Cerebrum diabeticum: существует ли диабетическая энцефалопатия?
- Аннотация
- Статья
- Ссылки
- English
Субстрат энцефалопатии остается неясным, а ее статус, критерии диагностики продолжают обсуждаться, в связи с чем в последние годы данный термин применяется существенно реже. При СД 1 типа ДЭ носит первичный характер и связана с нарушением действия инсулина и гипергликемией, тогда как при СД 2 типа в значительной части случаев она является следствием сосудистых осложнений.
Субстрат энцефалопатии остается неясным, а ее статус, критерии диагностики продолжают обсуждаться, в связи с чем в последние годы данный термин применяется существенно реже. При СД 1 типа ДЭ носит первичный характер и связана с нарушением действия инсулина и гипергликемией, тогда как при СД 2 типа в значительной части случаев она является следствием сосудистых осложнений.
Сахарный диабет (СД) – одно из самых частых хронических заболеваний, поражающее до 10% населения. Нервная система – одна из основных мишеней СД. Традиционно считалось, что при СД в патологический процесс может вовлекаться центральная нервная система (ЦНС), однако преимущественно страдает периферическая нервная система (ПНС). Перефразируя знаменитое изречение, можно сказать, что СД «лижет» ЦНС, но «кусает» ПНС.
В последние десятилетия представления изменились. Установлено, что дисфункция головного мозга – частое явление при СД. Однако ее патогенез, нозологический статус, клиника, прогноз, критерии диагностики, подходы к терапии плохо изучены и вызывают дискуссии [1–3].
Классификация поражений головного мозга при сахарном диабете
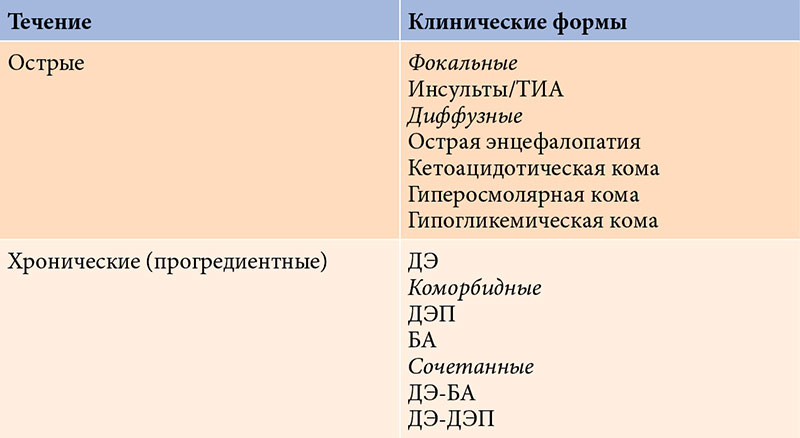
Поражение головного мозга при СД может быть острым или хроническим (см. таблицу). Острые поражения обусловлены метаболическими нарушениями (острые метаболические энцефалопатии, или комы: гипергликемическая, кетоацидотическая, гипогликемическая и т.д.). Как правило, они обратимы, однако могут оставлять после себя (особенно при гипогликемической коме) резидуальный дефект, который от эпизода к эпизоду может «накапливаться». Острым поражением также считается инсульт или транзиторная ишемическая атака (ТИА), частота которых у лиц с СД существенно выше, чем в популяции [2, 4, 5].
К хроническим, как правило, прогредиентным поражениям головного мозга относят диабетическую энцефалопатию (ДЭ) и так называемые коморбидные состояния (прежде всего дисциркуляторную энцефалопатию (ДЭП) и болезнь Альцгеймера (БА)), которые представляют собой самостоятельные нозологические формы, патогенетически связанные с СД.
Кроме того, как показывает клиническая практика, существуют переходные (сочетанные) формы. Они занимают промежуточное положение между ДЭ и БА или ДЭП.
Термин «диабетическая энцефалопатия» предложил R. de Jong в 1950 г., описавший когнитивное снижение у молодого человека с СД 1 типа. Под ДЭ понимают медленно прогрессирующие диффузные изменения в головном мозге, непосредственно связанные с метаболическими нарушениями и проявляющиеся когнитивным снижением, церебральной атрофией и/или диффузным поражением белого вещества головного мозга [1, 5–8].
