Современное состояние проблемы лечения диссеминированного рака поджелудочной железы и возможные перспективы
- Аннотация
- Статья
- Ссылки
Введение
Рак поджелудочной железы (РПЖ) – нерешенная проблема современной онкологии, так как ни один из методов лечения не позволяет длительное время контролировать болезнь. Из 230 000 больных, зарегистрированных в мире, умирают 98%. За последние 5 лет заболеваемость РПЖ возросла на 4,2% среди мужчин и на 12,1% среди женщин. В России в 2007 г. количество больных с впервые установленным диагнозом составило 14 037 (в структуре заболеваемости РПЖ занял 4-е место среди опухолей пищеварительной системы), а смертность от РПЖ – 14 473 случая (превалирование смертности над заболеваемостью обусловлено посмертной диагностикой болезни) [1, 2]. Европейские статистические данные по РПЖ аналогичны российским: в 2008 г. показатель заболеваемости (68 500 случаев) был меньше показателя смертности (70 200 случаев) [3].
Согласно данным Международного агентства по изучению рака (International Agency for Research on Cancer, IARC), РПЖ занимает 13-е место по заболеваемости в мире, а по смертности – 8-е. Так, в 2009 г. в США РПЖ заболели 42 470 человек (10-е место для мужчин и женщин), а умерли 35 240, соотношение смертности и заболеваемости составило 0,83 [4].
В целом пятилетняя выживаемость больных РПЖ не достигает 5%. После установления диагноза один год переживают 15,2% мужчин и 16,4% женщин. В США за 25 лет
(с 1975 по 2000 г.) показатель пятилетней выживаемости для пациентов с РПЖ увеличился с 3 до 4%. Для сравнения: за этот же период показатель пятилетней выживаемости при раке толстой кишки вырос на 12%, при раке пищевода – на 9%, раке желудка – на 8%.
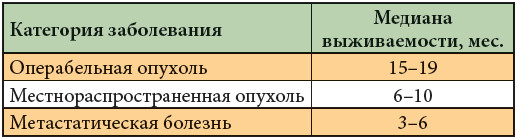
Лишь 10% больных РПЖ потенциально операбельны на момент первичной диагностики. Лимфатическая диссеминация выявляется в 45–70% случаев при опухолях размером менее 2 см в диаметре. До 30% вновь выявленных больных имеют локально распространенный процесс, а 60% – отдаленные метастазы [5]. Медианы выживаемости больных в зависимости от распространения опухоли представлены в табл. 1.
В настоящей статье мы остановимся на химиотерапии и таргетной терапии диссеминированного РПЖ.
Химиотерапия диссеминированного РПЖ
Важное значение для лекарственной терапии РПЖ имело открытие гемцитабина. Гемцитабин был включен в стандартные схемы лечения метастатического РПЖ в 1997 г., после публикации результатов североамериканского исследования с участием 126 пациентов. В этом исследовании II фазы на фоне лечения гемцитабином в сравнении с 5-фторурацилом клиническое улучшение отмечено у 23,8 и 4,8% больных, медиана выживаемости составила 5,65 и 4,41 месяца, годичная выживаемость – 18 и 2% соответственно; разница по всем показателям была статистически значимой; переносимость лечения удовлетворительной [6]. В том же году A.M. Storniolo и соавт. обобщили данные о монотерапии гемцитабином 3023 больных РПЖ, медиана продолжительности жизни пациентов составила 8 месяцев [7]. Применение гемцитабина допускается даже при оценке общего состояния пациента по шкале Карновского 60%.
После констатации эффективности гемцитабина начались широкие исследования его комбинаций с другими препаратами: 5-фторурацилом, капецитабином, UFT, S1, томудексом, цисплатином, оксалиплатином, иринотеканом и др. В современной практике среди химиотерапевтических дуплетов на основе гемцитабина чаще других используют его сочетание с производными платины и капецитабином.
