Поражения печени при гемохроматозе: клинические проявления и диагностика
- Аннотация
- Статья
- Ссылки
- English
Гемохроматоз – наследственно обусловленное или приобретенное заболевание из группы болезней накопления, характеризующееся нарушением обмена железа с его патологически высоким депонированием в жизненно важных органах, в частности печени [1]. Синдром перегрузки железом может самостоятельно вызывать патологию печени или способствовать прогрессированию уже имеющихся хронических диффузных заболеваний печени [2].
Перегрузка железом (гемосидероз) – патофизиологический процесс, связанный с формированием отложений гемосидерина (темно-желтого пигмента на основе оксида железа). Гемосидероз начинается прежде всего в тканях печени, впоследствии затрагивает другие ткани организма (почки, сердце, головной мозг и др.). Гемосидероз стимулирует развитие провоспалительных реакций, интенсификацию окислительного стресса (в том числе перекисного окисления липидов), повреждение паренхимы органов с развитием фиброза [3].
Диагноз чаще устанавливается на стадии сформированного гемохроматоза – при циррозе, кардиопатии и/или диабете, наличии фенотипических признаков перегрузки железом – гиперферритинемии (> 200 нг/мл у женщин и > 300 нг/мл у мужчин), сатурации трансферрина (> 45%).
Возможности лечения на стадии сформированного гемохроматоза, когда уровень ферритина превышает 1000 нг/мл, ограниченны, поскольку гемосидерин, представляющий собой деградированный ферритин, удалить из тканей крайне сложно [4].
Основная проблема дифференциальной диагностики синдрома перегрузки железом – установление первичного характера избыточного накопления железа. Помимо постановки диагноза у конкретного больного подтверждение первичного (наследственного) характера заболевания определяет необходимость проведения обследования и профилактики развития заболевания у родственников [5].
Эпидемиология
Высокая частота встречаемости наследственного гемохроматоза (по некоторым данным, до восьми случаев на 1000 населения) предполагает гетерозиготное носительство патологического гена у 10–13% населения. В России диагноз наследственного гемохроматоза устанавливают крайне редко либо не устанавливают вовсе, что объясняется значительной фенотипической гетерогенностью заболевания и отсутствием патогномоничных симптомов [5].
Гемохроматоз диагностируется у мужчин в 5–10 раз чаще, чем у женщин. Меньшая частота выявления заболевания у женщин обусловлена, в частности, менструальными кровопотерями. У мужчин заболевание обычно диагностируют в возрасте 40–60 лет, у женщин – после менопаузы [5]. Ювенильный гемохроматоз манифестирует в молодом возрасте (10–30 лет) и характеризуется выраженным синдромом перегрузки железом, сопровождается быстро прогрессирующими признаками поражения печени и сердца. Частота клинически манифестных форм поражения печени при гемохроматозе в популяции – два случая на 1000 жителей [6].
Этиопатогенез
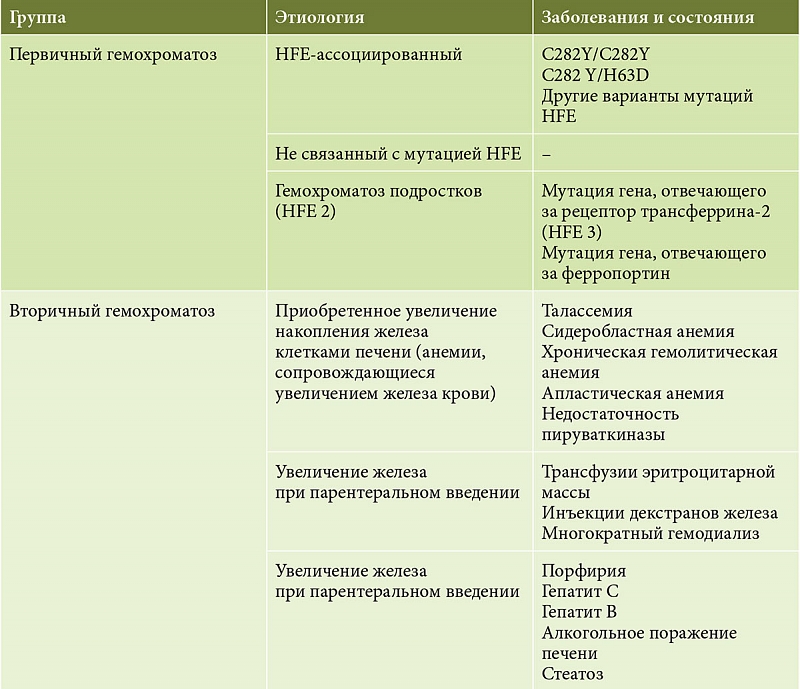
Первичный гемохроматоз (наследственный, классический, бронзовый диабет, пигментный цирроз печени, синдром Труазье – Ано – Шоффара) обусловлен многочисленными генетическими мутациями. Вторичный гемохроматоз диагностируется у 20% детей вследствие гемотрансфузий и длительного лечения препаратами железа, а также у 40% больных диффузными заболеваниями печени [1, 3, 7]. Синдром перегрузки железом усугубляет течение заболеваний печени, к которым железо имеет тропизм: вирусного гепатита, цирроза печени и портальной гипертензии, алкогольного и неалкогольного стеатогепатитов, болезни Вильсона и других хронических диффузных заболеваний печени (таблица) [1, 8].
Состояния, сопровождающиеся повышением накопления железа клетками печени
Болезни печени влияют на все метаболические процессы в организме, в том числе на обмен железа. В печени синтезируется большинство белков – переносчиков железа (трансферрин, aпoферритин, ферропортин), белков-регуляторов (пептидгепсидин). Кроме того, печень является основным депо железа. При перегрузке железом именно печень страдает в первую очередь, поскольку железо становится мощным эндогенным индуктором свободнорадикального окисления (реакция Фентона), которое приводит к разрушению клеточных мембран и гибели клеток. При этом повышенная экспрессия трансформирующего фактора роста бета-1 усиливает синтез коллагена, что способствует развитию соединительной ткани в печени, то есть фиброза и цирроза [6, 9].
Классификация и клинические проявления
Клинические проявления гемохроматоза полиморфны и неспецифичны, причем у всех больных независимо от формы выявляются патологические изменения в печени, приводящие к ее увеличению. Нередко у больных гемохроматозом при патологии печени ошибочно диагностируют реактивный и криптогенный гепатит [1].
Симптомы поражения печени обнаруживаются либо случайно, либо на стадии цирроза и его осложнений, что знаменует неблагоприятный исход заболевания [6, 10].
Преобладание признаков поражения определенных органов и систем послужило основанием для выделения четырех клинических форм наследственного гемохроматоза [5, 6, 11]:
- HFE (классическая форма) – наиболее распространенный тип гемохроматоза, связанный с мутациями гена HFE в 6-й хромосоме; наблюдается классическая триада признаков (сахарный диабет, цирроз печени и пигментация кожных покровов), часто в сочетании с симптомами поражения сердца и эндокринных желез, на фоне повышения сывороточных показателей обмена железа;
- HFE 2 (ювенильная форма). Данная форма встречается редко, наследуется по аутосомно-рецессивному типу. Мутации при этом типе гемохроматоза расположены в гене HAMP, ответственном за синтез в печени гепсидина, который модулирует метаболизм железа через уменьшение кишечной абсорбции железа, замедление его высвобождения из депо в печени и регулирование захвата железа ретикулоэндотелиальными клетками. Как правило, первые признаки болезни – упорная боль в животе в сочетании с отставанием полового развития и признаками поражения печени и миокарда (нарушения ритма и проводимости);
- HFE 3 (гемохроматоз 3-го типа). Генетическую основу гемохроматоза 3-го типа составляют мутации гена HJV, кодирующего синтез рецептора трансферрина 2-го типа, который является модулятором продукции гепсидина в ответ на избыток железа в организме. Наследуется по аутосомно-рецессивному типу, клинически мало отличается от классической формы;
- HFE 4 (аутосомно-доминантный гемохроматоз). При этом типе заболевания железо откладывается преимущественно в ретикулоэндотелиальной системе. Значительные отложения железа обнаруживают в клетках Купфера, что определяет наличие в клинической картине признаков поражения печени. Клинический дебют приходится обычно на пожилой возраст.
Отдельно выделяют 5-й тип гемохроматоза – неонатальный. Заболевание характеризуется задержкой внутриутробного развития и дебютирует быстро прогрессирующими симптомами печеночной недостаточности, приводящими к летальному исходу вскоре после рождения [6, 11].
Накопление железа проходит несколько этапов: от бессимптомного периода перегрузки железом до формирования полиорганной недостаточности [1].
Клинические стадии развития первичного гемохроматоза
Выделяют несколько стадий развития первичного гемохроматоза:
- латентную – наличие генетического дефекта в отсутствие синдрома перегрузки железом. На данном этапе происходит постепенное накопление железа;
- бессимптомную – отсутствие клинических проявлений заболевания с лабораторными признаками синдрома перегрузки железом;
- синдром перегрузки железом с ранними симптомами. Клиническая картина малоспецифична и характеризуется признаками астенического синдрома;
- синдром перегрузки железом с поражением органов-мишеней. Наблюдаются признаки поражения отдельных органов (признаки внешнесекреторной недостаточности поджелудочной железы, панкреатогенного сахарного диабета, сердечной недостаточности и аритмий, поражения гипофиза, надпочечников, гипогонадизм и др.).
Обычно гемохроматоз с выраженными симптомами диагностируют у мужчин старше 40 лет. Характерные жалобы: слабость, вялость, боль в животе и суставах, меланодермия, снижение либидо. HFE-ассоциированный гемохроматоз отличают симметричные артропатии, поражающие сложные суставы. Специфично повреждение проксимальных суставов фаланг, пястно-фаланговых сочленений, запястий, коленей и межпозвоночных суставов. К важным клиническим проявлениям заболевания относятся гепатомегалия, цирроз, диабет и поражение кожи [8].
Гемолитическая и ятрогенная трансфузионная перегрузка железом клинически протекает намного тяжелее, приводит к поражению сердца, печени (вплоть до цирроза) и полиорганной недостаточности уже в детском возрасте, требует назначения хелаторов железа и обычно не вызывает диагностических трудностей [9].
Диагностика
Последнее время наблюдается трансформация клинической картины гемохроматоза: реже встречаются больные с классической клинической триадой, описанной Dutournier в 1885 г., чаще заболевание диагностируют на доклинических стадиях, когда основные симптомы болезни отсутствуют [5].
Диагностика основана прежде всего на клинической симптоматике в сочетании с наследственной предрасположенностью. В процессе сбора анамнеза выявляют клинические признаки гемохроматоза. При их наличии проверяют биохимические параметры повышения уровня железа [8].
Обмен железа определяется при исследовании пяти основных диагностических маркеров [2, 9, 12]:
- железо сыворотки крови (уровень повышен в большинстве случаев, но не всегда);
- ферритин (у мужчин > 300 мкг/л, у женщин > 200 мкг/л);
- трансферрин;
- общая железосвязывающая способность сыворотки (ОЖССС) (< 28 мкмоль/л);
- степень насыщения трансферрина железом/сатурации трансферрина железом (СНТЖ).
Чувствительным тестом считается определение концентрации ферритина в сыворотке крови, которая прямо пропорциональна общему запасу железа в организме. Повышение уровня ферритина более 1000 нг/мл свидетельствует о тяжелой перегрузке железом [11].
Последний показатель расчетный и представляет собой отношение сывороточного железа к ОЖССС. В норме СНТЖ не превышает 40%. По мнению большинства исследователей, повышенной считается степень насыщения трансферрина более 42%. На доклинической стадии маркером избытка железа является СНТЖ > 45%. При клинически сформированном наследственном гемохроматозе СНТЖ приближается к 100% и даже превышает этот порог [9, 12].
Общеклиническое и биохимическое исследования крови также должны включать общий анализ, протеинограмму, определение билирубина и его фракций, анализ активности аспартатаминотрансферазы и аланинаминотрансферазы, гамма-глутамилтранспептидазы, щелочной фосфатазы, церулоплазмина.
Молекулярно-генетическое исследование с целью верификации диагноза наследственного гемохроматоза проводится больным [13], а также их близким родственникам с подтвержденным наследственным гемохроматозом. Диагноз можно считать установленным, когда пациент является гомозиготным носителем C282Y или сложным гетерозиготным носителем C282Y/H63D. В этих случаях проведение биопсии печени обычно не требуется.
Изолированные гетерозиготные мутации С282Y и Н63D в сочетании с признаками перегрузки железом, гепатодепрессии, повышенной активностью сывороточных аминотрансфераз требуют проведения пункционной биопсии печени с морфологическим исследованием биоптата и окраской реактивом Перлса (берлинской лазурью) на содержание железа в гепатоцитах, а также биохимическим анализом концентрации железа в ткани печени (Liver Iron Content, LIC) [6, 7, 13]. Степень активности патологического процесса в биоптате печени определятся по индексу гистологической активности по классификации Кноделя [1]. Уровень железа в биоптатах выше 70 мкг на 1 г нативной печени считается патологическим.
Несмотря на то что биопсия печени может предоставить весьма ценную информацию, она является инвазивным методом исследования и связана с определенным риском осложнений [8].
Важными инструментальными исследованиями остаются ультразвуковое исследование органов брюшной полости, радиоизотопное сканирование печени с технецием (99mTc), магнитно-резонансная томография печени и сердца в Т2-взвешенном режиме (выявляет накопление железа в этих органах на доклинической стадии) [7, 14, 15].
