Болезнь Фабри в практике ревматолога
- Аннотация
- Статья
- Ссылки
- English
Болезнь Фабри – прогрессирующая, мультисистемная, Х-сцепленная лизосомная болезнь накопления вследствие дефекта в гене, кодирующем фермент альфа-галактозидазу. Данная патология сопровождается высоким риском осложнений [1–3]. Кроме того, она ассоциируется с преждевременной смертью от почечной недостаточности, кардио- и цереброваскулярной патологий [4].
Основная причини болезни – неспособность расщеплять определенные гликосфинголипиды. Это приводит к прогрессирующему, внутрилизосомальному накоплению субстрата в разных типах клеток [3, 4].
Распространенность болезни Фабри варьируется от одного случая на 40 тыс. у мужчин до одного случая на 117 тыс. в общей популяции. Результаты недавно проведенного регионального пилотного скринингового исследования новорожденных свидетельствуют, что ее встречаемость среди живорожденных достигает одного случая на 3,1 тыс. Несмотря на Х-сцепленный тип наследования болезни Фабри, большая доля гетерозиготных женщин также страдает от выраженных симптомов болезни [5, 6].
Болезнь Фабри может манифестировать в разном возрасте, иметь разный набор симптомов и характер прогрессирования. Однако чаще патология проявляется в детском возрасте и у подростков [1, 5].
Клинические проявления
Одним из наиболее инвалидизирующих симптомов у детей признана периферическая нейропатическая боль. Она описывается как хроническая, ноющая, покалывающая, жгучая в ладонях и подошвах. Острая, мучительная боль в конечностях может быть вызвана болезнью, физическими упражнениями, лихорадкой, стрессом или изменениями погоды. Эти эпизоды продолжаются от нескольких минут до нескольких недель и, как правило, сопровождаются лихорадкой, болями в суставах, иногда повышением скорости оседания эритроцитов (СОЭ). Часто они ошибочно принимаются за суставные боли [7, 8].
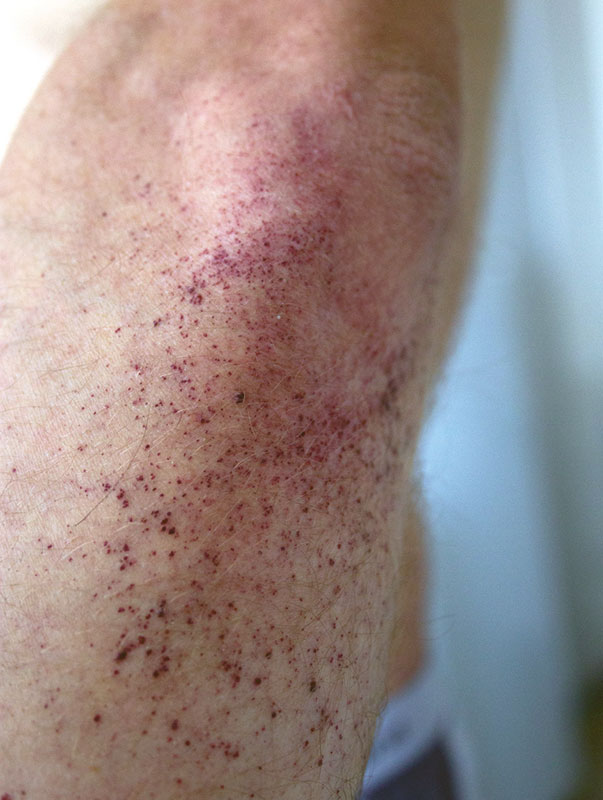
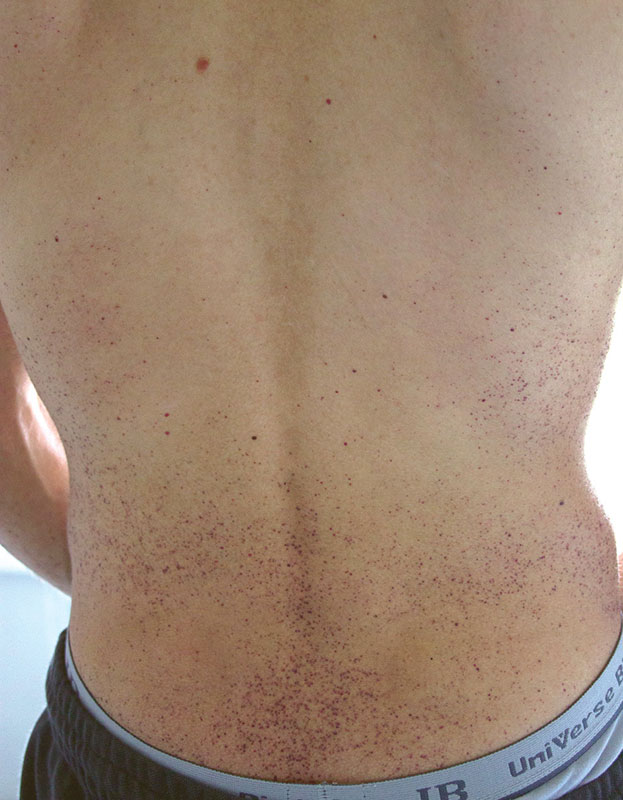
В детском возрасте отмечаются характерные для болезни Фабри поражения кожи – ангиокератомы, которые локализуются симметрично на бедрах, спине, ягодицах, в области коленных суставов и пупка, а также гипо- или ангидроз.