В европейское исследование III фазы GERCOR / GISCAD было рандомизировано 326 пациентов, которые получали комбинацию гемцитабина и оксалиплатина или монотерапию гемцитабином. В группе, получавшей терапию в режиме GEMOX, отмечены более высокая эффективность лечения (26,8 против 17,3%, р = 0,04) и более длительная продолжительность жизни без прогрессирования (5,8 против 3,7 месяца, р = 0,04) по сравнению с пациентами на гемцитабине в монорежиме. Также выявлено статистически незначимое увеличение общей выживаемости: 9 и 7,1 месяца соответственно (p = 0,13) [8].
На основании нескольких метаанализов, проведенных для сравнения эффективности гемцитабина в монотерапии и в сочетании с производными платины, было показано преимущество комбинированной терапии в отношении статистически значимого повышения эффективности лечения и медианы времени до прогрессирования, а также скромное достоверное увеличение общей выживаемости [9–12].
Другим препаратом, который чаще всего комбинируют с гемцитабином, является капецитабин. Сравнение комбинации гемцитабина и капецитабина с режимом монотерапии гемцитабином, выполненное исследователями из Великобритании, свидетельствовало в пользу комбинированной терапии: эффект получен в 14,2% случаев комбинации «гемцитабин + капецитабин» и в 7,1% случаев на одном гемцитабине (р = 0,008), однолетняя выживаемость составила 23 и 17%
(р = 0,023), медиана общей выживаемости – 7,4 и 6,0 месяца соответственно (p > 0,05) [13].
Швейцарская группа по клиническим исследованиям и группа ECOG (Eastern Cooperative Oncology Group – Восточная объединенная группа онкологов) сравнили монотерапию гемцитабином и комбинацию капецитабина (650 мг/м2 два раза в сутки с 1-го по 14-й день) с гемцитабином (1000 мг/м2 30-минутная инфузия в 1 и 8-й дни каждые 3 недели) в рандомизированном исследовании с участием 319 больных c распространенным РПЖ. Медиана общей выживаемости составила 8,4 и 7,2 месяца в пользу режима GEMCAP, но данные оказались статистически недостоверными (р = 0,234). Однако дополнительный анализ в подгруппе пациентов, общее состояние которых оценивалось как «хорошее» (статус по шкале Карновского от 90 до 100%), продемонстрировал значительное увеличение медианы общей выживаемости при использовании комбинации «гемцитабин + капецитабин» по сравнению с монотерапией гемцитабином (10,1 против 7,4 месяца соответственно, р = 0,014), при этом частота побочных эффектов 3-й или 4-й степени была одинаковой в обеих группах [14].
Японскими исследователями опубликованы результаты метаанализа, обобщающего данные 18 рандомизированных исследований, объединивших 4237 пациентов с распространенным РПЖ, которые были разделены на 5 подгрупп в зависимости от схемы лечения: «гемцитабин + капецитабин», «гемцитабин + цисплатин», «гемцитабин + 5-фторурацил», «гемцитабин + иринотекан» и «гемцитабин + оксалиплатин». Анализ данных по параметру общего состояния больного выполнен в 4 исследованиях (1325 больных). Выявлено, что применение комбинированной химиотерапии у больных с плохим общим состоянием увеличивает риск смерти в течение 6 месяцев (1,17, р = 0,04) и одного года (1,09, р = 0,04) и, напротив, использование комбинированной терапии у больных с хорошим общим состоянием снижает риск смерти в течение года (0,93, р = 0,08). Метаанализ продемонстрировал значительное увеличение выживаемости при сочетании гемцитабина с капецитабином или оксалиплатином [15].
