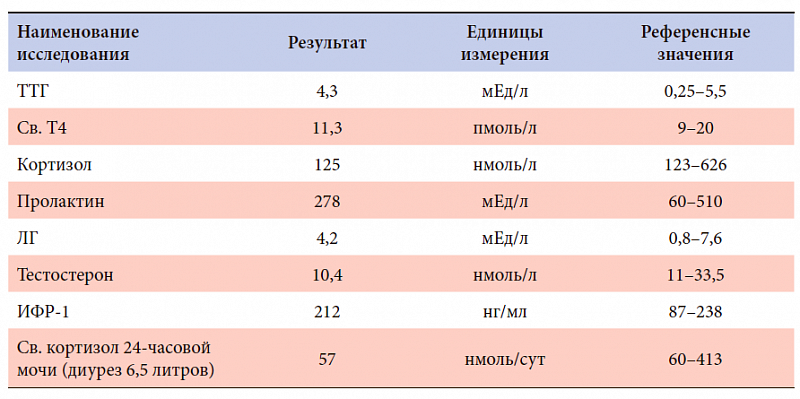


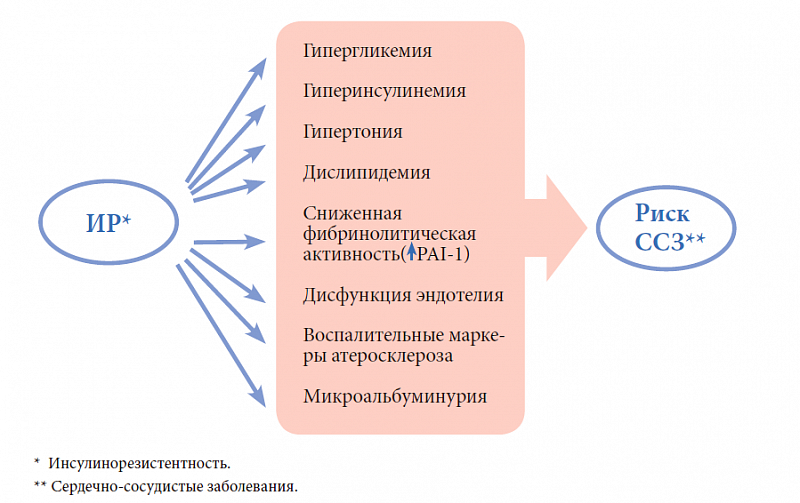


–í–ĺ–∑—Ä–į—Ā—ā
–í–ĺ–∑—Ä–į—Ā—ā –ī–Ķ–Ī—é—ā–į –°–Ē 2 —ā–ł–Ņ–į –Ņ–ĺ—Ā—ā–Ķ–Ņ–Ķ–Ĺ–Ĺ–ĺ —Ā–Ĺ–ł–∂–į–Ķ—ā—Ā—Ź. –Ę–į–ļ, –≤ —Ä–į–∑–≤–ł–≤–į—é—Č–ł—Ö—Ā—Ź —Ā—ā—Ä–į–Ĺ–į—Ö –ľ–į–ļ—Ā–ł–ľ–į–Ľ—Ć–Ĺ–į—Ź —á–į—Ā—ā–ĺ—ā–į –≤—č—Ź–≤–Ľ–Ķ–Ĺ–ł—Ź –ī–ł–į–Ī–Ķ—ā–į –Ņ—Ä–ł—Ö–ĺ–ī–ł—ā—Ā—Ź –Ĺ–į –≤–ĺ–∑—Ä–į—Ā—ā–Ĺ—É—é –≥—Ä—É–Ņ–Ņ—É 45‚Äď64 –≥–ĺ–ī–į, –≤ —ā–ĺ –≤—Ä–Ķ–ľ—Ź –ļ–į–ļ –≤ —Ä–į–∑–≤–ł—ā—č—Ö —Ā—ā—Ä–į–Ĺ–į—Ö ‚Äď 65 –Ľ–Ķ—ā –ł —Ā—ā–į—Ä—ą–Ķ. –ě—ā–ľ–Ķ—á–į–Ķ—ā—Ā—Ź —ā—Ä–Ķ–≤–ĺ–∂–Ĺ–į—Ź —ā–Ķ–Ĺ–ī–Ķ–Ĺ—Ü–ł—Ź –Ņ–ĺ–≤—č—ą–Ķ–Ĺ–ł—Ź —Ä–ł—Ā–ļ–į —Ä–į–∑–≤–ł—ā–ł—Ź –°–Ē 2 —ā–ł–Ņ–į —É –Ņ–ĺ–ī—Ä–ĺ—Ā—ā–ļ–ĺ–≤ –ł –ī–Ķ—ā–Ķ–Ļ [3]. –°–Ĺ–ł–∂–Ķ–Ĺ–ł–Ķ –≤–ĺ–∑—Ä–į—Ā—ā–Ĺ–ĺ–≥–ĺ –Ņ–ĺ—Ä–ĺ–≥–į –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł—Ź –≤–Ķ–ī–Ķ—ā –ļ —É–≤–Ķ–Ľ–ł—á–Ķ–Ĺ–ł—é —á–į—Ā—ā–ĺ—ā—č —Ä–į–Ĺ–Ĺ–Ķ–Ļ —Ā–ľ–Ķ—Ä—ā–Ĺ–ĺ—Ā—ā–ł, —Ä–į–∑–Ľ–ł—á–Ĺ—č—Ö –ĺ—Ā–Ľ–ĺ–∂–Ĺ–Ķ–Ĺ–ł–Ļ, –Ņ—Ä–ł–≤–ĺ–ī—Ź—Č–ł—Ö –ļ –ł–Ĺ–≤–į–Ľ–ł–ī–Ĺ–ĺ—Ā—ā–ł, —Ā–Ĺ–ł–∂–Ķ–Ĺ–ł—é —Ā–ĺ—Ü–ł–į–Ľ—Ć–Ĺ–ĺ–Ļ –į–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā–ł –ł —É—Ö—É–ī—ą–Ķ–Ĺ–ł—é –ļ–į—á–Ķ—Ā—ā–≤–į –∂–ł–∑–Ĺ–ł. –Ē–ĺ–Ľ–≥–ĺ—Ā—Ä–ĺ—á–Ĺ—č–Ķ –Ņ–ĺ—Ā–Ľ–Ķ–ī—Ā—ā–≤–ł—Ź —ć—ā–ĺ–Ļ —ć–Ņ–ł–ī–Ķ–ľ–ł–ł –≤—č–Ľ–ł–≤–į—é—ā—Ā—Ź –≤ –ĺ–≥—Ä–ĺ–ľ–Ĺ—č–Ķ —á–Ķ–Ľ–ĺ–≤–Ķ—á–Ķ—Ā–ļ–ł–Ķ —Ā—ā—Ä–į–ī–į–Ĺ–ł—Ź –ł —ć–ļ–ĺ–Ĺ–ĺ–ľ–ł—á–Ķ—Ā–ļ–ł–Ķ –∑–į—ā—Ä–į—ā—č.
–ě—Ā–Ľ–ĺ–∂–Ĺ–Ķ–Ĺ–ł—Ź
–ė—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź –Ņ–ĺ–ļ–į–∑–į–Ľ–ł, —á—ā–ĺ –Ĺ–į –ľ–ĺ–ľ–Ķ–Ĺ—ā –ī–Ķ–Ī—é—ā–į –°–Ē 2 —ā–ł–Ņ–į –ĺ–ļ–ĺ–Ľ–ĺ 50% –Ī–ĺ–Ľ—Ć–Ĺ—č—Ö —É–∂–Ķ –ł–ľ–Ķ—é—ā –ľ–į–ļ—Ä–ĺ- –ł –ľ–ł–ļ—Ä–ĺ—Ā–ĺ—Ā—É–ī–ł—Ā—ā—č–Ķ –ĺ—Ā–Ľ–ĺ–∂–Ĺ–Ķ–Ĺ–ł—Ź. –í–ĺ–∑–ľ–ĺ–∂–Ĺ–ĺ, —ć—ā–ĺ —Ä–Ķ–∑—É–Ľ—Ć—ā–į—ā —ā–ĺ–≥–ĺ, —á—ā–ĺ –ľ–Ķ—ā–į–Ī–ĺ–Ľ–ł—á–Ķ—Ā–ļ–ł–Ķ –Ĺ–į—Ä—É—ą–Ķ–Ĺ–ł—Ź –≤–ĺ–∑–Ĺ–ł–ļ–į—é—ā –≥–ĺ—Ä–į–∑–ī–ĺ —Ä–į–Ĺ—Ć—ą–Ķ –Ņ–Ķ—Ä–≤—č—Ö –ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ł—Ö –Ņ—Ä–ĺ—Ź–≤–Ľ–Ķ–Ĺ–ł–Ļ –°–Ē, –ł –ļ –ľ–ĺ–ľ–Ķ–Ĺ—ā—É –Ņ–ĺ—Ā—ā–į–Ĺ–ĺ–≤–ļ–ł –ī–ł–į–≥–Ĺ–ĺ–∑–į –Ņ—Ä–ł–≤–ĺ–ī—Ź—ā –ļ –Ĺ–Ķ–ĺ–Ī—Ä–į—ā–ł–ľ—č–ľ —Ā–ĺ—Ā—É–ī–ł—Ā—ā—č–ľ –ł–∑–ľ–Ķ–Ĺ–Ķ–Ĺ–ł—Ź–ľ. –Ě–į–ł–Ī–ĺ–Ľ–Ķ–Ķ –ī—Ä–į–ľ–į—ā–ł—á–Ĺ–ĺ —Ä–į–∑–≤–ł—ā–ł–Ķ —Ü–Ķ–Ĺ—ā—Ä–į–Ľ—Ć–Ĺ—č—Ö —Ą–ĺ—Ä–ľ –ī–ł–į–Ī–Ķ—ā–ł—á–Ķ—Ā–ļ–ĺ–Ļ –Ĺ–Ķ–Ļ—Ä–ĺ–Ņ–į—ā–ł–ł, –ļ –ļ–ĺ—ā–ĺ—Ä—č–ľ –ĺ—ā–Ĺ–ĺ—Ā—Ź—ā—Ā—Ź –ĺ—Ā—ā—Ä—č–Ķ –Ĺ–Ķ—Ä–≤–Ĺ–ĺ-–Ņ—Ā–ł—Ö–ł—á–Ķ—Ā–ļ–ł–Ķ —Ä–į—Ā—Ā—ā—Ä–ĺ–Ļ—Ā—ā–≤–į –Ĺ–į —Ą–ĺ–Ĺ–Ķ –ī–Ķ–ļ–ĺ–ľ–Ņ–Ķ–Ĺ—Ā–į—Ü–ł–ł –ľ–Ķ—ā–į–Ī–ĺ–Ľ–ł–∑–ľ–į (–ļ–Ķ—ā–ĺ–į—Ü–ł–ī–ĺ–∑–į, –Ľ–į–ļ—ā–į—ā–į—Ü–ł–ī–ĺ–∑–į, –≥–ł–Ņ–Ķ—Ä–ĺ—Ā–ľ–ĺ–Ľ—Ź—Ä–Ĺ–ĺ–≥–ĺ –ł –≥–ł–Ņ–ĺ–≥–Ľ–ł–ļ–Ķ–ľ–ł—á–Ķ—Ā–ļ–ĺ–≥–ĺ —Ā–ĺ—Ā—ā–ĺ—Ź–Ĺ–ł–Ļ); –ĺ—Ā—ā—Ä—č–Ķ –Ĺ–į—Ä—É—ą–Ķ–Ĺ–ł—Ź –ľ–ĺ–∑–≥–ĺ–≤–ĺ–≥–ĺ –ļ—Ä–ĺ–≤–ĺ–ĺ–Ī—Ä–į—Č–Ķ–Ĺ–ł—Ź ‚Äď –ł–Ĺ—Ā—É–Ľ—Ć—ā, –Ņ—Ä–Ķ—Ö–ĺ–ī—Ź—Č–ł–Ķ –Ĺ–į—Ä—É—ą–Ķ–Ĺ–ł—Ź –ľ–ĺ–∑–≥–ĺ–≤–ĺ–≥–ĺ –ļ—Ä–ĺ–≤–ĺ–ĺ–Ī—Ä–į—Č–Ķ–Ĺ–ł—Ź; –Ņ—Ä–ĺ–≥—Ä–Ķ—Ā—Ā–ł—Ä—É—é—Č–į—Ź –ī–ł–į–Ī–Ķ—ā–ł—á–Ķ—Ā–ļ–į—Ź —ć–Ĺ—Ü–Ķ—Ą–į–Ľ–ĺ–Ņ–į—ā–ł—Ź. –ö –ł—Ā—ā–ł–Ĺ–Ĺ–ĺ –ī–ł–į–Ī–Ķ—ā–ł—á–Ķ—Ā–ļ–ĺ–Ļ –Ņ—Ä–ł–Ĺ—Ź—ā–ĺ –ĺ—ā–Ĺ–ĺ—Ā–ł—ā—Ć –Ņ—Ä–ĺ–≥—Ä–Ķ–ī–ł–Ķ–Ĺ—ā–Ĺ–ĺ —Ä–į–∑–≤–ł–≤–į—é—Č—É—é—Ā—Ź –Ĺ–į —Ą–ĺ–Ĺ–Ķ –Ĺ–į—Ä—É—ą–Ķ–Ĺ–ł–Ļ —É–≥–Ľ–Ķ–≤–ĺ–ī–Ĺ–ĺ–≥–ĺ –ĺ–Ī–ľ–Ķ–Ĺ–į –ľ–Ķ—ā–į–Ī–ĺ–Ľ–ł—á–Ķ—Ā–ļ—É—é —ć–Ĺ—Ü–Ķ—Ą–į–Ľ–ĺ–Ņ–į—ā–ł—é [1]. –ě–ī–Ĺ–į–ļ–ĺ –≤—č–ī–Ķ–Ľ–Ķ–Ĺ–ł–Ķ ¬ę—á–ł—Ā—ā–ĺ–Ļ¬Ľ, –ľ–Ķ—ā–į–Ī–ĺ–Ľ–ł—á–Ķ—Ā–ļ–ĺ–Ļ —Ą–ĺ—Ä–ľ—č —ć–Ĺ—Ü–Ķ—Ą–į–Ľ–ĺ–Ņ–į—ā–ł–ł –Ņ—Ä–ł –°–Ē –≤–Ķ—Ā—Ć–ľ–į –Ņ—Ä–ĺ–Ī–Ľ–Ķ–ľ–į—ā–ł—á–Ĺ–ĺ, –Ņ–ĺ—Ā–ļ–ĺ–Ľ—Ć–ļ—É —Ā —ā–Ķ—á–Ķ–Ĺ–ł–Ķ–ľ –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł—Ź –Ņ—Ä–ĺ–≥—Ä–Ķ—Ā—Ā–ł—Ä—É—é—ā —Ü–Ķ—Ä–Ķ–Ī—Ä–į–Ľ—Ć–Ĺ—č–Ķ –Ĺ–į—Ä—É—ą–Ķ–Ĺ–ł—Ź, –ĺ–Ī—É—Ā–Ľ–ĺ–≤–Ľ–Ķ–Ĺ–Ĺ—č–Ķ —Ä–į–∑–≤–ł—ā–ł–Ķ–ľ —Ā–ĺ—Ā—É–ī–ł—Ā—ā—č—Ö –ł–∑–ľ–Ķ–Ĺ–Ķ–Ĺ–ł–Ļ, –į—Ä—ā–Ķ—Ä–ł–į–Ľ—Ć–Ĺ–ĺ–Ļ –≥–ł–Ņ–Ķ—Ä—ā–Ķ–Ĺ–∑–ł–ł –ł –į–≤—ā–ĺ–Ĺ–ĺ–ľ–Ĺ–ĺ–Ļ –Ĺ–Ķ–Ļ—Ä–ĺ–Ņ–į—ā–ł–ł. –Ē–ł–į–Ī–Ķ—ā–ł—á–Ķ—Ā–ļ–į—Ź —ć–Ĺ—Ü–Ķ—Ą–į–Ľ–ĺ–Ņ–į—ā–ł—Ź –ĺ–Ī—č—á–Ĺ–ĺ —Ä–į–∑–≤–ł–≤–į–Ķ—ā—Ā—Ź –Ņ–ĺ—Ā—ā–Ķ–Ņ–Ķ–Ĺ–Ĺ–ĺ, —É –ľ–ĺ–Ľ–ĺ–ī—č—Ö –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ –Ķ–Ķ —Ö—Ä–ĺ–Ĺ–ł—á–Ķ—Ā–ļ–ł–Ķ –Ņ—Ä–ĺ—Ź–≤–Ľ–Ķ–Ĺ–ł—Ź —É—Ā—É–≥—É–Ī–Ľ—Ź—é—ā—Ā—Ź –Ņ–ĺ—Ā–Ľ–Ķ–ī—Ā—ā–≤–ł—Ź–ľ–ł –Ņ–Ķ—Ä–Ķ–Ĺ–Ķ—Ā–Ķ–Ĺ–Ĺ—č—Ö –ĺ—Ā—ā—Ä—č—Ö –≥–ł–Ņ–Ķ—Ä- –ł –≥–ł–Ņ–ĺ–≥–Ľ–ł–ļ–Ķ–ľ–ł—á–Ķ—Ā–ļ–ł—Ö —ć–Ņ–ł–∑–ĺ–ī–ĺ–≤, —É –Ņ–ĺ–∂–ł–Ľ—č—Ö ‚Äď –Ĺ–į—Ä—É—ą–Ķ–Ĺ–ł—Ź–ľ–ł –ľ–ĺ–∑–≥–ĺ–≤–ĺ–≥–ĺ –ļ—Ä–ĺ–≤–ĺ–ĺ–Ī—Ä–į—Č–Ķ–Ĺ–ł—Ź.
–ö–ĺ–≥–Ĺ–ł—ā–ł–≤–Ĺ—č–Ļ –ī–Ķ—Ą–ł—Ü–ł—ā
–ö–į–ļ –ł –Ņ—Ä–ł –ī—Ä—É–≥–ł—Ö –ľ–Ķ—ā–į–Ī–ĺ–Ľ–ł—á–Ķ—Ā–ļ–ł—Ö —ć–Ĺ—Ü–Ķ—Ą–į–Ľ–ĺ–Ņ–į—ā–ł—Ź—Ö, –ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ł–Ķ –Ņ—Ä–ĺ—Ź–≤–Ľ–Ķ–Ĺ–ł—Ź –ī–ł–į–Ī–Ķ—ā–ł—á–Ķ—Ā–ļ–ĺ–Ļ —ć–Ĺ—Ü–Ķ—Ą–į–Ľ–ĺ–Ņ–į—ā–ł–ł –Ĺ–Ķ—Ā–Ņ–Ķ—Ü–ł—Ą–ł—á–Ĺ—č, –Ĺ–į–ł–Ī–ĺ–Ľ–Ķ–Ķ —á–į—Ā—ā–ĺ —Ä–į–∑–≤–ł–≤–į–Ķ—ā—Ā—Ź –Ĺ–į—Ä—É—ą–Ķ–Ĺ–ł–Ķ –ļ–ĺ–≥–Ĺ–ł—ā–ł–≤–Ĺ—č—Ö —Ą—É–Ĺ–ļ—Ü–ł–Ļ: —Ā–Ĺ–ł–∂–Ķ–Ĺ–ł–Ķ –Ņ–į–ľ—Ź—ā–ł –ł –≤–Ĺ–ł–ľ–į–Ĺ–ł—Ź, –∑–į–ľ–Ķ–ī–Ľ–Ķ–Ĺ–ł–Ķ –ľ—č—ą–Ľ–Ķ–Ĺ–ł—Ź, —Ā–Ĺ–ł–∂–Ķ–Ĺ–ł–Ķ —Ā–ļ–ĺ—Ä–ĺ—Ā—ā–ł –Ņ—Ā–ł—Ö–ł—á–Ķ—Ā–ļ–ł—Ö —Ä–Ķ–į–ļ—Ü–ł–Ļ –ł —Ā–Ņ–ĺ—Ā–ĺ–Ī–Ĺ–ĺ—Ā—ā–ł –ļ –ĺ–Ī—É—á–Ķ–Ĺ–ł—é. –£—Ā—ā–į–Ĺ–ĺ–≤–Ľ–Ķ–Ĺ–į –ĺ–Ī—Č–Ĺ–ĺ—Ā—ā—Ć –ľ–Ķ—Ö–į–Ĺ–ł–∑–ľ–ĺ–≤ —Ä–į–∑–≤–ł—ā–ł—Ź –ļ–ĺ–≥–Ĺ–ł—ā–ł–≤–Ĺ–ĺ–≥–ĺ –ī–Ķ—Ą–ł—Ü–ł—ā–į –Ņ—Ä–ł –°–Ē –ł –Ī–ĺ–Ľ–Ķ–∑–Ĺ–ł –ź–Ľ—Ć—Ü–≥–Ķ–Ļ–ľ–Ķ—Ä–į, –Ī–ĺ–Ľ—Ć–Ĺ—č–Ķ —Ā –°–Ē –≤—Ö–ĺ–ī—Ź—ā –≤ –≥—Ä—É–Ņ–Ņ—É —Ä–ł—Ā–ļ–į —Ä–į–∑–≤–ł—ā–ł—Ź –ī–Ķ–ľ–Ķ–Ĺ—Ü–ł–ł. –ė–∑–≤–Ķ—Ā—ā–Ĺ–ĺ, —á—ā–ĺ –≤ –Ņ—Ä–ĺ—Ü–Ķ—Ā—Ā–Ķ —Ā—ā–į—Ä–Ķ–Ĺ–ł—Ź –ľ–ĺ–∑–≥–į –Ņ—Ä–ł–Ĺ–ł–ľ–į—é—ā —É—á–į—Ā—ā–ł–Ķ —ā–Ķ –∂–Ķ –Ņ–į—ā–ĺ–≥–Ķ–Ĺ–Ķ—ā–ł—á–Ķ—Ā–ļ–ł–Ķ –ľ–Ķ—Ö–į–Ĺ–ł–∑–ľ—č, —á—ā–ĺ –ł –Ņ—Ä–ł —Ä–į–∑–≤–ł—ā–ł–ł –ī–ł–į–Ī–Ķ—ā–ł—á–Ķ—Ā–ļ–ł—Ö –ĺ—Ā–Ľ–ĺ–∂–Ĺ–Ķ–Ĺ–ł–Ļ, –ł –ļ–Ľ—é—á–Ķ–≤—č–ľ —Ą–į–ļ—ā–ĺ—Ä–ĺ–ľ –∑–ī–Ķ—Ā—Ć —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź –ĺ–ļ—Ā–ł–ī–į—ā–ł–≤–Ĺ—č–Ļ —Ā—ā—Ä–Ķ—Ā—Ā. –í—č—Ź–≤–Ľ—Ź–Ķ–ľ—č–Ķ –Ņ—Ä–ł –Ĺ–Ķ–Ļ—Ä–ĺ–Ņ—Ā–ł—Ö–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ĺ–ľ –ĺ–Ī—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–ł –ł–∑–ľ–Ķ–Ĺ–Ķ–Ĺ–ł—Ź —É –Ī–ĺ–Ľ—Ć–Ĺ—č—Ö –°–Ē 2 —ā–ł–Ņ–į –Ī–ĺ–Ľ–Ķ–Ķ —Ā—ā–ĺ–Ļ–ļ–ł–Ķ, —á–Ķ–ľ –Ņ—Ä–ł –°–Ē 1 —ā–ł–Ņ–į. –ß–į—Č–Ķ –≤—Ā–Ķ–≥–ĺ —ć—ā–ĺ –Ĺ–į—Ä—É—ą–Ķ–Ĺ–ł—Ź –≤–Ķ—Ä–Ī–į–Ľ—Ć–Ĺ–ĺ–Ļ –Ņ–į–ľ—Ź—ā–ł –ł –Ņ—Ä–ĺ—Ü–Ķ—Ā—Ā–į –ĺ–Ī—Ä–į–Ī–ĺ—ā–ļ–ł –ł–Ĺ—Ą–ĺ—Ä–ľ–į—Ü–ł–ł —Ā—Ä–Ķ–ī–Ĺ–Ķ–Ļ —Ā—ā–Ķ–Ņ–Ķ–Ĺ–ł –≤—č—Ä–į–∂–Ķ–Ĺ–Ĺ–ĺ—Ā—ā–ł. –ė–Ĺ–ĺ–≥–ī–į –Ņ—Ä–ł –°–Ē –≤—č—Ź–≤–Ľ—Ź—é—ā—Ā—Ź –Ĺ–į—Ä—É—ą–Ķ–Ĺ–ł—Ź –Ņ—Ä–į–ļ—Ā–ł—Ā–į, –≥–Ĺ–ĺ–∑–ł—Ā–į, —Ä–Ķ—á–Ķ–≤—č—Ö –ł –Ņ—Ä–ĺ—Ā—ā—Ä–į–Ĺ—Ā—ā–≤–Ķ–Ĺ–Ĺ—č—Ö —Ą—É–Ĺ–ļ—Ü–ł–Ļ, –∑—Ä–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ–Ļ –ł —Ā–Ľ—É—Ö–ĺ–≤–ĺ–Ļ –Ņ–į–ľ—Ź—ā–ł, –į —ā–į–ļ–∂–Ķ –Ĺ–į—Ä—É—ą–Ķ–Ĺ–ł—Ź –ľ–Ķ–∂–Ņ–ĺ–Ľ—É—ą–į—Ä–Ĺ—č—Ö –≤–∑–į–ł–ľ–ĺ–ī–Ķ–Ļ—Ā—ā–≤–ł–Ļ —Ā –ī–ł—Ā—Ą—É–Ĺ–ļ—Ü–ł–Ķ–Ļ –Ņ—Ä–į–≤–ĺ–≥–ĺ –Ņ–ĺ–Ľ—É—ą–į—Ä–ł—Ź [4].
–Ę–Ķ—á–Ķ–Ĺ–ł–Ķ –°–Ē —Ā–ĺ–Ņ—Ä–ĺ–≤–ĺ–∂–ī–į–Ķ—ā—Ā—Ź —á–į—Ā—ā—č–ľ–ł –ļ–ĺ–Ľ–Ķ–Ī–į–Ĺ–ł—Ź–ľ–ł —É—Ä–ĺ–≤–Ĺ—Ź —Ā–į—Ö–į—Ä–į –≤ –ļ—Ä–ĺ–≤–ł, –Ņ–ĺ—Ä–ĺ–Ļ —Ä–Ķ–∑–ļ–ł–ľ–ł, –ļ–ĺ—ā–ĺ—Ä—č–ľ –Ņ—Ä–ł–ī–į–Ķ—ā—Ā—Ź –ĺ—á–Ķ–Ĺ—Ć –Ī–ĺ–Ľ—Ć—ą–ĺ–Ķ –∑–Ĺ–į—á–Ķ–Ĺ–ł–Ķ –≤ —Ä–į–∑–≤–ł—ā–ł–ł –ľ–ĺ–∑–≥–ĺ–≤—č—Ö —Ä–į—Ā—Ā—ā—Ä–ĺ–Ļ—Ā—ā–≤. –ě—Ā–ĺ–Ī–Ķ–Ĺ–Ĺ–ĺ –ĺ–Ņ–į—Ā–Ĺ—č –≤ —ć—ā–ĺ–ľ –ĺ—ā–Ĺ–ĺ—ą–Ķ–Ĺ–ł–ł –≥–ł–Ņ–ĺ–≥–Ľ–ł–ļ–Ķ–ľ–ł—á–Ķ—Ā–ļ–ł–Ķ —ć–Ņ–ł–∑–ĺ–ī—č, –≤–ĺ–∑–Ĺ–ł–ļ–į—é—Č–ł–Ķ –≤–ĺ –≤—Ä–Ķ–ľ—Ź —ā–Ķ—Ä–į–Ņ–ł–ł –ł–Ĺ—Ā—É–Ľ–ł–Ĺ–ĺ–ľ –ł–Ľ–ł –Ņ–Ķ—Ä–ĺ—Ä–į–Ľ—Ć–Ĺ—č–ľ–ł —Ā–į—Ö–į—Ä–ĺ—Ā–Ĺ–ł–∂–į—é—Č–ł–ľ–ł —Ā—Ä–Ķ–ī—Ā—ā–≤–į–ľ–ł. –ö–ĺ–≥–Ĺ–ł—ā–ł–≤–Ĺ—č–Ļ –ī–Ķ—Ą–ł—Ü–ł—ā —á–į—Ā—ā–ĺ –ł–ľ–Ķ–Ķ—ā –ľ–Ķ—Ā—ā–ĺ –ł —É –Ī–ĺ–Ľ—Ć–Ĺ—č—Ö —Ā –≤–Ņ–Ķ—Ä–≤—č–Ķ –ī–ł–į–≥–Ĺ–ĺ—Ā—ā–ł—Ä–ĺ–≤–į–Ĺ–Ĺ—č–ľ –°–Ē 2 —ā–ł–Ņ–į, –Ķ—Č–Ķ –Ĺ–Ķ –Ņ–ĺ–Ľ—É—á–į–≤—ą–ł—Ö —Ā–į—Ö–į—Ä–ĺ—Ā–Ĺ–ł–∂–į—é—Č—É—é —ā–Ķ—Ä–į–Ņ–ł—é, –Ĺ–ĺ —É –ļ–ĺ—ā–ĺ—Ä—č—Ö —É–∂–Ķ –ī–ł–į–≥–Ĺ–ĺ—Ā—ā–ł—Ä—É–Ķ—ā—Ā—Ź —ć–Ĺ—Ü–Ķ—Ą–į–Ľ–ĺ–Ņ–į—ā–ł—Ź ‚Äď —Ā–ĺ—Ā—ā–ĺ—Ź–Ĺ–ł–Ķ, –ļ–ĺ—ā–ĺ—Ä–ĺ–Ķ, –Ņ–ĺ –ľ–Ĺ–Ķ–Ĺ–ł—é –ľ–Ĺ–ĺ–≥–ł—Ö —É—á–Ķ–Ĺ—č—Ö, –Ņ—Ä–ł –ī–ł–į–Ī–Ķ—ā–Ķ —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź –Ĺ–Ķ–ł–∑–Ī–Ķ–∂–Ĺ—č–ľ [5]. –í –Ņ–į—ā–ĺ–≥–Ķ–Ĺ–Ķ–∑–Ķ –ī–ł–į–Ī–Ķ—ā–ł—á–Ķ—Ā–ļ–ĺ–Ļ —ć–Ĺ—Ü–Ķ—Ą–į–Ľ–ĺ–Ņ–į—ā–ł–ł (–ļ–į–ļ –ł –ī–ł–į–Ī–Ķ—ā–ł—á–Ķ—Ā–ļ–ĺ–Ļ –Ņ–ĺ–Ľ–ł–Ĺ–Ķ–Ļ—Ä–ĺ–Ņ–į—ā–ł–ł) –Ņ—Ä–ł–Ĺ–ł–ľ–į—é—ā —É—á–į—Ā—ā–ł–Ķ —Ü–Ķ–Ľ—č–Ļ —Ä—Ź–ī —Ą–į–ļ—ā–ĺ—Ä–ĺ–≤: —Ā–ĺ—Ā—É–ī–ł—Ā—ā–į—Ź –ī–ł—Ā—Ą—É–Ĺ–ļ—Ü–ł—Ź, –Ņ—Ä–ł–≤–ĺ–ī—Ź—Č–į—Ź –ļ —É–ľ–Ķ–Ĺ—Ć—ą–Ķ–Ĺ–ł—é –ļ—Ä–ĺ–≤–ĺ—Ā–Ĺ–į–Ī–∂–Ķ–Ĺ–ł—Ź –Ĺ–Ķ—Ä–≤–ĺ–≤ –ł –ľ–ĺ–∑–≥–ĺ–≤–ĺ–Ļ —ā–ļ–į–Ĺ–ł, –Ĺ–į—Ä—É—ą–Ķ–Ĺ–ł–Ķ —ā—Ä–ĺ—Ą–ł–ļ–ł –ł –Ņ—Ä—Ź–ľ–ĺ–Ķ —ā–ĺ–ļ—Ā–ł—á–Ķ—Ā–ļ–ĺ–Ķ –≤–Ľ–ł—Ź–Ĺ–ł–Ķ –≥–ł–Ņ–Ķ—Ä–≥–Ľ–ł–ļ–Ķ–ľ–ł–ł –Ĺ–į –Ĺ–Ķ—Ä–≤—č. –í–Ľ–ł—Ź–Ĺ–ł–Ķ –ī–ł–į–Ī–Ķ—ā–į –Ĺ–į –ľ–ĺ–∑–≥ –Ī–ĺ–Ľ–Ķ–Ķ –≤—č—Ä–į–∂–Ķ–Ĺ–ĺ —É –Ņ–ĺ–∂–ł–Ľ—č—Ö –Ľ—é–ī–Ķ–Ļ: –Ņ—Ä–ĺ–ł—Ā—Ö–ĺ–ī–ł—ā —É—Ā–ļ–ĺ—Ä–Ķ–Ĺ–ł–Ķ –ĺ–Ī—É—Ā–Ľ–ĺ–≤–Ľ–Ķ–Ĺ–Ĺ–ĺ–≥–ĺ —Ā—ā–į—Ä–Ķ–Ĺ–ł–Ķ–ľ –ļ–ĺ–≥–Ĺ–ł—ā–ł–≤–Ĺ–ĺ–≥–ĺ —Ā–Ĺ–ł–∂–Ķ–Ĺ–ł—Ź –ł —Ä–į–∑–≤–ł—ā–ł—Ź —ć–Ĺ—Ü–Ķ—Ą–į–Ľ–ĺ–Ņ–į—ā–ł–ł [6].
