–Э–∞—А—Г—И–µ–љ–Є–µ –ї–Є–њ–Є–і–љ–Њ–≥–Њ –Њ–±–Љ–µ–љ–∞ —Г¬†–і–µ—В–µ–є —Б¬†–Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П–Љ–Є –њ–µ—З–µ–љ–Є. XXIII –Ї–Њ–љ–≥—А–µ—Б—Б –і–µ—В—Б–Ї–Є—Е –≥–∞—Б—В—А–Њ—Н–љ—В–µ—А–Њ–ї–Њ–≥–Њ–≤ –†–Њ—Б—Б–Є–Є –Є —Б—В—А–∞–љ –°–Э–У ¬Ђ–Р–Ї—В—Г–∞–ї—М–љ—Л–µ –≤–Њ–њ—А–Њ—Б—Л –∞–±–і–Њ–Љ–Є–љ–∞–ї—М–љ–Њ–є –њ–∞—В–Њ–ї–Њ–≥–Є–Є —Г –і–µ—В–µ–є¬ї
- –Р–љ–љ–Њ—В–∞—Ж–Є—П
- –°—В–∞—В—М—П
- –°—Б—Л–ї–Ї–Є

–Ю–±—Й–Є–µ –њ–Њ–ї–Њ–ґ–µ–љ–Є—П
–Ю¬†–љ–∞—А—Г—И–µ–љ–Є–Є –ї–Є–њ–Є–і–љ–Њ–≥–Њ –Њ–±–Љ–µ–љ–∞ —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ –ї—О–±–Њ–≥–Њ –≤–Њ–Ј—А–∞—Б—В–∞ –Љ–Њ–ґ–љ–Њ —Б—Г–і–Є—В—М –њ–Њ¬†—Б–Њ–і–µ—А–ґ–∞–љ–Є—О —Д–Њ—Б—Д–Њ–ї–Є–њ–Є–і–Њ–≤, –љ–µ—Н—Б—В–µ—А–Є—Д–Є—Ж–Є—А–Њ–≤–∞–љ–љ—Л—Е –ґ–Є—А–љ—Л—Е –Ї–Є—Б–ї–Њ—В, —В—А–Є–≥–ї–Є—Ж–µ—А–Є–і–Њ–≤, —Б–≤–Њ–±–Њ–і–љ–Њ–≥–Њ –Є¬†—Н—Б—В–µ—А–Є—Д–Є—Ж–Є—А–Њ–≤–∞–љ–љ–Њ–≥–Њ —Е–Њ–ї–µ—Б—В–µ—А–Є–љ–∞. –Ы–Є–њ–Є–і—Л –љ–∞—Е–Њ–і—П—В—Б—П –≤¬†–њ–ї–∞–Ј–Љ–µ –Ї—А–Њ–≤–Є –≤¬†—Б–≤—П–Ј–∞–љ–љ–Њ–є —Б¬†–±–µ–ї–Ї–∞–Љ–Є —Д–Њ—А–Љ–µ –Є¬†–Њ–±—А–∞–Ј—Г—О—В —Б–ї–µ–і—Г—О—Й–Є–µ –Ї–ї–∞—Б—Б—Л:
- —Е–Є–ї–Њ–Љ–Є–Ї—А–Њ–љ—Л;
- –ї–Є–њ–Њ–њ—А–Њ—В–µ–Є–љ—Л –Њ—З–µ–љ—М –љ–Є–Ј–Ї–Њ–є –њ–ї–Њ—В–љ–Њ—Б—В–Є (–Ы–Я–Ю–Э–Я);
- –ї–Є–њ–Њ–њ—А–Њ—В–µ–Є–љ—Л –љ–Є–Ј–Ї–Њ–є –њ–ї–Њ—В–љ–Њ—Б—В–Є (–Ы–Я–Э–Я)¬†вАУ –±–µ—В–∞-–ї–Є–њ–Њ–њ—А–Њ—В–µ–Є–љ—Л;
- –ї–Є–њ–Њ–њ—А–Њ—В–µ–Є–љ—Л –≤—Л—Б–Њ–Ї–Њ–є –њ–ї–Њ—В–љ–Њ—Б—В–Є (–Ы–Я–Т–Я)¬†вАУ –∞–ї—М—Д–∞-–ї–Є–њ–Њ¬≠–њ—А–Њ—В–µ–Є–љ—Л.
–†–∞–Ј–і–µ–ї–µ–љ–Є–µ –ї–Є–њ–Њ–њ—А–Њ—В–µ–Є–љ–Њ–≤ –љ–∞¬†–Ї–ї–∞—Б—Б—Л –Њ–±—Г—Б–ї–Њ–≤–ї–µ–љ–Њ —А–∞–Ј–ї–Є—З–Є–µ–Љ –≤¬†–њ–ї–Њ—В–љ–Њ—Б—В–Є –і–≤–Є–ґ–µ–љ–Є—П –њ—А–Є —Н–ї–µ–Ї—В—А–Њ—Д–Њ—А–µ–Ј–µ.
–Т¬†–Ј–∞–≤–Є—Б–Є–Љ–Њ—Б—В–Є –Њ—В¬†–њ–Њ–Ї–∞–Ј–∞—В–µ–ї–µ–є –њ—А–Є–Љ–µ–љ—П—О—В—Б—П —Д–Њ—А–Љ—Г–ї—Л –і–ї—П –≤—Л—З–Є—Б–ї–µ–љ–Є—П –Є–љ–і–µ–Ї—Б–∞ –∞—В–µ—А–Њ–≥–µ–љ–љ–Њ—Б—В–Є. –Т¬†–њ–µ–і–Є–∞—В—А–Є—З–µ—Б–Ї–Њ–є –њ—А–∞–Ї—В–Є–Ї–µ –Ї¬†–њ–Њ–і–Њ–±–љ–Њ–Љ—Г —А–∞—Б—З–µ—В—Г –њ—А–Є–±–µ–≥–∞—О—В –љ–µ—З–∞—Б—В–Њ, –њ–Њ—Б–Ї–Њ–ї—М–Ї—Г –Ї–Њ—А—А–µ–ї—П—Ж–Є–Є –Љ–µ–ґ–і—Г —Г—А–Њ–≤–љ–µ–Љ —Г–Ї–∞–Ј–∞–љ–љ–Њ–≥–Њ –Є–љ–і–µ–Ї—Б–∞ –Є¬†–∞—В–µ—А–Њ—Б–Ї–ї–µ—А–Њ—В–Є—З–µ—Б–Ї–Є–Љ –њ–Њ—А–∞–ґ–µ–љ–Є–µ–Љ —Г¬†–і–µ—В–µ–є –љ–µ¬†—Г—Б—В–∞–љ–Њ–≤–ї–µ–љ–Њ.
–Я–Њ–і –і–Є—Б–ї–Є–њ–Є–і–µ–Љ–Є–µ–є –њ–Њ–љ–Є–Љ–∞—О—В —Б–∞–Љ–Њ—Б—В–Њ—П—В–µ–ї—М–љ—Л–µ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П –Є¬†–њ–∞—В–Њ–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Є–µ —Б–Њ—Б—В–Њ—П–љ–Є—П, –Њ–±—Г—Б–ї–Њ–≤–ї–µ–љ–љ—Л–µ –Ї–Њ–ї–Є—З–µ—Б—В–≤–µ–љ–љ—Л–Љ–Є –Є¬†–Ї–∞—З–µ—Б—В–≤–µ–љ–љ—Л–Љ–Є –љ–∞—А—Г—И–µ–љ–Є—П–Љ–Є —Б–Њ—Б—В–∞–≤–∞ –ї–Є–њ–Є–і–Њ–≤ –Є¬†–ї–Є–њ–Њ–њ—А–Њ—В–µ–Є–љ–Њ–≤ –≤¬†–Ї—А–Њ–≤–Є.
–Т—Л–і–µ–ї—П—О—В –њ–µ—А–≤–Є—З–љ—Л–µ –Є¬†–≤—В–Њ—А–Є—З–љ—Л–µ –і–Є—Б–ї–Є–њ–Є–і–µ–Љ–Є–Є. –Я–µ—А–≤–Є—З–љ—Л–µ¬†–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Є –і–µ—В–µ—А–Љ–Є–љ–Є—А–Њ–≤–∞–љ—Л. –Т—Л—П–≤–ї—П—О—В—Б—П —Б—В–∞–±–Є–ї—М–љ—Л–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П –Њ–і–љ–Њ–≥–Њ –Є–ї–Є –љ–µ—Б–Ї–Њ–ї—М–Ї–Є—Е –њ–Њ–Ї–∞–Ј–∞—В–µ–ї–µ–є –ї–Є–њ–Є–і–љ–Њ–≥–Њ –Њ–±–Љ–µ–љ–∞ –≤¬†–Њ—В—Б—Г—В—Б—В–≤–Є–µ –Ї–∞–Ї–Є—Е-–ї–Є–±–Њ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є. –Т—В–Њ—А–Є—З–љ—Л–µ –і–Є—Б–ї–Є–њ–Є–і–µ–Љ–Є–Є¬†вАУ —Б–ї–µ–і—Б—В–≤–Є–µ —И–Є—А–Њ–Ї–Њ–≥–Њ —Б–њ–µ–Ї—В—А–∞ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є: –њ–∞–љ–Ї—А–µ–∞—В–Є—В–∞,¬†–≥–µ–њ–∞—В–Є—В–∞, —Е–Њ–ї–µ—Ж–Є—Б—В–Є—В–∞, –љ–µ—Д—А–Є—В–∞,¬†–≥–Є–њ–Њ—В–Є—А–µ–Њ–Ј–∞, –љ–∞—Б–ї–µ–і—Б—В–≤–µ–љ–љ—Л—Е –±–Њ–ї–µ–Ј–љ–µ–є –Њ–±–Љ–µ–љ–∞ –Є¬†—В.–і.
–Ф–Є—Б–ї–Є–њ–Є–і–µ–Љ–Є–Є –Ї–ї–∞—Б—Б–Є—Д–Є—Ж–Є—А—Г—О—В –љ–∞¬†–≤—А–Њ–ґ–і–µ–љ–љ—Л–µ –Є¬†–њ—А–Є–Њ–±—А–µ—В–µ–љ–љ—Л–µ. –Т—А–Њ–ґ–і–µ–љ–љ—Л–µ –љ–∞–±–ї—О–і–∞—О—В—Б—П —Г¬†—А–µ–±–µ–љ–Ї–∞ —Б¬†–њ–µ—А–≤—Л—Е –Љ–µ—Б—П—Ж–µ–≤ –ґ–Є–Ј–љ–Є, –љ–Њ¬†–љ–µ –Є–Љ–µ—О—В –љ–∞—Б–ї–µ–і—Б—В–≤–µ–љ–љ–Њ–≥–Њ –Є–ї–Є —Б–µ–Љ–µ–є–љ–Њ–≥–Њ —Е–∞—А–∞–Ї—В–µ—А–∞ –Є¬†–Њ–±—Г—Б–ї–Њ–≤–ї–µ–љ—Л –љ–µ–±–ї–∞–≥–Њ–њ—А–Є—П—В–љ—Л–Љ–Є —Д–∞–Ї—В–Њ—А–∞–Љ–Є –≤–Њ–Ј–і–µ–є—Б—В–≤–Є—П –љ–∞¬†–њ–ї–Њ–і¬†вАУ —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–є¬†–≥–Є–њ–Њ–Ї—Б–Є–µ–є, –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П–Љ–Є –Љ–∞—В–µ—А–Є, –≤¬†—З–∞—Б—В–љ–Њ—Б—В–Є —Б–∞—Е–∞—А–љ—Л–Љ –і–Є–∞–±–µ—В–Њ–Љ, –љ–µ—Д—А–Њ–њ–∞—В–Є–µ–є, –њ–Њ–Ј–і–љ–Є–Љ¬†–≥–µ—Б—В–Њ–Ј–Њ–Љ. –Я—А–Є–Њ–±—А–µ—В–µ–љ–љ—Л–µ –і–Є—Б–ї–Є–њ–Є–і–µ–Љ–Є–Є —А–∞–Ј–≤–Є–≤–∞—О—В—Б—П –≤¬†—А–∞–Ј–љ—Л–µ –њ–µ—А–Є–Њ–і—Л –ґ–Є–Ј–љ–Є —А–µ–±–µ–љ–Ї–∞ –љ–∞¬†—Д–Њ–љ–µ:
- –љ–∞—А—Г—И–µ–љ–Є—П —А–µ–ґ–Є–Љ–∞ –њ–Є—В–∞–љ–Є—П;
- –Є–Ј–Љ–µ–љ–µ–љ–Є—П —Е–Є–Љ–Є—З–µ—Б–Ї–Њ–≥–Њ —Б–Њ—Б—В–∞–≤–∞ —А–∞—Ж–Є–Њ–љ–∞ –њ–Є—В–∞–љ–Є—П;¬†–≥–Є–њ–Њ–і–Є–љ–∞–Љ–Є–Є;
- –і–Є—Б—Д—Г–љ–Ї—Ж–Є–Є –ґ–µ–ї—Г–і–Њ—З–љ–Њ-–Ї–Є—И–µ—З–љ–Њ–≥–Њ —В—А–∞–Ї—В–∞;
- —А–∞–Ј–ї–Є—З–љ—Л—Е —Б–Њ–њ—Г—В—Б—В–≤—Г—О—Й–Є—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є.
–°–Њ–≥–ї–∞—Б–љ–Њ –Ї–ї–∞—Б—Б–Є—Д–Є–Ї–∞—Ж–Є–Є –Т—Б–µ–Љ–Є—А–љ–Њ–є –Њ—А–≥–∞–љ–Є–Ј–∞—Ж–Є–Є –Ј–і—А–∞–≤–Њ–Њ—Е—А–∞–љ–µ–љ–Є—П, –≤¬†–Ј–∞–≤–Є—Б–Є–Љ–Њ—Б—В–Є –Њ—В¬†—Б–њ–µ–Ї—В—А–∞ –≤¬†–ї–Є–њ–Є–і–Њ–≥—А–∞–Љ–Љ–µ —А–∞–Ј–ї–Є—З–∞—О—В –њ—П—В—М —В–Є–њ–Њ–≤¬†–≥–Є–њ–µ—А–ї–Є–њ–Є–і–µ–Љ–Є–Є. –£¬†–і–µ—В–µ–є —З–∞—Й–µ –≤—Б—В—А–µ—З–∞—О—В—Б—П —В–Є–њ—Л IIa –Є¬†IIb.