В отечественной клинической практике все реже используют оригинальные препараты, заменяя их на воспроизводимые в Российской Федерации. Так, мы применяли оксалиплатин и гемцитабин (препараты компании ОАО «ВЕРОФАРМ») в качестве комбинации GEMOX для второй линии терапии при прогрессировании заболевания, но не раньше чем через 3 месяца после прекращения терапии гемцитабином (препарат Гемзар). Всего в исследование было включено 11 пациентов (7 женщин и 4 мужчин), медиана возраста – 68 лет. Общее состояние 8 больных оценивалось как 1 балл по шкале ECOG, троих – 2 балла. В результате лечения отмечены в одном случае частичная регрессия и в четырех – стабилизация
(у двух пациентов – более 5 месяцев). На основании полученных данных представляется возможным возобновление гемцитабинсодержащих комбинаций у больных в хорошем состоянии после монотерапии Гемзаром. Подчеркнем, что лечение Гемзаром должно быть прекращено не раньше чем за 3 месяца до начала терапии второй линии. Из особенностей применения отечественных препаратов следует обратить внимание на более частое (у половины больных) развитие лихорадки и астении по сравнению с применением оригинальных зарубежных препаратов.
Одним из направлений в лечении распространенного РПЖ, позволивших улучшить результаты терапии, стало введение в практику химиотерапевтических триплетов. В 2011 г. были опубликованы данные исследования комбинации FOLFIRINOX (фторурацил, лейковорин, иринотекан и оксалиплатин) в сравнении с монотерапией гемцитабином. Установлено достоверное увеличение медианы общей выживаемости (11,1 против 6,8 месяца, р = 0,001), медианы времени до прогрессирования (6,4 против
3,3 месяца, р = 0,001), частоты объективных ответов (33,6 против 9,4%), снижение риска смерти на 47%. При этом следует отметить, что проведение полихимиотерапии целесообразно и возможно у пациентов моложе 65 лет в хорошем общем состоянии [16].
Из-за низкой чувствительности аденокарциномы поджелудочной железы к классической химиотерапии в мире широко изучается эффективность таргетных препаратов при данном заболевании. Ниже рассмотрены основные пути патогенеза РПЖ и молекулярные мишени для таргетной терапии.
Таргетная терапия диссеминированного РПЖ
Мутация гена KRAS
При аденокарциноме поджелудочной железы мутации гена KRAS встречаются, по разным данным, в 74–100% случаев, при этом чаще всего локализуются в 12, 13 и 67-м кодонах [17–20]. Этот протоонкоген кодирует высокогомологичный мембраносвязанный белок (p21ras) с молекулярной массой 21 кДа, который функционально представляет собой G-белок. Связывая гуанозинтрифосфат (ГТФ), p21ras переходит в активную конформацию, осуществляющую передачу сигнала от рецепторов, расположенных на поверхности клетки, к ядру [21].
Точечные мутации в гене приводят к замещению аминокислотных остатков в белке, что ослабляет ГТФ-азную активность p21ras, в результате чего создается эффект постоянного проведения внутриклеточного сигнала [22].
Необходимым условием для функционирования белка Ras является его связь с внутренней поверхностью цитоплазматической мембраны. После синтеза про-Ras протеин в цитоплазме претерпевает ряд посттрансляционных изменений, одним из которых является фарнезилирование, в результате чего его C-домен становится гидрофобным и обеспечивает надежное сцепление с мембраной клетки. Таким образом, ингибирование фермента фарнезилтрансферазы приводит к инактивации p21ras. Кроме того, фарнезилирование необходимо для функционирования белка RhoB и белков сигнального пути PI3K/Akt2 [21].
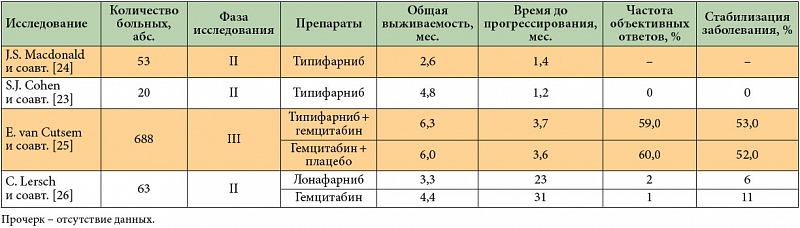
Типифарниб (R115777, Зарнестра) является селективным конкурентным ингибитором фарнезилтрансферазы. Несмотря на многообещающие результаты многих доклинических испытаний, применение типифарниба как в монотерапии, так и в комбинации с гемцитабином не привело к улучшению результатов лечения РПЖ ни в одном из клинических исследований II и III фазы [23–25]. Схожие данные были получены и в отношении ингибитора фарнезилтрансферазы второго поколения лонафарниба (SCH66336) [26] (табл. 2).
