1-–Ī–Ķ—ā–į –ł¬†18, —É—á–į—Ā—ā–≤—É—é—Č–ł—Ö –≤–ĺ –≤—Ä–ĺ–∂–ī–Ķ–Ĺ–Ĺ–ĺ–ľ –ł¬†–į–ī–į–Ņ—ā–ł–≤–Ĺ–ĺ–ľ –ł–ľ–ľ—É–Ĺ–ł—ā–Ķ—ā–Ķ [38‚Äď42]. –í¬†—Ā–≤—Ź–∑–ł —Ā¬†—ć—ā–ł–ľ –Ī–Ķ–Ľ–ļ–ł C3 –ł¬†C5 —Ä–į—Ā—Ā–ľ–į—ā—Ä–ł–≤–į—é—ā—Ā—Ź –ļ–į–ļ –ł–ī–Ķ–į–Ľ—Ć–Ĺ—č–Ķ –ľ–ł—ą–Ķ–Ĺ–ł –ī–Ľ—Ź —ā–Ķ—Ä–į–Ņ–Ķ–≤—ā–ł—á–Ķ—Ā–ļ–ĺ–≥–ĺ –ł–Ĺ–≥–ł–Ī–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź –Ņ—Ä–ł –ď–ź.
–ü—Ä–ł —Ä–į–∑—Ä–į–Ī–ĺ—ā–ļ–Ķ –ł–Ĺ–≥–ł–Ī–ł—ā–ĺ—Ä–ĺ–≤ –ļ–ĺ–ľ–Ņ–Ľ–Ķ–ľ–Ķ–Ĺ—ā–į –ī–Ľ—Ź –Ľ–Ķ—á–Ķ–Ĺ–ł—Ź –ď–ź —Ā–Ľ–Ķ–ī—É–Ķ—ā —É—á–ł—ā—č–≤–į—ā—Ć, —á—ā–ĺ —ć—ā–į —Ā–ł—Ā—ā–Ķ–ľ–į —É—á–į—Ā—ā–≤—É–Ķ—ā –Ĺ–Ķ¬†—ā–ĺ–Ľ—Ć–ļ–ĺ –≤¬†–Ņ–į—ā–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ł—Ö –Ņ—Ä–ĺ—Ü–Ķ—Ā—Ā–į—Ö, –Ŗ嬆–ł –≤¬†—ć–Ľ–ł–ľ–ł–Ĺ–į—Ü–ł–ł –ł–ľ–ľ—É–Ĺ–Ĺ—č—Ö –ļ–ĺ–ľ–Ņ–Ľ–Ķ–ļ—Ā–ĺ–≤ –ł¬†–į–Ņ–ĺ–Ņ—ā–ĺ—ā–ł—á–Ķ—Ā–ļ–ł—Ö –ļ–Ľ–Ķ—ā–ĺ–ļ, —Ā–ĺ—Ö—Ä–į–Ĺ—Ź—Ź —Ü–Ķ–Ľ–ĺ—Ā—ā–Ĺ–ĺ—Ā—ā—Ć —Ā–Ķ—ā—á–į—ā–ļ–ł. –Ę–į–ļ, –ī–Ľ–ł—ā–Ķ–Ľ—Ć–Ĺ–į—Ź –Ī–Ľ–ĺ–ļ–į–ī–į –°3¬†–ľ–ĺ–∂–Ķ—ā –Ņ—Ä–ł–≤–ĺ–ī–ł—ā—Ć –ļ¬†–Ĺ–Ķ–≥–į—ā–ł–≤–Ĺ—č–ľ –Ņ–ĺ—Ā–Ľ–Ķ–ī—Ā—ā–≤–ł—Ź–ľ, —Ā–≤—Ź–∑–į–Ĺ–Ĺ—č–ľ —Ā¬†–Ĺ–Ķ–ī–ĺ—Ā—ā–į—ā–ĺ—á–Ĺ–ĺ—Ā—ā—Ć—é –ļ–ĺ–ľ–Ņ–Ľ–Ķ–ľ–Ķ–Ĺ—ā–į [43]. –Ě–į¬†–ľ—č—ą–ł–Ĺ–ĺ–Ļ –ľ–ĺ–ī–Ķ–Ľ–ł –Ņ–ĺ–ļ–į–∑–į–Ĺ–ĺ, —á—ā–嬆–≥–Ķ–Ĺ–Ķ—ā–ł—á–Ķ—Ā–ļ–į—Ź –į–Ī–Ľ—Ź—Ü–ł—Ź –°3¬†—Ā–Ĺ–ł–∂–į–Ķ—ā –ļ–Ľ–ł—Ä–Ķ–Ĺ—Ā –į–Ņ–ĺ–Ņ—ā–ĺ—ā–ł—á–Ķ—Ā–ļ–ł—Ö –ļ–Ľ–Ķ—ā–ĺ–ļ –ł¬†—É—Ā–ļ–ĺ—Ä—Ź–Ķ—ā –ī–Ķ–≥–Ķ–Ĺ–Ķ—Ä–į—Ü–ł—é —Ą–ĺ—ā–ĺ—Ä–Ķ—Ü–Ķ–Ņ—ā–ĺ—Ä–ĺ–≤ [3]. –Ě–į–Ņ—Ä–ĺ—ā–ł–≤, –≤–ĺ–∑–ī–Ķ–Ļ—Ā—ā–≤–ł–Ķ –Ĺ–į¬†–ī–ł—Ā—ā–į–Ľ—Ć–Ĺ—É—é —á–į—Ā—ā—Ć –ļ–į—Ā–ļ–į–ī–į¬†‚Äď –Ī–Ķ–Ľ–ĺ–ļ –°5¬†‚Äď –Ņ–ĺ–∑–≤–ĺ–Ľ—Ź–Ķ—ā —É–ľ–Ķ–Ĺ—Ć—ą–ł—ā—Ć –ĺ–Ī—Ä–į–∑–ĺ–≤–į–Ĺ–ł–Ķ –ľ–Ķ–ľ–Ī—Ä–į–Ĺ–ĺ–į—ā–į–ļ—É—é—Č–ł—Ö –ļ–ĺ–ľ–Ņ–Ľ–Ķ–ļ—Ā–ĺ–≤ (–ú–ź–ö) –ł¬†–Ņ—Ä–ĺ–ī—É–ļ—Ü–ł—é –°5–į. –ü—Ä–ł —ć—ā–ĺ–ľ –į–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć –°3¬†—Ā–ĺ—Ö—Ä–į–Ĺ—Ź–Ķ—ā—Ā—Ź, –į¬†–∑–Ĺ–į—á–ł—ā, —Ā–ĺ—Ö—Ä–į–Ĺ—Ź—é—ā—Ā—Ź –Ņ–ĺ–Ľ–Ķ–∑–Ĺ—č–Ķ —Ą—É–Ĺ–ļ—Ü–ł–ł —Ā–ł—Ā—ā–Ķ–ľ—č –ļ–ĺ–ľ–Ņ–Ľ–Ķ–ľ–Ķ–Ĺ—ā–į –Ī–Ķ–∑ –Ķ–Ķ —á—Ä–Ķ–∑–ľ–Ķ—Ä–Ĺ–ĺ–Ļ –į–ļ—ā–ł–≤–į—Ü–ł–ł¬†[29, 44, 45] (—Ä–ł—Ā.¬†3).
–Ę–Ķ—Ä–į–Ņ–Ķ–≤—ā–ł—á–Ķ—Ā–ļ–ł–Ķ –Ņ–ĺ–ī—Ö–ĺ–ī—č
–Ę–į—Ä–≥–Ķ—ā–Ĺ–į—Ź —ā–Ķ—Ä–į–Ņ–ł—Ź –ď–ź —É–∂–Ķ —Ä–į–∑—Ä–į–Ī–ĺ—ā–į–Ĺ–į –ł¬†–ī–ĺ—Ā—ā—É–Ņ–Ĺ–į –≤¬†–Ĺ–Ķ–ļ–ĺ—ā–ĺ—Ä—č—Ö —Ā—ā—Ä–į–Ĺ–į—Ö –ľ–ł—Ä–į. –Ě–į–Ņ—Ä–ł–ľ–Ķ—Ä, –≤¬†–°–®–ź, –Į–Ņ–ĺ–Ĺ–ł–ł –ł¬†–ź–≤—Ā—ā—Ä–į–Ľ–ł–ł –ĺ–ī–ĺ–Ī—Ä–Ķ–Ĺ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā –į–≤–į—Ü–ł–Ĺ–ļ–į–Ņ—ā–į–ī –Ņ–Ķ–≥–ĺ–Ľ (–ź–ö–ü) (–ė–∑–Ķ—Ä–≤–Ķ–Ļ) [46].