При болезни Фабри происходит накопление сфинголипидов и гликопротеина в органах сердечно-сосудистой системы и почках. Поэтому отмечаются симптомы стенокардии, поражение клапанов сердца (митральная недостаточность и аортальный стеноз), гипертрофия левого желудочка, протеинурия, снижение фильтрационной способности почек, почечная недостаточность.
Артериальная гипертензия (скорее всего, почечного происхождения) усиливает нарушения со стороны сердца и способствует поражению сосудов мозга.
К изменениям со стороны желудочно-кишечного тракта относятся тошнота, рвота, диарея, боли в животе [8].
Нередко обнаруживаются деформации дистальных межфаланговых суставов, остеопороз позвонков и асептический некроз головки бедренной и таранных костей, что может стать причиной постановки неверного диагноза.
Возможна также задержка роста и полового развития.
Диагностика
Несмотря на то что болезнь Фабри, как правило, манифестирует в детском или подростковом возрасте, она часто остается незамеченной до совершеннолетия. Нередко пациенты живут с неверным диагнозом до 30-летнего возраста [1]. Данные регистра пациентов с болезнью Фабри свидетельствуют, что задержка в постановке диагноза составляет 14 лет для мужчин и 19 лет для женщин [1]. Это демонстрирует как неспецифический характер ранних жалоб, так и отсутствие распознавания врачами разных специальностей ранних симптомов патологии.
Для раннего выявления заболевания требуется взаимодействие генетиков, нефрологов, неврологов, педиатров, дерматологов, кардиологов, офтальмологов, ревматологов, гастроэнтерологов и специалистов первичного звена. Это чрезвычайно важно, поскольку эффективная патогенетическая терапия может предотвратить или замедлить его прогрессирование.
Исключить системное ревматическое заболевание в начале диагностики позволяет наличие таких признаков, как поражение суставов, кожи, почек и лихорадка.
При подозрении на болезнь Фабри проводят обследование на недостаточность фермента альфа-галактозидазы.
Лечение
В настоящее время при болезни Фабри используется фермент-заместительная терапия. Она способствует восстановлению физиологического уровня фермента, уменьшению накопления субстрата (Gb3) в тканях и нормализации функции внутренних органов.
Своевременно начатая патогенетическая терапия может значительно снизить интенсивность невропатической боли или полностью купировать ее, улучшить слух, положительно повлиять на состояние почек, сердечно-сосудистой системы, устранить гастроинтестинальные проявления болезни [9].
В настоящее время в России зарегистрированы два препарата для лечения болезни Фабри: агалсидаза альфа 0,2 мг/кг и агалсидаза бета 1 мг/кг. Препараты вводятся внутривенно один раз в две недели [9, 10].
Клинический случай
Пациент К. 29 лет доставлен бригадой скорой помощи в отделение ревматологии Пензенской областной клинической больницы с диагнозом «геморрагический васкулит».
В июне 2015 г. предъявлял жалобы на жгучие приступообразные боли в суставах кистей, локтевых и коленных суставах, онемение в руках и ногах, снижение чувствительности в ногах, высыпания без зуда на коже ног и живота, плохую переносимость жары, шум в ушах.