–í–Ľ–ł—Ź–Ĺ–ł–Ķ –≥–ł–Ņ–Ķ—Ä–ł–Ĺ—Ā—É–Ľ–ł–Ĺ–Ķ–ľ–ł–ł
–ě—Ā–ĺ–Ī—č–ľ –≤–ĺ–Ņ—Ä–ĺ—Ā–ĺ–ľ –≤ –ĺ–Ī—Č–Ķ–Ļ –Ņ—Ä–ĺ–Ī–Ľ–Ķ–ľ–Ķ –°–Ē —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź –≤–Ľ–ł—Ź–Ĺ–ł–Ķ –ł–Ĺ—Ā—É–Ľ–ł–Ĺ–į –Ĺ–į –ļ–ĺ–≥–Ĺ–ł—ā–ł–≤–Ĺ—č–Ķ —Ą—É–Ĺ–ļ—Ü–ł–ł. –ė–Ĺ—Ā—É–Ľ–ł–Ĺ, –ł–ī–Ķ–Ĺ—ā–ł—á–Ĺ—č–Ļ –Ņ–į–Ĺ–ļ—Ä–Ķ–į—ā–ł—á–Ķ—Ā–ļ–ĺ–ľ—É, –ł —ā–ł–Ņ–ł—á–Ĺ—č–Ķ –ł–Ĺ—Ā—É–Ľ–ł–Ĺ–ĺ–≤—č–Ķ —Ä–Ķ—Ü–Ķ–Ņ—ā–ĺ—Ä—č —ą–ł—Ä–ĺ–ļ–ĺ —ć–ļ—Ā–Ņ—Ä–Ķ—Ā—Ā–ł—Ä—É—é—ā—Ā—Ź –≤ —Ä–į–∑–Ĺ—č—Ö –ĺ—ā–ī–Ķ–Ľ–į—Ö –ľ–ĺ–∑–≥–į. –ė–Ĺ—Ā—É–Ľ–ł–Ĺ –Ņ—Ä–ĺ–Ĺ–ł–ļ–į–Ķ—ā –≤ –ľ–ĺ–∑–≥ –Ņ–ĺ—Ā—Ä–Ķ–ī—Ā—ā–≤–ĺ–ľ —Ä–Ķ—Ü–Ķ–Ņ—ā–ĺ—Ä–∑–į–≤–ł—Ā–ł–ľ–ĺ–≥–ĺ —ā—Ä–į–Ĺ—Ā–Ņ–ĺ—Ä—ā–į —á–Ķ—Ä–Ķ–∑ –≥–Ķ–ľ–į—ā–ĺ—ć–Ĺ—Ü–Ķ—Ą–į–Ľ–ł—á–Ķ—Ā–ļ–ł–Ļ –Ī–į—Ä—Ć–Ķ—Ä –ł –Ņ—Ä–ł–Ĺ–ł–ľ–į–Ķ—ā —É—á–į—Ā—ā–ł–Ķ –≤ —Ä–Ķ–≥—É–Ľ—Ź—Ü–ł–ł —ć–Ĺ–Ķ—Ä–≥–Ķ—ā–ł—á–Ķ—Ā–ļ–ĺ–≥–ĺ –≥–ĺ–ľ–Ķ–ĺ—Ā—ā–į–∑–į, —Ä–Ķ–Ņ—Ä–ĺ–ī—É–ļ—ā–ł–≤–Ĺ—č—Ö –ł –ļ–ĺ–≥–Ĺ–ł—ā–ł–≤–Ĺ—č—Ö —Ą—É–Ĺ–ļ—Ü–ł–Ļ –ĺ—Ä–≥–į–Ĺ–ł–∑–ľ–į. –í–ĺ–Ņ—Ä–ĺ—Ā—č, –ļ–į—Ā–į—é—Č–ł–Ķ—Ā—Ź –Ĺ–Ķ–Ļ—Ä–ĺ–Ĺ–į–Ľ—Ć–Ĺ—č—Ö —ć—Ą—Ą–Ķ–ļ—ā–ĺ–≤ –ł–Ĺ—Ā—É–Ľ–ł–Ĺ–į, –ĺ—Ā—ā–į—é—ā—Ā—Ź –≤–ĺ –ľ–Ĺ–ĺ–≥–ĺ–ľ –ī–ł—Ā–ļ—É—Ā—Ā–ł–ĺ–Ĺ–Ĺ—č–ľ–ł, –ĺ–ī–Ĺ–į–ļ–ĺ –≤ –Ņ–ĺ—Ā–Ľ–Ķ–ī–Ĺ–Ķ–Ķ –ī–Ķ—Ā—Ź—ā–ł–Ľ–Ķ—ā–ł–Ķ –Ņ–ĺ–Ľ—É—á–Ķ–Ĺ—č —É–Ī–Ķ–ī–ł—ā–Ķ–Ľ—Ć–Ĺ—č–Ķ –ī–ĺ–ļ–į–∑–į—ā–Ķ–Ľ—Ć—Ā—ā–≤–į —ā–ĺ–≥–ĺ, —á—ā–ĺ –ł–Ĺ—Ā—É–Ľ–ł–Ĺ –ł –ł–Ĺ—Ā—É–Ľ–ł–Ĺ—Ä–Ķ—Ü–Ķ–Ņ—ā–ĺ—Ä–Ĺ–į—Ź —Ā–ł–≥–Ĺ–į–Ľ—Ć–Ĺ–į—Ź —Ā–ł—Ā—ā–Ķ–ľ–į –ľ–ĺ–∑–≥–į –Ĺ–Ķ–ĺ–Ī—Ö–ĺ–ī–ł–ľ—č –ī–Ľ—Ź –Ĺ–ĺ—Ä–ľ–į–Ľ—Ć–Ĺ–ĺ–≥–ĺ —Ą—É–Ĺ–ļ—Ü–ł–ĺ–Ĺ–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź –Ĺ–Ķ–Ļ—Ä–ĺ–Ĺ–ĺ–≤. –Ē–ł—Ā—Ą—É–Ĺ–ļ—Ü–ł—Ź —ć—ā–ĺ–Ļ —Ā–ł—Ā—ā–Ķ–ľ—č –Ņ—Ä–ł–≤–ĺ–ī–ł—ā –ļ —Ä–į–∑–≤–ł—ā–ł—é –Ĺ–Ķ–Ļ—Ä–ĺ–ī–Ķ–≥–Ķ–Ĺ–Ķ—Ä–į—ā–ł–≤–Ĺ—č—Ö –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł–Ļ. –Ď—č–Ľ–ĺ –Ņ–ĺ–ļ–į–∑–į–Ĺ–ĺ, —á—ā–ĺ –ł–Ĺ—Ā—É–Ľ–ł–Ĺ –ł –ł–Ĺ—Ā—É–Ľ–ł–Ĺ–ĺ–≤—č–Ķ —Ä–Ķ—Ü–Ķ–Ņ—ā–ĺ—Ä—č –ł–≥—Ä–į—é—ā –≤–į–∂–Ĺ—É—é —Ä–ĺ–Ľ—Ć –≤ —Ā–ł–Ĺ–į–Ņ—ā–ł—á–Ķ—Ā–ļ–ĺ–Ļ –Ņ–Ķ—Ä–Ķ–ī–į—á–Ķ –ł –ľ–ĺ–≥—É—ā –Ī—č—ā—Ć —Ā–≤—Ź–∑–į–Ĺ—č —Ā —ā–į–ļ–ł–ľ–ł –≤–į–∂–Ĺ–Ķ–Ļ—ą–ł–ľ–ł —Ą—É–Ĺ–ļ—Ü–ł—Ź–ľ–ł –ľ–ĺ–∑–≥–į, –ļ–į–ļ –Ņ–ł—Č–Ķ–≤–ĺ–Ķ –Ņ–ĺ–≤–Ķ–ī–Ķ–Ĺ–ł–Ķ, –ĺ–Ī—É—á–Ķ–Ĺ–ł–Ķ –ł –Ņ–į–ľ—Ź—ā—Ć [7].
–ė–ľ–Ķ—é—ā—Ā—Ź –ī–į–Ĺ–Ĺ—č–Ķ –ĺ —ā–ĺ–ľ, —á—ā–ĺ –ł–Ĺ—Ā—É–Ľ–ł–Ĺ –ľ–ĺ–∂–Ķ—ā –Ņ—Ä–ł–Ĺ–ł–ľ–į—ā—Ć –Ĺ–Ķ–Ņ–ĺ—Ā—Ä–Ķ–ī—Ā—ā–≤–Ķ–Ĺ–Ĺ–ĺ–Ķ —É—á–į—Ā—ā–ł–Ķ –≤ —Ä–Ķ–į–Ľ–ł–∑–į—Ü–ł–ł –Ĺ–Ķ–ļ–ĺ—ā–ĺ—Ä—č—Ö –ļ–ĺ–≥–Ĺ–ł—ā–ł–≤–Ĺ—č—Ö —Ą—É–Ĺ–ļ—Ü–ł–Ļ, –į –Ĺ–į—Ä—É—ą–Ķ–Ĺ–ł—Ź –Ķ–≥–ĺ –ľ–Ķ—ā–į–Ī–ĺ–Ľ–ł–∑–ľ–į –ľ–ĺ–≥—É—ā —Ā–ĺ–Ņ—Ä–ĺ–≤–ĺ–∂–ī–į—ā—Ć—Ā—Ź —Ä–į–∑–≤–ł—ā–ł–Ķ–ľ —Ä—Ź–ī–į –Ĺ–Ķ–≤—Ä–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ł—Ö —Ā–ł–Ĺ–ī—Ä–ĺ–ľ–ĺ–≤ –ł –ļ–ĺ–≥–Ĺ–ł—ā–ł–≤–Ĺ—č—Ö —Ä–į—Ā—Ā—ā—Ä–ĺ–Ļ—Ā—ā–≤. –ü–ĺ–ľ–ł–ľ–ĺ –≤—č–Ņ–ĺ–Ľ–Ĺ–Ķ–Ĺ–ł—Ź –≤ –≥–ĺ–Ľ–ĺ–≤–Ĺ–ĺ–ľ –ľ–ĺ–∑–≥–Ķ –ľ–Ķ–ī–ł–į—ā–ĺ—Ä–Ĺ—č—Ö —Ą—É–Ĺ–ļ—Ü–ł–Ļ, –ł–Ĺ—Ā—É–Ľ–ł–Ĺ –Ņ—Ä–ł–Ĺ–ł–ľ–į–Ķ—ā —É—á–į—Ā—ā–ł–Ķ –≤ —Ä–Ķ–≥—É–Ľ—Ź—Ü–ł–ł —Ā–ł–Ĺ—ā–Ķ–∑–į –Ī–Ķ–Ľ–ļ–į-–Ņ—Ä–Ķ–ī—ą–Ķ—Ā—ā–≤–Ķ–Ĺ–Ĺ–ł–ļ–į –į–ľ–ł–Ľ–ĺ–ł–ī–į –ł –Ņ—Ä–ĺ–ī—É–ļ—ā–į –Ķ–≥–ĺ –ľ–Ķ—ā–į–Ī–ĺ–Ľ–ł–∑–ľ–į ő≤-–į–ľ–ł–Ľ–ĺ–ł–ī–į ‚Äď –ĺ—Ā–Ĺ–ĺ–≤–Ĺ–ĺ–≥–ĺ –ļ–ĺ–ľ–Ņ–ĺ–Ĺ–Ķ–Ĺ—ā–į –į–ľ–ł–Ľ–ĺ–ł–ī–Ĺ—č—Ö –ĺ—ā–Ľ–ĺ–∂–Ķ–Ĺ–ł–Ļ, –į —ā–į–ļ–∂–Ķ —Ä–Ķ–≥—É–Ľ–ł—Ä—É–Ķ—ā —Ą–ĺ—Ā—Ą–ĺ—Ä–ł–Ľ–ł—Ä–ĺ–≤–į–Ĺ–ł–Ķ —ā–į—É-–Ņ—Ä–ĺ—ā–Ķ–ł–Ĺ–į, —Ā–ĺ—Ā—ā–į–≤–Ľ—Ź—é—Č–Ķ–≥–ĺ –ĺ—Ā–Ĺ–ĺ–≤—É –Ĺ–Ķ–Ļ—Ä–ĺ—Ą–ł–Ī—Ä–ł–Ľ–Ľ—Ź—Ä–Ĺ—č—Ö –ĺ–Ī—Ä–į–∑–ĺ–≤–į–Ĺ–ł–Ļ [8]. –Ę–į–ļ–ł–ľ –ĺ–Ī—Ä–į–∑–ĺ–ľ, –ľ–ĺ–∂–Ĺ–ĺ –≥–ĺ–≤–ĺ—Ä–ł—ā—Ć –ĺ —Ā–≤—Ź–∑–ł –°–Ē 2 –Ĺ–Ķ —ā–ĺ–Ľ—Ć–ļ–ĺ —Ā —Ā–ĺ—Ā—É–ī–ł—Ā—ā—č–ľ –Ņ–ĺ—Ä–į–∂–Ķ–Ĺ–ł–Ķ–ľ –≥–ĺ–Ľ–ĺ–≤–Ĺ–ĺ–≥–ĺ –ľ–ĺ–∑–≥–į, –Ĺ–ĺ –ł —Ā –Ĺ–Ķ–Ļ—Ä–ĺ–ī–Ķ–≥–Ķ–Ĺ–Ķ—Ä–į—ā–ł–≤–Ĺ—č–ľ –Ņ—Ä–ĺ—Ü–Ķ—Ā—Ā–ĺ–ľ, –Ľ–Ķ–∂–į—Č–ł–ľ –≤ –ĺ—Ā–Ĺ–ĺ–≤–Ķ –ī–Ķ–ľ–Ķ–Ĺ—Ü–ł–ł, —á—ā–ĺ –Ņ–ĺ–ī—ā–≤–Ķ—Ä–∂–ī–į–Ķ—ā—Ā—Ź –≤ —Ü–Ķ–Ľ–ĺ–ľ —Ä—Ź–ī–Ķ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–Ļ, –≤—č—Ź–≤–ł–≤—ą–ł—Ö —ć—ā—É —Ā–≤—Ź–∑—Ć [9]. –ď–ł–Ņ–Ķ—Ä–ł–Ĺ—Ā—É–Ľ–ł–Ĺ–Ķ–ľ–ł—Ź –ľ–ĺ–∂–Ķ—ā –ĺ–Ņ—Ä–Ķ–ī–Ķ–Ľ—Ź—ā—Ć –ļ–ĺ–≥–Ĺ–ł—ā–ł–≤–Ĺ–ĺ–Ķ —Ā–Ĺ–ł–∂–Ķ–Ĺ–ł–Ķ, –į –Ĺ–į—Ä—É—ą–Ķ–Ĺ–ł—Ź –≤ —Ā–ł—Ā—ā–Ķ–ľ–Ķ —Ā–ł–Ĺ—ā–Ķ–∑–į –ł —Ā–Ķ–ļ—Ä–Ķ—Ü–ł–ł –ł–Ĺ—Ā—É–Ľ–ł–Ĺ–į ‚Äď –Ĺ–Ķ–≥–į—ā–ł–≤–Ĺ–ĺ –≤–Ľ–ł—Ź—ā—Ć –Ĺ–į –ļ–ĺ–≥–Ĺ–ł—ā–ł–≤–Ĺ—č–Ķ —Ą—É–Ĺ–ļ—Ü–ł–ł [10]. –£—Ā—ā–į–Ĺ–ĺ–≤–Ľ–Ķ–Ĺ–ĺ —ā–į–ļ–∂–Ķ, —á—ā–ĺ —É –Ī–ĺ–Ľ—Ć–Ĺ—č—Ö –°–Ē 2 —ā–ł–Ņ–į, –Ņ–ĺ–Ľ—É—á–į—é—Č–ł—Ö –ł–Ĺ—Ā—É–Ľ–ł–Ĺ, –≤—č—Ā–ĺ–ļ —Ä–ł—Ā–ļ —Ä–į–∑–≤–ł—ā–ł—Ź –ī–Ķ–ľ–Ķ–Ĺ—Ü–ł–ł, –ļ–ĺ—ā–ĺ—Ä–į—Ź –Ĺ–Ķ –Ņ—Ä–ĺ—Ā—ā–ĺ –ĺ—ā—Ä–į–∂–į–Ķ—ā —ā—Ź–∂–Ķ—Ā—ā—Ć –ī–ł–į–Ī–Ķ—ā–į, –Ĺ–ĺ –ł –ľ–ĺ–∂–Ķ—ā –Ī—č—ā—Ć –Ĺ–Ķ–Ņ–ĺ—Ā—Ä–Ķ–ī—Ā—ā–≤–Ķ–Ĺ–Ĺ–ĺ —Ā–≤—Ź–∑–į–Ĺ–į —Ā –Ĺ–į–∑–Ĺ–į—á–Ķ–Ĺ–ł–Ķ–ľ –ł–Ĺ—Ā—É–Ľ–ł–Ĺ–ĺ—ā–Ķ—Ä–į–Ņ–ł–ł [11]. –Ē–į–Ĺ–Ĺ—č–Ķ –Ņ–ĺ—Ā–Ľ–Ķ–ī–Ĺ–ł—Ö –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–Ļ –≤ –ĺ–Ī–Ľ–į—Ā—ā–ł –ł–∑—É—á–Ķ–Ĺ–ł—Ź –ļ–ĺ–≥–Ĺ–ł—ā–ł–≤–Ĺ–ĺ–≥–ĺ –∑–ī–ĺ—Ä–ĺ–≤—Ć—Ź –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ —Ā –°–Ē 2 —ā–ł–Ņ–į –Ņ–ĺ–ļ–į–∑—č–≤–į—é—ā, —á—ā–ĺ —Ä–Ķ–∑–ļ–ĺ –≤—č—Ä–į–∂–Ķ–Ĺ–Ĺ–ĺ–Ķ —Ā–Ĺ–ł–∂–Ķ–Ĺ–ł–Ķ –ļ–ĺ–≥–Ĺ–ł—ā–ł–≤–Ĺ—č—Ö —Ā–Ņ–ĺ—Ā–ĺ–Ī–Ĺ–ĺ—Ā—ā–Ķ–Ļ –ī–Ķ–ľ–ĺ–Ĺ—Ā—ā—Ä–ł—Ä—É—é—ā –ī–į–∂–Ķ –Ņ–į—Ü–ł–Ķ–Ĺ—ā—č —Ā—Ä–Ķ–ī–Ĺ–Ķ–≥–ĺ –≤–ĺ–∑—Ä–į—Ā—ā–į –ļ–į–ļ –≤ –ī–Ķ–Ī—é—ā–Ķ –ī–ł–į–Ī–Ķ—ā–į, —ā–į–ļ –ł —Ā –Ĺ–Ķ–Ī–ĺ–Ľ—Ć—ą–ĺ–Ļ –ī–Ľ–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ—Ā—ā—Ć—é –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł—Ź [12].
–Ę–į–ļ–ł–ľ –ĺ–Ī—Ä–į–∑–ĺ–ľ, –ł–Ĺ—Ā—É–Ľ–ł–Ĺ–ĺ—ā–Ķ—Ä–į–Ņ–ł—Ź –ł–Ľ–ł –ł—Ā–Ņ–ĺ–Ľ—Ć–∑–ĺ–≤–į–Ĺ–ł–Ķ —Ā—Ä–Ķ–ī—Ā—ā–≤, —á—Ä–Ķ–∑–ľ–Ķ—Ä–Ĺ–ĺ —Ā—ā–ł–ľ—É–Ľ–ł—Ä—É—é—Č–ł—Ö —Ā–Ķ–ļ—Ä–Ķ—Ü–ł—é –ł–Ĺ—Ā—É–Ľ–ł–Ĺ–į, —Ā –ĺ–ī–Ĺ–ĺ–Ļ —Ā—ā–ĺ—Ä–ĺ–Ĺ—č –ĺ—Ā–Ľ–į–Ī–Ľ—Ź—é—ā —ā–ĺ–ļ—Ā–ł—á–Ķ—Ā–ļ–ł–Ļ —ć—Ą—Ą–Ķ–ļ—ā —Ö—Ä–ĺ–Ĺ–ł—á–Ķ—Ā–ļ–ĺ–Ļ –≥–ł–Ņ–Ķ—Ä–≥–Ľ–ł–ļ–Ķ–ľ–ł–ł, —Ā –ī—Ä—É–≥–ĺ–Ļ —Ā—ā–ĺ—Ä–ĺ–Ĺ—č –Ņ—Ä–ł–≤–ĺ–ī—Ź—ā –ļ –≤—č—Ä–į–∂–Ķ–Ĺ–Ĺ–ĺ–Ļ –≥–ł–Ņ–Ķ—Ä–ł–Ĺ—Ā—É–Ľ–ł–Ĺ–Ķ–ľ–ł–ł. –ü–ĺ—Ā–Ľ–Ķ–ī—Ā—ā–≤–ł—Ź–ľ–ł –≥–ł–Ņ–Ķ—Ä–ł–Ĺ—Ā—É–Ľ–ł–Ĺ–Ķ–ľ–ł–ł –ľ–ĺ–≥—É—ā –Ī—č—ā—Ć –Ņ—Ä–ĺ–≥—Ä–Ķ—Ā—Ā–ł—Ä–ĺ–≤–į–Ĺ–ł–Ķ –ĺ–∂–ł—Ä–Ķ–Ĺ–ł—Ź –ł –ľ–į–ļ—Ä–ĺ—Ā–ĺ—Ā—É–ī–ł—Ā—ā—č—Ö –ĺ—Ā–Ľ–ĺ–∂–Ĺ–Ķ–Ĺ–ł–Ļ, –į —ā–į–ļ–∂–Ķ —Ā–≤—Ź–∑–į–Ĺ–Ĺ—č–Ķ —Ā –Ĺ–ł–ľ–ł –Ĺ–į—Ä–į—Ā—ā–į—é—Č–ł–Ķ –ł–∑–ľ–Ķ–Ĺ–Ķ–Ĺ–ł—Ź –≤ —Ā–ł–Ĺ–į–Ņ—ā–ł—á–Ķ—Ā–ļ–ł—Ö —Ā—ā—Ä—É–ļ—ā—É—Ä–į—Ö –ł –ļ–Ľ–Ķ—ā–ĺ—á–Ĺ—č—Ö –ľ–Ķ–ľ–Ī—Ä–į–Ĺ–į—Ö —Ü–Ķ–Ĺ—ā—Ä–į–Ľ—Ć–Ĺ–ĺ–Ļ –Ĺ–Ķ—Ä–≤–Ĺ–ĺ–Ļ —Ā–ł—Ā—ā–Ķ–ľ—č, —á—ā–ĺ —Ā–Ņ–ĺ—Ā–ĺ–Ī—Ā—ā–≤—É–Ķ—ā —Ä–į–∑–≤–ł—ā–ł—é –ī–Ķ–ľ–Ķ–Ĺ—Ü–ł–ł. –í –Ĺ–į—Ā—ā–ĺ—Ź—Č–Ķ–Ķ –≤—Ä–Ķ–ľ—Ź –Ĺ–Ķ—ā –ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ł—Ö –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–Ļ, –≥–ī–Ķ –Ī—č–Ľ–į –Ī—č –ī–ĺ–ļ–į–∑–į–Ĺ–į —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć —Ā–į—Ö–į—Ä–ĺ—Ā–Ĺ–ł–∂–į—é—Č–ł—Ö –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–ĺ–≤, –Ņ—Ä–ł–ľ–Ķ–Ĺ—Ź–Ķ–ľ—č—Ö –≤ —Ā–ĺ–≤—Ä–Ķ–ľ–Ķ–Ĺ–Ĺ–ĺ–Ļ —ā–Ķ—Ä–į–Ņ–ł–ł –°–Ē (–ĺ—Ā–ĺ–Ī–Ķ–Ĺ–Ĺ–ĺ 2 —ā–ł–Ņ–į), –≤ –ĺ—ā–Ĺ–ĺ—ą–Ķ–Ĺ–ł–ł –Ņ—Ä–ĺ—Ą–ł–Ľ–į–ļ—ā–ł–ļ–ł —Ä–į–∑–≤–ł—ā–ł—Ź –ļ–ĺ–≥–Ĺ–ł—ā–ł–≤–Ĺ—č—Ö –Ĺ–į—Ä—É—ą–Ķ–Ĺ–ł–Ļ [13].
–ė–∑—É—á–Ķ–Ĺ–ł–Ķ –Ņ–į—ā–ĺ—Ą–ł–∑–ł–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ł—Ö –ĺ—Ā–Ĺ–ĺ–≤ —Ä–į–∑–≤–ł—ā–ł—Ź –°–Ē –ľ–ĺ–∂–Ķ—ā –ī–į—ā—Ć –Ņ—Ä–Ķ–ī—Ā—ā–į–≤–Ľ–Ķ–Ĺ–ł–Ķ –ĺ –ľ–į—Ā—ą—ā–į–Ī–į—Ö —Ä–į—Ā–Ņ—Ä–ĺ—Ā—ā—Ä–į–Ĺ–Ķ–Ĺ–Ĺ–ĺ—Ā—ā–ł —Ā–≤—Ź–∑–į–Ĺ–Ĺ—č—Ö —Ā –Ĺ–ł–ľ —Ā–ĺ—Ā—ā–ĺ—Ź–Ĺ–ł–Ļ –ł –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł–Ļ, –į —ā–į–ļ–∂–Ķ —Ä–į—Ā—ą–ł—Ä–ł—ā—Ć –≤–ĺ–∑–ľ–ĺ–∂–Ĺ–ĺ—Ā—ā–ł –ł—Ö —Ā–≤–ĺ–Ķ–≤—Ä–Ķ–ľ–Ķ–Ĺ–Ĺ–ĺ–Ļ –ļ–ĺ—Ä—Ä–Ķ–ļ—Ü–ł–ł. –†–į–∑–≤–ł—ā–ł–Ķ –Ĺ–į—Ä—É—ą–Ķ–Ĺ–ł–Ļ —É–≥–Ľ–Ķ–≤–ĺ–ī–Ĺ–ĺ–≥–ĺ –ĺ–Ī–ľ–Ķ–Ĺ–į –ł –ł—Ö —Ā–≤—Ź–∑—Ć —Ā –Ĺ–į—Ä—É—ą–Ķ–Ĺ–ł—Ź–ľ–ł –∂–ł—Ä–ĺ–≤–ĺ–≥–ĺ –ĺ–Ī–ľ–Ķ–Ĺ–į –í –ļ—Ä—É–Ņ–Ĺ—č—Ö —ć–Ņ–ł–ī–Ķ–ľ–ł–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ł—Ö –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź—Ö –Ī—č–Ľ–ĺ —É–Ī–Ķ–ī–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ –Ņ–ĺ–ļ–į–∑–į–Ĺ–ĺ, —á—ā–ĺ –ī–ł–į–≥–Ĺ–ĺ—Ā—ā–ł–ļ–į –Ņ–į—ā–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ł—Ö —Ā–ĺ—Ā—ā–ĺ—Ź–Ĺ–ł–Ļ –≥–ł–Ņ–Ķ—Ä–≥–Ľ–ł–ļ–Ķ–ľ–ł–ł —ā–ĺ–Ľ—Ć–ļ–ĺ –Ņ–ĺ —É—Ä–ĺ–≤–Ĺ—é –≥–Ľ—é–ļ–ĺ–∑—č –ļ—Ä–ĺ–≤–ł –Ĺ–į—ā–ĺ—Č–į–ļ (—Ä–Ķ–ļ–ĺ–ľ–Ķ–Ĺ–ī–į—Ü–ł–ł –ź–ľ–Ķ—Ä–ł–ļ–į–Ĺ—Ā–ļ–ĺ–Ļ –ī–ł–į–Ī–Ķ—ā–ł—á–Ķ—Ā–ļ–ĺ–Ļ –į—Ā—Ā–ĺ—Ü–ł–į—Ü–ł–ł ‚Äď ADA) –Ņ—Ä–ł–≤–ĺ–ī–ł—ā –ļ –Ĺ–Ķ–ī–ĺ–ĺ—Ü–Ķ–Ĺ–ļ–Ķ –ł—Ā—ā–ł–Ĺ–Ĺ–ĺ–Ļ —Ä–į—Ā–Ņ—Ä–ĺ—Ā—ā—Ä–į–Ĺ–Ķ–Ĺ–Ĺ–ĺ—Ā—ā–ł –ī–ł–į–Ī–Ķ—ā–į –Ī–ĺ–Ľ–Ķ–Ķ —á–Ķ–ľ –Ĺ–į —ā—Ä–Ķ—ā—Ć. –í –Ĺ–į—Ā—ā–ĺ—Ź—Č–Ķ–Ķ –≤—Ä–Ķ–ľ—Ź –ĺ–Ī—Č–Ķ–Ņ—Ä–ł–Ĺ—Ź—ā—č–ľ–ł –ī–ł–į–≥–Ĺ–ĺ—Ā—ā–ł—á–Ķ—Ā–ļ–ł–ľ–ł –Ľ–į–Ī–ĺ—Ä–į—ā–ĺ—Ä–Ĺ—č–ľ–ł –ļ—Ä–ł—ā–Ķ—Ä–ł—Ź–ľ–ł –°–Ē —Ź–≤–Ľ—Ź—é—ā—Ā—Ź –Ņ—Ä–Ķ–ī—Ā—ā–į–≤–Ľ–Ķ–Ĺ–Ĺ—č–Ķ –≤ —ā–į–Ī–Ľ–ł—Ü–Ķ 1 –Ņ–ĺ—Ä–ĺ–≥–ĺ–≤—č–Ķ —É—Ä–ĺ–≤–Ĺ–ł –≥–Ľ–ł–ļ–Ķ–ľ–ł–ł –Ĺ–į—ā–ĺ—Č–į–ļ –ł —á–Ķ—Ä–Ķ–∑ 2 —á–į—Ā–į –Ņ–ĺ—Ā–Ľ–Ķ –Ĺ–į–≥—Ä—É–∑–ļ–ł –≥–Ľ—é–ļ–ĺ–∑–ĺ–Ļ.