–ź–ö–ü¬†‚Äď –Ņ–Ķ–≥–ł–Ľ–ł—Ä–ĺ–≤–į–Ĺ–Ĺ—č–Ļ –†–Ě–ö-–į–Ņ—ā–į–ľ–Ķ—Ä. –ě–Ĺ –į–ļ—ā–ł–≤–Ĺ–ĺ —Ā–≤—Ź–∑—č–≤–į–Ķ—ā—Ā—Ź —Ā¬†C5, –Ī–Ľ–ĺ–ļ–ł—Ä—É–Ķ—ā –Ķ–≥–ĺ, –Ņ—Ä–Ķ–ī–ĺ—ā–≤—Ä–į—Č–į—Ź —Ä–į—Ā—Č–Ķ–Ņ–Ľ–Ķ–Ĺ–ł–Ķ –Ĺ–į¬†–Ī–ł–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ł –į–ļ—ā–ł–≤–Ĺ—č–Ķ —Ą—Ä–į–≥–ľ–Ķ–Ĺ—ā—č C5a (–ľ–ĺ—Č–Ĺ—č–Ļ –Ņ—Ä–ĺ–≤–ĺ—Ā–Ņ–į–Ľ–ł—ā–Ķ–Ľ—Ć–Ĺ—č–Ļ –į–Ĺ–į—Ą–ł–Ľ–į—ā–ĺ–ļ—Ā–ł–Ĺ) –ł¬†C5b, —á—ā–ĺ –ī–Ķ–Ľ–į–Ķ—ā –Ĺ–Ķ–≤–ĺ–∑–ľ–ĺ–∂–Ĺ—č–ľ –ĺ–Ī—Ä–į–∑–ĺ–≤–į–Ĺ–ł–Ķ –ú–ź–ö (C5b‚Äď9). –ú–ź–ö —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź –ĺ—Ā–Ĺ–ĺ–≤–Ĺ—č–ľ —Ü–ł—ā–ĺ—ā–ĺ–ļ—Ā–ł—á–Ķ—Ā–ļ–ł–ľ —Ą–į–ļ—ā–ĺ—Ä–ĺ–ľ, –Ņ–ĺ–≤—Ä–Ķ–∂–ī–į—é—Č–ł–ľ –ļ–Ľ–Ķ—ā–ļ–ł –Ņ–ł–≥–ľ–Ķ–Ĺ—ā–Ĺ–ĺ–≥–ĺ —ć–Ņ–ł—ā–Ķ–Ľ–ł—Ź. –Ę–į–ļ–ł–ľ –ĺ–Ī—Ä–į–∑–ĺ–ľ, –ł–Ĺ–≥–ł–Ī–ł—Ä–ĺ–≤–į–Ĺ–ł–Ķ C5 —Ā–Ņ–ĺ—Ā–ĺ–Ī–Ĺ–ĺ –∑–į–ľ–Ķ–ī–Ľ—Ź—ā—Ć –Ņ—Ä–ĺ–≥—Ä–Ķ—Ā—Ā–ł—Ä–ĺ–≤–į–Ĺ–ł–Ķ –ī–Ķ–≥–Ķ–Ĺ–Ķ—Ä–į—Ü–ł–ł –ļ–Ľ–Ķ—ā–ĺ–ļ —Ā–Ķ—ā—á–į—ā–ļ–ł –Ņ—Ä–ł –í–ú–Ē [47].
–ė—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź II –ł¬†III —Ą–į–∑ –ź–ö–ü –Ņ–ĺ–ļ–į–∑–į–Ľ–ł –ľ–Ĺ–ĺ–≥–ĺ–ĺ–Ī–Ķ—Č–į—é—Č–ł–Ķ —Ä–Ķ–∑—É–Ľ—Ć—ā–į—ā—č —É¬†–Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ —Ā¬†–ď–ź. –ü–嬆–ī–į–Ĺ–Ĺ—č–ľ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź GATHER1, —á–Ķ—Ä–Ķ–∑ 12 –ľ–Ķ—Ā—Ź—Ü–Ķ–≤ —ā–Ķ—Ä–į–Ņ–ł–ł –ź–ö–ü –≤¬†–ī–ĺ–∑–į—Ö 2 –ł¬†4 –ľ–≥ (–ł–Ĺ—ā—Ä–į–≤–ł—ā—Ä–Ķ–į–Ľ—Ć–Ĺ–ĺ –ĺ–ī–ł–Ĺ —Ä–į–∑ –≤¬†–ľ–Ķ—Ā—Ź—Ü) –Ĺ–į–Ī–Ľ—é–ī–į–Ľ–ĺ—Ā—Ć –∑–Ĺ–į—á–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ–Ķ –∑–į–ľ–Ķ–ī–Ľ–Ķ–Ĺ–ł–Ķ —Ä–ĺ—Ā—ā–į –ĺ—á–į–≥–ĺ–≤ –ď–ź –Ņ–嬆—Ā—Ä–į–≤–Ĺ–Ķ–Ĺ–ł—é —Ā¬†–≥—Ä—É–Ņ–Ņ–ĺ–Ļ –ł–ľ–ł—ā–į—Ü–ł–ł –Ľ–Ķ—á–Ķ–Ĺ–ł—Ź (–Ĺ–į¬†27,4 –ł¬†27,8% —Ā–ĺ–ĺ—ā–≤–Ķ—ā—Ā—ā–≤–Ķ–Ĺ–Ĺ–ĺ; p < 0,05) –Ņ—Ä–ł –Ņ—Ä–ł–Ķ–ľ–Ľ–Ķ–ľ–ĺ–ľ –Ņ—Ä–ĺ—Ą–ł–Ľ–Ķ –Ī–Ķ–∑–ĺ–Ņ–į—Ā–Ĺ–ĺ—Ā—ā–ł [48]. –ß–Ķ—Ä–Ķ–∑ 12 –ľ–Ķ—Ā—Ź—Ü–Ķ–≤ –Ľ–Ķ—á–Ķ–Ĺ–ł—Ź –≤¬†–≥—Ä—É–Ņ–Ņ–Ķ –ź–ö–ü –≤¬†–ī–ĺ–∑–Ķ 2 –ľ–≥ –ĺ—ā–ľ–Ķ—á–į–Ľ–ĺ—Ā—Ć —Ā–Ĺ–ł–∂–Ķ–Ĺ–ł–Ķ —Ä–ł—Ā–ļ–į —Ā—ā–ĺ–Ļ–ļ–ĺ–Ļ –Ņ–ĺ—ā–Ķ—Ä–ł –ĺ—Ā—ā—Ä–ĺ—ā—č –∑—Ä–Ķ–Ĺ–ł—Ź –Ī–ĺ–Ľ–Ķ–Ķ —á–Ķ–ľ –Ĺ–į¬†15 –Ī—É–ļ–≤ –Ņ–嬆—ą–ļ–į–Ľ–Ķ ETDRS (Early Treatment Diabetic Retinopathy Study) –ĺ—ā¬†–ł—Ā—Ö–ĺ–ī–Ĺ–ĺ–≥–ĺ —É—Ä–ĺ–≤–Ĺ—Ź BCVA (Best Corrected Visual Acuity, –Ĺ–į–ł–Ľ—É—á—ą–į—Ź –ļ–ĺ—Ä—Ä–ł–≥–ł—Ä–ĺ–≤–į–Ĺ–Ĺ–į—Ź –ĺ—Ā—ā—Ä–ĺ—ā–į –∑—Ä–Ķ–Ĺ–ł—Ź) –Ņ–嬆—Ā—Ä–į–≤–Ĺ–Ķ–Ĺ–ł—é —Ā¬†–ļ–ĺ–Ĺ—ā—Ä–ĺ–Ľ—Ć–Ĺ–ĺ–Ļ¬†–≥—Ä—É–Ņ–Ņ–ĺ–Ļ: –≤¬†—Ā—Ä–Ķ–ī–Ĺ–Ķ–ľ —Ā–Ĺ–ł–∂–Ķ–Ĺ–ł–Ķ –ĺ—Ā—ā—Ä–ĺ—ā—č –∑—Ä–Ķ–Ĺ–ł—Ź —Ā–ĺ—Ā—ā–į–≤–ł–Ľ–ĺ 3,4 –ł¬†7,8% —Ā–ĺ–ĺ—ā–≤–Ķ—ā—Ā—ā–≤–Ķ–Ĺ–Ĺ–ĺ [49]. –ß–Ķ—Ä–Ķ–∑ 18 –ľ–Ķ—Ā—Ź—Ü–Ķ–≤ –∑–į–ľ–Ķ–ī–Ľ–Ķ–Ĺ–ł–Ķ —Ä–ĺ—Ā—ā–į –ĺ—á–į–≥–ĺ–≤ –į—ā—Ä–ĺ—Ą–ł–ł –ī–ĺ—Ā—ā–ł–≥–Ľ–ĺ 28,1% (–ī–Ľ—Ź –ī–ĺ–∑—č 2 –ľ–≥) –ł¬†30,0% (–ī–Ľ—Ź –ī–ĺ–∑—č 4 –ľ–≥) –Ņ–嬆—Ā—Ä–į–≤–Ĺ–Ķ–Ĺ–ł—é —Ā¬†–ļ–ĺ–Ĺ—ā—Ä–ĺ–Ľ—Ć–Ĺ–ĺ–Ļ¬†–≥—Ä—É–Ņ–Ņ–ĺ–Ļ¬†[50]. –£–≤–Ķ–Ľ–ł—á–Ķ–Ĺ–ł–Ķ –Ņ—Ä–ĺ–ī–ĺ–Ľ–∂–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ—Ā—ā–ł –Ľ–Ķ—á–Ķ–Ĺ–ł—Ź (–ī–ĺ 18¬†–ľ–Ķ—Ā—Ź—Ü–Ķ–≤) –Ĺ–Ķ¬†–≤—č—Ź–≤–ł–Ľ–ĺ –ī–ĺ–Ņ–ĺ–Ľ–Ĺ–ł—ā–Ķ–Ľ—Ć–Ĺ—č—Ö —Ā–ł–≥–Ĺ–į–Ľ–ĺ–≤ –Ī–Ķ–∑–ĺ–Ņ–į—Ā–Ĺ–ĺ—Ā—ā–ł. –Ď–ĺ–Ľ—Ć—ą–ł–Ĺ—Ā—ā–≤–ĺ –Ĺ–Ķ–∂–Ķ–Ľ–į—ā–Ķ–Ľ—Ć–Ĺ—č—Ö —Ź–≤–Ľ–Ķ–Ĺ–ł–Ļ —Ā–嬆—Ā—ā–ĺ—Ä–ĺ–ŗ謆–≥–Ľ–į–∑–į –Ī—č–Ľ–ĺ —Ā–≤—Ź–∑–į–Ĺ–ĺ —Ā¬†–Ņ—Ä–ĺ—Ü–Ķ–ī—É—Ä–ĺ–Ļ –ł–Ĺ—ä–Ķ–ļ—Ü–ł–ł. –ó–į 18 –ľ–Ķ—Ā—Ź—Ü–Ķ–≤ –Ĺ–į–Ī–Ľ—é–ī–Ķ–Ĺ–ł—Ź –∑–į—Ä–Ķ–≥–ł—Ā—ā—Ä–ł—Ä–ĺ–≤–į–Ĺ –ĺ–ī–ł–Ĺ —Ā–Ľ—É—á–į–Ļ –ł—ą–Ķ–ľ–ł—á–Ķ—Ā–ļ–ĺ–Ļ –Ĺ–Ķ–Ļ—Ä–ĺ–Ņ–į—ā–ł–ł –∑—Ä–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ–≥–ĺ –Ĺ–Ķ—Ä–≤–į (–≤¬†–≥—Ä—É–Ņ–Ņ–Ķ –ź–ö–ü 2 –ľ–≥) –ł¬†–ĺ–ī–ł–Ĺ —Ā–Ľ—É—á–į–Ļ –ĺ—ā—Ā–Ľ–ĺ–Ļ–ļ–ł —Ā–Ķ—ā—á–į—ā–ļ–ł (–≤¬†–≥—Ä—É–Ņ–Ņ–Ķ –ź–ö–ü 4 –ľ–≥). –°–Ľ—É—á–į–Ķ–≤ —ć–Ĺ–ī–ĺ—Ą—ā–į–Ľ—Ć–ľ–ł—ā–į –Ĺ–Ķ¬†–∑–į—Ą–ł–ļ—Ā–ł—Ä–ĺ–≤–į–Ĺ–ĺ. –£¬†–Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤, –Ņ–ĺ–Ľ—É—á–į–≤—ą–ł—Ö –ź–ö–ü, —á–į—Č–Ķ –ĺ—ā–ľ–Ķ—á–į–Ľ–į—Ā—Ć –ľ–į–ļ—É–Ľ—Ź—Ä–Ĺ–į—Ź –Ĺ–Ķ–ĺ–≤–į—Ā–ļ—É–Ľ—Ź—Ä–ł–∑–į—Ü–ł—Ź (—ɬ†11,9 –ł¬†15,7% –≤¬†–≥—Ä—É–Ņ–Ņ–į—Ö –ź–ö–ü 2¬† –ł¬†4 –ľ–≥ —Ā–ĺ–ĺ—ā–≤–Ķ—ā—Ā—ā–≤–Ķ–Ĺ–Ĺ–ĺ –Ņ—Ä–ĺ—ā–ł–≤ 2,7% –≤¬†–ļ–ĺ–Ĺ—ā—Ä–ĺ–Ľ—Ć–Ĺ–ĺ–Ļ¬†–≥—Ä—É–Ņ–Ņ–Ķ) [50]. –ü—Ä–ł—á–ł–Ĺ—č —ć—ā–ĺ–≥–ĺ —Ź–≤–Ľ–Ķ–Ĺ–ł—Ź –ĺ—Ā—ā–į—é—ā—Ā—Ź –Ĺ–Ķ–ł–∑–≤–Ķ—Ā—ā–Ĺ—č–ľ–ł.
–†–Ķ–∑—É–Ľ—Ć—ā–į—ā—č –ĺ—ā–ļ—Ä—č—ā–ĺ–≥–ĺ —Ä–į—Ā—ą–ł—Ä–Ķ–Ĺ–Ĺ–ĺ–≥–ĺ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź GATHER2 –Ņ—Ä–ĺ–ī–Ķ–ľ–ĺ–Ĺ—Ā—ā—Ä–ł—Ä–ĺ–≤–į–Ľ–ł –ī–į–Ľ—Ć–Ĺ–Ķ–Ļ—ą–Ķ–Ķ –∑–į–ľ–Ķ–ī–Ľ–Ķ–Ĺ–ł–Ķ —Ä–ĺ—Ā—ā–į –ĺ—á–į–≥–ĺ–≤ –ď–ź –Ņ—Ä–ł —É–≤–Ķ–Ľ–ł—á–Ķ–Ĺ–ł–ł –ī–Ľ–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ—Ā—ā–ł —ā–Ķ—Ä–į–Ņ–ł–ł (–ī–ĺ –ī–≤—É—Ö –Ľ–Ķ—ā). –ß–Ķ—Ä–Ķ–∑ 48 –ľ–Ķ—Ā—Ź—Ü–Ķ–≤ –ĺ—ā¬†–Ĺ–į—á–į–Ľ–į –Ľ–Ķ—á–Ķ–Ĺ–ł—Ź –Ņ—Ä–ł –Ņ—Ä–ĺ–ī–ĺ–Ľ–∂–Ķ–Ĺ–ł–ł –Ņ—Ä–ł–Ķ–ľ–į –ź–ö–ü –≤¬†–ī–ĺ–∑–Ķ 2 –ľ–≥ —É–≤–Ķ–Ľ–ł—á–Ķ–Ĺ–ł–Ķ —Ä–į–∑–ľ–Ķ—Ä–į –ĺ—á–į–≥–ĺ–≤ –Ņ–ĺ—Ä–į–∂–Ķ–Ĺ–ł—Ź –Ī—č–Ľ–ĺ –Ĺ–į¬†40,5% (–ľ–ľ¬≤/–≥–ĺ–ī) –ľ–Ķ–Ĺ—Ć—ą–Ķ –Ņ–嬆—Ā—Ä–į–≤–Ĺ–Ķ–Ĺ–ł—é —Ā¬†–Ņ—Ä–ĺ–≥–Ĺ–ĺ–∑–ł—Ä—É–Ķ–ľ—č–ľ –≤¬†–ļ–ĺ–Ĺ—ā—Ä–ĺ–Ľ—Ć–Ĺ–ĺ–Ļ¬†–≥—Ä—É–Ņ–Ņ–Ķ –≤¬†–Ņ–Ķ—Ä–ł–ĺ–ī —Ā¬†24-–≥–ĺ –Ņ–嬆42-–Ļ –ľ–Ķ—Ā—Ź—Ü (p < 0,001) [51]. –°—Ä–Ķ–ī–ł –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ –ļ–ĺ–Ĺ—ā—Ä–ĺ–Ľ—Ć–Ĺ–ĺ–Ļ¬†–≥—Ä—É–Ņ–Ņ—č, –Ņ–Ķ—Ä–Ķ–≤–Ķ–ī–Ķ–Ĺ–Ĺ—č—Ö –Ĺ–į¬†—ā–Ķ—Ä–į–Ņ–ł—é –ź–ö–ü 2 –ľ–≥ –ĺ–ī–ł–Ĺ —Ä–į–∑ –≤¬†–ľ–Ķ—Ā—Ź—Ü, —ā–į–ļ–∂–Ķ –ĺ—ā–ľ–Ķ—á–į–Ľ–ĺ—Ā—Ć –∑–į–ľ–Ķ–ī–Ľ–Ķ–Ĺ–ł–Ķ —Ä–ĺ—Ā—ā–į –į—ā—Ä–ĺ—Ą–ł–ł: –ļ¬†–ļ–ĺ–Ĺ—Ü—É –Ĺ–į–Ī–Ľ—é–ī–Ķ–Ĺ–ł—Ź —Ä–į–∑–ľ–Ķ—Ä –ĺ—á–į–≥–ĺ–≤ –Ī—č–Ľ –≤¬†—Ā—Ä–Ķ–ī–Ĺ–Ķ–ľ –Ĺ–į¬†37,1% –ľ–Ķ–Ĺ—Ć—ą–Ķ –Ņ—Ä–ĺ–≥–Ĺ–ĺ–∑–ł—Ä—É–Ķ–ľ–ĺ–≥–ĺ [51].
–Ę–į–ļ–ł–ľ –ĺ–Ī—Ä–į–∑–ĺ–ľ, —ā–Ķ—Ä–į–Ņ–ł—Ź –ź–ö–ü —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–į –ī–Ľ—Ź –∑–į–ľ–Ķ–ī–Ľ–Ķ–Ĺ–ł—Ź –Ņ—Ä–ĺ–≥—Ä–Ķ—Ā—Ā–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź –ď–ź –ł¬†–Ī–Ķ–∑–ĺ–Ņ–į—Ā–Ĺ–į –≤¬†–ī–ĺ–Ľ–≥–ĺ—Ā—Ä–ĺ—á–Ĺ–ĺ–Ļ –Ņ–Ķ—Ä—Ā–Ņ–Ķ–ļ—ā–ł–≤–Ķ –Ņ–嬆—Ā—Ä–į–≤–Ĺ–Ķ–Ĺ–ł—é —Ā¬†–ĺ—ā—Ā—É—ā—Ā—ā–≤–ł–Ķ–ľ –Ľ–Ķ—á–Ķ–Ĺ–ł—Ź (–ł–Ľ–ł –ł–ľ–ł—ā–į—Ü–ł–Ķ–Ļ), –į¬†—Ä–į–Ĺ–Ĺ–Ķ–Ķ –Ĺ–į—á–į–Ľ–ĺ —ā–Ķ—Ä–į–Ņ–ł–ł –ĺ–Ī–Ķ—Ā–Ņ–Ķ—á–ł–≤–į–Ķ—ā –Ī–ĺ–Ľ–Ķ–Ķ –≤—č—Ä–į–∂–Ķ–Ĺ–Ĺ–ĺ–Ķ –ł¬†—É—Ā—ā–ĺ–Ļ—á–ł–≤–ĺ–Ķ –∑–į–ľ–Ķ–ī–Ľ–Ķ–Ĺ–ł–Ķ –Ņ—Ä–ĺ–≥—Ä–Ķ—Ā—Ā–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł—Ź.