–Х—Й–µ —Б–Њ–≤—Б–µ–Љ –љ–µ–і–∞–≤–љ–Њ –њ—А–Њ–≤–µ–і–µ–љ–Є–µ –Љ–Њ–ї–µ–Ї—Г–ї—П—А–љ–Њ-–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Є—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є –і–ї—П –Њ–њ—А–µ–і–µ–ї–µ–љ–Є—П —В–Є–њ–∞ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П –Ї–∞–Ј–∞–ї–Њ—Б—М –љ–µ–≤–Њ–Ј–Љ–Њ–ґ–љ—Л–Љ. –°–µ–≥–Њ–і–љ—П —Г¬†–Љ–љ–Њ–≥–Є—Е –њ–∞—Ж–Є–µ–љ—В–Њ–≤ –Њ–±–љ–∞—А—Г–ґ–µ–љ—Л¬†–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Є–µ –ї–Њ–Ї—Г—Б—Л, –њ–Њ–і—В–≤–µ—А–ґ–і–∞—О—Й–Є–µ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–µ. –С–Є–Њ—Е–Є–Љ–Є—З–µ—Б–Ї–Њ–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ –ї–Є–њ–Є–і–Њ–≥—А–∞–Љ–Љ—Л —Б—З–Є—В–∞–µ—В—Б—П –Њ—Б–љ–Њ–≤–љ—Л–Љ –Љ–µ—В–Њ–і–Њ–Љ –Њ–њ—А–µ–і–µ–ї–µ–љ–Є—П —В–Є–њ–∞ –і–Є—Б–ї–Є–њ–Є–і–µ–Љ–Є–Є.
–Я—А–Є —В–Є–њ–µ I¬†–≥–Є–њ–µ—А—Е–Є–ї–Њ–Љ–Є–Ї—А–Њ–љ–µ–Љ–Є–Є –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є–µ —Б–Є–Љ–њ—В–Њ–Љ—Л –≤–Ї–ї—О—З–∞—О—В —Б–њ–ї–µ–љ–Њ–Љ–µ–≥–∞–ї–Є—О, –∞–±–і–Њ–Љ–Є–љ–∞–ї—М–љ—Л–µ –Ї–Њ–ї–Є–Ї–Є, –њ–∞–љ–Ї—А–µ–∞—В–Є—В. –£¬†–і–µ—В–µ–є —А–∞–љ–љ–µ–≥–Њ –≤–Њ–Ј—А–∞—Б—В–∞ –і–Њ—Б—В–∞—В–Њ—З–љ–Њ –±—Л—Б—В—А–Њ —Д–Њ—А–Љ–Є—А—Г—О—В—Б—П –∞—В–µ—А–Њ—Б–Ї–ї–µ—А–Њ—В–Є—З–µ—Б–Ї–Є–µ –±–ї—П—И–Ї–Є. –У–Є–њ–µ—А—Е–Њ–ї–µ—Б—В–µ—А–Є–љ–µ–Љ–Є—П —В–Є–њ–∞ IIa –њ—А–µ–і–њ–Њ–ї–∞–≥–∞–µ—В –љ–∞–ї–Є—З–Є–µ –∞—В–µ—А–Њ—Б–Ї–ї–µ—А–Њ–Ј–∞ –Ї–Њ—А–Њ–љ–∞—А–љ—Л—Е –∞—А—В–µ—А–Є–є –Є¬†—Б—Г—Б—В–∞–≤–љ–Њ–≥–Њ —Б–Є–љ–і—А–Њ–Љ–∞. –Я—А–Є —В–Є–њ–µ IIb –Є–Љ–µ—О—В –Љ–µ—Б—В–Њ –Є–Ј–±—Л—В–Њ—З–љ–∞—П –Љ–∞—Б—Б–∞ —В–µ–ї–∞, –љ–µ–∞–ї–Ї–Њ–≥–Њ–ї—М–љ–∞—П –ґ–Є—А–Њ–≤–∞—П –±–Њ–ї–µ–Ј–љ—М –њ–µ—З–µ–љ–Є (–Э–Р–Ц–С–Я), —Б–∞—Е–∞—А–љ—Л–є –і–Є–∞–±–µ—В –Є¬†—А–∞–љ–љ–Є–µ —Д–Њ—А–Љ—Л –∞—В–µ—А–Њ—Б–Ї–ї–µ—А–Њ–Ј–∞. –Я—А–Є —В–Є–њ–µ III¬†–≥–Є–њ–µ—А–ї–Є–њ–Є–і–µ–Љ–Є–Є –і–µ–±—О—В –њ—А–Њ–Є—Б—Е–Њ–і–Є—В –≤¬†—А–∞–љ–љ–µ–Љ –≤–Њ–Ј—А–∞—Б—В–µ, –њ–Њ—П–≤–ї—П—О—В—Б—П –Ї—Б–∞–љ—В–Њ–Љ—Л –љ–∞¬†–ї–∞–і–Њ–љ—П—Е –Є¬†–Љ–µ—Б—В–∞—Е –і–∞–≤–ї–µ–љ–Є—П –Њ–і–µ–ґ–і—Л, —Д–Њ—А–Љ–Є—А—Г–µ—В—Б—П –Э–Р–Ц–С–Я. –Я—А–Є —В–Є–њ–µ IV –љ–∞—З–∞–ї–Њ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П, –Ї–∞–Ї –њ—А–∞–≤–Є–ї–Њ, –њ—А–Є—Е–Њ–і–Є—В—Б—П –љ–∞¬†–Љ–Њ–ї–Њ–і–Њ–є –Є¬†—Б—А–µ–і–љ–Є–є –≤–Њ–Ј—А–∞—Б—В, —А–∞–Ј–≤–Є–≤–∞—О—В—Б—П¬†–≥–µ–њ–∞—В–Њ–Љ–µ–≥–∞–ї–Є—П, —Б–∞—Е–∞—А–љ—Л–є –і–Є–∞–±–µ—В –Є¬†–Њ–ґ–Є—А–µ–љ–Є–µ. –Я—А–Є —В–Є–њ–µ V —Д–Њ—А–Љ–Є—А—Г—О—В—Б—П –∞—В–µ—А–Њ—Б–Ї–ї–µ—А–Њ–Ј —Б–Њ—Б—Г–і–Њ–≤, –Њ–ґ–Є—А–µ–љ–Є–µ, –њ–∞–љ–Ї—А–µ–∞—В–Є—В.
–Э–µ¬†–Љ–µ–љ—М—И—Г—О —А–Њ–ї—М –≤¬†—Б—В—А—Г–Ї—В—Г—А–µ –і–Є—Б–ї–Є–њ–Є–і–µ–Љ–Є–є –Є–≥—А–∞—О—В –≤—Л—П–≤–ї—П–µ–Љ—Л–µ —Г¬†—З–∞—Б—В–Є –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —А–∞–Ј–ї–Є—З–љ—Л–µ —Д–Њ—А–Љ—Л¬†–≥–Є–њ–Њ–ї–Є–њ–Є–і–µ–Љ–Є–Є.
–У–Є–њ–Њ-–∞–ї—М—Д–∞-–ї–Є–њ–Њ–њ—А–Њ—В–µ–Є–љ–µ–Љ–Є—П (–±–Њ–ї–µ–Ј–љ—М –Ґ–∞–љ–ґ–µ—А–∞) —Е–∞—А–∞–Ї—В–µ—А–Є–Ј—Г–µ—В—Б—П –Њ—В—Б—Г—В—Б—В–≤–Є–µ–Љ –Є–ї–Є —Б–љ–Є–ґ–µ–љ–Є–µ–Љ –і–Њ 1вАУ4% –≤¬†–њ–ї–∞–Ј–Љ–µ –Ї—А–Њ–≤–Є —Г—А–Њ–≤–љ–µ–є –Ы–Я–Т–Я –Є¬†–Њ–±—Й–µ–≥–Њ —Е–Њ–ї–µ—Б—В–µ—А–Є–љ–∞. –Ъ–ї–Є–љ–Є—З–µ—Б–Ї–Є –љ–∞–±–ї—О–і–∞—О—В—Б—П —Г–≤–µ–ї–Є—З–µ–љ–Є–µ –Љ–Є–љ–і–∞–ї–Є–љ —П—А–Ї–Њ–≥–Њ –ґ–µ–ї—В–Њ-–Њ—А–∞–љ–ґ–µ–≤–Њ–≥–Њ —Ж–≤–µ—В–∞ (–њ–∞—В–Њ–≥–љ–Њ–Љ–Њ–љ–Є—З–љ—Л–є –њ—А–Є–Ј–љ–∞–Ї), —Б–њ–ї–µ–љ–Њ–Љ–µ–≥–∞–ї–Є—П, —Г–≤–µ–ї–Є—З–µ–љ–Є–µ –ї–Є–Љ—Д–∞—В–Є—З–µ—Б–Ї–Є—Е —Г–Ј–ї–Њ–≤, –Љ—Л—И–µ—З–љ–∞—П —Б–ї–∞–±–Њ—Б—В—М –≤¬†–≤–µ—А—Е–љ–Є—Е –Є¬†–љ–Є–ґ–љ–Є—Е –Ї–Њ–љ–µ—З–љ–Њ—Б—В—П—Е, –Њ—Б–ї–∞–±–ї–µ–љ–Є–µ —А–µ—Д–ї–µ–Ї—Б–Њ–≤, –њ–Њ—В–µ—А—П —З—Г–≤—Б—В–≤–Є—В–µ–ї—М–љ–Њ—Б—В–Є. –Т¬†–њ—Г–љ–Ї—В–∞—В–µ –Ї–Њ—Б—В–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞ –Њ–±–љ–∞—А—Г–ґ–Є–≤–∞—О—В—Б—П ¬Ђ–њ–µ–љ–Є—Б—В—Л–µ¬ї –Ї–ї–µ—В–Ї–Є.
–°–µ–Љ–µ–є–љ–∞—П –∞–±–µ—В–∞–ї–Є–њ–Њ–њ—А–Њ—В–µ–Є–љ–µ–Љ–Є—П (—Б–Є–љ–і—А–Њ–Љ –С–∞—Б—Б–µ–љ–∞¬†вАУ –Ъ–Њ—А–љ—Ж–≤–µ–є–≥–∞) –њ—А–µ–і—Б—В–∞–≤–ї—П–µ—В —Б–Њ–±–Њ–є –љ–∞—Б–ї–µ–і—Б—В–≤–µ–љ–љ–Њ–µ –∞—Г—В–Њ—Б–Њ–Љ–љ–Њ-—А–µ—Ж–µ—Б—Б–Є–≤–љ–Њ–µ –љ–∞—А—Г—И–µ–љ–Є–µ –Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є—П –Р–њ–Њ-–Т-—Б–Њ–і–µ—А–ґ–∞—Й–Є—Е –ї–Є–њ–Њ–њ—А–Њ—В–µ–Є–љ–Њ–≤. –Ч–∞–±–Њ–ї–µ–≤–∞–љ–Є–µ —Е–∞—А–∞–Ї—В–µ—А–Є–Ј—Г–µ—В—Б—П —Б–Є–љ–і—А–Њ–Љ–Њ–Љ –Љ–∞–ї—М–∞–±—Б–Њ—А–±—Ж–Є–Є, —Б—В–µ–∞—В–Њ—А–µ–µ–є, –њ—А–Њ–≥—А–µ—Б—Б–Є—А—Г—О—Й–µ–є –і–Є—Б—В—А–Њ—Д–Є–µ–є, –Ј–∞–і–µ—А–ґ–Ї–Њ–є —А–∞–Ј–≤–Є—В–Є—П, —Г–Љ—Б—В–≤–µ–љ–љ–Њ–є –Њ—В—Б—В–∞–ї–Њ—Б—В—М—О, –њ–Є–≥–Љ–µ–љ—В–љ—Л–Љ —А–µ—В–Є–љ–Є—В–Њ–Љ, –љ–∞–ї–Є—З–Є–µ–Љ –∞–Ї–∞–љ—В–Њ—Ж–Є—В–Њ–Ј–∞, –∞—В–∞–Ї—Б–Є—З–µ—Б–Ї–Њ–є –љ–µ–≤—А–Њ–њ–∞—В–Є–µ–є.