–°–ĺ–≥–Ľ–į—Ā–Ĺ–ĺ —Ā–ĺ–≤—Ä–Ķ–ľ–Ķ–Ĺ–Ĺ—č–ľ –Ņ—Ä–Ķ–ī—Ā—ā–į–≤–Ľ–Ķ–Ĺ–ł—Ź–ľ, –Ĺ–į—Ä—É—ą–Ķ–Ĺ–Ĺ–į—Ź —ā–ĺ–Ľ–Ķ—Ä–į–Ĺ—ā–Ĺ–ĺ—Ā—ā—Ć –ļ –≥–Ľ—é–ļ–ĺ–∑–Ķ (–Ě–Ę–ď) –ł –Ĺ–į—Ä—É—ą–Ķ–Ĺ–Ĺ–į—Ź –≥–Ľ–ł–ļ–Ķ–ľ–ł—Ź –Ĺ–į—ā–ĺ—Č–į–ļ (–Ě–ď–Ě) —Ā–Ņ–ĺ—Ā–ĺ–Ī–Ĺ—č –Ņ—Ä–ĺ–≥—Ä–Ķ—Ā—Ā–ł—Ä–ĺ–≤–į—ā—Ć —Ā –ł—Ā—Ö–ĺ–ī–ĺ–ľ –≤ –°–Ē 2 —ā–ł–Ņ–į. –í 1990-—Ö –≥–≥. —ć–ļ—Ā–Ņ–Ķ—Ä—ā—č –í–ě–ó –Ņ—Ä–Ķ–ī–Ľ–ĺ–∂–ł–Ľ–ł –ł—Ā–Ņ–ĺ–Ľ—Ć–∑–ĺ–≤–į—ā—Ć –Ĺ–ĺ–≤—č–Ļ —ā–Ķ—Ä–ľ–ł–Ĺ ‚Äď ¬ę–Ņ—Ä–Ķ–ī–ł–į–Ī–Ķ—ā¬Ľ, –ļ–ĺ—ā–ĺ—Ä—č–Ļ –ĺ–Ī—ä–Ķ–ī–ł–Ĺ–ł–Ľ –Ě–Ę–ď –ł –Ě–ď–Ě. –°–Ķ–≥–ĺ–ī–Ĺ—Ź –≤ –ľ–ł—Ä–Ķ –ĺ–ļ–ĺ–Ľ–ĺ 314 –ľ–Ľ–Ĺ —á–Ķ–Ľ–ĺ–≤–Ķ–ļ –ł–ľ–Ķ—é—ā –Ņ—Ä–Ķ–ī–ł–į–Ī–Ķ—ā, —á–Ķ—Ä–Ķ–∑ 20 –Ľ–Ķ—ā –ł—Ö —á–ł—Ā–Ľ–ĺ —É–≤–Ķ–Ľ–ł—á–ł—ā—Ā—Ź –≤ 1,5 —Ä–į–∑–į –ł —Ā–ĺ—Ā—ā–į–≤–ł—ā –ĺ–ļ–ĺ–Ľ–ĺ 500 –ľ–Ľ–Ĺ —á–Ķ–Ľ–ĺ–≤–Ķ–ļ, –ļ–ĺ—ā–ĺ—Ä—č–Ķ –Ņ–ĺ–Ņ–ĺ–Ľ–Ĺ—Ź—ā –ľ–Ĺ–ĺ–≥–ĺ–ľ–ł–Ľ–Ľ–ł–ĺ–Ĺ–Ĺ—É—é –į—Ä–ľ–ł—é –Ī–ĺ–Ľ—Ć–Ĺ—č—Ö —Ā–į—Ö–į—Ä–Ĺ—č–ľ –ī–ł–į–Ī–Ķ—ā–ĺ–ľ 2 —ā–ł–Ņ–į.
–£—Ā—ā–į–Ĺ–ĺ–≤–Ľ–Ķ–Ĺ–ĺ, —á—ā–ĺ —á–į—Ā—ā–ĺ—ā–į —Ä–į–∑–≤–ł—ā–ł—Ź –°–Ē —É –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ —Ā –Ě–Ę–ď –ł –Ě–ď–Ě –Ņ—Ä–ł–ľ–Ķ—Ä–Ĺ–ĺ –ĺ–ī–ł–Ĺ–į–ļ–ĺ–≤–į. –ü–ĺ—Ā–ļ–ĺ–Ľ—Ć–ļ—É –Ě–Ę–ď –Ī–ĺ–Ľ–Ķ–Ķ —Ä–į—Ā–Ņ—Ä–ĺ—Ā—ā—Ä–į–Ĺ–Ķ–Ĺ–į –≤ –Ņ–ĺ–Ņ—É–Ľ—Ź—Ü–ł—Ź—Ö, —á–Ķ–ľ –Ě–ď–Ě, –ł–ľ–Ķ–Ĺ–Ĺ–ĺ —Ā –Ņ–Ķ—Ä–≤—č–ľ —Ā–ĺ—Ā—ā–ĺ—Ź–Ĺ–ł–Ķ–ľ —Ā–≤—Ź–∑–į–Ĺ–ĺ –Ī–ĺ–Ľ—Ć—ą–Ķ–Ķ —á–ł—Ā–Ľ–ĺ –Ĺ–ĺ–≤—č—Ö —Ā–Ľ—É—á–į–Ķ–≤ –°–Ē. –ü–ĺ –ī–į–Ĺ–Ĺ—č–ľ —ć–Ņ–ł–ī–Ķ–ľ–ł–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ł—Ö –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–Ļ, –≤–Ķ—Ä–ĺ—Ź—ā–Ĺ–ĺ—Ā—ā—Ć –Ņ–Ķ—Ä–Ķ—Ö–ĺ–ī–į –Ņ—Ä–Ķ–ī–ł–į–Ī–Ķ—ā–į –≤ –°–Ē 2 —ā–ł–Ņ–į –∑–į–≤–ł—Ā–ł—ā –ĺ—ā –≤–ĺ–∑—Ä–į—Ā—ā–į –Ī–ĺ–Ľ—Ć–Ĺ–ĺ–≥–ĺ, —Ä–į—Ā–ĺ–≤–ĺ–Ļ –Ņ—Ä–ł–Ĺ–į–ī–Ľ–Ķ–∂–Ĺ–ĺ—Ā—ā–ł, —Ā—ā–Ķ–Ņ–Ķ–Ĺ–ł –ĺ–∂–ł—Ä–Ķ–Ĺ–ł—Ź. –í —Ā—Ä–Ķ–ī–Ĺ–Ķ–ľ —á–į—Ā—ā–ĺ—ā–į —Ä–į–∑–≤–ł—ā–ł—Ź –°–Ē 2 —ā–ł–Ņ–į —É –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ —Ā –Ņ—Ä–Ķ–ī–ł–į–Ī–Ķ—ā–ĺ–ľ —Ā–ĺ—Ā—ā–į–≤–Ľ—Ź–Ķ—ā 5% –≤ –≥–ĺ–ī (–ĺ—ā 3,6 –ī–ĺ 8,7%). –ü—Ä–ł –Ņ—Ź—ā–ł–Ľ–Ķ—ā–Ĺ–Ķ–ľ –Ĺ–į–Ī–Ľ—é–ī–Ķ–Ĺ–ł–ł –∑–į —ā–į–ļ–ł–ľ–ł –Ī–ĺ–Ľ—Ć–Ĺ—č–ľ–ł –°–Ē 2 —ā–ł–Ņ–į —Ä–į–∑–≤–ł–≤–į–Ķ—ā—Ā—Ź —É 35‚Äď40%, –į –Ņ—Ä–ł —Ā–ĺ—á–Ķ—ā–į–Ĺ–ł–ł –Ě–Ę–ď –ł –Ě–ď–Ę ‚Äď —É 65% –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ [6]. –í —ć—ā–ĺ–Ļ —Ā–≤—Ź–∑–ł –ł–∑—É—á–Ķ–Ĺ–ł—é –ĺ—Ā–ĺ–Ī–Ķ–Ĺ–Ĺ–ĺ—Ā—ā–Ķ–Ļ —ā–Ķ—á–Ķ–Ĺ–ł—Ź –Ņ—Ä–Ķ–ī–ł–į–Ī–Ķ—ā–į, –Ķ–≥–ĺ —Ä–į–Ĺ–Ĺ–Ķ–Ļ –ī–ł–į–≥–Ĺ–ĺ—Ā—ā–ł–ļ–Ķ –ł –≤–ĺ–∑–ľ–ĺ–∂–Ĺ–ĺ—Ā—ā–Ķ–Ļ –Ņ–Ķ—Ä–≤–ł—á–Ĺ–ĺ–Ļ –ł –≤—ā–ĺ—Ä–ł—á–Ĺ–ĺ–Ļ –Ņ—Ä–ĺ—Ą–ł–Ľ–į–ļ—ā–ł–ļ–ł –Ņ—Ä–ł–ī–į–Ķ—ā—Ā—Ź –Ī–ĺ–Ľ—Ć—ą–ĺ–Ķ –∑–Ĺ–į—á–Ķ–Ĺ–ł–Ķ. –í—Ä–į—á—É –Ľ—é–Ī–ĺ–Ļ —Ā–Ņ–Ķ—Ü–ł–į–Ľ—Ć–Ĺ–ĺ—Ā—ā–ł —Ä–Ķ–ļ–ĺ–ľ–Ķ–Ĺ–ī—É–Ķ—ā—Ā—Ź —É–ī–Ķ–Ľ—Ź—ā—Ć –ĺ—Ā–ĺ–Ī–ĺ–Ķ –≤–Ĺ–ł–ľ–į–Ĺ–ł–Ķ —Ą–į–ļ—ā–ĺ—Ä–į–ľ —Ä–ł—Ā–ļ–į —Ä–į–∑–≤–ł—ā–ł—Ź –°–Ē 2 —ā–ł–Ņ–į (—Ä–ł—Ā. 1) –ł –Ņ—Ä–ĺ–≤–ĺ–ī–ł—ā—Ć —Ā–ļ—Ä–ł–Ĺ–ł–Ĺ–≥ (—ā–į–Ī–Ľ. 2), –Ņ–ĺ—Ā–ļ–ĺ–Ľ—Ć–ļ—É —Ä–į–Ĺ–ĺ –≤—č—Ź–≤–Ľ–Ķ–Ĺ–Ĺ—č–Ļ –Ņ—Ä–Ķ–ī–ł–į–Ī–Ķ—ā –ł –Ņ—Ä–į–≤–ł–Ľ—Ć–Ĺ—č–Ķ –Ņ—Ä–ĺ—Ą–ł–Ľ–į–ļ—ā–ł—á–Ķ—Ā–ļ–ł–Ķ –ľ–Ķ—Ä—č –ľ–ĺ–≥—É—ā –Ņ–ĺ–≤–Ľ–ł—Ź—ā—Ć –Ĺ–į —Ā—É–ī—Ć–Ī—É –Ņ–į—Ü–ł–Ķ–Ĺ—ā–į.
–ď–Ľ–į–≤–Ĺ—č–ľ —Ą–į–ļ—ā–ĺ—Ä–ĺ–ľ —Ä–ł—Ā–ļ–į —Ä–į–∑–≤–ł—ā–ł—Ź –°–Ē 2 —ā–ł–Ņ–į —É –Ņ–į—Ü–ł–Ķ–Ĺ—ā–į —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź –Ĺ–į–Ľ–ł—á–ł–Ķ –ł–∑–Ī—č—ā–ĺ—á–Ĺ–ĺ–≥–ĺ –≤–Ķ—Ā–į, –ĺ—Ā–ĺ–Ī–Ķ–Ĺ–Ĺ–ĺ –į–Ī–ī–ĺ–ľ–ł–Ĺ–į–Ľ—Ć–Ĺ–ĺ–Ķ –ĺ–∂–ł—Ä–Ķ–Ĺ–ł–Ķ. –í –Ņ–ĺ—Ā–Ľ–Ķ–ī–Ĺ–ł–Ķ –ī–Ķ—Ā—Ź—ā–ł–Ľ–Ķ—ā–ł—Ź —É—á–Ķ–Ĺ—č–Ķ –≤—č—Ā–ļ–į–∑—č–≤–į—é—ā –Ņ—Ä–Ķ–ī–Ņ–ĺ–Ľ–ĺ–∂–Ķ–Ĺ–ł—Ź –ĺ–Ī –ĺ–Ī—Č–Ĺ–ĺ—Ā—ā–ł –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł–Ļ, –į—Ā—Ā–ĺ—Ü–ł–ł—Ä–ĺ–≤–į–Ĺ–Ĺ—č—Ö —Ā –ł–∑–Ī—č—ā–ĺ—á–Ĺ—č–ľ –≤–Ķ—Ā–ĺ–ľ, –ł —Ä–į–∑–Ľ–ł—á–Ĺ—č—Ö –ľ–Ķ—ā–į–Ī–ĺ–Ľ–ł—á–Ķ—Ā–ļ–ł—Ö –Ĺ–į—Ä—É—ą–Ķ–Ĺ–ł–Ļ. –í 1960-–Ķ –≥–≥. –ī–Ķ–Ľ–į–Ľ–ł—Ā—Ć –Ņ–ĺ–Ņ—č—ā–ļ–ł –ĺ–Ī—ä–Ķ–ī–ł–Ĺ–Ķ–Ĺ–ł—Ź –≤–∑–į–ł–ľ–ĺ—Ā–≤—Ź–∑–į–Ĺ–Ĺ—č—Ö –ľ–Ķ—ā–į–Ī–ĺ–Ľ–ł—á–Ķ—Ā–ļ–ł—Ö –Ĺ–į—Ä—É—ą–Ķ–Ĺ–ł–Ļ, —É—Ā–ļ–ĺ—Ä—Ź—é—Č–ł—Ö —Ä–į–∑–≤–ł—ā–ł–Ķ –ľ–į–ļ—Ä–ĺ—Ā–ĺ—Ā—É–ī–ł—Ā—ā—č—Ö –į—ā–Ķ—Ä–ĺ—Ā–ļ–Ľ–Ķ—Ä–ĺ—ā–ł—á–Ķ—Ā–ļ–ł—Ö –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł–Ļ –ł –°–Ē 2 —ā–ł–Ņ–į. –í 1988 –≥. –į–ľ–Ķ—Ä–ł–ļ–į–Ĺ—Ā–ļ–ł–Ļ —É—á–Ķ–Ĺ—č–Ļ Gerald Reaven, –ĺ–Ī—ä–Ķ–ī–ł–Ĺ–ł–≤ –Ĺ–į—Ä—É—ą–Ķ–Ĺ–ł—Ź —É–≥–Ľ–Ķ–≤–ĺ–ī–Ĺ–ĺ–≥–ĺ –ĺ–Ī–ľ–Ķ–Ĺ–į, –į—Ä—ā–Ķ—Ä–ł–į–Ľ—Ć–Ĺ—É—é –≥–ł–Ņ–Ķ—Ä—ā–Ķ–Ĺ–∑–ł—é –ł –ī–ł—Ā–Ľ–ł–Ņ–ł–ī–Ķ–ľ–ł—é –Ņ–ĺ–Ĺ—Ź—ā–ł–Ķ–ľ ¬ę—Ā–ł–Ĺ–ī—Ä–ĺ–ľ¬†–•¬Ľ, –≤–Ņ–Ķ—Ä–≤—č–Ķ –≤—č—Ā–ļ–į–∑–į–Ľ –Ņ—Ä–Ķ–ī–Ņ–ĺ–Ľ–ĺ–∂–Ķ–Ĺ–ł–Ķ –ĺ —ā–ĺ–ľ, —á—ā–ĺ –ĺ–Ī—Č–Ķ–Ļ –ĺ—Ā–Ĺ–ĺ–≤–ĺ–Ļ —ć—ā–ł—Ö –Ĺ–į—Ä—É—ą–Ķ–Ĺ–ł–Ļ –ľ–ĺ–∂–Ķ—ā –Ī—č—ā—Ć –ł–Ĺ—Ā—É–Ľ–ł–Ĺ–ĺ—Ä–Ķ–∑–ł—Ā—ā–Ķ–Ĺ—ā–Ĺ–ĺ—Ā—ā—Ć –ł –ļ–ĺ–ľ–Ņ–Ķ–Ĺ—Ā–į—ā–ĺ—Ä–Ĺ–į—Ź –≥–ł–Ņ–Ķ—Ä–ł–Ĺ—Ā—É–Ľ–ł–Ĺ–Ķ–ľ–ł—Ź [14]. –í –ļ–ĺ–Ĺ—Ü–Ķ –Ņ—Ä–ĺ—ą–Ľ–ĺ–≥–ĺ –≤–Ķ–ļ–į –ľ–Ķ—ā–į–Ī–ĺ–Ľ–ł—á–Ķ—Ā–ļ–ł–Ķ –Ĺ–į—Ä—É—ą–Ķ–Ĺ–ł—Ź –ł –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł—Ź, —Ä–į–∑–≤–ł–≤–į—é—Č–ł–Ķ—Ā—Ź —É –Ľ–ł—Ü —Ā –ĺ–∂–ł—Ä–Ķ–Ĺ–ł–Ķ–ľ, –Ĺ–į–∑–≤–į–Ľ–ł –ľ–Ķ—ā–į–Ī–ĺ–Ľ–ł—á–Ķ—Ā–ļ–ł–ľ —Ā–ł–Ĺ–ī—Ä–ĺ–ľ–ĺ–ľ.
- –į–Ī–ī–ĺ–ľ–ł–Ĺ–į–Ľ—Ć–Ĺ–ĺ–Ķ –ĺ–∂–ł—Ä–Ķ–Ĺ–ł–Ķ;
- –ł–Ĺ—Ā—É–Ľ–ł–Ĺ–ĺ—Ä–Ķ–∑–ł—Ā—ā–Ķ–Ĺ—ā–Ĺ–ĺ—Ā—ā—Ć –ł –≥–ł–Ņ–Ķ—Ä–ł–Ĺ—Ā—É–Ľ–ł–Ĺ–Ķ–ľ–ł—Ź;
- –į—Ä—ā–Ķ—Ä–ł–į–Ľ—Ć–Ĺ–į—Ź –≥–ł–Ņ–Ķ—Ä—ā–Ķ–Ĺ–∑–ł—Ź;
- –≥–ł–Ņ–Ķ—Ä—ā—Ä–ł–≥–Ľ–ł—Ü–Ķ—Ä–ł–ī–Ķ–ľ–ł—Ź;
- –Ņ–ĺ–≤—č—ą–Ķ–Ĺ–ł–Ķ —Ö–ĺ–Ľ–Ķ—Ā—ā–Ķ—Ä–ł–Ĺ–į –Ľ–ł–Ņ–ĺ–Ņ—Ä–ĺ—ā–Ķ–ł–ī–ĺ–≤ –Ĺ–ł–∑–ļ–ĺ–Ļ –Ņ–Ľ–ĺ—ā–Ĺ–ĺ—Ā—ā–ł (–•–° –õ–ü–Ě–ü) –ł —Ā–Ĺ–ł–∂–Ķ–Ĺ–ł–Ķ —Ö–ĺ–Ľ–Ķ—Ā—ā–Ķ—Ä–ł–Ĺ–į –Ľ–ł–Ņ–ĺ–Ņ—Ä–ĺ—ā–Ķ–ł–ī–ĺ–≤ –≤—č—Ā–ĺ–ļ–ĺ–Ļ –Ņ–Ľ–ĺ—ā–Ĺ–ĺ—Ā—ā–ł (–•–° –õ–ü–í–ü);
- –Ĺ–į—Ä—É—ą–Ķ–Ĺ–ł–Ķ —ā–ĺ–Ľ–Ķ—Ä–į–Ĺ—ā–Ĺ–ĺ—Ā—ā–ł –ļ –≥–Ľ—é–ļ–ĺ–∑–Ķ –ł/–ł–Ľ–ł –Ĺ–į—Ä—É—ą–Ķ–Ĺ–Ĺ–į—Ź –≥–Ľ–ł–ļ–Ķ–ľ–ł—Ź –Ĺ–į—ā–ĺ—Č–į–ļ;
- –Ĺ–į—Ä—É—ą–Ķ–Ĺ–ł—Ź —Ā–ł—Ā—ā–Ķ–ľ—č –≥–Ķ–ľ–ĺ—Ā—ā–į–∑–į;
- —Ö—Ä–ĺ–Ĺ–ł—á–Ķ—Ā–ļ–ĺ–Ķ —Ā–ĺ—Ā—É–ī–ł—Ā—ā–ĺ–Ķ –≤–ĺ—Ā–Ņ–į–Ľ–Ķ–Ĺ–ł–Ķ.
–í –Ĺ–į—Ā—ā–ĺ—Ź—Č–Ķ–Ķ –≤—Ä–Ķ–ľ—Ź —Ā—É—Č–Ķ—Ā—ā–≤—É—é—ā 5 –≥—Ä—É–Ņ–Ņ –ī–ł–į–≥–Ĺ–ĺ—Ā—ā–ł—á–Ķ—Ā–ļ–ł—Ö –ļ—Ä–ł—ā–Ķ—Ä–ł–Ķ–≤ –ú–°. –ě—ā–Ķ—á–Ķ—Ā—ā–≤–Ķ–Ĺ–Ĺ—č–ľ–ł —É—á–Ķ–Ĺ—č–ľ–ł, —ć–ļ—Ā–Ņ–Ķ—Ä—ā–į–ľ–ł –í—Ā–Ķ—Ä–ĺ—Ā—Ā–ł–Ļ—Ā–ļ–ĺ–≥–ĺ –Ĺ–į—É—á–Ĺ–ĺ–≥–ĺ –ĺ–Ī—Č–Ķ—Ā—ā–≤–į –ļ–į—Ä–ī–ł–ĺ–Ľ–ĺ–≥–ĺ–≤ (–í–Ě–ě–ö) –Ī—č–Ľ–ł —Ä–į–∑—Ä–į–Ī–ĺ—ā–į–Ĺ—č –ł –ĺ–Ņ—É–Ī–Ľ–ł–ļ–ĺ–≤–į–Ĺ—č –ļ—Ä–ł—ā–Ķ—Ä–ł–ł –ľ–Ķ—ā–į–Ī–ĺ–Ľ–ł—á–Ķ—Ā–ļ–ĺ–≥–ĺ —Ā–ł–Ĺ–ī—Ä–ĺ–ľ–į [25]. –ě—Ā–Ĺ–ĺ–≤–Ĺ–ĺ–Ļ –ļ—Ä–ł—ā–Ķ—Ä–ł–Ļ: —Ü–Ķ–Ĺ—ā—Ä–į–Ľ—Ć–Ĺ—č–Ļ (–į–Ī–ī–ĺ–ľ–ł–Ĺ–į–Ľ—Ć–Ĺ—č–Ļ) —ā–ł–Ņ –ĺ–∂–ł—Ä–Ķ–Ĺ–ł—Ź ‚Äď –ĺ–ļ—Ä—É–∂–Ĺ–ĺ—Ā—ā—Ć —ā–į–Ľ–ł–ł (–ě–Ę) –Ī–ĺ–Ľ–Ķ–Ķ 80 —Ā–ľ —É –∂–Ķ–Ĺ—Č–ł–Ĺ –ł –Ī–ĺ–Ľ–Ķ–Ķ 94 —Ā–ľ —É –ľ—É–∂—á–ł–Ĺ.
–Ē–ĺ–Ņ–ĺ–Ľ–Ĺ–ł—ā–Ķ–Ľ—Ć–Ĺ—č–Ķ –ļ—Ä–ł—ā–Ķ—Ä–ł–ł:
- –į—Ä—ā–Ķ—Ä–ł–į–Ľ—Ć–Ĺ–į—Ź –≥–ł–Ņ–Ķ—Ä—ā–ĺ–Ĺ–ł—Ź (–ź–Ē ‚Č• 130/85 –ľ–ľ —Ä—ā. —Ā—ā.);
- –Ņ–ĺ–≤—č—ą–Ķ–Ĺ–ł–Ķ —É—Ä–ĺ–≤–Ĺ—Ź —ā—Ä–ł–≥–Ľ–ł—Ü–Ķ—Ä–ł–ī–ĺ–≤ (‚Č• 1,7 –ľ–ľ–ĺ–Ľ—Ć/–Ľ);
- —Ā–Ĺ–ł–∂–Ķ–Ĺ–ł–Ķ —É—Ä–ĺ–≤–Ĺ—Ź –•–° –õ–ü–í–ü (< 1,0 –ľ–ľ–ĺ–Ľ—Ć/–Ľ —É –ľ—É–∂—á–ł–Ĺ; < 1,2 –ľ–ľ–ĺ–Ľ—Ć/–Ľ —É –∂–Ķ–Ĺ—Č–ł–Ĺ);
- –Ņ–ĺ–≤—č—ą–Ķ–Ĺ–ł–Ķ —É—Ä–ĺ–≤–Ĺ—Ź –•–° –õ–ü–Ě–ü > 3,0 –ľ–ľ–ĺ–Ľ—Ć/–Ľ;
- –≥–ł–Ņ–Ķ—Ä–≥–Ľ–ł–ļ–Ķ–ľ–ł—Ź –Ĺ–į—ā–ĺ—Č–į–ļ (–≥–Ľ—é–ļ–ĺ–∑–į –≤ –Ņ–Ľ–į–∑–ľ–Ķ –ļ—Ä–ĺ–≤–ł –Ĺ–į—ā–ĺ—Č–į–ļ ‚Č• 6,1 –ľ–ľ–ĺ–Ľ—Ć/–Ľ);
- –Ĺ–į—Ä—É—ą–Ķ–Ĺ–ł–Ķ —ā–ĺ–Ľ–Ķ—Ä–į–Ĺ—ā–Ĺ–ĺ—Ā—ā–ł –ļ –≥–Ľ—é–ļ–ĺ–∑–Ķ (–≥–Ľ—é–ļ–ĺ–∑–į –≤ –Ņ–Ľ–į–∑–ľ–Ķ –ļ—Ä–ĺ–≤–ł —á–Ķ—Ä–Ķ–∑ 2 —á–į—Ā–į –Ņ–ĺ—Ā–Ľ–Ķ –Ĺ–į–≥—Ä—É–∑–ļ–ł –≥–Ľ—é–ļ–ĺ–∑–ĺ–Ļ –≤ –Ņ—Ä–Ķ–ī–Ķ–Ľ–į—Ö ‚Č• 7,8 –ł ‚ȧ 11,1 –ľ–ľ–ĺ–Ľ—Ć/–Ľ).