Из анамнеза: с десяти лет стал отмечать темно-бордовые мелкоточечные высыпания в области пупка, затем в области локтей, бедер и поясницы, на губах, веках. Получил консультацию дерматолога, поставлен диагноз «гемангиоматоз». С 12 лет – приступообразные жгучие боли в крупных и мелких суставах, без отечности, но с повышением температуры до субфебрильной. Приступы боли провоцировались пребыванием на солнце. Лечение преднизолоном и нестероидными противовоспалительными препаратами не было эффективным.
С 20 лет пациент отмечал онемение и снижение чувствительности в верхних и нижних конечностях, их похолодание.
С 28 лет присоединились головные боли, шум в ушах, с 29 лет – боли в области сердца.
За три дня до госпитализации жаловался на сильные жгучие боли в суставах.
Объективно: астеническое телосложение, кожа сухая, гипогидроз, на коже обоих локтей, поясницы, коленных суставов, ягодиц, бедрах, нижних отделах живота множественные мелкоточечные темно-бордового цвета высыпания (рис. 1 и 2), не исчезающие при надавливании, телеангиэктазии на верхних веках и слизистой оболочке губ. Болезненность при пальпации и умеренное ограничение в объеме движений в коленных и плечевых суставах, небольшая дефигурация проксимальных суставов обеих кистей. В легких везикулярное дыхание, частота дыхания – 18 в минуту, тоны сердца ясные, ритм правильный, частота сердечных сокращений – 80 в минуту, артериальное давление – 120/80 мм рт. ст. Живот безболезненный.
Данные лабораторного обследования: в общем анализе крови гемоглобин – 133 г/л (норма – 120–140 г/л), эритроциты – 4,4 × 1012/л, лейкоциты – 8,8 × 109/л, тромбоциты – 224 × 109/л, палочкоядерные нейтрофилы – 6%, сегментоядерные нейтрофилы – 59%, лимфоциты – 32%, моноциты – 7%, базофилы – 2%, СОЭ – 28 мм/ч, в биохимическом: С-реактивный белок отрицательный, аланинаминотрансфераза – 8 Ед/л, аспартатаминотрансфераза – 15 Ед/л, креатинин – 87 мкмоль/л, билирубин – 12 мкмоль/л, фибриноген – 3,2 г/л, иммунологическом: циркулирующих иммунных комплексов – 50 опт. ед., ревматоидный фактор отрицательный. В общем анализе мочи: плотность – 1021 г/л, рН – 6,0, белок – 0,5 г/л, лейкоциты – 2–4 в п/зр, суточная протеинурия – 0,4 г/л.
На рентгенограмме кистей и стоп – остеопороз, высота суставных щелей не изменена, костей таза – высота суставных щелей тазобедренных суставов не изменена, ширина сакроилеальных сочленений сохранена, органов грудной клетки – легкие без очаговых и инфильтративных теней, плевральные синусы свободные, сердце в размерах не увеличено. Исходя из анамнеза, клиники, показателей лабораторных анализов, не подтверждающих ревматологическую природу поражения суставов, сделано предположение о наличии болезни Фабри.
Взят образец сухого пятна крови на активность фермента альфа-галактозидазы (проводится в России бесплатно). Выявлено снижение уровня активности фермента альфа-галактозидазы в плазме крови до 8,46 пмоль/диск*20 ч (норма – 160–2000 пмоль/диск*20 ч), что характерно для данной патологии.
В июле 2015 г. пациент направлен в отделение ревматологии Клиники нефрологии, внутренних и профессиональных заболеваний им. Е.М. Тареева Первого Московского государственного медицинского университета им. И.М. Сеченова.
Дополнительно проведено холтеровское мониторирование (суточное мониторирование электрокардиограммы (ЭКГ)). Обнаружены суправентрикулярные экстрасистолы, укорочение интервала PQ. На эхокардиографии – без выраженной патологии, при магнитно-резонансной томографии (МРТ) сердца с контрастированием – митральная регургитация первой степени.
Микроальбуминурия – 349 мг/л (норма – 30 мг/л).
Консультация офтальмолога: признаки воронковидной кератопатии.
На МРТ головного мозга – в правой задневисочной области очаговые изменения, которые могут быть расценены как химический сдвиг из-за микрокальцинатов.