MAPK-опосредованный внутриклеточный путь передачи сигнала
При связывании ряда факторов роста (EGF, PDGF), цитокинов (интерлейкинов-2,-3; G-CSF), инсулина с экстрацеллюлярными доменами рецепторов к ним происходит активация белка Ras, который в свою очередь приводит к запуску нескольких внутриклеточных ферментативных каскадов, передающих сигнал к генам, ответственным за клеточную пролиферацию. Одним из таких сигнальных путей является Raf-1/MEK/MAPK/ERK/c-fos. К настоящему времени уже синтезировано несколько молекул, блокирующих митогенактивированные протеинкиназы (AZD6244, CI-1040, PD0325901), ведутся клинические исследования по изучению их эффективности при РПЖ [27]. Опубликованы результаты только одного исследования II фазы, в котором изучалась эффективность препарата CI-1040 при различных типах солидных опухолей, в том числе и среди пациентов с аденокарциномой поджелудочной железы. Каких-либо значительных результатов не получено: ни у одного из пациентов не зарегистрировано объективного ответа на лечение, только у 2 (13%) из 15 больных РПЖ удалось достичь стабилизации опухолевого процесса, при этом выраженных побочных эффектов не отмечено [28].
Мультитаргетным препаратом, ингибирующим Raf-киназы, тирозинкиназные домены VEGFR-2, VEGFR-3 и PDGFR-бета, является сорафениб. В исследовании I фазы была продемонстрирована хорошая переносимость пациентами комбинации препаратов сорафениб и гемцитабин [29]. Однако в клиническом испытании II фазы не выявлено ее преимуществ по сравнению с химиотерапией [30].
Src-опосредованный внутриклеточный путь передачи сигнала
Src является одним из 9 представителей семейства не связанных с рецептором тирозинкиназ, повышенная экспрессия Src характерна для опухолевых клеток РПЖ [31]. Src в норме – важнейший медиатор внутриклеточных сигнальных путей, активирующихся при связывании эпидермального и других факторов роста с рецепторами. Src активирует киназу фокальной адгезии (FAK), фосфорилирует STAT-3 и связанные с рецепторами G-белки, взаимодействует с MAPK [32]. Повышение активности Src способствует метастазированию опухоли за счет утраты клетками межклеточных контактов, перестройки актинового цитоскелета, образования фокальных контактов [33].
Саракатиниб (AZD0530) является малой молекулой, ингибитором тирозинкиназ Src-семейства. В двух исследованиях II фазы, где саракатиниб применялся как в комбинации с гемцитабином в качестве первой линии терапии [34], так и в монотерапии после прогрессирования на фоне гемцитабина [35], не получено каких-либо данных об улучшении результатов по сравнению со стандартными схемами химиотерапии.
В настоящее время идет набор пациентов в исследование II фазы по изучению влияния ингибитора Src-киназ второго поколения дазатиниба как фактора, препятствующего диссеминации при местнораспространенном РПЖ [36].
Ингибиторы матриксных металлопротеиназ
Еще одним направлением в лечении РПЖ было включение в противоопухолевые комбинации ингибиторов матриксных металлопротеиназ (MMPs). MMPs представляют собой семейство цинк- и кальцийзависимых эндопептидаз, которые за счет деградации экстрацеллюлярного матрикса и стромы способствуют миграции злокачественных клеток. Семейство матриксных металлопротеиназ состоит из 20 ферментов, способных расщеплять практически все компоненты внеклеточного матрикса.