–Ě–į–Ľ–ł—á–ł–Ķ —É –Ņ–į—Ü–ł–Ķ–Ĺ—ā–į —Ü–Ķ–Ĺ—ā—Ä–į–Ľ—Ć–Ĺ–ĺ–≥–ĺ –ĺ–∂–ł—Ä–Ķ–Ĺ–ł—Ź –ł –ī–≤—É—Ö –ī–ĺ–Ņ–ĺ–Ľ–Ĺ–ł—ā–Ķ–Ľ—Ć–Ĺ—č—Ö –ļ—Ä–ł—ā–Ķ—Ä–ł–Ķ–≤ —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź –ĺ—Ā–Ĺ–ĺ–≤–į–Ĺ–ł–Ķ–ľ –ī–Ľ—Ź –ī–ł–į–≥–Ĺ–ĺ—Ā—ā–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź —É –Ĺ–Ķ–≥–ĺ –ľ–Ķ—ā–į–Ī–ĺ–Ľ–ł—á–Ķ—Ā–ļ–ĺ–≥–ĺ —Ā–ł–Ĺ–ī—Ä–ĺ–ľ–į. –°–ĺ—á–Ķ—ā–į–Ĺ–ł–Ķ –Ĺ–į—Ä—É—ą–Ķ–Ĺ–ł–Ļ, –ĺ–Ī—ä–Ķ–ī–ł–Ĺ–Ķ–Ĺ–Ĺ—č—Ö –Ņ–ĺ–Ĺ—Ź—ā–ł–Ķ–ľ ¬ę–ľ–Ķ—ā–į–Ī–ĺ–Ľ–ł—á–Ķ—Ā–ļ–ł–Ļ —Ā–ł–Ĺ–ī—Ä–ĺ–ľ¬Ľ, –≤ –∑–Ĺ–į—á–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ–Ļ —Ā—ā–Ķ–Ņ–Ķ–Ĺ–ł —É—Ā–ļ–ĺ—Ä—Ź–Ķ—ā —Ä–į–∑–≤–ł—ā–ł–Ķ –ł –Ņ—Ä–ĺ–≥—Ä–Ķ—Ā—Ā–ł—Ä–ĺ–≤–į–Ĺ–ł–Ķ –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł–Ļ, —Ā–≤—Ź–∑–į–Ĺ–Ĺ—č—Ö —Ā –į—ā–Ķ—Ä–ĺ—Ā–ļ–Ľ–Ķ—Ä–ĺ–∑–ĺ–ľ. –ö—Ä–ĺ–ľ–Ķ —ć—ā–ĺ–≥–ĺ, –ľ–Ĺ–ĺ–≥–ł–Ķ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į—ā–Ķ–Ľ–ł —Ä–į—Ā—Ā–ľ–į—ā—Ä–ł–≤–į—é—ā –ú–° –ļ–į–ļ –Ņ—Ä–Ķ–Ľ—é–ī–ł—é —Ā–į—Ö–į—Ä–Ĺ–ĺ–≥–ĺ –ī–ł–į–Ī–Ķ—ā–į 2 —ā–ł–Ņ–į: —Ä–ł—Ā–ļ —Ä–į–∑–≤–ł—ā–ł—Ź –°–Ē 2 —ā–ł–Ņ–į —É –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ —Ā –ú–° –≤ —Ā—Ä–Ķ–ī–Ĺ–Ķ–ľ –≤ 5‚Äď9 —Ä–į–∑ –≤—č—ą–Ķ, —á–Ķ–ľ —É –Ľ–ł—Ü –Ī–Ķ–∑ –Ĺ–Ķ–≥–ĺ [15]. –ü–ĺ –ī–į–Ĺ–Ĺ—č–ľ —Ā—ā–į—ā–ł—Ā—ā–ł–ļ–ł, —É –Ī–ĺ–Ľ—Ć–Ĺ—č—Ö —Ā –≤–Ņ–Ķ—Ä–≤—č–Ķ –ī–ł–į–≥–Ĺ–ĺ—Ā—ā–ł—Ä–ĺ–≤–į–Ĺ–Ĺ—č–ľ –°–Ē 2 —ā–ł–Ņ–į —É–∂–Ķ –Ņ—Ä–ł –Ņ–Ķ—Ä–≤–ĺ–ľ –ĺ–Ī—Ä–į—Č–Ķ–Ĺ–ł–ł –ļ –≤—Ä–į—á—É –≤—č—Ź–≤–Ľ—Ź—é—ā—Ā—Ź –ľ–į–ļ—Ä–ĺ- –ł –ľ–ł–ļ—Ä–ĺ—Ā–ĺ—Ā—É–ī–ł—Ā—ā—č–Ķ –ĺ—Ā–Ľ–ĺ–∂–Ĺ–Ķ–Ĺ–ł—Ź —ć—ā–ĺ–≥–ĺ –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł—Ź: –Ĺ–į—Ä—É—ą–Ķ–Ĺ–ł–Ķ –∑—Ä–Ķ–Ĺ–ł—Ź (–ī–ł–į–Ī–Ķ—ā–ł—á–Ķ—Ā–ļ–į—Ź —Ä–Ķ—ā–ł–Ĺ–ĺ–Ņ–į—ā–ł—Ź), –Ĺ–į—Ä—É—ą–Ķ–Ĺ–ł–Ķ —Ą—É–Ĺ–ļ—Ü–ł–ł –Ņ–ĺ—á–Ķ–ļ (–ī–ł–į–Ī–Ķ—ā–ł—á–Ķ—Ā–ļ–į—Ź –Ĺ–Ķ—Ą—Ä–ĺ–Ņ–į—ā–ł—Ź), –Ņ–ĺ—Ä–į–∂–Ķ–Ĺ–ł–Ķ —Ā–ĺ—Ā—É–ī–ĺ–≤ —Ā–Ķ—Ä–ī—Ü–į, –ľ–ĺ–∑–≥–į, —Ā–ĺ—Ā—É–ī–ĺ–≤ –Ĺ–ł–∂–Ĺ–ł—Ö –ļ–ĺ–Ĺ–Ķ—á–Ĺ–ĺ—Ā—ā–Ķ–Ļ –ł –ī—Ä. –ź –Ņ—Ä–ł —Ä–į–∑–≤–ł–≤—ą–Ķ–ľ—Ā—Ź –°–Ē 2 —ā–ł–Ņ–į —Ä–ł—Ā–ļ —Ä–į–∑–≤–ł—ā–ł—Ź —Ā–Ķ—Ä–ī–Ķ—á–Ĺ–ĺ-—Ā–ĺ—Ā—É–ī–ł—Ā—ā—č—Ö –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł–Ļ (–°–°–ó) –≤ 3‚Äď4 —Ä–į–∑–į –≤—č—ą–Ķ, —á–Ķ–ľ –Ī–Ķ–∑ –Ĺ–Ķ–≥–ĺ [16]. –ė–ľ–Ķ–Ĺ–Ĺ–ĺ —ć—ā–ł –ĺ—Ā–Ľ–ĺ–∂–Ĺ–Ķ–Ĺ–ł—Ź —Ź–≤–Ľ—Ź—é—ā—Ā—Ź –ĺ—Ā–Ĺ–ĺ–≤–Ĺ–ĺ–Ļ –Ņ—Ä–ł—á–ł–Ĺ–ĺ–Ļ –≤—č—Ā–ĺ–ļ–ĺ–Ļ –ł–Ĺ–≤–į–Ľ–ł–ī–ł–∑–į—Ü–ł–ł –ł —Ā–ľ–Ķ—Ä—ā–Ĺ–ĺ—Ā—ā–ł –Ī–ĺ–Ľ—Ć–Ĺ—č—Ö –°–Ē 2 —ā–ł–Ņ–į (–ī–ĺ 70% –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ —É–ľ–ł—Ä–į—é—ā –ĺ—ā –ł–Ĺ—Ą–į—Ä–ļ—ā–į –ľ–ł–ĺ–ļ–į—Ä–ī–į –ł–Ľ–ł –ł–Ĺ—Ā—É–Ľ—Ć—ā–į –ł –ł—Ö –Ņ–ĺ—Ā–Ľ–Ķ–ī—Ā—ā–≤–ł–Ļ).
–ú–Ķ—Ö–į–Ĺ–ł–∑–ľ—č —Ä–į–∑–≤–ł—ā–ł—Ź –°–°–󬆖ł –°–Ē 2 —ā–ł–Ņ–į —É –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ —Ā –ú–°
–ö–Ľ—é—á–Ķ–≤—č–ľ –∑–≤–Ķ–Ĺ–ĺ–ľ –Ņ–į—ā–ĺ–≥–Ķ–Ĺ–Ķ–∑–į –ľ–Ķ—ā–į–Ī–ĺ–Ľ–ł—á–Ķ—Ā–ļ–ĺ–≥–ĺ —Ā–ł–Ĺ–ī—Ä–ĺ–ľ–į —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź –Ņ–Ķ—Ä–≤–ł—á–Ĺ–į—Ź –ł–Ĺ—Ā—É–Ľ–ł–Ĺ–ĺ—Ä–Ķ–∑–ł—Ā—ā–Ķ–Ĺ—ā–Ĺ–ĺ—Ā—ā—Ć (–ė–†) –ł –ļ–ĺ–ľ–Ņ–Ķ–Ĺ—Ā–į—ā–ĺ—Ä–Ĺ–į—Ź –≥–ł–Ņ–Ķ—Ä–ł–Ĺ—Ā—É–Ľ–ł–Ĺ–Ķ–ľ–ł—Ź (–ď–ė). –ė–Ĺ—Ā—É–Ľ–ł–Ĺ–ĺ—Ä–Ķ–∑–ł—Ā—ā–Ķ–Ĺ—ā–Ĺ–ĺ—Ā—ā—Ć ‚Äď —ć—ā–ĺ –Ĺ–į—Ä—É—ą–Ķ–Ĺ–ł–Ķ –ł–Ĺ—Ā—É–Ľ–ł–Ĺ–ĺ–Ņ–ĺ—Ā—Ä–Ķ–ī–ĺ–≤–į–Ĺ–Ĺ–ĺ–Ļ —É—ā–ł–Ľ–ł–∑–į—Ü–ł–ł –≥–Ľ—é–ļ–ĺ–∑—č –ļ–Ľ–Ķ—ā–ļ–į–ľ–ł. –ė–† ‚Äď —Ā–ĺ—Ā—ā–ĺ—Ź–Ĺ–ł–Ķ, —Ā–ĺ–Ņ—Ä–ĺ–≤–ĺ–∂–ī–į—é—Č–Ķ–Ķ —Ü–Ķ–Ľ—č–Ļ —Ä—Ź–ī —Ą–ł–∑–ł–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ł—Ö –ł –Ņ–į—ā–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ł—Ö –Ņ—Ä–ĺ—Ü–Ķ—Ā—Ā–ĺ–≤. –§–ł–∑–ł–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–į—Ź –ė–† –≤—č—Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź –≤ –Ņ—É–Ī–Ķ—Ä—ā–į—ā–Ĺ–ĺ–ľ –Ņ–Ķ—Ä–ł–ĺ–ī–Ķ, –Ņ—Ä–ł –Ī–Ķ—Ä–Ķ–ľ–Ķ–Ĺ–Ĺ–ĺ—Ā—ā–ł, –≤ –ļ–Ľ–ł–ľ–į–ļ—ā–Ķ—Ä–ł—á–Ķ—Ā–ļ–ĺ–ľ –Ņ–Ķ—Ä–ł–ĺ–ī–Ķ, –≤–ĺ –≤—Ä–Ķ–ľ—Ź –Ĺ–ĺ—á–Ĺ–ĺ–≥–ĺ —Ā–Ĺ–į, –Ņ—Ä–ł –Ī–ĺ–≥–į—ā–ĺ–Ļ –∂–ł—Ä–ĺ–ľ –ī–ł–Ķ—ā–Ķ. –ú–Ķ—ā–į–Ī–ĺ–Ľ–ł—á–Ķ—Ā–ļ–į—Ź –ė–† —Ö–į—Ä–į–ļ—ā–Ķ—Ä–Ĺ–į –ī–Ľ—Ź –ľ–Ķ—ā–į–Ī–ĺ–Ľ–ł—á–Ķ—Ā–ļ–ĺ–≥–ĺ —Ā–ł–Ĺ–ī—Ä–ĺ–ľ–į, –°–Ē 2 —ā–ł–Ņ–į, –ī–Ķ–ļ–ĺ–ľ–Ņ–Ķ–Ĺ—Ā–ł—Ä–ĺ–≤–į–Ĺ–Ĺ–ĺ–≥–ĺ –°–Ē 1 —ā–ł–Ņ–į, –ī–ł–į–Ī–Ķ—ā–ł—á–Ķ—Ā–ļ–ĺ–≥–ĺ –ļ–Ķ—ā–ĺ–į—Ü–ł–ī–ĺ–∑–į, –ĺ–∂–ł—Ä–Ķ–Ĺ–ł—Ź, –≤—č—Ä–į–∂–Ķ–Ĺ–Ĺ–ĺ–Ļ –Ĺ–Ķ–ī–ĺ—Ā—ā–į—ā–ĺ—á–Ĺ–ĺ—Ā—ā–ł –Ņ–ł—ā–į–Ĺ–ł—Ź, –≥–ł–Ņ–Ķ—Ä—É—Ä–ł–ļ–Ķ–ľ–ł–ł, –ł–Ĺ–ī—É—Ü–ł—Ä–ĺ–≤–į–Ĺ–Ĺ–ĺ–Ļ –ł–Ĺ—Ā—É–Ľ–ł–Ĺ–ĺ–ľ –≥–ł–Ņ–ĺ–≥–Ľ–ł–ļ–Ķ–ľ–ł–ł, –į–Ľ–ļ–ĺ–≥–ĺ–Ľ–ł–∑–ľ–į. –≠–Ĺ–ī–ĺ–ļ—Ä–ł–Ĺ–Ĺ–į—Ź –ė–† –ĺ—ā–ľ–Ķ—á–į–Ķ—ā—Ā—Ź –Ņ—Ä–ł —ā–ł—Ä–Ķ–ĺ—ā–ĺ–ļ—Ā–ł–ļ–ĺ–∑–Ķ, –≥–ł–Ņ–ĺ—ā–ł—Ä–Ķ–ĺ–∑–Ķ, —Ā–ł–Ĺ–ī—Ä–ĺ–ľ–Ķ –ö—É—ą–ł–Ĺ–≥–į, –į–ļ—Ä–ĺ–ľ–Ķ–≥–į–Ľ–ł–ł, —Ą–Ķ–ĺ—Ö—Ä–ĺ–ľ–ĺ—Ü–ł—ā–ĺ–ľ–Ķ. –Ě–Ķ—ć–Ĺ–ī–ĺ–ļ—Ä–ł–Ĺ–Ĺ–į—Ź –ė–† —ā–ł–Ņ–ł—á–Ĺ–į –ī–Ľ—Ź –≥–ł–Ņ–Ķ—Ä—ā–ĺ–Ĺ–ł—á–Ķ—Ā–ļ–ĺ–Ļ –Ī–ĺ–Ľ–Ķ–∑–Ĺ–ł, —Ö—Ä–ĺ–Ĺ–ł—á–Ķ—Ā–ļ–ĺ–Ļ –Ņ–ĺ—á–Ķ—á–Ĺ–ĺ–Ļ –Ĺ–Ķ–ī–ĺ—Ā—ā–į—ā–ĺ—á–Ĺ–ĺ—Ā—ā–ł, —Ü–ł—Ä—Ä–ĺ–∑–į –Ņ–Ķ—á–Ķ–Ĺ–ł, —Ā–Ķ—Ä–ī–Ķ—á–Ĺ–ĺ–Ļ –Ĺ–Ķ–ī–ĺ—Ā—ā–į—ā–ĺ—á–Ĺ–ĺ—Ā—ā–ł, —Ä–Ķ–≤–ľ–į—ā–ĺ–ł–ī–Ĺ–ĺ–≥–ĺ –į—Ä—ā—Ä–ł—ā–į, —á–Ķ—Ä–Ĺ–ĺ–≥–ĺ –į–ļ–į–Ĺ—ā–ĺ–∑–į, –ľ–ł–ĺ—ā–ĺ–Ĺ–ł—á–Ķ—Ā–ļ–ĺ–Ļ –ī–ł—Ā—ā—Ä–ĺ—Ą–ł–ł, —Ä–į–ļ–ĺ–≤–ĺ–Ļ –ļ–į—Ö–Ķ–ļ—Ā–ł–ł, –į —ā–į–ļ–∂–Ķ –Ĺ–į–Ī–Ľ—é–ī–į–Ķ—ā—Ā—Ź –Ņ—Ä–ł —ā—Ä–į–≤–ľ–į—Ö, –ĺ–∂–ĺ–≥–į—Ö, —Ā–Ķ–Ņ—Ā–ł—Ā–Ķ, —Ā–ĺ—Ā—ā–ĺ—Ź–Ĺ–ł—Ź—Ö –Ņ–ĺ—Ā–Ľ–Ķ —Ö–ł—Ä—É—Ä–≥–ł—á–Ķ—Ā–ļ–ł—Ö –≤–ľ–Ķ—ą–į—ā–Ķ–Ľ—Ć—Ā—ā–≤.
–Ě–į–ł–Ī–ĺ–Ľ—Ć—ą–Ķ–Ķ –ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ĺ–Ķ –∑–Ĺ–į—á–Ķ–Ĺ–ł–Ķ –ł–ľ–Ķ–Ķ—ā –Ņ–ĺ—ā–Ķ—Ä—Ź —á—É–≤—Ā—ā–≤–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ—Ā—ā–ł –ļ –ł–Ĺ—Ā—É–Ľ–ł–Ĺ—É –ľ—č—ą–Ķ—á–Ĺ–ĺ–Ļ, –∂–ł—Ä–ĺ–≤–ĺ–Ļ –ł –Ņ–Ķ—á–Ķ–Ĺ–ĺ—á–Ĺ–ĺ–Ļ —ā–ļ–į–Ĺ–Ķ–Ļ. –ü—Ä–Ķ–ī–Ņ–ĺ–Ľ–į–≥–į—é—ā, —á—ā–ĺ –Ņ—Ä–ł—á–ł–Ĺ–ĺ–Ļ —É—Ā–ļ–ĺ—Ä–Ķ–Ĺ–Ĺ–ĺ–≥–ĺ –į—ā–Ķ—Ä–ĺ–≥–Ķ–Ĺ–Ķ–∑–į –ł –≤—č—Ā–ĺ–ļ–ĺ–Ļ –Ľ–Ķ—ā–į–Ľ—Ć–Ĺ–ĺ—Ā—ā–ł –ĺ—ā –ł—ą–Ķ–ľ–ł—á–Ķ—Ā–ļ–ĺ–Ļ –Ī–ĺ–Ľ–Ķ–∑–Ĺ–ł —Ā–Ķ—Ä–ī—Ü–į –ł –ł–Ĺ—Ā—É–Ľ—Ć—ā–ĺ–≤ —É –Ī–ĺ–Ľ—Ć–Ĺ—č—Ö –°–Ē 2 —ā–ł–Ņ–į —ā–į–ļ–∂–Ķ –ľ–ĺ–≥—É—ā –Ī—č—ā—Ć –ė–† –ł —Ā–ĺ–Ņ—É—ā—Ā—ā–≤—É—é—Č–į—Ź –Ķ–Ļ –ď–ė. –£ –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ —Ā –ė–† –ł–ľ–Ķ—é—ā—Ā—Ź –ī–Ķ—Ą–Ķ–ļ—ā—č –≥–Ķ–Ĺ–ĺ–≤, –ĺ—ā–≤–Ķ—ā—Ā—ā–≤–Ķ–Ĺ–Ĺ—č—Ö –∑–į –Ņ–Ķ—Ä–Ķ–ī–į—á—É —Ā–ł–≥–Ĺ–į–Ľ–į –Ņ–ĺ—Ā–Ľ–Ķ —Ā–ĺ–Ķ–ī–ł–Ĺ–Ķ–Ĺ–ł—Ź –ł–Ĺ—Ā—É–Ľ–ł–Ĺ–į —Ā–ĺ —Ā–≤–ĺ–ł–ľ —Ä–Ķ—Ü–Ķ–Ņ—ā–ĺ—Ä–ĺ–ľ (–Ņ–ĺ—Ā—ā—Ä–Ķ—Ü–Ķ–Ņ—ā–ĺ—Ä–Ĺ—č–Ķ –ī–Ķ—Ą–Ķ–ļ—ā—č): –Ņ—Ä–Ķ–∂–ī–Ķ –≤—Ā–Ķ–≥–ĺ —É –Ĺ–ł—Ö –Ĺ–į—Ä—É—ą–į–Ķ—ā—Ā—Ź —ā—Ä–į–Ĺ—Ā–Ľ–ĺ–ļ–į—Ü–ł—Ź –ł —Ā–ł–Ĺ—ā–Ķ–∑ –≤–Ĺ—É—ā—Ä–ł–ļ–Ľ–Ķ—ā–ĺ—á–Ĺ–ĺ–≥–ĺ —ā—Ä–į–Ĺ—Ā–Ņ–ĺ—Ä—ā–Ķ—Ä–į –≥–Ľ—é–ļ–ĺ–∑—č GLUT-4. –ö—Ä–ĺ–ľ–Ķ —ā–ĺ–≥–ĺ, —É –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ —Ā –ė–† –ľ–ĺ–≥—É—ā –Ī—č—ā—Ć –ĺ–Ī–Ĺ–į—Ä—É–∂–Ķ–Ĺ—č –ī–Ķ—Ą–Ķ–ļ—ā—č –≥–Ķ–Ĺ–ĺ–≤, –ļ–ĺ–ī–ł—Ä—É—é—Č–ł—Ö —Ā—É–Ī—Ā—ā—Ä–į—ā —Ä–Ķ—Ü–Ķ–Ņ—ā–ĺ—Ä–į –ł–Ĺ—Ā—É–Ľ–ł–Ĺ–į —ā–ł–Ņ–į 1 –ł/–ł–Ľ–ł —Ą–ĺ—Ā—Ą–į—ā–ł–ī–ł–Ľ–ł–Ĺ–ĺ–∑–ł—ā–ĺ–Ľ-3-–ļ–ł–Ĺ–į–∑—É, –į —ā–į–ļ–∂–Ķ –Ĺ–į—Ä—É—ą–Ķ–Ĺ–ł–Ķ —ć–ļ—Ā–Ņ—Ä–Ķ—Ā—Ā–ł–ł –ī—Ä—É–≥–ł—Ö –≥–Ķ–Ĺ–ĺ–≤, –ĺ–Ī–Ķ—Ā–Ņ–Ķ—á–ł–≤–į—é—Č–ł—Ö –ľ–Ķ—ā–į–Ī–ĺ–Ľ–ł–∑–ľ –≥–Ľ—é–ļ–ĺ–∑—č –ł –Ľ–ł–Ņ–ł–ī–ĺ–≤¬†‚Äď –≥–Ľ—é–ļ–ĺ–∑–ĺ-6-—Ą–ĺ—Ā—Ą–į—ā–ī–Ķ–≥–ł–ī—Ä–ĺ–≥–Ķ–Ĺ–į–∑—č, –≥–Ľ—é–ļ–ĺ–ļ–ł–Ĺ–į–∑—č, –Ľ–ł–Ņ–ĺ–Ņ—Ä–ĺ—ā–Ķ–ł–Ĺ–Ľ–ł–Ņ–į–∑—č, —Ā–ł–Ĺ—ā–į–∑—č –∂–ł—Ä–Ĺ—č—Ö –ļ–ł—Ā–Ľ–ĺ—ā –ł –ī—Ä.
–ď–Ķ–Ĺ–Ķ—ā–ł—á–Ķ—Ā–ļ–į—Ź –Ņ—Ä–Ķ–ī—Ä–į—Ā–Ņ–ĺ–Ľ–ĺ–∂–Ķ–Ĺ–Ĺ–ĺ—Ā—ā—Ć –ļ –ė–† –ľ–ĺ–∂–Ķ—ā –Ĺ–Ķ —Ä–Ķ–į–Ľ–ł–∑–ĺ–≤–į—ā—Ć—Ā—Ź –ł –Ĺ–Ķ –Ņ—Ä–ĺ—Ź–≤–ł—ā—Ć—Ā—Ź –ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ł (–≤¬†–≤–ł–ī–Ķ –ľ–Ķ—ā–į–Ī–ĺ–Ľ–ł—á–Ķ—Ā–ļ–ĺ–≥–ĺ —Ā–ł–Ĺ–ī—Ä–ĺ–ľ–į –ł/–ł–Ľ–ł –°–Ē 2 —ā–ł–Ņ–į) –Ņ—Ä–ł –ĺ—ā—Ā—É—ā—Ā—ā–≤–ł–ł –Ĺ–Ķ–ĺ–Ī—Ö–ĺ–ī–ł–ľ—č—Ö –ī–Ľ—Ź —ć—ā–ĺ–≥–ĺ –≤–Ĺ–Ķ—ą–Ĺ–ł—Ö —Ą–į–ļ—ā–ĺ—Ä–ĺ–≤ ‚Äď –ł–∑–Ī—č—ā–ĺ—á–Ĺ–ĺ–≥–ĺ –ļ–į–Ľ–ĺ—Ä–ł–Ļ–Ĺ–ĺ–≥–ĺ –Ņ–ł—ā–į–Ĺ–ł—Ź (–ĺ—Ā–ĺ–Ī–Ķ–Ĺ–Ĺ–ĺ –∂–ł—Ä–Ĺ–ĺ–Ļ –Ņ–ł—Č–ł) –ł –Ĺ–ł–∑–ļ–ĺ–Ļ —Ą–ł–∑–ł—á–Ķ—Ā–ļ–ĺ–Ļ –į–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā–ł. –≠—ā–ł —Ą–į–ļ—ā–ĺ—Ä—č —Ā–į–ľ–ł –Ņ–ĺ —Ā–Ķ–Ī–Ķ —Ā–Ņ–ĺ—Ā–ĺ–Ī—Ā—ā–≤—É—é—ā —Ä–į–∑–≤–ł—ā–ł—é –į–Ī–ī–ĺ–ľ–ł–Ĺ–į–Ľ—Ć–Ĺ–ĺ–≥–ĺ –ĺ–∂–ł—Ä–Ķ–Ĺ–ł—Ź, –Ĺ–į–ļ–ĺ–Ņ–Ľ–Ķ–Ĺ–ł—é —Ā–≤–ĺ–Ī–ĺ–ī–Ĺ—č—Ö –∂–ł—Ä–Ĺ—č—Ö –ļ–ł—Ā–Ľ–ĺ—ā –ł, —Ā–Ľ–Ķ–ī–ĺ–≤–į—ā–Ķ–Ľ—Ć–Ĺ–ĺ, —É—Ā–ł–Ľ–Ķ–Ĺ–ł—é –ł–ľ–Ķ—é—Č–Ķ–Ļ—Ā—Ź –ė–†. –í—č–∑–≤–į–Ĺ–Ĺ–į—Ź –ł–Ĺ—Ā—É–Ľ–ł–Ĺ–ĺ—Ä–Ķ–∑–ł—Ā—ā–Ķ–Ĺ—ā–Ĺ–ĺ—Ā—ā—Ć—é –ļ–ĺ–ľ–Ņ–Ķ–Ĺ—Ā–į—ā–ĺ—Ä–Ĺ–į—Ź –ď–ė –Ņ–ĺ–∑–≤–ĺ–Ľ—Ź–Ķ—ā –ļ–į–ļ–ĺ–Ķ-—ā–ĺ –≤—Ä–Ķ–ľ—Ź –Ņ–ĺ–ī–ī–Ķ—Ä–∂–ł–≤–į—ā—Ć —É–≥–Ľ–Ķ–≤–ĺ–ī–Ĺ—č–Ļ –ĺ–Ī–ľ–Ķ–Ĺ –≤ –Ĺ–ĺ—Ä–ľ–Ķ, –Ĺ–ĺ –≤–ľ–Ķ—Ā—ā–Ķ —Ā —ā–Ķ–ľ —Ā–Ņ–ĺ—Ā–ĺ–Ī—Ā—ā–≤—É–Ķ—ā —Ä–į–∑–≤–ł—ā–ł—é –ľ–Ķ—ā–į–Ī–ĺ–Ľ–ł—á–Ķ—Ā–ļ–ł—Ö, –≥–Ķ–ľ–ĺ–ī–ł–Ĺ–į–ľ–ł—á–Ķ—Ā–ļ–ł—Ö –ł –ĺ—Ä–≥–į–Ĺ–Ĺ—č—Ö –Ĺ–į—Ä—É—ą–Ķ–Ĺ–ł–Ļ, —á—ā–ĺ –≤ –ļ–ĺ–Ĺ–Ķ—á–Ĺ–ĺ–ľ –ł—ā–ĺ–≥–Ķ –Ņ—Ä–ł–≤–ĺ–ī–ł—ā –ļ —Ā–Ķ—Ä–ī–Ķ—á–Ĺ–ĺ-—Ā–ĺ—Ā—É–ī–ł—Ā—ā—č–ľ –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł—Ź–ľ –ł –°–Ē 2 —ā–ł–Ņ–į.