Частота встречаемости ДЭ среди пациентов с СД колеблется от 5 до 80%. Столь значительный разброс объясняется отсутствием общепринятых критериев диагностики заболевания. В чистом виде ДЭ как дисметаболическое расстройство наблюдается преимущественно у больных СД 1 типа. У данной категории пациентов заболевание обусловлено прежде всего неэффективным контролем гликемии (гипергликемией или гипогликемическими эпизодами). У пациентов с СД 2 типа ДЭ более чем в 80% случаев сопровождается цереброваскулярной патологией, что объясняется более старшим возрастом и широким распространением сосудистых факторов риска (артериальной гипертензии, дислипидемии). Кроме того, если при СД 1 типа решающую роль в развитии метаболических нарушений и дисфункции головного мозга играет дефицит инсулина, при СД 2 типа – инсулинорезистентность [3, 6, 8].
Болезнь Альцгеймера – коморбидное состояние или диабет 3 типа?
Существует параллелизм в развитии СД и БА, который во многом объясняется нарушением функции инсулина. В отличие от периферических тканей в головном мозге инсулин не участвует в утилизации глюкозы, однако участвует в регуляции когнитивных процессов.
Рецепторы инсулина обнаружены в коре головного мозга и гиппокампе. Их активация приводит к усилению когнитивных и прежде всего мнестических функций.
Снижение с возрастом в мозге концентрации инсулина и количества инсулиновых рецепторов частично объясняет возрастную когнитивную дисфункцию. Кроме того, при тяжелой БА в мозге отмечено снижение уровня инсулина и промежуточных продуктов инсулинового сигнального пути. Этому может способствовать нарушение транспорта инсулина в мозг через гематоэнцефалический барьер и снижение синтеза инсулина в мозге. Таким образом, у пациентов с БА при нормальном или высоком уровне периферического инсулина уровень инсулина в цереброспинальной жидкости обычно снижен [9–12]. При БА и СД также выявлено уменьшение уровня инсулизина (инсулин-деградирующего фермента), расщепляющего как инсулин, так и амилоидный белок, что нарушает клиренс амилоидного белка из вещества головного мозга.
Кроме того, инсулин инактивирует киназу-3 гликогенсинтетазы, препятствуя фосфорилированию тау-протеина и тем самым тормозя накопление и агрегацию указанного белка.
Инсулинорезистентность связана с нарушением обмена амилоида, атрофией гиппокампа, нарушением памяти и повышением риска развития БА в полтора – два раза. О роли инсулина в регулировании когнитивных процессов свидетельствует улучшение процессов памяти при его внутривенном или интраназальном введении [1, 7, 13].
Общими факторами патогенеза СД и БА являются окислительный стресс и повышенное образование конечных продуктов гликирования. Наблюдается также сходство в дизрегуляции метаболизма глюкозы, нарушении обмена аполипопротеина Е, митохондриальной дисфункции, нарушении гомеостаза кальция. На экспериментальной модели показано, что СД сопровождается поражением нейронов гиппокампа и миндалины – зон, которые страдают на раннем этапе развития БА. Кроме того, пациенты с БА предрасположены к развитию СД 2 типа и чаще имеют гиперинсулинемию и гипергликемию по сравнению со здоровыми лицами того же возраста [8].
Соотношение роли метаболических нарушений и сосудистых факторов риска в поражении головного мозга при сахарном диабете
Сложно сопоставить значимость собственно метаболических расстройств и сосудистых факторов риска, часто наблюдающихся у пациентов с СД 2 типа, в развитии атрофии головного мозга или лейкоэнцефалопатии, выявляемых при нейровизуализации, и связанных с ними когнитивных нарушений.
Отправной точкой метаболического пути поражения головного мозга, вероятно, является гипергликемия, однако конкретные механизмы могут быть связаны с накоплением конечных продуктов гликирования, активных форм кислорода и свободных радикалов (окислительный стресс), дисфункцией инсулинового сигнального пути. Токсическое воздействие избытка глюкозы может быть связано с активацией полиолового и гексозаминового путей. При улучшении контроля СД когнитивная дисфункция, отражающая поражение мозга, частично обратима.