Генетическое исследование. Прямое секвенирование гена выявило мутацию с.550Т>С в гемизиготном состоянии (молекулярно-генетическое исследование на болезнь Фабри проводится в России бесплатно).
Диагноз по результатам консилиума: болезнь Фабри (мутация в гене GLA) с поражением кожи (ангиокератомы), центральной (очаговые изменения в тканях головного мозга) и периферической (акропарестезии, сенсорные нарушения) нервной системы, мышц и суставов (миалгии, артралгии), потовых желез (гипогидроз), почек (протеинурия до 0,5 г/сут), органа зрения (воронковидная кератопатия), лихорадочный синдром.
Таким образом, у пациента очевидные ревматологические причины данной клинической картины исключены. Симптоматика расценена в рамках болезни Фабри, подтверждена результатами генетических и биохимических тестов.
Назначено лечение: агалсидаза бета 1 мг/кг каждые две недели пожизненно.
С февраля 2016 г. пациент еженедельно в стационаре получает препарат 1 мг/кг внутривенно капельно.
В результате лечения самочувствие больного улучшилось: отсутствие мучительных болей в суставах, лихорадки, уменьшение общей слабости, повышение аппетита.
Были взяты анализы крови у родственников больного.
У матери, бабушки по материнской линии, двоюродных сестер подтверждено носительство гена болезни Фабри.
В июле 2015 г. у брата пациента и дяди диагностирована болезнь Фабри. У дяди (45 лет) по линии матери четыре года отмечались мучительные боли в кистях и стопах длительностью до двух дней, которые усиливались при физической нагрузке и в жару, повышение температуры тела до 39 ºС, высыпания на коже без зуда (в настоящее время пациент обследуется). У родного брата пациента отмечаются перебои в работе сердца после физической нагрузки и высыпания на коже в паховой области и области пупка. Из анамнеза: с 18 лет высыпания бурового цвета в области пупка и на слизистой оболочке верхней губы, перебои в работе сердца. При обследовании пациента в отделении ревматологии Клиники нефрологии, внутренних и профессиональных болезней им. Е.М. Тареева проведено генетическое исследование, подтвердившее мутацию. Объективно: в паховых областях и околопупочной области обнаружены ангиокератомы, суставы не изменены, тоны сердца ясные, ритм правильный, частота сердечных сокращений – 76 в минуту, артериальное давление – 110/70 мм рт. ст. В общем анализе мочи: белок – 0,09 г/л, суточная протеинурия – 0,04 г/л, микроальбуминурия – 76 мг/л. На ЭКГ – синдром преждевременного возбуждения желудочков, МРТ сердца с контрастированием – митральная регургитация первой степени. Консультация офтальмолога: признаки воронковидной кератопатии. Диагностирована болезнь Фабри с поражением кожи (ангиокератомы), почек (микроальбуминурия), органа зрения (воронкообразная кератопатия).
С февраля 2016 г. по прибытии на место жительства пациент еженедельно получает в стационаре агалсидазу бета 1 мг/кг внутривенно капельно. Эффект терапии положительный.
Заключение
В своей практике ревматологи могут столкнуться с ревматологическими проявлениями болезни Фабри. Своевременная диагностика этой патологии позволит назначить адекватное лечение, улучшить прогноз и качество жизни пациентов.
Прозрачность исследований. Исследование не имело спонсорской поддержки. Авторы несут полную ответственность за предоставление окончательной версии рукописи в печать.
Декларация о финансовых и других взаимоотношениях. Все авторы принимали участие в разработке концепции статьи в написании рукописи. Окончательная версия рукописи была одобрена всеми авторами. Авторы не получали гонораров за статью.
V.T. Komarov, N.S. Hichina
Burdenko Regional Clinical Hospital of Penza
Contact person: Nataliya Serafimovna Hichina, nataliyahichina@rambler.ru
Pain in distal interphalangeal joints of the upper and lower limbs is usually regarded as a sign of rheumatic pathologies. However, it may be the result of diseases of other etiologies. In the article on the specific example the similar case is considered. For a long time a patient with arthralgia was observed with regard to the different rheumatic diseases. In 17 years the correct diagnosis of Fabry disease (a rare genetic disorder) was made. Its timely diagnosis will allow providing adequate treatment which as a result will improve the prognosis and quality of life of patients.