–í–ĺ–∑–ľ–ĺ–∂–Ĺ–ĺ—Ā—ā–ł –ļ–ĺ—Ä—Ä–Ķ–ļ—Ü–ł–ł —Ü–Ķ—Ä–Ķ–Ī—Ä–į–Ľ—Ć–Ĺ–ĺ–Ļ –ł—ą–Ķ–ľ–ł–ł —Ā –Ņ–ĺ–ľ–ĺ—Č—Ć—é –ļ–Ľ—é—á–Ķ–≤–ĺ–≥–ĺ –ľ–Ķ—ā–į–Ī–ĺ–Ľ–ł—ā–į –ī—č—Ö–į—ā–Ķ–Ľ—Ć–Ĺ–ĺ–Ļ —Ü–Ķ–Ņ–ł –ľ–ł—ā–ĺ—Ö–ĺ–Ĺ–ī—Ä–ł–Ļ ‚Äď —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ–į –°
- –ź–Ĺ–Ĺ–ĺ—ā–į—Ü–ł—Ź
- –°—ā–į—ā—Ć—Ź
- –°—Ā—č–Ľ–ļ–ł
- English
–í —Ā—ā–į—ā—Ć–Ķ —Ä–į—Ā—Ā–ľ–ĺ—ā—Ä–Ķ–Ĺ—č —Ą—É–Ĺ–ī–į–ľ–Ķ–Ĺ—ā–į–Ľ—Ć–Ĺ—č–Ķ –ľ–Ķ—Ö–į–Ĺ–ł–∑–ľ—č —Ü–Ķ—Ä–Ķ–Ī—Ä–į–Ľ—Ć–Ĺ–ĺ–≥–ĺ –ł—ą–Ķ–ľ–ł—á–Ķ—Ā–ļ–ĺ–≥–ĺ –Ņ–ĺ–≤—Ä–Ķ–∂–ī–Ķ–Ĺ–ł—Ź, –≤ –ĺ—Ā–Ĺ–ĺ–≤–Ķ –ļ–ĺ—ā–ĺ—Ä—č—Ö –Ľ–Ķ–∂–ł—ā —Ä–į–∑–≤–ł—ā–ł–Ķ –ľ–ł—ā–ĺ—Ö–ĺ–Ĺ–ī—Ä–ł–į–Ľ—Ć–Ĺ–ĺ–Ļ –Ĺ–Ķ–ī–ĺ—Ā—ā–į—ā–ĺ—á–Ĺ–ĺ—Ā—ā–ł. –ě—Ā–ĺ–Ī–ĺ–Ķ –≤–Ĺ–ł–ľ–į–Ĺ–ł–Ķ —É–ī–Ķ–Ľ–Ķ–Ĺ–ĺ —Ä–ĺ–Ľ–ł —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ–į –°. –ě–Ņ–ł—Ā–į–Ĺ—č –ľ–Ķ—Ö–į–Ĺ–ł–∑–ľ—č —É—á–į—Ā—ā–ł—Ź —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ–į –° –≤ –Ņ—Ä–ĺ—Ü–Ķ—Ā—Ā–į—Ö –Ņ–ĺ–≤—Ä–Ķ–∂–ī–Ķ–Ĺ–ł—Ź –ļ–Ľ–Ķ—ā–ļ–ł, –į —ā–į–ļ–∂–Ķ –Ķ–≥–ĺ –∑–į—Č–ł—ā–Ĺ—č–Ļ (–į–Ĺ—ā–ł–ĺ–ļ—Ā–ł–ī–į–Ĺ—ā–Ĺ—č–Ļ) –Ņ–ĺ—ā–Ķ–Ĺ—Ü–ł–į–Ľ –Ņ—Ä–ł —Ä—Ź–ī–Ķ –Ņ–į—ā–ĺ–Ľ–ĺ–≥–ł–Ļ, –≤–ļ–Ľ—é—á–į—Ź —Ü–Ķ—Ä–Ķ–Ī—Ä–į–Ľ—Ć–Ĺ–ĺ–Ķ –ł—ą–Ķ–ľ–ł—á–Ķ—Ā–ļ–ĺ–Ķ –Ņ–ĺ–≤—Ä–Ķ–∂–ī–Ķ–Ĺ–ł–Ķ. –ě–Ī–ĺ–∑–Ĺ–į—á–Ķ–Ĺ—č –Ņ–ĺ—ā–Ķ–Ĺ—Ü–ł–į–Ľ—Ć–Ĺ—č–Ķ –≤–ĺ–∑–ľ–ĺ–∂–Ĺ–ĺ—Ā—ā–ł —ā–Ķ—Ä–į–Ņ–Ķ–≤—ā–ł—á–Ķ—Ā–ļ–ĺ–≥–ĺ –ł—Ā–Ņ–ĺ–Ľ—Ć–∑–ĺ–≤–į–Ĺ–ł—Ź —ć–ļ–∑–ĺ–≥–Ķ–Ĺ–Ĺ–ĺ–≥–ĺ —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ–į –° –Ņ—Ä–ł –ĺ—Ā—ā—Ä—č—Ö –ł —Ö—Ä–ĺ–Ĺ–ł—á–Ķ—Ā–ļ–ł—Ö —Ü–Ķ—Ä–Ķ–Ī—Ä–ĺ–≤–į—Ā–ļ—É–Ľ—Ź—Ä–Ĺ—č—Ö –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł—Ź—Ö.
–í —Ā—ā–į—ā—Ć–Ķ —Ä–į—Ā—Ā–ľ–ĺ—ā—Ä–Ķ–Ĺ—č —Ą—É–Ĺ–ī–į–ľ–Ķ–Ĺ—ā–į–Ľ—Ć–Ĺ—č–Ķ –ľ–Ķ—Ö–į–Ĺ–ł–∑–ľ—č —Ü–Ķ—Ä–Ķ–Ī—Ä–į–Ľ—Ć–Ĺ–ĺ–≥–ĺ –ł—ą–Ķ–ľ–ł—á–Ķ—Ā–ļ–ĺ–≥–ĺ –Ņ–ĺ–≤—Ä–Ķ–∂–ī–Ķ–Ĺ–ł—Ź, –≤ –ĺ—Ā–Ĺ–ĺ–≤–Ķ –ļ–ĺ—ā–ĺ—Ä—č—Ö –Ľ–Ķ–∂–ł—ā —Ä–į–∑–≤–ł—ā–ł–Ķ –ľ–ł—ā–ĺ—Ö–ĺ–Ĺ–ī—Ä–ł–į–Ľ—Ć–Ĺ–ĺ–Ļ –Ĺ–Ķ–ī–ĺ—Ā—ā–į—ā–ĺ—á–Ĺ–ĺ—Ā—ā–ł. –ě—Ā–ĺ–Ī–ĺ–Ķ –≤–Ĺ–ł–ľ–į–Ĺ–ł–Ķ —É–ī–Ķ–Ľ–Ķ–Ĺ–ĺ —Ä–ĺ–Ľ–ł —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ–į –°. –ě–Ņ–ł—Ā–į–Ĺ—č –ľ–Ķ—Ö–į–Ĺ–ł–∑–ľ—č —É—á–į—Ā—ā–ł—Ź —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ–į –° –≤ –Ņ—Ä–ĺ—Ü–Ķ—Ā—Ā–į—Ö –Ņ–ĺ–≤—Ä–Ķ–∂–ī–Ķ–Ĺ–ł—Ź –ļ–Ľ–Ķ—ā–ļ–ł, –į —ā–į–ļ–∂–Ķ –Ķ–≥–ĺ –∑–į—Č–ł—ā–Ĺ—č–Ļ (–į–Ĺ—ā–ł–ĺ–ļ—Ā–ł–ī–į–Ĺ—ā–Ĺ—č–Ļ) –Ņ–ĺ—ā–Ķ–Ĺ—Ü–ł–į–Ľ –Ņ—Ä–ł —Ä—Ź–ī–Ķ –Ņ–į—ā–ĺ–Ľ–ĺ–≥–ł–Ļ, –≤–ļ–Ľ—é—á–į—Ź —Ü–Ķ—Ä–Ķ–Ī—Ä–į–Ľ—Ć–Ĺ–ĺ–Ķ –ł—ą–Ķ–ľ–ł—á–Ķ—Ā–ļ–ĺ–Ķ –Ņ–ĺ–≤—Ä–Ķ–∂–ī–Ķ–Ĺ–ł–Ķ. –ě–Ī–ĺ–∑–Ĺ–į—á–Ķ–Ĺ—č –Ņ–ĺ—ā–Ķ–Ĺ—Ü–ł–į–Ľ—Ć–Ĺ—č–Ķ –≤–ĺ–∑–ľ–ĺ–∂–Ĺ–ĺ—Ā—ā–ł —ā–Ķ—Ä–į–Ņ–Ķ–≤—ā–ł—á–Ķ—Ā–ļ–ĺ–≥–ĺ –ł—Ā–Ņ–ĺ–Ľ—Ć–∑–ĺ–≤–į–Ĺ–ł—Ź —ć–ļ–∑–ĺ–≥–Ķ–Ĺ–Ĺ–ĺ–≥–ĺ —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ–į –° –Ņ—Ä–ł –ĺ—Ā—ā—Ä—č—Ö –ł —Ö—Ä–ĺ–Ĺ–ł—á–Ķ—Ā–ļ–ł—Ö —Ü–Ķ—Ä–Ķ–Ī—Ä–ĺ–≤–į—Ā–ļ—É–Ľ—Ź—Ä–Ĺ—č—Ö –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł—Ź—Ö.
–í–≤–Ķ–ī–Ķ–Ĺ–ł–Ķ
–ú–ĺ–∑–≥ —á–Ķ–Ľ–ĺ–≤–Ķ–ļ–į —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź —Ā–į–ľ—č–ľ –ľ–ĺ—Č–Ĺ—č–ľ –Ņ–ĺ—ā—Ä–Ķ–Ī–ł—ā–Ķ–Ľ–Ķ–ľ —ć–Ĺ–Ķ—Ä–≥–ł–ł –≤¬†–ĺ—Ä–≥–į–Ĺ–ł–∑–ľ–Ķ, –Ĺ–į¬†–Ĺ–Ķ–≥–ĺ –Ņ—Ä–ł—Ö–ĺ–ī–ł—ā—Ā—Ź –ĺ–ļ–ĺ–Ľ–ĺ 25% –≤—Ā–Ķ—Ö –ľ–Ķ—ā–į–Ī–ĺ–Ľ–ł—á–Ķ—Ā–ļ–ł—Ö –∑–į—ā—Ä–į—ā. –ü—Ä–ł —ć—ā–ĺ–ľ –≤–Ķ—Ā –ľ–ĺ–∑–≥–į, –ļ–į–ļ –Ņ—Ä–į–≤–ł–Ľ–ĺ, –Ĺ–Ķ¬†–Ņ—Ä–Ķ–≤—č—ą–į–Ķ—ā 2,5% –ľ–į—Ā—Ā—č —ā–Ķ–Ľ–į. –°¬†—É—á–Ķ—ā–ĺ–ľ –≤—č—Ā–ĺ—á–į–Ļ—ą–ł—Ö —ć–Ĺ–Ķ—Ä–≥–Ķ—ā–ł—á–Ķ—Ā–ļ–ł—Ö –Ņ–ĺ—ā—Ä–Ķ–Ī–Ĺ–ĺ—Ā—ā–Ķ–Ļ –ľ–ĺ–∑–≥ –ļ—Ä–į–Ļ–Ĺ–Ķ —á—É–≤—Ā—ā–≤–ł—ā–Ķ–Ľ–Ķ–Ĺ –ļ¬†–Ĺ–į—Ä—É—ą–Ķ–Ĺ–ł—Ź–ľ –ļ—Ä–ĺ–≤–ĺ—ā–ĺ–ļ–į. –Ē–Ľ—Ź –Ņ–ĺ–ī–ī–Ķ—Ä–∂–į–Ĺ–ł—Ź –Ĺ–ĺ—Ä–ľ–į–Ľ—Ć–Ĺ–ĺ–Ļ —Ä–į–Ī–ĺ—ā—č —Ā—Ä–Ķ–ī–Ĺ–ł–Ļ —É—Ä–ĺ–≤–Ķ–Ĺ—Ć —Ü–Ķ—Ä–Ķ–Ī—Ä–į–Ľ—Ć–Ĺ–ĺ–≥–ĺ –ļ—Ä–ĺ–≤–ĺ—ā–ĺ–ļ–į –ī–ĺ–Ľ–∂–Ķ–Ĺ —Ā–ĺ—Ā—ā–į–≤–Ľ—Ź—ā—Ć –ĺ–ļ–ĺ–Ľ–ĺ 50 –ľ–Ľ/100¬†–≥ —ā–ļ–į–Ĺ–ł –ľ–ĺ–∑–≥–į –≤¬†–ľ–ł–Ĺ—É—ā—É. –Ē–į–Ĺ–Ĺ—č–Ļ –Ņ–ĺ–ļ–į–∑–į—ā–Ķ–Ľ—Ć –ĺ–Ī–Ķ—Ā–Ņ–Ķ—á–ł–≤–į–Ķ—ā—Ā—Ź –∑–į —Ā—á–Ķ—ā —Ą—É–Ĺ–ļ—Ü–ł–ĺ–Ĺ–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź —Ä—Ź–ī–į –ľ–Ķ—Ö–į–Ĺ–ł–∑–ľ–ĺ–≤, –ĺ–Ī—ä–Ķ–ī–ł–Ĺ—Ź–Ķ–ľ—č—Ö —ā–Ķ—Ä–ľ–ł–Ĺ–ĺ–ľ ¬ę—Ü–Ķ—Ä–Ķ–Ī—Ä–į–Ľ—Ć–Ĺ–į—Ź –į—É—ā–ĺ—Ä–Ķ–≥—É–Ľ—Ź—Ü–ł—Ź¬Ľ [1]. –ü—Ä–ł –Ĺ–į—Ä—É—ą–Ķ–Ĺ–ł–ł —Ü–Ķ—Ä–Ķ–Ī—Ä–į–Ľ—Ć–Ĺ–ĺ–≥–ĺ –ļ—Ä–ĺ–≤–ĺ—ā–ĺ–ļ–į, —Ā–Ĺ–ł–∂–Ķ–Ĺ–ł–ł –ł–Ľ–ł, –Ĺ–į–ĺ–Ī–ĺ—Ä–ĺ—ā, –ł–∑–Ī—č—ā–ĺ—á–Ĺ–ĺ–ľ —É–≤–Ķ–Ľ–ł—á–Ķ–Ĺ–ł–ł, –Ĺ–į–Ņ—Ä–ł–ľ–Ķ—Ä –Ĺ–į¬†—Ą–ĺ–Ĺ–Ķ —Ā–ĺ—Ā—É–ī–ł—Ā—ā–ĺ–≥–ĺ —Ā–Ņ–į–∑–ľ–į, –Ņ—Ä–ĺ–ł—Ā—Ö–ĺ–ī—Ź—ā —Ā–Ĺ–ł–∂–Ķ–Ĺ–ł–Ķ –Ņ–Ķ—Ä—Ą—É–∑–ł–ł –ľ–ĺ–∑–≥–į –ł¬†—Ä–į–∑–≤–ł—ā–ł–Ķ —Ü–Ķ—Ä–Ķ–Ī—Ä–į–Ľ—Ć–Ĺ–ĺ–Ļ –ł—ą–Ķ–ľ–ł–ł. –ü—Ä–ł –ĺ—Ā—ā—Ä–Ķ–Ļ—ą–Ķ–ľ —Ä–į–∑–≤–ł—ā–ł–ł —Ü–Ķ—Ä–Ķ–Ī—Ä–į–Ľ—Ć–Ĺ–ĺ–Ļ –ł—ą–Ķ–ľ–ł–ł, –į¬†–ł–ľ–Ķ–Ĺ–Ĺ–ĺ —Ā–Ĺ–ł–∂–Ķ–Ĺ–ł–ł –ĺ–Ī—ä–Ķ–ľ–Ĺ–ĺ–Ļ —Ā–ļ–ĺ—Ä–ĺ—Ā—ā–ł —Ü–Ķ—Ä–Ķ–Ī—Ä–į–Ľ—Ć–Ĺ–ĺ–≥–ĺ –ļ—Ä–ĺ–≤–ĺ—ā–ĺ–ļ–į –ľ–Ķ–Ĺ–Ķ–Ķ 30 –ľ–Ľ/100¬†–≥ –≤¬†–ľ–ł–Ĺ—É—ā—É –ĺ–Ī—č—á–Ŗ嬆–≥–ĺ–≤–ĺ—Ä—Ź—ā –嬆—Ä–į–∑–≤–ł—ā–ł–ł –ł—ą–Ķ–ľ–ł—á–Ķ—Ā–ļ–ĺ–≥–ĺ –ł–Ĺ—Ā—É–Ľ—Ć—ā–į. –í¬†—ā–ĺ –∂–Ķ –≤—Ä–Ķ–ľ—Ź –ī–Ľ–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ–Ķ, –Ŗ嬆–Ĺ–Ķ –ļ—Ä–ł—ā–ł—á–Ķ—Ā–ļ–ĺ–Ķ —Ā–Ĺ–ł–∂–Ķ–Ĺ–ł–Ķ —Ā–ļ–ĺ—Ä–ĺ—Ā—ā–Ĺ—č—Ö –Ņ–ĺ–ļ–į–∑–į—ā–Ķ–Ľ–Ķ–Ļ –Ņ–ĺ–ī –≤–Ľ–ł—Ź–Ĺ–ł–Ķ–ľ —Ā–ĺ—Ā—É–ī–ł—Ā—ā—č—Ö —Ą–į–ļ—ā–ĺ—Ä–ĺ–≤ —Ä–ł—Ā–ļ–į —Ā–Ņ–ĺ—Ā–ĺ–Ī—Ā—ā–≤—É–Ķ—ā —Ä–į–∑–≤–ł—ā–ł—é —Ö—Ä–ĺ–Ĺ–ł—á–Ķ—Ā–ļ–ĺ–Ļ —Ü–Ķ—Ä–Ķ–Ī—Ä–į–Ľ—Ć–Ĺ–ĺ–Ļ –ł—ą–Ķ–ľ–ł–ł [2]. –Ě–Ķ–∑–į–≤–ł—Ā–ł–ľ–ĺ –ĺ—ā¬†—ć—ā–ł–ĺ–Ľ–ĺ–≥–ł–ł –ł—ą–Ķ–ľ–ł—鬆–≥–ĺ–Ľ–ĺ–≤–Ĺ–ĺ–≥–ĺ –ľ–ĺ–∑–≥–į –≤—Ā–Ķ–≥–ī–į —Ā–ĺ–Ņ—Ä–ĺ–≤–ĺ–∂–ī–į–Ķ—ā –ļ–į—Ā–ļ–į–ī –Ņ–į—ā–ĺ–Ī–ł–ĺ—Ö–ł–ľ–ł—á–Ķ—Ā–ļ–ł—Ö –ł–∑–ľ–Ķ–Ĺ–Ķ–Ĺ–ł–Ļ, –ł–Ľ–ł –ł—ą–Ķ–ľ–ł—á–Ķ—Ā–ļ–ł–Ļ –ļ–į—Ā–ļ–į–ī, –ĺ–Ī—É—Ā–Ľ–ĺ–≤–Ľ–Ķ–Ĺ–Ĺ—č–Ļ —Ā–Ĺ–ł–∂–Ķ–Ĺ–ł–Ķ–ľ –ľ–ĺ–∑–≥–ĺ–≤–ĺ–≥–ĺ –ļ—Ä–ĺ–≤–ĺ—ā–ĺ–ļ–į, –ļ–ĺ—ā–ĺ—Ä—č–Ļ –Ņ—Ä–ł –Ĺ–Ķ–Ī–Ľ–į–≥–ĺ–Ņ—Ä–ł—Ź—ā–Ĺ–ĺ–ľ —ā–Ķ—á–Ķ–Ĺ–ł–ł –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł—Ź –ł/–ł–Ľ–ł –Ĺ–Ķ–ī–ĺ—Ā—ā–į—ā–ĺ—á–Ĺ–ĺ–Ļ –ļ–ĺ—Ä—Ä–Ķ–ļ—Ü–ł–ł –∑–į–≤–Ķ—Ä—ą–į–Ķ—ā—Ā—Ź –Ĺ–Ķ–ĺ–Ī—Ä–į—ā–ł–ľ—č–ľ –Ņ–ĺ–≤—Ä–Ķ–∂–ī–Ķ–Ĺ–ł–Ķ–ľ –Ĺ–Ķ—Ä–≤–Ĺ–ĺ–Ļ —ā–ļ–į–Ĺ–ł –Ņ–嬆–ľ–Ķ—Ö–į–Ĺ–ł–∑–ľ–į–ľ –Ĺ–Ķ–ļ—Ä–ĺ–∑–į –ł¬†–į–Ņ–ĺ–Ņ—ā–ĺ–∑–į. –í—Ā–Ķ —ć—ā–į–Ņ—č –ł—ą–Ķ–ľ–ł—á–Ķ—Ā–ļ–ĺ–≥–ĺ –ļ–į—Ā–ļ–į–ī–į —Ä–į–∑–≤–ł–≤–į—é—ā—Ā—Ź –≤¬†–Ņ–Ķ—Ä–≤—č–Ķ –ľ–ł–Ĺ—É—ā—č –ł¬†—á–į—Ā—č –ł—ą–Ķ–ľ–ł—á–Ķ—Ā–ļ–ĺ–≥–ĺ –Ņ–ĺ—Ä–į–∂–Ķ–Ĺ–ł—Ź¬†–≥–ĺ–Ľ–ĺ–≤–Ĺ–ĺ–≥–ĺ –ľ–ĺ–∑–≥–į –ł, –≤–∑–į–ł–ľ–Ĺ–ĺ –Ņ–ĺ—ā–Ķ–Ĺ—Ü–ł—Ä—É—Ź –ī–Ķ–Ļ—Ā—ā–≤–ł–Ķ –ī—Ä—É–≥ –ī—Ä—É–≥–į, –Ņ—Ä–ł–≤–ĺ–ī—Ź—ā –ļ¬†—ā—Ź–∂–Ķ–Ľ–ĺ–ľ—É —Ą—É–Ĺ–ļ—Ü–ł–ĺ–Ĺ–į–Ľ—Ć–Ĺ–ĺ-–ľ–ĺ—Ä—Ą–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ĺ–ľ—É –Ņ–ĺ–≤—Ä–Ķ–∂–ī–Ķ–Ĺ–ł—é –≤–Ķ—Č–Ķ—Ā—ā–≤–į –ľ–ĺ–∑–≥–į. –ü–Ķ—Ä–≤—č–ľ –≤–į–∂–Ĺ–Ķ–Ļ—ą–ł–ľ –ľ–Ķ—Ö–į–Ĺ–ł–∑–ľ–ĺ–ľ –∑–į–Ņ—É—Ā–ļ–į –ł—ą–Ķ–ľ–ł—á–Ķ—Ā–ļ–ĺ–≥–ĺ –ļ–į—Ā–ļ–į–ī–į —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź —Ā–Ĺ–ł–∂–Ķ–Ĺ–ł–Ķ –ľ–ĺ–∑–≥–ĺ–≤–ĺ–≥–ĺ –ļ—Ä–ĺ–≤–ĺ—ā–ĺ–ļ–į —Ā¬†—Ä–į–∑–≤–ł—ā–ł–Ķ–ľ –ī–Ķ—Ą–ł—Ü–ł—ā–į –ļ–ł—Ā–Ľ–ĺ—Ä–ĺ–ī–į, –į¬†—Ā–Ľ–Ķ–ī–ĺ–≤–į—ā–Ķ–Ľ—Ć–Ĺ–ĺ, –ī–Ķ—Ą–ł—Ü–ł—ā–į —ć–Ĺ–Ķ—Ä–≥–ł–ł. –í¬†—É—Ā–Ľ–ĺ–≤–ł—Ź—Ö –Ĺ–Ķ–ī–ĺ—Ā—ā–į—ā–ļ–į –Ņ–Ķ—Ä—Ą—É–∑–ł–ł –Ņ–ĺ–Ľ—É—á–Ķ–Ĺ–ł–Ķ —ć–Ĺ–Ķ—Ä–≥–ł–ł –ĺ—Ā—É—Č–Ķ—Ā—ā–≤–Ľ—Ź–Ķ—ā—Ā—Ź –∑–į —Ā—á–Ķ—ā –į–Ĺ–į—ć—Ä–ĺ–Ī–Ĺ–ĺ–≥–嬆–≥–Ľ–ł–ļ–ĺ–Ľ–ł–∑–į, —Ä–Ķ–į–ļ—Ü–ł–ł –ļ–ĺ—ā–ĺ—Ä–ĺ–≥–ĺ –∑–į–≤–Ķ—Ä—ą–į—é—ā—Ā—Ź –ĺ–Ī—Ä–į–∑–ĺ–≤–į–Ĺ–ł–Ķ–ľ —ā–ĺ–Ľ—Ć–ļ–ĺ –ī–≤—É—Ö –ľ–ĺ–Ľ–Ķ–ļ—É–Ľ –į–ī–Ķ–Ĺ–ĺ–∑–ł–Ĺ—ā—Ä–ł—Ą–ĺ—Ā—Ą–ĺ—Ä–Ĺ–ĺ–Ļ –ļ–ł—Ā–Ľ–ĺ—ā—č (–ź–Ę–§) –ł¬†–Ĺ–į–ļ–ĺ–Ņ–Ľ–Ķ–Ĺ–ł–Ķ–ľ –Ľ–į–ļ—ā–į—ā–į. –Ě–į¬†—Ä–į–Ĺ–Ĺ–ł—Ö —ć—ā–į–Ņ–į—Ö –ł—ą–Ķ–ľ–ł–ł –ļ–Ľ–Ķ—ā–ĺ—á–Ĺ—č–Ļ –į—Ü–ł–ī–ĺ–∑ –ľ–ĺ–∂–Ĺ–ĺ —Ä–į—Ā—Ā–ľ–į—ā—Ä–ł–≤–į—ā—Ć –ļ–į–ļ –∑–į—Č–ł—ā–Ĺ—É—é —Ä–Ķ–į–ļ—Ü–ł—é, –Ņ–ĺ—Ā–ļ–ĺ–Ľ—Ć–ļ—É —Ā–Ĺ–ł–∂–Ķ–Ĺ–ł–Ķ —Ä–Ě –ĺ–ļ–į–∑—č–≤–į–Ķ—ā —Ā—ā–į–Ī–ł–Ľ–ł–∑–ł—Ä—É—é—Č–Ķ–Ķ –ī–Ķ–Ļ—Ā—ā–≤–ł–Ķ –Ĺ–į¬†–ļ–Ľ–Ķ—ā–ĺ—á–Ĺ—č–Ķ –ľ–Ķ–ľ–Ī—Ä–į–Ĺ—č. –ě–ī–Ĺ–į–ļ–ĺ –Ņ—Ä–ĺ–≥—Ä–Ķ—Ā—Ā–ł—Ä–ĺ–≤–į–Ĺ–ł–Ķ –į—Ü–ł–ī–ĺ–∑–į –≤—č–∑—č–≤–į–Ķ—ā –ī–Ķ–Ĺ–į—ā—É—Ä–į—Ü–ł—é —Ä—Ź–ī–į –ļ–Ľ–Ķ—ā–ĺ—á–Ĺ—č—Ö –Ī–Ķ–Ľ–ļ–ĺ–≤. –í¬†–ī–į–Ľ—Ć–Ĺ–Ķ–Ļ—ą–Ķ–ľ –ł–∑-–∑–į –Ĺ–į—Ä–į—Ā—ā–į–Ĺ–ł—Ź –Ľ–į–ļ—ā–į—ā–į—Ü–ł–ī–ĺ–∑–į –Ī–Ľ–ĺ–ļ–ł—Ä—É—é—ā—Ā—Ź –ł¬†–Ņ—Ä–ĺ—Ü–Ķ—Ā—Ā—č –į–Ĺ–į—ć—Ä–ĺ–Ī–Ĺ–ĺ–≥–嬆–≥–Ľ–ł–ļ–ĺ–Ľ–ł–∑–į, –≤¬†–ļ–Ľ–Ķ—ā–ļ–Ķ —Ą–ĺ—Ä–ľ–ł—Ä—É–Ķ—ā—Ā—Ź –ł—Ā—ā–ł–Ĺ–Ĺ—č–Ļ –ī–Ķ—Ą–ł—Ü–ł—ā –ź–Ę–§. –Ě–į¬†–≤—ā–ĺ—Ä–ĺ–Ļ —Ā—ā–į–ī–ł–ł –ł—ą–Ķ–ľ–ł—á–Ķ—Ā–ļ–ĺ–≥–ĺ –ļ–į—Ā–ļ–į–ī–į, —á–Ķ—Ä–Ķ–∑ 10‚Äď30 –ľ–ł–Ĺ—É—ā –ĺ—ā¬†–ľ–ĺ–ľ–Ķ–Ĺ—ā–į –Ķ–≥–ĺ –≤–ĺ–∑–Ĺ–ł–ļ–Ĺ–ĺ–≤–Ķ–Ĺ–ł—Ź, –Ņ—Ä–ĺ–ł—Ā—Ö–ĺ–ī–ł—ā –≤—č–Ī—Ä–ĺ—Ā¬†–≥–Ľ—É—ā–į–ľ–į—ā–į —Ā¬†—Ä–į–∑–≤–ł—ā–ł–Ķ–ľ¬†–≥–Ľ—É—ā–į–ľ–į—ā–Ĺ–ĺ–Ļ —ć–ļ—Ā–į–Ļ—ā–ĺ—ā–ĺ–ļ—Ā–ł—á–Ĺ–ĺ—Ā—ā–ł. –í–ĺ–∑–Ī—É–∂–ī–Ķ–Ĺ–ł–Ķ¬†–≥–Ľ—É—ā–į–ľ–į—ā–Ĺ—č—Ö NMDA-—Ä–Ķ—Ü–Ķ–Ņ—ā–ĺ—Ä–ĺ–≤, —Ä–Ķ–≥—É–Ľ–ł—Ä—É—é—Č–ł—Ö —Ā–ĺ–ī–Ķ—Ä–∂–į–Ĺ–ł–Ķ K+, Na+, –°–į2+, Cl –≤–ĺ –≤–Ĺ–Ķ- –ł¬†–≤–Ĺ—É—ā—Ä–ł–ļ–Ľ–Ķ—ā–ĺ—á–Ĺ–ĺ–ľ –Ņ—Ä–ĺ—Ā—ā—Ä–į–Ĺ—Ā—ā–≤–Ķ, –į–ļ—ā–ł–≤–ł—Ä—É–Ķ—ā Ca-–ļ–į–Ĺ–į–Ľ—č, —á—ā–ĺ –Ņ—Ä–ł–≤–ĺ–ī–ł—ā –ļ¬†—É—Ā–ł–Ľ–Ķ–Ĺ–ł—é –Ņ–ĺ—Ā—ā—É–Ņ–Ľ–Ķ–Ĺ–ł—Ź –≤–Ĺ–Ķ–ļ–Ľ–Ķ—ā–ĺ—á–Ĺ–ĺ–≥–ĺ –°–į2+ –≤¬†–ļ–Ľ–Ķ—ā–ļ—É –ł¬†–≤—č—Ā–≤–ĺ–Ī–ĺ–∂–ī–Ķ–Ĺ–ł—é –≤–Ĺ—É—ā—Ä–ł–ļ–Ľ–Ķ—ā–ĺ—á–Ĺ–ĺ–≥–嬆–°–į2+ –ł–∑¬†–ī–Ķ–Ņ–ĺ –ł, –ļ–į–ļ —Ā–Ľ–Ķ–ī—Ā—ā–≤–ł–Ķ, –į–ļ—ā–ł–≤–į—Ü–ł–ł —Ä–į–∑–Ľ–ł—á–Ĺ—č—Ö —Ą–Ķ—Ä–ľ–Ķ–Ĺ—ā–Ĺ—č—Ö —Ā–ł—Ā—ā–Ķ–ľ. –í¬†—Ä–Ķ–∑—É–Ľ—Ć—ā–į—ā–Ķ –Ĺ–į—Ä—É—ą–į–Ķ—ā—Ā—Ź —Ą–ĺ—Ā—Ą–ĺ—Ä–ł–Ľ–ł—Ä–ĺ–≤–į–Ĺ–ł–Ķ –Ī–Ķ–Ľ–ļ–ĺ–≤, —Ä–į—Ā—Č–Ķ–Ņ–Ľ—Ź—é—ā—Ā—Ź —Ą–ĺ—Ā—Ą–ĺ–Ľ–ł–Ņ–ł–ī—č –ł¬†–≤—č—Ā–≤–ĺ–Ī–ĺ–∂–ī–į–Ķ—ā—Ā—Ź –į—Ä–į—Ö–ł–ī–ĺ–Ĺ–ĺ–≤–į—Ź –ļ–ł—Ā–Ľ–ĺ—ā–į, –ĺ–Ī—Ä–į–∑—É—é—ā—Ā—Ź —ā–ĺ–ļ—Ā–ł—á–Ĺ—č–Ķ –Ņ—Ä–ĺ–ī—É–ļ—ā—č, —Ā–≤–ĺ–Ī–ĺ–ī–Ĺ—č–Ķ —Ä–į–ī–ł–ļ–į–Ľ—č, –ĺ–ļ–į–∑—č–≤–į—é—Č–ł–Ķ —Ü–ł—ā–ĺ—ā–ĺ–ļ—Ā–ł—á–Ķ—Ā–ļ–ĺ–Ķ, –ł–ľ–ľ—É–Ĺ–ĺ–≥–Ķ–Ĺ–Ĺ–ĺ–Ķ –ł¬†–ľ—É—ā–į–≥–Ķ–Ĺ–Ĺ–ĺ–Ķ –ī–Ķ–Ļ—Ā—ā–≤–ł–Ķ, –Ņ–ĺ–≤—Ä–Ķ–∂–ī–į—é—Č–ł–Ķ –ľ–ł—ā–ĺ—Ö–ĺ–Ĺ–ī—Ä–ł–ł, –ļ–Ľ–Ķ—ā–ĺ—á–Ĺ—É—é –Ē–Ě–ö –ł¬†–†–Ě–ö [3].