–Э–∞—Б–ї–µ–і—Б—В–≤–µ–љ–љ–∞—П¬†–≥–Є–њ–Њ-–±–µ—В–∞-–ї–Є–њ–Њ–њ—А–Њ—В–µ–Є–љ–µ–Љ–Є—П –Њ—В–ї–Є—З–∞–µ—В—Б—П –∞—Г—В–Њ—Б–Њ–Љ–љ–Њ-–і–Њ–Љ–Є–љ–∞–љ—В–љ—Л–Љ —В–Є–њ–Њ–Љ –љ–∞—Б–ї–µ–і–Њ–≤–∞–љ–Є—П, –і–µ—Д–µ–Ї—В–Њ–Љ –Р–њ–Њ-–Т, –њ—А–Є–≤–Њ–і—П—Й–Є–Љ –Ї¬†–љ–Є–Ј–Ї–Њ–Љ—Г —Г—А–Њ–≤–љ—О –±–µ—В–∞-–ї–Є–њ–Њ–њ—А–Њ—В–µ–Є–љ–Њ–≤ –≤¬†–Ї—А–Њ–≤–Є. –£¬†–±–Њ–ї—М–љ—Л—Е —Н—В–Њ–є —Д–Њ—А–Љ–Њ–є¬†–≥–Є–њ–Њ–ї–Є–њ–Є–і–µ–Љ–Є–Є –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ —Б–љ–Є–ґ–∞–µ—В—Б—П —Г—А–Њ–≤–µ–љ—М —Е–Њ–ї–µ—Б—В–µ—А–Є–љ–∞ –Ы–Я–Э–Я. –Ъ–ї–Є–љ–Є—З–µ—Б–Ї–∞—П —Б–Є–Љ–њ—В–Њ–Љ–∞—В–Є–Ї–∞ –њ–Њ—З—В–Є —В–∞–Ї–∞—П –ґ–µ, –Ї–∞–Ї –≤¬†–њ—А–µ–і—Л–і—Г—Й–µ–Љ —Б–ї—Г—З–∞–µ, –љ–Њ¬†–Є–Љ–µ–µ—В –±–Њ–ї–µ–µ –Љ—П–≥–Ї–Њ–µ —В–µ—З–µ–љ–Є–µ. –Ф–Є–∞–≥–љ–Њ–Ј –њ–Њ–і—В–≤–µ—А–ґ–і–∞–µ—В—Б—П –њ—А–Є –Њ–±–љ–∞—А—Г–ґ–µ–љ–Є–Є –≤¬†–Ї—А–Њ–≤–Є –љ–Є–Ј–Ї–Њ–≥–Њ —Б–Њ–і–µ—А–ґ–∞–љ–Є—П —Е–Њ–ї–µ—Б—В–µ—А–Є–љ–∞, —В—А–Є–≥–ї–Є—Ж–µ—А–Є–і–Њ–≤, –Р–њ–Њ-–Т. –Ъ–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є–Є —Е–Є–ї–Њ–Љ–Є–Ї—А–Њ–љ–Њ–≤, –Ы–Я–Ю–Э–Я –Є¬†–Ы–Я–Э–Я —В–∞–Ї–ґ–µ –љ–Є–Ј–Ї–Є–µ.
–£¬†–і–µ—В–µ–є —Б¬†—Е—А–Њ–љ–Є—З–µ—Б–Ї–Є–Љ–Є –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П–Љ–Є –њ–µ—З–µ–љ–Є –љ–µ—А–µ–і–Ї–Њ —А–µ–≥–Є—Б—В—А–Є—А—Г—О—В—Б—П –љ–∞—А—Г—И–µ–љ–Є—П –ї–Є–њ–Є–і–љ–Њ–≥–Њ –Љ–µ—В–∞–±–Њ–ї–Є–Ј–Љ–∞.
–Я–Њ¬†–і–∞–љ–љ—Л–Љ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є, –Є–Ј¬†2393 –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†—Е—А–Њ–љ–Є—З–µ—Б–Ї–Є–Љ–Є –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П–Љ–Є –њ–µ—З–µ–љ–Є –љ–∞—А—Г—И–µ–љ–Є–µ –ї–Є–њ–Є–і–љ–Њ–≥–Њ –Њ–±–Љ–µ–љ–∞ –Ј–∞—Д–Є–Ї—Б–Є—А–Њ–≤–∞–љ–Њ –≤¬†15% —Б–ї—Г—З–∞–µ–≤. –Я—А–Є—З–µ–Љ —Г¬†–±–Њ–ї—М—И–Є–љ—Б—В–≤–∞ –њ–∞—Ж–Є–µ–љ—В–Њ–≤ –Є–Љ–µ–ї–Є –Љ–µ—Б—В–Њ¬†–≥–ї–Є–Ї–Њ–≥–µ–љ–Њ–≤–∞—П –±–Њ–ї–µ–Ј–љ—М, –њ—А–µ–Є–Љ—Г—Й–µ—Б—В–≤–µ–љ–љ–Њ —В–Є–њ–∞ I, —Е—А–Њ–љ–Є—З–µ—Б–Ї–Є–µ –≤–Є—А—Г—Б–љ—Л–µ –Є¬†—Е—А–Њ–љ–Є—З–µ—Б–Ї–Є–µ –Ї—А–Є–њ—В–Њ–≥–µ–љ–љ—Л–µ¬†–≥–µ–њ–∞—В–Є—В—Л.
–І–∞—Б—В–Њ—В–∞ –≤—Л—П–≤–ї–µ–љ–Є—П –і–Є—Б–ї–Є–њ–Є–і–µ–Љ–Є–є —Б–Њ—Б—В–∞–≤–Є–ї–∞: –њ—А–Є —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–Љ –≤–Є—А—Г—Б–љ–Њ–Љ¬†–≥–µ–њ–∞—В–Є—В–µ¬†вАУ 7%, –∞—Г—В–Њ–Є–Љ–Љ—Г–љ–љ—Л—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П—Е¬†вАУ 23%, —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–Љ –Ї—А–Є–њ—В–Њ–≥–µ–љ–љ–Њ–Љ¬†–≥–µ–њ–∞—В–Є—В–µ¬†вАУ 24%,¬†–≥–ї–Є–Ї–Њ–≥–µ–љ–Њ–≤–Њ–є –±–Њ–ї–µ–Ј–љ–Є¬†вАУ 65%, —Д—А—Г–Ї—В–Њ–Ј–µ–Љ–Є–Є¬†вАУ 23%, –љ–∞—Б–ї–µ–і—Б—В–≤–µ–љ–љ–Њ–Љ —Е–Њ–ї–µ—Б—В–∞–Ј–µ¬†вАУ 80%, –±–Њ–ї–µ–Ј–љ–Є –Т–Є–ї—М—Б–Њ–љ–∞¬†вАУ 10%. –С–Њ–ї–µ–µ –≤—Л—Б–Њ–Ї–Є–µ —Г—А–Њ–≤–љ–Є —Е–Њ–ї–µ—Б—В–µ—А–Є–љ–∞ –Њ—В–Љ–µ—З–∞–ї–Є—Б—М –њ—А–Є –љ–∞—Б–ї–µ–і—Б—В–≤–µ–љ–љ–Њ–Љ —Е–Њ–ї–µ—Б—В–∞–Ј–µ¬†вАУ 9,3 –Љ–Љ–Њ–ї—М/–ї, –∞–љ–Њ–Љ–∞–ї–Є–Є –ґ–µ–ї—З–љ—Л—Е –њ—А–Њ—В–Њ–Ї–Њ–≤¬†вАУ 8,8 –Љ–Љ–Њ–ї—М/–ї,¬†–≥–ї–Є–Ї–Њ–≥–µ–љ–Њ–≤–Њ–є –±–Њ–ї–µ–Ј–љ–Є¬†вАУ 7,5 –Љ–Љ–Њ–ї—М/–ї.
–Я—А–Є¬†–≥–ї–Є–Ї–Њ–≥–µ–љ–Њ–≤–Њ–є –±–Њ–ї–µ–Ј–љ–Є –љ–∞–Є–±–Њ–ї–µ–µ —В—П–ґ–µ–ї—Л–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П –ї–Є–њ–Є–і–љ–Њ–≥–Њ —Б–њ–µ–Ї—В—А–∞ –Њ—В–љ–Њ—Б—П—В—Б—П –Ї¬†–њ–µ—А–≤–Њ–Љ—Г —В–Є–њ—Г –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П¬†вАУ —Г—А–Њ–≤–µ–љ—М —Е–Њ–ї–µ—Б—В–µ—А–Є–љ–∞ –њ—А–µ–≤—Л—И–∞–µ—В 7 –Љ–Љ–Њ–ї—М/–ї.
–Э–∞¬†—Б–µ–≥–Њ–і–љ—П—И–љ–Є–є –і–µ–љ—М –љ–∞–Ї–Њ–њ–ї–µ–љ —Б–Њ–ї–Є–і–љ—Л–є –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є–є –Њ–њ—Л—В –≤–µ–і–µ–љ–Є—П –њ–∞—Ж–Є–µ–љ—В–Њ–≤ –і–µ—В—Б–Ї–Њ–≥–Њ –≤–Њ–Ј—А–∞—Б—В–∞ —Б¬†–Э–Р–Ц–С–Я¬†вАУ —Б–≤—Л—И–µ 700 —З–µ–ї–Њ–≤–µ–Ї. –Т¬†–Њ—Б–љ–Њ–≤–љ–Њ–Љ —Н—В–Њ –і–µ—В–Є —Б¬†–Є–Ј–±—Л—В–Њ—З–љ–Њ–є –Љ–∞—Б—Б–Њ–є —В–µ–ї–∞ –Є¬†–Њ–ґ–Є—А–µ–љ–Є–µ–Љ. –Э–∞–Є–±–Њ–ї—М—И–Є–є –Є–љ—В–µ—А–µ—Б –њ—А–µ–і—Б—В–∞–≤–ї—П—О—В —Г—З–∞—Б—В–Є–≤—И–Є–µ—Б—П —Б–ї—Г—З–∞–Є –њ–Њ—П–≤–ї–µ–љ–Є—П –ї–Њ–Ї–∞–ї—М–љ–Њ–≥–Њ –љ–µ–∞–ї–Ї–Њ–≥–Њ–ї—М–љ–Њ–≥–Њ¬†–≥–µ–њ–∞—В–Њ–Ј–∞. –£¬†–љ–∞–Ј–≤–∞–љ–љ–Њ–є –Ї–∞—В–µ–≥–Њ—А–Є–Є –±–Њ–ї—М–љ—Л—Е —З–∞—Б—В–Њ –≤—Л—П–≤–ї—П—О—В—Б—П –љ–∞—А—Г—И–µ–љ–Є—П –ї–Є–њ–Є–і–љ–Њ–≥–Њ –Њ–±–Љ–µ–љ–∞.
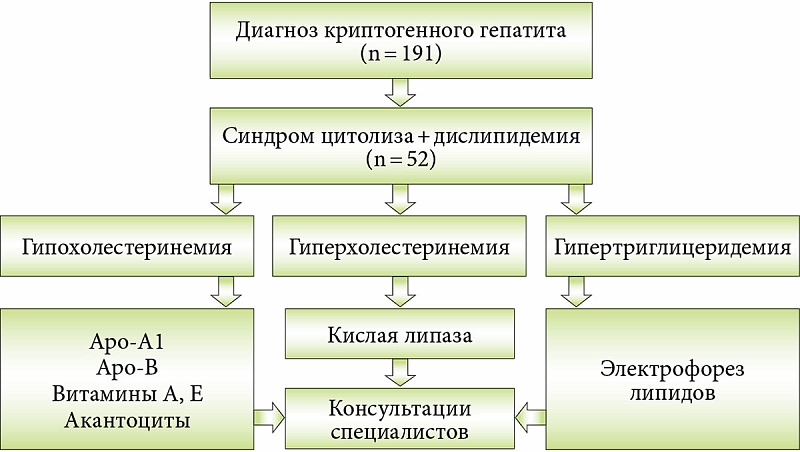
–Ю–њ—А–µ–і–µ–ї–Є—В—М –љ–∞–ї–Є—З–Є–µ –і–Є—Б–ї–Є–њ–Є–і–µ–Љ–Є–Є —Г¬†–і–µ—В–µ–є —Б¬†–Ї—А–Є–њ—В–Њ–≥–µ–љ–љ—Л–Љ¬†–≥–µ–њ–∞—В–Є—В–Њ–Љ –њ–Њ–Ј–≤–Њ–ї—П–µ—В –і–Є–∞–≥–љ–Њ—Б—В–Є—З–µ—Б–Ї–Є–є –∞–ї–≥–Њ—А–Є—В–Љ. –Ф–Њ–Ї–ї–∞–і—З–Є–Ї –њ—А–Є–≤–µ–ї–∞ –і–∞–љ–љ—Л–µ —Б–Њ–±—Б—В–≤–µ–љ–љ–Њ–≥–Њ –љ–∞–±–ї—О–і–µ–љ–Є—П –Ј–∞ 191 —А–µ–±–µ–љ–Ї–Њ–Љ —Б¬†–і–Є–∞–≥–љ–Њ–Ј–Њ–Љ —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–≥–Њ –Ї—А–Є–њ—В–Њ–≥–µ–љ–љ–Њ–≥–Њ¬†–≥–µ–њ–∞—В–Є—В–∞. –£¬†52 (27,2%) –Є–Ј¬†–љ–Є—Е —Б–Є–љ–і—А–Њ–Љ —Ж–Є—В–Њ–ї–Є–Ј–∞ —Б–Њ—З–µ—В–∞–ї—Б—П —Б¬†–і–Є—Б–ї–Є–њ–Є–і–µ–Љ–Є–µ–є. –Т¬†—Б–Њ–Њ—В–≤–µ—В—Б—В–≤–Є–Є —Б¬†—А–∞–Ј—А–∞–±–Њ—В–∞–љ–љ—Л–Љ –∞–ї–≥–Њ—А–Є—В–Љ–Њ–Љ –њ–Њ¬†—А–µ–Ј—Г–ї—М—В–∞—В–∞–Љ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П –ї–Є–њ–Є–і–Њ–≥—А–∞–Љ–Љ—Л —Г—Б—В–∞–љ–Њ–≤–ї–µ–љ–Њ –љ–∞–ї–Є—З–Є–µ¬†–≥–Є–њ–Њ—Е–Њ–ї–µ—Б—В–µ—А–Є–љ–µ–Љ–Є–Є,¬†–≥–Є–њ–µ—А—Е–Њ–ї–µ—Б—В–µ—А–Є–љ–µ–Љ–Є–Є –Є–ї–Є¬†–≥–Є–њ–µ—А—В—А–Є–≥–ї–Є—Ж–µ—А–Є–і–µ–Љ–Є–Є. –Я—А–Є –љ–∞–ї–Є—З–Є–Є¬†–≥–Є–њ–Њ—Е–Њ–ї–µ—Б—В–µ—А–Є–љ–µ–Љ–Є–Є –њ—А–Њ–≤–Њ–і–Є–ї–Є –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ —Г—А–Њ–≤–љ—П –Р–њ–Њ-–Р1, –Р–њ–Њ-–Т, —Б–Њ–і–µ—А–ґ–∞–љ–Є—П –≤–Є—В–∞–Љ–Є–љ–Њ–≤ A, –Х –Є¬†–Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ –∞–Ї–∞–љ—В–Њ—Ж–Є—В–Њ–≤, –њ–Њ—Б–Ї–Њ–ї—М–Ї—Г –∞–Ї–∞–љ—В–Њ—Ж–Є—В–Њ–Ј —А–∞–Ј–≤–Є–≤–∞–µ—В—Б—П —Г¬†30% –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–∞–±–µ—В–∞–ї–Є–њ–Њ–њ—А–Њ—В–µ–Є–љ–µ–Љ–Є–µ–є. –Я—А–Є¬†–≥–Є–њ–µ—А—Е–Њ–ї–µ—Б—В–µ—А–Є–љ–µ–Љ–Є–Є –Є–Ј—Г—З–∞–ї–Є –∞–Ї—В–Є–≤–љ–Њ—Б—В—М –Ї–Є—Б–ї–Њ–є –ї–Є–њ–∞–Ј—Л. –Я—А–Є¬†–≥–Є–њ–µ—А—В—А–Є–≥–ї–Є—Ж–µ—А–Є–і–µ–Љ–Є–Є –љ–µ–Њ–±—Е–Њ–і–Є–Љ—Л–Љ —Б—З–Є—В–∞–ї–Њ—Б—М –њ—А–Њ–≤–µ–і–µ–љ–Є–µ —Н–ї–µ–Ї—В—А–Њ—Д–Њ—А–µ–Ј–∞ –ї–Є–њ–Є–і–Њ–≤. –Т–∞–ґ–љ—Л–є –Љ–Њ–Љ–µ–љ—В: –њ–∞—Ж–Є–µ–љ—В—Л —Б¬†–≤—Л—П–≤–ї–µ–љ–љ–Њ–є –і–Є—Б–ї–Є–њ–Є–і–µ–Љ–Є–µ–є —В—А–µ–±—Г—О—В –Ї–Њ–љ—Б—Г–ї—М—В–∞—Ж–Є–Є —А—П–і–∞ —Б–њ–µ—Ж–Є–∞–ї–Є—Б—В–Њ–≤: –Њ—Д—В–∞–ї—М–Љ–Њ–ї–Њ–≥–∞, –љ–µ–≤—А–Њ–њ–∞—В–Њ–ї–Њ–≥–∞, –Ї–∞—А–і–Є–Њ–ї–Њ–≥–∞ –Є¬†–і—А. (—А–Є—Б.¬†1).