Роль гипогликемии в развитии атрофии головного мозга или лейкоэнцефалопатии подтверждена не во всех исследованиях, что, возможно, отражает позитивное влияние инсулина на когнитивные функции [11, 14, 15].
Атрофия мозга и диффузное поражение белого вещества, выявляемые при магнитно-резонансной томографии, ассоциированы с нарушением миелинизации нервных волокон. Рецепторы конечных продуктов гликирования экспрессированы как в сером веществе (гиппокампе и коре головного мозга), так и в белом, особенно в мозолистом теле и внутренней капсуле. Повышенное количество этих рецепторов объясняется устранением торможения под влиянием нуклеарного (ядерного) фактора κB, что связано с дисфункцией инсулинового сигнального пути. Этот же фактор влияет на продукцию фактора некроза опухоли альфа, который также участвует в развитии диабетической лейкоэнцефалопатии. Однако лейкоэнцефалопатия при СД может быть вызвана и артериальной гипертензией. Тем не менее результаты исследований показывают, что метаболические нарушения, связанные с СД, теснее коррелируют с развитием церебральной атрофии и изменением белого вещества, тогда как артериальная гипертензия ассоциирована лишь с легким изменением численности нейронов гиппокампа и утратой синапсов. Кроме того, когнитивные нарушения при СД выражены в большей степени и наступают быстрее, чем при наличии только артериальной гипертензии. Таким образом, как клинические, так и экспериментальные данные указывают на синергизм метаболических нарушений и гипертензии в поражении мозга [6, 8, 11, 13, 16].
Роль гиперлипидемии в развитии метаболического поражения мозга не определена. Известно, что повышенный уровень триглицеридов способствует прогрессированию диабетической невропатии. Нарушение метаболизма липидов может вносить существенный вклад в звенья патогенеза, связанные с конечными продуктами гликирования. Оксидация липопротеинов низкой плотности может играть решающую роль в развитии атеросклероза, окислительного стресса и воспалительных процессов [15, 16].
Роль сосудистого фактора в развитии когнитивных нарушений при СД может преувеличиваться в силу более простого выявления сосудистых изменений мозга при визуализации. У больных СД диффузные изменения белого вещества, немые инфаркты и церебральная атрофия могут отражать возрастные изменения, которые не обязательно сопровождаются когнитивным снижением. На экспериментальных моделях ДЭ не выявлено снижения перфузии мозга. Изменений перфузии не установлено и в клинических исследованиях [7, 9, 17].
В то же время у больных СД 2 типа, при котором чаще отмечаются атрофия подкорковых структур и патология белого вещества, как правило, отмечается дисрегуляторный (подкорково-лобный) профиль когнитивных нарушений. Таким образом, корреляция между характером когнитивных нарушений и нейровизуализационными изменениями может отражать вклад сосудистых, дисметаболических и дегенеративных изменений в формирование клинической картины [11, 15, 17].
Когнитивная дисфункция – основное проявление диабетической энцефалопатии
Основным проявлением нарушений головного мозга при СД служат когнитивные и аффективные изменения, что отражает преимущественную дисфункцию фронтостриарных кругов, регулирующих соответствующие нейропсихологические процессы. При СД нарушаются память, внимание, зрительно-пространственные и регуляторные функции.
О прогрессировании заболевания свидетельствует прежде всего снижение памяти (особенно при проведении тестов на отсроченное воспроизведение). В повседневной жизни в большей степени проявляется дисрегуляторный дефицит, предопределяющий неспособность пациентов обрабатывать неструктурированную информацию, особенно в ситуациях, требующих немедленных решений.
Большинство пациентов с СД 2 типа не страдают деменцией, однако демонстрируют определенные когнитивные затруднения, которые можно характеризовать как умеренные когнитивные расстройства. В то же время у пациентов с умеренными когнитивными расстройствами повышен риск конверсии их в деменцию [1, 8, 9].