–Ę–į–ļ–ł–ľ –ĺ–Ī—Ä–į–∑–ĺ–ľ, –Ņ—Ä–ł —Ü–Ķ—Ä–Ķ–Ī—Ä–į–Ľ—Ć–Ĺ–ĺ–Ļ –ł—ą–Ķ–ľ–ł–ł –Ņ–ĺ–≤—Ä–Ķ–∂–ī–į—é—ā—Ā—Ź –≤—Ā–Ķ –ļ–Ľ–Ķ—ā–ĺ—á–Ĺ—č–Ķ —ć–Ľ–Ķ–ľ–Ķ–Ĺ—ā—č, –Ŗ嬆–ļ–Ľ—é—á–Ķ–≤–ĺ–Ļ —Ā—ā—Ä—É–ļ—ā—É—Ä–ĺ–Ļ, —Ä–į–∑—Ä—É—ą–Ķ–Ĺ–ł–Ķ –ļ–ĺ—ā–ĺ—Ä–ĺ–Ļ –Ņ—Ä–ł–≤–ĺ–ī–ł—ā –ļ¬†—Ä–į–∑–≤–ł—ā–ł—é –≤—Ā–Ķ—Ö –Ņ–ĺ—Ā–Ľ–Ķ–ī—É—é—Č–ł—Ö –Ņ–į—ā–ĺ—Ą–ł–∑–ł–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ł—Ö —Ä–Ķ–į–ļ—Ü–ł–Ļ, —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź –ľ–ł—ā–ĺ—Ö–ĺ–Ĺ–ī—Ä–ł—Ź.
–£–Ĺ–ł–ļ–į–Ľ—Ć–Ĺ–ĺ—Ā—ā—Ć –ľ–ł—ā–ĺ—Ö–ĺ–Ĺ–ī—Ä–ł–ł –ľ–Ľ–Ķ–ļ–ĺ–Ņ–ł—ā–į—é—Č–ł—Ö –≤¬†–ĺ—ā–Ľ–ł—á–ł–Ķ –ĺ—ā¬†–ī—Ä—É–≥–ł—Ö –ĺ—Ä–≥–į–Ĺ–Ķ–Ľ–Ľ –ļ–Ľ–Ķ—ā–ļ–ł –∑–į–ļ–Ľ—é—á–į–Ķ—ā—Ā—Ź –≤¬†–ĺ–Ī–Ľ–į–ī–į–Ĺ–ł–ł —Ā–ĺ–Ī—Ā—ā–≤–Ķ–Ĺ–Ĺ–ĺ–Ļ –Ē–Ě–ö, –ļ–ĺ–ī–ł—Ä—É—é—Č–Ķ–Ļ 13 —Ā—É–Ī—ä–Ķ–ī–ł–Ĺ–ł—Ü —Ą–Ķ—Ä–ľ–Ķ–Ĺ—ā–ĺ–≤ –ī—č—Ö–į—ā–Ķ–Ľ—Ć–Ĺ–ĺ–Ļ —Ü–Ķ–Ņ–ł, 22 —ā—Ä–į–Ĺ—Ā–Ņ–ĺ—Ä—ā–Ĺ—č–Ķ –†–Ě–ö –ł¬†–ī–≤–Ķ —Ä–ł–Ī–ĺ—Ā–ĺ–ľ–į–Ľ—Ć–Ĺ—č–Ķ –†–Ě–ö [4]. –í¬†–ľ–ł—ā–ĺ—Ö–ĺ–Ĺ–ī—Ä–ł–ł –Ņ—Ä–ĺ—ā–Ķ–ļ–į—é—ā –≤—Ā–Ķ —Ä–Ķ–į–ļ—Ü–ł–ł —Ü–ł–ļ–Ľ–į –ö—Ä–Ķ–Ī—Ā–į, –≤¬†—Ö–ĺ–ī–Ķ –ļ–ĺ—ā–ĺ—Ä–ĺ–≥–ĺ –∑–į —Ā—á–Ķ—ā —É—ā–ł–Ľ–ł–∑–į—Ü–ł–ł —Ā–ĺ–ĺ—ā–≤–Ķ—ā—Ā—ā–≤—É—é—Č–ł—Ö —Ā—É–Ī—Ā—ā—Ä–į—ā–ĺ–≤ –Ņ—Ä–ĺ–ł—Ā—Ö–ĺ–ī—Ź—ā —ā—Ä–į–Ĺ—Ā–Ņ–ĺ—Ä—ā —ć–Ľ–Ķ–ļ—ā—Ä–ĺ–Ĺ–ĺ–≤ –ł¬†–ĺ–Ī—Ä–į–∑–ĺ–≤–į–Ĺ–ł–Ķ –≤–Ĺ—É—ā—Ä–ł–ļ–Ľ–Ķ—ā–ĺ—á–Ĺ—č—Ö –∑–į–Ņ–į—Ā–ĺ–≤ —ć–Ĺ–Ķ—Ä–≥–ł–ł –≤¬†–≤–ł–ī–Ķ –ź–Ę–§. –ú–ł—ā–ĺ—Ö–ĺ–Ĺ–ī—Ä–ł—Ź –Ī–ĺ–Ľ–Ķ–Ķ —á–Ķ–ľ –Ĺ–į¬†90% —É–ī–ĺ–≤–Ľ–Ķ—ā–≤–ĺ—Ä—Ź–Ķ—ā —ć–Ĺ–Ķ—Ä–≥–Ķ—ā–ł—á–Ķ—Ā–ļ–ł–Ķ –Ņ–ĺ—ā—Ä–Ķ–Ī–Ĺ–ĺ—Ā—ā–ł —Ā–≤–ĺ–Ķ–Ļ –ļ–Ľ–Ķ—ā–ļ–ł. –ě–ī–Ĺ–į–ļ–ĺ —Ą—É–Ĺ–ļ—Ü–ł—Ź –ľ–ł—ā–ĺ—Ö–ĺ–Ĺ–ī—Ä–ł–ł –Ĺ–Ķ¬†–ĺ–≥—Ä–į–Ĺ–ł—á–ł–≤–į–Ķ—ā—Ā—Ź —ā–ĺ–Ľ—Ć–ļ–ĺ –Ņ—Ä–ĺ–ł–∑–≤–ĺ–ī—Ā—ā–≤–ĺ–ľ —ć–Ĺ–Ķ—Ä–≥–ł–ł. –Ē—Ä—É–≥–ł–ľ–ł –≤–į–∂–Ĺ–Ķ–Ļ—ą–ł–ľ–ł —Ā–ł—Ā—ā–Ķ–ľ–į–ľ–ł –Ņ–ĺ–ī–ī–Ķ—Ä–∂–į–Ĺ–ł—Ź –≤–Ĺ—É—ā—Ä–ł–ļ–Ľ–Ķ—ā–ĺ—á–Ĺ–ĺ–≥–嬆–≥–ĺ–ľ–Ķ–ĺ—Ā—ā–į–∑–į —Ź–≤–Ľ—Ź—é—ā—Ā—Ź —É—á–į—Ā—ā–ł–Ķ –≤¬†—ā—Ä–į–Ĺ—Ā–Ņ–ĺ—Ä—ā–Ķ –ł–ĺ–Ĺ–ĺ–≤ –ļ–į–Ľ—Ć—Ü–ł—Ź –ł¬†–ĺ–Ī—Ä–į–∑–ĺ–≤–į–Ĺ–ł–Ķ –į–ļ—ā–ł–≤–Ĺ—č—Ö —Ą–ĺ—Ä–ľ –ļ–ł—Ā–Ľ–ĺ—Ä–ĺ–ī–į (–ź–§–ö) [5, 6]. –Ď–į–Ľ–į–Ĺ—Ā –ľ–Ķ–∂–ī—É –Ņ—Ä–ĺ—Ü–Ķ—Ā—Ā–ĺ–ľ –Ņ—Ä–ĺ–ł–∑–≤–ĺ–ī—Ā—ā–≤–į —ć–Ĺ–Ķ—Ä–≥–ł–ł –ł¬†–Ņ—Ä–ĺ—Ü–Ķ—Ā—Ā–ĺ–ľ –ĺ–Ī—Ä–į–∑–ĺ–≤–į–Ĺ–ł—Ź –ź–§–ö –≤¬†–ľ–ł—ā–ĺ—Ö–ĺ–Ĺ–ī—Ä–ł–ł –ĺ–Ī–Ķ—Ā–Ņ–Ķ—á–ł–≤–į–Ķ—ā –Ĺ–ĺ—Ä–ľ–į–Ľ—Ć–Ĺ—É—é —Ä–į–Ī–ĺ—ā—É –ļ–Ľ–Ķ—ā–ļ–ł. –°–ī–≤–ł–≥ –ī–į–Ĺ–Ĺ–ĺ–≥–ĺ —Ä–į–≤–Ĺ–ĺ–≤–Ķ—Ā–ł—Ź –≤¬†—Ā—ā–ĺ—Ä–ĺ–Ĺ—É —É–≤–Ķ–Ľ–ł—á–Ķ–Ĺ–ł—Ź –ĺ–Ī—Ä–į–∑–ĺ–≤–į–Ĺ–ł—Ź –ź–§–ö –Ľ–Ķ–∂–ł—ā –≤¬†–ĺ—Ā–Ĺ–ĺ–≤–Ķ —Ä–į–∑–≤–ł—ā–ł—Ź –Ĺ–Ķ¬†—ā–ĺ–Ľ—Ć–ļ–ĺ –ł—ą–Ķ–ľ–ł—á–Ķ—Ā–ļ–ĺ–≥–ĺ –Ņ–ĺ—Ä–į–∂–Ķ–Ĺ–ł—Ź¬†–≥–ĺ–Ľ–ĺ–≤–Ĺ–ĺ–≥–ĺ –ľ–ĺ–∑–≥–į, –Ŗ嬆–ł –ľ–Ĺ–ĺ–≥–ł—Ö –ī—Ä—É–≥–ł—Ö –Ņ–į—ā–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ł—Ö —Ā–ĺ—Ā—ā–ĺ—Ź–Ĺ–ł–Ļ, –≤–ļ–Ľ—é—á–į—Ź –Ĺ–Ķ–Ļ—Ä–ĺ–ī–Ķ–≥–Ķ–Ĺ–Ķ—Ä–į—ā–ł–≤–Ĺ—č–Ķ, –ĺ–Ĺ–ļ–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ł–Ķ –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł—Ź, —Ā–į—Ö–į—Ä–Ĺ—č–Ļ –ī–ł–į–Ī–Ķ—ā [7, 8]. –ú–ł—ā–ĺ—Ö–ĺ–Ĺ–ī—Ä–ł—Ź –ł–≥—Ä–į–Ķ—ā –ļ–Ľ—é—á–Ķ–≤—É—é —Ä–ĺ–Ľ—Ć –≤¬†–Ņ—Ä–ĺ—Ü–Ķ—Ā—Ā–į—Ö —Ā—ā–į—Ä–Ķ–Ĺ–ł—Ź, —á—ā–ĺ —Ā–ĺ–Ņ—Ä–ĺ–≤–ĺ–∂–ī–į–Ķ—ā—Ā—Ź —Ā–Ĺ–ł–∂–Ķ–Ĺ–ł–Ķ–ľ —Ā–Ņ–ĺ—Ā–ĺ–Ī–Ĺ–ĺ—Ā—ā–ł –Ņ—Ä–ĺ–ł–∑–≤–ĺ–ī–ł—ā—Ć —ć–Ĺ–Ķ—Ä–≥–ł—é –ł¬†–Ĺ–į–ļ–ĺ–Ņ–Ľ–Ķ–Ĺ–ł–Ķ–ľ –Ņ–ĺ–≤—Ä–Ķ–∂–ī–Ķ–Ĺ–Ĺ—č—Ö –ľ–ł—ā–ĺ—Ö–ĺ–Ĺ–ī—Ä–ł–Ļ, –≤¬†—ā–ĺ–ľ —á–ł—Ā–Ľ–Ķ –ľ–ł—ā–ĺ—Ö–ĺ–Ĺ–ī—Ä–ł–Ļ —Ā¬†–Ī–ĺ–Ľ—Ć—ą–ł–ľ –ļ–ĺ–Ľ–ł—á–Ķ—Ā—ā–≤–ĺ–ľ –ľ—É—ā–į—Ü–ł–Ļ –Ē–Ě–ö [9]. –í¬†–Ņ—Ä–ĺ—Ü–Ķ—Ā—Ā–Ķ —Ä–į–Ī–ĺ—ā—č —ā—Ä–į–Ĺ—Ā–Ņ–ĺ—Ä—ā–Ĺ–ĺ–Ļ —Ü–Ķ–Ņ–ł —ć–Ľ–Ķ–ļ—ā—Ä–ĺ–Ĺ–ĺ–≤ –Ĺ–į—Ä—Ź–ī—É —Ā¬†–ĺ–Ī—Ä–į–∑–ĺ–≤–į–Ĺ–ł–Ķ–ľ –ź–Ę–§ –Ņ—Ä–ĺ–ł—Ā—Ö–ĺ–ī–ł—ā –ĺ–Ī—Ä–į–∑–ĺ–≤–į–Ĺ–ł–Ķ –ź–§–ö. –ü–ĺ–ļ–į–∑–į–Ĺ–ĺ, —á—ā–ĺ –ī–ĺ 2% –ł—Ā–Ņ–ĺ–Ľ—Ć–∑—É–Ķ–ľ–ĺ–≥–ĺ –ļ–ł—Ā–Ľ–ĺ—Ä–ĺ–ī–į –ľ–ĺ–∂–Ķ—ā —ā—Ä–į–Ĺ—Ā—Ą–ĺ—Ä–ľ–ł—Ä–ĺ–≤–į—ā—Ć—Ā—Ź –≤¬†—Ā—É–Ņ–Ķ—Ä–ĺ–ļ—Ā–ł–ī–Ĺ—č–Ļ –į–Ĺ–ł–ĺ–Ĺ (O2‚ÄĘ-) [10]. –Ę–į–ļ–ł–ľ –ĺ–Ī—Ä–į–∑–ĺ–ľ, –ĺ—ā¬†–Ņ—Ä–į–≤–ł–Ľ—Ć–Ĺ–ĺ–Ļ —Ä–į–Ī–ĺ—ā—č –ľ–ł—ā–ĺ—Ö–ĺ–Ĺ–ī—Ä–ł–ł –∑–į–≤–ł—Ā–ł—ā –ļ–į–ļ –≤—č–∂–ł–≤–į–Ķ–ľ–ĺ—Ā—ā—Ć –ĺ—ā–ī–Ķ–Ľ—Ć–Ĺ–ĺ–Ļ –ļ–Ľ–Ķ—ā–ļ–ł, —ā–į–ļ –ł¬†–∑–ī–ĺ—Ä–ĺ–≤—Ć–Ķ –∂–ł–≤—č—Ö –ĺ—Ä–≥–į–Ĺ–ł–∑–ľ–ĺ–≤ –≤¬†—Ü–Ķ–Ľ–ĺ–ľ. –í¬†—Ā–Ľ—É—á–į–Ķ –Ņ—Ä–Ķ–ĺ–Ī–Ľ–į–ī–į–Ĺ–ł—Ź –Ĺ–Ķ–≥–į—ā–ł–≤–Ĺ—č—Ö –≤–Ĺ—É—ā—Ä–ł–ļ–Ľ–Ķ—ā–ĺ—á–Ĺ—č—Ö —Ā–ł–≥–Ĺ–į–Ľ–ĺ–≤ –≤–ĺ–∑–ľ–ĺ–∂–Ķ–Ĺ –∑–į–Ņ—É—Ā–ļ —ā–į–ļ –Ĺ–į–∑—č–≤–į–Ķ–ľ–ĺ–≥–ĺ –≤–Ĺ—É—ā—Ä–Ķ–Ĺ–Ĺ–Ķ–≥–ĺ –Ņ—É—ā–ł –į–Ņ–ĺ–Ņ—ā–ĺ–∑–į II —ā–ł–Ņ–į. –Ē–į–Ĺ–Ĺ—č–Ļ —ā–ł–Ņ –į–Ņ–ĺ–Ņ—ā–ĺ–∑–į –Ĺ–į–∑—č–≤–į–Ķ—ā—Ā—Ź —ā–į–ļ–∂–Ķ –ľ–ł—ā–ĺ—Ö–ĺ–Ĺ–ī—Ä–ł–į–Ľ—Ć–Ĺ—č–ľ, –Ņ–ĺ—Ā–ļ–ĺ–Ľ—Ć–ļ—É –∑–į–Ņ—É—Ā–ļ–į–Ķ—ā—Ā—Ź –Ņ—Ä–ł –Ņ–ĺ–≤—Ä–Ķ–∂–ī–Ķ–Ĺ–ł–ł –ľ–ł—ā–ĺ—Ö–ĺ–Ĺ–ī—Ä–ł–į–Ľ—Ć–Ĺ–ĺ–Ļ –ľ–Ķ–ľ–Ī—Ä–į–Ĺ—č –ł¬†–≤–ļ–Ľ—é—á–į–Ķ—ā —Ü–ł–ļ–Ľ —Ä–Ķ–į–ļ—Ü–ł–Ļ, –≤¬†–ļ–ĺ—ā–ĺ—Ä—č—Ö —É—á–į—Ā—ā–≤—É—é—ā —Ą–Ķ—Ä–ľ–Ķ–Ĺ—ā—č –ī—č—Ö–į—ā–Ķ–Ľ—Ć–Ĺ–ĺ–Ļ —Ü–Ķ–Ņ–ł, —á—ā–ĺ –≤¬†–ļ–ĺ–Ĺ–Ķ—á–Ĺ–ĺ–ľ –ł—ā–ĺ–≥–Ķ –Ņ—Ä–ł–≤–ĺ–ī–ł—ā –ļ¬†–≥–ł–Ī–Ķ–Ľ–ł –ļ–Ľ–Ķ—ā–ļ–ł [11]. –í¬†—É—Ā–Ľ–ĺ–≤–ł—Ź—Ö –ł—ą–Ķ–ľ–ł—á–Ķ—Ā–ļ–ĺ–≥–ĺ –Ņ–ĺ–≤—Ä–Ķ–∂–ī–Ķ–Ĺ–ł—Ź –ľ–ĺ–∑–≥–į –ľ–ł—ā–ĺ—Ö–ĺ–Ĺ–ī—Ä–ł—Ź —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź –ĺ–ī–Ĺ–ĺ–Ļ –ł–∑¬†–Ĺ–į–ł–Ī–ĺ–Ľ–Ķ–Ķ —É—Ź–∑–≤–ł–ľ—č—Ö –≤–Ĺ—É—ā—Ä–ł–ļ–Ľ–Ķ—ā–ĺ—á–Ĺ—č—Ö —Ā—ā—Ä—É–ļ—ā—É—Ä. –Ě–į—Ä—É—ą–Ķ–Ĺ–ł–Ķ –Ķ–Ķ —Ą—É–Ĺ–ļ—Ü–ł–ĺ–Ĺ–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź, —Ā¬†–ĺ–ī–Ĺ–ĺ–Ļ —Ā—ā–ĺ—Ä–ĺ–Ĺ—č, –Ņ—Ä–ł–≤–ĺ–ī–ł—ā –ļ¬†–ļ—Ä–ł—ā–ł—á–Ķ—Ā–ļ–ĺ–ľ—É –ī–Ķ—Ą–ł—Ü–ł—ā—É —ć–Ĺ–Ķ—Ä–≥–ł–ł, —Ā¬†–ī—Ä—É–≥–ĺ–Ļ¬†‚Äď –∑–į–Ņ—É—Ā–ļ–į–Ķ—ā –Ņ—Ä–ĺ—Ü–Ķ—Ā—Ā –Ņ—Ä–ĺ–≥—Ä–į–ľ–ľ–ł—Ä—É–Ķ–ľ–ĺ–Ļ –ļ–Ľ–Ķ—ā–ĺ—á–Ĺ–ĺ–Ļ —Ā–ľ–Ķ—Ä—ā–ł.