–Ґ–∞–Ї–Є–Љ –Њ–±—А–∞–Ј–Њ–Љ, –Є–Ј¬†52 –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†—Б–Є–љ–і—А–Њ–Љ–Њ–Љ –і–Є—Б–ї–Є–њ–Є–і–µ–Љ–Є–Є 34 —Г–і–∞–ї–Њ—Б—М –њ–Њ—Б—В–∞–≤–Є—В—М —В–Њ—З–љ—Л–є –і–Є–∞–≥–љ–Њ–Ј. –Ф–µ—Д–Є—Ж–Є—В –ї–Є–Ј–Њ—Б–Њ–Љ–љ–Њ–є –Ї–Є—Б–ї–Њ–є –ї–Є–њ–∞–Ј—Л (–Ф–Ы–Ъ–Ы) –Њ–±–љ–∞—А—Г–ґ–µ–љ –≤¬†38% —Б–ї—Г—З–∞–µ–≤,¬†–≥–Є–њ–µ—А—Е–Є–ї–Њ–Љ–Є–Ї—А–Њ–љ–µ–Љ–Є—П¬†вАУ –≤¬†6%, —Б–µ–Љ–µ–є–љ–∞—П¬†–≥–Є–њ–µ—А—Е–Њ–ї–µ—Б—В–µ—А–Є–љ–µ–Љ–Є—П¬†вАУ –≤¬†47%, –±–Њ–ї–µ–Ј–љ—М –Ґ–∞–љ–ґ–µ—А–∞ –Є¬†–∞–±–µ—В–∞–ї–Є–њ–Њ–њ—А–Њ—В–µ–Є–љ–µ–Љ–Є—П¬†вАУ –≤¬†3%.
–Ф–Є–∞–≥–љ–Њ—Б—В–Є–Ї–∞ –±–Њ–ї–µ–Ј–љ–µ–є –љ–∞—А—Г—И–µ–љ–Є–є –ї–Є–њ–Є–і–љ–Њ–≥–Њ –Њ–±–Љ–µ–љ–∞ –Њ—Б–љ–Њ–≤–∞–љ–∞ –љ–∞¬†—А–µ–Ј—Г–ї—М—В–∞—В–∞—Е –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Њ–≥–Њ –Є¬†–њ–∞—А–∞¬≠–Ї–ї–Є–љ–Є—З–µ—Б–Ї–Њ–≥–Њ –Њ–±—Б–ї–µ–і–Њ–≤–∞–љ–Є—П, –∞¬†—В–∞–Ї–ґ–µ –і–∞–љ–љ—Л—Е —Б–µ–Љ–µ–є–љ–Њ–≥–Њ –∞–љ–∞–Љ–љ–µ–Ј–∞. –Ю–±—А–∞—В–Є—В–µ –≤–љ–Є–Љ–∞–љ–Є–µ: –љ–∞—Б–ї–µ–і—Б—В–≤–µ–љ–љ—Л–µ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П –Љ–Њ–≥—Г—В –њ—А–Њ—В–µ–Ї–∞—В—М –њ–Њ¬†–∞–љ–∞–ї–Њ–≥–Є–Є —Б¬†–љ–µ–љ–∞—Б–ї–µ–і—Б—В–≤–µ–љ–љ—Л–Љ–Є. –Э–µ—А–µ–і–Ї–Њ –љ–∞—Б–ї–µ–і—Б—В–≤–µ–љ–љ–Њ–µ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–µ —Г¬†–і–µ—В–µ–є —Б–Њ–њ—Г—В—Б—В–≤—Г–µ—В –Њ—Б–љ–Њ–≤–љ–Њ–Љ—Г, –љ–µ–љ–∞—Б–ї–µ–і—Б—В–≤–µ–љ–љ–Њ–Љ—Г –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—О, —Б¬†–Ї–Њ—В–Њ—А—Л–Љ —А–Њ–і–Є—В–µ–ї–Є —А–µ–±–µ–љ–Ї–∞ –Њ–±—А–∞—В–Є–ї–Є—Б—М –Ј–∞ –Љ–µ–і–Є—Ж–Є–љ—Б–Ї–Њ–є –њ–Њ–Љ–Њ—Й—М—О. –Я–Њ—Н—В–Њ–Љ—Г –≤—Л—П–≤–ї–µ–љ–љ—Л–µ –љ–∞—А—Г—И–µ–љ–Є—П –ї–Є–њ–Є–і–љ–Њ–≥–Њ –Њ–±–Љ–µ–љ–∞ —В—А–µ–±—Г—О—В –Њ–±—П–Ј–∞—В–µ–ї—М–љ–Њ–≥–Њ –і–Њ–њ–Њ–ї–љ–Є—В–µ–ї—М–љ–Њ–≥–Њ –Њ–±—Б–ї–µ–і–Њ–≤–∞–љ–Є—П —Б¬†—Ж–µ–ї—М—О —Б–≤–Њ–µ–≤—А–µ–Љ–µ–љ–љ–Њ–є –њ–Њ—Б—В–∞–љ–Њ–≤–Ї–Є –і–Є–∞–≥–љ–Њ–Ј–∞ –Є¬†–љ–∞—З–∞–ї–∞ —Н—В–Є–Њ–њ–∞—В–Њ–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Њ–є —В–µ—А–∞–њ–Є–Є –і–ї—П –њ—А–µ–і—Г–њ—А–µ–ґ–і–µ–љ–Є—П –Њ—Б–ї–Њ–ґ–љ–µ–љ–Є–є –і–Є—Б–ї–Є–њ–Є–і–µ–Љ–Є–є. –Т–Љ–µ—Б—В–µ —Б¬†—В–µ–Љ —Б–ї–µ–і—Г–µ—В –њ–Њ–Љ–љ–Є—В—М, —З—В–Њ –і–Є—Б–ї–Є–њ–Є–і–µ–Љ–Є—П –љ–µ—А–µ–і–Ї–Њ –њ—А–Є–≤–Њ–і–Є—В –Ї¬†–≤—Л—А–∞–ґ–µ–љ–љ–Њ–Љ—Г —Е–Є–ї–µ–Ј—Г —Б—Л–≤–Њ—А–Њ—В–Ї–Є –Є¬†–Є—Б–Ї–∞–ґ–µ–љ–Є—О –ї–∞–±–Њ—А–∞—В–Њ—А–љ—Л—Е —А–µ–Ј—Г–ї—М—В–∞—В–Њ–≤ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П.
–Ъ–ї–Є–љ–Є—З–µ—Б–Ї–Є–є —Б–ї—Г—З–∞–є
–Ь–∞–ї—М—З–Є–Ї 2002¬†–≥–Њ–і–∞ —А–Њ–ґ–і–µ–љ–Є—П –Њ—В¬†—В—А–µ—В—М–µ–є –љ–Њ—А–Љ–∞–ї—М–љ–Њ –њ—А–Њ—В–µ–Ї–∞–≤—И–µ–є –±–µ—А–µ–Љ–µ–љ–љ–Њ—Б—В–Є (—Б—В–∞—А—И–Є–µ –і–µ—В–Є –Ј–і–Њ—А–Њ–≤—Л). –Э–∞—Б–ї–µ–і—Б—В–≤–µ–љ–љ–Њ—Б—В—М –њ–Њ¬†–Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П–Љ –ї–Є–њ–Є–і–љ–Њ–≥–Њ –Њ–±–Љ–µ–љ–∞ –љ–µ¬†–Њ—В—П–≥–Њ—Й–µ–љ–∞. –Т–µ—Б –њ—А–Є —А–Њ–ґ–і–µ–љ–Є–Є¬†вАУ 4900¬†–≥,¬†—А–Њ—Б—В¬†вАУ 56 —Б–Љ. –Я—А–Њ—Д–Є–ї–∞–Ї—В–Є—З–µ—Б–Ї–Є–µ –њ—А–Є–≤–Є–≤–Ї–Є –њ—А–Њ–≤–µ–і–µ–љ—Л —Б–Њ–≥–ї–∞—Б–љ–Њ –Э–∞—Ж–Є–Њ–љ–∞–ї—М–љ–Њ–Љ—Г –Ї–∞–ї–µ–љ–і–∞—А—О –≤–∞–Ї—Ж–Є–љ–∞—Ж–Є–Є. –Я—Б–Є—Е–Њ–Љ–Њ—В–Њ—А–љ–Њ–µ —А–∞–Ј–≤–Є—В–Є–µ —Б–Њ–Њ—В–≤–µ—В—Б—В–≤—Г–µ—В –≤–Њ–Ј—А–∞—Б—В—Г. –Э–∞—Е–Њ–і–Є–ї—Б—П –љ–∞¬†–≥—А—Г–і–љ–Њ–Љ –≤—Б–Ї–∞—А–Љ–ї–Є–≤–∞–љ–Є–Є –і–Њ –≤–Њ—Б—М–Љ–Є –Љ–µ—Б—П—Ж–µ–≤. –°¬†—В—А–µ—Е–Љ–µ—Б—П—З–љ–Њ–≥–Њ –≤–Њ–Ј—А–∞—Б—В–∞ –Є–Љ–µ–ї–Є –Љ–µ—Б—В–Њ –њ—А–Њ—П–≤–ї–µ–љ–Є—П –∞—В–Њ–њ–Є—З–µ—Б–Ї–Њ–≥–Њ –і–µ—А–Љ–∞—В–Є—В–∞. –Т¬†–љ–∞—Б—В–Њ—П—Й–µ–µ –≤—А–µ–Љ—П —Г¬†—А–µ–±–µ–љ–Ї–∞ –љ–∞–±–ї—О–і–∞—О—В—Б—П –±—А–Њ–љ—Е–Є–∞–ї—М–љ–∞—П –∞—Б—В–Љ–∞, –Ї—А—Г–≥–ї–Њ–≥–Њ–і–Є—З–љ—Л–є –∞–ї–ї–µ—А–≥–Є—З–µ—Б–Ї–Є–є —А–Є–љ–Є—В.