При СД 1 типа, дебютирующем в детском возрасте, отмечаются замедление интеллектуального развития и трудности обучения в школе. Этот дефект персистирует и в более зрелом возрасте, нередко сопровождаясь дальнейшим когнитивным снижением и более выраженной церебральной атрофией. Важную роль играют повторяющиеся эпизоды гипогликемии, которые способствуют усугублению церебральной атрофии.
При СД 2 типа снижение когнитивных функций может происходить параллельно с переходом состояния пациента от нарушения толерантности к глюкозе к развитию СД, что также сопровождается нарастающим влиянием сосудистых факторов риска, таких как артериосклероз и артериальная гипертензия. У лиц пожилого возраста вероятность когнитивного снижения выше, что объясняется возрастными изменениями головного мозга [6, 18, 19].
Риск развития ДЭ не зависит от пола, этнической принадлежности. Однако он повышается при увеличении длительности заболевания, неадекватном контроле гипергликемии, ассоциированном с более значительным микроваскулярным поражением, повышенным уровнем гликированного гемоглобина, отражающего степень персистирования гипергликемии в течение дня.
Конверсия умеренных когнитивных расстройств в деменцию и присоединение коморбидных заболеваний
Развитие деменции, риск которой при ДЭ повышен, вероятно, становится возможным не в рамках самой ДЭ, а в результате присоединения коморбидных заболеваний – БА и/или выраженной цереброваскулярной патологии. Как развитие инсульта, так и хроническая цереброваскулярная патология усугубляют когнитивный статус пациента с ДЭ. Наличие немых инфарктов мозга у больных СД удваивает риск развития деменции в течение четырех лет [3].
Предикторами конверсии умеренных когнитивных расстройств в деменцию могут выступать генетическая предрасположенность, например аллель АРОЕ4, а также депрессия, которая может отражать как психологические проблемы пациентов, так и нейромедиаторные и структурные изменения в веществе головного мозга.
По некоторым данным, у больных СД чаще диагностируется смешанная форма деменции. Комбинация цереброваскулярного и дегенеративного процессов сопровождается более тяжелой эндотелиальной дисфункцией, нарушением целостности гематоэнцефалического барьера, более тяжелым окислительным стрессом, более интенсивным накоплением бета-амилоида и нейровоспалительным процессом. Кроме того, при сочетании БА и СД возможно развитие церебральной амилоидной ангиопатии. СД ухудшает когнитивный статус пациентов с развивающейся БА. Однако не доказано, что СД усугубляет патоморфологические изменения, свойственные БА.
Лечение диабетической энцефалопатии
Лечение ДЭ должно включать адекватную физическую активность и диету, коррекцию и тщательный контроль уровня гликемии и сосудистых факторов риска [13, 20–22].
Препараты для лечения ДЭ должны быть направлены на усиление активности инсулизина, повышение чувствительности рецепторов к инсулину, что может способствовать повышению клиренса бета-амилоида. Модуляторы инсулизина в форме небольших пептидов могут усилить гидролиз бета-амилоида, не повышая распад самого инсулина.
Применение пиоглитазона – агониста рецептора пролифератора пероксисом гамма – приводит к улучшению чувствительности к инсулину. Системное использование препаратов инсулина для лечения заболеваний головного мозга возможно, так как инсулин способен проникать через гематоэнцефалический барьер с помощью специальной транспортной системы. Однако он должен вводиться в количествах, не вызывающих гипогликемию.
Интраназальное введение инсулина, проникающего в мозг через систему обонятельного и тройничного нервов, может стать альтернативой системному введению. Экспериментальные данные свидетельствуют, что интраназальное введение инсулина способно положительно влиять на когнитивное снижение, степень атрофии головного мозга и изменения белого вещества, но лишь при условии относительного дефицита инсулина, что может наблюдаться как при СД 1 типа, так и при СД 2 типа [4, 6, 13, 14, 20].
Важен поиск эффективных и безопасных препаратов, противодействующих окислительному стрессу, нейровоспалительному процессу, а также модулирующих глутаматергическую, холинергическую и катехоламинергическую передачу.