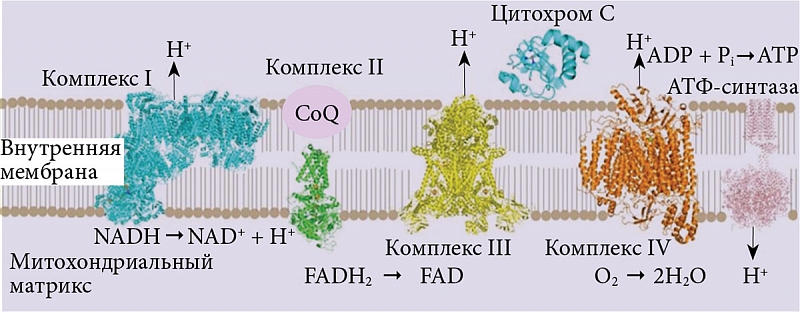
–í–į–∂–Ĺ–Ķ–Ļ—ą–ł–ľ —Ą–Ķ—Ä–ľ–Ķ–Ĺ—ā–ĺ–ľ –ī—č—Ö–į—ā–Ķ–Ľ—Ć–Ĺ–ĺ–Ļ —Ü–Ķ–Ņ–ł –ľ–ł—ā–ĺ—Ö–ĺ–Ĺ–ī—Ä–ł–Ļ, —Ā¬†–ļ–ĺ—ā–ĺ—Ä—č–ľ —Ā–≤—Ź–∑–į–Ĺ—č –Ņ—Ä–ĺ—Ü–Ķ—Ā—Ā—č –ĺ–Ī—Ä–į–∑–ĺ–≤–į–Ĺ–ł—Ź —ć–Ĺ–Ķ—Ä–≥–ł–ł –ł¬†—Ā–ĺ–ĺ—ā–≤–Ķ—ā—Ā—ā–≤–Ķ–Ĺ–Ĺ–ĺ –≤—č–∂–ł–≤–į–Ĺ–ł—Ź –ļ–Ľ–Ķ—ā–ļ–ł, –į¬†—ā–į–ļ–∂–Ķ –Ņ—Ä–ĺ—Ü–Ķ—Ā—Ā—č –į–Ņ–ĺ–Ņ—ā–ĺ—ā–ł—á–Ķ—Ā–ļ–ĺ–Ļ¬†–≥–ł–Ī–Ķ–Ľ–ł –ļ–Ľ–Ķ—ā–ļ–ł, —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ –°.
–¶–ł—ā–ĺ—Ö—Ä–ĺ–ľ –°¬†
–¶–ł—ā–ĺ—Ö—Ä–ĺ–ľ –°¬†(Cytc)¬†‚Äď –Ņ–Ķ–Ņ—ā–ł–ī, —Ā–ĺ—Ā—ā–ĺ—Ź—Č–ł–Ļ –ł–∑¬†104 –į–ľ–ł–Ĺ–ĺ–ļ–ł—Ā–Ľ–ĺ—ā, —Ā¬†–ļ–ĺ—ā–ĺ—Ä—č–ľ–ł –ļ–ĺ–≤–į–Ľ–Ķ–Ĺ—ā–Ĺ–ĺ —á–Ķ—Ä–Ķ–∑ —ā–ł–ĺ—ć—Ą–ł—Ä–Ĺ—č–Ķ –ľ–ĺ—Ā—ā–ł–ļ–ł —Ā–≤—Ź–∑–į–Ĺ–į –∂–Ķ–Ľ–Ķ–∑–ĺ—Ā–ĺ–ī–Ķ—Ä–∂–į—Č–į—Ź¬†–≥—Ä—É–Ņ–Ņ–į¬†–≥–Ķ–ľ (Heme). –ď—Ä—É–Ņ–Ņ–į Heme —Ā–≤—Ź–∑–į–Ĺ–į —Ā¬†–ĺ—Ā–Ĺ–ĺ–≤–Ĺ—č–ľ –Ņ–Ķ–Ņ—ā–ł–ī–ĺ–ľ –≤¬†His18- –ł¬†Met80-–Ņ–ĺ–∑–ł—Ü–ł—Ź—Ö, –ĺ–Ī—Ä–į–∑—É—Ź –į–ľ–ł–Ĺ–ĺ–ļ–ł—Ā–Ľ–ĺ—ā–Ĺ—č–Ķ –Ľ–ł–≥–į–Ĺ–ī—č. –ú–ĺ—Č–Ĺ—č–Ļ –ĺ–ļ–ł—Ā–Ľ–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ-–≤–ĺ—Ā—Ā—ā–į–Ĺ–ĺ–≤–ł—ā–Ķ–Ľ—Ć–Ĺ—č–Ļ –Ņ–ĺ—ā–Ķ–Ĺ—Ü–ł–į–Ľ Cytc —Ā–≤—Ź–∑–į–Ĺ –ł–ľ–Ķ–Ĺ–Ĺ–ĺ —Ā–嬆—Ā–Ņ–Ķ—Ü–ł—Ą–ł—á–Ķ—Ā–ļ–ł–ľ —Ä–į—Ā–Ņ–ĺ–Ľ–ĺ–∂–Ķ–Ĺ–ł–Ķ–ľ¬†–≥—Ä—É–Ņ–Ņ—č Heme –≤–Ĺ—É—ā—Ä–ł –Ņ–Ķ–Ņ—ā–ł–ī–į. –ě—Ā–Ĺ–ĺ–≤–Ĺ–į—Ź —á–į—Ā—ā—Ƭ†–≥—Ä—É–Ņ–Ņ—č Heme¬†–≥–ł–ī—Ä–ĺ—Ą–ĺ–Ī–Ĺ–į, –Ľ–ł—ą—Ć 7,5% –Ņ–ĺ–≤–Ķ—Ä—Ö–Ĺ–ĺ—Ā—ā–ł –ĺ–Ī–Ľ–į–ī–į–Ķ—ā¬†–≥–ł–ī—Ä–ĺ—Ą–ł–Ľ—Ć–Ĺ–ĺ–Ļ —Ā–Ņ–ĺ—Ā–ĺ–Ī–Ĺ–ĺ—Ā—ā—Ć—é. –≠—ā–į —á–į—Ā—ā—Ć –ł¬†–ĺ—ā–≤–Ķ—ā—Ā—ā–≤–Ķ–Ĺ–Ĺ–į –∑–į –Ņ–Ķ—Ä–Ķ–Ĺ–ĺ—Ā —ć–Ľ–Ķ–ļ—ā—Ä–ĺ–Ĺ–ĺ–≤ —Ā¬†Q-—Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ –°-–ĺ–ļ—Ā–ł–ī–ĺ—Ä–Ķ–ī—É–ļ—ā–į–∑—謆‚Äď –ļ–ĺ–ľ–Ņ–Ľ–Ķ–ļ—Ā–į, —Ä–į—Ā–Ņ–ĺ–Ľ–ĺ–∂–Ķ–Ĺ–Ĺ–ĺ–≥–ĺ –Ĺ–į¬†–≤–Ĺ—É—ā—Ä–Ķ–Ĺ–Ĺ–Ķ–Ļ –ľ–Ķ–ľ–Ī—Ä–į–Ĺ–Ķ –ľ–ł—ā–ĺ—Ö–ĺ–Ĺ–ī—Ä–ł–ł,¬†‚Äď –Ĺ–į¬†—Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ –°-–ĺ–ļ—Ā–ł–ī–į–∑—É [12]. –ü—Ä–ĺ—Ü–Ķ—Ā—Ā –ĺ–ļ–ł—Ā–Ľ–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ–≥–ĺ —Ą–ĺ—Ā—Ą–ĺ—Ä–ł–Ľ–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź, –Ņ—Ä–ĺ—ā–Ķ–ļ–į—é—Č–ł–Ļ –Ĺ–į¬†–≤–Ĺ—É—ā—Ä–Ķ–Ĺ–Ĺ–Ķ–Ļ –ľ–Ķ–ľ–Ī—Ä–į–Ĺ–Ķ –ľ–ł—ā–ĺ—Ö–ĺ–Ĺ–ī—Ä–ł–ł –ł¬†–≤–ļ–Ľ—é—á–į—é—Č–ł–Ļ —ć—ā–į–Ņ—č —ć–Ľ–Ķ–ļ—ā—Ä–ĺ–Ĺ–Ĺ–ĺ–≥–ĺ —ā—Ä–į–Ĺ—Ā–Ņ–ĺ—Ä—ā–į, –Ņ—Ä–ł–≤–ĺ–ī–ł—ā –ļ¬†—Ā–ł–Ĺ—ā–Ķ–∑—É –ź–Ę–§ (—Ä–ł—Ā.¬†1) [13]. –í¬†—ć—ā–ĺ–ľ –Ņ—Ä–ĺ—Ü–Ķ—Ā—Ā–Ķ —É—á–į—Ā—ā–≤—É–Ķ—ā —Ä—Ź–ī —Ā—É–Ī—Ā—ā–į–Ĺ—Ü–ł–Ļ¬†‚Äď –ī–ĺ–Ĺ–ĺ—Ä–ĺ–≤ —ć–Ľ–Ķ–ļ—ā—Ä–ĺ–Ĺ–ĺ–≤, —ā–į–ļ–ł—Ö –ļ–į–ļ NADH –ł¬†FADH2, –į¬†—ā–į–ļ–∂–Ķ —Ä—Ź–ī –Ī–Ķ–Ľ–ļ–ĺ–≤, –ĺ–Ī–Ľ–į–ī–į—é—Č–ł—Ö –ľ–Ĺ–ĺ–≥–ĺ–Ņ–Ľ–į–Ĺ–ĺ–≤–ĺ–Ļ –Ī–ł–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ĺ–Ļ –į–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć—é –ł¬†–Ĺ–į–∑—č–≤–į–Ķ–ľ—č—Ö –ļ–ĺ–ľ–Ņ–Ľ–Ķ–ļ—Ā–į–ľ–ł: –ļ–ĺ–ľ–Ņ–Ľ–Ķ–ļ—Ā I¬†‚Äď —É–Ī–ł—Ö–ł–Ĺ–ĺ–Ĺ-–ĺ–ļ—Ā–ł–ī–ĺ—Ä–Ķ–ī—É–ļ—ā–į–∑–į, –ļ–ĺ–ľ–Ņ–Ľ–Ķ–ļ—Ā II¬†‚Äď —Ā—É–ļ—Ü–ł–Ĺ–į—ā-—Ä–Ķ–ī—É–ļ—ā–į–∑–į, –ļ–ĺ–ľ–Ņ–Ľ–Ķ–ļ—Ā III¬†‚Äď Q-—Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ –°-–ĺ–ļ—Ā–ł–ī–ĺ—Ä–Ķ–ī—É–ļ—ā–į–∑–į (bc1), –ļ–ĺ–ľ–Ņ–Ľ–Ķ–ļ—Ā IV¬†‚Äď —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ –°-–ĺ–ļ—Ā–ł–ī–į–∑–į –ł¬†–ļ–ĺ–ľ–Ņ–Ľ–Ķ–ļ—Ā V¬†‚Äď –ź–Ę–§-—Ā–ł–Ĺ—ā–į–∑–į. –í¬†–Ņ—Ä–ĺ—Ü–Ķ—Ā—Ā–Ķ —ā—Ä–į–Ĺ—Ā–Ņ–ĺ—Ä—ā–į —ć–Ľ–Ķ–ļ—ā—Ä–ĺ–Ĺ–ĺ–≤ –Ĺ–į¬†–ľ–ł—ā–ĺ—Ö–ĺ–Ĺ–ī—Ä–ł–į–Ľ—Ć–Ĺ–ĺ–Ļ –ľ–Ķ–ľ–Ī—Ä–į–Ĺ–Ķ¬†–≥–Ķ–Ĺ–Ķ—Ä–ł—Ä—É–Ķ—ā—Ā—Ź —ā–į–ļ –Ĺ–į–∑—č–≤–į–Ķ–ľ—č–Ļ –ľ–ł—ā–ĺ—Ö–ĺ–Ĺ–ī—Ä–ł–į–Ľ—Ć–Ĺ—č–Ļ –ľ–Ķ–ľ–Ī—Ä–į–Ĺ–Ĺ—č–Ļ –Ņ–ĺ—ā–Ķ–Ĺ—Ü–ł–į–Ľ (őĒő®m). NADH-–ī–Ķ–≥–ł–ī—Ä–ĺ–≥–Ķ–Ĺ–į–∑–į, –ļ–ĺ–ľ–Ņ–Ľ–Ķ–ļ—Ā III –ł¬†—Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ –°-–ĺ–ļ—Ā–ł–ī–į–∑–į –Ņ—Ä–ĺ–ļ–į—á–ł–≤–į—é—ā –Ņ—Ä–ĺ—ā–ĺ–Ĺ—č –ł–∑¬†–ľ–ł—ā–ĺ—Ö–ĺ–Ĺ–ī—Ä–ł–į–Ľ—Ć–Ĺ–ĺ–≥–ĺ –ľ–į—ā—Ä–ł–ļ—Ā–į –≤¬†–ľ–Ķ–∂–ľ–Ķ–ľ–Ī—Ä–į–Ĺ–Ĺ–ĺ–Ķ –Ņ—Ä–ĺ—Ā—ā—Ä–į–Ĺ—Ā—ā–≤–ĺ. –ö–ĺ–Ĺ–Ķ—á–Ĺ—č–ľ —ć—ā–į–Ņ–ĺ–ľ –ĺ–Ī—Ä–į–∑–ĺ–≤–į–Ĺ–ł—Ź –ź–Ę–§ —Ā—á–ł—ā–į–Ķ—ā—Ā—Ź —É—ā–ł–Ľ–ł–∑–į—Ü–ł—Ź –ľ–Ķ–ľ–Ī—Ä–į–Ĺ–Ĺ–ĺ–≥–ĺ –Ņ–ĺ—ā–Ķ–Ĺ—Ü–ł–į–Ľ–į –ź–Ę–§-—Ā–ł–Ĺ—ā–į–∑–ĺ–Ļ, —Ź–≤–Ľ—Ź—é—Č–Ķ–Ļ—Ā—Ź –Ņ–嬆—Ā—É—ā–ł ¬ę–ľ–ł–ļ—Ä–ĺ–ľ–ĺ—ā–ĺ—Ä–ĺ–ľ¬Ľ, –Ņ—Ä–Ķ–ĺ–Ī—Ä–į–∑—É—é—Č–ł–ľ –ľ–Ķ–ľ–Ī—Ä–į–Ĺ–Ĺ—č–Ļ –Ņ–ĺ—ā–Ķ–Ĺ—Ü–ł–į–Ľ –≤¬†—Ö–ł–ľ–ł—á–Ķ—Ā–ļ—É—é –ł¬†–ļ–ł–Ĺ–Ķ—ā–ł—á–Ķ—Ā–ļ—É—é —ć–Ĺ–Ķ—Ä–≥–ł—é [14]. Cytc ¬ę—Ä–į–Ī–ĺ—ā–į–Ķ—ā¬Ľ –Ĺ–į¬†–ļ–ĺ–Ĺ–Ķ—á–Ĺ–ĺ–ľ —ć—ā–į–Ņ–Ķ –Ņ–Ķ—Ä–Ķ–Ĺ–ĺ—Ā–į —ć–Ľ–Ķ–ļ—ā—Ä–ĺ–Ĺ–ĺ–≤ —Ā¬†bc1-–ļ–ĺ–ľ–Ņ–Ľ–Ķ–ļ—Ā–į –Ĺ–į¬†—Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ –°-–ĺ–ļ—Ā–ł–ī–į–∑—É. –Ę–į–ļ–ł–ľ –ĺ–Ī—Ä–į–∑–ĺ–ľ, Cytc¬†‚Äď –ļ–Ľ—é—á–Ķ–≤–ĺ–Ļ –ľ–ł—ā–ĺ—Ö–ĺ–Ĺ–ī—Ä–ł–į–Ľ—Ć–Ĺ—č–Ļ –Ī–Ķ–Ľ–ĺ–ļ, –≤—č–Ņ–ĺ–Ľ–Ĺ—Ź—é—Č–ł–Ļ –≤–į–∂–Ĺ–Ķ–Ļ—ą—É—é —Ä–ĺ–Ľ—Ć –≤¬†–Ņ—Ä–ĺ—Ü–Ķ—Ā—Ā–Ķ —Ā–ł–Ĺ—ā–Ķ–∑–į —ć–Ĺ–Ķ—Ä–≥–ł–ł.
–Ď–ĺ–Ľ—Ć—ą–ł–Ĺ—Ā—ā–≤–ĺ –ľ–ł—ā–ĺ—Ö–ĺ–Ĺ–ī—Ä–ł–į–Ľ—Ć–Ĺ—č—Ö –Ī–Ķ–Ľ–ļ–ĺ–≤ —ć–ļ—Ā–Ņ—Ä–Ķ—Ā—Ā–ł—Ä—É—é—ā—Ā—Ź –≤–Ĺ–Ķ –ľ–ł—ā–ĺ—Ö–ĺ–Ĺ–ī—Ä–ł–ł. –°–ļ–į–∑–į–Ĺ–Ĺ–ĺ–Ķ –ĺ—ā–Ĺ–ĺ—Ā–ł—ā—Ā—Ź –ł¬†–ļ —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ—É –°.¬†–°–ĺ–ĺ—ā–≤–Ķ—ā—Ā—ā–≤–Ķ–Ĺ–Ĺ–ĺ –ĺ–Ĺ–ł –ī–ĺ–Ľ–∂–Ĺ—č –ĺ–Ņ—Ä–Ķ–ī–Ķ–Ľ–Ķ–Ĺ–Ĺ—č–ľ –ĺ–Ī—Ä–į–∑–ĺ–ľ –Ņ–ĺ—Ā—ā—É–Ņ–į—ā—Ć –≤¬†–ľ–ł—ā–ĺ—Ö–ĺ–Ĺ–ī—Ä–ł—é. –Ē–į–Ĺ–Ĺ—č–Ļ –Ņ—Ä–ĺ—Ü–Ķ—Ā—Ā —Ä–Ķ–į–Ľ–ł–∑—É–Ķ—ā—Ā—Ź —Ā¬†–Ņ–ĺ–ľ–ĺ—Č—Ć—é —Ā–Ņ–Ķ—Ü–ł—Ą–ł—á–Ķ—Ā–ļ–ł—Ö –Ī–Ķ–Ľ–ļ–ĺ–≤¬†‚Äď —ā—Ä–į–Ĺ—Ā–Ľ–ĺ–ļ–į–∑, —Ā–≤—Ź–∑—č–≤–į—é—Č–ł—Ö –≤–Ĺ—É—ā—Ä–Ķ–Ĺ–Ĺ—é—é –ł¬†–Ĺ–į—Ä—É–∂–Ĺ—É—é –ľ–Ķ–ľ–Ī—Ä–į–Ĺ—č –ľ–ł—ā–ĺ—Ö–ĺ–Ĺ–ī—Ä–ł–ł. –°–Ľ–Ķ–ī—É–Ķ—ā –ĺ—ā–ľ–Ķ—ā–ł—ā—Ć, —á—ā–ĺ –≤¬†–Ņ—Ä–ĺ—Ü–Ķ—Ā—Ā–Ķ –Ņ–Ķ—Ä–Ķ–Ĺ–ĺ—Ā–į –≤–Ĺ—É—ā—Ä—Ć –ľ–ł—ā–ĺ—Ö–ĺ–Ĺ–ī—Ä–ł–ł –Ī–Ķ–Ľ–ļ–ł, –ļ–į–ļ –Ņ—Ä–į–≤–ł–Ľ–ĺ, —ā—Ä–į–Ĺ—Ā—Ą–ĺ—Ä–ľ–ł—Ä—É—é—ā—Ā—Ź –ł–∑¬†–Ĺ–Ķ–į–ļ—ā–ł–≤–Ĺ—č—Ö –į–Ņ–ĺ—Ą–ĺ—Ä–ľ –≤¬†–į–ļ—ā–ł–≤–Ĺ—č–Ķ, —Ā–≤—Ź–∑–į–Ĺ–Ĺ—č–Ķ —Ā¬†–ļ–ĺ-—Ą–į–ļ—ā–ĺ—Ä–į–ľ–ł —Ö–ĺ–Ľ–ĺ—Ą–ĺ—Ä–ľ—č. –̖嬆–ī–Ľ—Ź —ć—ā–ĺ–≥–ĺ –Ņ—Ä–ĺ—Ü–Ķ—Ā—Ā–į —ā–į–ļ–∂–Ķ —ā—Ä–Ķ–Ī—É–Ķ—ā—Ā—Ź —Ä—Ź–ī —Ą–Ķ—Ä–ľ–Ķ–Ĺ—ā–ĺ–≤. –í¬†—á–į—Ā—ā–Ĺ–ĺ—Ā—ā–ł, Cytc —ā—Ä–į–Ĺ—Ā–Ņ–ĺ—Ä—ā–ł—Ä—É–Ķ—ā—Ā—Ź –≤–Ĺ—É—ā—Ä—Ć –ľ–ł—ā–ĺ—Ö–ĺ–Ĺ–ī—Ä–ł–ł –≤¬†–≤–ł–ī–Ķ –į–Ņ–ĺ—Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ–į –°.¬†–ü—Ä–ł—Ā–ĺ–Ķ–ī–ł–Ĺ–Ķ–Ĺ–ł–Ķ¬†–≥—Ä—É–Ņ–Ņ—č Heme –Ņ—Ä–ł–≤–ĺ–ī–ł—ā –ļ¬†—ā—Ä–į–Ĺ—Ā—Ą–ĺ—Ä–ľ–į—Ü–ł–ł –≤¬†–∑—Ä–Ķ–Ľ—É—é —Ą–ĺ—Ä–ľ—É —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ–į –°.¬†–Ē–į–Ĺ–Ĺ—č–Ļ –Ņ—Ä–ĺ—Ü–Ķ—Ā—Ā –ļ–į—ā–į–Ľ–ł–∑–ł—Ä—É–Ķ—ā—Ā—Ź —Ą–Ķ—Ä–ľ–Ķ–Ĺ—ā–ĺ–ľ —Ö–ĺ–Ľ–ĺ—Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ –°-—Ā–ł–Ĺ—ā–į–∑–ĺ–Ļ [15]. –í¬†–ľ–Ķ–∂–ľ–Ķ–ľ–Ī—Ä–į–Ĺ–Ĺ–ĺ–ľ –Ņ—Ä–ĺ—Ā—ā—Ä–į–Ĺ—Ā—ā–≤–Ķ –ľ–ł—ā–ĺ—Ö–ĺ–Ĺ–ī—Ä–ł–ł –Ĺ–į—Ö–ĺ–ī–ł—ā—Ā—Ź —Ä—Ź–ī –≤–į–∂–Ĺ—č—Ö, —Ą—É–Ĺ–ļ—Ü–ł–ĺ–Ĺ–į–Ľ—Ć–Ĺ–ĺ —Ā–≤—Ź–∑–į–Ĺ–Ĺ—č—Ö —Ā¬†—Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ–ĺ–ľ –°¬†–Ī–Ķ–Ľ–ļ–ĺ–≤¬†‚Äď Erv1 –ł¬†Mia40. –í¬†—á–į—Ā—ā–Ĺ–ĺ—Ā—ā–ł, Mia40 —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź —É—á–į—Ā—ā–Ĺ–ł–ļ–ĺ–ľ –Ņ—Ä–ĺ—Ü–Ķ—Ā—Ā–į –Ņ–Ķ—Ä–Ķ–ī–į—á–ł —ć–Ľ–Ķ–ļ—ā—Ä–ĺ–Ĺ–ĺ–≤ –ī–Ľ—Ź —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ–į –°¬†[16]. –°–ł–Ĺ—ā–Ķ–∑ –ź–Ę–§ —Ä–Ķ–≥—É–Ľ–ł—Ä—É–Ķ—ā—Ā—Ź –∑–į —Ā—á–Ķ—ā –ļ–ĺ–Ĺ—ā—Ä–ĺ–Ľ—Ź –į–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā–ł –Ņ—Ä–ĺ—Ü–Ķ—Ā—Ā–ĺ–≤ –Ņ–Ķ—Ä–Ķ–Ĺ–ĺ—Ā–į —ć–Ľ–Ķ–ļ—ā—Ä–ĺ–Ĺ–ĺ–≤ –ł¬†–Ī–Ľ–ĺ–ļ–ł—Ä—É–Ķ—ā—Ā—Ź –Ņ–ĺ—Ā—Ä–Ķ–ī—Ā—ā–≤–ĺ–ľ —Ā–≤—Ź–∑—č–≤–į–Ĺ–ł—Ź –ź–Ę–§ —Ā¬†—Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ–ĺ–ľ –°¬†–ł —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ –°-–ĺ–ļ—Ā–ł–ī–į–∑–ĺ–Ļ. –Ē–į–Ĺ–Ĺ—č–Ļ –ľ–Ķ—Ö–į–Ĺ–ł–∑–ľ —Ä–į–Ī–ĺ—ā–į–Ķ—ā –Ņ–嬆–Ņ—Ä–ł–Ĺ—Ü–ł–Ņ—É –ĺ–Ī—Ä–į—ā–Ĺ–ĺ–Ļ —Ā–≤—Ź–∑–ł [17]. –ē—Č–Ķ –ĺ–ī–Ĺ–ł–ľ –ľ–Ķ—Ö–į–Ĺ–ł–∑–ľ–ĺ–ľ —Ä–Ķ–≥—É–Ľ—Ź—Ü–ł–ł –į–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā–ł —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ–į –°¬†—Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź –Ņ—Ä–ĺ—Ü–Ķ—Ā—Ā —Ą–ĺ—Ā—Ą–ĺ—Ä–ł–Ľ–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź –į–ľ–ł–Ĺ–ĺ–ļ–ł—Ā–Ľ–ĺ—ā—č —ā–ł—Ä–ĺ–∑–ł–Ĺ–į, —á—ā–ĺ –Ņ—Ä–ł–≤–ĺ–ī–ł—ā –ļ¬†–∑–Ĺ–į—á–ł–ľ–ĺ–ľ—É —Ā–Ĺ–ł–∂–Ķ–Ĺ–ł—é —Ā–Ņ–ĺ—Ā–ĺ–Ī–Ĺ–ĺ—Ā—ā–ł –Ņ–Ķ—Ä–Ķ–Ĺ–ĺ—Ā–į —ć–Ľ–Ķ–ļ—ā—Ä–ĺ–Ĺ–ĺ–≤ –Ĺ–į¬†—Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ –°-–ĺ–ļ—Ā–ł–ī–į–∑—É –ł —Ā–ĺ–ĺ—ā–≤–Ķ—ā—Ā—ā–≤–Ķ–Ĺ–Ĺ–ĺ –Ņ–ĺ–ī–į–≤–Ľ–Ķ–Ĺ–ł—é –Ņ—Ä–ĺ—Ü–Ķ—Ā—Ā–ĺ–≤ –ľ–ł—ā–ĺ—Ö–ĺ–Ĺ–ī—Ä–ł–į–Ľ—Ć–Ĺ–ĺ–≥–ĺ –ī—č—Ö–į–Ĺ–ł—Ź [8]. –í¬†—É—Ā–Ľ–ĺ–≤–ł—Ź—Ö –ł—ą–Ķ–ľ–ł—á–Ķ—Ā–ļ–ĺ–≥–ĺ –Ņ–ĺ–≤—Ä–Ķ–∂–ī–Ķ–Ĺ–ł—Ź –Ĺ–į—Ä—É—ą–į–Ķ—ā—Ā—Ź –Ņ—Ä–ĺ—Ü–Ķ—Ā—Ā –Ņ–Ķ—Ä–Ķ–Ĺ–ĺ—Ā–į –į–Ņ–ĺ—Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ–į –°¬†–≤–Ĺ—É—ā—Ä—Ć –ľ–ł—ā–ĺ—Ö–ĺ–Ĺ–ī—Ä–ł–ł, –į¬†—ā–į–ļ–∂–Ķ —Ä–Ķ–≥—É–Ľ—Ź—Ü–ł—Ź –į–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā–ł Cytc –∑–į —Ā—á–Ķ—ā –Ņ—Ä–ĺ—Ü–Ķ—Ā—Ā–ĺ–≤ —Ā–≤—Ź–∑—č–≤–į–Ĺ–ł—Ź —Ā¬†–ź–Ę–§ –ł¬†—Ą–ĺ—Ā—Ą–ĺ—Ä–ł–Ľ–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź —ā–ł—Ä–ĺ–∑–ł–Ĺ–į, —á—ā–ĺ —É—Ā—É–≥—É–Ī–Ľ—Ź–Ķ—ā –Ņ–ĺ–≤—Ä–Ķ–∂–ī–Ķ–Ĺ–ł–Ķ –ļ–Ľ–Ķ—ā–ļ–ł.