–Т¬†–≤–Њ–Ј—А–∞—Б—В–µ —З–µ—В—Л—А–µ—Е –ї–µ—В –≤¬†–Љ–∞–µ 2006¬†–≥.¬†–њ—А–Є –њ–Њ–і–≥–Њ—В–Њ–≤–Ї–µ –Ї¬†–∞–і–µ–љ–Њ—В–Њ–Љ–Є–Є —Г¬†—А–µ–±–µ–љ–Ї–∞ –≤—Л—П–≤–ї–µ–љ—Л —Б–Є–љ–і—А–Њ–Љ —Ж–Є—В–Њ–ї–Є–Ј–∞ (—Г—А–Њ–≤–µ–љ—М –∞–ї–∞–љ–Є–љ–∞–Љ–Є–љ–Њ—В—А–∞–љ—Б—Д–µ—А–∞–Ј—Л, –∞—Б–њ–∞—А—В–∞—В–∞–Љ–Є–љ–Њ—В—А–∞–љ—Б—Д–µ—А–∞–Ј—Л¬†вАУ 67 –Х–і/–ї), –њ–Њ–≤—Л—И–µ–љ–љ–Њ–µ —Б–Њ–і–µ—А–ґ–∞–љ–Є–µ —В—А–Є–≥–ї–Є—Ж–µ—А–Є–і–Њ–≤, —Е–Њ–ї–µ—Б—В–µ—А–Є–љ–∞, –Ы–Я–Э–Я –≤¬†–Ї—А–Њ–≤–Є. –Я—А–Є –њ—А–Њ–≤–µ–і–µ–љ–Є–Є —Г–ї—М—В—А–∞–Ј–≤—Г–Ї–Њ–≤–Њ–≥–Њ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П (–£–Ч–Ш) –±—А—О—И–љ–Њ–є –њ–Њ–ї–Њ—Б—В–Є –≤—Л—П–≤–ї–µ–љ—Л¬†–≥–µ–њ–∞—В–Њ–Љ–µ–≥–∞–ї–Є—П –Є¬†–і–Є—Д—Д—Г–Ј–љ—Л–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П –њ–∞—А–µ–љ—Е–Є–Љ—Л –њ–µ—З–µ–љ–Є. –Ш—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ –Љ–∞—А–Ї–µ—А–Њ–≤ –≤–Є—А—Г—Б–љ—Л—Е¬†–≥–µ–њ–∞—В–Є—В–Њ–≤ –і–∞–ї–Њ –Њ—В—А–Є—Ж–∞—В–µ–ї—М–љ—Л–є —А–µ–Ј—Г–ї—М—В–∞—В. –Я–Њ—Б–ї–µ –Ї–Њ–љ—Б—Г–ї—М—В–∞—Ж–Є–Є¬†–≥–µ–љ–µ—В–Є–Ї–∞ –њ–Њ—Б—В–∞–≤–ї–µ–љ –њ—А–µ–і–≤–∞—А–Є—В–µ–ї—М–љ—Л–є –і–Є–∞–≥–љ–Њ–Ј —Б–µ–Љ–µ–є–љ–Њ–є¬†–≥–Є–њ–µ—А—Е–Њ–ї–µ—Б—В–µ—А–Є–љ–µ–Љ–Є–Є —В–Є–њ–∞ II–∞. –Т¬†–∞–њ—А–µ–ї–µ 2007¬†–≥.¬†–њ—А–Є –Њ–њ—А–µ–і–µ–ї–µ–љ–Є–Є¬†–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Є—Е –њ–Њ–ї–Є–Љ–Њ—А—Д–Є–Ј–Љ–Њ–≤ —Г—Б—В–∞–љ–Њ–≤–ї–µ–љ–Њ –љ–∞–ї–Є—З–Є–µ –Љ–Њ–љ–Њ–Ј–Є–≥–Њ—В–љ–Њ–≥–Њ –≤–∞—А–Є–∞–љ—В–∞ –ї–Є–њ–Њ–њ—А–Њ—В–µ–Є–љ–Њ–≤–Њ–є –ї–Є–њ–∞–Ј—Л —Б¬†–њ–Њ–≤—Л—И–µ–љ–љ—Л–Љ —Г—А–Њ–≤–љ–µ–Љ —В—А–Є–≥–ї–Є—Ж–µ—А–Є–і–Њ–≤, —Б–љ–Є–ґ–µ–љ–љ—Л–Љ —Г—А–Њ–≤–љ–µ–Љ –Ы–Я–Т–Я –≤¬†–Ї—А–Њ–≤–Є, —З—В–Њ —Б–≤–Є–і–µ—В–µ–ї—М—Б—В–≤–Њ–≤–∞–ї–Њ –Њ¬†–њ–Њ–≤—Л—И–µ–љ–љ–Њ–Љ —А–Є—Б–Ї–µ —А–∞–Ј–≤–Є—В–Є—П –∞—В–µ—А–Њ—Б–Ї–ї–µ—А–Њ—В–Є—З–µ—Б–Ї–Є—Е –њ–Њ—А–∞–ґ–µ–љ–Є–є —Б–Њ—Б—Г–і–Њ–≤. –Т¬†—В–Њ–Љ –ґ–µ¬†–≥–Њ–і—Г –њ—А–Њ–≤–µ–і–µ–љ—Л¬†–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Є–µ –∞–љ–∞–ї–Є–Ј—Л.
–Т¬†–Ї–ї–Є–љ–Є–Ї—Г –§–Ш–¶ –њ–Є—В–∞–љ–Є—П –Є¬†–±–Є–Њ—В–µ—Е–љ–Њ–ї–Њ–≥–Є–Є —А–µ–±–µ–љ–Њ–Ї –њ–Њ—Б—В—Г–њ–Є–ї —Б¬†–і–Є–∞–≥–љ–Њ–Ј–Њ–Љ —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–≥–Њ –Ї—А–Є–њ—В–Њ–≥–µ–љ–љ–Њ–≥–Њ¬†–≥–µ–њ–∞—В–Є—В–∞. –£¬†–љ–µ–≥–Њ –Њ–њ—А–µ–і–µ–ї—П–ї–∞—Б—М¬†–≥–µ–њ–∞—В–Њ–Љ–µ–≥–∞–ї–Є—П: –њ–µ—З–µ–љ—М –≤—Л—Б—В—Г–њ–∞–ї–∞ –Є–Ј-–њ–Њ–і –Ї—А–∞—П —А–µ–±–µ—А–љ–Њ–є –і—Г–≥–Є –љ–∞¬†4 —Б–Љ, —З—В–Њ –њ–Њ–і—В–≤–µ—А–ґ–і–∞–ї–Њ—Б—М –і–∞–љ–љ—Л–Љ–Є –£–Ч–Ш. –Я—А–Є –љ–µ–Њ–і–љ–Њ–Ї—А–∞—В–љ–Њ–Љ –Њ–±—Б–ї–µ–і–Њ–≤–∞–љ–Є–Є —Г¬†–Љ–∞–ї—М—З–Є–Ї–∞ —А–µ–≥–Є—Б—В—А–Є—А–Њ–≤–∞–ї–Є—Б—М —Б–љ–Є–ґ–µ–љ–љ—Л–є —Г—А–Њ–≤–µ–љ—М –∞–ї—М—Д–∞-1-–∞–љ—В–Є—В—А–Є–њ—Б–Є–љ–∞ (–љ–∞¬†30% –Њ—В¬†–љ–Њ—А–Љ—Л), –њ–Њ–≤—Л—И–µ–љ–љ–Њ–µ —Б–Њ–і–µ—А–ґ–∞–љ–Є–µ —Д–µ—А—А–Є—В–Є–љ–∞ (646 –Љ–≥/–ї). –Я–µ—А–Є–Њ–і–Є—З–µ—Б–Ї–Є –Њ—В–Љ–µ—З–∞–ї–Є—Б—М¬†–≥–Є–њ–Њ–≥–ї–Є–Ї–µ–Љ–Є—П (3,3вАУ2,8 –Љ–Љ–Њ–ї—М/–ї), –Ї–Њ—В–Њ—А–∞—П –Њ–њ—А–µ–і–µ–ї—П–ї–∞—Б—М –њ—А–Є –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–Є –±–Є–Њ—Е–Є–Љ–Є—З–µ—Б–Ї–Њ–≥–Њ –∞–љ–∞–ї–Є–Ј–∞ –Ї—А–Њ–≤–Є –Є¬†–≤ –њ—А–Њ–±–µ —Б¬†–љ–∞–≥—А—Г–Ј–Ї–Њ–є¬†–≥–ї—О–Ї–Њ–Ј–Њ–є, –ї–∞–Ї—В–∞—В-–∞—Ж–Є–і–Њ–Ј. –£¬†–њ–∞—Ж–Є–µ–љ—В–∞ –њ—А–Њ–≤–Њ–і–Є–ї—Б—П –і–Є—Д—Д–µ—А–µ–љ—Ж–Є–∞–ї—М–љ—Л–є –і–Є–∞–≥–љ–Њ–Ј –Љ–µ–ґ–і—Г –і–µ—Д–Є—Ж–Є—В–Њ–Љ –∞–ї—М—Д–∞-1-–∞–љ—В–Є—В—А–Є–њ—Б–Є–љ–∞, –±–Њ–ї–µ–Ј–љ—П–Љ–Є –љ–∞–Ї–Њ–њ–ї–µ–љ–Є—П¬†–≥–ї–Є–Ї–Њ–≥–µ–љ–∞,¬†–≥–µ–Љ–Њ—Е—А–Њ–Љ–∞—В–Њ–Ј–Њ–Љ, –љ–∞—Б–ї–µ–і—Б—В–≤–µ–љ–љ—Л–Љ–Є –љ–∞—А—Г—И–µ–љ–Є—П–Љ–Є –ї–Є–њ–Є–і–љ–Њ–≥–Њ –Њ–±–Љ–µ–љ–∞.
–Я—А–Є –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–Є¬†–≥–µ–љ–∞ –∞–ї—М—Д–∞-1-–∞–љ—В–Є—В—А–Є–њ—Б–Є–љ–∞ –±—Л–ї–∞ –Њ–±–љ–∞—А—Г–ґ–µ–љ–∞ –Љ—Г—В–∞—Ж–Є—П –Х34–Ъ (Z-–∞–ї–ї–µ–ї—М) –≤¬†–≥–µ—В–µ—А–Њ–Ј–Є–≥–Њ—В–љ–Њ–Љ —Б–Њ—Б—В–Њ—П–љ–Є–Є, —Е–∞—А–∞–Ї—В–µ—А–љ–∞—П –і–ї—П —Д–µ–љ–Њ—В–Є–њ–∞ PiMZ (—А–Є—Б–Ї¬†—А–∞–Ј–≤–Є—В–Є—П –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П¬†вАУ 20%). –Я—А–Є –і–∞–ї—М–љ–µ–є—И–Є—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П—Е —Г—А–Њ–≤–µ–љ—М –∞–ї—М—Д–∞-1-–∞–љ—В–Є—В—А–Є–њ—Б–Є–љ–∞ –љ–Њ—А–Љ–∞–ї–Є–Ј–Њ–≤–∞–ї—Б—П –Є¬†–њ—А–Є¬†–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Њ–Љ –∞–љ–∞–ї–Є–Ј–µ –≤¬†–∞–ї—М—В–µ—А–љ–∞—В–Є–≤–љ–Њ–є –ї–∞–±–Њ—А–∞—В–Њ—А–Є–Є –≤¬†–≥–µ–љ–µ –∞–ї—М—Д–∞-1-–∞–љ—В–Є—В—А–Є–њ—Б–Є–љ–∞ –љ–µ¬†–Њ–±–љ–∞—А—Г–ґ–µ–љ–Њ –Љ—Г—В–∞—Ж–Є–є (—Д–µ–љ–Њ—В–Є–њ PiMM¬†вАУ –љ–Њ—А–Љ–∞–ї—М–љ–∞—П –∞–Ї—В–Є–≤–љ–Њ—Б—В—М –∞–ї—М—Д–∞-1-–∞–љ—В–Є—В—А–Є–њ—Б–Є–љ–∞). –†–µ–Ј—Г–ї—М—В–∞—В—Л –Љ–Њ–ї–µ–Ї—Г–ї—П—А–љ–Њ-–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Њ–≥–Њ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П¬†–≥–µ–љ–∞ HFE –љ–∞¬†–≤—Л—П–≤–ї–µ–љ–Є–µ —З–∞—Б—В—Л—Е –Љ—Г—В–∞—Ж–Є–є, –Њ—В–≤–µ—В—Б—В–≤–µ–љ–љ—Л—Е –Ј–∞ —А–∞–Ј–≤–Є—В–Є–µ¬†–≥–µ–Љ–Њ—Е—А–Њ–Љ–∞—В–Њ–Ј–∞ (C282Y, H63D), –љ–µ¬†–њ–Њ–і—В–≤–µ—А–і–Є–ї–Є –Є—Е –љ–∞–ї–Є—З–Є—П.
–°¬†—Ж–µ–ї—М—О –≤–µ—А–Є—Д–Є–Ї–∞—Ж–Є–Є –і–Є–∞–≥–љ–Њ–Ј–∞, –∞¬†—В–∞–Ї–ґ–µ –Њ–њ—А–µ–і–µ–ї–µ–љ–Є—П —Б—В–∞–і–Є–Є –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П –≤¬†—Б–µ–љ—В—П–±—А–µ 2010¬†–≥.¬†—А–µ–±–µ–љ–Ї—Г –њ—А–Њ–≤–µ–ї–Є –њ—Г–љ–Ї—Ж–Є–Њ–љ–љ—Г—О –±–Є–Њ–њ—Б–Є—О –њ–µ—З–µ–љ–Є. –Ш—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ –±–Є–Њ–њ—В–∞—В–∞ –њ–µ—З–µ–љ–Є –њ–Њ–Ї–∞–Ј–∞–ї–Њ —Г–Љ–µ—А–µ–љ–љ—Л–є —Д–Є–±—А–Њ–Ј –њ–Њ—А—В–∞–ї—М–љ—Л—Е —В—А–∞–Ї—В–Њ–≤, —Д–Є–±—А–Њ–Ј —Б—В–µ–љ–Њ–Ї —Ж–µ–љ—В—А–∞–ї—М–љ—Л—Е –≤–µ–љ, –љ–∞–ї–Є—З–Є–µ –њ–Њ—А—В–Њ-–њ–Њ—А—В–∞–ї—М–љ—Л—Е —Б–µ–њ—В –Є¬†–Ј–Њ–љ—Л –њ–µ—А–Є–≥–µ–њ–∞—В–Њ—Ж–µ–ї–ї—О–ї—П—А–љ–Њ–≥–Њ —Д–Є–±—А–Њ–Ј–∞. –Т¬†–≥–µ–њ–∞—В–Њ—Ж–Є—В–∞—Е –Њ–Ї—А—Г–≥–ї–Њ–є —Д–Њ—А–Љ—Л —Б–Њ¬†—Б–≤–µ—В–ї–Њ–є —Ж–Є—В–Њ–њ–ї–∞–Ј–Љ–Њ–є –Њ–њ—А–µ–і–µ–ї—П–ї–Є—Б—М –Љ–љ–Њ–ґ–µ—Б—В–≤–µ–љ–љ—Л–µ –Љ–µ–ї–Ї–Є–µ –≤–∞–Ї—Г–Њ–ї–Є, —З—В–Њ —Г–Ї–∞–Ј—Л–≤–∞–ї–Њ –љ–∞¬†–Љ–Є–Ї—А–Њ–≤–µ–Ј–Є–Ї—Г–ї—П—А–љ—Л–є —Б—В–µ–∞—В–Њ–Ј. –°–Њ–≥–ї–∞—Б–љ–Њ –Љ–Њ—А—Д–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–Љ—Г –Ј–∞–Ї–ї—О—З–µ–љ–Є—О, –њ—А–Є–Ј–љ–∞–Ї–Њ–≤, —Е–∞—А–∞–Ї—В–µ—А–љ—Л—Е –і–ї—П –і–µ—Д–Є—Ж–Є—В–∞ –∞–ї—М—Д–∞-1-–∞–љ—В–Є—В—А–Є–њ—Б–Є–љ–∞, –±–Њ–ї–µ–Ј–љ–µ–є –љ–∞–Ї–Њ–њ–ї–µ–љ–Є—П¬†–≥–ї–Є–Ї–Њ–≥–µ–љ–∞, –љ–µ—В. –Ш–љ–і–µ–Ї—Б —Б–Ї–ї–µ—А–Њ–Ј–∞ –њ–Њ¬†Desmet¬†вАУ 2вАУ3 –±–∞–ї–ї–∞ (—Г–Љ–µ—А–µ–љ–љ—Л–є).