–ó–į–ļ–Ľ—é—á–Ķ–Ĺ–ł–Ķ
–Ē–ĺ –Ĺ–į—Ā—ā–ĺ—Ź—Č–Ķ–≥–ĺ –≤—Ä–Ķ–ľ–Ķ–Ĺ–ł –Ĺ–Ķ¬†—Ā—É—Č–Ķ—Ā—ā–≤–ĺ–≤–į–Ľ–ĺ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–ĺ–≤, —Ā–Ņ–ĺ—Ā–ĺ–Ī–Ĺ—č—Ö –ĺ—Ā—ā–į–Ĺ–ĺ–≤–ł—ā—Ć –ł–Ľ–ł –∑–į–ľ–Ķ–ī–Ľ–ł—ā—Ć –Ņ—Ä–ĺ–≥—Ä–Ķ—Ā—Ā–ł—Ä–ĺ–≤–į–Ĺ–ł–Ķ –ď–ź. –õ–Ķ—á–Ķ–Ĺ–ł–Ķ –ĺ—Ā—ā–į–≤–į–Ľ–ĺ—Ā—Ć —Ā–ł–ľ–Ņ—ā–ĺ–ľ–į—ā–ł—á–Ķ—Ā–ļ–ł–ľ, —á—ā–ĺ –Ņ—Ä–ł–≤–ĺ–ī–ł–Ľ–ĺ –ļ¬†–Ĺ–į—Ä–į—Ā—ā–į–Ĺ–ł—é –ł–Ĺ–≤–į–Ľ–ł–ī–ł–∑–į—Ü–ł–ł –ł¬†–Ĺ–Ķ—É–ļ–Ľ–ĺ–Ĺ–Ĺ–ĺ–ľ—É —Ā–Ĺ–ł–∂–Ķ–Ĺ–ł—é –ļ–į—á–Ķ—Ā—ā–≤–į –∂–ł–∑–Ĺ–ł –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤.
–ü—Ä–ł–ľ–Ķ–Ĺ–Ķ–Ĺ–ł–Ķ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–ĺ–≤, –Ī–Ľ–ĺ–ļ–ł—Ä—É—é—Č–ł—Ö –Ī–Ķ–Ľ–ĺ–ļ –ļ–ĺ–ľ–Ņ–Ľ–Ķ–ľ–Ķ–Ĺ—ā–į –°5, –ĺ—Ā–Ĺ–ĺ–≤–į–Ĺ–ĺ –Ĺ–į¬†–≥–Ľ—É–Ī–ĺ–ļ–ĺ–ľ –ł–∑—É—á–Ķ–Ĺ–ł–ł –Ņ–į—ā–ĺ—Ą–ł–∑–ł–ĺ–Ľ–ĺ–≥–ł–ł –ď–ź –ł¬†–Ņ—Ä–Ķ–ī—Ā—ā–į–≤–Ľ—Ź–Ķ—ā —Ā–ĺ–Ī–ĺ–Ļ –Ĺ–ĺ–≤—č–Ļ –Ņ–Ķ—Ä—Ā–Ņ–Ķ–ļ—ā–ł–≤–Ĺ—č–Ļ –ľ–Ķ—ā–ĺ–ī –Ľ–Ķ—á–Ķ–Ĺ–ł—Ź, —Ā–Ņ–ĺ—Ā–ĺ–Ī–Ĺ—č–Ļ –∑–į—ā–ĺ—Ä–ľ–ĺ–∑–ł—ā—Ć –Ņ—Ä–ĺ–≥—Ä–Ķ—Ā—Ā–ł—Ä–ĺ–≤–į–Ĺ–ł–Ķ –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł—Ź. –£—á–ł—ā—č–≤–į—Ź —Ä–ĺ—Ā—ā –Ņ—Ä–ĺ–ī–ĺ–Ľ–∂–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ—Ā—ā–ł –∂–ł–∑–Ĺ–ł, —É–≤–Ķ–Ľ–ł—á–Ķ–Ĺ–ł–Ķ —á–ł—Ā–Ľ–Ķ–Ĺ–Ĺ–ĺ—Ā—ā–ł –Ī–ĺ–Ľ—Ć–Ĺ—č—Ö –ď–ź –ł¬†–Ķ–Ķ –ł–Ĺ–≤–į–Ľ–ł–ī–ł–∑–ł—Ä—É—é—Č–ł–Ļ —Ö–į—Ä–į–ļ—ā–Ķ—Ä, –≤–į–∂–Ĺ–ĺ –ĺ–Ī—Ä–į—ā–ł—ā—Ć –≤–Ĺ–ł–ľ–į–Ĺ–ł–Ķ –Ĺ–į¬†–≤–ĺ–∑–ľ–ĺ–∂–Ĺ–ĺ—Ā—ā—Ć –≤–Ĺ–Ķ–ī—Ä–Ķ–Ĺ–ł—Ź –ź–ö–ü –≤¬†–ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ—É—é –Ņ—Ä–į–ļ—ā–ł–ļ—É –ł¬†–≤–ļ–Ľ—é—á–Ķ–Ĺ–ł—Ź –Ķ–≥–ĺ –≤¬†—Ā—ā–į–Ĺ–ī–į—Ä—ā—č –ĺ–ļ–į–∑–į–Ĺ–ł—Ź –ľ–Ķ–ī–ł—Ü–ł–Ĺ—Ā–ļ–ĺ–Ļ –Ņ–ĺ–ľ–ĺ—Č–ł –Ņ–į—Ü–ł–Ķ–Ĺ—ā–į–ľ —Ā¬†–ď–ź –≤¬†–†–ĺ—Ā—Ā–ł–Ļ—Ā–ļ–ĺ–Ļ –§–Ķ–ī–Ķ—Ä–į—Ü–ł–ł.