Cytc –≤—č–Ņ–ĺ–Ľ–Ĺ—Ź–Ķ—ā –≤–į–∂–Ĺ—É—é —Ä–ĺ–Ľ—Ć –≤¬†–Ņ—Ä–ĺ—Ü–Ķ—Ā—Ā–į—Ö —ā–ļ–į–Ĺ–Ķ–≤–ĺ–≥–ĺ –ī—č—Ö–į–Ĺ–ł—Ź. –£—Ā—ā–į–Ĺ–ĺ–≤–Ľ–Ķ–Ĺ–ĺ –Ķ–≥–ĺ —É—á–į—Ā—ā–ł–Ķ –≤¬†–Ņ—Ä–ĺ—Ü–Ķ—Ā—Ā–į—Ö –ĺ–Ī—Ä–į–∑–ĺ–≤–į–Ĺ–ł—Ź –ł¬†—É—ā–ł–Ľ–ł–∑–į—Ü–ł–ł –į–ļ—ā–ł–≤–Ĺ—č—Ö —Ą–ĺ—Ä–ľ –ļ–ł—Ā–Ľ–ĺ—Ä–ĺ–ī–į (–ź–§–ö). –Ę—Ä–į–ī–ł—Ü–ł–ĺ–Ĺ–Ĺ–ĺ –ļ¬†–į–ļ—ā–ł–≤–Ĺ—č–ľ —Ą–ĺ—Ä–ľ–į–ľ –ļ–ł—Ā–Ľ–ĺ—Ä–ĺ–ī–į –Ņ—Ä–ł—á–ł—Ā–Ľ—Ź—é—ā —Ā—É–Ņ–Ķ—Ä–ĺ–ļ—Ā–ł–ī–Ĺ—č–Ļ –į–Ĺ–ł–ĺ–Ŭ†‚Äď –ł–ĺ–Ĺ –ľ–ĺ–Ľ–Ķ–ļ—É–Ľ—č –ļ–ł—Ā–Ľ–ĺ—Ä–ĺ–ī–į —Ā¬†–Ĺ–Ķ—Ā–Ņ–į—Ä–Ķ–Ĺ–Ĺ—č–ľ —ć–Ľ–Ķ–ļ—ā—Ä–ĺ–Ĺ–ĺ–ľ (O2‚ÄĘ-), –≤—č—Ā–ĺ–ļ–ĺ—Ä–Ķ–į–ļ—Ü–ł–ĺ–Ĺ–Ĺ—č–Ļ –ł¬†–ļ–ĺ—Ä–ĺ—ā–ļ–ĺ–∂–ł–≤—É—Č–ł–Ļ —Ä–į–ī–ł–ļ–į–Ľ (‚ÄĘOH), –ĺ–Ī—Ä–į–∑–ĺ–≤–į–Ĺ–Ĺ—č–Ļ —Ā–ĺ–Ķ–ī–ł–Ĺ–Ķ–Ĺ–ł–Ķ–ľ –į—ā–ĺ–ľ–ĺ–≤ –ļ–ł—Ā–Ľ–ĺ—Ä–ĺ–ī–į –ł¬†–≤–ĺ–ī–ĺ—Ä–ĺ–ī–į, –į¬†—ā–į–ļ–∂–Ķ –Ņ–Ķ—Ä–Ķ–ļ–ł—Ā—Ć –≤–ĺ–ī–ĺ—Ä–ĺ–ī–į (H2O2), –Ĺ–Ķ¬†—Ź–≤–Ľ—Ź—é—Č—É—é—Ā—Ź —Ā–≤–ĺ–Ī–ĺ–ī–Ĺ—č–ľ —Ä–į–ī–ł–ļ–į–Ľ–ĺ–ľ. –Ę–Ķ—Ä–ľ–ł–Ĺ ¬ę—Ā–≤–ĺ–Ī–ĺ–ī–Ĺ—č–Ļ —Ä–į–ī–ł–ļ–į–Ľ¬Ľ –ĺ—ā–Ĺ–ĺ—Ā–ł—ā—Ā—Ź –ļ¬†–Ľ—é–Ī–ĺ–Ļ –ľ–ĺ–Ľ–Ķ–ļ—É–Ľ–Ķ, —Ā–ĺ–ī–Ķ—Ä–∂–į—Č–Ķ–Ļ –į—ā–ĺ–ľ –ļ–ł—Ā–Ľ–ĺ—Ä–ĺ–ī–į, —Ā—ā—Ä–Ķ–ľ—Ź—Č–ł–Ļ—Ā—Ź –∑–į–Ņ–ĺ–Ľ–Ĺ–ł—ā—Ć –Ĺ–Ķ–ī–ĺ—Ā—ā–į—é—Č–ł–Ļ —ć–Ľ–Ķ–ļ—ā—Ä–ĺ–Ĺ –Ĺ–į¬†—Ā–≤–ĺ–Ķ–Ļ –ĺ—Ä–Ī–ł—ā–Ķ –∑–į —Ā—á–Ķ—ā –ī—Ä—É–≥–ł—Ö –ľ–ĺ–Ľ–Ķ–ļ—É–Ľ. –Ě–į–Ľ–ł—á–ł–Ķ –≤¬†—Ā—ā—Ä—É–ļ—ā—É—Ä–Ķ –Ņ–Ķ—Ä–Ķ—Ö–ĺ–ī–Ĺ—č—Ö –ľ–Ķ—ā–į–Ľ–Ľ–ĺ–≤ —Ö–į—Ä–į–ļ—ā–Ķ—Ä–Ĺ–ĺ –ī–Ľ—Ź —Ā–≤–ĺ–Ī–ĺ–ī–Ĺ—č—Ö —Ä–į–ī–ł–ļ–į–Ľ–ĺ–≤. –°–≤–ĺ–Ī–ĺ–ī–Ĺ—č–Ķ —Ä–į–ī–ł–ļ–į–Ľ—謆‚Äď —É–Ĺ–ł–≤–Ķ—Ä—Ā–į–Ľ—Ć–Ĺ—č–Ķ –ĺ–ļ–ł—Ā–Ľ–ł—ā–Ķ–Ľ–ł, –Ņ–ĺ–≤—Ä–Ķ–∂–ī–į—é—Č–ł–Ķ –Ē–Ě–ö –ł¬†–ī—Ä—É–≥–ł–Ķ —Ā—ā—Ä—É–ļ—ā—É—Ä—č –ļ–Ľ–Ķ—ā–ļ–ł.
–ź–§–ö –≤¬†–ł–∑–Ī—č—ā–ļ–Ķ –ĺ–Ī—Ä–į–∑—É—é—ā—Ā—Ź –≤¬†–ľ–ł—ā–ĺ—Ö–ĺ–Ĺ–ī—Ä–ł–ł –≤¬†—Ä–Ķ–∑—É–Ľ—Ć—ā–į—ā–Ķ —Ä–į–Ī–ĺ—ā—č —Ü–Ķ–Ņ–ł –Ņ–Ķ—Ä–Ķ–Ĺ–ĺ—Ā–į —ć–Ľ–Ķ–ļ—ā—Ä–ĺ–Ĺ–ĺ–≤. –ě—Ā–Ĺ–ĺ–≤–Ĺ—č–ľ–ł ¬ę–Ņ—É–Ĺ–ļ—ā–į–ľ–ł¬Ľ –ĺ–Ī—Ä–į–∑–ĺ–≤–į–Ĺ–ł—Ź –ź–§–ö —Ź–≤–Ľ—Ź—é—ā—Ā—Ź NADH (–ļ–ĺ–ľ–Ņ–Ľ–Ķ–ļ—Ā I), –ļ–ĺ–ľ–Ņ–Ľ–Ķ–ļ—Ā bc1 (–ļ–ĺ–ľ–Ņ–Ľ–Ķ–ļ—Ā III) –ł¬†—É–Ī–ł—Ö–ł–Ĺ–ĺ–Ĺ. –ö–ĺ–ľ–Ņ–Ľ–Ķ–ļ—Ā¬†I¬†–≥–Ķ–Ĺ–Ķ—Ä–ł—Ä—É–Ķ—ā –ź–§–ö –Ĺ–į¬†–ľ–ł—ā–ĺ—Ö–ĺ–Ĺ–ī—Ä–ł–į–Ľ—Ć–Ĺ–ĺ–ľ –ľ–į—ā—Ä–ł–ļ—Ā–Ķ, –ļ–ĺ–ľ–Ņ–Ľ–Ķ–ļ—Ā¬†III –≤—č—Ā–≤–ĺ–Ī–ĺ–∂–ī–į–Ķ—ā –ź–§–ö –≤¬†–ľ–ł—ā–ĺ—Ö–ĺ–Ĺ–ī—Ä–ł–į–Ľ—Ć–Ĺ—č–Ļ –ľ–į—ā—Ä–ł–ļ—Ā –ł¬†–ľ–Ķ–∂–ľ–Ķ–ľ–Ī—Ä–į–Ĺ–Ĺ–ĺ–Ķ –Ņ—Ä–ĺ—Ā—ā—Ä–į–Ĺ—Ā—ā–≤–ĺ. –ė–∑–Ī—č—ā–ĺ—á–Ĺ–į—Ź –į–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć –ī–į–Ĺ–Ĺ—č—Ö –Ņ—Ä–ĺ—Ü–Ķ—Ā—Ā–ĺ–≤ –≤¬†–ł—ā–ĺ–≥–Ķ –Ņ—Ä–ł–≤–ĺ–ī–ł—ā –ļ¬†–į–ļ—ā–ł–≤–į—Ü–ł–ł —Ä–Ķ–į–ļ—Ü–ł–Ļ –į–Ņ–ĺ–Ņ—ā–ĺ–∑–į [9]. –ě–Ī—Ä–į–∑–ĺ–≤–į–Ĺ–ł–Ķ —Ā–≤–ĺ–Ī–ĺ–ī–Ĺ—č—Ö —Ä–į–ī–ł–ļ–į–Ľ–ĺ–≤¬†‚Äď –Ĺ–Ķ–Ņ—Ä–Ķ—Ä—č–≤–Ĺ—č–Ļ –Ņ—Ä–ĺ—Ü–Ķ—Ā—Ā, –Ņ—Ä–ĺ—ā–Ķ–ļ–į—é—Č–ł–Ļ –≤¬†–ļ–į–∂–ī–ĺ–Ļ –ļ–Ľ–Ķ—ā–ļ–Ķ –ł¬†—ā—Ä–Ķ–Ī—É—é—Č–ł–Ļ –ĺ–Ņ—Ä–Ķ–ī–Ķ–Ľ–Ķ–Ĺ–Ĺ—č—Ö —Ä–Ķ–≥—É–Ľ—Ź—ā–ĺ—Ä–Ĺ—č—Ö –ľ–Ķ—Ö–į–Ĺ–ł–∑–ľ–ĺ–≤. –Ě–į–Ņ—Ä–ł–ľ–Ķ—Ä, —Ą–Ķ—Ä–ľ–Ķ–Ĺ—ā —Ā—É–Ņ–Ķ—Ä–ĺ–ļ—Ā–ł–ī–ī–ł—Ā–ľ—É—ā–į–∑–į –ĺ–Ī–Ķ–∑–≤—Ä–Ķ–∂–ł–≤–į–Ķ—ā —Ā—É–Ņ–Ķ—Ä–ĺ–ļ—Ā–ł–ī–Ĺ—č–Ķ —Ä–į–ī–ł–ļ–į–Ľ—č –ī–ĺ –Ņ–Ķ—Ä–Ķ–ļ–ł—Ā–ł –≤–ĺ–ī–ĺ—Ä–ĺ–ī–į, –≤¬†–ī–į–Ľ—Ć–Ĺ–Ķ–Ļ—ą–Ķ–ľ —É—ā–ł–Ľ–ł–∑–ł—Ä—É–Ķ–ľ–ĺ–Ļ –ļ–į—ā–į–Ľ–į–∑–ĺ–Ļ. –ě–ī–Ĺ–ĺ–Ļ –ł–∑¬†—Ą—É–Ĺ–ļ—Ü–ł–ĺ–Ĺ–į–Ľ—Ć–Ĺ—č—Ö –ĺ—Ā–ĺ–Ī–Ķ–Ĺ–Ĺ–ĺ—Ā—ā–Ķ–Ļ —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ–į¬†–°¬†—ā–į–ļ–∂–Ķ —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź —Ā–Ņ–ĺ—Ā–ĺ–Ī–Ĺ–ĺ—Ā—ā—Ć –ĺ–Ī–Ķ–∑–≤—Ä–Ķ–∂–ł–≤–į—ā—Ć —Ā–≤–ĺ–Ī–ĺ–ī–Ĺ—č–Ķ —Ä–į–ī–ł–ļ–į–Ľ—č –Ĺ–į¬†–≤–Ĺ—É—ā—Ä–Ķ–Ĺ–Ĺ–Ķ–Ļ –ľ–Ķ–ľ–Ī—Ä–į–Ĺ–Ķ (–≤—č—Ā—ā—É–Ņ–į–Ķ—ā –ī–ĺ–Ĺ–į—ā–ĺ—Ä–ĺ–ľ —ć–Ľ–Ķ–ļ—ā—Ä–ĺ–Ĺ–į –ł¬†–≤–ĺ—Ā—Ā—ā–į–Ĺ–į–≤–Ľ–ł–≤–į–Ķ—ā —Ā—É–Ņ–Ķ—Ä–ĺ–ļ—Ā–ł–ī–Ĺ—č–Ļ —Ä–į–ī–ł–ļ–į–Ľ –ī–ĺ –ě2). –í–Ņ–ĺ—Ā–Ľ–Ķ–ī—Ā—ā–≤–ł–ł —ć—ā–ĺ—ā —ć–Ľ–Ķ–ļ—ā—Ä–ĺ–Ĺ –ľ–ĺ–∂–Ķ—ā —É—á–į—Ā—ā–≤–ĺ–≤–į—ā—Ć –≤¬†—Ā–ł–Ĺ—ā–Ķ–∑–Ķ —ć–Ĺ–Ķ—Ä–≥–ł–ł, –≤–ĺ—Ā—Ā—ā–į–Ĺ–į–≤–Ľ–ł–≤–į—Ź –ĺ–ļ–ł—Ā–Ľ–Ķ–Ĺ–Ĺ—É—é —Ą–ĺ—Ä–ľ—É —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ–į –°¬†–Ņ–ĺ—Ā—Ä–Ķ–ī—Ā—ā–≤–ĺ–ľ —ā—Ä–į–Ĺ—Ā–Ľ–ĺ–ļ–į—Ü–ł–ł —á–Ķ—Ä–Ķ–∑ —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ –°-–ĺ–ļ—Ā–ł–ī–į–∑—É [20]. –ö—Ä–ĺ–ľ–Ķ —ā–ĺ–≥–ĺ, –≤¬†–ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–ł Z.B.¬†Wang –ł¬†—Ā–ĺ–į–≤—ā. (2003) –Ņ–ĺ–ļ–į–∑–į–Ĺ–ĺ, —á—ā–ĺ —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ –°¬†—Ā–Ņ–ĺ—Ā–ĺ–Ī–Ķ–Ĺ —É–Ľ–į–≤–Ľ–ł–≤–į—ā—Ć –Ě2–ě2 [21]. –Ę–į–ļ–ł–ľ –ĺ–Ī—Ä–į–∑–ĺ–ľ, –Ņ–ĺ—Ā–Ľ–Ķ–ī–ĺ–≤–į—ā–Ķ–Ľ—Ć–Ĺ–ĺ –Ņ—Ä–ĺ—Ö–ĺ–ī—Ź —Ü–ł–ļ–Ľ—č –ĺ–ļ–ł—Ā–Ľ–Ķ–Ĺ–ł—Ź/–≤–ĺ—Ā—Ā—ā–į–Ĺ–ĺ–≤–Ľ–Ķ–Ĺ–ł—Ź, —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ –°¬†—Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź —É–Ĺ–ł–≤–Ķ—Ä—Ā–į–Ľ—Ć–Ĺ—č–ľ –≤–Ĺ—É—ā—Ä–ł–ļ–Ľ–Ķ—ā–ĺ—á–Ĺ—č–ľ –į–Ĺ—ā–ł–ĺ–ļ—Ā–ł–ī–į–Ĺ—ā–ĺ–ľ, —Ā–ĺ—á–Ķ—ā–į—Ź –≤¬†—Ā–Ķ–Ī–Ķ –≤–ĺ–∑–ľ–ĺ–∂–Ĺ–ĺ—Ā—ā–ł –ī–ĺ–Ĺ–į—ā–ĺ—Ä–į —ć–Ľ–Ķ–ļ—ā—Ä–ĺ–Ĺ–ĺ–≤ –ł¬†–ĺ–ļ–ł—Ā–Ľ–ł—ā–Ķ–Ľ—Ź.
–¶–ł—ā–ĺ—Ö—Ä–ĺ–ľ –°¬†—É—á–į—Ā—ā–≤—É–Ķ—ā –≤¬†–Ņ—Ä–ĺ—Ü–Ķ—Ā—Ā–į—Ö —Ā—ā–į—Ä–Ķ–Ĺ–ł—Ź –ļ–Ľ–Ķ—ā–ļ–ł. –í¬†—á–į—Ā—ā–Ĺ–ĺ—Ā—ā–ł, –≤¬†—Ä—Ź–ī–Ķ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–Ļ –Ņ–ĺ–ļ–į–∑–į–Ĺ–ĺ, —á—ā–ĺ —Ā¬†–≤–ĺ–∑—Ä–į—Ā—ā–ĺ–ľ —É–≤–Ķ–Ľ–ł—á–ł–≤–į–Ķ—ā—Ā—Ź —Ą–ĺ—Ā—Ą–ĺ—Ä–ł–Ľ–ł—Ä–ĺ–≤–į–Ĺ–ł–Ķ –ĺ–ī–Ĺ–ĺ–≥–ĺ –ł–∑¬†—Ä–Ķ–≥—É–Ľ—Ź—ā–ĺ—Ä–Ĺ—č—Ö –≤–Ĺ—É—ā—Ä–ł–ľ–ł—ā–ĺ—Ö–ĺ–Ĺ–ī—Ä–ł–į–Ľ—Ć–Ĺ—č—Ö –Ī–Ķ–Ľ–ļ–ĺ–≤ p66shc, —á—ā–ĺ –Ņ—Ä–ł–≤–ĺ–ī–ł—ā –ļ¬†—É–≤–Ķ–Ľ–ł—á–Ķ–Ĺ–ł—é –ļ–ĺ–Ľ–ł—á–Ķ—Ā—ā–≤–į —Ā–≤–ĺ–Ī–ĺ–ī–Ĺ—č—Ö —Ä–į–ī–ł–ļ–į–Ľ–ĺ–≤ –ł¬†–ł–Ĺ—ā–Ķ–Ĺ—Ā–ł–≤–Ĺ–ĺ—Ā—ā–ł –ĺ–ļ–ł—Ā–Ľ–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ–≥–ĺ —Ā—ā—Ä–Ķ—Ā—Ā–į [22]. –ě–ļ–ł—Ā–Ľ–Ķ–Ĺ–ł–Ķ p66shc –Ņ—Ä–ĺ–ł—Ā—Ö–ĺ–ī–ł—ā –∑–į —Ā—á–Ķ—ā –Ņ–ĺ–Ľ—É—á–Ķ–Ĺ–ł—Ź —ć–Ľ–Ķ–ļ—ā—Ä–ĺ–Ĺ–į –ĺ—ā¬†—Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ–į –°¬†[23]. –í¬†–ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź—Ö –ļ–Ľ–Ķ—ā–ĺ—á–Ĺ—č—Ö –Ľ–ł–Ĺ–ł–Ļ, –Ľ–ł—ą–Ķ–Ĺ–Ĺ—č—Ö p66shc, –ļ–ĺ–Ľ–ł—á–Ķ—Ā—ā–≤–ĺ —Ā–≤–ĺ–Ī–ĺ–ī–Ĺ—č—Ö —Ä–į–ī–ł–ļ–į–Ľ–ĺ–≤ –Ī—č–Ľ–ĺ –Ĺ–ł–∂–Ķ. –≠—ā–ł –ļ–Ľ–Ķ—ā–ļ–ł –Ī—č–Ľ–ł –≤¬†–ľ–Ķ–Ĺ—Ć—ą–Ķ–Ļ —Ā—ā–Ķ–Ņ–Ķ–Ĺ–ł –Ņ–ĺ–ī–≤–Ķ—Ä–∂–Ķ–Ĺ—č –ĺ–ļ–ł—Ā–Ľ–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ–ľ—É –Ņ–ĺ–≤—Ä–Ķ–∂–ī–Ķ–Ĺ–ł—é –Ņ–ĺ—Ā–Ľ–Ķ –ĺ–Ī—Ä–į–Ī–ĺ—ā–ļ–ł –Ņ–Ķ—Ä–Ķ–ļ–ł—Ā—Ć—é –≤–ĺ–ī–ĺ—Ä–ĺ–ī–į –ł–Ľ–ł —É–Ľ—Ć—ā—Ä–į—Ą–ł–ĺ–Ľ–Ķ—ā–ĺ–≤–ĺ–≥–ĺ –ĺ–Ī–Ľ—É—á–Ķ–Ĺ–ł—Ź¬†[24]. –£–≤–Ķ–Ľ–ł—á–Ķ–Ĺ–ł–Ķ –ļ–ĺ–Ľ–ł—á–Ķ—Ā—ā–≤–į —Ā–≤–ĺ–Ī–ĺ–ī–Ĺ—č—Ö —Ä–į–ī–ł–ļ–į–Ľ–ĺ–≤ –Ņ–嬆–ľ–Ķ—Ö–į–Ĺ–ł–∑–ľ—É —Ą–ĺ—Ā—Ą–ĺ—Ä–ł–Ľ–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź p66shc —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź –ĺ–ī–Ĺ–ł–ľ –ł–∑¬†–ľ–Ķ—Ö–į–Ĺ–ł–∑–ľ–ĺ–≤ –∑–į–Ņ—É—Ā–ļ–į –į–Ņ–ĺ–Ņ—ā–ĺ–∑–į –ł¬†–ĺ–Ī—É—Ā–Ľ–ĺ–≤–Ľ–Ķ–Ĺ–ĺ –Ĺ–į—Ä–į—Ā—ā–į–Ĺ–ł–Ķ–ľ –ĺ–ļ–ł—Ā–Ľ–Ķ–Ĺ–ł—Ź —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ–ĺ–ľ –°¬†–ļ–į—Ä–ī–ł–ĺ–Ľ–ł–Ņ–ł–Ĺ–į¬†‚Äď –ĺ–ī–Ĺ–ĺ–≥–ĺ –ł–∑¬†–ļ–Ľ—é—á–Ķ–≤—č—Ö —Ą–ĺ—Ā—Ą–ĺ–Ľ–ł–Ņ–ł–ī–ĺ–≤ –≤–Ĺ—É—ā—Ä–Ķ–Ĺ–Ĺ–Ķ–Ļ –ľ–Ķ–ľ–Ī—Ä–į–Ĺ—č –ľ–ł—ā–ĺ—Ö–ĺ–Ĺ–ī—Ä–ł–ł [25]. Cytc –ł–≥—Ä–į–Ķ—ā –ļ–Ľ—é—á–Ķ–≤—É—é —Ä–ĺ–Ľ—Ć –≤¬†–Ņ—Ä–ĺ—Ü–Ķ—Ā—Ā–į—Ö –į–Ņ–ĺ–Ņ—ā–ĺ–∑–į. Cytc, –≤—č—Ā–≤–ĺ–Ī–ĺ–∂–ī–į—Ź—Ā—Ć –≤¬†—Ü–ł—ā–ĺ–∑–ĺ–Ľ—Ć –ļ–Ľ–Ķ—ā–ļ–ł, —Ā–≤—Ź–∑—č–≤–į–Ķ—ā—Ā—Ź —Ā¬†–Ī–Ķ–Ľ–ļ–ĺ–≤—č–ľ —Ą–į–ļ—ā–ĺ—Ä–ĺ–ľ, –į–ļ—ā–ł–≤–ł—Ä—É—é—Č–ł–ľ –į–Ņ–ĺ–Ņ—ā–ĺ–∑ (Apaf-1). –Ē–į–Ĺ–Ĺ—č–Ļ –ļ–ĺ–ľ–Ņ–Ľ–Ķ–ļ—Ā –≤¬†—Ā–≤–ĺ—é –ĺ—á–Ķ—Ä–Ķ–ī—Ć –ĺ–Ī—ä–Ķ–ī–ł–Ĺ—Ź–Ķ—ā—Ā—Ź —Ā¬†–ī–Ķ–ĺ–ļ—Ā–ł–į–ī–Ķ–Ĺ–ĺ–∑–ł–Ĺ—ā—Ä–ł—Ą–ĺ—Ā—Ą–į—ā–ĺ–ľ (dATF), —á—ā–ĺ –Ĺ–Ķ–ĺ–Ī—Ö–ĺ–ī–ł–ľ–ĺ –ī–Ľ—Ź —Ą–ĺ—Ä–ľ–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź –į–Ņ–ĺ–Ņ—ā–ĺ—Ā–ĺ–ľ—č [26]. –¶–ł—ā–ĺ—Ö—Ä–ĺ–ľ –°¬†—ā–į–ļ–∂–Ķ —É—á–į—Ā—ā–≤—É–Ķ—ā –≤¬†–Ņ—Ä–ĺ—Ü–Ķ—Ā—Ā–Ķ –į–ļ—ā–ł–≤–į—Ü–ł–ł –ļ–į—Ā–Ņ–į–∑. –í¬†–ĺ–ī–Ĺ–ĺ–ľ –ł–∑¬†–ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–Ļ –≤¬†–ļ–Ľ–Ķ—ā–ĺ—á–Ĺ—č—Ö –Ľ–ł–Ĺ–ł—Ź—Ö, –Ľ–ł—ą–Ķ–Ĺ–Ĺ—č—Ö¬†–≥–Ķ–Ĺ–į Cytc, –į–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć –ļ–į—Ā–Ņ–į–∑—č-3 –Ī—č–Ľ–į —Ā—É—Č–Ķ—Ā—ā–≤–Ķ–Ĺ–Ĺ–ĺ –Ĺ–ł–∂–Ķ –Ņ—Ä–ł —Ā—ā–ł–ľ—É–Ľ—Ź—Ü–ł–ł –ļ–Ľ–Ķ—ā–ĺ–ļ –į–ļ—ā–ł–≤–į—ā–ĺ—Ä–ĺ–ľ –į–Ņ–ĺ–Ņ—ā–ĺ–∑–į [27]. –í¬†–ī—Ä—É–≥–ł—Ö –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź—Ö Cytc –∑–į–Ņ—É—Ā–ļ–į–Ľ –į–Ņ–ĺ–Ņ—ā–ĺ–∑, —Ā–≤—Ź–∑—č–≤–į—Ź—Ā—Ć —Ā¬†—Ä–Ķ—Ü–Ķ–Ņ—ā–ĺ—Ä–ĺ–ľ IP3, —á—ā–ĺ —Ā–Ņ–ĺ—Ā–ĺ–Ī—Ā—ā–≤–ĺ–≤–į–Ľ–ĺ –Ĺ–į–ļ–ĺ–Ņ–Ľ–Ķ–Ĺ–ł—é –ļ–į–Ľ—Ć—Ü–ł—Ź –≤–Ĺ—É—ā—Ä–ł –ļ–Ľ–Ķ—ā–ļ–ł, –į–ļ—ā–ł–≤–į—Ü–ł–ł –ļ–į–Ľ–Ņ–į–ł–Ĺ–į (–ļ–į–Ľ—Ć—Ü–ł–Ļ-–į–ļ—ā–ł–≤–ł—Ä—É–Ķ–ľ–į—Ź —Ü–ł—Ā—ā–Ķ–ł–Ĺ–ĺ–≤–į—Ź –Ņ—Ä–ĺ—ā–Ķ–į–∑–į) –ł¬†–≤—č—Ā–≤–ĺ–Ī–ĺ–∂–ī–Ķ–Ĺ–ł—é –į–Ņ–ĺ–Ņ—ā–ĺ–∑-–ł–Ĺ–ī—É—Ü–ł—Ä—É—é—Č–Ķ–≥–ĺ —Ą–į–ļ—ā–ĺ—Ä–į (AIF) (–Ī–Ķ–Ľ–ĺ–ļ, –∑–į–Ņ—É—Ā–ļ–į—é—Č–ł–Ļ –Ņ–ĺ–≤—Ä–Ķ–∂–ī–Ķ–Ĺ–ł–Ķ —Ö—Ä–ĺ–ľ–į—ā–ł–Ĺ–į –ł¬†—Ą—Ä–į–≥–ľ–Ķ–Ĺ—ā–į—Ü–ł—é –Ē–Ě–ö) [8].