Перспективы мультимодальной терапии диабетической энцефалопатии
Терапия, одновременно направленная на несколько ключевых патофизиологических звеньев ДЭ, по-видимому, является наиболее перспективной. В этой связи представляется целесообразным применение комбинаций препаратов или многокомпонентных препаратов. Примером последнего может служить Актовегин [12, 14, 22, 23].
Актовегин – депротеинизированный гемодериват, получаемый из крови телят путем ультрафильтрации, в состав которого входят около 200 низкомолекулярных соединений весом до 5000 дальтон. Актовегин содержит инозитолфосфоолигосахариды, которые активируют транспорт глюкозы и стимулируют активность некоторых ферментов, включая пируватдегидрогеназу – ключевой фермент цикла Кребса. Инсулиннезависимая активация переносчиков глюкозы может ослаблять метаболические последствия инсулинорезистентности, являющейся одним из факторов альцгеймеровских изменений в мозге.
Установлено, что Актовегин увеличивает утилизацию кислорода, что приводит к увеличению синтеза аденозинтрифосфата и креатинфосфата и, как следствие, защите клеток от гипоксического повреждения. Актовегин оказывает нейропротективный и нейротрофический эффекты, способствуя торможению апоптоза (за счет блокирования каспазы-3), увеличению численности нейронов и синаптических связей. Препарат способен модулировать активность нуклеарного (ядерного) фактора κB, играющего важную роль в регуляции процессов апоптоза и воспаления. Кроме того, он способен улучшать микроциркуляцию в тканях, позитивно воздействуя на эндотелий сосудов [14, 16, 24–28].
В плацебоконтролируемых исследованиях продемонстрирована способность препарата Актовегин улучшать когнитивные функции и степень повседневной активности у пациентов с сосудистой деменцией и БА [27, 29–32].
В ряде открытых исследований показано положительное воздействие препарата Актовегин на состояние когнитивных функций у пациентов с ДЭ [2, 14]. В крупном многоцентровом плацебоконтролируемом исследовании у пациентов с СД 2 типа на фоне приема препарата Актовегин отмечено улучшение со стороны ПНС и психологического состояния (по шкале качества жизни) [12].
Результаты недавно завершившегося многоцентрового рандомизированного исследования ARTEMIDA, в котором изучалась эффективность препарата у пациентов с постинсультными когнитивными нарушениями при назначении в остром периоде инсульта, показали, что через шесть месяцев препаратом Актовегин достоверно улучшаются когнитивные функции по сравнению с приемом плацебо. Кроме того, снижается количество пациентов с диагнозом «деменция». Указанный эффект отмечался как через шесть месяцев терапии, так и через шесть месяцев последующего наблюдения [12].
Таким образом, Актовегин может применяться как при легких и умеренных когнитивных нарушениях, так и на стадии деменции в виде монотерапии или в комбинации с препаратами, воздействующими на нейромедиаторные системы. Учитывая дозозависимый характер действия, препарат необходимо применять в адекватных дозах: внутривенно капельно по 0,4–2 г (на курс до 20 инфузий), перорально – по 400 мг три раза в день. Длительность приема зависит от выраженности симптоматики. В среднем она составляет от четырех – шести недель до шести месяцев.
O.S. Levin, O.V. Babkina
Russian Medical Academy of Postgraduate Education
Contact person: Oleg Semyonovich Levin, oslevin@mail.ru
Impact of diabetes mellitus (DM) on nervous system is routinely associated with injury of peripheral nerves and nervous fibers. However, recently more interest was risen by dysfunction of the brain related to development of cognitive impairment known as diabetic encephalopathy (DE) implying slowly deteriorating cognitive impairment that usually does not reach (within DE) dementia, and caused by metabolic disorders directly triggered by DM.
A substrate of encephalopathy remains unclear, and its state, diagnostic criteria are still debated explaining why this term has been much rarely used in last years. DE develops primary to type 1 DM, which is related to impaired action of insulin and hyperglycemia, whereas during type 2 DM it is a consequence of vascular complications in substantial proportions of cases.