–ė–† –ľ—č—ą–Ķ—á–Ĺ–ĺ–Ļ —ā–ļ–į–Ĺ–ł –Ņ—Ä–ĺ—Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź –≤ —Ā–Ĺ–ł–∂–Ķ–Ĺ–ł–ł –Ņ–ĺ—Ā—ā—É–Ņ–Ľ–Ķ–Ĺ–ł—Ź –≥–Ľ—é–ļ–ĺ–∑—č –ł–∑ –ļ—Ä–ĺ–≤–ł –ł —É—ā–ł–Ľ–ł–∑–į—Ü–ł–ł –Ķ–Ķ –ľ–ł–ĺ—Ü–ł—ā–į–ľ–ł. –ė–† –∂–ł—Ä–ĺ–≤–ĺ–Ļ —ā–ļ–į–Ĺ–ł –≤—č—Ä–į–∂–į–Ķ—ā—Ā—Ź –≤ —ā–ĺ–ľ, —á—ā–ĺ –į–ī–ł–Ņ–ĺ—Ü–ł—ā—č —Ā—ā–į–Ĺ–ĺ–≤—Ź—ā—Ā—Ź –Ĺ–Ķ—á—É–≤—Ā—ā–≤–ł—ā–Ķ–Ľ—Ć–Ĺ—č–ľ–ł –ļ –į–Ĺ—ā–ł–Ľ–ł–Ņ–ĺ–Ľ–ł—ā–ł—á–Ķ—Ā–ļ–ĺ–ľ—É –ī–Ķ–Ļ—Ā—ā–≤–ł—é –ł–Ĺ—Ā—É–Ľ–ł–Ĺ–į, —á—ā–ĺ —Ā–Ņ–ĺ—Ā–ĺ–Ī—Ā—ā–≤—É–Ķ—ā –≤—č—Ö–ĺ–ī—É –≤ –ļ—Ä–ĺ–≤—Ć —Ā–≤–ĺ–Ī–ĺ–ī–Ĺ—č—Ö –∂–ł—Ä–Ĺ—č—Ö –ļ–ł—Ā–Ľ–ĺ—ā –ł –≥–Ľ–ł—Ü–Ķ—Ä–ł–Ĺ–į. –°–≤–ĺ–Ī–ĺ–ī–Ĺ—č–Ķ –∂–ł—Ä–Ĺ—č–Ķ –ļ–ł—Ā–Ľ–ĺ—ā—č –Ņ–ĺ—Ā—ā—É–Ņ–į—é—ā –≤ –Ņ–Ķ—á–Ķ–Ĺ—Ć, –≥–ī–Ķ —Ā—ā–į–Ĺ–ĺ–≤—Ź—ā—Ā—Ź –ĺ—Ā–Ĺ–ĺ–≤–Ĺ—č–ľ –ł—Ā—ā–ĺ—á–Ĺ–ł–ļ–ĺ–ľ —Ą–ĺ—Ä–ľ–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź –į—ā–Ķ—Ä–ĺ–≥–Ķ–Ĺ–Ĺ—č—Ö –Ľ–ł–Ņ–ĺ–Ņ—Ä–ĺ—ā–Ķ–ł–Ĺ–ĺ–≤ –ĺ—á–Ķ–Ĺ—Ć –Ĺ–ł–∑–ļ–ĺ–Ļ –Ņ–Ľ–ĺ—ā–Ĺ–ĺ—Ā—ā–ł. –ė–† —ā–ļ–į–Ĺ–ł –Ņ–Ķ—á–Ķ–Ĺ–ł —Ö–į—Ä–į–ļ—ā–Ķ—Ä–ł–∑—É–Ķ—ā—Ā—Ź —Ā–Ĺ–ł–∂–Ķ–Ĺ–ł–Ķ–ľ —Ā–ł–Ĺ—ā–Ķ–∑–į –≥–Ľ–ł–ļ–ĺ–≥–Ķ–Ĺ–į –ł –į–ļ—ā–ł–≤–į—Ü–ł–Ķ–Ļ –Ņ—Ä–ĺ—Ü–Ķ—Ā—Ā–ĺ–≤ —Ä–į—Ā–Ņ–į–ī–į –≥–Ľ–ł–ļ–ĺ–≥–Ķ–Ĺ–į –ī–ĺ –≥–Ľ—é–ļ–ĺ–∑—č (–≥–Ľ–ł–ļ–ĺ–≥–Ķ–Ĺ–ĺ–Ľ–ł–∑) –ł —Ā–ł–Ĺ—ā–Ķ–∑–į –≥–Ľ—é–ļ–ĺ–∑—č de novo –ł–∑ –į–ľ–ł–Ĺ–ĺ–ļ–ł—Ā–Ľ–ĺ—ā, –Ľ–į–ļ—ā–į—ā–į, –Ņ–ł—Ä—É–≤–į—ā–į, –≥–Ľ–ł—Ü–Ķ—Ä–ł–Ĺ–į (–≥–Ľ—é–ļ–ĺ–Ĺ–Ķ–ĺ–≥–Ķ–Ĺ–Ķ–∑), –≤ —Ä–Ķ–∑—É–Ľ—Ć—ā–į—ā–Ķ —á–Ķ–≥–ĺ –≥–Ľ—é–ļ–ĺ–∑–į –ł–∑ –Ņ–Ķ—á–Ķ–Ĺ–ł –Ņ–ĺ—Ā—ā—É–Ņ–į–Ķ—ā –≤ –ļ—Ä–ĺ–≤–ĺ—ā–ĺ–ļ. –í —Ü–Ķ–Ľ–ĺ–ľ, –ė–† ‚Äď —ć—ā–ĺ —ć–≤–ĺ–Ľ—é—Ü–ł–ĺ–Ĺ–Ĺ–ĺ –∑–į–ļ—Ä–Ķ–Ņ–Ľ–Ķ–Ĺ–Ĺ—č–Ļ –ľ–Ķ—Ö–į–Ĺ–ł–∑–ľ –≤—č–∂–ł–≤–į–Ĺ–ł—Ź –≤ –Ĺ–Ķ–Ī–Ľ–į–≥–ĺ–Ņ—Ä–ł—Ź—ā–Ĺ—č—Ö —É—Ā–Ľ–ĺ–≤–ł—Ź—Ö, –ļ–ĺ–≥–ī–į –Ņ–Ķ—Ä–ł–ĺ–ī—č –ł–∑–ĺ–Ī–ł–Ľ–ł—Ź —á–Ķ—Ä–Ķ–ī–ĺ–≤–į–Ľ–ł—Ā—Ć —Ā –Ņ–Ķ—Ä–ł–ĺ–ī–į–ľ–ł –≥–ĺ–Ľ–ĺ–ī–į. –Ě–į–Ľ–ł—á–ł–Ķ –ė–† –ĺ–Ī–Ķ—Ā–Ņ–Ķ—á–ł–≤–į–Ľ–ĺ –Ĺ–į–ļ–ĺ–Ņ–Ľ–Ķ–Ĺ–ł–Ķ —ć–Ĺ–Ķ—Ä–≥–ł–ł –≤ –≤–ł–ī–Ķ –ĺ—ā–Ľ–ĺ–∂–Ķ–Ĺ–ł–Ļ –∂–ł—Ä–į, –∑–į–Ņ–į—Ā–ĺ–≤ –ļ–ĺ—ā–ĺ—Ä–ĺ–≥–ĺ —Ö–≤–į—ā–į–Ľ–ĺ –Ĺ–į —ā–ĺ, —á—ā–ĺ–Ī—č –Ņ–Ķ—Ä–Ķ–∂–ł—ā—Ć –≥–ĺ–Ľ–ĺ–ī [17]. –í —Ā—ā—Ä–į–Ĺ–į—Ö —Ā –≤—č—Ā–ĺ–ļ–ł–ľ —ć–ļ–ĺ–Ĺ–ĺ–ľ–ł—á–Ķ—Ā–ļ–ł–ľ —Ä–į–∑–≤–ł—ā–ł–Ķ–ľ, –≥–ī–Ķ —Ā–ĺ–∑–ī–į–Ĺ—č —É—Ā–Ľ–ĺ–≤–ł—Ź –Ņ–ł—Č–Ķ–≤–ĺ–≥–ĺ –ł–∑–ĺ–Ī–ł–Ľ–ł—Ź –ł –Ľ—é–ī–ł –Ĺ–Ķ—Ä–Ķ–ī–ļ–ĺ –≤–Ķ–ī—É—ā –ľ–į–Ľ–ĺ–Ņ–ĺ–ī–≤–ł–∂–Ĺ—č–Ļ –ĺ–Ī—Ä–į–∑ –∂–ł–∑–Ĺ–ł, —Ā–ĺ—Ö—Ä–į–Ĺ–ł–≤—ą–ł–Ķ—Ā—Ź –≤ –ł—Ö –≥–Ķ–Ĺ–Ķ—ā–ł—á–Ķ—Ā–ļ–ĺ–Ļ –Ņ–į–ľ—Ź—ā–ł –ľ–Ķ—Ö–į–Ĺ–ł–∑–ľ—č –ė–† –Ņ—Ä–ĺ–ī–ĺ–Ľ–∂–į—é—ā —Ä–į–Ī–ĺ—ā–į—ā—Ć –Ĺ–į –Ĺ–į–ļ–ĺ–Ņ–Ľ–Ķ–Ĺ–ł–Ķ —ć–Ĺ–Ķ—Ä–≥–ł–ł, —á—ā–ĺ —Ā–Ņ–ĺ—Ā–ĺ–Ī—Ā—ā–≤—É–Ķ—ā —Ä–į–∑–≤–ł—ā–ł—é –į–Ī–ī–ĺ–ľ–ł–Ĺ–į–Ľ—Ć–Ĺ–ĺ–≥–ĺ –ĺ–∂–ł—Ä–Ķ–Ĺ–ł—Ź, –ī–ł—Ā–Ľ–ł–Ņ–ł–ī–Ķ–ľ–ł–ł, —Ä–į–Ĺ–Ĺ–Ķ–ľ—É –į—ā–Ķ—Ä–ĺ—Ā–ļ–Ľ–Ķ—Ä–ĺ–∑—É, –į—Ä—ā–Ķ—Ä–ł–į–Ľ—Ć–Ĺ–ĺ–Ļ –≥–ł–Ņ–Ķ—Ä—ā–Ķ–Ĺ–∑–ł–ł –ł –°–Ē 2 —ā–ł–Ņ–į.
–ö –Ĺ–į—Ā—ā–ĺ—Ź—Č–Ķ–ľ—É –≤—Ä–Ķ–ľ–Ķ–Ĺ–ł –Ņ—Ä–ĺ–≤–Ķ–ī–Ķ–Ĺ–ĺ –Ī–ĺ–Ľ–Ķ–Ķ 10 –ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ł—Ö –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–Ļ —Ā —É—á–į—Ā—ā–ł–Ķ–ľ –Ĺ–Ķ –ľ–Ķ–Ĺ–Ķ–Ķ 15 —ā—č—Ā. —á–Ķ–Ľ–ĺ–≤–Ķ–ļ. –†–Ķ–∑—É–Ľ—Ć—ā–į—ā—č –ĺ–Ņ—É–Ī–Ľ–ł–ļ–ĺ–≤–į–Ĺ–Ĺ—č—Ö —Ä–į–Ī–ĺ—ā –Ņ–ĺ–∑–≤–ĺ–Ľ—Ź—é—ā —É—ā–≤–Ķ—Ä–∂–ī–į—ā—Ć, —á—ā–ĺ –ł–Ĺ—Ā—É–Ľ–ł–Ĺ–ĺ—Ä–Ķ–∑–ł—Ā—ā–Ķ–Ĺ—ā–Ĺ–ĺ—Ā—ā—Ć –ł —Ā–ĺ–Ņ—É—ā—Ā—ā–≤—É—é—Č–į—Ź –Ķ–Ļ –≥–ł–Ņ–Ķ—Ä–ł–Ĺ—Ā—É–Ľ–ł–Ĺ–Ķ–ľ–ł—Ź —Ź–≤–Ľ—Ź—é—ā—Ā—Ź —Ą–į–ļ—ā–ĺ—Ä–į–ľ–ł —Ä–ł—Ā–ļ–į —É—Ā–ļ–ĺ—Ä–Ķ–Ĺ–Ĺ–ĺ–≥–ĺ –į—ā–Ķ—Ä–ĺ–≥–Ķ–Ĺ–Ķ–∑–į –ł –≤—č—Ā–ĺ–ļ–ĺ–Ļ –Ľ–Ķ—ā–į–Ľ—Ć–Ĺ–ĺ—Ā—ā–ł –ĺ—ā –ł—ą–Ķ–ľ–ł—á–Ķ—Ā–ļ–ĺ–Ļ –Ī–ĺ–Ľ–Ķ–∑–Ĺ–ł —Ā–Ķ—Ä–ī—Ü–į (–ė–Ď–°). –ė–ľ–Ķ–Ķ—ā—Ā—Ź –ī–ĺ—Ā—ā–į—ā–ĺ—á–Ĺ–ĺ –ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ł—Ö –ī–ĺ–ļ–į–∑–į—ā–Ķ–Ľ—Ć—Ā—ā–≤ —ā–ĺ–≥–ĺ, —á—ā–ĺ –≥–ł–Ņ–Ķ—Ä–ł–Ĺ—Ā—É–Ľ–ł–Ĺ–Ķ–ľ–ł—Ź —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź –Ĺ–Ķ–∑–į–≤–ł—Ā–ł–ľ—č–ľ —Ą–į–ļ—ā–ĺ—Ä–ĺ–ľ —Ä–ł—Ā–ļ–į —Ä–į–∑–≤–ł—ā–ł—Ź –ė–Ď–° —É –Ľ–ł—Ü –Ī–Ķ–∑ –°–Ē 2 —ā–ł–Ņ–į: Paris prospective study (–ĺ–ļ–ĺ–Ľ–ĺ 7 000 –ĺ–Ī—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–Ĺ—č—Ö), Busselton (–Ī–ĺ–Ľ–Ķ–Ķ 1 000 –ĺ–Ī—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–Ĺ—č—Ö) –ł Helsinki Policemen Study (982 –ĺ–Ī—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–Ĺ—č—Ö) [18]. –í –Ņ–ĺ—Ā–Ľ–Ķ–ī–Ĺ–ł–Ķ –≥–ĺ–ī—č —ć—ā–ĺ –Ņ–ĺ–ī—ā–≤–Ķ—Ä–∂–ī–Ķ–Ĺ–ĺ –ł –≤ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź—Ö —Ā —É—á–į—Ā—ā–ł–Ķ–ľ –Ī–ĺ–Ľ—Ć–Ĺ—č—Ö –°–Ē 2 —ā–ł–Ņ–į. Robert W. Stout –Ņ–ĺ–ļ–į–∑–į–Ľ, —á—ā–ĺ –ł–Ĺ—Ā—É–Ľ–ł–Ĺ –ĺ–ļ–į–∑—č–≤–į–Ķ—ā –Ņ—Ä—Ź–ľ–ĺ–Ķ –į—ā–Ķ—Ä–ĺ–≥–Ķ–Ĺ–Ĺ–ĺ–Ķ –ī–Ķ–Ļ—Ā—ā–≤–ł–Ķ –Ĺ–į —Ā—ā–Ķ–Ĺ–ļ–ł —Ā–ĺ—Ā—É–ī–ĺ–≤, –≤—č–∑—č–≤–į—Ź –Ņ—Ä–ĺ–Ľ–ł—Ą–Ķ—Ä–į—Ü–ł—é –ł –ľ–ł–≥—Ä–į—Ü–ł—é –≥–Ľ–į–ī–ļ–ĺ–ľ—č—ą–Ķ—á–Ĺ—č—Ö –ļ–Ľ–Ķ—ā–ĺ–ļ, —Ā–ł–Ĺ—ā–Ķ–∑ –Ľ–ł–Ņ–ł–ī–ĺ–≤ –≤ –Ĺ–ł—Ö –ł –Ņ—Ä–ĺ–Ľ–ł—Ą–Ķ—Ä–į—Ü–ł—é —Ą–ł–Ī—Ä–ĺ–Ī–Ľ–į—Ā—ā–ĺ–≤. –Ę–į–ļ–ł–ľ –ĺ–Ī—Ä–į–∑–ĺ–ľ, –ė–† –ł –ď–ė –≤–Ĺ–ĺ—Ā—Ź—ā –≤–Ķ—Ā–ĺ–ľ—č–Ļ –≤–ļ–Ľ–į–ī –≤ –Ņ—Ä–ĺ–≥—Ä–Ķ—Ā—Ā–ł—Ä–ĺ–≤–į–Ĺ–ł–Ķ –į—ā–Ķ—Ä–ĺ—Ā–ļ–Ľ–Ķ—Ä–ĺ–∑–į –ļ–į–ļ —É –Ľ–ł—Ü –Ī–Ķ–∑ –°–Ē, —ā–į–ļ –ł —É –Ī–ĺ–Ľ—Ć–Ĺ—č—Ö –°–Ē 2 —ā–ł–Ņ–į [19].
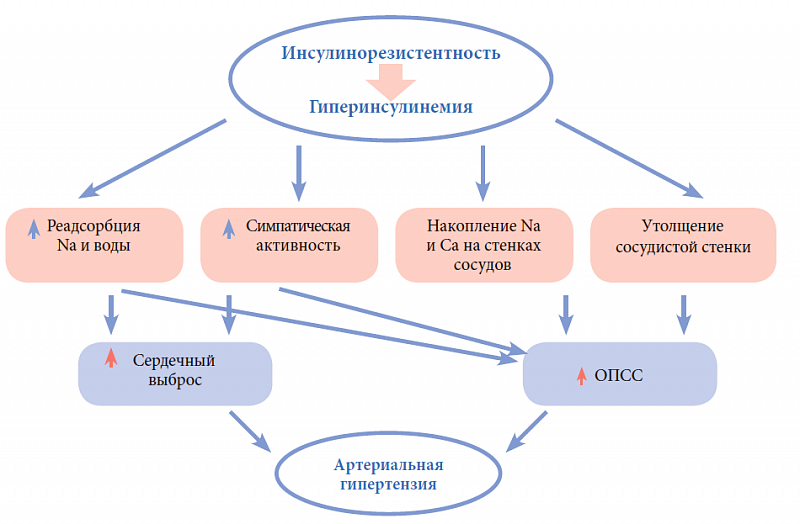
–ė–† –ł–≥—Ä–į–Ķ—ā —Ā—É—Č–Ķ—Ā—ā–≤–Ķ–Ĺ–Ĺ—É—é —Ä–ĺ–Ľ—Ć –≤ —Ä–į–∑–≤–ł—ā–ł–ł –į—Ä—ā–Ķ—Ä–ł–į–Ľ—Ć–Ĺ–ĺ–Ļ –≥–ł–Ņ–Ķ—Ä—ā–Ķ–Ĺ–∑–ł–ł (–ź–ď). –í–∑–į–ł–ľ–ĺ—Ā–≤—Ź–∑—Ć –≥–ł–Ņ–Ķ—Ä–ł–Ĺ—Ā—É–Ľ–ł–Ĺ–Ķ–ľ–ł–ł –ł —ć—Ā—Ā–Ķ–Ĺ—Ü–ł–į–Ľ—Ć–Ĺ–ĺ–Ļ –ź–ď –Ĺ–į—Ā—ā–ĺ–Ľ—Ć–ļ–ĺ –Ņ—Ä–ĺ—á–Ĺ–į, —á—ā–ĺ –Ņ—Ä–ł –≤—č—Ā–ĺ–ļ–ĺ–Ļ –ļ–ĺ–Ĺ—Ü–Ķ–Ĺ—ā—Ä–į—Ü–ł–ł –ł–Ĺ—Ā—É–Ľ–ł–Ĺ–į –Ņ–Ľ–į–∑–ľ—č –ľ–ĺ–∂–Ĺ–ĺ –Ņ—Ä–ĺ–≥–Ĺ–ĺ–∑–ł—Ä–ĺ–≤–į—ā—Ć —É –Ī–ĺ–Ľ—Ć–Ĺ–ĺ–≥–ĺ —Ä–į–∑–≤–ł—ā–ł–Ķ –≤ —Ā–ļ–ĺ—Ä–ĺ–ľ –≤—Ä–Ķ–ľ–Ķ–Ĺ–ł –ź–ď. –≠—ā–į —Ā–≤—Ź–∑—Ć –Ņ—Ä–ĺ—Ā–Ľ–Ķ–∂–ł–≤–į–Ķ—ā—Ā—Ź –ļ–į–ļ —É –Ī–ĺ–Ľ—Ć–Ĺ—č—Ö —Ā –ĺ–∂–ł—Ä–Ķ–Ĺ–ł–Ķ–ľ, —ā–į–ļ –ł —É –Ľ–ł—Ü —Ā –Ĺ–ĺ—Ä–ľ–į–Ľ—Ć–Ĺ–ĺ–Ļ –ľ–į—Ā—Ā–ĺ–Ļ —ā–Ķ–Ľ–į.
–°—É—Č–Ķ—Ā—ā–≤—É—é—ā –Ĺ–Ķ—Ā–ļ–ĺ–Ľ—Ć–ļ–ĺ –ľ–Ķ—Ö–į–Ĺ–ł–∑–ľ–ĺ–≤, –ĺ–Ī—ä—Ź—Ā–Ĺ—Ź—é—Č–ł—Ö –Ņ–ĺ–≤—č—ą–Ķ–Ĺ–ł–Ķ –ź–Ē –Ņ—Ä–ł –≥–ł–Ņ–Ķ—Ä–ł–Ĺ—Ā—É–Ľ–ł–Ĺ–Ķ–ľ–ł–ł (—Ä–ł—Ā. 3). –ė–Ĺ—Ā—É–Ľ–ł–Ĺ —Ā–Ņ–ĺ—Ā–ĺ–Ī—Ā—ā–≤—É–Ķ—ā –į–ļ—ā–ł–≤–į—Ü–ł–ł —Ā–ł–ľ–Ņ–į—ā–ł—á–Ķ—Ā–ļ–ĺ–Ļ –Ĺ–Ķ—Ä–≤–Ĺ–ĺ–Ļ —Ā–ł—Ā—ā–Ķ–ľ—č, –Ņ–ĺ–≤—č—ą–Ķ–Ĺ–ł—é —Ä–Ķ–į–Ī—Ā–ĺ—Ä–Ī—Ü–ł–ł Na –ł –≤–ĺ–ī—č –≤ –Ņ–ĺ—á–Ķ—á–Ĺ—č—Ö –ļ–į–Ĺ–į–Ľ—Ć—Ü–į—Ö, –≤–Ĺ—É—ā—Ä–ł–ļ–Ľ–Ķ—ā–ĺ—á–Ĺ–ĺ–ľ—É –Ĺ–į–ļ–ĺ–Ņ–Ľ–Ķ–Ĺ–ł—é Na –ł Ca; –ļ–į–ļ –ľ–ł—ā–ĺ–≥–Ķ–Ĺ–Ĺ—č–Ļ —Ą–į–ļ—ā–ĺ—Ä –ł–Ĺ—Ā—É–Ľ–ł–Ĺ –į–ļ—ā–ł–≤–ł—Ä—É–Ķ—ā –Ņ—Ä–ĺ–Ľ–ł—Ą–Ķ—Ä–į—Ü–ł—é –≥–Ľ–į–ī–ļ–ĺ-–ľ—č—ą–Ķ—á–Ĺ—č—Ö –ļ–Ľ–Ķ—ā–ĺ–ļ —Ā–ĺ—Ā—É–ī–ĺ–≤, —á—ā–ĺ –≤–Ķ–ī–Ķ—ā –ļ —É—ā–ĺ–Ľ—Č–Ķ–Ĺ–ł—é —Ā–ĺ—Ā—É–ī–ł—Ā—ā–ĺ–Ļ —Ā—ā–Ķ–Ĺ–ļ–ł. –ú–Ķ—Ö–į–Ĺ–ł–∑–ľ –≤–Ľ–ł—Ź–Ĺ–ł—Ź –ł–Ĺ—Ā—É–Ľ–ł–Ĺ–į –Ĺ–į —Ā–ł–ľ–Ņ–į—ā–ł—á–Ķ—Ā–ļ—É—é –Ĺ–Ķ—Ä–≤–Ĺ—É—é —Ā–ł—Ā—ā–Ķ–ľ—É (–°–Ě–°) –ī–ĺ –ļ–ĺ–Ĺ—Ü–į –Ĺ–Ķ —Ź—Ā–Ķ–Ĺ. –ü—Ä–Ķ–ī–Ņ–ĺ–Ľ–į–≥–į—é—ā, —á—ā–ĺ –ł–Ĺ—Ā—É–Ľ–ł–Ĺ –ľ–ĺ–∂–Ķ—ā –į–ļ—ā–ł–≤–ł—Ä–ĺ–≤–į—ā—Ć –°–Ě–° –Ņ—É—ā–Ķ–ľ –Ņ—Ä—Ź–ľ–ĺ–≥–ĺ –≤–ĺ–∑–ī–Ķ–Ļ—Ā—ā–≤–ł—Ź –Ĺ–į —Ü–Ķ–Ĺ—ā—Ä–į–Ľ—Ć–Ĺ—É—é –Ĺ–Ķ—Ä–≤–Ĺ—É—é —Ā–ł—Ā—ā–Ķ–ľ—É, –Ņ—Ä–ĺ–Ĺ–ł–ļ–į—Ź —á–Ķ—Ä–Ķ–∑ –≥–Ķ–ľ–į—ā–ĺ—ć–Ĺ—Ü–Ķ—Ą–į–Ľ–ł—á–Ķ—Ā–ļ–ł–Ļ –Ī–į—Ä—Ć–Ķ—Ä –≤ –Ņ–Ķ—Ä–ł–≤–Ķ–Ĺ—ā—Ä–ł–ļ—É–Ľ—Ź—Ä–Ĺ—É—é –ĺ–Ī–Ľ–į—Ā—ā—Ć –≥–ł–Ņ–ĺ—ā–į–Ľ–į–ľ—É—Ā–į, –≥–ī–Ķ, —Ā–≤—Ź–∑—č–≤–į—Ź—Ā—Ć —Ā–ĺ —Ā–≤–ĺ–ł–ľ–ł —Ä–Ķ—Ü–Ķ–Ņ—ā–ĺ—Ä–į–ľ–ł –Ĺ–į –Ņ–ĺ–≤–Ķ—Ä—Ö–Ĺ–ĺ—Ā—ā–ł –Ĺ–Ķ–Ļ—Ä–ĺ–Ĺ–ĺ–≤, –Ī–Ľ–ĺ–ļ–ł—Ä—É–Ķ—ā –į–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć –Ņ–į—Ä–į—Ā–ł–ľ–Ņ–į—ā–ł—á–Ķ—Ā–ļ–ĺ–Ļ –Ĺ–Ķ—Ä–≤–Ĺ–ĺ–Ļ —Ā–ł—Ā—ā–Ķ–ľ—č –ł, –Ĺ–į–Ņ—Ä–ĺ—ā–ł–≤, –į–ļ—ā–ł–≤–ł—Ä—É–Ķ—ā –°–Ě–°. Gerald M. Reavean –Ņ—Ä–Ķ–ī–Ņ–ĺ–Ľ–ĺ–∂–ł–Ľ, —á—ā–ĺ –Ņ—Ä–ł—á–ł–Ĺ–ĺ–Ļ –≥–ł–Ņ–Ķ—Ä–į–ļ—ā–ł–≤–į—Ü–ł–ł –°–Ě–° –≤ —É—Ā–Ľ–ĺ–≤–ł—Ź—Ö –≥–ł–Ņ–Ķ—Ä–≥–Ľ–ł–ļ–Ķ–ľ–ł–ł –ľ–ĺ–∂–Ķ—ā –Ī—č—ā—Ć –Ņ–ĺ–≤—č—ą–Ķ–Ĺ–Ĺ—č–Ļ –ľ–Ķ—ā–į–Ī–ĺ–Ľ–ł–∑–ľ –≥–Ľ—é–ļ–ĺ–∑—č –≤ —Ź–ī—Ä–į—Ö –≥–ł–Ņ–ĺ—ā–į–Ľ–į–ľ—É—Ā–į, —á—ā–ĺ —ā–ĺ—Ä–ľ–ĺ–∑–ł—ā –Ņ–Ķ—Ä–Ķ–ī–į—á—É –Ī–Ľ–ĺ–ļ–ł—Ä—É—é—Č–ł—Ö –ł–ľ–Ņ—É–Ľ—Ć—Ā–ĺ–≤ –Ĺ–į —Ā–ł–ľ–Ņ–į—ā–ł—á–Ķ—Ā–ļ–ł–Ķ —Ü–Ķ–Ĺ—ā—Ä—č –Ņ—Ä–ĺ–ī–ĺ–Ľ–≥–ĺ–≤–į—ā–ĺ–≥–ĺ –ľ–ĺ–∑–≥–į.
–°—ā–ł–ľ—É–Ľ—Ź—Ü–ł—Ź –°–Ě–° –Ņ—Ä–ł –≥–ł–Ņ–Ķ—Ä–ł–Ĺ—Ā—É–Ľ–ł–Ĺ–Ķ–ľ–ł–ł —Ā–ĺ–Ņ—Ä–ĺ–≤–ĺ–∂–ī–į–Ķ—ā—Ā—Ź —É–≤–Ķ–Ľ–ł—á–Ķ–Ĺ–ł–Ķ–ľ —Ā–Ķ—Ä–ī–Ķ—á–Ĺ–ĺ–≥–ĺ –≤—č–Ī—Ä–ĺ—Ā–į, –Ņ–ĺ–≤—č—ą–Ķ–Ĺ–ł–Ķ–ľ –ĺ–Ī—Č–Ķ–≥–ĺ –Ņ–Ķ—Ä–ł—Ą–Ķ—Ä–ł—á–Ķ—Ā–ļ–ĺ–≥–ĺ —Ā–ĺ—Ā—É–ī–ł—Ā—ā–ĺ–≥–ĺ —Ā–ĺ–Ņ—Ä–ĺ—ā–ł–≤–Ľ–Ķ–Ĺ–ł—Ź, —á—ā–ĺ –Ĺ–Ķ–ł–∑–Ī–Ķ–∂–Ĺ–ĺ –Ņ—Ä–ł–≤–ĺ–ī–ł—ā –ļ –Ņ–ĺ–≤—č—ą–Ķ–Ĺ–ł—é –ź–Ē. –ě–ī–Ĺ–ĺ–≤—Ä–Ķ–ľ–Ķ–Ĺ–Ĺ–ĺ–Ķ —Ā–Ĺ–ł–∂–Ķ–Ĺ–ł–Ķ –į–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā–ł –Ņ–į—Ä–į—Ā–ł–ľ–Ņ–į—ā–ł—á–Ķ—Ā–ļ–ĺ–Ļ —Ā–ł—Ā—ā–Ķ–ľ—č, –≤—č–∑–≤–į–Ĺ–Ĺ–ĺ–Ķ –≥–ł–Ņ–Ķ—Ä–ł–Ĺ—Ā—É–Ľ–ł–Ĺ–Ķ–ľ–ł–Ķ–Ļ, —É–≤–Ķ–Ľ–ł—á–ł–≤–į–Ķ—ā —á–į—Ā—ā–ĺ—ā—É —Ā–Ķ—Ä–ī–Ķ—á–Ĺ—č—Ö —Ā–ĺ–ļ—Ä–į—Č–Ķ–Ĺ–ł–Ļ. –ė–Ĺ—Ā—É–Ľ–ł–Ĺ –ĺ–ļ–į–∑—č–≤–į–Ķ—ā –Ņ—Ä—Ź–ľ–ĺ–Ķ –≤–ĺ–∑–ī–Ķ–Ļ—Ā—ā–≤–ł–Ķ –Ĺ–į –Ņ—Ä–ĺ–ļ—Ā–ł–ľ–į–Ľ—Ć–Ĺ—č–Ķ –ļ–į–Ĺ–į–Ľ—Ć—Ü—č –Ĺ–Ķ—Ą—Ä–ĺ–Ĺ–ĺ–≤, –Ņ–ĺ—ć—ā–ĺ–ľ—É –≤ —É—Ā–Ľ–ĺ–≤–ł—Ź—Ö –≥–ł–Ņ–Ķ—Ä–ł–Ĺ—Ā—É–Ľ–ł–Ĺ–Ķ–ľ–ł–ł –Ņ—Ä–ĺ–ł—Ā—Ö–ĺ–ī–ł—ā –Ņ–ĺ–≤—č—ą–Ķ–Ĺ–ł–Ķ —Ä–Ķ–į–Ī—Ā–ĺ—Ä–Ī—Ü–ł–ł –Ĺ–į—ā—Ä–ł—Ź, –ļ–į–Ľ–ł—Ź, —É—Ä–į—ā–ĺ–≤ –ł –≤–ĺ–ī—č. –í —Ä–Ķ–∑—É–Ľ—Ć—ā–į—ā–Ķ —É–≤–Ķ–Ľ–ł—á–ł–≤–į–Ķ—ā—Ā—Ź –ĺ–Ī—ä–Ķ–ľ —Ü–ł—Ä–ļ—É–Ľ–ł—Ä—É—é—Č–Ķ–Ļ –∂–ł–ī–ļ–ĺ—Ā—ā–ł, —á—ā–ĺ –Ņ—Ä–ł–≤–ĺ–ī–ł—ā –ļ –Ņ–ĺ–≤—č—ą–Ķ–Ĺ–ł—é —Ā–Ķ—Ä–ī–Ķ—á–Ĺ–ĺ–≥–ĺ –≤—č–Ī—Ä–ĺ—Ā–į.