–í—č—Ā–≤–ĺ–Ī–ĺ–∂–ī–Ķ–Ĺ–ł–Ķ Cytc –ł–∑¬†–ľ–ł—ā–ĺ—Ö–ĺ–Ĺ–ī—Ä–ł–Ļ –Ĺ–ĺ—Ā–ł—ā –ĺ–Ī—Ä–į—ā–ł–ľ—č–Ļ —Ö–į—Ä–į–ļ—ā–Ķ—Ä. –í¬†–Ņ–į—ā–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ł—Ö —É—Ā–Ľ–ĺ–≤–ł—Ź—Ö, —Ö–į—Ä–į–ļ—ā–Ķ—Ä–ł–∑—É—é—Č–ł—Ö—Ā—Ź –ł–Ĺ—ā–Ķ–Ĺ—Ā–ł–≤–Ĺ—č–ľ –ĺ–Ī—Ä–į–∑–ĺ–≤–į–Ĺ–ł–Ķ–ľ –į–ļ—ā–ł–≤–Ĺ—č—Ö —Ą–ĺ—Ä–ľ –ļ–ł—Ā–Ľ–ĺ—Ä–ĺ–ī–į, –≤—č—Ö–ĺ–ī —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ–į –°¬†–≤ —Ü–ł—ā–ĺ–∑–ĺ–Ľ—Ć —É–≤–Ķ–Ľ–ł—á–ł–≤–į–Ķ—ā—Ā—Ź. –í¬†–ĺ–ī–Ĺ–ĺ–ľ –ł–∑¬†—ć–ļ—Ā–Ņ–Ķ—Ä–ł–ľ–Ķ–Ĺ—ā–į–Ľ—Ć–Ĺ—č—Ö –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–Ļ –Ī—č–Ľ–ł –Ņ–ĺ–Ľ—É—á–Ķ–Ĺ—č –ł–Ĺ—ā–Ķ—Ä–Ķ—Ā–Ĺ—č–Ķ –ī–į–Ĺ–Ĺ—č–Ķ. –í¬†—É—Ā–Ľ–ĺ–≤–ł—Ź—Ö —ć–ļ—Ā–Ņ–Ķ—Ä–ł–ľ–Ķ–Ĺ—ā–į–Ľ—Ć–Ĺ–ĺ–≥–ĺ —Ā–Ķ–Ņ—Ā–ł—Ā–į –≤–Ĺ—É—ā—Ä–ł–≤–Ķ–Ĺ–Ĺ–ĺ–Ķ –≤–≤–Ķ–ī–Ķ–Ĺ–ł–Ķ —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ–į¬†–°¬†—É–≤–Ķ–Ľ–ł—á–ł–≤–į–Ľ–ĺ –ĺ–Ī—Ä–į—ā–Ĺ—č–Ļ –∑–į—Ö–≤–į—ā –ľ–ł—ā–ĺ—Ö–ĺ–Ĺ–ī—Ä–ł—Ź–ľ–ł –ļ–į—Ä–ī–ł–ĺ–ľ–ł–ĺ—Ü–ł—ā–ĺ–≤ Cytc, —á—ā–ĺ —Ā–Ņ–ĺ—Ā–ĺ–Ī—Ā—ā–≤–ĺ–≤–į–Ľ–ĺ –Ĺ–ĺ—Ä–ľ–į–Ľ–ł–∑–į—Ü–ł–ł –ī—č—Ö–į—ā–Ķ–Ľ—Ć–Ĺ–ĺ–Ļ —Ą—É–Ĺ–ļ—Ü–ł–ł –ľ–ł—ā–ĺ—Ö–ĺ–Ĺ–ī—Ä–ł–Ļ –ł¬†–ī–ĺ—Ā—ā–ĺ–≤–Ķ—Ä–Ĺ–ĺ–ľ—É —É–≤–Ķ–Ľ–ł—á–Ķ–Ĺ–ł—é –ļ–ĺ–Ľ–ł—á–Ķ—Ā—ā–≤–į –≤—č–∂–ł–≤—ą–ł—Ö –∂–ł–≤–ĺ—ā–Ĺ—č—Ö –Ņ–嬆—Ā—Ä–į–≤–Ĺ–Ķ–Ĺ–ł—é —Ā¬†–∂–ł–≤–ĺ—ā–Ĺ—č–ľ–ł, –Ĺ–Ķ¬†–Ņ–ĺ–Ľ—É—á–į–≤—ą–ł–ľ–ł —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ¬†–°¬†[29]. –ě–Ī—Ä–į—ā–Ĺ—č–Ļ –∑–į—Ö–≤–į—ā, –≤–Ķ—Ä–ĺ—Ź—ā–Ĺ–ĺ, –ĺ–Ī—É—Ā–Ľ–ĺ–≤–Ľ–Ķ–Ĺ –Ĺ–į–Ľ–ł—á–ł–Ķ–ľ –≤¬†—Ā—ā—Ä—É–ļ—ā—É—Ä–Ķ Cytc —ć–Ņ–ł—ā–ĺ–Ņ–ĺ–≤ –Ņ–Ķ–Ņ—ā–ł–ī–į, –ĺ—ā–≤–Ķ—ā—Ā—ā–≤–Ķ–Ĺ–Ĺ–ĺ–≥–ĺ –∑–į –Ņ–Ķ–Ĺ–Ķ—ā—Ä–į—Ü–ł—é (CPP) [30]. –ü—Ä–ł –į–Ņ–ĺ–Ņ—ā–ĺ–∑–Ķ –Ņ—Ä–ĺ–ł—Ā—Ö–ĺ–ī–ł—ā –Ņ–Ķ—Ä–Ķ—Ä–į—Ā–Ņ—Ä–Ķ–ī–Ķ–Ľ–Ķ–Ĺ–ł–Ķ Cytc. –ě–ļ–ĺ–Ľ–ĺ 40% —Ā¬†–≤–Ĺ—É—ā—Ä–Ķ–Ĺ–Ĺ–Ķ–Ļ –ľ–Ķ–ľ–Ī—Ä–į–Ĺ—č –ľ–ł—ā–ĺ—Ö–ĺ–Ĺ–ī—Ä–ł–ł –Ņ–Ķ—Ä–Ķ–ľ–Ķ—Č–į–Ķ—ā—Ā—Ź –Ĺ–į¬†–Ĺ–į—Ä—É–∂–Ĺ—É—é –ľ–Ķ–ľ–Ī—Ä–į–Ĺ—É,¬†–≥–ī–Ķ –ĺ–Ī—Ä–į–∑—É–Ķ—ā—Ā—Ź –ļ–ĺ–ľ–Ņ–Ľ–Ķ–ļ—Ā ¬ę—Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ –°¬†‚Äď –ļ–į—Ä–ī–ł–ĺ–Ľ–ł–Ņ–ł–Ĺ-–Ņ–Ķ—Ä–ĺ–ļ—Ā–ł–ī–į–∑–į¬Ľ –ł¬†–∑–į—ā–Ķ–ľ –Ņ—Ä–ĺ–ł—Ā—Ö–ĺ–ī–ł—ā –ĺ–ļ–ł—Ā–Ľ–Ķ–Ĺ–ł–Ķ –ļ–į—Ä–ī–ł–ĺ–Ľ–ł–Ņ–ł–Ĺ–į. –ü—Ä–ł —ć—ā–ĺ–ľ –≤¬†–ľ–Ķ–ľ–Ī—Ä–į–Ĺ–Ķ –ľ–ł—ā–ĺ—Ö–ĺ–Ĺ–ī—Ä–ł–ł —Ą–ĺ—Ä–ľ–ł—Ä—É—é—ā—Ā—Ź –Ņ–ĺ—Ä—č, —á–Ķ—Ä–Ķ–∑ –ļ–ĺ—ā–ĺ—Ä—č–Ķ —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ –°¬†–ł –ī—Ä—É–≥–ł–Ķ —É—á–į—Ā—ā–Ĺ–ł–ļ–ł —Ä–Ķ–į–ļ—Ü–ł–Ļ –į–Ņ–ĺ–Ņ—ā–ĺ–∑–į –≤—č—Ö–ĺ–ī—Ź—ā –≤¬†—Ü–ł—ā–ĺ–∑–ĺ–Ľ—Ć –ļ–Ľ–Ķ—ā–ļ–ł [31]. –ö–į–ļ —É–∂–Ķ –ĺ—ā–ľ–Ķ—á–į–Ľ–ĺ—Ā—Ć, –ļ–ĺ–ľ–Ņ–Ľ–Ķ–ļ—Ā ¬ęCytc¬†‚Äď Apaf-1¬Ľ —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź –ļ–Ľ—é—á–Ķ–≤—č–ľ –≤¬†–ļ–į—Ā–ļ–į–ī–Ķ –į–Ņ–ĺ–Ņ—ā–ĺ–∑–į. –ě–Ī—Ä–į–∑–ĺ–≤–į–Ĺ–ł–Ķ –ī–į–Ĺ–Ĺ–ĺ–≥–ĺ –ļ–ĺ–ľ–Ņ–Ľ–Ķ–ļ—Ā–į —Ä–Ķ–≥—É–Ľ–ł—Ä—É–Ķ—ā—Ā—Ź —á–Ķ—Ä–Ķ–∑ —Ä–Ķ–į–ļ—Ü–ł—é —Ą–ĺ—Ā—Ą–ĺ—Ä–ł–Ľ–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ–į –°.¬†–§–ĺ—Ā—Ą–ĺ—Ä–ł–Ľ–ł—Ä–ĺ–≤–į–Ĺ–ł–Ķ –į–ľ–ł–Ĺ–ĺ–ļ–ł—Ā–Ľ–ĺ—ā—č Tyr97 —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ–į¬†–°¬†–Ņ—Ä–ł–≤–ĺ–ī–ł—ā –ļ¬†–ĺ–Ī—Ä–į–∑–ĺ–≤–į–Ĺ–ł—é –ł–ĺ–Ĺ–Ĺ–ĺ–≥–ĺ –ľ–ĺ—Ā—ā–ł–ļ–į —Ā¬†—Ā–ĺ—Ā–Ķ–ī–Ĺ–Ķ–Ļ –į–ľ–ł–Ĺ–ĺ–ļ–ł—Ā–Ľ–ĺ—ā–ĺ–Ļ Lys7 –ł¬†–Ī–Ľ–ĺ–ļ–ł—Ä–ĺ–≤–į–Ĺ–ł—é –Ņ—Ä–ĺ—Ü–Ķ—Ā—Ā–į —Ą–ĺ—Ä–ľ–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź –į–Ņ–ĺ–Ņ—ā–ĺ—Ā–ĺ–ľ—č. –Ē–Ľ—Ź —Ą–ĺ—Ä–ľ–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź –į–Ņ–ĺ–Ņ—ā–ĺ—Ā–ĺ–ľ—č –Ĺ–Ķ–ĺ–Ī—Ö–ĺ–ī–ł–ľ —Ä—Ź–ī –į–ľ–ł–Ĺ–ĺ–ļ–ł—Ā–Ľ–ĺ—ā. Lys7 –ĺ–ī–Ĺ–į –ł–∑¬†–Ĺ–ł—Ö [32]. –Ę–į–ļ–ł–ľ –ĺ–Ī—Ä–į–∑–ĺ–ľ, –≤¬†–Ņ–į—ā–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ł—Ö —É—Ā–Ľ–ĺ–≤–ł—Ź—Ö, –≤–ļ–Ľ—é—á–į—Ź —Ü–Ķ—Ä–Ķ–Ī—Ä–į–Ľ—Ć–Ĺ—É—é –ł—ą–Ķ–ľ–ł—é, Cytc –ł–≥—Ä–į–Ķ—ā –ļ–Ľ—é—á–Ķ–≤—É—é —Ä–ĺ–Ľ—Ć –≤¬†–ł–Ĺ–ī—É–ļ—Ü–ł–ł –į–Ņ–ĺ–Ņ—ā–ĺ–∑–į. –ě–ī–Ĺ–į–ļ–ĺ –ł–ľ–Ķ—é—ā—Ā—Ź —ć–ļ—Ā–Ņ–Ķ—Ä–ł–ľ–Ķ–Ĺ—ā–į–Ľ—Ć–Ĺ—č–Ķ –ī–į–Ĺ–Ĺ—č–Ķ, —Ā–≤–ł–ī–Ķ—ā–Ķ–Ľ—Ć—Ā—ā–≤—É—é—Č–ł–Ķ –嬆—ā–ĺ–ľ, —á—ā–ĺ Cytc, –≤—č—Ā–≤–ĺ–Ī–ĺ–∂–ī–į—Ź—Ā—Ć –≤¬†—Ü–ł—ā–ĺ–∑–ĺ–Ľ—Ć –ļ–Ľ–Ķ—ā–ļ–ł, —Ā–Ņ–ĺ—Ā–ĺ–Ī–Ķ–Ĺ –ł–Ĺ–ł—Ü–ł–ł—Ä–ĺ–≤–į—ā—Ć –į–Ĺ—ā–ł–į–Ņ–ĺ–Ņ—ā–ĺ—ā–ł—á–Ķ—Ā–ļ–ł–Ļ –Ņ—É—ā—Ć, —Ā–≤—Ź–∑—č–≤–į—Ź—Ā—Ć —Ā¬†—Ü–ł—ā–ĺ–Ņ–Ľ–į–∑–ľ–į—ā–ł—á–Ķ—Ā–ļ–ł–ľ –Ī–Ķ–Ľ–ļ–ĺ–ľ —ā–Ķ–Ņ–Ľ–ĺ–≤–ĺ–≥–ĺ —ą–ĺ–ļ–į 27 [33]. –ö—Ä–ĺ–ľ–Ķ —ā–ĺ–≥–ĺ, –į–Ĺ—ā–ł–ĺ–ļ–ł—Ā–Ľ–ł—ā–Ķ–Ľ—Ć–Ĺ—č–Ļ –Ņ–ĺ—ā–Ķ–Ĺ—Ü–ł–į–Ľ Cytc –∑–į —Ā—á–Ķ—ā –ĺ–ļ–ł—Ā–Ľ–Ķ–Ĺ–ł—Ź —Ā—É–Ņ–Ķ—Ä–ĺ–ļ—Ā–ł–ī–Ĺ–ĺ–≥–ĺ —Ä–į–ī–ł–ļ–į–Ľ–į –ĺ–Ī—É—Ā–Ľ–ĺ–≤–Ľ–ł–≤–į–Ķ—ā –Ņ–ĺ–ī–į–≤–Ľ–Ķ–Ĺ–ł–Ķ –Ņ—Ä–ĺ—Ü–Ķ—Ā—Ā–ĺ–≤ –į–Ņ–ĺ–Ņ—ā–ĺ–∑–į [34]. –í¬†—ā–ĺ –∂–Ķ –≤—Ä–Ķ–ľ—Ź —Ä–į—Ā—Ā–ľ–į—ā—Ä–ł–≤–į—ā—Ć –į–Ņ–ĺ–Ņ—ā–ĺ–∑ –ļ–į–ļ –ĺ–ī–Ĺ–ĺ–∑–Ĺ–į—á–Ĺ–ĺ –Ņ–į—ā–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ł–Ļ –Ņ—Ä–ĺ—Ü–Ķ—Ā—Ā –Ĺ–Ķ–≤–Ķ—Ä–Ĺ–ĺ, —ā–į–ļ –ļ–į–ļ –Ņ–ĺ—Ā—Ä–Ķ–ī—Ā—ā–≤–ĺ–ľ –ł–Ĺ–ī—É–ļ—Ü–ł–ł –ł¬†–ľ–ĺ–ī—É–Ľ—Ź—Ü–ł–ł –į–Ņ–ĺ–Ņ—ā–ĺ–∑–į —á–Ķ—Ä–Ķ–∑ –∑–į–Ņ—É—Ā–ļ —Ä–į–∑–Ľ–ł—á–Ĺ—č—Ö –≤–Ĺ—É—ā—Ä–ł–ļ–Ľ–Ķ—ā–ĺ—á–Ĺ—č—Ö —Ā–ł–≥–Ĺ–į–Ľ—Ć–Ĺ—č—Ö –Ņ—É—ā–Ķ–Ļ —Ä–Ķ–≥—É–Ľ–ł—Ä—É—é—ā—Ā—Ź –Ņ—Ä–ĺ—Ü–Ķ—Ā—Ā—č –≤—č–∂–ł–≤–į–Ĺ–ł—Ź –ł¬†–ĺ–Ī–Ĺ–ĺ–≤–Ľ–Ķ–Ĺ–ł—Ź –ļ–Ľ–Ķ—ā–ĺ–ļ, –≤–ļ–Ľ—é—á–į—Ź –Ņ—Ä–ĺ—Ü–Ķ—Ā—Ā—č –Ĺ–Ķ–Ļ—Ä–ĺ–Ņ–Ľ–į—Ā—ā–ł—á–Ĺ–ĺ—Ā—ā–ł [35].
–†–ĺ–Ľ—Ć —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ–į –°¬†–≤ —Ä–į–∑–≤–ł—ā–ł–ł —Ä—Ź–ī–į –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł–Ļ
–†–Ķ–į–ļ—Ü–ł–ł –į–Ņ–ĺ–Ņ—ā–ĺ–∑–į –ł–≥—Ä–į—é—ā –ļ–Ľ—é—á–Ķ–≤—É—é —Ä–ĺ–Ľ—Ć –≤¬†–Ņ–į—ā–ĺ–≥–Ķ–Ĺ–Ķ–∑–Ķ –ľ–Ĺ–ĺ–≥–ł—Ö –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł–Ļ. –ú–ł—ā–ĺ—Ö–ĺ–Ĺ–ī—Ä–ł–į–Ľ—Ć–Ĺ–į—Ź –ī–ł—Ā—Ą—É–Ĺ–ļ—Ü–ł—Ź, –≤—č—Ā–≤–ĺ–Ī–ĺ–∂–ī–Ķ–Ĺ–ł–Ķ —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ–į –°¬†–≤ —Ü–ł—ā–ĺ–Ņ–Ľ–į–∑–ľ—É –ļ–Ľ–Ķ—ā–ļ–ł –ł¬†–Ņ–ĺ—Ā–Ľ–Ķ–ī—É—é—Č–ł–Ļ –į–Ņ–ĺ–Ņ—ā–ĺ–∑ –ĺ–Ņ–ł—Ā–į–Ĺ—č –Ņ—Ä–ł –ł–Ĺ—Ā—É–Ľ—Ć—ā–Ķ, —ā—Ä–į–≤–ľ–Ķ —Ü–Ķ–Ĺ—ā—Ä–į–Ľ—Ć–Ĺ–ĺ–Ļ –Ĺ–Ķ—Ä–≤–Ĺ–ĺ–Ļ —Ā–ł—Ā—ā–Ķ–ľ—č, –Ī–ĺ–ļ–ĺ–≤–ĺ–ľ –į–ľ–ł–ĺ—ā—Ä–ĺ—Ą–ł—á–Ķ—Ā–ļ–ĺ–ľ —Ā–ļ–Ľ–Ķ—Ä–ĺ–∑–Ķ, —Ö–ĺ—Ä–Ķ–Ķ –ď–Ķ–Ĺ—ā–ł–Ĺ–≥—ā–ĺ–Ĺ–į, –Ī–ĺ–Ľ–Ķ–∑–Ĺ–ł –ü–į—Ä–ļ–ł–Ĺ—Ā–ĺ–Ĺ–į –ł¬†–Ī–ĺ–Ľ–Ķ–∑–Ĺ–ł –ź–Ľ—Ć—Ü–≥–Ķ–Ļ–ľ–Ķ—Ä–į [36]. –ě–Ņ—É–Ī–Ľ–ł–ļ–ĺ–≤–į–Ĺ–ĺ —ā–į–ļ–∂–Ķ –Ī–ĺ–Ľ—Ć—ą–ĺ–Ķ –ļ–ĺ–Ľ–ł—á–Ķ—Ā—ā–≤–ĺ —ć–ļ—Ā–Ņ–Ķ—Ä–ł–ľ–Ķ–Ĺ—ā–į–Ľ—Ć–Ĺ—č—Ö –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–Ļ —Ä–ĺ–Ľ–ł —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ–į –°¬†–Ņ—Ä–ł —Ä—Ź–ī–Ķ –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł–Ļ. –ē—Č–Ķ –≤¬†1999¬†–≥.¬†M.A.¬†P√©rez-Pinz√≥n –ł¬†—Ā–ĺ–į–≤—ā. –≤¬†—É—Ā–Ľ–ĺ–≤–ł—Ź—Ö —ć–ļ—Ā–Ņ–Ķ—Ä–ł–ľ–Ķ–Ĺ—ā–į–Ľ—Ć–Ĺ–ĺ–Ļ —Ü–Ķ—Ä–Ķ–Ī—Ä–į–Ľ—Ć–Ĺ–ĺ–Ļ –ł—ą–Ķ–ľ–ł–ł –≤¬†–Ņ–ĺ–≤—Ä–Ķ–∂–ī–Ķ–Ĺ–Ĺ–ĺ–Ļ —ā–ļ–į–Ĺ–ł –ĺ—ā–ľ–Ķ—á–į–Ľ–ł –ł–∑–Ī—č—ā–ĺ—á–Ĺ—č–Ļ —É—Ä–ĺ–≤–Ķ–Ĺ—Ć —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ–į –°.¬†–ö—Ä–ĺ–ľ–Ķ —ā–ĺ–≥–ĺ, –Ņ—Ä–ĺ–≤–Ķ–ī–Ķ–Ĺ–ł–Ķ —Ä–Ķ–Ņ–Ķ—Ä—Ą—É–∑–ł–ł –į—Ā—Ā–ĺ—Ü–ł–ł—Ä–ĺ–≤–į–Ľ–ĺ—Ā—Ć —Ā¬†—É–≤–Ķ–Ľ–ł—á–Ķ–Ĺ–ł–Ķ–ľ —É—Ä–ĺ–≤–Ĺ—Ź —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ–į –°¬†[37].