–†–∞–љ–µ–µ –≤—Л—П–≤–ї–µ–љ–љ—Л–µ —Г¬†–њ–∞—Ж–Є–µ–љ—В–∞ —Г—Б—В–Њ–є—З–Є–≤–Њ–µ –њ–Њ–≤—Л—И–µ–љ–Є–µ —Г—А–Њ–≤–љ–µ–є —В—А–∞–љ—Б–∞–Љ–Є–љ–∞–Ј, —В—А–Є–≥–ї–Є—Ж–µ—А–Є–і–Њ–≤, —Е–Њ–ї–µ—Б—В–µ—А–Є–љ–∞ –Є¬†–Ы–Я–Э–Я, –њ—А–Њ–≥—А–µ—Б—Б–Є—А—Г—О—Й–∞—П¬†–≥–µ–њ–∞—В–Њ–Љ–µ–≥–∞–ї–Є—П —Б¬†–њ—А–Є–Ј–љ–∞–Ї–∞–Љ–Є —Д–Є–±—А–Њ–Ј–∞, –∞¬†—В–∞–Ї–ґ–µ –Љ–Є–Ї—А–Њ–≤–µ–Ј–Є–Ї—Г–ї—П—А–љ—Л–є —Б—В–µ–∞—В–Њ–Ј¬†–≥–µ–њ–∞—В–Њ—Ж–Є—В–Њ–≤¬†–≥–Њ–≤–Њ—А–Є–ї–Є –Њ¬†–≤—Л—Б–Њ–Ї–Њ–є –≤–µ—А–Њ—П—В–љ–Њ—Б—В–Є –љ–∞–ї–Є—З–Є—П –±–Њ–ї–µ–Ј–љ–Є –љ–∞–Ї–Њ–њ–ї–µ–љ–Є—П —Н—Д–Є—А–Њ–≤ —Е–Њ–ї–µ—Б—В–µ—А–Є–љ–∞ (–Њ–і–љ–∞ –Є–Ј¬†—Д–µ–љ–Њ—В–Є–њ–Є—З–µ—Б–Ї–Є—Е —Д–Њ—А–Љ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П, –Ї–Њ—В–Њ—А—Л–µ –≤¬†—Б–Њ–≤—А–µ–Љ–µ–љ–љ–Њ–є –ї–Є—В–µ—А–∞—В—Г—А–µ –Њ–±—К–µ–і–Є–љ–µ–љ—Л –њ–Њ–і –љ–∞–Ј–≤–∞–љ–Є–µ–Љ –Ф–Ы–Ъ–Ы).
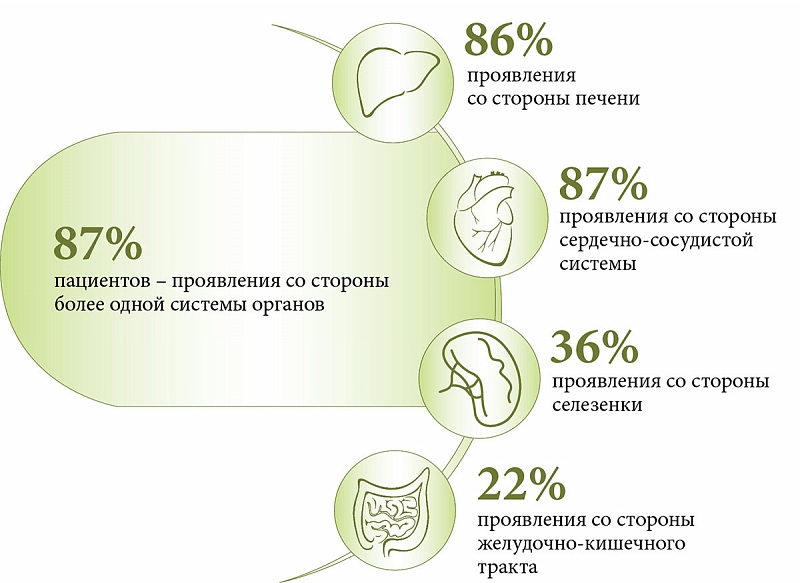
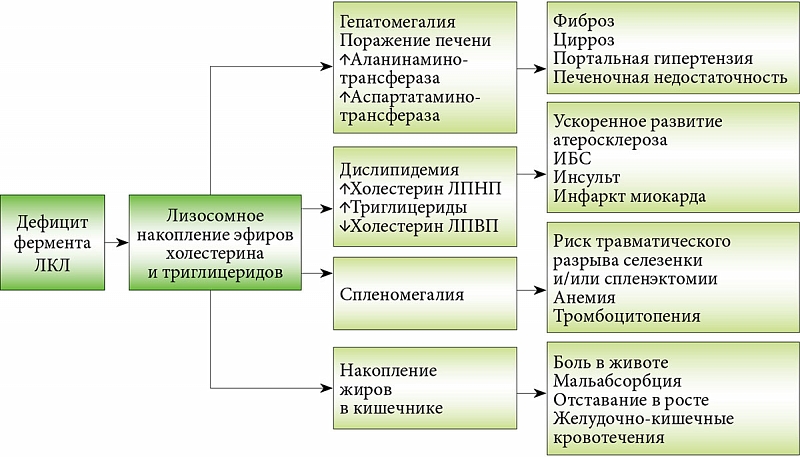
–Ф–Ы–Ъ–Ы –њ—А–µ–і—Б—В–∞–≤–ї—П–µ—В —Б–Њ–±–Њ–є –Љ–Њ–љ–Њ–≥–µ–љ–љ–Њ–µ –љ–∞—А—Г—И–µ–љ–Є–µ, –њ—А–Є –Ї–Њ—В–Њ—А–Њ–Љ –Љ—Г—В–∞—Ж–Є–Є –≤—Л–Ј—Л–≤–∞—О—В –і–µ—Д–Є—Ж–Є—В –Ы–Ъ–Ы, —З—В–Њ —Б–њ–Њ—Б–Њ–±—Б—В–≤—Г–µ—В –ї–Є–Ј–Њ—Б–Њ–Љ–љ–Њ–Љ—Г –љ–∞–Ї–Њ–њ–ї–µ–љ–Є—О —Н—Д–Є—А–Њ–≤ —Е–Њ–ї–µ—Б—В–µ—А–Є–љ–∞ –Є¬†—В—А–Є–≥–ї–Є—Ж–µ—А–Є–і–Њ–≤. –°–Є—Б—В–µ–Љ–љ–Њ–µ –ї–Є–Ј–Њ—Б–Њ–Љ–љ–Њ–µ –љ–∞–Ї–Њ–њ–ї–µ–љ–Є–µ –ї–Є–њ–Є–і–Њ–≤ –њ—А–Є–≤–Њ–і–Є—В –Ї¬†–њ—А–Њ–≥—А–µ—Б—Б–Є—А—Г—О—Й–µ–Љ—Г –њ–Њ—А–∞–ґ–µ–љ–Є—О —А–∞–Ј–ї–Є—З–љ—Л—Е –Њ—А–≥–∞–љ–Њ–≤. –Я–Њ¬†–і–∞–љ–љ—Л–Љ –ї–Є—В–µ—А–∞—В—Г—А—Л, —Б—А–µ–і–Є –і–µ—В–µ–є –Є¬†–≤–Ј—А–Њ—Б–ї—Л—Е —Б¬†–Ф–Ы–Ъ–Ы –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є–µ –њ—А–Њ—П–≤–ї–µ–љ–Є—П —Б–Њ¬†—Б—В–Њ—А–Њ–љ—Л –±–Њ–ї–µ–µ –Њ–і–љ–Њ–є —Б–Є—Б—В–µ–Љ—Л –Њ—А–≥–∞–љ–Њ–≤ –љ–∞–±–ї—О–і–∞—О—В—Б—П –≤¬†87% —Б–ї—Г—З–∞–µ–≤ (—А–Є—Б.¬†2 –Є¬†3).
–Т¬†–Ь–µ–і–Є–Ї–Њ-–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Њ–Љ –љ–∞—Г—З–љ–Њ–Љ —Ж–µ–љ—В—А–µ (–Ь–У–Э–¶) –†–Р–Э –≤–њ–µ—А–≤—Л–µ –≤¬†–†–Њ—Б—Б–Є–Є –±—Л–ї–Њ –љ–∞–ї–∞–ґ–µ–љ–Њ –Є–Ј–Љ–µ—А–µ–љ–Є–µ –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є –њ—Г–ї–∞ –ї–Є–њ–∞–Ј –≤¬†–ї–µ–є–Ї–Њ—Ж–Є—В–∞—Е –Ї—А–Њ–≤–Є –і–ї—П –і–Є–∞–≥–љ–Њ—Б—В–Є–Ї–Є –Ф–Ы–Ъ–Ы. –Ф–∞–љ–љ—Л–є –Љ–µ—В–Њ–і –њ–Њ–Ј–≤–Њ–ї—П–µ—В –Њ–њ—А–µ–і–µ–ї—П—В—М –Њ–±—Й—Г—О –ї–Є–њ–∞–Ј–љ—Г—О –∞–Ї—В–Є–≤–љ–Њ—Б—В—М –≤¬†–ї–µ–є–Ї–Њ—Ж–Є—В–∞—Е –њ–µ—А–Є—Д–µ—А–Є—З–µ—Б–Ї–Њ–є –Ї—А–Њ–≤–Є, –≤¬†—В–Њ–Љ —З–Є—Б–ї–µ –∞–Ї—В–Є–≤–љ–Њ—Б—В—М –Ы–Ъ–Ы. –Э–µ¬†—Б–ї—Г—З–∞–є–љ–Њ –Њ–±—А–∞–Ј–µ—Ж –Ї—А–Њ–≤–Є –њ–∞—Ж–Є–µ–љ—В–∞ –±—Л–ї –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ –Є–Љ–µ–љ–љ–Њ –≤¬†—Н—В–Њ–є –ї–∞–±–Њ—А–∞—В–Њ—А–Є–Є. –Я–Њ–Ї–∞–Ј–∞—В–µ–ї—М –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є —Б–Њ—Б—В–∞–≤–Є–ї 43,8 –љ–Ь/–Љ–≥/—З, —З—В–Њ –њ—А–µ–≤—Л—И–∞–ї–Њ –љ–Є–ґ–љ—О—О¬†–≥—А–∞–љ–Є—Ж—Г –љ–Њ—А–Љ—Л (30вАУ118 –љ–Ь/–Љ–≥/—З).
–Э–µ—Б–Љ–Њ—В—А—П –љ–∞¬†–±–∞–Ј–Є—Б–љ—Г—О —В–µ—А–∞–њ–Є—О –Є¬†—Б—В—А–Њ–≥–Њ–µ —Б–Њ–±–ї—О–і–µ–љ–Є–µ –і–Є–µ—В—Л, –њ—А–Є –Њ—З–µ—А–µ–і–љ—Л—Е –Њ–±—Б–ї–µ–і–Њ–≤–∞–љ–Є—П—Е —А–µ–±–µ–љ–Ї–∞ –Ј–∞—Д–Є–Ї—Б–Є—А–Њ–≤–∞–љ—Л –њ—А–Є–Ј–љ–∞–Ї–Є –љ–∞—А–∞—Б—В–∞–љ–Є—П¬†–≥–µ–њ–∞—В–Њ–Љ–µ–≥–∞–ї–Є–Є (–і–Њ +7 —Б–Љ –Є–Ј-–њ–Њ–і –Ї—А–∞—П —А–µ–±–µ—А–љ–Њ–є –і—Г–≥–Є), —Б–Є–љ–і—А–Њ–Љ–∞ —Ж–Є—В–Њ–ї–Є–Ј–∞ (–і–Њ 180 –Х–і/–ї) –Є¬†–і–Є—Б–ї–Є–њ–Є–і–µ–Љ–Є–Є –Ј–∞ —Б—З–µ—В¬†–≥–Є–њ–µ—А—Е–Њ–ї–µ—Б—В–µ—А–Є–љ–µ–Љ–Є–Є (–і–Њ 9,9 –Љ–Љ–Њ–ї—М/–ї), –њ–Њ–≤—Л—И–µ–љ–Є—П –Ы–Я–Э–Я (–і–Њ 8,14 –Љ–Љ–Њ–ї—М/–ї).