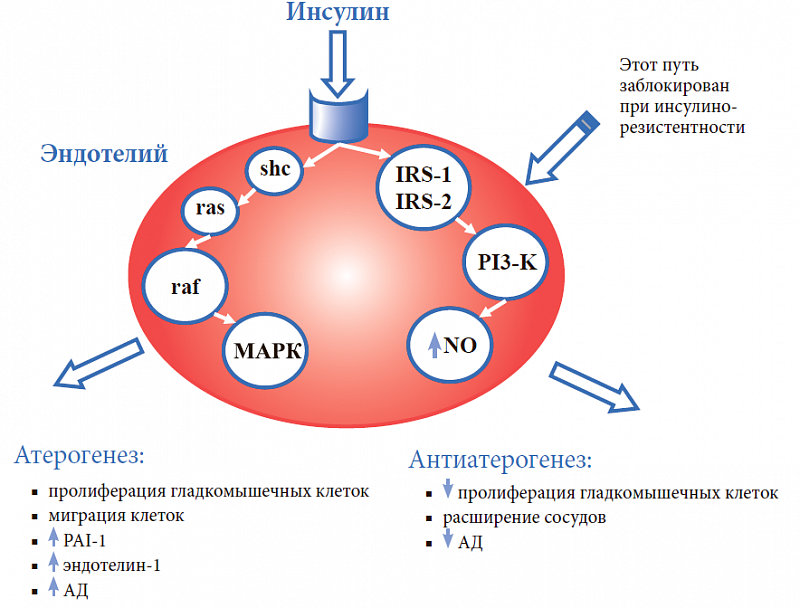
–í–Ĺ—É—ā—Ä–ł–ļ–Ľ–Ķ—ā–ĺ—á–Ĺ–ĺ–Ķ –Ĺ–į–ļ–ĺ–Ņ–Ľ–Ķ–Ĺ–ł–Ķ Na+ –ł Ca2+ ‚Äď —ć—Ą—Ą–Ķ–ļ—ā –ī–Ķ–Ļ—Ā—ā–≤–ł—Ź –ł–Ĺ—Ā—É–Ľ–ł–Ĺ–į. –ė–Ĺ—Ā—É–Ľ–ł–Ĺ –Ī–Ľ–ĺ–ļ–ł—Ä—É–Ķ—ā –į–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć –Ĺ–į—ā—Ä–ł–Ļ-–ļ–į–Ľ–ł–Ļ-–ź–Ę–§-–į–∑—č –ł –ļ–į–Ľ—Ć—Ü–ł–Ļ-–ľ–į–≥–Ĺ–ł–Ļ-–ź–Ę–§-–į–∑—č –ļ–Ľ–Ķ—ā–ĺ—á–Ĺ—č—Ö –ľ–Ķ–ľ–Ī—Ä–į–Ĺ, —á—ā–ĺ –Ņ—Ä–ł–≤–ĺ–ī–ł—ā –ļ –Ņ–ĺ–≤—č—ą–Ķ–Ĺ–ł—é –≤–Ĺ—É—ā—Ä–ł–ļ–Ľ–Ķ—ā–ĺ—á–Ĺ–ĺ–≥–ĺ —Ā–ĺ–ī–Ķ—Ä–∂–į–Ĺ–ł—Ź Na+ –ł Ca2+. –í—Ā–Ľ–Ķ–ī—Ā—ā–≤–ł–Ķ –Ĺ–į–ļ–ĺ–Ņ–Ľ–Ķ–Ĺ–ł—Ź —ć—ā–ł—Ö —ć–Ľ–Ķ–ļ—ā—Ä–ĺ–Ľ–ł—ā–ĺ–≤ –≤ —Ā—ā–Ķ–Ĺ–ļ–Ķ —Ā–ĺ—Ā—É–ī–ĺ–≤ –Ņ–ĺ–≤—č—ą–į–Ķ—ā—Ā—Ź —á—É–≤—Ā—ā–≤–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ—Ā—ā—Ć —Ā–ĺ—Ā—É–ī–ł—Ā—ā—č—Ö —Ä–Ķ—Ü–Ķ–Ņ—ā–ĺ—Ä–ĺ–≤ –ļ –ī–Ķ–Ļ—Ā—ā–≤–ł—é —Ā–ĺ—Ā—É–ī–ĺ—Ā—É–∂–ł–≤–į—é—Č–ł—Ö —Ą–į–ļ—ā–ĺ—Ä–ĺ–≤. –ü–ĺ–ī –≤–Ľ–ł—Ź–Ĺ–ł–Ķ–ľ –ł–Ĺ—Ā—É–Ľ–ł–Ĺ–į –Ņ—Ä–ĺ–ł—Ā—Ö–ĺ–ī–ł—ā —É—ā–ĺ–Ľ—Č–Ķ–Ĺ–ł–Ķ —Ā—ā–Ķ–Ĺ–ļ–ł —Ā–ĺ—Ā—É–ī–ĺ–≤. –ú–ł—ā–ĺ–≥–Ķ–Ĺ–Ĺ—č–Ķ —Ā–≤–ĺ–Ļ—Ā—ā–≤–į –ł–Ĺ—Ā—É–Ľ–ł–Ĺ–į –ĺ–Ī–Ĺ–į—Ä—É–∂–Ķ–Ĺ—č –ī–ĺ—Ā—ā–į—ā–ĺ—á–Ĺ–ĺ –ī–į–≤–Ĺ–ĺ –≤ —Ā–Ķ—Ä–ł–ł —ć–ļ—Ā–Ņ–Ķ—Ä–ł–ľ–Ķ–Ĺ—ā–į–Ľ—Ć–Ĺ—č—Ö —Ä–į–Ī–ĺ—ā, –≥–ī–Ķ –Ī—č–Ľ–ĺ –Ņ–ĺ–ļ–į–∑–į–Ĺ–ĺ, —á—ā–ĺ –ĺ–Ĺ —Ā—ā–ł–ľ—É–Ľ–ł—Ä—É–Ķ—ā –ļ–Ľ–Ķ—ā–ĺ—á–Ĺ—č–Ļ —Ä–ĺ—Ā—ā, –Ņ—Ä–ĺ–Ľ–ł—Ą–Ķ—Ä–į—Ü–ł—é –ł –ľ–ł–≥—Ä–į—Ü–ł—é –≥–Ľ–į–ī–ļ–ĺ–ľ—č—ą–Ķ—á–Ĺ—č—Ö –ļ–Ľ–Ķ—ā–ĺ–ļ —Ā–ĺ—Ā—É–ī–ĺ–≤, –Ņ—Ä–ł–≤–ĺ–ī–ł—ā –ļ —É—ā–ĺ–Ľ—Č–Ķ–Ĺ–ł—é –ł—Ö —Ā—ā–Ķ–Ĺ–ļ–ł. –í –Ĺ–ĺ—Ä–ľ–Ķ –ł–Ĺ—Ā—É–Ľ–ł–Ĺ, —Ā–≤—Ź–∑—č–≤–į—Ź—Ā—Ć —Ā —Ä–Ķ—Ü–Ķ–Ņ—ā–ĺ—Ä–į–ľ–ł –Ĺ–į –Ņ–ĺ–≤–Ķ—Ä—Ö–Ĺ–ĺ—Ā—ā–ł –ļ–Ľ–Ķ—ā–ĺ–ļ —ć–Ĺ–ī–ĺ—ā–Ķ–Ľ–ł—Ź, –ľ–ĺ–∂–Ķ—ā –ī–Ķ–Ļ—Ā—ā–≤–ĺ–≤–į—ā—Ć –ī–≤—É–ľ—Ź —Ä–į–∑–Ľ–ł—á–Ĺ—č–ľ–ł –Ņ—É—ā—Ź–ľ–ł (—Ä–ł—Ā. 4).
–ü–Ķ—Ä–≤—č–Ļ –Ņ—É—ā—Ć ‚Äď —ć—ā–ĺ –į–ļ—ā–ł–≤–į—Ü–ł—Ź —Ā–Ķ–ļ—Ä–Ķ—Ü–ł–ł –ĺ–ļ—Ā–ł–ī–į –į–∑–ĺ—ā–į (NO) —á–Ķ—Ä–Ķ–∑ —Ā—É–Ī—Ā—ā—Ä–į—ā—č –ł–Ĺ—Ā—É–Ľ–ł–Ĺ–ĺ–≤—č—Ö —Ä–Ķ—Ü–Ķ–Ņ—ā–ĺ—Ä–ĺ–≤ 1 –ł 2 (IRS-1 –ł IRS-2) –ł —Ą–ĺ—Ā—Ą–į—ā–ł–ī–ł–Ľ–ł–Ĺ–ĺ–∑–ł—ā–ĺ–Ľ-3-–ļ–ł–Ĺ–į–∑—É (PI3-K). –≠—ā–ĺ—ā –ľ–Ķ—Ö–į–Ĺ–ł–∑–ľ –ĺ–Ī–Ķ—Ā–Ņ–Ķ—á–ł–≤–į–Ķ—ā —Ā–ĺ—Ā—É–ī–ĺ—Ä–į—Ā—ą–ł—Ä—Ź—é—Č–ł–Ķ –ł –į–Ĺ—ā–ł–į—ā–Ķ—Ä–ĺ–≥–Ķ–Ĺ–Ĺ—č–Ķ —Ā–≤–ĺ–Ļ—Ā—ā–≤–į –ł–Ĺ—Ā—É–Ľ–ł–Ĺ–į, —É—á–į—Ā—ā–≤—É–Ķ—ā –≤ –ł–Ĺ—Ā—É–Ľ–ł–Ĺ–∑–į–≤–ł—Ā–ł–ľ–ĺ–ľ —ā—Ä–į–Ĺ—Ā–Ņ–ĺ—Ä—ā–Ķ –≥–Ľ—é–ļ–ĺ–∑—č –≤ –ļ–Ľ–Ķ—ā–ļ–ł. –í—ā–ĺ—Ä–ĺ–Ļ –Ņ—É—ā—Ć ‚Äď —Ä–Ķ–į–Ľ–ł–∑–į—Ü–ł—Ź –ľ–ł—ā–ĺ–≥–Ķ–Ĺ–Ĺ—č—Ö —Ā–≤–ĺ–Ļ—Ā—ā–≤ –ł–Ĺ—Ā—É–Ľ–ł–Ĺ–į —á–Ķ—Ä–Ķ–∑ –ļ–į—Ā–ļ–į–ī –Ņ–ĺ—Ā—Ä–Ķ–ī–Ĺ–ł–ļ–ĺ–≤ (–į–ī–į–Ņ—ā–ł–≤–Ĺ—č–Ļ –Ī–Ķ–Ľ–ĺ–ļ (shc), –Ī–Ķ–Ľ–ĺ–ļ, —Ā–≤—Ź–∑—č–≤–į—é—Č–ł–Ļ shc (ras), –ļ–ł–Ĺ–į–∑–į —Ā–Ķ—Ä–ł–Ĺ-—ā—Ä–Ķ–ĺ–Ĺ–ł–Ĺ–į (raf)), –Ņ–ĺ–≤—č—ą–į—é—Č–ł—Ö –į–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć –ľ–ł—ā–ĺ–≥–Ķ–Ĺ-–į–ļ—ā–ł–≤–ł—Ä–ĺ–≤–į–Ĺ–Ĺ–ĺ–Ļ –Ņ—Ä–ĺ—ā–Ķ–ł–Ĺ–ļ–ł–Ĺ–į–∑—č (–ú–ź–†–ö), —á—ā–ĺ –∑–į–≤–Ķ—Ä—ą–į–Ķ—ā—Ā—Ź –Ņ—Ä–ĺ–Ľ–ł—Ą–Ķ—Ä–į—Ü–ł–Ķ–Ļ –ł –ľ–ł–≥—Ä–į—Ü–ł–Ķ–Ļ –≥–Ľ–į–ī–ļ–ĺ–ľ—č—ą–Ķ—á–Ĺ—č—Ö –ļ–Ľ–Ķ—ā–ĺ–ļ, –į–ļ—ā–ł–≤–į—Ü–ł–Ķ–Ļ —Ā–ł–Ĺ—ā–Ķ–∑–į —Ā–ĺ—Ā—É–ī–ĺ—Ā—É–∂–ł–≤–į—é—Č–Ķ–≥–ĺ —Ą–į–ļ—ā–ĺ—Ä–į —ć–Ĺ–ī–ĺ—ā–Ķ–Ľ–ł–Ĺ–į-1 –ł –Ņ–ĺ–≤—č—ą–Ķ–Ĺ–ł–Ķ–ľ –ź–Ē. –ě–ļ–į–∑–į–Ľ–ĺ—Ā—Ć, —á—ā–ĺ –≤ —É—Ā–Ľ–ĺ–≤–ł—Ź—Ö –ė–† –Ņ–Ķ—Ä–≤—č–Ļ –ľ–Ķ—Ö–į–Ĺ–ł–∑–ľ –Ĺ–Ķ —Ä–į–Ī–ĺ—ā–į–Ķ—ā ‚Äď –ł–ľ–Ķ–Ĺ–Ĺ–ĺ —ć—ā–ĺ—ā –Ņ—É—ā—Ć —Ä–Ķ–∑–ł—Ā—ā–Ķ–Ĺ—ā–Ķ–Ĺ –ļ –ī–Ķ–Ļ—Ā—ā–≤–ł—é –ł–Ĺ—Ā—É–Ľ–ł–Ĺ–į, —Ā–Ľ–Ķ–ī–ĺ–≤–į—ā–Ķ–Ľ—Ć–Ĺ–ĺ, –ľ–ĺ–Ľ–Ķ–ļ—É–Ľ–į NO –Ĺ–Ķ —Ā–ł–Ĺ—ā–Ķ–∑–ł—Ä—É–Ķ—ā—Ā—Ź. –í —ā–ĺ –∂–Ķ –≤—Ä–Ķ–ľ—Ź –≤—ā–ĺ—Ä–ĺ–Ļ –ľ–Ķ—Ö–į–Ĺ–ł–∑–ľ —Ā–ĺ—Ö—Ä–į–Ĺ—Ź–Ķ—ā —Ā–≤–ĺ—é –≤—č—Ā–ĺ–ļ—É—é –į–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć. –ü–ĺ —ć—ā–ĺ–Ļ –Ņ—Ä–ł—á–ł–Ĺ–Ķ –≥–ł–Ņ–Ķ—Ä–ł–Ĺ—Ā—É–Ľ–ł–Ĺ–Ķ–ľ–ł—Ź, —Ä–į–∑–≤–ł–≤–į—é—Č–į—Ź—Ā—Ź –≤—Ā–Ľ–Ķ–ī—Ā—ā–≤–ł–Ķ –ė–† (–Ņ—Ä–ł –ľ–Ķ—ā–į–Ī–ĺ–Ľ–ł—á–Ķ—Ā–ļ–ĺ–ľ —Ā–ł–Ĺ–ī—Ä–ĺ–ľ–Ķ, –°–Ē 2 —ā–ł–Ņ–į, –≤–ł—Ā—Ü–Ķ—Ä–į–Ľ—Ć–Ĺ–ĺ–ľ –ĺ–∂–ł—Ä–Ķ–Ĺ–ł–ł), –Ĺ–Ķ —ā–ĺ–Ľ—Ć–ļ–ĺ –Ĺ–Ķ —Ā–Ĺ–ł–∂–į–Ķ—ā –ź–Ē, –į –Ĺ–į–Ņ—Ä–ĺ—ā–ł–≤, –ĺ–ļ–į–∑—č–≤–į–Ķ—ā –≥–ł–Ņ–Ķ—Ä—ā–Ķ–Ĺ–∑–ł–≤–Ĺ–ĺ–Ķ –ł –į—ā–Ķ—Ä–ĺ–≥–Ķ–Ĺ–Ĺ–ĺ–Ķ –ī–Ķ–Ļ—Ā—ā–≤–ł–Ķ.
–°—É—Č–Ķ—Ā—ā–≤—É–Ķ—ā –≤–∑–į–ł–ľ–ĺ—Ā–≤—Ź–∑—Ć –ľ–Ķ–∂–ī—É –į–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć—é —Ä–Ķ–Ĺ–ł–Ĺ-–į–Ĺ–≥–ł–ĺ—ā–Ķ–Ĺ–∑–ł–Ĺ–ĺ–≤–ĺ–Ļ —Ā–ł—Ā—ā–Ķ–ľ—č, —É—Ä–ĺ–≤–Ĺ–Ķ–ľ –ź–Ē –ł —á—É–≤—Ā—ā–≤–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ—Ā—ā—Ć—é —ā–ļ–į–Ĺ–Ķ–Ļ –ļ –ł–Ĺ—Ā—É–Ľ–ł–Ĺ—É. –•–ĺ—Ä–ĺ—ą–ĺ –ł–∑–≤–Ķ—Ā—ā–Ĺ–ĺ, —á—ā–ĺ –≥–ł–Ņ–Ķ—Ä–į–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć —Ä–Ķ–Ĺ–ł–Ĺ-–į–Ĺ–≥–ł–ĺ—ā–Ķ–Ĺ–∑–ł–Ĺ–ĺ–≤–ĺ–Ļ —Ā–ł—Ā—ā–Ķ–ľ—č —Ā—ā–ĺ–Ļ–ļ–ĺ –Ņ–ĺ–ī–ī–Ķ—Ä–∂–ł–≤–į–Ķ—ā –≤—č—Ā–ĺ–ļ–ĺ–Ķ –ź–Ē. –ě–ī–Ĺ–į–ļ–ĺ –Ľ–ł—ą—Ć –Ĺ–Ķ–ī–į–≤–Ĺ–ĺ –≤ —ć–ļ—Ā–Ņ–Ķ—Ä–ł–ľ–Ķ–Ĺ—ā–į–Ľ—Ć–Ĺ—č—Ö —É—Ā–Ľ–ĺ–≤–ł—Ź—Ö –Ņ–ĺ–Ľ—É—á–Ķ–Ĺ—č —É–Ī–Ķ–ī–ł—ā–Ķ–Ľ—Ć–Ĺ—č–Ķ –ī–į–Ĺ–Ĺ—č–Ķ –ĺ —ā–ĺ–ľ, —á—ā–ĺ –į–Ĺ–≥–ł–ĺ—ā–Ķ–Ĺ–∑–ł–Ĺ II (–ź–Ę II) –ī–ĺ–∑–ĺ–∑–į–≤–ł—Ā–ł–ľ–ĺ –ł–Ĺ–≥–ł–Ī–ł—Ä—É–Ķ—ā –Ņ–ĺ—Ā—ā—Ä–Ķ—Ü–Ķ–Ņ—ā–ĺ—Ä–Ĺ—É—é —Ā–ł–≥–Ĺ–į–Ľ—Ć–Ĺ—É—é —Ā–ł—Ā—ā–Ķ–ľ—É –ł–Ĺ—Ā—É–Ľ–ł–Ĺ–į (–ļ–ĺ–ľ–Ņ–Ľ–Ķ–ļ—Ā IRS-1 –ł IRS-2, —Ā–≤—Ź–∑–į–Ĺ–Ĺ—č–Ļ —Ā PI3-K), —Ä–Ķ–į–Ľ–ł–∑—É—é—Č—É—é —ā—Ä–į–Ĺ—Ā–Ņ–ĺ—Ä—ā –≥–Ľ—é–ļ–ĺ–∑—č –≤ –ļ–Ľ–Ķ—ā–ļ–ł –ł –Ņ—Ä–ĺ–ī—É–ļ—Ü–ł—é NO [18]. –ě–ī–Ĺ–ĺ–≤—Ä–Ķ–ľ–Ķ–Ĺ–Ĺ–ĺ –ź–ʬ†II —Ā—ā–ł–ľ—É–Ľ–ł—Ä—É–Ķ—ā –ú–ź–†–ö, –∑–į–ī–Ķ–Ļ—Ā—ā–≤–ĺ–≤–į–Ĺ–Ĺ—É—é –≤ –ĺ—Ā—É—Č–Ķ—Ā—ā–≤–Ľ–Ķ–Ĺ–ł–ł –ľ–ł—ā–ĺ–≥–Ķ–Ĺ–Ĺ–ĺ–Ļ –ł –Ņ—Ä–ĺ–Ľ–ł—Ą–Ķ—Ä–į—ā–ł–≤–Ĺ–ĺ–Ļ –į–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā–ł –ł–Ĺ—Ā—É–Ľ–ł–Ĺ–į. –Ę–į–ļ–ł–ľ –ĺ–Ī—Ä–į–∑–ĺ–ľ, –≥–ł–Ņ–Ķ—Ä–į–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć —Ä–Ķ–Ĺ–ł–Ĺ-–į–Ĺ–≥–ł–ĺ—ā–Ķ–Ĺ–∑–ł–Ĺ–ĺ–≤–ĺ–Ļ —Ā–ł—Ā—ā–Ķ–ľ—č –ł –ź–Ę II –≤—č–∑—č–≤–į–Ķ—ā —Ä–Ķ–∑–ł—Ā—ā–Ķ–Ĺ—ā–Ĺ–ĺ—Ā—ā—Ć —ā–ļ–į–Ĺ–Ķ–Ļ –ļ –į–Ĺ—ā–ł–į—ā–Ķ—Ä–ĺ–≥–Ķ–Ĺ–Ĺ–ĺ–ľ—É –ł –≥–ł–Ņ–ĺ—ā–Ķ–Ĺ–∑–ł–≤–Ĺ–ĺ–ľ—É –ī–Ķ–Ļ—Ā—ā–≤–ł—é –ł–Ĺ—Ā—É–Ľ–ł–Ĺ–į, —á—ā–ĺ –Ņ—Ä–ł–≤–ĺ–ī–ł—ā –ļ —Ä–į–∑–≤–ł—ā–ł—é —Ā–Ķ—Ä–ī–Ķ—á–Ĺ–ĺ-—Ā–ĺ—Ā—É–ī–ł—Ā—ā—č—Ö –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł–Ļ, –į —ā–į–ļ–∂–Ķ –Ī–Ľ–ĺ–ļ–ł—Ä—É–Ķ—ā —ā—Ä–į–Ĺ—Ā–Ņ–ĺ—Ä—ā –≥–Ľ—é–ļ–ĺ–∑—č –≤ –ļ–Ľ–Ķ—ā–ļ–ł, —Ā–Ņ–ĺ—Ā–ĺ–Ī—Ā—ā–≤—É—Ź —Ä–į–∑–≤–ł—ā–ł—é –Ņ—Ä–Ķ–ī–ł–į–Ī–Ķ—ā–į, –į –∑–į—ā–Ķ–ľ –ł –°–Ē 2 —ā–ł–Ņ–į.
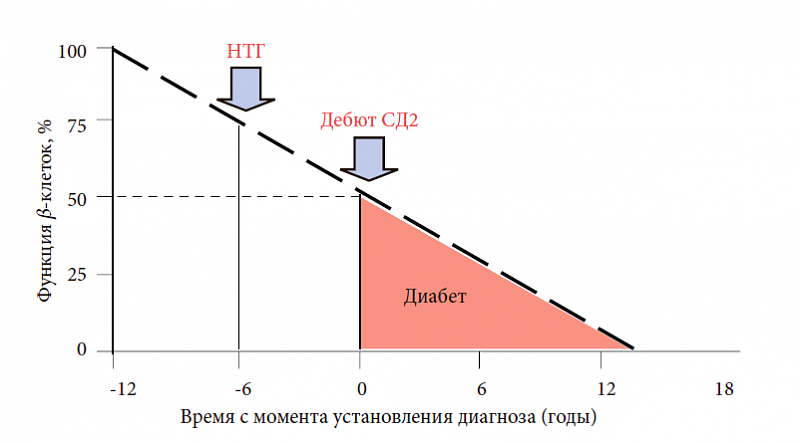
–í–ĺ–∑–ľ–ĺ–∂–Ĺ–ĺ—Ā—ā—Ć –Ņ—Ä–ĺ—Ą–ł–Ľ–į–ļ—ā–ł–ļ–ł —Ä–į–∑–≤–ł—ā–ł—Ź –°–Ē 2 —ā–ł–Ņ–į
–ü—Ä–ĺ—Ā–Ņ–Ķ–ļ—ā–ł–≤–Ĺ–ĺ–Ķ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–Ķ UKPDS (U.K. Prospective Diabetes Study) –Ņ–ĺ–∑–≤–ĺ–Ľ–ł–Ľ–ĺ –Ņ—Ä–ĺ–į–Ĺ–į–Ľ–ł–∑–ł—Ä–ĺ–≤–į—ā—Ć –Ņ—Ä–ĺ–≥—Ä–Ķ—Ā—Ā–ł—Ä–ĺ–≤–į–Ĺ–ł–Ķ –°–Ē 2 —ā–ł–Ņ–į –≤ —ā–Ķ—á–Ķ–Ĺ–ł–Ķ –Ī–ĺ–Ľ–Ķ–Ķ 10 –Ľ–Ķ—ā. –Ď—č–Ľ–ĺ —É—Ā—ā–į–Ĺ–ĺ–≤–Ľ–Ķ–Ĺ–ĺ, —á—ā–ĺ –ļ –ľ–ĺ–ľ–Ķ–Ĺ—ā—É –ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ĺ–≥–ĺ –ī–Ķ–Ī—é—ā–į –°–Ē 2 —ā–ł–Ņ–į —ā–ĺ–Ľ—Ć–ļ–ĺ 50‚Äď60% –ĺ—ā –≤—Ā–Ķ–Ļ –ľ–į—Ā—Ā—č ő≤-–ļ–Ľ–Ķ—ā–ĺ–ļ –Ņ–ĺ–ī–∂–Ķ–Ľ—É–ī–ĺ—á–Ĺ–ĺ–Ļ –∂–Ķ–Ľ–Ķ–∑—č –Ņ—Ä–ĺ–ī–ĺ–Ľ–∂–į—é—ā –į–ļ—ā–ł–≤–Ĺ–ĺ —Ā–Ķ–ļ—Ä–Ķ—ā–ł—Ä–ĺ–≤–į—ā—Ć –ł–Ĺ—Ā—É–Ľ–ł–Ĺ ‚Äď —ć—ā–ĺ –Ņ–Ķ—Ä–≤–į—Ź –ł–∑ –Ņ—Ä–ł—á–ł–Ĺ –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł—Ź (—Ä–ł—Ā. 5) [20]. –Ę–į–ļ–ł–ľ –ĺ–Ī—Ä–į–∑–ĺ–ľ, –ľ–Ķ—ā–į–Ī–ĺ–Ľ–ł—á–Ķ—Ā–ļ–ł–Ķ –Ĺ–į—Ä—É—ą–Ķ–Ĺ–ł—Ź, –≤–Ķ–ī—É—Č–ł–Ķ –ļ —Ä–į–∑–≤–ł—ā–ł—é –°–Ē 2 —ā–ł–Ņ–į, —Ä–Ķ–į–Ľ—Ć–Ĺ–ĺ —Ä–į–∑–≤–ł–≤–į—é—ā—Ā—Ź –∑–į–ī–ĺ–Ľ–≥–ĺ –ī–ĺ –ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ĺ–≥–ĺ –ī–Ķ–Ī—é—ā–į –ī–ł–į–Ī–Ķ—ā–į. –ü—Ä–ł–ľ–Ķ—Ä–Ĺ–ĺ –∑–į 5‚Äď6 –Ľ–Ķ—ā –ī–ĺ –ľ–į–Ĺ–ł—Ą–Ķ—Ā—ā–į—Ü–ł–ł –ī–ł–į–Ī–Ķ—ā–į (–Ņ—Ä–ł —Ā–Ĺ–ł–∂–Ķ–Ĺ–ł–ł —Ą—É–Ĺ–ļ—Ü–ł–ĺ–Ĺ–į–Ľ—Ć–Ĺ–ĺ–Ļ —Ā–Ņ–ĺ—Ā–ĺ–Ī–Ĺ–ĺ—Ā—ā–ł ő≤-–ļ–Ľ–Ķ—ā–ĺ–ļ –ī–ĺ 75%) –ľ–ĺ–∂–Ĺ–ĺ –ī–ł–į–≥–Ĺ–ĺ—Ā—ā–ł—Ä–ĺ–≤–į—ā—Ć –Ņ—Ä–Ķ–ī–ł–į–Ī–Ķ—ā ‚Äď –Ĺ–į—Ä—É—ą–Ķ–Ĺ–Ĺ—É—é —ā–ĺ–Ľ–Ķ—Ä–į–Ĺ—ā–Ĺ–ĺ—Ā—ā—Ć –ļ –≥–Ľ—é–ļ–ĺ–∑–Ķ –ł–Ľ–ł —Ā–ĺ—á–Ķ—ā–į–Ĺ–ł–Ķ –Ě–Ę–ď –ł –Ě–ď–Ě.
–í—ā–ĺ—Ä–ĺ–Ļ –ĺ—Ā–Ĺ–ĺ–≤–Ĺ–ĺ–Ļ –Ņ—Ä–ł—á–ł–Ĺ–ĺ–Ļ —Ä–į–∑–≤–ł—ā–ł—Ź –°–Ē 2 —ā–ł–Ņ–į —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź —Ā–Ĺ–ł–∂–Ķ–Ĺ–Ĺ–į—Ź —á—É–≤—Ā—ā–≤–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ—Ā—ā—Ć –ľ—č—ą–Ķ—á–Ĺ–ĺ–Ļ –ł –∂–ł—Ä–ĺ–≤–ĺ–Ļ —ā–ļ–į–Ĺ–Ķ–Ļ, –į —ā–į–ļ–∂–Ķ –Ņ–Ķ—á–Ķ–Ĺ–ł –ļ –ī–Ķ–Ļ—Ā—ā–≤–ł—é —ć–Ĺ–ī–ĺ–≥–Ķ–Ĺ–Ĺ–ĺ–≥–ĺ –ł–Ĺ—Ā—É–Ľ–ł–Ĺ–į. –ė–Ĺ—Ā—É–Ľ–ł–Ĺ–ĺ—Ä–Ķ–∑–ł—Ā—ā–Ķ–Ĺ—ā–Ĺ–ĺ—Ā—ā—Ć —ā–ļ–į–Ĺ–Ķ–Ļ, —Ā—É—Č–Ķ—Ā—ā–≤—É—é—Č–į—Ź –∑–į–ī–ĺ–Ľ–≥–ĺ –ī–ĺ –ī–Ķ–Ī—é—ā–į –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł—Ź, –Ņ—Ä–ł–≤–ĺ–ī–ł—ā –ļ –ł—Ā—ā–ĺ—Č–Ķ–Ĺ–ł—é —ć–Ĺ–ī–ĺ–≥–Ķ–Ĺ–Ĺ—č—Ö –∑–į–Ņ–į—Ā–ĺ–≤ –ł–Ĺ—Ā—É–Ľ–ł–Ĺ–į. –§—É–Ĺ–ļ—Ü–ł–ĺ–Ĺ–į–Ľ—Ć–Ĺ–į—Ź –į–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć ő≤-–ļ–Ľ–Ķ—ā–ĺ–ļ –Ņ–ĺ–ī–∂–Ķ–Ľ—É–ī–ĺ—á–Ĺ–ĺ–Ļ –∂–Ķ–Ľ–Ķ–∑—č –ļ –ľ–ĺ–ľ–Ķ–Ĺ—ā—É –ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ĺ–Ļ –ľ–į–Ĺ–ł—Ą–Ķ—Ā—ā–į—Ü–ł–ł –°–Ē 2 —ā–ł–Ņ–į —Ā–Ĺ–ł–∂–į–Ķ—ā—Ā—Ź –Ĺ–į 50%, —á—ā–ĺ –ł –Ņ—Ä–ł–≤–ĺ–ī–ł—ā –ļ –Ņ–ĺ–≤—č—ą–Ķ–Ĺ–ł—é —É—Ä–ĺ–≤–Ĺ—Ź –≥–Ľ–ł–ļ–Ķ–ľ–ł–ł. –Ē–ĺ–Ľ–≥–ĺ–Ķ –≤—Ä–Ķ–ľ—Ź (–ł–Ĺ–ĺ–≥–ī–į –≤ —ā–Ķ—á–Ķ–Ĺ–ł–Ķ –Ĺ–Ķ—Ā–ļ–ĺ–Ľ—Ć–ļ–ł—Ö –Ľ–Ķ—ā) –Ī–ĺ–Ľ—Ć–Ĺ–ĺ–Ļ —Ā—É–Ī—ä–Ķ–ļ—ā–ł–≤–Ĺ–ĺ –ľ–ĺ–∂–Ķ—ā –Ĺ–Ķ –ĺ—Č—É—Č–į—ā—Ć –Ņ—Ä–ł–∑–Ĺ–į–ļ–ĺ–≤ –≥–ł–Ņ–Ķ—Ä–≥–Ľ–ł–ļ–Ķ–ľ–ł–ł, —ā–ĺ –Ķ—Ā—ā—Ć –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł–Ķ –Ņ—Ä–ĺ—ā–Ķ–ļ–į–Ķ—ā –Ī–Ķ—Ā—Ā–ł–ľ–Ņ—ā–ĺ–ľ–Ĺ–ĺ. –í —ā–ĺ –∂–Ķ –≤—Ä–Ķ–ľ—Ź —Ā–ĺ—Ā—É–ī–ł—Ā—ā—č–Ķ –ĺ—Ā–Ľ–ĺ–∂–Ĺ–Ķ–Ĺ–ł—Ź —Ā–į—Ö–į—Ä–Ĺ–ĺ–≥–ĺ –ī–ł–į–Ī–Ķ—ā–į, –≤ —ā–ĺ–ľ —á–ł—Ā–Ľ–Ķ –ł —ć–Ĺ—Ü–Ķ—Ą–į–Ľ–ĺ–Ņ–į—ā–ł—Ź, —É–∂–Ķ –Ĺ–į—á–ł–Ĺ–į—é—ā —Ä–į–∑–≤–ł–≤–į—ā—Ć—Ā—Ź, —á—ā–ĺ –ĺ–Ī—É—Ā–Ľ–ĺ–≤–Ľ–Ķ–Ĺ–ĺ –Ī—č—Ā—ā—Ä—č–ľ –Ņ—Ä–ĺ–≥—Ä–Ķ—Ā—Ā–ł—Ä–ĺ–≤–į–Ĺ–ł–Ķ–ľ –Ņ–ĺ—Ā—ā–Ņ—Ä–į–Ĺ–ī–ł–į–Ľ—Ć–Ĺ–ĺ–Ļ –≥–ł–Ņ–Ķ—Ä–≥–Ľ–ł–ļ–Ķ–ľ–ł–ł, –ļ–ĺ—ā–ĺ—Ä–į—Ź –ł–ľ–Ķ–Ķ—ā –Ī–ĺ–Ľ—Ć—ą–ĺ–Ķ –∑–Ĺ–į—á–Ķ–Ĺ–ł–Ķ –≤ —Ä–į–∑–≤–ł—ā–ł–ł —Ā–Ķ—Ä–ī–Ķ—á–Ĺ–ĺ-—Ā–ĺ—Ā—É–ī–ł—Ā—ā–ĺ–Ļ –Ņ–į—ā–ĺ–Ľ–ĺ–≥–ł–ł.