H. Hu –ł¬†—Ā–ĺ–į–≤—ā. (2020) –≤¬†—É—Ā–Ľ–ĺ–≤–ł—Ź—Ö —ć–ļ—Ā–Ņ–Ķ—Ä–ł–ľ–Ķ–Ĺ—ā–į–Ľ—Ć–Ĺ–ĺ–Ļ –ł—ą–Ķ–ľ–ł–ł –ł–∑—É—á–į–Ľ–ł –ľ–Ķ—Ö–į–Ĺ–ł–∑–ľ—č, –Ľ–Ķ–∂–į—Č–ł–Ķ –≤¬†–ĺ—Ā–Ĺ–ĺ–≤–Ķ –Ņ–ĺ–≤—č—ą–Ķ–Ĺ–ł—Ź –Ņ—Ä–ĺ–Ĺ–ł—Ü–į–Ķ–ľ–ĺ—Ā—ā–ł¬†–≥–Ķ–ľ–į—ā–ĺ—ć–Ĺ—Ü–Ķ—Ą–į–Ľ–ł—á–Ķ—Ā–ļ–ĺ–≥–ĺ –Ī–į—Ä—Ć–Ķ—Ä–į (–ď–≠–Ď). –ź–≤—ā–ĺ—Ä—č –Ņ–ĺ–ļ–į–∑–į–Ľ–ł, —á—ā–ĺ —É—Ä–ĺ–≤–Ķ–Ĺ—Ć —Ä–Ķ–≥—É–Ľ—Ź—ā–ĺ—Ä–Ĺ–ĺ–Ļ –ľ–ł–ļ—Ä–ĺ–†–Ě–ö miR-34a –≤¬†–ł—ą–Ķ–ľ–ł–∑–ł—Ä–ĺ–≤–į–Ĺ–Ĺ–ĺ–Ļ —ā–ļ–į–Ĺ–ł –Ĺ–į–Ņ—Ä—Ź–ľ—É—é —Ā–≤—Ź–∑–į–Ĺ —Ā¬†–Ņ–ĺ–≤—č—ą–Ķ–Ĺ–ł–Ķ–ľ –Ņ—Ä–ĺ–Ĺ–ł—Ü–į–Ķ–ľ–ĺ—Ā—ā–ł –ď–≠–Ď. –ö—Ä–ĺ–ľ–Ķ —ā–ĺ–≥–ĺ, —É—Ä–ĺ–≤–Ķ–Ĺ—Ć —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ–į –°¬†–Ī—č–Ľ –Ņ–ĺ–≤—č—ą–Ķ–Ĺ –≤¬†–ĺ–Ī–Ľ–į—Ā—ā–ł —Ü–Ķ—Ä–Ķ–Ī—Ä–į–Ľ—Ć–Ĺ–ĺ–Ļ –ł—ą–Ķ–ľ–ł–ł –ł¬†–Ņ—Ä—Ź–ľ–ĺ –ļ–ĺ—Ä—Ä–Ķ–Ľ–ł—Ä–ĺ–≤–į–Ľ —Ā¬†—É—Ä–ĺ–≤–Ĺ–Ķ–ľ miR-34a. –£¬†—ā—Ä–į–Ĺ—Ā–≥–Ķ–Ĺ–Ĺ—č—Ö –∂–ł–≤–ĺ—ā–Ĺ—č—Ö —Ā¬†–≤—č–ļ–Ľ—é—á–Ķ–Ĺ–Ĺ—č–ľ¬†–≥–Ķ–Ĺ–ĺ–ľ miR-34a —É—Ä–ĺ–≤–Ķ–Ĺ—Ć —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ–į –°¬†–Ī—č–Ľ –≤¬†—Ä–į–∑—č –Ĺ–ł–∂–Ķ –ł¬†–∑–ĺ–Ĺ–į –ł—ą–Ķ–ľ–ł–ł —Ā—É—Č–Ķ—Ā—ā–≤–Ķ–Ĺ–Ĺ–ĺ –ľ–Ķ–Ĺ—Ć—ą–Ķ [38]. –Ę–į–ļ–ł–ľ –ĺ–Ī—Ä–į–∑–ĺ–ľ, –Ņ–ĺ—ā–Ķ—Ä—Ź —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ–į –°¬†–ľ–ł—ā–ĺ—Ö–ĺ–Ĺ–ī—Ä–ł—Ź–ľ–ł –Ņ—Ä–ł–≤–ĺ–ī–ł—ā –ļ¬†–Ĺ–į—Ä—É—ą–Ķ–Ĺ–ł—é —Ą—É–Ĺ–ļ—Ü–ł–ĺ–Ĺ–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź –ī—č—Ö–į—ā–Ķ–Ľ—Ć–Ĺ–ĺ–Ļ —Ü–Ķ–Ņ–ł, —Ā¬†–ĺ–ī–Ĺ–ĺ–Ļ —Ā—ā–ĺ—Ä–ĺ–Ĺ—č, –ł¬†—É–ľ–Ķ–Ĺ—Ć—ą–į–Ķ—ā –į–Ĺ—ā–ł–ĺ–ļ–ł—Ā–Ľ–ł—ā–Ķ–Ľ—Ć–Ĺ—č–Ķ –≤–ĺ–∑–ľ–ĺ–∂–Ĺ–ĺ—Ā—ā–ł —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ –°-–ĺ–ļ—Ā–ł–ī–į–∑—謆‚Äď –≤–į–∂–Ĺ–ĺ–≥–ĺ —Ä–Ķ–≥—É–Ľ—Ź—ā–ĺ—Ä–į —É—Ä–ĺ–≤–Ĺ—Ź —Ā–≤–ĺ–Ī–ĺ–ī–Ĺ—č—Ö —Ä–į–ī–ł–ļ–į–Ľ–ĺ–≤¬†‚Äď —Ā¬†–ī—Ä—É–≥–ĺ–Ļ [39].
G. Yang –ł¬†—Ā–ĺ–į–≤—ā. (2009) –ĺ–Ī–Ĺ–į—Ä—É–∂–ł–Ľ–ł, —á—ā–ĺ –Ņ–ĺ—ā–Ķ—Ä—Ź –ľ–ł—ā–ĺ—Ö–ĺ–Ĺ–ī—Ä–ł—Ź–ľ–ł —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ–į –°¬†–Ņ—Ä–ł —Ä–į–ī–ł–į—Ü–ł–ĺ–Ĺ–Ĺ–ĺ–ľ –Ņ–ĺ–≤—Ä–Ķ–∂–ī–Ķ–Ĺ–ł–ł –Ņ—Ä–ł–≤–ĺ–ī–ł—ā –ļ¬†–ł—Ö –≤—č—Ä–į–∂–Ķ–Ĺ–Ĺ–ĺ–Ļ –ī–ł—Ā—Ą—É–Ĺ–ļ—Ü–ł–ł [40], —á—ā–ĺ –ľ–ĺ–∂–Ķ—ā –Ņ–ĺ–ī—ā–≤–Ķ—Ä–∂–ī–į—ā—Ć –Ĺ–Ķ–ĺ–Ī—Ö–ĺ–ī–ł–ľ–ĺ—Ā—ā—Ć –ł—Ā–Ņ–ĺ–Ľ—Ć–∑–ĺ–≤–į–Ĺ–ł—Ź Cytc –≤¬†–Ľ–Ķ—á–Ķ–Ī–Ĺ—č—Ö —Ü–Ķ–Ľ—Ź—Ö.
P.¬†Pasdois –ł¬†—Ā–ĺ–į–≤—ā. (2011) —É—Ā—ā–į–Ĺ–ĺ–≤–ł–Ľ–ł, —á—ā–ĺ –Ņ—Ä–ł –ł—ą–Ķ–ľ–ł–ł –ľ–ł–ĺ–ļ–į—Ä–ī–į –∑–Ĺ–į—á–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ —Ā–Ĺ–ł–∂–į–Ķ—ā—Ā—Ź —É—Ä–ĺ–≤–Ķ–Ĺ—Ć Cytc –≤¬†–ľ–ł—ā–ĺ—Ö–ĺ–Ĺ–ī—Ä–ł—Ź—Ö, –≤¬†—Ä–Ķ–∑—É–Ľ—Ć—ā–į—ā–Ķ —á–Ķ–≥–ĺ —Ā—É—Č–Ķ—Ā—ā–≤–Ķ–Ĺ–Ĺ–ĺ —É–≤–Ķ–Ľ–ł—á–ł–≤–į–Ķ—ā—Ā—Ź –ļ–ĺ–Ľ–ł—á–Ķ—Ā—ā–≤–ĺ —Ā–≤–ĺ–Ī–ĺ–ī–Ĺ—č—Ö —Ä–į–ī–ł–ļ–į–Ľ–ĺ–≤ [41].
–Ę–į–ļ–ł–ľ –ĺ–Ī—Ä–į–∑–ĺ–ľ, –ľ–ĺ–∂–Ĺ–ĺ –Ņ—Ä–Ķ–ī–Ņ–ĺ–Ľ–ĺ–∂–ł—ā—Ć, —á—ā–ĺ –≤¬†—É—Ā–Ľ–ĺ–≤–ł—Ź—Ö –ł—ą–Ķ–ľ–ł—á–Ķ—Ā–ļ–ĺ–≥–ĺ –Ņ–ĺ–≤—Ä–Ķ–∂–ī–Ķ–Ĺ–ł—Ź –ľ–ĺ–∑–≥–į —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ –°¬†—É—á–į—Ā—ā–≤—É–Ķ—ā –≤–ĺ –ľ–Ĺ–ĺ–∂–Ķ—Ā—ā–≤–Ķ —Ä–į–∑–Ĺ–ĺ–Ĺ–į–Ņ—Ä–į–≤–Ľ–Ķ–Ĺ–Ĺ—č—Ö –Ņ–į—ā–ĺ–Ī–ł–ĺ—Ö–ł–ľ–ł—á–Ķ—Ā–ļ–ł—Ö —Ä–Ķ–į–ļ—Ü–ł–Ļ, –∑–į–Ņ—É—Ā–ļ–į—Ź –į–Ņ–ĺ–Ņ—ā–ĺ–∑ –≤¬†—Ü–ł—ā–ĺ–∑–ĺ–Ľ–Ķ –ļ–Ľ–Ķ—ā–ļ–ł. –í¬†—ā–ĺ –∂–Ķ –≤—Ä–Ķ–ľ—Ź –Ņ–ĺ—ā–Ķ—Ä—Ź —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ–į –°¬†–ľ–ł—ā–ĺ—Ö–ĺ–Ĺ–ī—Ä–ł—Ź–ľ–ł –Ņ—Ä–ł–≤–ĺ–ī–ł—ā –ļ¬†—Ā–Ĺ–ł–∂–Ķ–Ĺ–ł—é –į–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā–ł –Ņ—Ä–ĺ—Ü–Ķ—Ā—Ā–ĺ–≤ –ļ–Ľ–Ķ—ā–ĺ—á–Ĺ–ĺ–≥–ĺ –ī—č—Ö–į–Ĺ–ł—Ź –ł¬†–Ĺ–į—Ä–į—Ā—ā–į–Ĺ–ł—é –≤–Ĺ—É—ā—Ä–ł–ľ–ł—ā–ĺ—Ö–ĺ–Ĺ–ī—Ä–ł–į–Ľ—Ć–Ĺ–ĺ–≥–ĺ –ĺ–ļ–ł—Ā–Ľ–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ–≥–ĺ —Ā—ā—Ä–Ķ—Ā—Ā–į.
–í¬†–Ņ–ĺ—Ā–Ľ–Ķ–ī–Ĺ–ł–Ķ¬†–≥–ĺ–ī—č –Ņ–ĺ—Ź–≤–ł–Ľ–ł—Ā—Ć –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź —Ä–ĺ–Ľ–ł —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ–į –°¬†–ł –ī—Ä—É–≥–ł—Ö —É—á–į—Ā—ā–Ĺ–ł–ļ–ĺ–≤ —Ü–Ķ–Ņ–ł –Ņ–Ķ—Ä–Ķ–Ĺ–ĺ—Ā–į —ć–Ľ–Ķ–ļ—ā—Ä–ĺ–Ĺ–ĺ–≤ –≤¬†—Ä–į–∑–≤–ł—ā–ł–ł —Ö—Ä–ĺ–Ĺ–ł—á–Ķ—Ā–ļ–ł—Ö —Ü–Ķ—Ä–Ķ–Ī—Ä–ĺ–≤–į—Ā–ļ—É–Ľ—Ź—Ä–Ĺ—č—Ö –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł–Ļ. –í¬†—ć–ļ—Ā–Ņ–Ķ—Ä–ł–ľ–Ķ–Ĺ—ā–į–Ľ—Ć–Ĺ–ĺ–ľ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–ł D.S.¬†Martin –ł¬†—Ā–ĺ–į–≤—ā. (2002) –ĺ—Ü–Ķ–Ĺ–ł–≤–į–Ľ–į—Ā—Ć —Ā–≤—Ź–∑—Ć —Ā—ā–į—Ä–Ķ–Ĺ–ł—Ź –ľ–ĺ–∑–≥–į —Ā¬†—É—Ä–ĺ–≤–Ĺ–Ķ–ľ —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ–į –°.¬†–ź–≤—ā–ĺ—Ä—č –Ņ–ĺ–ļ–į–∑–į–Ľ–ł, —á—ā–ĺ —É¬†—Ā—ā–į—Ä—č—Ö –Ľ–į–Ī–ĺ—Ä–į—ā–ĺ—Ä–Ĺ—č—Ö –∂–ł–≤–ĺ—ā–Ĺ—č—Ö –≤¬†—ā–ļ–į–Ĺ–ł –ľ–ĺ–∑–≥–į –≤–ĺ–∑—Ä–į—Ā—ā–į–Ķ—ā –ļ–ĺ–Ĺ—Ü–Ķ–Ĺ—ā—Ä–į—Ü–ł—Ź –≤–į–∂–Ĺ–ĺ–≥–ĺ –Ņ—Ä–ĺ–≤–ĺ—Ā–Ņ–į–Ľ–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ–≥–ĺ —Ü–ł—ā–ĺ–ļ–ł–Ĺ–į¬†‚Äď –ł–Ĺ—ā–Ķ—Ä–Ľ–Ķ–Ļ–ļ–ł–Ĺ–į (–ė–õ) 1-–Ī–Ķ—ā–į, –į–ļ—ā–ł–≤–ł—Ä—É–Ķ—ā—Ā—Ź –Ņ—Ä–ĺ—ā–Ķ–ł–Ĺ —Ä38, —Ź–≤–Ľ—Ź—é—Č–ł–Ļ—Ā—Ź –ĺ–ī–Ĺ–ĺ–Ļ –ł–∑¬†–ľ–ł—ā–ĺ–≥–Ķ–Ĺ-–į–ļ—ā–ł–≤–ł—Ä—É—é—Č–ł—Ö –ļ–ł–Ĺ–į–∑ (–ú–ź–†). –ü—Ä–ł —ć—ā–ĺ–ľ —É–≤–Ķ–Ľ–ł—á–ł–≤–į–Ķ—ā—Ā—Ź –≤—č—Ö–ĺ–ī —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ–į –°¬†–ł–∑ –ľ–ł—ā–ĺ—Ö–ĺ–Ĺ–ī—Ä–ł–Ļ. –õ–Ķ—á–Ķ–Ĺ–ł–Ķ —Ā–Ņ–Ķ—Ü–ł—Ą–ł—á–Ķ—Ā–ļ–ł–ľ –Ī–Ľ–ĺ–ļ–į—ā–ĺ—Ä–ĺ–ľ —Ä38 (SB203580) —Ā–Ņ–ĺ—Ā–ĺ–Ī—Ā—ā–≤—É–Ķ—ā –Ĺ–ĺ—Ä–ľ–į–Ľ–ł–∑–į—Ü–ł–ł —É—Ä–ĺ–≤–Ĺ—Ź —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ–į –≤–Ĺ—É—ā—Ä–ł –ľ–ł—ā–ĺ—Ö–ĺ–Ĺ–ī—Ä–ł–Ļ [42].
–í¬†—ć–ļ—Ā–Ņ–Ķ—Ä–ł–ľ–Ķ–Ĺ—ā–į–Ľ—Ć–Ĺ–ĺ–Ļ —Ä–į–Ī–ĺ—ā–Ķ A. Auchter –ł¬†—Ā–ĺ–į–≤—ā. (2020) –ľ–ĺ–ī–Ķ–Ľ–ł—Ä–ĺ–≤–į–Ľ–ł –Ľ–į–Ī–ĺ—Ä–į—ā–ĺ—Ä–Ĺ—č–ľ –∂–ł–≤–ĺ—ā–Ĺ—č–ľ —É—Ā–Ľ–ĺ–≤–ł—Ź —Ö—Ä–ĺ–Ĺ–ł—á–Ķ—Ā–ļ–ĺ–Ļ —Ü–Ķ—Ä–Ķ–Ī—Ä–į–Ľ—Ć–Ĺ–ĺ–Ļ –ł—ą–Ķ–ľ–ł–ł¬†‚Äď —Ā¬†–ĺ–Ī–Ķ–ł—Ö —Ā—ā–ĺ—Ä–ĺ–Ĺ –Ņ–Ķ—Ä–Ķ–≤—Ź–∑—č–≤–į–Ľ–ł—Ā—Ć —Ā–ĺ–Ĺ–Ĺ—č–Ķ –į—Ä—ā–Ķ—Ä–ł–ł. –ü–ĺ–ī–ĺ–Ī–Ĺ–į—Ź –Ņ—Ä–ĺ—Ü–Ķ–ī—É—Ä–į –Ĺ–Ķ¬†–ļ—Ä–ł—ā–ł—á–Ĺ–į –ī–Ľ—Ź –ļ—Ä—č—Ā, –Ņ–ĺ—Ā–ļ–ĺ–Ľ—Ć–ļ—É —ɬ†–Ī–ĺ–Ľ—Ć—ą–ł–Ĺ—Ā—ā–≤–į –Ņ–ĺ—Ä–ĺ–ī –ĺ—ā–ľ–Ķ—á–į–Ķ—ā—Ā—Ź –∑–į–ľ–ļ–Ĺ—É—ā–ĺ—Ā—ā—Ć –≤–ł–Ľ–Ľ–ł–∑–ł–Ķ–≤–į –ļ—Ä—É–≥–į. –ź–≤—ā–ĺ—Ä—č –≤—č—Ź–≤–ł–Ľ–ł —Ā–Ĺ–ł–∂–Ķ–Ĺ–ł–Ķ –≤–ĺ –ľ–Ĺ–ĺ–≥–ł—Ö –ĺ—ā–ī–Ķ–Ľ–į—Ö –ľ–ĺ–∑–≥–ĺ–≤–ĺ–Ļ –ļ–ĺ—Ä—č —É—Ä–ĺ–≤–Ĺ—Ź —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ –°-–ĺ–ļ—Ā–ł–ī–į–∑—č. –Ě–į¬†–Ņ—Ä–ĺ—ā—Ź–∂–Ķ–Ĺ–ł–ł –ľ–Ķ—Ā—Ź—Ü–į –Ņ–ĺ—Ā–Ľ–Ķ –ĺ–Ņ–Ķ—Ä–į—Ü–ł–ł –∂–ł–≤–ĺ—ā–Ĺ—č–ľ –Ĺ–į¬†—Ą–ĺ–Ĺ–Ķ –≤–≤–Ķ–ī–Ķ–Ĺ–ł—Ź –ľ–Ķ—ā–ł–Ľ—ā–ł–ĺ–Ĺ–ł–Ĺ–ł—Ź —Ö–Ľ–ĺ—Ä–ł–ī–į –≤—č–Ņ–ĺ–Ľ–Ĺ—Ź–Ľ–ł –ļ–ĺ–≥–Ĺ–ł—ā–ł–≤–Ĺ–ĺ–Ķ —ā–Ķ—Ā—ā–ł—Ä–ĺ–≤–į–Ĺ–ł–Ķ. –ě–ī–Ĺ–ĺ–Ļ –ł–∑¬†—Ą–į—Ä–ľ–į–ļ–ĺ–ī–ł–Ĺ–į–ľ–ł—á–Ķ—Ā–ļ–ł—Ö –ĺ—Ā–ĺ–Ī–Ķ–Ĺ–Ĺ–ĺ—Ā—ā–Ķ–Ļ –ī–į–Ĺ–Ĺ–ĺ–≥–ĺ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź —Ā–Ņ–ĺ—Ā–ĺ–Ī–Ĺ–ĺ—Ā—ā—Ć —Ā—ā–ł–ľ—É–Ľ–ł—Ä–ĺ–≤–į—ā—Ć –į–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ –°-–ĺ–ļ—Ā–ł–ī–į–∑—č. –í¬†–ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–ł –Ī—č–Ľ–ĺ –≤—č—Ź–≤–Ľ–Ķ–Ĺ–ĺ, —á—ā–ĺ –∂–ł–≤–ĺ—ā–Ĺ—č–Ķ, –Ņ–ĺ–Ľ—É—á–į–≤—ą–ł–Ķ –ľ–Ķ—ā–ł–Ľ—ā–ł–ĺ–Ĺ–ł–Ĺ–ł—Ź —Ö–Ľ–ĺ—Ä–ł–ī, –ł–ľ–Ķ–Ľ–ł –ī–ĺ—Ā—ā–ĺ–≤–Ķ—Ä–Ĺ–ĺ –ľ–Ķ–Ĺ–Ķ–Ķ –∑–Ĺ–į—á–ł–ľ–ĺ–Ķ —Ā–Ĺ–ł–∂–Ķ–Ĺ–ł–Ķ –į–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā–ł —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ –°-–ĺ–ļ—Ā–ł–ī–į–∑—č –ł¬†–ľ–Ķ–Ĺ–Ķ–Ķ –∑–Ĺ–į—á–ł–ľ–ĺ–Ķ —Ā–Ĺ–ł–∂–Ķ–Ĺ–ł–Ķ –ļ–ĺ–≥–Ĺ–ł—ā–ł–≤–Ĺ—č—Ö —Ā–Ņ–ĺ—Ā–ĺ–Ī–Ĺ–ĺ—Ā—ā–Ķ–Ļ. –ė—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į—ā–Ķ–Ľ–ł –Ņ—Ä–Ķ–ī–Ņ–ĺ–Ľ–ĺ–∂–ł–Ľ–ł, —á—ā–ĺ –ĺ–ī–Ĺ–ł–ľ –ł–∑¬†–Ĺ–į–Ņ—Ä–į–≤–Ľ–Ķ–Ĺ–ł–Ļ –Ĺ–Ķ–Ļ—Ä–ĺ–Ņ—Ä–ĺ—ā–Ķ–ļ—Ü–ł–ł –ľ–ĺ–∂–Ķ—ā –Ī—č—ā—Ć –ł–ľ–Ķ–Ĺ–Ĺ–ĺ –ľ–Ķ–ī–ł–ļ–į–ľ–Ķ–Ĺ—ā–ĺ–∑–Ĺ–į—Ź –į–ļ—ā–ł–≤–į—Ü–ł—Ź –ļ–ĺ–ľ–Ņ–Ľ–Ķ–ļ—Ā–ĺ–≤, —É—á–į—Ā—ā–≤—É—é—Č–ł—Ö –≤¬†—Ü–Ķ–Ņ–ł –Ņ–Ķ—Ä–Ķ–Ĺ–ĺ—Ā–į —ć–Ľ–Ķ–ļ—ā—Ä–ĺ–Ĺ–ĺ–≤ [43].
–£—á–Ķ–Ĺ—č–Ķ –ĺ—Ü–Ķ–Ĺ–ł–≤–į–Ľ–ł —É—Ä–ĺ–≤–Ķ–Ĺ—Ć –ľ–į—Ä–ļ–Ķ—Ä–į –į–Ņ–ĺ–Ņ—ā–ĺ–∑–į¬†‚Äď –Ņ—Ä–ĺ—ā–Ķ–ł–Ĺ–į Bax –ł¬†—Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ–į –°¬†—É –Ľ–į–Ī–ĺ—Ä–į—ā–ĺ—Ä–Ĺ—č—Ö –∂–ł–≤–ĺ—ā–Ĺ—č—Ö —Ā¬†–ľ–ĺ–ī–Ķ–Ľ—Ć—é —ć–ļ—Ā–Ņ–Ķ—Ä–ł–ľ–Ķ–Ĺ—ā–į–Ľ—Ć–Ĺ–ĺ–Ļ —Ü–Ķ—Ä–Ķ–Ī—Ä–į–Ľ—Ć–Ĺ–ĺ–Ļ¬†–≥–ł–Ņ–ĺ–Ņ–Ķ—Ä—Ą—É–∑–ł–ł. –ź–≤—ā–ĺ—Ä—č –ĺ–Ī–Ĺ–į—Ä—É–∂–ł–Ľ–ł, —á—ā–ĺ —É—Ä–ĺ–≤–Ķ–Ĺ—Ć Bax –ł¬†—Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ–į –°¬†–Ī—č–Ľ —Ā—É—Č–Ķ—Ā—ā–≤–Ķ–Ĺ–Ĺ–ĺ –≤—č—ą–Ķ –≤¬†–ľ–ĺ–∑–≥–Ķ –∂–ł–≤–ĺ—ā–Ĺ—č—Ö —Ā¬†–ľ–ĺ–ī–Ķ–Ľ—Ć—é —Ö—Ä–ĺ–Ĺ–ł—á–Ķ—Ā–ļ–ĺ–Ļ –ł—ą–Ķ–ľ–ł–ł, –Ņ—Ä–ł—á–Ķ–ľ —ć–ļ—Ā–Ņ–Ķ—Ä–ł–ľ–Ķ–Ĺ—ā–į–Ľ—Ć–Ĺ–ĺ–Ķ –Ľ–Ķ—á–Ķ–Ĺ–ł–Ķ –∂–ł–≤–ĺ—ā–Ĺ—č—Ö —ć–ļ—Ā—ā—Ä–į–ļ—ā–ĺ–ľ –ļ–ĺ—Ä–Ĺ—Ź –Ņ–ł–ĺ–Ĺ–į (–Ī–ł–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ł –į–ļ—ā–ł–≤–Ĺ–į—Ź –ī–ĺ–Ī–į–≤–ļ–į, –ł—Ā–Ņ–ĺ–Ľ—Ć–∑—É–Ķ–ľ–į—Ź –≤¬†—ā—Ä–į–ī–ł—Ü–ł–ĺ–Ĺ–Ĺ–ĺ–Ļ –ļ–ł—ā–į–Ļ—Ā–ļ–ĺ–Ļ –ľ–Ķ–ī–ł—Ü–ł–Ĺ–Ķ) —Ā–Ņ–ĺ—Ā–ĺ–Ī—Ā—ā–≤–ĺ–≤–į–Ľ–ĺ –Ĺ–ĺ—Ä–ľ–į–Ľ–ł–∑–į—Ü–ł–ł —É—Ä–ĺ–≤–Ĺ—Ź –ł—Ā—Ā–Ľ–Ķ–ī—É–Ķ–ľ—č—Ö —Ā—É–Ī—Ā—ā–į–Ĺ—Ü–ł–Ļ [44].