Лечение
Целями лечения гемохроматоза являются удаление из организма избыточного количества железа и профилактика осложнений заболевания (сахарный диабет, печеночная недостаточность, кардиомиопатия) [5].
Пациенты с гемохроматозом должны соблюдать определенный диетический режим, предполагающий ограничение потребления продуктов, богатых железом (мясо, гречневая крупа, яблоки, гранаты) и витамином C, исключение из рациона алкоголя, особенно красного вина. Кроме того, не рекомендуется принимать поливитаминно-минеральные комплексы и биологически активные добавки, содержащие железо и витамин C [5, 6, 10, 16].
Флеботомия (кровопускание, венесекция) позволяет удалить избыток железа без значительных побочных эффектов. Начало лечения кровопусканием до развития цирроза способно снизить заболеваемость и смертность [8]. Регулярная флеботомия считается самым эффективным и безопасным методом лечения гемохроматоза. Лечебное кровопускание (500 мл крови), позволяющее вывести 250 мг железа с каждой процедурой, должно проводиться еженедельно. Снижение гематокрита после каждого сеанса флеботомии не должно превышать 20% (рекомендации Американской ассоциации заболеваний печени). С учетом того что лечение носит пожизненный характер, венесекции проводят 4–6 раз в год [5, 6, 8, 10].
Эффективность флеботомии оценивают по уменьшению астенического синдрома, гепатомегалии, пигментации кожи, улучшению лабораторных показателей (снижение гиперферментемии, компенсация углеводного обмена).
Среди других методов лечения гемохроматоза – плазмаферез, цитаферез и гемосорбция. Они также направлены на выведение из организма избытка железа [6].
Хелаторная терапия
Хелаторы – лекарственные средства, способные связывать и выводить из организма избыточное железо. Согласно современной концепции, пациенты, получающие систематические заместительные трансфузии эритроцитарной массы, нуждаются в адекватной хелаторной терапии, направленной на снижение уровня токсичного железа внутри клеток и во внеклеточном пространстве, общих запасов железа в организме и, как следствие, предотвращение токсических эффектов свободного железа [5, 7, 8, 17].
Терапия хелаторами (дефероксамина мезилат) играет менее важную роль при лечении наследственного гемохроматоза, чем кровопускание, и может сопровождаться побочными эффектами. Применение хелаторов позволяет удалять значительно меньшее количество железа (не более 100 мг в неделю).
Дефероксамин (Десферал) применяют в дозе 1 г/сут внутримышечно. Наиболее рациональным считается использование деферазирокса (Эксиджада). Препарат выпускается в форме таблеток для перорального применения и содержит 125, 250 или 500 мг деферазирокса. Эксиджад является пероральным препаратом, селективно связывающим железо (например, радикалы Fe3+) [5, 8, 17].
Прогноз
У пациентов, которым была проведена ранняя диагностика гемохроматоза и начато своевременное адекватное лечение, прогноз благоприятный. В случае поздней диагностики заболевания, при наличии цирроза печени, сахарного диабета, кардиомиопатии прогноз определяется тяжестью этих необратимых осложнений. Пятилетняя выживаемость пациентов с гемохроматозом достигает 93–72% (18% в отсутствие лечения), десятилетняя – 77–47% (0–6% в отсутствие лечения) [6].
Почти у 30% больных гемохроматозом развивается рак печени. Риск его появления у пациентов с гемохроматозом в 200 раз выше, чем в среднем в популяции, не коррелирует ни со степенью поражения печени, ни с эффективностью проводимого лечения. Данное обстоятельство определяет необходимость проведения скрининга гепатоцеллюлярной карциномы один раз в шесть месяцев (ультразвуковое исследование органов брюшной полости, компьютерная томография, концентрация альфа-фетопротеина) [5, 10].
V.V. Skvortsov, MD, PhD, A.N. Gorbach
Volgograd State Medical University
Contact person: Vsevolod V. Skvortsov, vskvortsov1@ya.ru
This article provides information on the etiopathogenesis of hemochromatosis, presents the clinical symptoms of the disease, as well as methods of laboratory and instrumental diagnostics, basic approaches to therapy.
Уважаемый посетитель uMEDp!
Уведомляем Вас о том, что здесь содержится информация, предназначенная исключительно для специалистов здравоохранения.
Если Вы не являетесь специалистом здравоохранения, администрация не несет ответственности за возможные отрицательные последствия, возникшие в результате самостоятельного использования Вами информации с портала без предварительной консультации с врачом.
Нажимая на кнопку «Войти», Вы подтверждаете, что являетесь врачом или студентом медицинского вуза.