Уровень экспрессии MPPs в опухоли значительно выше, чем в нормальной ткани поджелудочной железы. Основными протеиназами, вовлеченными в патогенез РПЖ, являются MMP-2 и MMP-9 [37]. Высокий уровень экспрессии MMP-2 коррелирует с плохим прогнозом и быстрым прогрессированием заболевания [38].
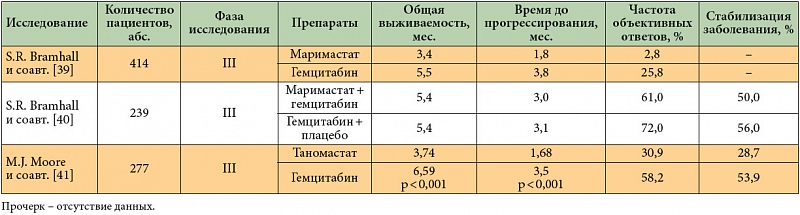
Маримастат – первый и наиболее изученный при солидных опухолях ингибитор MMPs 1, 2, 3, 7 и 9-го типа. Однако в двух рандомизированных исследованиях III фазы применение маримастата в монотерапии или в комбинации с гемцитабином не показало никаких преимуществ перед стандартной химиотерапией РПЖ [39, 40]. Кроме того, таргетный препарат дает специфические побочные эффекты (артралгии, мышечно-костные боли, появление скованности движений).
Набор пациентов в исследование III фазы по изучению ингибитора MMPs второго поколения таномастата (BAY 12-9566) был досрочно завершен в связи с тем, что применение данного препарата приводило к статистически значимому сокращению продолжительности жизни пациентов [41].
Селективный ингибитор металлопротеиназ 2 и 9-го типа Ro 28-2653 проходит в настоящее время доклинические испытания [42]. Результаты исследований ингибиторов MMPs при РПЖ обобщены в табл. 3.
Рецепторы эпидермального фактора роста
Семейство рецепторов эпидермального фактора роста (EGFR) представляет собой группу трансмембранных белков, ответственных за клеточную пролиферацию. Известно 4 вида таких рецепторов: EGFR (HER1, или ErbB1), HER2/neu (ErbB2), HER3 (ErbB3) и HER4 (ErbB4). Повышенная экспрессия EGFR и его лигандов EGF и TGF-альфа характерна для РПЖ [43]. Высокий уровень EGFR в опухоли является неблагоприятным прогностическим фактором, связан с более агрессивным течением заболевания, резистентностью к лучевой терапии [44]. Сейчас в клинической практике применяются две группы таргетных препаратов, блокирующих EGFR-путь: моноклональные антитела непосредственно к внеклеточному домену рецептора (цетуксимаб, панитумумаб) и малые молекулы, блокирующие активность тирозинкиназного домена рецептора (гефитиниб, эрлотиниб).
Эрлотиниб на сегодняшний день – единственный таргетный препарат, одобренный Управлением по контролю пищевых продуктов и лекарственных препаратов США (Food and Drug Administration, FDA) для лечения больных РПЖ. В 2007 г. были опубликованы результаты рандомизированного двойного слепого плацебоконтролируемого исследования III фазы NCIC CTG с участием 569 пациентов, доказавшего эффективность эрлотиниба при РПЖ. Пациенты получали комбинацию эрлотиниба (по 100–150 мг/сут) с гемцитабином (7 еженедельных доз по 1 г/м2) или монотерапию гемцитабином. Медиана общей продолжительности жизни больных, получавших оба препарата, оказалась выше: 6,24 и 5,9 месяца соответственно. Разница составила всего лишь
0,33 месяца, однако оказалась статистически достоверной (p = 0,038), что позволило FDA разрешить применение эрлотиниба при метастатическом РПЖ.