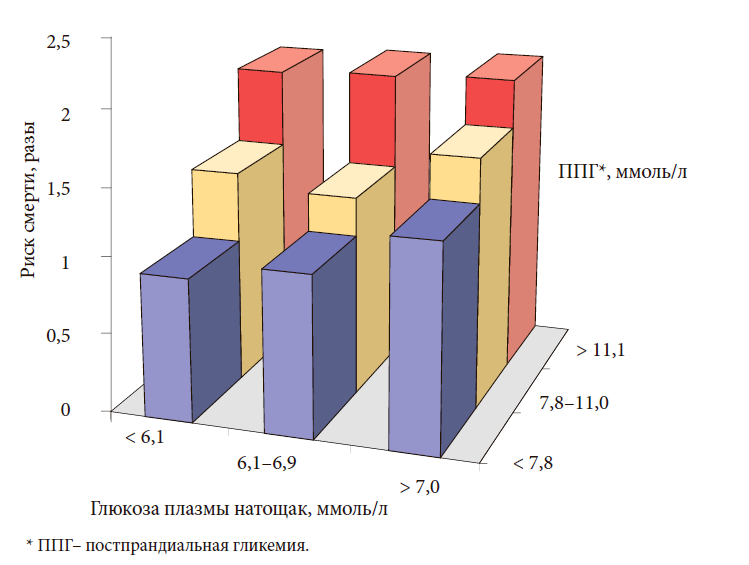
–ü–ĺ—Ā—ā–Ņ—Ä–į–Ĺ–ī–ł–į–Ľ—Ć–Ĺ–į—Ź –≥–ł–Ņ–Ķ—Ä–≥–Ľ–ł–ļ–Ķ–ľ–ł—Ź (—É—Ä–ĺ–≤–Ķ–Ĺ—Ć –≥–Ľ—é–ļ–ĺ–∑—č –Ņ–Ľ–į–∑–ľ—č –ļ—Ä–ĺ–≤–ł –Ņ–ĺ—Ā–Ľ–Ķ –Ņ—Ä–ł–Ķ–ľ–į –Ņ–ł—Č–ł) —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź —Ā–į–ľ–ĺ—Ā—ā–ĺ—Ź—ā–Ķ–Ľ—Ć–Ĺ—č–ľ —Ą–į–ļ—ā–ĺ—Ä–ĺ–ľ —Ä–ł—Ā–ļ–į —Ä–į–∑–≤–ł—ā–ł—Ź —Ā–Ķ—Ä–ī–Ķ—á–Ĺ–ĺ-—Ā–ĺ—Ā—É–ī–ł—Ā—ā—č—Ö –ĺ—Ā–Ľ–ĺ–∂–Ĺ–Ķ–Ĺ–ł–Ļ –ł –Ņ—Ä–Ķ–∂–ī–Ķ–≤—Ä–Ķ–ľ–Ķ–Ĺ–Ĺ–ĺ–Ļ —Ā–ľ–Ķ—Ä—ā–ł, –ļ–į–ļ –Ņ–ĺ–ļ–į–∑–į–Ľ–ł —Ä–Ķ–∑—É–Ľ—Ć—ā–į—ā—č –ļ—Ä—É–Ņ–Ĺ—č—Ö –ľ–Ķ–∂–ī—É–Ĺ–į—Ä–ĺ–ī–Ĺ—č—Ö –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–Ļ [21]. –í –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–ł DECODE –ł—Ā–Ņ–ĺ–Ľ—Ć–∑–ĺ–≤–į–Ľ–ł—Ā—Ć –ī–į–Ĺ–Ĺ—č–Ķ —Ä—Ź–ī–į –Ņ—Ä–ĺ—Ā–Ņ–Ķ–ļ—ā–ł–≤–Ĺ—č—Ö –ļ—Ä—É–Ņ–Ĺ–ĺ–ľ–į—Ā—ą—ā–į–Ī–Ĺ—č—Ö –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–Ļ, –Ņ—Ä–ĺ–≤–ĺ–ī–ł–≤—ą–ł—Ö—Ā—Ź –≤ –ē–≤—Ä–ĺ–Ņ–Ķ. –Ě–į –ĺ—Ā–Ĺ–ĺ–≤–į–Ĺ–ł–ł –ł—Ö –į–Ĺ–į–Ľ–ł–∑–į –Ī—č–Ľ–ĺ –Ņ–ĺ–ļ–į–∑–į–Ĺ–ĺ, —á—ā–ĺ –Ī–ĺ–Ľ–Ķ–Ķ –≤—č—Ā–ĺ–ļ–ł–Ļ —Ä–ł—Ā–ļ —Ā–Ķ—Ä–ī–Ķ—á–Ĺ–ĺ-—Ā–ĺ—Ā—É–ī–ł—Ā—ā–ĺ–Ļ —Ā–ľ–Ķ—Ä—ā–Ĺ–ĺ—Ā—ā–ł –Ĺ–į–Ī–Ľ—é–ī–į–Ľ—Ā—Ź —É –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ —Ā –Ņ–ĺ–≤—č—ą–Ķ–Ĺ–ł–Ķ–ľ –Ņ–ĺ—Ā—ā–Ņ—Ä–į–Ĺ–ī–ł–į–Ľ—Ć–Ĺ–ĺ–≥–ĺ —É—Ä–ĺ–≤–Ĺ—Ź –≥–Ľ—é–ļ–ĺ–∑—č –Ī–ĺ–Ľ–Ķ–Ķ —á–Ķ–ľ 11,1 –ľ–ľ–ĺ–Ľ—Ć/–Ľ, –Ĺ–Ķ–∑–į–≤–ł—Ā–ł–ľ–ĺ –ĺ—ā —É—Ä–ĺ–≤–Ĺ—Ź –≥–Ľ–ł–ļ–Ķ–ľ–ł–ł –Ĺ–į—ā–ĺ—Č–į–ļ (—Ä–ł—Ā. 6).
–ö–į–ļ –Ī—č–Ľ–ĺ –ĺ—ā–ľ–Ķ—á–Ķ–Ĺ–ĺ –≤—č—ą–Ķ, –Ĺ–į—á–į–Ľ–ĺ –°–Ē 2 —ā–ł–Ņ–į —Ö–į—Ä–į–ļ—ā–Ķ—Ä–ł–∑—É–Ķ—ā—Ā—Ź –Ĺ–Ķ —ā–ĺ–Ľ—Ć–ļ–ĺ –≤—č—Ä–į–∂–Ķ–Ĺ–Ĺ—č–ľ–ł –Ĺ–į—Ä—É—ą–Ķ–Ĺ–ł—Ź–ľ–ł —á—É–≤—Ā—ā–≤–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ—Ā—ā–ł —ā–ļ–į–Ĺ–Ķ–Ļ –ļ –ł–Ĺ—Ā—É–Ľ–ł–Ĺ—É, –Ĺ–ĺ –ł –Ĺ–Ķ–ī–ĺ—Ā—ā–į—ā–ĺ—á–Ĺ–ĺ—Ā—ā—Ć—é —Ą—É–Ĺ–ļ—Ü–ł–ł ő≤-–ļ–Ľ–Ķ—ā–ĺ–ļ –Ņ–ĺ–ī–∂–Ķ–Ľ—É–ī–ĺ—á–Ĺ–ĺ–Ļ –∂–Ķ–Ľ–Ķ–∑—č, –ļ–ĺ—ā–ĺ—Ä–į—Ź –Ĺ–Ķ –ľ–ĺ–∂–Ķ—ā –ļ–ĺ–ľ–Ņ–Ķ–Ĺ—Ā–ł—Ä–ĺ–≤–į—ā—Ć –ł–Ĺ—Ā—É–Ľ–ł–Ĺ–ĺ—Ä–Ķ–∑–ł—Ā—ā–Ķ–Ĺ—ā–Ĺ–ĺ—Ā—ā—Ć –Ņ—É—ā–Ķ–ľ –Ņ–ĺ–≤—č—ą–Ķ–Ĺ–ł—Ź —Ā–Ķ–ļ—Ä–Ķ—Ü–ł–ł –ł–Ĺ—Ā—É–Ľ–ł–Ĺ–į. –≠—ā–ł –ł–∑–ľ–Ķ–Ĺ–Ķ–Ĺ–ł—Ź –Ĺ–į —Ā—ā–į–ī–ł–ł –ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ł—Ö –Ņ—Ä–ĺ—Ź–≤–Ľ–Ķ–Ĺ–ł–Ļ –°–Ē –Ī–Ķ–∑—É—Ā–Ľ–ĺ–≤–Ĺ–ĺ –Ĺ–Ķ–ĺ–Ī—Ä–į—ā–ł–ľ—č, –Ņ–ĺ—ć—ā–ĺ–ľ—É –ī–į–∂–Ķ —Ä–į–Ĺ–Ĺ–Ķ–Ķ –Ĺ–į—á–į–Ľ–ĺ –Ľ–Ķ—á–Ķ–Ĺ–ł—Ź –°–Ē¬†2 —ā–ł–Ņ–į –Ĺ–Ķ –ľ–ĺ–∂–Ķ—ā –Ņ—Ä–ł–≤–Ķ—Ā—ā–ł –ļ –Ņ–ĺ–Ľ–Ĺ–ĺ–ľ—É –≤—č–∑–ī–ĺ—Ä–ĺ–≤–Ľ–Ķ–Ĺ–ł—é, –ĺ–ī–Ĺ–į–ļ–ĺ –Ņ—Ä–ł–ľ–Ķ–Ĺ–Ķ–Ĺ–ł–Ķ –Ľ–Ķ–ļ–į—Ä—Ā—ā–≤–Ķ–Ĺ–Ĺ—č—Ö —Ā—Ä–Ķ–ī—Ā—ā–≤, —É—Ā—ā—Ä–į–Ĺ—Ź—é—Č–ł—Ö –≤–Ľ–ł—Ź–Ĺ–ł–Ķ –ĺ—Ā–Ĺ–ĺ–≤–Ĺ—č—Ö –Ņ–į—ā–ĺ–≥–Ķ–Ĺ–Ķ—ā–ł—á–Ķ—Ā–ļ–ł—Ö –ľ–Ķ—Ö–į–Ĺ–ł–∑–ľ–ĺ–≤ ‚Äď –≥–Ľ–į–≤–Ĺ—č–ľ –ĺ–Ī—Ä–į–∑–ĺ–ľ –ė–† ‚Äď –Ĺ–Ķ–ĺ–Ī—Ö–ĺ–ī–ł–ľ–ĺ –Ĺ–į –Ľ—é–Ī–ĺ–ľ —ć—ā–į–Ņ–Ķ —Ä–į–∑–≤–ł—ā–ł—Ź —É–≥–Ľ–Ķ–≤–ĺ–ī–Ĺ—č—Ö –Ĺ–į—Ä—É—ą–Ķ–Ĺ–ł–Ļ. –í —ā–ĺ –∂–Ķ –≤—Ä–Ķ–ľ—Ź, –Ķ—Ā–Ľ–ł –Ĺ–į—á–į—ā—Ć –Ņ—Ä–ĺ—Ą–ł–Ľ–į–ļ—ā–ł—á–Ķ—Ā–ļ–ł–Ķ –ľ–Ķ—Ä–ĺ–Ņ—Ä–ł—Ź—ā–ł—Ź –ī–ĺ –ī–Ķ–Ī—é—ā–į –°–Ē 2 —ā–ł–Ņ–į ‚Äď –Ĺ–į —ć—ā–į–Ņ–Ķ –Ņ—Ä–Ķ–ī–ł–į–Ī–Ķ—ā–į, —ā–ĺ –ľ–ĺ–∂–Ĺ–ĺ –Ņ—Ä–Ķ–ī–ĺ—ā–≤—Ä–į—ā–ł—ā—Ć —Ä–į–∑–≤–ł—ā–ł–Ķ –ł –Ņ—Ä–ĺ–≥—Ä–Ķ—Ā—Ā–ł—Ä–ĺ–≤–į–Ĺ–ł–Ķ –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł—Ź –ł —Ā–ĺ–Ņ—É—ā—Ā—ā–≤—É—é—Č–ł—Ö –Ķ–ľ—É —ā—Ź–∂–Ķ–Ľ—č—Ö –ĺ—Ā–Ľ–ĺ–∂–Ĺ–Ķ–Ĺ–ł–Ļ. –ü—Ä–ĺ—Ą–ł–Ľ–į–ļ—ā–ł–ļ–į –ł —Ā–ĺ–≤—Ä–Ķ–ľ–Ķ–Ĺ–Ĺ–į—Ź —ā–Ķ—Ä–į–Ņ–ł—Ź –°–Ē –ĺ–Ī—É—Ā–Ľ–ĺ–≤–Ľ–Ķ–Ĺ–į –Ņ–į—ā–ĺ–≥–Ķ–Ĺ–Ķ–∑–ĺ–ľ, –Ņ–ĺ—ć—ā–ĺ–ľ—É –ĺ—Ā–Ĺ–ĺ–≤—č–≤–į–Ķ—ā—Ā—Ź –Ĺ–į –Ņ—Ä–ł–ľ–Ķ–Ĺ–Ķ–Ĺ–ł–ł –ĺ–ī–Ĺ–ł—Ö –ł —ā–Ķ—Ö –∂–Ķ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–ĺ–≤ –ł –ľ–Ķ—ā–ĺ–ī–ĺ–≤.
–†–Ķ–ļ–ĺ–ľ–Ķ–Ĺ–ī–į—Ü–ł–ł –Ņ–ĺ –Ĺ–Ķ–ľ–Ķ–ī–ł–ļ–į–ľ–Ķ–Ĺ—ā–ĺ–∑–Ĺ–ĺ–ľ—É –Ľ–Ķ—á–Ķ–Ĺ–ł—é –≤–ļ–Ľ—é—á–į—é—ā –ļ–ĺ—Ä—Ä–Ķ–ļ—Ü–ł—é –ĺ–Ī—Ä–į–∑–į –∂–ł–∑–Ĺ–ł: —Ā–ĺ–Ī–Ľ—é–ī–Ķ–Ĺ–ł–Ķ –ī–ł–Ķ—ā—č, —Ä–Ķ–≥—É–Ľ—Ź—Ä–Ĺ—č–Ķ —Ą–ł–∑–ł—á–Ķ—Ā–ļ–ł–Ķ –Ĺ–į–≥—Ä—É–∑–ļ–ł ‚Äď –ľ–Ķ—Ä—č, –Ĺ–į–Ņ—Ä–į–≤–Ľ–Ķ–Ĺ–Ĺ—č–Ķ –Ĺ–į —Ā–Ĺ–ł–∂–Ķ–Ĺ–ł–Ķ –ľ–į—Ā—Ā—č —ā–Ķ–Ľ–į, –ł–Ĺ—Ā—É–Ľ–ł–Ĺ–ĺ—Ä–Ķ–∑–ł—Ā—ā–Ķ–Ĺ—ā–Ĺ–ĺ—Ā—ā–ł, –Ĺ–ĺ—Ä–ľ–į–Ľ–ł–∑–į—Ü–ł—é –į—Ä—ā–Ķ—Ä–ł–į–Ľ—Ć–Ĺ–ĺ–≥–ĺ –ī–į–≤–Ľ–Ķ–Ĺ–ł—Ź, –Ľ–ł–Ņ–ł–ī–Ĺ—č—Ö –Ĺ–į—Ä—É—ą–Ķ–Ĺ–ł–Ļ –ł –≤–ĺ—Ā—Ā—ā–į–Ĺ–ĺ–≤–Ľ–Ķ–Ĺ–ł–Ķ —É–≥–Ľ–Ķ–≤–ĺ–ī–Ĺ–ĺ–≥–ĺ –ĺ–Ī–ľ–Ķ–Ĺ–į. –í —Ā–Ľ—É—á–į–Ķ –Ĺ–ł–∑–ļ–ĺ–Ļ –Ņ—Ä–ł–≤–Ķ—Ä–∂–Ķ–Ĺ–Ĺ–ĺ—Ā—ā–ł –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ –ļ –Ĺ–Ķ–ľ–Ķ–ī–ł–ļ–į–ľ–Ķ–Ĺ—ā–ĺ–∑–Ĺ—č–ľ –ľ–Ķ—ā–ĺ–ī–į–ľ –ļ–ĺ—Ä—Ä–Ķ–ļ—Ü–ł–ł, –į —ā–į–ļ–∂–Ķ –Ņ—Ä–ł –ł—Ö –Ĺ–Ķ—ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā–ł –ĺ–Ņ—Ä–į–≤–ī–į–Ĺ–ĺ –Ņ—Ä–ł–ľ–Ķ–Ĺ–Ķ–Ĺ–ł–Ķ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–ĺ–≤, —É–Ľ—É—á—ą–į—é—Č–ł—Ö —á—É–≤—Ā—ā–≤–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ—Ā—ā—Ć —ā–ļ–į–Ĺ–Ķ–Ļ –ļ –ł–Ĺ—Ā—É–Ľ–ł–Ĺ—É, —á—ā–ĺ –ĺ–Ī–Ľ–Ķ–≥—á–į–Ķ—ā –Ņ–ĺ—Ā—ā—É–Ņ–Ľ–Ķ–Ĺ–ł–Ķ –≥–Ľ—é–ļ–ĺ–∑—č –≤ –ļ–Ľ–Ķ—ā–ļ–ł –Ī–Ķ–∑ –ī–ĺ–Ņ–ĺ–Ľ–Ĺ–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ–Ļ –ď–ė. –ö —ā–į–ļ–ł–ľ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į–ľ –≤ –Ņ–Ķ—Ä–≤—É—é –ĺ—á–Ķ—Ä–Ķ–ī—Ć –ĺ—ā–Ĺ–ĺ—Ā–ł—ā—Ā—Ź –ľ–Ķ—ā—Ą–ĺ—Ä–ľ–ł–Ĺ, —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć –ļ–ĺ—ā–ĺ—Ä–ĺ–≥–ĺ –≤ —Ā–Ĺ–ł–∂–Ķ–Ĺ–ł–ł —Ä–ł—Ā–ļ–į —Ä–į–∑–≤–ł—ā–ł—Ź –°–Ē 2 —ā–ł–Ņ–į –Ī—č–Ľ–į –ī–ĺ–ļ–į–∑–į–Ĺ–į –≤ –ļ—Ä—É–Ņ–Ĺ—č—Ö –ľ–Ķ–∂–ī—É–Ĺ–į—Ä–ĺ–ī–Ĺ—č—Ö –Ņ–Ľ–į—Ü–Ķ–Ī–ĺ-–ļ–ĺ–Ĺ—ā—Ä–ĺ–Ľ–ł—Ä—É–Ķ–ľ—č—Ö –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź—Ö. –í–į–∂–Ĺ–ĺ –Ņ–ĺ–ľ–Ĺ–ł—ā—Ć, —á—ā–ĺ —Ā–į–ľ—č–ľ —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ—č–ľ –ľ–Ķ—ā–ĺ–ī–ĺ–ľ –Ľ–Ķ—á–Ķ–Ĺ–ł—Ź –°–Ē 2 —ā–ł–Ņ–į —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź —Ā–ĺ—á–Ķ—ā–į–Ĺ–ł–Ķ –ľ–Ķ–ī–ł–ļ–į–ľ–Ķ–Ĺ—ā–ĺ–∑–Ĺ–ĺ–Ļ –ļ–ĺ—Ä—Ä–Ķ–ļ—Ü–ł–ł —Ā –ł–∑–ľ–Ķ–Ĺ–Ķ–Ĺ–ł–Ķ–ľ –ĺ–Ī—Ä–į–∑–į –∂–ł–∑–Ĺ–ł.
–ü—Ä–ĺ—Ą–ł–Ľ–į–ļ—ā–ł–ļ–į —Ä–į–∑–≤–ł—ā–ł—Ź –ī–Ķ–ľ–Ķ–Ĺ—Ü–ł–ł —É –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ —Ā ¬†–Ĺ–į—Ä—É—ą–Ķ–Ĺ–ł—Ź–ľ–ł —É–≥–Ľ–Ķ–≤–ĺ–ī–Ĺ–ĺ–≥–ĺ –ł ¬†–∂–ł—Ä–ĺ–≤–ĺ–≥–ĺ –ĺ–Ī–ľ–Ķ–Ĺ–ĺ–≤
–ü—Ä–ĺ—Ą–ł–Ľ–į–ļ—ā–ł–ļ–į –Ľ—é–Ī–ĺ–≥–ĺ –ĺ—Ā–Ľ–ĺ–∂–Ĺ–Ķ–Ĺ–ł—Ź, —Ā–≤—Ź–∑–į–Ĺ–Ĺ–ĺ–≥–ĺ —Ā —Ā–į—Ö–į—Ä–Ĺ—č–ľ –ī–ł–į–Ī–Ķ—ā–ĺ–ľ, –ĺ—Ā–ĺ–Ī–Ķ–Ĺ–Ĺ–ĺ 2 —ā–ł–Ņ–į, —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź –Ĺ–Ķ–Ņ—Ä–ĺ—Ā—ā–ĺ–Ļ –∑–į–ī–į—á–Ķ–Ļ, –Ņ–ĺ—Ā–ļ–ĺ–Ľ—Ć–ļ—É –∑–ī–Ķ—Ā—Ć —Ā–≤—Ź–∑–į–Ĺ—č –≤–ĺ–Ķ–ī–ł–Ĺ–ĺ –ľ–Ĺ–ĺ–∂–Ķ—Ā—ā–≤–ĺ —Ą–į–ļ—ā–ĺ—Ä–ĺ–≤: —Ā–Ľ–ĺ–∂–Ĺ—č–Ļ –Ņ–į—ā–ĺ–≥–Ķ–Ĺ–Ķ–∑ –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł—Ź –ł –ī–Ľ–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ–Ķ –Ľ–į—ā–Ķ–Ĺ—ā–Ĺ–ĺ–Ķ —ā–Ķ—á–Ķ–Ĺ–ł–Ķ –°–Ē, –Ĺ–Ķ –Ņ—Ä–ĺ—Ź–≤–Ľ—Ź—é—Č–Ķ–≥–ĺ—Ā—Ź –ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ł –Ņ–Ķ—Ä–≤—č–Ķ 7‚Äď8 –Ľ–Ķ—ā; —ā—Ä–Ķ–Ī—É—é—Č–ł–Ķ—Ā—Ź –ĺ—ā –≤—Ä–į—á–į (—á–į—Č–Ķ –≤—Ā–Ķ–≥–ĺ –Ĺ–Ķ–≤—Ä–ĺ–Ľ–ĺ–≥–į –ł–Ľ–ł —ā–Ķ—Ä–į–Ņ–Ķ–≤—ā–į) –≥–Ľ—É–Ī–ĺ–ļ–ł–Ķ –∑–Ĺ–į–Ĺ–ł—Ź –Ņ—Ä–ĺ–Ī–Ľ–Ķ–ľ—č –ī–Ľ—Ź –Ņ—Ä–ĺ–≤–Ķ–ī–Ķ–Ĺ–ł—Ź –ī–ĺ–Ņ–ĺ–Ľ–Ĺ–ł—ā–Ķ–Ľ—Ć–Ĺ—č—Ö –ľ–Ķ—ā–ĺ–ī–ĺ–≤ –ĺ–Ī—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź –ł —Ä–į–Ĺ–Ĺ–Ķ–Ļ –ī–ł–į–≥–Ĺ–ĺ—Ā—ā–ł–ļ–ł –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł—Ź –ł–Ľ–ł –Ĺ–į–Ņ—Ä–į–≤–Ľ–Ķ–Ĺ–ł—Ź –ļ —ć–Ĺ–ī–ĺ–ļ—Ä–ł–Ĺ–ĺ–Ľ–ĺ–≥—É. –Ę–į–ļ–ł–ľ –ĺ–Ī—Ä–į–∑–ĺ–ľ, –≤ —Ā–ĺ–≤—Ä–Ķ–ľ–Ķ–Ĺ–Ĺ–ĺ–Ļ –Ņ—Ä–į–ļ—ā–ł–ļ–Ķ –ī–ł–į–≥–Ĺ–ĺ–∑ –°–Ē 2 —ā–ł–Ņ–į —á–į—Č–Ķ –≤—Ā–Ķ–≥–ĺ —É—Ā—ā–į–Ĺ–į–≤–Ľ–ł–≤–į–Ķ—ā—Ā—Ź –ĺ–ī–Ĺ–ĺ–≤—Ä–Ķ–ľ–Ķ–Ĺ–Ĺ–ĺ —Ā –Ņ–ĺ—Ź–≤–Ľ–Ķ–Ĺ–ł–Ķ–ľ —É –Ņ–į—Ü–ł–Ķ–Ĺ—ā–į –Ņ–Ķ—Ä–≤–ĺ–≥–ĺ –ľ–į–ļ—Ä–ĺ–≤–į—Ā–ļ—É–Ľ—Ź—Ä–Ĺ–ĺ–≥–ĺ –ĺ—Ā–Ľ–ĺ–∂–Ĺ–Ķ–Ĺ–ł—Ź ‚Äď –ĺ—Ā—ā—Ä–ĺ–≥–ĺ –ł–Ĺ—Ą–į—Ä–ļ—ā–į –ł–Ľ–ł –ł–Ĺ—Ā—É–Ľ—Ć—ā–į, –ļ–ĺ—ā–ĺ—Ä–ĺ–Ķ, —Ā–ĺ–Ī—Ā—ā–≤–Ķ–Ĺ–Ĺ–ĺ, –ł —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź –Ņ–ĺ–≤–ĺ–ī–ĺ–ľ –ī–Ľ—Ź –Ī–ĺ–Ľ–Ķ–Ķ —Ā–Ķ—Ä—Ć–Ķ–∑–Ĺ–ĺ–≥–ĺ –ĺ–Ī—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź.