–Я–Њ–Ј–і–љ–µ–µ, —Б¬†–љ–∞–Ї–Њ–њ–ї–µ–љ–Є–µ–Љ –Њ–њ—Л—В–∞ –і–Є–∞–≥–љ–Њ—Б—В–Є–Ї–Є —В–∞–Ї–Њ–≥–Њ —А–µ–і–Ї–Њ–≥–Њ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П, –Ї–∞–Ї –Ф–Ы–Ъ–Ы, –≤—Л—П—Б–љ–Є–ї–Є—Б—М –љ–µ–Ї–Њ—В–Њ—А—Л–µ –Њ—Б–Њ–±–µ–љ–љ–Њ—Б—В–Є –Є–Ј–Љ–µ—А–µ–љ–Є—П –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є –Ы–Ъ–Ы –≤¬†–ї–µ–є–Ї–Њ—Ж–Є—В–∞—Е –њ–µ—А–Є—Д–µ—А–Є—З–µ—Б–Ї–Њ–є –Ї—А–Њ–≤–Є. –Я—А–Є –≤—Л—Б–Њ–Ї–Њ–є –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є –њ—А–Њ—З–Є—Е –ї–Є–њ–∞–Ј –Ї—А–Њ–≤–Є –Є—В–Њ–≥–Њ–≤–∞—П –∞–Ї—В–Є–≤–љ–Њ—Б—В—М –Љ–Њ–ґ–µ—В –Љ–∞—Б–Ї–Є—А–Њ–≤–∞—В—М –∞–Ї—В–Є–≤–љ–Њ—Б—В—М –Ы–Ъ–Ы –≤¬†—Б–ї—Г—З–∞–µ –µ–µ –њ–∞—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–≥–Њ —Б–љ–Є–ґ–µ–љ–Є—П. –Т¬†—В–∞–Ї–Њ–є —Б–Є—В—Г–∞—Ж–Є–Є —Г¬†–њ–∞—Ж–Є–µ–љ—В–∞ —Б¬†–Ф–Ы–Ъ–Ы –Є—В–Њ–≥–Њ–≤–∞—П –∞–Ї—В–Є–≤–љ–Њ—Б—В—М –±—Г–і–µ—В –≤—Л—И–µ –љ–Њ—А–Љ—Л (–ї–Њ–ґ–љ–Њ–Њ—В—А–Є—Ж–∞—В–µ–ї—М–љ—Л–є —А–µ–Ј—Г–ї—М—В–∞—В). –Х—Й–µ –Њ–і–љ–∞ –Њ—Б–Њ–±–µ–љ–љ–Њ—Б—В—М –Љ–µ—В–Њ–і–∞¬†вАУ —З—Г–≤—Б—В–≤–Є—В–µ–ї—М–љ–Њ—Б—В—М –Ї¬†–Ї–∞—З–µ—Б—В–≤—Г –Њ–±—А–∞–Ј—Ж–∞. –Т¬†—А—П–і–µ —Б–ї—Г—З–∞–µ–≤, –љ–∞–њ—А–Є–Љ–µ—А –њ—А–Є –љ–µ—Б–Њ–±–ї—О–і–µ–љ–Є–Є –љ–µ–Њ–±—Е–Њ–і–Є–Љ—Л—Е —Г—Б–ї–Њ–≤–Є–є —В—А–∞–љ—Б–њ–Њ—А—В–Є—А–Њ–≤–Ї–Є, –ї–Є–њ–∞–Ј–љ–∞—П –∞–Ї—В–Є–≤–љ–Њ—Б—В—М –≤¬†–Њ–±—А–∞–Ј—Ж–µ —Б–љ–Є–ґ–∞–µ—В—Б—П, —З—В–Њ –Љ–Њ–ґ–µ—В –і–∞—В—М –ї–Њ–ґ–љ–Њ–њ–Њ–ї–Њ–ґ–Є—В–µ–ї—М–љ—Л–є —А–µ–Ј—Г–ї—М—В–∞—В. –Ґ–∞–Ї–Є–Љ –Њ–±—А–∞–Ј–Њ–Љ, –љ–µ—Б–Љ–Њ—В—А—П –љ–∞¬†–љ–∞–ї–Є—З–Є–µ –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є—Е, –ї–∞–±–Њ—А–∞—В–Њ—А–љ—Л—Е –Є¬†–Љ–Њ—А—Д–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є—Е –і–∞–љ–љ—Л—Е, —Г–Ї–∞–Ј—Л–≤–∞—О—Й–Є—Е –љ–∞¬†–≤—Л—Б–Њ–Ї—Г—О –≤–µ—А–Њ—П—В–љ–Њ—Б—В—М –Ф–Ы–Ъ–Ы, –і–∞–љ–љ—Л–є –і–Є–∞–≥–љ–Њ–Ј –љ–µ¬†–±—Л–ї –Є—Б–Ї–ї—О—З–µ–љ –Є–ї–Є –њ–Њ–і—В–≤–µ—А–ґ–і–µ–љ.
–Т¬†2016¬†–≥.¬†–≤¬†–Ы–∞–±–Њ—А–∞—В–Њ—А–Є–Є –љ–∞—Б–ї–µ–і—Б—В–≤–µ–љ–љ—Л—Е –±–Њ–ї–µ–Ј–љ–µ–є –Њ–±–Љ–µ–љ–∞ –≤–µ—Й–µ—Б—В–≤ –Ь–У–Э–¶ –†–Р–Э –±—Л–ї –≤–љ–µ–і—А–µ–љ —Б–Њ–≤—А–µ–Љ–µ–љ–љ—Л–є –Љ–µ—В–Њ–і –Є–Ј–Љ–µ—А–µ–љ–Є—П —Б–њ–µ—Ж–Є—Д–Є—З–µ—Б–Ї–Њ–є –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є –Ы–Ъ–Ы –≤¬†—Б—Г—Е–Є—Е –њ—П—В–љ–∞—Е –Ї—А–Њ–≤–Є. –Ґ–∞–Ї–Њ–є –Љ–µ—В–Њ–і —Е–∞—А–∞–Ї—В–µ—А–Є–Ј—Г–µ—В—Б—П –Њ—З–µ–љ—М –≤—Л—Б–Њ–Ї–Њ–є —З—Г–≤—Б—В–≤–Є—В–µ–ї—М–љ–Њ—Б—В—М—О, –њ–Њ—Б–Ї–Њ–ї—М–Ї—Г –њ—А–Є–Љ–µ–љ—П–µ—В—Б—П —Б–њ–µ—Ж–Є—Д–Є—З–µ—Б–Ї–Є–є –Є–љ–≥–Є–±–Є—В–Њ—А –Ы–Ъ–Ы. –Ш–љ–≥–Є–±–Є—В–Њ—А –ї–∞–ї–Є—Б—В–∞—В 2 –њ—А–∞–Ї—В–Є—З–µ—Б–Ї–Є –њ–Њ–ї–љ–Њ—Б—В—М—О –Є–љ–≥–Є–±–Є—А—Г–µ—В –∞–Ї—В–Є–≤–љ–Њ—Б—В—М –Ы–Ъ–Ы, –њ—А–Є —Н—В–Њ–Љ –љ–µ¬†–≤–ї–Є—П–µ—В –љ–∞¬†–і—А—Г–≥–Є–µ –ї–Є–њ–∞–Ј–љ—Л–µ –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є –≤¬†—Б—Г—Е–Є—Е –њ—П—В–љ–∞—Е –Ї—А–Њ–≤–Є. –≠—В–Њ –њ–Њ–Ј–≤–Њ–ї—П–µ—В –њ—А–∞–Ї—В–Є—З–µ—Б–Ї–Є –Њ–і–љ–Њ–Ј–љ–∞—З–љ–Њ –Ї–ї–∞—Б—Б–Є—Д–Є—Ж–Є—А–Њ–≤–∞—В—М –Ј–і–Њ—А–Њ–≤—Л—Е –ї—О–і–µ–є, –љ–Њ—Б–Є—В–µ–ї–µ–є –Є¬†–±–Њ–ї—М–љ—Л—Е –Ф–Ы–Ъ–Ы.
–Т¬†—П–љ–≤–∞—А–µ 2016¬†–≥.¬†—А–µ–±–µ–љ–Ї—Г –±—Л–ї–Њ –њ—А–Њ–≤–µ–і–µ–љ–Њ –Њ–њ—А–µ–і–µ–ї–µ–љ–Є–µ –Ы–Ъ–Ы –≤¬†–њ—П—В–љ–µ –Ї—А–Њ–≤–Є. –Т¬†—А–µ–Ј—Г–ї—М—В–∞—В–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П –Њ–±–љ–∞—А—Г–ґ–µ–љ–∞ –Њ—З–µ–љ—М –љ–Є–Ј–Ї–∞—П —Б–њ–µ—Ж–Є—Д–Є—З–µ—Б–Ї–∞—П –∞–Ї—В–Є–≤–љ–Њ—Б—В—М –Ы–Ъ–Ы, —Е–∞—А–∞–Ї—В–µ—А–љ–∞—П —В–Њ–ї—М–Ї–Њ –і–ї—П –Ф–Ы–Ъ–Ы,¬†вАУ 0,02 –љ–Љ–Њ–ї—М/–Љ–≥/—З (–љ–Њ—А–Љ–∞ 0,37вАУ2,3 –љ–Љ–Њ–ї—М/–Љ–≥/—З). –Ф–Є–∞–≥–љ–Њ–Ј –Ф–Ы–Ъ–Ы –±—Л–ї –њ–Њ–і—В–≤–µ—А–ґ–і–µ–љ —Б–њ–µ—Ж–Є—Д–Є—З–µ—Б–Ї–Є–Љ –Љ–µ—В–Њ–і–Њ–Љ –Є–Ј–Љ–µ—А–µ–љ–Є—П –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є –Ы–Ъ–Ы. –Ъ—А–Њ–Љ–µ —В–Њ–≥–Њ, –≤¬†—Е–Њ–і–µ –і–Њ–њ–Њ–ї–љ–Є—В–µ–ї—М–љ—Л—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є –њ–∞—Ж–Є–µ–љ—В—Г –њ—А–Њ–≤–µ–ї–Є —Б–µ–Ї–≤–µ–љ–Є—А–Њ–≤–∞–љ–Є–µ —Н–Ї–Ј–Њ–љ–Њ–≤¬†–≥–µ–љ–∞ LIPA, –Ї–Њ–і–Є—А—Г—О—Й–µ–≥–Њ –Ы–Ъ–Ы. –Т¬†–Ї–Њ–і–Є—А—Г—О—Й–µ–є —З–∞—Б—В–Є¬†–≥–µ–љ–∞ –Љ—Г—В–∞—Ж–Є–Є –≤—Л—П–≤–ї–µ–љ–∞ –Ї–Њ–Љ–њ–∞—Г–љ–і-–≥–µ—В–µ—А–Њ–Ј–Є–≥–Њ—В–љ–∞—П –Љ—Г—В–∞—Ж–Є—П, –≤¬†—Н–Ї–Ј–Њ–љ–µ 8¬†вАУ –Љ—Г—В–∞—Ж–Є—П c.894G>A –≤¬†–≥–µ—В–µ—А–Њ–Ј–Є–≥–Њ—В–љ–Њ–Љ —Б–Њ—Б—В–Њ—П–љ–Є–Є, –њ—А–Є–≤–Њ–і—П—Й–∞—П –Ї¬†–љ–∞—А—Г—И–µ–љ–Є—О —Б–њ–ї–∞–є—Б–Є–љ–≥–∞. –Ь—Г—В–∞—Ж–Є—П –Њ–њ–Є—Б–∞–љ–∞ S.A. Scott –Є¬†—Б–Њ–∞–≤—В. —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–±–Њ–ї–µ–Ј–љ—М—О –Т–Њ–ї—М–Љ–∞–љ–∞1. –Т¬†—Н–Ї–Ј–Њ–љ–µ 4 –Њ–±–љ–∞—А—Г–ґ–µ–љ–∞ –і–µ–ї–µ—Ж–Є—П c.421del –≤¬†–≥–µ—В–µ—А–Њ–Ј–Є–≥–Њ—В–љ–Њ–Љ —Б–Њ—Б—В–Њ—П–љ–Є–Є, –њ—А–Є–≤–Њ–і—П—Й–∞—П –Ї¬†—Б–і–≤–Є–≥—Г —А–∞–Љ–Ї–Є —Б—З–Є—В—Л–≤–∞–љ–Є—П p.Ala141Leufs*20. –Т—Л—П–≤–ї–µ–љ–љ–∞—П –і–µ–ї–µ—Ж–Є—П —А–∞–љ–µ–µ –љ–µ¬†–±—Л–ї–∞ –Њ–њ–Є—Б–∞–љ–∞ –Є, –њ–Њ¬†–і–∞–љ–љ—Л–Љ –Ї–Њ–Љ–њ—М—О—В–µ—А–љ–Њ–≥–Њ –∞–љ–∞–ї–Є–Ј–∞ (Alamut Visual), —П–≤–ї—П–µ—В—Б—П –њ–∞—В–Њ–≥–µ–љ–љ–Њ–є.
–Ґ–∞–Ї–Є–Љ –Њ–±—А–∞–Ј–Њ–Љ, —Б¬†2006 –њ–Њ¬†2016¬†–≥.¬†–Њ—Б–љ–Њ–≤–љ—Л–Љ –і–Є–∞–≥–љ–Њ–Ј–Њ–Љ –њ–∞—Ж–Є–µ–љ—В–∞ –±—Л–ї ¬Ђ—Е—А–Њ–љ–Є—З–µ—Б–Ї–Є–є –Ї—А–Є–њ—В–Њ–≥–µ–љ–љ—Л–є¬†–≥–µ–њ–∞—В–Є—В¬ї, —Б–Њ–њ—А–Њ–≤–Њ–ґ–і–∞–≤—И–Є–є—Б—П¬†–≥–µ–њ–∞—В–Њ–Љ–µ–≥–∞–ї–Є–µ–є, –њ–Њ–≤—Л—И–µ–љ–Є–µ–Љ —Г—А–Њ–≤–љ–µ–є —В—А–∞–љ—Б–∞–Љ–Є–љ–∞–Ј, –і–Є—Б–ї–Є–њ–Є–і–µ–Љ–Є–µ–є (—Г–≤–µ–ї–Є—З–µ–љ–Є–µ–Љ —Б–Њ–і–µ—А–ґ–∞–љ–Є—П —Е–Њ–ї–µ—Б—В–µ—А–Є–љ–∞, –Ы–Я–Э–Я). –Т¬†2010¬†–≥.¬†–±—Л–ї–Є –Њ–±–љ–∞—А—Г–ґ–µ–љ—Л —Е–∞—А–∞–Ї—В–µ—А–љ—Л–µ –і–ї—П —А–µ–і–Ї–Њ–≥–Њ –љ–∞—Б–ї–µ–і—Б—В–≤–µ–љ–љ–Њ–≥–Њ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П –Ф–Ы–Ъ–Ы —Д–Є–±—А–Њ—В–Є—З–µ—Б–Ї–Є–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П –Є¬†–Љ–Є–Ї—А–Њ–≤–µ–Ј–Є–Ї—Г–ї—П—А–љ—Л–є —Б—В–µ–∞—В–Њ–Ј –≤¬†–Њ–±—А–∞–Ј—Ж–µ –њ–µ—З–µ–љ–Є –њ–∞—Ж–Є–µ–љ—В–∞. –Ю–і–љ–∞–Ї–Њ –і–∞–љ–љ—Л–є –і–Є–∞–≥–љ–Њ–Ј –љ–µ¬†—Г–і–∞–ї–Њ—Б—М —Б–≤–Њ–µ–≤—А–µ–Љ–µ–љ–љ–Њ –њ–Њ–і—В–≤–µ—А–і–Є—В—М –Є–ї–Є –Њ—В–≤–µ—А–≥–љ—Г—В—М –Є–Ј-–Ј–∞ –Њ–≥—А–∞–љ–Є—З–µ–љ–Є–є –Љ–µ—В–Њ–і–∞ –і–Є–∞–≥–љ–Њ—Б—В–Є–Ї–Є –Ф–Ы–Ъ–Ы –≤¬†–њ–µ—А–Є–Њ–і –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П –њ–∞—Ж–Є–µ–љ—В–∞. –Ґ–Њ–ї—М–Ї–Њ –≤¬†2016¬†–≥., –±–ї–∞–≥–Њ–і–∞—А—П –≤–љ–µ–і—А–µ–љ–Є—О –љ–∞–Є–±–Њ–ї–µ–µ –≤—Л—Б–Њ–Ї–Њ—З—Г–≤—Б—В–≤–Є—В–µ–ї—М–љ–Њ–≥–Њ –Є–Ј–±–Є—А–∞—В–µ–ї—М–љ–Њ–≥–Њ –Љ–µ—В–Њ–і–∞ –Є–Ј–Љ–µ—А–µ–љ–Є—П –Ы–Ъ–Ы –≤¬†–њ—П—В–љ–∞—Е –Ї—А–Њ–≤–Є, —Г–і–∞–ї–Њ—Б—М –Њ–і–љ–Њ–Ј–љ–∞—З–љ–Њ –њ–Њ–і—В–≤–µ—А–і–Є—В—М –і–Є–∞–≥–љ–Њ–Ј –Ф–Ы–Ъ–Ы.