–í¬†—Ä—Ź–ī–Ķ —Ä–į–Ī–ĺ—ā –Ņ–ĺ–ļ–į–∑–į–Ĺ–į —Ä–ĺ–Ľ—Ć —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ–į –°¬†–≤ —Ä–į–∑–≤–ł—ā–ł–ł –Ĺ–Ķ–ļ–ĺ—ā–ĺ—Ä—č—Ö –Ĺ–Ķ–Ļ—Ä–ĺ–ī–Ķ–≥–Ķ–Ĺ–Ķ—Ä–į—ā–ł–≤–Ĺ—č—Ö –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł–Ļ. –Ę–į–ļ, –≤¬†–ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–ł K. Ashutosh –ł¬†—Ā–ĺ–į–≤—ā. (2016) –Ĺ–į¬†–ľ–ĺ–ī–Ķ–Ľ–ł —ć–ļ—Ā–Ņ–Ķ—Ä–ł–ľ–Ķ–Ĺ—ā–į–Ľ—Ć–Ĺ–ĺ–Ļ –Ī–ĺ–Ľ–Ķ–∑–Ĺ–ł –ü–į—Ä–ļ–ł–Ĺ—Ā–ĺ–Ĺ–į –Ĺ–į–Ī–Ľ—é–ī–į–Ľ–ĺ—Ā—Ć —É–≤–Ķ–Ľ–ł—á–Ķ–Ĺ–ł–Ķ —É—Ä–ĺ–≤–Ĺ—Ź —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ–į –°¬†–≤ –Ņ–ĺ—Ä–į–∂–Ķ–Ĺ–Ĺ—č—Ö –Ĺ–Ķ–Ļ—Ä–ĺ–Ĺ–į—Ö —á–Ķ—Ä–Ĺ–ĺ–Ļ —Ā—É–Ī—Ā—ā–į–Ĺ—Ü–ł–ł. –ö—Ä–ĺ–ľ–Ķ —ā–ĺ–≥–ĺ, –≤¬†–Ĺ–ł—Ö –Ī—č–Ľ–ł –≤—č—Ź–≤–Ľ–Ķ–Ĺ—č –ļ–ĺ–ľ–Ņ–Ľ–Ķ–ļ—Ā—č, —Ā–ĺ—Ā—ā–ĺ—Ź—Č–ł–Ķ –ł–∑¬†—Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ–į –°¬†–ł –į–Ľ—Ć—Ą–į-—Ā–ł–Ĺ—É–ļ–Ľ–Ķ–ł–Ĺ–į. –í¬†—á–į—Ā—ā–Ĺ–ĺ—Ā—ā–ł, –≤¬†–Ņ—Ä–ł—Ā—É—ā—Ā—ā–≤–ł–ł —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ–į –°¬†–ĺ—ā–ľ–Ķ—á–į–Ľ–į—Ā—Ć –∑–Ĺ–į—á–ł–ľ–į—Ź –ĺ–Ľ–ł–≥–ĺ–ľ–Ķ—Ä–ł–∑–į—Ü–ł—Ź –į–Ľ—Ć—Ą–į-—Ā–ł–Ĺ—É–ļ–Ľ–Ķ–ł–Ĺ–į [45].
–ė–ľ–Ķ—é—ā—Ā—Ź –ī–į–Ĺ–Ĺ—č–Ķ –嬆—Ä–ĺ–Ľ–ł —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ–į –°¬†–≤ –Ņ—Ä–ĺ—Ü–Ķ—Ā—Ā–į—Ö –Ĺ–Ķ–Ļ—Ä–ĺ–≤–ĺ—Ā–Ņ–į–Ľ–Ķ–Ĺ–ł—Ź, –ł–≥—Ä–į—é—Č–ł—Ö –ļ–Ľ—é—á–Ķ–≤—É—é —Ä–ĺ–Ľ—Ć –≤¬†—Ä–į–∑–≤–ł—ā–ł–ł –Ĺ–Ķ–Ļ—Ä–ĺ–ī–Ķ–≥–Ķ–Ĺ–Ķ—Ä–į—Ü–ł–ł. A.K. Au –ł¬†—Ā–ĺ–į–≤—ā. (2012) –Ņ–ĺ–ļ–į–∑–į–Ľ–ł, —á—ā–ĺ –≤¬†–Ņ–į—ā–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ł—Ö —É—Ā–Ľ–ĺ–≤–ł—Ź—Ö —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ –°¬†–≤—č—Ā–≤–ĺ–Ī–ĺ–∂–ī–į–Ķ—ā—Ā—Ź –Ĺ–Ķ¬†—ā–ĺ–Ľ—Ć–ļ–ĺ –≤¬†—Ü–ł—ā–ĺ–∑–ĺ–Ľ—Ć –ļ–Ľ–Ķ—ā–ļ–ł,¬†–≥–ī–Ķ –ł–Ĺ–ł—Ü–ł–ł—Ä—É–Ķ—ā —Ä–Ķ–į–ļ—Ü–ł–ł –į–Ņ–ĺ–Ņ—ā–ĺ–∑–į, –Ŗ嬆–ł –≤¬†–ľ–Ķ–∂–ļ–Ľ–Ķ—ā–ĺ—á–Ĺ–ĺ–Ķ –Ņ—Ä–ĺ—Ā—ā—Ä–į–Ĺ—Ā—ā–≤–ĺ. –£—á–Ķ–Ĺ—č–Ķ –ĺ–Ī–Ĺ–į—Ä—É–∂–ł–Ľ–ł –ł–∑–Ī—č—ā–ĺ—á–Ĺ—É—é –ļ–ĺ–Ĺ—Ü–Ķ–Ĺ—ā—Ä–į—Ü–ł—é —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ–į –° –≤¬†—Ü–Ķ—Ä–Ķ–Ī—Ä–ĺ—Ā–Ņ–ł–Ĺ–į–Ľ—Ć–Ĺ–ĺ–Ļ –∂–ł–ī–ļ–ĺ—Ā—ā–ł —ɬ†–Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ —Ā¬†—á–Ķ—Ä–Ķ–Ņ–Ĺ–ĺ-–ľ–ĺ–∑–≥–ĺ–≤–ĺ–Ļ —ā—Ä–į–≤–ľ–ĺ–Ļ [46].
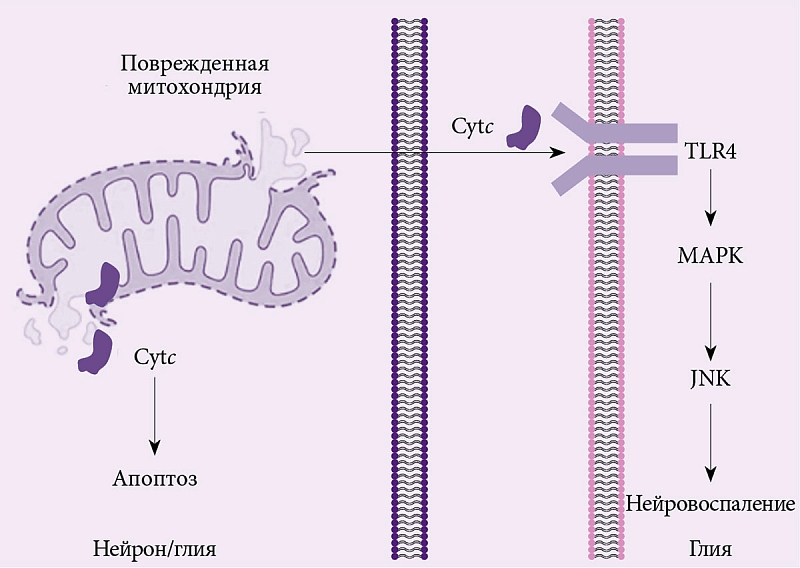
–í—č—Ā–≤–ĺ–Ī–ĺ–∂–ī–į—Ź—Ā—Ć –≤¬†–ľ–Ķ–∂–ļ–Ľ–Ķ—ā–ĺ—á–Ĺ–ĺ–Ķ –Ņ—Ä–ĺ—Ā—ā—Ä–į–Ĺ—Ā—ā–≤–ĺ, —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ –°¬†—Ā–≤—Ź–∑—č–≤–į–Ķ—ā—Ā—Ź —Ā¬†—ā–ĺ–Ľ–Ľ-–Ņ–ĺ–ī–ĺ–Ī–Ĺ—č–ľ–ł —Ä–Ķ—Ü–Ķ–Ņ—ā–ĺ—Ä–į–ľ–ł (TLR4), –≤¬†—Ä–Ķ–∑—É–Ľ—Ć—ā–į—ā–Ķ —á–Ķ–≥–ĺ –∑–į–Ņ—É—Ā–ļ–į–Ķ—ā—Ā—Ź —Ź–Ĺ—É—Ā-–ļ–ł–Ĺ–į–∑–Ĺ—č–Ļ —Ā–ł–≥–Ĺ–į–Ľ—Ć–Ĺ—č–Ļ –Ņ—É—ā—Ć (JNK), —á–Ķ—Ä–Ķ–∑ –ļ–ĺ—ā–ĺ—Ä—č–Ļ —Ä–Ķ–≥—É–Ľ–ł—Ä—É–Ķ—ā—Ā—Ź –į–ļ—ā–ł–≤–į—Ü–ł—Ź¬†–≥–Ľ–ł–į–Ľ—Ć–Ĺ—č—Ö –ļ–Ľ–Ķ—ā–ĺ–ļ (—Ä–ł—Ā.¬†2) [47, 48]. –í¬†—Ä–Ķ–∑—É–Ľ—Ć—ā–į—ā–Ķ –į–ļ—ā–ł–≤–ł—Ä–ĺ–≤–į–Ĺ–Ĺ—č–Ķ –į—Ā—ā—Ä–ĺ—Ü–ł—ā—č –Ĺ–į—á–ł–Ĺ–į—é—ā —ć–ļ—Ā–Ņ—Ä–Ķ—Ā—Ā–ł—Ä–ĺ–≤–į—ā—Ć –Ņ—Ä–ĺ–≤–ĺ—Ā–Ņ–į–Ľ–ł—ā–Ķ–Ľ—Ć–Ĺ—č–Ķ —Ü–ł—ā–ĺ–ļ–ł–Ĺ—č –ė–õ-1-–Ī–Ķ—ā–į –ł¬†–ė–õ-8, –≤—č–Ņ–ĺ–Ľ–Ĺ—Ź—é—Č–ł–Ķ –≤–į–∂–Ĺ—É—é —Ą—É–Ĺ–ļ—Ü–ł—é –≤¬†–į–ļ—ā–ł–≤–į—Ü–ł–ł –Ĺ–Ķ–Ļ—Ä–ĺ–≤–ĺ—Ā–Ņ–į–Ľ–Ķ–Ĺ–ł—Ź [48, 49]. –ė–ľ–Ķ—é—ā—Ā—Ź –ī–į–Ĺ–Ĺ—č–Ķ –嬆—Ä–ĺ–Ľ–ł —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ–į –° –≤¬†–Ņ–į—ā–ĺ–≥–Ķ–Ĺ–Ķ–∑–Ķ —Ä–į–∑–≤–ł—ā–ł—Ź –Ī–ĺ–Ľ–Ķ–∑–Ĺ–ł –ź–Ľ—Ć—Ü–≥–Ķ–Ļ–ľ–Ķ—Ä–į. –Ę–į–ļ, J.Y.¬†Lee –ł¬†—Ā–ĺ–į–≤—ā. (2015) –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ľ–ł –≤–ĺ–∑–ľ–ĺ–∂–Ĺ—č–Ķ —Ā–≤—Ź–∑–ł –ľ–Ķ–∂–ī—É –į–ľ–ł–Ľ–ĺ–ł–ī–ĺ–ľ –Ī–Ķ—ā–į –ł¬†—Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ–ĺ–ľ –°.¬†–í¬†—Ä–Ķ–∂–ł–ľ–Ķ —Ä–Ķ–į–Ľ—Ć–Ĺ–ĺ–≥–ĺ –≤—Ä–Ķ–ľ–Ķ–Ĺ–ł –ĺ—Ü–Ķ–Ĺ–ł–≤–į–Ľ–ł —É—Ä–ĺ–≤–Ķ–Ĺ—Ć —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ–į –°¬†–≤ –∂–ł–≤–ĺ–Ļ –ļ—É–Ľ—Ć—ā—É—Ä–Ķ –Ĺ–Ķ–Ļ—Ä–ĺ–Ĺ–ĺ–≤ –Ĺ–į¬†—Ą–ĺ–Ĺ–Ķ –≤–ĺ–∑–ī–Ķ–Ļ—Ā—ā–≤–ł—Ź –Ĺ–į¬†—ć—ā–ł –Ĺ–Ķ–Ļ—Ä–ĺ–Ĺ—č –į–ľ–ł–Ľ–ĺ–ł–ī–į –Ī–Ķ—ā–į. –ė—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į—ā–Ķ–Ľ–ł –≤—č—Ź–≤–ł–Ľ–ł –ī–ĺ—Ā—ā–į—ā–ĺ—á–Ĺ–ĺ –≤—č—Ā–ĺ–ļ–ł–Ļ —É—Ä–ĺ–≤–Ķ–Ĺ—Ć –≤—č—Ā–≤–ĺ–Ī–ĺ–∂–ī–Ķ–Ĺ–ł—Ź —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ–į –°¬†–≤ –ļ–Ľ–Ķ—ā–ļ–į—Ö, –ł–Ĺ–ļ—É–Ī–ł—Ä–ĺ–≤–į–Ĺ–Ĺ—č—Ö —Ā¬†–į–ľ–ł–Ľ–ĺ–ł–ī–ĺ–ľ –Ī–Ķ—ā–į [50].
–ú–Ķ–∂–ī—É —ā–Ķ–ľ –ī–ĺ—Ā—ā–į—ā–ĺ—á–Ĺ–ĺ –Ī–ĺ–Ľ—Ć—ą–ĺ–Ķ –ļ–ĺ–Ľ–ł—á–Ķ—Ā—ā–≤–ĺ –ļ–į–ļ –ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ł—Ö, —ā–į–ļ –ł¬†—ć–ļ—Ā–Ņ–Ķ—Ä–ł–ľ–Ķ–Ĺ—ā–į–Ľ—Ć–Ĺ—č—Ö —Ä–į–Ī–ĺ—ā –Ņ–ĺ—Ā–≤—Ź—Č–Ķ–Ĺ–ĺ –ł–∑—É—á–Ķ–Ĺ–ł—é —Ä–ĺ–Ľ–ł –ī—Ä—É–≥–ĺ–≥–ĺ —Ā–≤—Ź–∑–į–Ĺ–Ĺ–ĺ–≥–ĺ —Ā¬†—Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ–ĺ–ľ –°¬†–ļ–ĺ–ľ–Ņ–ĺ–Ĺ–Ķ–Ĺ—ā–į –ī—č—Ö–į—ā–Ķ–Ľ—Ć–Ĺ–ĺ–Ļ —Ü–Ķ–Ņ–ł –ľ–ł—ā–ĺ—Ö–ĺ–Ĺ–ī—Ä–ł–Ļ¬†‚Äď —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ –°-–ĺ–ļ—Ā–ł–ī–į–∑—č. –í¬†2021¬†–≥.¬†–Ī—č–Ľ –ĺ–Ņ—É–Ī–Ľ–ł–ļ–ĺ–≤–į–Ĺ –ļ—Ä—É–Ņ–Ĺ—č–Ļ –ľ–Ķ—ā–į–į–Ĺ–į–Ľ–ł–∑, –≤–ļ–Ľ—é—á–ł–≤—ą–ł–Ļ 1372 —Ā—ā–į—ā—Ć–ł. –†–Ķ–∑—É–Ľ—Ć—ā–į—ā—č –ľ–Ķ—ā–į–į–Ĺ–į–Ľ–ł–∑–į –Ņ–ĺ–ļ–į–∑–į–Ľ–ł, —á—ā–ĺ –Ņ—Ä–ł –Ī–ĺ–Ľ–Ķ–∑–Ĺ–ł –ź–Ľ—Ć—Ü–≥–Ķ–Ļ–ľ–Ķ—Ä–į –∑–Ĺ–į—á–ł–ľ–ĺ —Ā–Ĺ–ł–∂–į–Ķ—ā—Ā—Ź —É—Ä–ĺ–≤–Ķ–Ĺ—Ć —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ –°-–ĺ–ļ—Ā–ł–ī–į–∑—č, —á—ā–ĺ –Ņ–ĺ—ā–Ķ–Ĺ—Ü–ł–į–Ľ—Ć–Ĺ–ĺ –ľ–ĺ–∂–Ķ—ā —Ā–Ľ—É–∂–ł—ā—Ć –ľ–ł—ą–Ķ–Ĺ—Ć—é —ā–Ķ—Ä–į–Ņ–ł–ł –ī–į–Ĺ–Ĺ–ĺ–≥–ĺ –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł—Ź [51].
–ö–į–ļ —É–∂–Ķ –ĺ—ā–ľ–Ķ—á–į–Ľ–ĺ—Ā—Ć, –≤¬†–Ĺ–ĺ—Ä–ľ–į–Ľ—Ć–Ĺ—č—Ö —É—Ā–Ľ–ĺ–≤–ł—Ź—Ö –ĺ–ī–Ĺ–ł–ľ –ł–∑¬†–ľ–Ķ—Ö–į–Ĺ–ł–∑–ľ–ĺ–≤ —Ą—É–Ĺ–ļ—Ü–ł–ĺ–Ĺ–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ–į –°¬†—Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź —É—ā–ł–Ľ–ł–∑–į—Ü–ł—Ź –į–ļ—ā–ł–≤–Ĺ—č—Ö —Ą–ĺ—Ä–ľ –ļ–ł—Ā–Ľ–ĺ—Ä–ĺ–ī–į –∑–į —Ā—á–Ķ—ā –ī–ĺ–Ĺ–į—Ü–ł–ł —ć–Ľ–Ķ–ļ—ā—Ä–ĺ–Ĺ–į, —á—ā–ĺ –ĺ–Ņ—Ä–Ķ–ī–Ķ–Ľ—Ź–Ķ—ā –Ķ–≥–ĺ –į–Ĺ—ā–ł–ĺ–ļ–ł—Ā–Ľ–ł—ā–Ķ–Ľ—Ć–Ĺ—č–Ļ –Ņ–ĺ—ā–Ķ–Ĺ—Ü–ł–į–Ľ. –Ě–į—Ä—É—ą–Ķ–Ĺ–ł–Ķ –Ī–į–Ľ–į–Ĺ—Ā–į –ĺ–Ī—Ä–į–∑–ĺ–≤–į–Ĺ–ł—Ź –ł¬†—É—ā–ł–Ľ–ł–∑–į—Ü–ł–ł —Ā–≤–ĺ–Ī–ĺ–ī–Ĺ—č—Ö —Ä–į–ī–ł–ļ–į–Ľ–ĺ–≤ –Ľ–Ķ–∂–ł—ā –≤¬†–ĺ—Ā–Ĺ–ĺ–≤–Ķ —Ä–į–∑–≤–ł—ā–ł—Ź –ľ–Ĺ–ĺ–≥–ł—Ö –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł–Ļ. –°–ĺ–ĺ—ā–≤–Ķ—ā—Ā—ā–≤–Ķ–Ĺ–Ĺ–ĺ –į–Ĺ—ā–ł–ĺ–ļ–ł—Ā–Ľ–ł—ā–Ķ–Ľ—Ć–Ĺ–į—Ź —Ā–Ņ–ĺ—Ā–ĺ–Ī–Ĺ–ĺ—Ā—ā—Ć —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ–į –°¬†–Ņ–ĺ—ā–Ķ–Ĺ—Ü–ł–į–Ľ—Ć–Ĺ–ĺ –ľ–ĺ–≥–Ľ–į –Ī—č –Ī—č—ā—Ć –ł—Ā–Ņ–ĺ–Ľ—Ć–∑–ĺ–≤–į–Ĺ–į –Ņ—Ä–ł –Ľ–Ķ—á–Ķ–Ĺ–ł–ł —Ä—Ź–ī–į –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł–Ļ.
–í¬†—Ö–ĺ–ī–Ķ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–Ļ –ī–ĺ–ļ–į–∑–į–Ĺ–į —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į –¶–ł—ā–ĺ—Ö—Ä–ĺ–ľ –°¬†–Ņ—Ä–ł –ĺ–Ĺ–ļ–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ĺ–Ļ –Ņ–į—ā–ĺ–Ľ–ĺ–≥–ł–ł. –Ę–į–ļ–ĺ–Ļ —ć—Ą—Ą–Ķ–ļ—ā —Ā–ļ–ĺ—Ä–Ķ–Ķ –≤—Ā–Ķ–≥–ĺ —Ā–≤—Ź–∑–į–Ĺ —Ā¬†–ľ–ĺ–ī—É–Ľ–ł—Ä–ĺ–≤–į–Ĺ–ł–Ķ–ľ –į–Ĺ—ā–ł–ĺ–ļ–ł—Ā–Ľ–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ–Ļ –į–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā–ł, —Ā¬†–ĺ–ī–Ĺ–ĺ–Ļ —Ā—ā–ĺ—Ä–ĺ–Ĺ—č, –ł¬†–≤–ĺ–∑–ľ–ĺ–∂–Ĺ–ĺ–Ļ –į–ļ—ā–ł–≤–į—Ü–ł–Ķ–Ļ –į–Ņ–ĺ–Ņ—ā–ĺ–∑–į –≤¬†–ļ–Ľ–Ķ—ā–ļ–į—Ö –ĺ–Ņ—É—Ö–ĺ–Ľ–ł¬†‚Äď —Ā¬†–ī—Ä—É–≥–ĺ–Ļ [13].
–†–į–Ī–ĺ—ā, –Ņ–ĺ—Ā–≤—Ź—Č–Ķ–Ĺ–Ĺ—č—Ö –ĺ—Ü–Ķ–Ĺ–ļ–Ķ –≤–ĺ–∑–ľ–ĺ–∂–Ĺ–ĺ—Ā—ā–ł –ļ–ĺ—Ä—Ä–Ķ–ļ—Ü–ł–ł —Ü–Ķ—Ä–Ķ–Ī—Ä–į–Ľ—Ć–Ĺ–ĺ–Ļ –ł—ą–Ķ–ľ–ł–ł —Ā¬†–Ņ–ĺ–ľ–ĺ—Č—Ć—é —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ–į –°,¬†–Ņ—Ä–į–ļ—ā–ł—á–Ķ—Ā–ļ–ł –Ĺ–Ķ¬†–≤—Ā—ā—Ä–Ķ—á–į–Ķ—ā—Ā—Ź. –í¬†—ā–ĺ –∂–Ķ –≤—Ä–Ķ–ľ—Ź –ł–ľ–Ķ—é—ā—Ā—Ź —Ä–Ķ–∑—É–Ľ—Ć—ā–į—ā—č –ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ł—Ö –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–Ļ –ł¬†—ć–ļ—Ā–Ņ–Ķ—Ä–ł–ľ–Ķ–Ĺ—ā–į–Ľ—Ć–Ĺ—č–Ķ –ī–į–Ĺ–Ĺ—č–Ķ, –Ņ–ĺ–ī—ā–≤–Ķ—Ä–∂–ī–į—é—Č–ł–Ķ —Ā–Ĺ–ł–∂–Ķ–Ĺ–ł–Ķ –į–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā–ł —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ –°-–ĺ–ļ—Ā–ł–ī–į–∑—č. –£—Ā—ā–į–Ĺ–ĺ–≤–Ľ–Ķ–Ĺ–ĺ, —á—ā–ĺ —É—Ä–ĺ–≤–Ķ–Ĺ—Ć —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ¬†–°-–ĺ–ļ—Ā–ł–ī–į–∑—č –≤¬†—Ü–Ķ—Ä–Ķ–Ī—Ä–ĺ—Ā–Ņ–ł–Ĺ–į–Ľ—Ć–Ĺ–ĺ–Ļ –∂–ł–ī–ļ–ĺ—Ā—ā–ł –∑–Ĺ–į—á–ł–ľ–ĺ —Ā–Ĺ–ł–∂–į–Ķ—ā—Ā—Ź —ɬ†–Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ —Ā¬†–ł—ą–Ķ–ľ–ł—á–Ķ—Ā–ļ–ł–ľ –ł–Ĺ—Ā—É–Ľ—Ć—ā–ĺ–ľ. –ü—Ä–ł —ć—ā–ĺ–ľ —Ā—ā–Ķ–Ņ–Ķ–Ĺ—Ć –Ņ–ĺ–ī–ĺ–Ī–Ĺ–ĺ–≥–ĺ —Ā–Ĺ–ł–∂–Ķ–Ĺ–ł—Ź –Ņ—Ä—Ź–ľ–ĺ –ļ–ĺ—Ä—Ä–Ķ–Ľ–ł—Ä—É–Ķ—ā —Ā¬†—ā—Ź–∂–Ķ—Ā—ā—Ć—é –Ņ–ĺ—Ä–į–∂–Ķ–Ĺ–ł—Ź [52].