I.A. Loskutov, PhD, M.P. Yugay, PhD, A.R. Zotova
Moscow Regional Research and Clinical Institute
Contact person: Igor A. Loskutov, loskoutigor@mail.ru
Geographic atrophy (GA) is an outcome of the late stage of age related macular degeneration and leads to irreversible vision loss. The pathogenesis of GA is driven by the loss of photoreceptors and retinal pigment epithelium, as well as degeneration of the choriocapillaris, which results in the formation of atrophic areas in the macula. The disease is characterized by gradual painless loss of central vision and is associated with a significant decline in patients' quality of life. Due to increasing life expectancy, the number of such patients is steadily rising. A deeper understanding of the fundamental mechanisms of GA pathogenesis and the active implementation of modern therapeutic strategies will not only slow disease progression and improve clinical outcomes, but also prevent its manifestation at early stages, thereby preserving visual function and overall quality of life.



–£–≤–į–∂–į–Ķ–ľ—č–Ļ –Ņ–ĺ—Ā–Ķ—ā–ł—ā–Ķ–Ľ—Ć uMEDp!
–£–≤–Ķ–ī–ĺ–ľ–Ľ—Ź–Ķ–ľ –í–į—Ā –ĺ —ā–ĺ–ľ, —á—ā–ĺ –∑–ī–Ķ—Ā—Ć —Ā–ĺ–ī–Ķ—Ä–∂–ł—ā—Ā—Ź –ł–Ĺ—Ą–ĺ—Ä–ľ–į—Ü–ł—Ź, –Ņ—Ä–Ķ–ī–Ĺ–į–∑–Ĺ–į—á–Ķ–Ĺ–Ĺ–į—Ź –ł—Ā–ļ–Ľ—é—á–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ –ī–Ľ—Ź —Ā–Ņ–Ķ—Ü–ł–į–Ľ–ł—Ā—ā–ĺ–≤ –∑–ī—Ä–į–≤–ĺ–ĺ—Ö—Ä–į–Ĺ–Ķ–Ĺ–ł—Ź.
–ē—Ā–Ľ–ł –í—č –Ĺ–Ķ —Ź–≤–Ľ—Ź–Ķ—ā–Ķ—Ā—Ć —Ā–Ņ–Ķ—Ü–ł–į–Ľ–ł—Ā—ā–ĺ–ľ –∑–ī—Ä–į–≤–ĺ–ĺ—Ö—Ä–į–Ĺ–Ķ–Ĺ–ł—Ź, –į–ī–ľ–ł–Ĺ–ł—Ā—ā—Ä–į—Ü–ł—Ź –Ĺ–Ķ –Ĺ–Ķ—Ā–Ķ—ā –ĺ—ā–≤–Ķ—ā—Ā—ā–≤–Ķ–Ĺ–Ĺ–ĺ—Ā—ā–ł –∑–į –≤–ĺ–∑–ľ–ĺ–∂–Ĺ—č–Ķ –ĺ—ā—Ä–ł—Ü–į—ā–Ķ–Ľ—Ć–Ĺ—č–Ķ –Ņ–ĺ—Ā–Ľ–Ķ–ī—Ā—ā–≤–ł—Ź, –≤–ĺ–∑–Ĺ–ł–ļ—ą–ł–Ķ –≤ —Ä–Ķ–∑—É–Ľ—Ć—ā–į—ā–Ķ —Ā–į–ľ–ĺ—Ā—ā–ĺ—Ź—ā–Ķ–Ľ—Ć–Ĺ–ĺ–≥–ĺ –ł—Ā–Ņ–ĺ–Ľ—Ć–∑–ĺ–≤–į–Ĺ–ł—Ź –í–į–ľ–ł –ł–Ĺ—Ą–ĺ—Ä–ľ–į—Ü–ł–ł —Ā –Ņ–ĺ—Ä—ā–į–Ľ–į –Ī–Ķ–∑ –Ņ—Ä–Ķ–ī–≤–į—Ä–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ–Ļ –ļ–ĺ–Ĺ—Ā—É–Ľ—Ć—ā–į—Ü–ł–ł —Ā –≤—Ä–į—á–ĺ–ľ.
–Ě–į–∂–ł–ľ–į—Ź –Ĺ–į –ļ–Ĺ–ĺ–Ņ–ļ—É ¬ę–í–ĺ–Ļ—ā–ł¬Ľ, –í—č –Ņ–ĺ–ī—ā–≤–Ķ—Ä–∂–ī–į–Ķ—ā–Ķ, —á—ā–ĺ —Ź–≤–Ľ—Ź–Ķ—ā–Ķ—Ā—Ć –≤—Ä–į—á–ĺ–ľ –ł–Ľ–ł —Ā—ā—É–ī–Ķ–Ĺ—ā–ĺ–ľ –ľ–Ķ–ī–ł—Ü–ł–Ĺ—Ā–ļ–ĺ–≥–ĺ –≤—É–∑–į.