–Р–љ–∞–ї–Є–Ј –њ–Њ—Б–ї–µ–і–Њ–≤–∞—В–µ–ї—М–љ–Њ—Б—В–Є¬†–≥–µ–љ–∞ LIPA –њ–Њ–Ј–≤–Њ–ї–Є–ї –Њ–њ—А–µ–і–µ–ї–Є—В—М¬†–≥–µ–љ–µ—В–Є—З–µ—Б–Ї—Г—О –њ–Њ–ї–Њ–Љ–Ї—Г, –Ї–Њ—В–Њ—А–∞—П —Б—В–∞–ї–∞ –њ—А–Є—З–Є–љ–Њ–є –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П —Г¬†–і–∞–љ–љ–Њ–≥–Њ –њ–∞—Ж–Є–µ–љ—В–∞. –Я—А–Є –Ф–Ы–Ъ–Ы –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ¬†–≥–µ–љ–∞ —Б—В–∞–љ–і–∞—А—В–љ—Л–Љ–Є –Љ–µ—В–Њ–і–∞–Љ–Є –Љ–Њ–ґ–µ—В –±—Л—В—М —В–Њ–ї—М–Ї–Њ –њ–Њ–і—В–≤–µ—А–ґ–і–∞—О—Й–Є–Љ –і–Є–∞–≥–љ–Њ—Б—В–Є—З–µ—Б–Ї–Є–Љ —Б–њ–Њ—Б–Њ–±–Њ–Љ, –њ–Њ—Б–Ї–Њ–ї—М–Ї—Г –Є–Ј–≤–µ—Б—В–љ—Л —Б–ї—Г—З–∞–Є, –Ї–Њ–≥–і–∞ –Љ—Г—В–∞—Ж–Є—П –≤¬†–Ї–Њ–і–Є—А—Г—О—Й–Є—Е –Њ–±–ї–∞—Б—В—П—Е –љ–µ¬†–≤—Л—П–≤–ї—П–ї–∞—Б—М. –≠—В–Њ –Љ–Њ–ґ–µ—В –±—Л—В—М –Њ–±—Г—Б–ї–Њ–≤–ї–µ–љ–Њ –љ–∞–ї–Є—З–Є–µ–Љ –Љ—Г—В–∞—Ж–Є–є –≤¬†–љ–µ–Ї–Њ–і–Є—А—Г—О—Й–Є—Е —Г—З–∞—Б—В–Ї–∞—Е¬†–≥–µ–љ–∞ LIPA, –љ–∞–ї–Є—З–Є–µ–Љ –Ї—А—Г–њ–љ—Л—Е –њ–µ—А–µ—Б—В—А–Њ–µ–Ї, –∞¬†—В–∞–Ї–ґ–µ –і—А—Г–≥–Є–Љ–Є –Љ–µ—Е–∞–љ–Є–Ј–Љ–∞–Љ–Є —А–µ–≥—Г–ї–Є—А–Њ–≤–∞–љ–Є—П –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є –Ы–Ъ–Ы, –љ–∞–њ—А–Є–Љ–µ—А —Н–њ–Є–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Є–Љ–Є, –Ї–Њ—В–Њ—А—Л–µ –љ–µ¬†–Њ–±–љ–∞—А—Г–ґ–Є–≤–∞—О—В—Б—П –њ—А–Є —Б—В–∞–љ–і–∞—А—В–љ—Л—Е¬†–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Є—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П—Е. –Ґ–∞–Ї–Є–Љ –Њ–±—А–∞–Ј–Њ–Љ, –Њ—Б–љ–Њ–≤–љ—Л–Љ –Ї—А–Є—В–µ—А–Є–µ–Љ –і–Є–∞–≥–љ–Њ—Б—В–Є–Ї–Є –Ф–Ы–Ъ–Ы —П–≤–ї—П–µ—В—Б—П —Б–љ–Є–ґ–µ–љ–Є–µ –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є –Ы–Ъ–Ы –≤¬†—Б—Г—Е–Є—Е –њ—П—В–љ–∞—Е –Ї—А–Њ–≤–Є.
–Ч–∞–Ї–ї—О—З–µ–љ–Є–µ
–Ф–∞–љ–љ—Л–є –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є–є –њ—А–Є–Љ–µ—А –і–µ–Љ–Њ–љ—Б—В—А–Є—А—Г–µ—В —Ж–µ–ї–µ—Б–Њ–Њ–±—А–∞–Ј–љ–Њ—Б—В—М –Є–љ–і–Є–≤–Є–і—Г–∞–ї—М–љ–Њ–≥–Њ –њ–Њ–і—Е–Њ–і–∞ –Ї¬†–≤–µ–і–µ–љ–Є—О –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–љ–∞—А—Г—И–µ–љ–Є—П–Љ–Є –ї–Є–њ–Є–і–љ–Њ–≥–Њ –Њ–±–Љ–µ–љ–∞, –∞¬†—В–∞–Ї–ґ–µ –љ–µ–Њ–±—Е–Њ–і–Є–Љ–Њ—Б—В—М –Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–љ–Є—П —Б–Њ–≤—А–µ–Љ–µ–љ–љ—Л—Е –і–Є–∞–≥–љ–Њ—Б—В–Є—З–µ—Б–Ї–Є—Е –Љ–µ—В–Њ–і–Њ–≤.
–Я—А–Є –і–Є—Б–ї–Є–њ–Є–і–µ–Љ–Є–Є —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ –њ–Њ–≤—Л—И–∞–µ—В—Б—П —А–Є—Б–Ї¬†—А–∞–Ј–≤–Є—В–Є—П –Э–Р–Ц–С–Я, –њ–∞—В–Њ–ї–Њ–≥–Є–Є —Б–Њ—Б—Г–і–Њ–≤, –∞—В–µ—А–Њ—Б–Ї–ї–µ—А–Њ—В–Є—З–µ—Б–Ї–Є—Е –Є–Ј–Љ–µ–љ–µ–љ–Є–є. –Ш–Љ–µ–љ–љ–Њ –њ–Њ—Н—В–Њ–Љ—Г –њ—А–Є –љ–∞—А—Г—И–µ–љ–Є—П—Е –ї–Є–њ–Є–і–љ–Њ–≥–Њ –Љ–µ—В–∞–±–Њ–ї–Є–Ј–Љ–∞ —Г¬†–і–µ—В–µ–є –љ–µ–Њ–±—Е–Њ–і–Є–Љ –Ї–Њ–Љ–њ–ї–µ–Ї—Б–љ—Л–є —В–µ—А–∞–њ–µ–≤—В–Є—З–µ—Б–Ї–Є–є –њ–Њ–і—Е–Њ–і –≤—А–∞—З–µ–є —А–∞–Ј–љ—Л—Е —Б–њ–µ—Ж–Є–∞–ї—М–љ–Њ—Б—В–µ–є.
–Я—А–Є –љ–∞–ї–Є—З–Є–Є –њ—А–Є–Ј–љ–∞–Ї–Њ–≤¬†–≥–µ–њ–∞—В–Њ–Љ–µ–≥–∞–ї–Є–Є, —Б–Є–љ–і—А–Њ–Љ–∞ —Ж–Є—В–Њ–ї–Є–Ј–∞ –Є¬†–і–Є—Б–ї–Є–њ–Є–і–µ–Љ–Є–Є —А–∞–Ј–ї–Є—З–љ–Њ–є —Б—В–µ–њ–µ–љ–Є –≤—Л—А–∞–ґ–µ–љ–љ–Њ—Б—В–Є –њ–∞—Ж–Є–µ–љ—В–∞–Љ —Б–ї–µ–і—Г–µ—В –њ—А–Њ–≤–Њ–і–Є—В—М –і–Є–∞–≥–љ–Њ—Б—В–Є–Ї—Г –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є –ї–Є–Ј–Њ—Б–Њ–Љ–љ–Њ–є –Ї–Є—Б–ї–Њ–є –ї–Є–њ–∞–Ј—Л –≤¬†–њ—П—В–љ–∞—Е –Ї—А–Њ–≤–Є –і–ї—П –Є—Б–Ї–ї—О—З–µ–љ–Є—П –Ф–Ы–Ъ–Ы, –Њ–њ–∞—Б–љ–Њ–≥–Њ —В—П–ґ–µ–ї—Л–Љ–Є –њ–Њ—А–∞–ґ–µ–љ–Є—П–Љ–Є –њ–µ—З–µ–љ–Є, —Б–µ–ї–µ–Ј–µ–љ–Ї–Є, —Б–Њ—Б—Г–і–Њ–≤, –∞¬†—В–∞–Ї–ґ–µ –Є–љ–≤–∞–ї–Є–і–Є–Ј–∞—Ж–Є–µ–є –Є¬†—Б–љ–Є–ґ–µ–љ–Є–µ–Љ –Ї–∞—З–µ—Б—В–≤–∞ –ґ–Є–Ј–љ–Є –њ–∞—Ж–Є–µ–љ—В–Њ–≤. ¬†
–£–≤–∞–ґ–∞–µ–Љ—Л–є –њ–Њ—Б–µ—В–Є—В–µ–ї—М uMEDp!
–£–≤–µ–і–Њ–Љ–ї—П–µ–Љ –Т–∞—Б –Њ —В–Њ–Љ, —З—В–Њ –Ј–і–µ—Б—М —Б–Њ–і–µ—А–ґ–Є—В—Б—П –Є–љ—Д–Њ—А–Љ–∞—Ж–Є—П, –њ—А–µ–і–љ–∞–Ј–љ–∞—З–µ–љ–љ–∞—П –Є—Б–Ї–ї—О—З–Є—В–µ–ї—М–љ–Њ –і–ї—П —Б–њ–µ—Ж–Є–∞–ї–Є—Б—В–Њ–≤ –Ј–і—А–∞–≤–Њ–Њ—Е—А–∞–љ–µ–љ–Є—П.
–Х—Б–ї–Є –Т—Л –љ–µ —П–≤–ї—П–µ—В–µ—Б—М —Б–њ–µ—Ж–Є–∞–ї–Є—Б—В–Њ–Љ –Ј–і—А–∞–≤–Њ–Њ—Е—А–∞–љ–µ–љ–Є—П, –∞–і–Љ–Є–љ–Є—Б—В—А–∞—Ж–Є—П –љ–µ –љ–µ—Б–µ—В –Њ—В–≤–µ—В—Б—В–≤–µ–љ–љ–Њ—Б—В–Є –Ј–∞ –≤–Њ–Ј–Љ–Њ–ґ–љ—Л–µ –Њ—В—А–Є—Ж–∞—В–µ–ї—М–љ—Л–µ –њ–Њ—Б–ї–µ–і—Б—В–≤–Є—П, –≤–Њ–Ј–љ–Є–Ї—И–Є–µ –≤ —А–µ–Ј—Г–ї—М—В–∞—В–µ —Б–∞–Љ–Њ—Б—В–Њ—П—В–µ–ї—М–љ–Њ–≥–Њ –Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–љ–Є—П –Т–∞–Љ–Є –Є–љ—Д–Њ—А–Љ–∞—Ж–Є–Є —Б –њ–Њ—А—В–∞–ї–∞ –±–µ–Ј –њ—А–µ–і–≤–∞—А–Є—В–µ–ї—М–љ–Њ–є –Ї–Њ–љ—Б—Г–ї—М—В–∞—Ж–Є–Є —Б –≤—А–∞—З–Њ–Љ.
–Э–∞–ґ–Є–Љ–∞—П –љ–∞ –Ї–љ–Њ–њ–Ї—Г ¬Ђ–Т–Њ–є—В–Є¬ї, –Т—Л –њ–Њ–і—В–≤–µ—А–ґ–і–∞–µ—В–µ, —З—В–Њ —П–≤–ї—П–µ—В–µ—Б—М –≤—А–∞—З–Њ–Љ –Є–ї–Є —Б—В—Г–і–µ–љ—В–Њ–Љ –Љ–µ–і–Є—Ж–Є–љ—Б–Ї–Њ–≥–Њ –≤—Г–Ј–∞.