–ė–Ĺ—ā–Ķ—Ä–Ķ—Ā–Ĺ–ĺ–Ķ —ć–ļ—Ā–Ņ–Ķ—Ä–ł–ľ–Ķ–Ĺ—ā–į–Ľ—Ć–Ĺ–ĺ–Ķ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–Ķ –Ī—č–Ľ–ĺ –ĺ–Ņ—É–Ī–Ľ–ł–ļ–ĺ–≤–į–Ĺ–ĺ –≤¬†2018¬†–≥.¬†–ź–≤—ā–ĺ—Ä—č –ł–∑—É—á–į–Ľ–ł –≤–ĺ–∑–ľ–ĺ–∂–Ĺ–ĺ—Ā—ā—Ć –Ņ–ĺ–ī–į–≤–Ľ–Ķ–Ĺ–ł—Ź –į–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā–ł —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ –°-–ĺ–ļ—Ā–ł–ī–į–∑—č —Ā¬†–Ņ–ĺ–ľ–ĺ—Č—Ć—é –ļ–ĺ—Ä–ĺ—ā–ļ–ĺ–≤–ĺ–Ľ–Ĺ–ĺ–≤–ĺ–≥–ĺ –ł–Ĺ—Ą—Ä–į–ļ—Ä–į—Ā–Ĺ–ĺ–≥–ĺ –ł–∑–Ľ—É—á–Ķ–Ĺ–ł—Ź —Ā¬†–ī–Ľ–ł–Ĺ–ĺ–Ļ –≤–ĺ–Ľ–Ĺ—č 750 –ł¬†950 –Ĺ–ľ –ī–Ľ—Ź —É–ľ–Ķ–Ĺ—Ć—ą–Ķ–Ĺ–ł—Ź —Ä–Ķ–Ņ–Ķ—Ä—Ą—É–∑–ł–ĺ–Ĺ–Ĺ–ĺ–≥–ĺ –ĺ–ļ–ł—Ā–Ľ–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ–≥–ĺ –Ņ–ĺ–≤—Ä–Ķ–∂–ī–Ķ–Ĺ–ł—Ź. –ü–ĺ–ļ–į–∑–į–Ĺ–ĺ, —á—ā–ĺ –ī–į–Ĺ–Ĺ—č–Ļ –ľ–Ķ—ā–ĺ–ī —ć–ļ—Ā–Ņ–Ķ—Ä–ł–ľ–Ķ–Ĺ—ā–į–Ľ—Ć–Ĺ–ĺ–Ļ —ā–Ķ—Ä–į–Ņ–ł–ł –Ņ–ĺ–∑–≤–ĺ–Ľ—Ź–Ķ—ā —Ā–Ĺ–ł–∑–ł—ā—Ć –į–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ –°-–ĺ–ļ—Ā–ł–ī–į–∑—č, –į¬†—ā–į–ļ–∂–Ķ –∑–Ĺ–į—á–ł–ľ–ĺ —É–ľ–Ķ–Ĺ—Ć—ą–ł—ā—Ć –ļ–ĺ–Ľ–ł—á–Ķ—Ā—ā–≤–ĺ –Ņ–ĺ–≥–ł–Ī—ą–ł—Ö –Ĺ–Ķ–Ļ—Ä–ĺ–Ĺ–ĺ–≤ [53].
–ē—Č–Ķ –≤¬†–ĺ–ī–Ĺ–ĺ–Ļ —Ä–į–Ī–ĺ—ā–Ķ –Ĺ–Ķ–Ī–ĺ–Ľ—Ć—ą–ł–Ķ –ī–ĺ–∑—č —ć—ā–į–Ĺ–ĺ–Ľ–į —Ä–Ķ–≥—É–Ľ–ł—Ä–ĺ–≤–į–Ľ–ł –į–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ –°-–ĺ–ļ—Ā–ł–ī–į–∑—č, —Ā–Ĺ–ł–∑–ł–Ľ–ł –ĺ–Ī—Ä–į–∑–ĺ–≤–į–Ĺ–ł–Ķ –į–ļ—ā–ł–≤–Ĺ—č—Ö —Ą–ĺ—Ä–ľ –ļ–ł—Ā–Ľ–ĺ—Ä–ĺ–ī–į –ł¬†–ĺ–Ī–Ķ—Ā–Ņ–Ķ—á–ł–Ľ–ł –į–ī–Ķ–ļ–≤–į—ā–Ĺ—č–Ļ —É—Ä–ĺ–≤–Ķ–Ĺ—Ć –ź–Ę–§ –≤¬†–ł—ą–Ķ–ľ–ł–∑–ł—Ä–ĺ–≤–į–Ĺ–Ĺ—č—Ö –Ĺ–Ķ–Ļ—Ä–ĺ–Ĺ–į—Ö, –Ņ—Ä–ĺ–ī–Ķ–ľ–ĺ–Ĺ—Ā—ā—Ä–ł—Ä–ĺ–≤–į–≤ —ā–Ķ–ľ —Ā–į–ľ—č–ľ –Ĺ–Ķ–Ļ—Ä–ĺ–Ņ—Ä–ĺ—ā–Ķ–ļ—ā–ł–≤–Ĺ—É—é –į–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć [54].
–°—Ä–Ķ–ī–ł –ĺ—ā–Ķ—á–Ķ—Ā—ā–≤–Ķ–Ĺ–Ĺ—č—Ö —Ä–į–Ī–ĺ—ā –≤—Ā—ā—Ä–Ķ—á–į—é—ā—Ā—Ź –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā–ł –Ľ–Ķ–ļ–į—Ä—Ā—ā–≤–Ķ–Ĺ–Ĺ–ĺ–≥–ĺ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į –¶–ł—ā–ĺ—Ö—Ä–ĺ–ľ¬†–°¬†–Ņ—Ä–ł —Ä—Ź–ī–Ķ –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł–Ļ. –í¬†–ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ĺ–ľ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–ł –í.–ě. –°–ĺ–ļ–ĺ–Ľ–ĺ–≤ –ł¬†—Ā–ĺ–į–≤—ā. (2017) –Ĺ–į–Ī–Ľ—é–ī–į–Ľ–ł —É–Ľ—É—á—ą–Ķ–Ĺ–ł–Ķ –Ĺ–į¬†—Ą–ĺ–Ĺ–Ķ –Ņ—Ä–ł–Ķ–ľ–į –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į –¶–ł—ā–ĺ—Ö—Ä–ĺ–ľ –°¬†–≤ –≤–ł–ī–Ķ¬†–≥–Ľ–į–∑–Ĺ—č—Ö –ļ–į–Ņ–Ķ–Ľ—Ć –∑—Ä–ł—ā–Ķ–Ľ—Ć–Ĺ—č—Ö –Ņ–ĺ–ļ–į–∑–į—ā–Ķ–Ľ–Ķ–Ļ —ɬ†–Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ —Ā¬†—Ā—É–Ī—ć–Ņ–ł—ā–Ķ–Ľ–ł–į–Ľ—Ć–Ĺ—č–ľ–ł –Ņ–ĺ–ľ—É—ā–Ĺ–Ķ–Ĺ–ł—Ź–ľ–ł —Ä–ĺ–≥–ĺ–≤–ł—Ü—č –Ņ–ĺ—Ā–Ľ–Ķ –Ņ–Ķ—Ä–Ķ–Ĺ–Ķ—Ā–Ķ–Ĺ–Ĺ—č—Ö –ĺ—Ā—ā—Ä—č—Ö –ļ–Ķ—Ä–į—ā–ł—ā–ĺ–≤ [55].
–í¬†–ī–ł—Ā—Ā–Ķ—Ä—ā–į—Ü–ł–ĺ–Ĺ–Ĺ–ĺ–Ļ —Ä–į–Ī–ĺ—ā–Ķ –Ě.–í¬†–Ē–ĺ—Ä–ĺ–≥–ĺ–≤–į (2004) –≤¬†—ć–ļ—Ā–Ņ–Ķ—Ä–ł–ľ–Ķ–Ĺ—ā–į–Ľ—Ć–Ĺ—č—Ö —É—Ā–Ľ–ĺ–≤–ł—Ź—Ö –Ņ–ĺ–ļ–į–∑–į–Ĺ–į —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ–į –°¬†–Ņ—Ä–ł —ć–ļ—Ā–Ņ–Ķ—Ä–ł–ľ–Ķ–Ĺ—ā–į–Ľ—Ć–Ĺ–ĺ–Ļ —Ü–Ķ—Ä–Ķ–Ī—Ä–į–Ľ—Ć–Ĺ–ĺ–Ļ –ł—ą–Ķ–ľ–ł–ł. –ź–≤—ā–ĺ—Ä –≤—č—Ź–≤–ł–Ľ, —á—ā–ĺ —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ –°¬†–Ņ–ĺ–≤—č—ą–į–Ķ—ā –į–Ĺ—ā–ł–ĺ–ļ—Ā–ł–ī–į–Ĺ—ā–Ĺ—É—é –∑–į—Č–ł—ā—É –ł¬†–ĺ–≥—Ä–į–Ĺ–ł—á–ł–≤–į–Ķ—ā –į–ļ—ā–ł–≤–į—Ü–ł—é –Ņ—Ä–ĺ—Ü–Ķ—Ā—Ā–ĺ–≤ –Ņ–Ķ—Ä–Ķ–ļ–ł—Ā–Ĺ–ĺ–≥–ĺ –ĺ–ļ–ł—Ā–Ľ–Ķ–Ĺ–ł—Ź –Ľ–ł–Ņ–ł–ī–ĺ–≤ –≤¬†—ā–ļ–į–Ĺ–ł¬†–≥–ĺ–Ľ–ĺ–≤–Ĺ–ĺ–≥–ĺ –ľ–ĺ–∑–≥–į –ł¬†–Ņ–Ľ–į–∑–ľ–Ķ –ļ—Ä–ĺ–≤–ł. –ü—Ä–ł —Ä–Ķ–Ņ–Ķ—Ä—Ą—É–∑–ł–ĺ–Ĺ–Ĺ–ĺ–ľ –Ņ–ĺ—Ä–į–∂–Ķ–Ĺ–ł–ł¬†–≥–ĺ–Ľ–ĺ–≤–Ĺ–ĺ–≥–ĺ –ľ–ĺ–∑–≥–į —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ –°¬†—Ā–Ņ–ĺ—Ā–ĺ–Ī—Ā—ā–≤—É–Ķ—ā —Ä–ĺ—Ā—ā—É —Ā–ĺ–ī–Ķ—Ä–∂–į–Ĺ–ł—Ź –ľ–į–Ľ–ĺ–Ĺ–ĺ–≤–ĺ–≥–ĺ –ī–ł–į–Ľ—Ć–ī–Ķ–≥–ł–ī–į (–ú–Ē–ź) –≤¬†—ā–ļ–į–Ĺ–ł –ľ–ĺ–∑–≥–į –ł¬†–Ņ–Ľ–į–∑–ľ–Ķ –ļ—Ä–ĺ–≤–ł, —Ā–Ĺ–ł–∂–Ķ–Ĺ–ł—é –Ņ–ĺ–ļ–į–∑–į—ā–Ķ–Ľ—Ź –≤¬†—ć—Ä–ł—ā—Ä–ĺ—Ü–ł—ā–į—Ö [56].
–ē—Č–Ķ –≤¬†–ĺ–ī–Ĺ–ĺ–ľ —ć–ļ—Ā–Ņ–Ķ—Ä–ł–ľ–Ķ–Ĺ—ā–į–Ľ—Ć–Ĺ–ĺ–ľ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–ł –Ņ—Ä–ł¬†–≥–ł–Ņ–ĺ–ļ—Ā–ł—á–Ķ—Ā–ļ–ĺ–ľ –≤–ĺ–∑–ī–Ķ–Ļ—Ā—ā–≤–ł–ł –Ĺ–į¬†–ľ–ĺ–∑–≥ –Ņ—Ä–ĺ—Ą–ł–Ľ–į–ļ—ā–ł—á–Ķ—Ā–ļ–ĺ–Ķ –≤–≤–Ķ–ī–Ķ–Ĺ–ł–Ķ —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ–į –°¬†–≤ –ī–ĺ–∑–Ķ 0,3 –ľ–≥/–ļ–≥ –ĺ–ļ–į–∑–į–Ľ–ĺ —Ü–Ķ—Ä–Ķ–Ī—Ä–ĺ–Ņ—Ä–ĺ—ā–Ķ–ļ—ā–ł–≤–Ĺ—č–Ļ —ć—Ą—Ą–Ķ–ļ—ā, –Ņ—Ä–Ķ–ī–ĺ—ā–≤—Ä–į—ā–ł–≤ –ł–Ľ–ł —É–ľ–Ķ–Ĺ—Ć—ą–ł–≤ –Ņ–į—ā–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ł–Ķ —Ā–ī–≤–ł–≥–ł, —Ä–į–∑–≤–ł–≤—ą–ł–Ķ—Ā—Ź –≤¬†–ĺ—ā—Ā—É—ā—Ā—ā–≤–ł–Ķ –Ņ—Ä–ł–ľ–Ķ–Ĺ–Ķ–Ĺ–ł—Ź –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į. –≠—Ą—Ą–Ķ–ļ—ā –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į —Ö–į—Ä–į–ļ—ā–Ķ—Ä–ł–∑–ĺ–≤–į–Ľ—Ā—Ź —Ā–Ĺ–ł–∂–Ķ–Ĺ–ł–Ķ–ľ –≤—č—Ä–į–∂–Ķ–Ĺ–Ĺ–ĺ—Ā—ā–ł¬†–≥–ł–Ņ–ĺ–ļ—Ā–ł–ł –≤¬†–ļ–ĺ—Ä–Ķ –Ī–ĺ–Ľ—Ć—ą–ł—Ö –Ņ–ĺ–Ľ—É—ą–į—Ä–ł–Ļ –ł¬†–Ņ—Ä–Ķ–ī—É–Ņ—Ä–Ķ–∂–ī–Ķ–Ĺ–ł–Ķ–ľ –ł–Ľ–ł –ĺ—Ā–Ľ–į–Ī–Ľ–Ķ–Ĺ–ł–Ķ–ľ —Ä–į–∑–≤–ł—ā–ł—Ź –≤–Ĺ—É—ā—Ä–ł–ļ–Ľ–Ķ—ā–ĺ—á–Ĺ–ĺ–≥–ĺ –ł¬†–≤–Ĺ–Ķ–ļ–Ľ–Ķ—ā–ĺ—á–Ĺ–ĺ–≥–ĺ –ĺ—ā–Ķ–ļ–į —ā–ļ–į–Ĺ–ł –ľ–ĺ–∑–≥–į [57].
–ó–į–ļ–Ľ—é—á–Ķ–Ĺ–ł–Ķ
–°¬†—É—á–Ķ—ā–ĺ–ľ —Ą–ł–∑–ł–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ł—Ö –ł¬†–Ņ–į—ā–ĺ—Ą–ł–∑–ł–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ł—Ö –ĺ—Ā–ĺ–Ī–Ķ–Ĺ–Ĺ–ĺ—Ā—ā–Ķ–Ļ —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ–į –°,¬†–į¬†—ā–į–ļ–∂–Ķ –Ĺ–į–Ľ–ł—á–ł—Ź –Ĺ–Ķ–Ī–ĺ–Ľ—Ć—ą–ĺ–≥–ĺ –ļ–ĺ–Ľ–ł—á–Ķ—Ā—ā–≤–į —ć–ļ—Ā–Ņ–Ķ—Ä–ł–ľ–Ķ–Ĺ—ā–į–Ľ—Ć–Ĺ—č—Ö —Ä–į–Ī–ĺ—ā, –≤¬†–ļ–ĺ—ā–ĺ—Ä—č—Ö –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ľ–ł—Ā—Ć —Ą–į—Ä–ľ–į–ļ–ĺ–ī–ł–Ĺ–į–ľ–ł—á–Ķ—Ā–ļ–ł–Ķ –ľ–Ķ—Ö–į–Ĺ–ł–∑–ľ—č –ī–Ķ–Ļ—Ā—ā–≤–ł—Ź —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ–į –°,¬†–Ņ—Ä–Ķ–ī—Ā—ā–į–≤–Ľ—Ź–Ķ—ā—Ā—Ź —Ü–Ķ–Ľ–Ķ—Ā–ĺ–ĺ–Ī—Ä–į–∑–Ĺ—č–ľ –Ņ—Ä–ĺ–ī–ĺ–Ľ–∂–Ķ–Ĺ–ł–Ķ –ļ–į–ļ —ć–ļ—Ā–Ņ–Ķ—Ä–ł–ľ–Ķ–Ĺ—ā–į–Ľ—Ć–Ĺ—č—Ö, —ā–į–ļ –ł¬†–ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ł—Ö –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–Ļ –≤–ĺ–∑–ľ–ĺ–∂–Ĺ–ĺ—Ā—ā–ł –ł—Ā–Ņ–ĺ–Ľ—Ć–∑–ĺ–≤–į–Ĺ–ł—Ź –ī–į–Ĺ–Ĺ–ĺ–≥–ĺ –≤–Ķ—Č–Ķ—Ā—ā–≤–į –Ņ—Ä–ł –ĺ—Ā—ā—Ä—č—Ö –ł¬†—Ö—Ä–ĺ–Ĺ–ł—á–Ķ—Ā–ļ–ł—Ö —Ü–Ķ—Ä–Ķ–Ī—Ä–ĺ–≤–į—Ā–ļ—É–Ľ—Ź—Ä–Ĺ—č—Ö –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł—Ź—Ö. –ź–Ĺ—ā–ł–ĺ–ļ—Ā–ł–ī–į–Ĺ—ā–Ĺ—č–Ļ –ł¬†–Ņ—Ä–ĺ–į–Ņ–ĺ–Ņ—ā–ĺ—ā–ł—á–Ķ—Ā–ļ–ł–Ļ –Ņ—Ä–ĺ—Ą–ł–Ľ–ł —Ü–ł—ā–ĺ—Ö—Ä–ĺ–ľ–į –°¬†–≤ –ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ł—Ö —É—Ā–Ľ–ĺ–≤–ł—Ź—Ö –Ĺ–Ķ–ī–ĺ—Ā—ā–į—ā–ĺ—á–Ĺ–ĺ –ł–∑—É—á–Ķ–Ĺ—č. –ú–ĺ–∂–Ĺ–ĺ –Ņ—Ä–Ķ–ī–Ņ–ĺ–Ľ–ĺ–∂–ł—ā—Ć, —á—ā–ĺ —ā–Ķ—Ä–į–Ņ–Ķ–≤—ā–ł—á–Ķ—Ā–ļ–ł–Ļ –Ņ–ĺ—ā–Ķ–Ĺ—Ü–ł–į–Ľ –Ľ–Ķ–ļ–į—Ä—Ā—ā–≤–Ķ–Ĺ–Ĺ–ĺ–≥–ĺ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į –¶–ł—ā–ĺ—Ö—Ä–ĺ–ľ –°¬†—É –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ —Ā¬†—Ü–Ķ—Ä–Ķ–Ī—Ä–ĺ–≤–į—Ā–ļ—É–Ľ—Ź—Ä–Ĺ—č–ľ–ł –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł—Ź–ľ–ł –ī–ĺ—Ā—ā–į—ā–ĺ—á–Ĺ–ĺ –≤—č—Ā–ĺ–ļ, –Ņ–ĺ—Ā–ļ–ĺ–Ľ—Ć–ļ—É –Ĺ–į—Ä—É—ą–Ķ–Ĺ–ł–Ķ –Ņ—Ä–ĺ—Ü–Ķ—Ā—Ā–ĺ–≤ –ļ–Ľ–Ķ—ā–ĺ—á–Ĺ–ĺ–≥–ĺ –ī—č—Ö–į–Ĺ–ł—Ź —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź –ļ–Ľ—é—á–Ķ–≤—č–ľ –Ņ–į—ā–ĺ—Ą–ł–∑–ł–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ł–ľ –Ņ—Ä–ĺ—Ü–Ķ—Ā—Ā–ĺ–ľ —ɬ†–ī–į–Ĺ–Ĺ–ĺ–Ļ¬†–≥—Ä—É–Ņ–Ņ—č –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤. –ü–ĺ—Ā–Ľ–Ķ–ī—É—é—Č–ł–Ķ –∑–≤–Ķ–Ĺ—Ć—Ź –Ņ–ĺ–≤—Ä–Ķ–∂–ī–Ķ–Ĺ–ł—Ź –ľ–ĺ–∑–≥–į, –į¬†–ł–ľ–Ķ–Ĺ–Ĺ–ĺ –į–Ņ–ĺ–Ņ—ā–ĺ–∑ –Ĺ–Ķ–Ļ—Ä–ĺ–Ĺ–ĺ–≤ –ł¬†–≥–Ľ–ł–ł, –Ĺ–Ķ–Ļ—Ä–ĺ–≤–ĺ—Ā–Ņ–į–Ľ–Ķ–Ĺ–ł–Ķ, —ć–ļ—Ā–į–Ļ—ā–ĺ—ā–ĺ–ļ—Ā–ł—á–Ĺ–ĺ—Ā—ā—Ć, —Ź–≤–Ľ—Ź—é—ā—Ā—Ź –Ľ–ł—ą—Ć —Ä–Ķ–∑—É–Ľ—Ć—ā–į—ā–ĺ–ľ –ĺ—Ā—ā—Ä–ĺ–Ļ –ł–Ľ–ł —Ö—Ä–ĺ–Ĺ–ł—á–Ķ—Ā–ļ–ĺ–Ļ¬†–≥–ł–Ņ–ĺ–ļ—Ā–ł–ł, –Ņ–ĺ—ā–Ķ–Ĺ—Ü–ł–į–Ľ—Ć–Ĺ—č–ľ –ł¬†—ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ—č–ľ –ļ–ĺ—Ä—Ä–Ķ–ļ—ā–ĺ—Ä–ĺ–ľ –ļ–ĺ—ā–ĺ—Ä–ĺ–Ļ –ľ–ĺ–∂–Ķ—ā –Ī—č—ā—Ć –¶–ł—ā–ĺ—Ö—Ä–ĺ–ľ –°.
I.A. Shchukin, PhD
N.I. Pirogov Russian National Research Medical University
Contact person: Ivan A. Shchukin, ivashchukin@gmail.com
The key pathophysiological mechanism underlying the development of both acute ischemic stroke and chronic cerebral ischemia is a violation of cerebral blood flow, triggering a cascade of reactions leading to damage to the brain substance. These processes lead to irreversible damage to the nervous tissue by the mechanisms of necrosis and apoptosis. As a result of excessive excitation of glutamate NMDA receptors, intracellular accumulation of Ca2+ ions occurs, which subsequently contributes to the activation of various enzyme systems, phosphorylation of proteins, cleavage of phospholipids and release of arachidonic acid, the formation of toxic products, free radicals that have cytotoxic, immunogenic and mutagenic effects, damaging mitochondria, cellular DNA and RNA. In conditions of ischemic brain damage, the mitochondria is considered one of the most vulnerable intracellular structures, the disruption of which, on the one hand, leads to a critical energy deficit, on the other hand, triggers the process of programmed cell death. The most important enzyme of the mitochondrial respiratory chain, which is associated with the processes of energy formation and, accordingly, cell survival, as well as the processes of apoptotic cell death, is cytochrome C. The release of cytochrome C from damaged mitochondria leads to disruption of the functioning of the respiratory chain of the cell, the development of hypoxic damage to the neuron and activates apoptosis reactions.
The article discusses the fundamental mechanisms of cerebral ischemic injury, which are based on the development of mitochondrial insufficiency. Special attention is paid to the role of cytochrome C. The mechanisms of participation of cytochrome C in the processes of cell damage, as well as its protective (antioxidant) potential in a number of pathologies, including cerebral ischemic damage, are described. The potential therapeutic use of exogenous cytochrome C in acute and chronic cerebrovascular diseases is indicated.
–£–≤–į–∂–į–Ķ–ľ—č–Ļ –Ņ–ĺ—Ā–Ķ—ā–ł—ā–Ķ–Ľ—Ć uMEDp!
–£–≤–Ķ–ī–ĺ–ľ–Ľ—Ź–Ķ–ľ –í–į—Ā –ĺ —ā–ĺ–ľ, —á—ā–ĺ –∑–ī–Ķ—Ā—Ć —Ā–ĺ–ī–Ķ—Ä–∂–ł—ā—Ā—Ź –ł–Ĺ—Ą–ĺ—Ä–ľ–į—Ü–ł—Ź, –Ņ—Ä–Ķ–ī–Ĺ–į–∑–Ĺ–į—á–Ķ–Ĺ–Ĺ–į—Ź –ł—Ā–ļ–Ľ—é—á–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ –ī–Ľ—Ź —Ā–Ņ–Ķ—Ü–ł–į–Ľ–ł—Ā—ā–ĺ–≤ –∑–ī—Ä–į–≤–ĺ–ĺ—Ö—Ä–į–Ĺ–Ķ–Ĺ–ł—Ź.
–ē—Ā–Ľ–ł –í—č –Ĺ–Ķ —Ź–≤–Ľ—Ź–Ķ—ā–Ķ—Ā—Ć —Ā–Ņ–Ķ—Ü–ł–į–Ľ–ł—Ā—ā–ĺ–ľ –∑–ī—Ä–į–≤–ĺ–ĺ—Ö—Ä–į–Ĺ–Ķ–Ĺ–ł—Ź, –į–ī–ľ–ł–Ĺ–ł—Ā—ā—Ä–į—Ü–ł—Ź –Ĺ–Ķ –Ĺ–Ķ—Ā–Ķ—ā –ĺ—ā–≤–Ķ—ā—Ā—ā–≤–Ķ–Ĺ–Ĺ–ĺ—Ā—ā–ł –∑–į –≤–ĺ–∑–ľ–ĺ–∂–Ĺ—č–Ķ –ĺ—ā—Ä–ł—Ü–į—ā–Ķ–Ľ—Ć–Ĺ—č–Ķ –Ņ–ĺ—Ā–Ľ–Ķ–ī—Ā—ā–≤–ł—Ź, –≤–ĺ–∑–Ĺ–ł–ļ—ą–ł–Ķ –≤ —Ä–Ķ–∑—É–Ľ—Ć—ā–į—ā–Ķ —Ā–į–ľ–ĺ—Ā—ā–ĺ—Ź—ā–Ķ–Ľ—Ć–Ĺ–ĺ–≥–ĺ –ł—Ā–Ņ–ĺ–Ľ—Ć–∑–ĺ–≤–į–Ĺ–ł—Ź –í–į–ľ–ł –ł–Ĺ—Ą–ĺ—Ä–ľ–į—Ü–ł–ł —Ā –Ņ–ĺ—Ä—ā–į–Ľ–į –Ī–Ķ–∑ –Ņ—Ä–Ķ–ī–≤–į—Ä–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ–Ļ –ļ–ĺ–Ĺ—Ā—É–Ľ—Ć—ā–į—Ü–ł–ł —Ā –≤—Ä–į—á–ĺ–ľ.
–Ě–į–∂–ł–ľ–į—Ź –Ĺ–į –ļ–Ĺ–ĺ–Ņ–ļ—É ¬ę–í–ĺ–Ļ—ā–ł¬Ľ, –í—č –Ņ–ĺ–ī—ā–≤–Ķ—Ä–∂–ī–į–Ķ—ā–Ķ, —á—ā–ĺ —Ź–≤–Ľ—Ź–Ķ—ā–Ķ—Ā—Ć –≤—Ä–į—á–ĺ–ľ –ł–Ľ–ł —Ā—ā—É–ī–Ķ–Ĺ—ā–ĺ–ľ –ľ–Ķ–ī–ł—Ü–ł–Ĺ—Ā–ļ–ĺ–≥–ĺ –≤—É–∑–į.