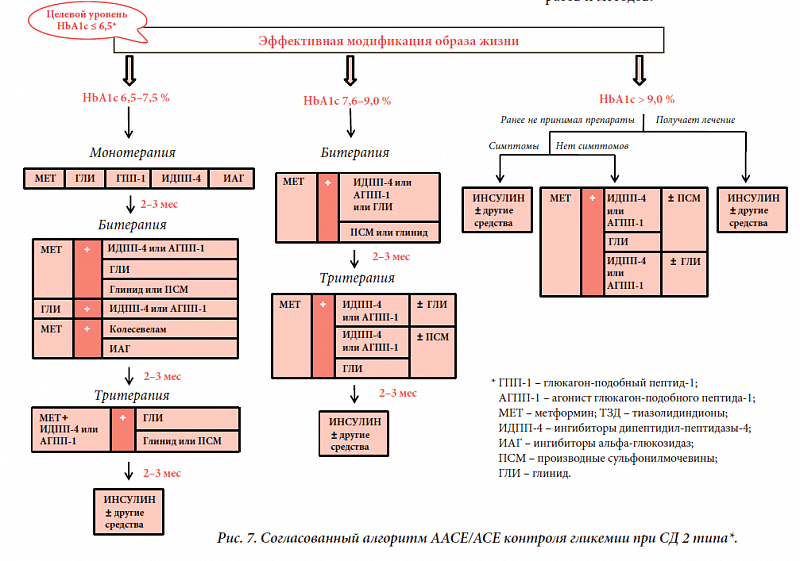
–ü—Ä–ł –°–Ē –ĺ–ī–Ĺ–ĺ–Ļ –ł–∑ –Ņ–Ķ—Ä–≤—č—Ö –Ņ–ĺ—Ä–į–∂–į–Ķ—ā—Ā—Ź —Ü–Ķ–Ĺ—ā—Ä–į–Ľ—Ć–Ĺ–į—Ź –ł –Ņ–Ķ—Ä–ł—Ą–Ķ—Ä–ł—á–Ķ—Ā–ļ–į—Ź –Ĺ–Ķ—Ä–≤–Ĺ–į—Ź —Ā–ł—Ā—ā–Ķ–ľ–į. –ü–Ķ—Ä–ł—Ą–Ķ—Ä–ł—á–Ķ—Ā–ļ–į—Ź –Ņ–ĺ–Ľ–ł–Ĺ–Ķ–Ļ—Ä–ĺ–Ņ–į—ā–ł—Ź –ł —ć–Ĺ—Ü–Ķ—Ą–į–Ľ–ĺ–Ņ–į—ā–ł—Ź —Ā–ľ–Ķ—ą–į–Ĺ–Ĺ–ĺ–≥–ĺ –≥–Ķ–Ĺ–Ķ–∑–į (—Ā–ĺ—Ā—É–ī–ł—Ā—ā–ĺ–≥–ĺ –ł –ľ–Ķ—ā–į–Ī–ĺ–Ľ–ł—á–Ķ—Ā–ļ–ĺ–≥–ĺ) –Ņ—Ä–ł–≤–ĺ–ī–ł—ā –ļ –Ņ—Ä–ĺ–≥—Ä–Ķ—Ā—Ā–ł—Ä—É—é—Č–Ķ–ľ—É —Ā–Ĺ–ł–∂–Ķ–Ĺ–ł—é –ļ–ĺ–≥–Ĺ–ł—ā–ł–≤–Ĺ–ĺ–Ļ —Ą—É–Ĺ–ļ—Ü–ł–ł –ł —Ä–į–∑–≤–ł—ā–ł—é –ī–Ķ–ľ–Ķ–Ĺ—Ü–ł–ł. –ź –Ķ—Ā–Ľ–ł —É—á–Ķ—Ā—ā—Ć, —á—ā–ĺ —ā–Ķ—Ä–į–Ņ–ł—Ź –°–Ē —ā–ĺ–∂–Ķ –ľ–ĺ–∂–Ķ—ā –Ĺ–Ķ–Ī–Ľ–į–≥–ĺ–Ņ—Ä–ł—Ź—ā–Ĺ–ĺ –≤–ĺ–∑–ī–Ķ–Ļ—Ā—ā–≤–ĺ–≤–į—ā—Ć –Ĺ–į –ļ–ĺ–≥–Ĺ–ł—ā–ł–≤–Ĺ—č–Ķ —Ą—É–Ĺ–ļ—Ü–ł–ł —É –Ņ–į—Ü–ł–Ķ–Ĺ—ā–į, –ľ–Ķ—Ä—č –Ņ–ĺ –Ņ—Ä–ĺ—Ą–ł–Ľ–į–ļ—ā–ł–ļ–Ķ —Ü–Ķ—Ä–Ķ–Ī—Ä–į–Ľ—Ć–Ĺ—č—Ö –Ĺ–į—Ä—É—ą–Ķ–Ĺ–ł–Ļ –ī–ĺ–Ľ–∂–Ĺ—č –Ī—č—ā—Ć –Ī–ĺ–Ľ–Ķ–Ķ –į–ļ—ā–ł–≤–Ĺ—č–ľ–ł. –ü–ĺ —ć—ā–ĺ–Ļ –Ņ—Ä–ł—á–ł–Ĺ–Ķ –Ņ—Ä–Ķ–ī—É–Ņ—Ä–Ķ–∂–ī–Ķ–Ĺ–ł–Ķ —Ä–į–∑–≤–ł—ā–ł—Ź –ĺ—Ā–Ľ–ĺ–∂–Ĺ–Ķ–Ĺ–ł–Ļ —É —ā–į–ļ–ł—Ö –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ –Ņ—Ä–Ķ–ī–Ņ–ĺ–Ľ–į–≥–į–Ķ—ā —Ä–į–Ĺ–Ĺ—é—é –ī–ł–į–≥–Ĺ–ĺ—Ā—ā–ł–ļ—É —Ā–į–ľ–ĺ–≥–ĺ –ī–ł–į–Ī–Ķ—ā–į –ł –Ņ—Ä–ł–ľ–Ķ–Ĺ–Ķ–Ĺ–ł–Ķ –Ņ–į—ā–ĺ—Ą–ł–∑–ł–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ĺ–≥–ĺ –Ņ–ĺ–ī—Ö–ĺ–ī–į –ļ –Ľ–Ķ—á–Ķ–Ĺ–ł—é. –Ě–į —Ā—ä–Ķ–∑–ī–Ķ –ú–Ķ–∂–ī—É–Ĺ–į—Ä–ĺ–ī–Ĺ–ĺ–Ļ —Ą–Ķ–ī–Ķ—Ä–į—Ü–ł–ł –ī–ł–į–Ī–Ķ—ā–į (2009) –ź–ľ–Ķ—Ä–ł–ļ–į–Ĺ—Ā–ļ–į—Ź –į—Ā—Ā–ĺ—Ü–ł–į—Ü–ł—Ź –ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ł—Ö —ć–Ĺ–ī–ĺ–ļ—Ä–ł–Ĺ–ĺ–Ľ–ĺ–≥–ĺ–≤ (American Association of Clinical Endocrinologists ‚Äď AACE) –Ņ—Ä–Ķ–ī—Ā—ā–į–≤–ł–Ľ–į –Ĺ–ĺ–≤—č–Ļ –į–Ľ–≥–ĺ—Ä–ł—ā–ľ —ā–Ķ—Ä–į–Ņ–ł–ł –°–Ē 2 —ā–ł–Ņ–į, –ł—Ā–Ņ–ĺ–Ľ—Ć–∑—É—é—Č–ł–Ļ —Ä–į–∑—É–ľ–Ĺ—č–Ļ –Ņ–į—ā–ĺ–≥–Ķ–Ĺ–Ķ—ā–ł—á–Ķ—Ā–ļ–ł–Ļ –Ņ–ĺ–ī—Ö–ĺ–ī –ļ –Ľ–Ķ—á–Ķ–Ĺ–ł—é —ā–į–ļ–ł—Ö –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ (—Ä–ł—Ā. 7) [22].¬†
–Ē–į–Ĺ–Ĺ–į—Ź —Ā—ā—Ä–į—ā–Ķ–≥–ł—Ź –≤–ļ–Ľ—é—á–į–Ķ—ā –ĺ–Ī—Ź–∑–į—ā–Ķ–Ľ—Ć–Ĺ–ĺ–Ķ –Ĺ–į –Ņ–Ķ—Ä–≤–ĺ–ľ —ć—ā–į–Ņ–Ķ —ā–Ķ—Ä–į–Ņ–ł–ł —Ā–Ĺ–ł–∂–Ķ–Ĺ–ł–Ķ –ľ–į—Ā—Ā—č —ā–Ķ–Ľ–į —É –≤—Ā–Ķ—Ö –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ —Ā –°–Ē 2 —ā–ł–Ņ–į –Ĺ–į 5‚Äď10% –ĺ—ā –ł—Ā—Ö–ĺ–ī–Ĺ–ĺ–≥–ĺ –∑–Ĺ–į—á–Ķ–Ĺ–ł—Ź. –ü—Ä–Ķ–Ņ–į—Ä–į—ā–į–ľ–ł –Ņ–Ķ—Ä–≤–ĺ–Ļ –Ľ–ł–Ĺ–ł–ł –ī–Ľ—Ź –Ĺ–ĺ—Ä–ľ–į–Ľ–ł–∑–į—Ü–ł–ł —É—Ä–ĺ–≤–Ĺ—Ź –≥–Ľ–ł–ļ–Ķ–ľ–ł–ł —Ź–≤–Ľ—Ź—é—ā—Ā—Ź –į–Ĺ—ā–ł–≥–ł–Ņ–Ķ—Ä–≥–Ľ–ł–ļ–Ķ–ľ–ł—á–Ķ—Ā–ļ–ł–Ķ —Ā—Ä–Ķ–ī—Ā—ā–≤–į —Ā –ĺ—ā—Ā—É—ā—Ā—ā–≤–ł–Ķ–ľ —Ä–ł—Ā–ļ–į –Ĺ–į–Ī–ĺ—Ä–į –≤–Ķ—Ā–į –ł –≥–ł–Ņ–ĺ–≥–Ľ–ł–ļ–Ķ–ľ–ł–ł: –ľ–Ķ—ā—Ą–ĺ—Ä–ľ–ł–Ĺ, –≥–Ľ–ł—ā–į–∑–ĺ–Ĺ—č, –ł–Ĺ–≥–ł–Ī–ł—ā–ĺ—Ä—č őĪ-–≥–Ľ—é–ļ–ĺ–∑–ł–ī–į–∑—č, –į–Ĺ–į–Ľ–ĺ–≥–ł –≥–Ľ—é–ļ–į–≥–ĺ–Ĺ–ĺ–Ņ–ĺ–ī–ĺ–Ī–Ĺ–ĺ–≥–ĺ –Ņ–Ķ–Ņ—ā–ł–ī–į 1 –ł –ł–Ĺ–≥–ł–Ī–ł—ā–ĺ—Ä—č –ī–ł–Ņ–Ķ–Ņ—ā–ł–ī–ł–Ľ–Ņ–Ķ–Ņ—ā–ł–ī–į–∑—č 4. –†–į–Ĺ–Ĺ–Ķ–Ķ –Ĺ–į–∑–Ĺ–į—á–Ķ–Ĺ–ł–Ķ –ł–Ĺ—Ā—É–Ľ–ł–Ĺ–ĺ—ā–Ķ—Ä–į–Ņ–ł–ł, —Ā–ĺ–≥–Ľ–į—Ā–Ĺ–ĺ —ć—ā–ĺ–ľ—É –į–Ľ–≥–ĺ—Ä–ł—ā–ľ—É, –Ĺ–Ķ–≤–ĺ–∑–ľ–ĺ–∂–Ĺ–ĺ –Ī–Ķ–∑ —ć—ā–į–Ņ–į –Ņ—Ä–į–≤–ł–Ľ—Ć–Ĺ–ĺ–Ļ –į–Ĺ—ā–ł–≥–ł–Ņ–Ķ—Ä–≥–Ľ–ł–ļ–Ķ–ľ–ł—á–Ķ—Ā–ļ–ĺ–Ļ —ā–Ķ—Ä–į–Ņ–ł–ł, —Ā–ĺ—Ā—ā–ĺ—Ź—Č–Ķ–Ļ –ł–∑ –ī–≤—É—Ö –ł–Ľ–ł —ā—Ä–Ķ—Ö –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–ĺ–≤, –≤–Ľ–ł—Ź—é—Č–ł—Ö –Ĺ–į —Ä–į–∑–Ĺ—č–Ķ —ć—ā–į–Ņ—č —Ä–į–∑–≤–ł—ā–ł—Ź –ī–ł–į–Ī–Ķ—ā–į. –ö–į–ļ –į–Ľ—Ć—ā–Ķ—Ä–Ĺ–į—ā–ł–≤–į –ł–Ĺ—Ā—É–Ľ–ł–Ĺ–ĺ—ā–Ķ—Ä–į–Ņ–ł–ł (–Ņ—Ä–ł –ĺ—ā—Ā—É—ā—Ā—ā–≤–ł–ł —Ā–ł–ľ–Ņ—ā–ĺ–ľ–ĺ–≤ –į–Ī—Ā–ĺ–Ľ—é—ā–Ĺ–ĺ–Ļ –Ĺ–Ķ–ī–ĺ—Ā—ā–į—ā–ĺ—á–Ĺ–ĺ—Ā—ā–ł –ł–Ĺ—Ā—É–Ľ–ł–Ĺ–į) –≤–ĺ–∑–ľ–ĺ–∂–Ĺ–į —Ā—ā–į—Ä—ā–ĺ–≤–į—Ź –ļ–ĺ–ľ–Ī–ł–Ĺ–ł—Ä–ĺ–≤–į–Ĺ–Ĺ–į—Ź —ā–Ķ—Ä–į–Ņ–ł—Ź –į–Ĺ—ā–ł–≥–ł–Ņ–Ķ—Ä–≥–Ľ–ł–ļ–Ķ–ľ–ł—á–Ķ—Ā–ļ–ł–ľ–ł –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į–ľ–ł, –Ņ—Ä–ł—á–Ķ–ľ –ļ–į–ļ —É –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ —Ā —Ā—É–Ī–ļ–ĺ–ľ–Ņ–Ķ–Ĺ—Ā–į—Ü–ł–Ķ–Ļ, —ā–į–ļ –ł —É –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ —Ā —ā—Ź–∂–Ķ–Ľ–ĺ–Ļ –ī–Ķ–ļ–ĺ–ľ–Ņ–Ķ–Ĺ—Ā–į—Ü–ł–Ķ–Ļ –ł —É—Ä–ĺ–≤–Ĺ–Ķ–ľ –≥–Ľ–ł–ļ–ł—Ä–ĺ–≤–į–Ĺ–Ĺ–ĺ–≥–ĺ –≥–Ķ–ľ–ĺ–≥–Ľ–ĺ–Ī–ł–Ĺ–į –Ī–ĺ–Ľ–Ķ–Ķ 9%.
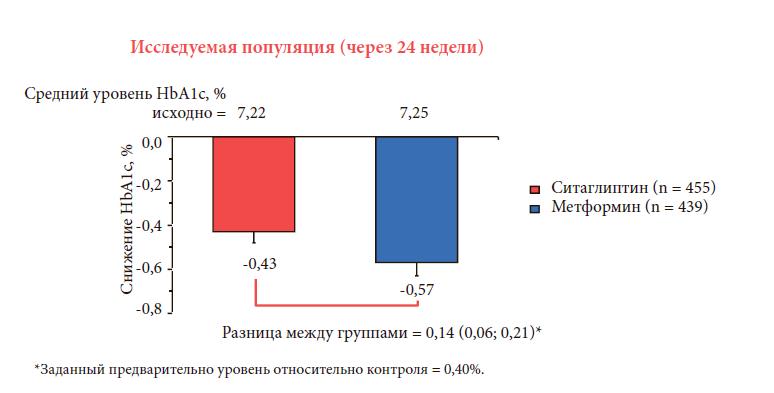
–í –Ĺ–į—Ā—ā–ĺ—Ź—Č–Ķ–Ķ –≤—Ä–Ķ–ľ—Ź –Ņ–ĺ–Ľ—É—á–Ķ–Ĺ—č –ī–į–Ĺ–Ĺ—č–Ķ –Ī–ĺ–Ľ—Ć—ą–ĺ–≥–ĺ –ļ–ĺ–Ľ–ł—á–Ķ—Ā—ā–≤–į –ľ–Ķ–∂–ī—É–Ĺ–į—Ä–ĺ–ī–Ĺ—č—Ö –ľ–Ĺ–ĺ–≥–ĺ—Ü–Ķ–Ĺ—ā—Ä–ĺ–≤—č—Ö –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–Ļ, —Ā—Ä–į–≤–Ĺ–ł–≤–į—é—Č–ł—Ö —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć –ł –Ī–Ķ–∑–ĺ–Ņ–į—Ā–Ĺ–ĺ—Ā—ā—Ć —Ä–į–∑–Ĺ—č—Ö –ļ–Ľ–į—Ā—Ā–ĺ–≤ —Ā–į—Ö–į—Ä–ĺ—Ā–Ĺ–ł–∂–į—é—Č–ł—Ö –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–ĺ–≤. –Ę–į–ļ, –≤ —Ä–į–Ĺ–ī–ĺ–ľ–ł–∑–ł—Ä–ĺ–≤–į–Ĺ–Ĺ–ĺ–ľ, –ī–≤–ĺ–Ļ–Ĺ–ĺ–ľ —Ā–Ľ–Ķ–Ņ–ĺ–ľ, –į–ļ—ā–ł–≤–Ĺ–ĺ –ļ–ĺ–Ĺ—ā—Ä–ĺ–Ľ–ł—Ä—É–Ķ–ľ–ĺ–ľ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–ł –≤ –Ņ–į—Ä–į–Ľ–Ľ–Ķ–Ľ—Ć–Ĺ—č—Ö –≥—Ä—É–Ņ–Ņ–į—Ö —É –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ —Ā –°–Ē 2 —ā–ł–Ņ–į (n = 1172), –Ĺ–Ķ –ī–ĺ—Ā—ā–ł–≥—ą–ł—Ö —Ü–Ķ–Ľ–Ķ–≤–ĺ–≥–ĺ —É—Ä–ĺ–≤–Ĺ—Ź –≥–Ľ–ł–ļ–Ķ–ľ–ł–ł, —Ā—Ä–į–≤–Ĺ–ł–≤–į–Ľ–į—Ā—Ć —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć –ł –Ī–Ķ–∑–ĺ–Ņ–į—Ā–Ĺ–ĺ—Ā—ā—Ć –ļ–ĺ–ľ–Ī–ł–Ĺ–ł—Ä–ĺ–≤–į–Ĺ–Ĺ–ĺ–Ļ —ā–Ķ—Ä–į–Ņ–ł–ł —Ā –ī–ĺ–Ī–į–≤–Ľ–Ķ–Ĺ–ł–Ķ–ľ –ļ —ā–Ķ–ļ—É—Č–Ķ–Ļ —ā–Ķ—Ä–į–Ņ–ł–ł –ľ–Ķ—ā—Ą–ĺ—Ä–ľ–ł–Ĺ–ĺ–ľ ‚Č•1500 –ľ–≥/—Ā—É—ā –Ņ—Ä–ĺ–ł–∑–≤–ĺ–ī–Ĺ—č—Ö —Ā—É–Ľ—Ć—Ą–ĺ–Ĺ–ł–Ľ–ľ–ĺ—á–Ķ–≤–ł–Ĺ—č (–ü–°–ú) (–≥–Ľ–ł–Ņ–ł–∑–ł–ī) —Ā —ā–ł—ā—Ä–į—Ü–ł–Ķ–Ļ –ī–ĺ 20 –ľ–≥/—Ā—É—ā –ł–Ľ–ł —Ā–ł—ā–į–≥–Ľ–ł–Ņ—ā–ł–Ĺ–į 100 –ľ–≥/—Ā—É—ā [23].
–Ē–Ľ–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ—Ā—ā—Ć —ā–Ķ—Ä–į–Ņ–ł–ł —Ā–ĺ—Ā—ā–į–≤–ł–Ľ–į 52 –Ĺ–Ķ–ī–Ķ–Ľ–ł. –í –ĺ—ā–Ĺ–ĺ—ą–Ķ–Ĺ–ł–ł —Ā–Ĺ–ł–∂–Ķ–Ĺ–ł—Ź —É—Ä–ĺ–≤–Ĺ—Ź –≥–Ľ–ł–ļ–ł—Ä–ĺ–≤–į–Ĺ–Ĺ–ĺ–≥–ĺ –≥–Ķ–ľ–ĺ–≥–Ľ–ĺ–Ī–ł–Ĺ–į (-0,7%) –Ī—č–Ľ–į –ī–ĺ–ļ–į–∑–į–Ĺ–į —Ā–ĺ–Ņ–ĺ—Ā—ā–į–≤–ł–ľ–į—Ź —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć –ĺ–Ī–Ķ–ł—Ö –ļ–ĺ–ľ–Ī–ł–Ĺ–į—Ü–ł–Ļ, –Ņ—Ä–ł —ć—ā–ĺ–ľ –Ņ—Ä–ĺ—Ą–ł–Ľ—Ć –Ī–Ķ–∑–ĺ–Ņ–į—Ā–Ĺ–ĺ—Ā—ā–ł –Ī—č–Ľ –ī–ĺ—Ā—ā–ĺ–≤–Ķ—Ä–Ĺ–ĺ –ł —Ā—ā–į—ā–ł—Ā—ā–ł—á–Ķ—Ā–ļ–ł –∑–Ĺ–į—á–ł–ľ–ĺ –Ľ—É—á—ą–Ķ –≤ –≥—Ä—É–Ņ–Ņ–Ķ —ā–Ķ—Ä–į–Ņ–ł–ł —Ā–ł—ā–į–≥–Ľ–ł–Ņ—ā–ł–Ĺ–ĺ–ľ¬†+ –ľ–Ķ—ā—Ą–ĺ—Ä–ľ–ł–Ĺ. –°–Ľ—É—á–į–ł –≥–ł–Ņ–ĺ–≥–Ľ–ł–ļ–Ķ–ľ–ł–ł –Ī—č–Ľ–ł –≤—č—Ź–≤–Ľ–Ķ–Ĺ—č –≤ 32% –≤ –≥—Ä—É–Ņ–Ņ–Ķ –ī–ĺ–Ī–į–≤–Ľ–Ķ–Ĺ–ł—Ź –ü–°–ú –Ņ–ĺ —Ā—Ä–į–≤–Ĺ–Ķ–Ĺ–ł—é —Ā 5% –≤ –≥—Ä—É–Ņ–Ņ–Ķ —Ā–ł—ā–į–≥–Ľ–ł–Ņ—ā–ł–Ĺ–į. –ü—Ä–ł–Ī–į–≤–ļ–į –ľ–į—Ā—Ā—č —ā–Ķ–Ľ–į +1,1 –ļ–≥ –Ī—č–Ľ–į –∑–į—Ä–Ķ–≥–ł—Ā—ā—Ä–ł—Ä–ĺ–≤–į–Ĺ–į –≤ –≥—Ä—É–Ņ–Ņ–Ķ –ü–°–ú –Ņ–ĺ —Ā—Ä–į–≤–Ĺ–Ķ–Ĺ–ł—é —Ā -1,5 –ļ–≥ –≤ –≥—Ä—É–Ņ–Ņ–Ķ —Ā–ł—ā–į–≥–Ľ–ł–Ņ—ā–ł–Ĺ–į, –ĺ–Ī—Č–į—Ź —Ä–į–∑–Ĺ–ł—Ü–į –ľ–Ķ–∂–ī—É –≥—Ä—É–Ņ–Ņ–į–ľ–ł –≤ –ľ–į—Ā—Ā–Ķ —ā–Ķ–Ľ–į —Ā–ĺ—Ā—ā–į–≤–ł–Ľ–į 2,6 –ļ–≥ (—Ä–ł—Ā. 8) [23]. –ü—Ä–ł –Ĺ–Ķ–Ņ–Ķ—Ä–Ķ–Ĺ–ĺ—Ā–ł–ľ–ĺ—Ā—ā–ł –Ņ–į—Ü–ł–Ķ–Ĺ—ā–į–ľ–ł –ľ–Ķ—ā—Ą–ĺ—Ä–ľ–ł–Ĺ–į –ł–Ľ–ł –≤–ĺ–∑–Ĺ–ł–ļ–Ĺ–ĺ–≤–Ķ–Ĺ–ł–ł –Ņ–ĺ–Ī–ĺ—á–Ĺ—č—Ö —ć—Ą—Ą–Ķ–ļ—ā–ĺ–≤ –Ņ—Ä–ł –Ķ–≥–ĺ –Ņ—Ä–ł–ľ–Ķ–Ĺ–Ķ–Ĺ–ł–ł –ľ–ĺ–∂–Ĺ–ĺ –Ĺ–į–∑–Ĺ–į—á–ł—ā—Ć —Ā—ā–į—Ä—ā–ĺ–≤—É—é —ā–Ķ—Ä–į–Ņ–ł—é –°–Ē 2 —ā–ł–Ņ–į —Ā–ł—ā–į–≥–Ľ–ł–Ņ—ā–ł–Ĺ–ĺ–ľ. –°–ĺ–≥–Ľ–į—Ā–Ĺ–ĺ –ī–į–Ĺ–Ĺ—č–ľ 24-–Ĺ–Ķ–ī–Ķ–Ľ—Ć–Ĺ–ĺ–≥–ĺ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź —Ā —É—á–į—Ā—ā–ł–Ķ–ľ –Ī–ĺ–Ľ–Ķ–Ķ 1 000 –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤, –≤ –ļ–ĺ—ā–ĺ—Ä–ĺ–ľ —Ā—Ä–į–≤–Ĺ–ł–≤–į–Ľ–į—Ā—Ć —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć –ł –Ņ–Ķ—Ä–Ķ–Ĺ–ĺ—Ā–ł–ľ–ĺ—Ā—ā—Ć —Ā–ł—ā–į–≥–Ľ–ł–Ņ—ā–ł–Ĺ–į 100 –ľ–≥/—Ā—É—ā –Ņ–ĺ —Ā—Ä–į–≤–Ĺ–Ķ–Ĺ–ł—é —Ā –ľ–Ķ—ā—Ą–ĺ—Ä–ľ–ł–Ĺ–ĺ–ľ 2000 –ľ–≥/—Ā—É—ā, –Ī—č–Ľ–ĺ –Ņ–ĺ–ļ–į–∑–į–Ĺ–ĺ —Ā–ĺ–Ņ–ĺ—Ā—ā–į–≤–ł–ľ–ĺ–Ķ —Ā–Ĺ–ł–∂–Ķ–Ĺ–ł–Ķ —É—Ä–ĺ–≤–Ĺ—Ź –≥–Ľ–ł–ļ–ł—Ä–ĺ–≤–į–Ĺ–Ĺ–ĺ–≥–ĺ –≥–Ķ–ľ–ĺ–≥–Ľ–ĺ–Ī–ł–Ĺ–į (—Ä–į–∑–Ĺ–ł—Ü–į –ľ–Ķ–∂–ī—É –≥—Ä—É–Ņ–Ņ–į–ľ–ł –Ĺ–Ķ –Ī—č–Ľ–į —Ā—ā–į—ā–ł—Ā—ā–ł—á–Ķ—Ā–ļ–ł –ī–ĺ—Ā—ā–ĺ–≤–Ķ—Ä–Ĺ–ĺ–Ļ) (—Ä–ł—Ā. 9) [24].
–Ę–Ķ—Ä–į–Ņ–ł—Ź –ļ–į–ļ —Ā–ł—ā–į–≥–Ľ–ł–Ņ—ā–ł–Ĺ–ĺ–ľ, —ā–į–ļ –ł –ľ–Ķ—ā—Ą–ĺ—Ä–ľ–ł–Ĺ–ĺ–ľ –≤ —Ü–Ķ–Ľ–ĺ–ľ –Ņ–Ķ—Ä–Ķ–Ĺ–ĺ—Ā–ł–Ľ–į—Ā—Ć —Ö–ĺ—Ä–ĺ—ą–ĺ: —á–į—Ā—ā–ĺ—ā–į —Ä–į–∑–≤–ł—ā–ł—Ź –≥–ł–Ņ–ĺ–≥–Ľ–ł–ļ–Ķ–ľ–ł–ł –Ī—č–Ľ–į –Ĺ–ł–∑–ļ–ĺ–Ļ –Ņ—Ä–ł –Ņ—Ä–ł–ľ–Ķ–Ĺ–Ķ–Ĺ–ł–ł –ĺ–Ī–ĺ–ł—Ö –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–ĺ–≤, –Ĺ–ĺ –Ņ—Ä–ł –ł—Ā–Ņ–ĺ–Ľ—Ć–∑–ĺ–≤–į–Ĺ–ł–ł —Ā–ł—ā–į–≥–Ľ–ł–Ņ—ā–ł–Ĺ–į –ĺ—ā–ľ–Ķ—á–į–Ľ–į—Ā—Ć –Ī–ĺ–Ľ–Ķ–Ķ –Ĺ–ł–∑–ļ–į—Ź —á–į—Ā—ā–ĺ—ā–į —Ä–į–∑–≤–ł—ā–ł—Ź –Ĺ–Ķ–∂–Ķ–Ľ–į—ā–Ķ–Ľ—Ć–Ĺ—č—Ö —Ź–≤–Ľ–Ķ–Ĺ–ł–Ļ —Ā–ĺ —Ā—ā–ĺ—Ä–ĺ–Ĺ—č –∂–Ķ–Ľ—É–ī–ĺ—á–Ĺ–ĺ-–ļ–ł—ą–Ķ—á–Ĺ–ĺ–≥–ĺ —ā—Ä–į–ļ—ā–į –Ņ–ĺ —Ā—Ä–į–≤–Ĺ–Ķ–Ĺ–ł—é —Ā –ľ–Ķ—ā—Ą–ĺ—Ä–ľ–ł–Ĺ–ĺ–ľ, —á—ā–ĺ –Ņ—Ä–ĺ—Ź–≤–Ľ—Ź–Ľ–ĺ—Ā—Ć –≤ —Ā–Ĺ–ł–∂–Ķ–Ĺ–ł–ł —á–į—Ā—ā–ĺ—ā—č –ī–ł–į—Ä–Ķ–ł –ł —ā–ĺ—ą–Ĺ–ĺ—ā—č (—Ä–ł—Ā. 10) [24]. –Ę–į–ļ–ł–ľ –ĺ–Ī—Ä–į–∑–ĺ–ľ, —Ā–Ķ–≥–ĺ–ī–Ĺ—Ź —É –≤—Ä–į—á–į –Ķ—Ā—ā—Ć –≤—č–Ī–ĺ—Ä ‚Äď –Ĺ–į–∑–Ĺ–į—á–ł—ā—Ć –Ņ–į—Ü–ł–Ķ–Ĺ—ā—É —Ā –°–Ē 2 —ā–ł–Ņ–į —Ä–į–Ĺ–Ĺ—é—é –ł–Ĺ—Ā—É–Ľ–ł–Ĺ–ĺ—ā–Ķ—Ä–į–Ņ–ł—é –ł–Ľ–ł –Ņ—Ä–ĺ–ł–∑–≤–ĺ–ī–Ĺ—č–Ķ —Ā—É–Ľ—Ć—Ą–ĺ–Ĺ–ł–Ľ–ľ–ĺ—á–Ķ–≤–ł–Ĺ—č –Ņ–Ķ—Ä–≤–ĺ–≥–ĺ –Ņ–ĺ–ļ–ĺ–Ľ–Ķ–Ĺ–ł—Ź, –Ņ–ĺ–Ľ—É—á–ł–≤ —Ā—Ä–į–∑—É —Ü–Ķ–Ľ—č–Ļ —Ā–Ņ–Ķ–ļ—ā—Ä –Ĺ–Ķ–∂–Ķ–Ľ–į—ā–Ķ–Ľ—Ć–Ĺ—č—Ö –ľ–Ķ—ā–į–Ī–ĺ–Ľ–ł—á–Ķ—Ā–ļ–ł—Ö –Ņ–ĺ—Ā–Ľ–Ķ–ī—Ā—ā–≤–ł–Ļ ‚Äď –ĺ—ā –Ī—č—Ā—ā—Ä–ĺ–≥–ĺ –Ĺ–į–Ī–ĺ—Ä–į –≤–Ķ—Ā–į –ī–ĺ –Ņ—Ä–ĺ–≥—Ä–Ķ—Ā—Ā–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź —ć–Ĺ—Ü–Ķ—Ą–į–Ľ–ĺ–Ņ–į—ā–ł–ł, –ł–Ľ–ł –ł—Ā–Ņ–ĺ–Ľ—Ć–∑–ĺ–≤–į—ā—Ć —Ä–į–∑—É–ľ–Ĺ—č–Ļ –Ņ–į—ā–ĺ–≥–Ķ–Ĺ–Ķ—ā–ł—á–Ķ—Ā–ļ–ł–Ļ –Ņ–ĺ–ī—Ö–ĺ–ī —Ā —É—á–Ķ—ā–ĺ–ľ –ł–Ĺ–ī–ł–≤–ł–ī—É–į–Ľ—Ć–Ĺ–ĺ–≥–ĺ —Ä–ł—Ā–ļ–į –ī–Ľ—Ź –ļ–į–∂–ī–ĺ–≥–ĺ –Ņ–į—Ü–ł–Ķ–Ĺ—ā–į.¬†
NB