Отметим, что в ходе данного исследования не было выявлено корреляции между уровнем экспрессии EGFR и ответом на терапию эрлотинибом. Развитие у пациентов сыпи 2-й степени выраженности и выше являлось благоприятным прогностическим фактором. Медиана выживаемости пациентов с сыпью 0, 1 и ≥ 2-й степени составила 5,3, 5,8 и 10,5 месяца соответственно, а однолетняя выживаемость – 16, 9 и 43% [45].
Комбинацию эрлотиниба и капецитабина изучали в исследовании II фазы (30 пациентов) в качестве режима второй линии терапии РПЖ, резистентном к гемцитабину. Объективный ответ был зарегистрирован у 10% пациентов, медиана выживаемости составила 6,5 месяца [46].
В рандомизированном исследовании III фазы S0205, проведенном Юго-западной группой онкологов (Southwest Oncology Group, SWOG), 745 пациентов с местнораспространенной нерезектабельной опухолью или метастатическим РПЖ получали гемцитабин в монотерапии или в сочетании с цетуксимабом. Статистически значимой разницы в медиане общей выживаемости (6,9 и 5,9 месяца соответственно, р = 0,14), медиане времени до прогрессирования (3,0 и 3,5 месяца, р = 0,058) и частоте объективных ответов между двумя группами не выявлено. При этом в 90% случаев была обнаружена экспрессия EGFR в опухоли, что никак не повлияло на эффективность лечения [47].
Существует несколько гипотез, объясняющих резистентность аденокарциномы поджелудочной железы к анти-EGFR-терапии. На примере немелкоклеточного рака легкого было показано, что наличие мутации в тирозинкиназном домене EGFR – прогностический фактор в отношении ответа на лечение эрлотинибом. Судя по последним данным, мутация в гене EGFR крайне редко встречается при РПЖ [48, 49].
При блокировании тирозинкиназы EGFR остается возможным активизировать 2 главных внутриклеточных сигнальных пути (PI3K/Akt и MAPK) через тирозинкиназы рецепторов к другим ростовым факторам.
Как упоминалось выше, активирующая мутация гена KRAS определяется уже на ранних стадиях заболевания в 85% случаев [50]. При наличии мутации в гене KRAS анти-EGFR-терапия становится неэффективной [51, 52].
Цетуксимаб оказывает цитотоксическое действие на злокачественную клетку также за счет иммуноопосредованного противоопухолевого эффекта, реализующегося за счет связывания Fc-фрагмента химерного моноклонального антитела с Fc-рецепторами (Fc-гамма-Rs) на поверхности иммунных эффекторных клеток. При полиморфизме рецепторов (Fc-гамма-RIIa и Fc-гамма-RIIIa) лимфоцитов уменьшается их аффинность к Ig G1 (цетуксимаб), что приводит к снижению эффективности лечения [53].
Существует гипотеза, согласно которой опухолевые клетки могут переключать различные молекулярные пути передачи митогенного сигнала. В соответствии с этой концепцией эрлотиниб полностью блокирует EGFR-опосредованный путь, но при этом не оказывает влияния на пролиферацию клетки, активированную инсулиноподобным фактором роста [54].
Сосудистый эндотелиальный фактор роста
Важным условием формирования первичной опухоли и метастазов является неоангиогенез – формирование дополнительной капиллярной сети, обеспечивающей рост опухолевого узла. Белки семейства сосудистого эндотелиального фактора роста (VEGF) и рецепторы к ним обеспечивают образование новых сосудов, что необходимо для роста опухоли. Гиперэкспрессия VEGF наблюдается в 64% клеток РПЖ [55]. Продолжительность жизни пациентов, в опухоли которых определяется высокий уровень экспрессии VEGF, достоверно меньше, чем у пациентов без высокого уровня экспрессии VEGF [56]. Несмотря на многообещающие результаты доклинических испытаний бевацизумаба, в рандомизированном исследовании III фазы CALGB 80303 медианы общей продолжительности жизни в группах пациентов, получавших гемцитабин в сочетании с плацебо или с бевацизумабом, практически не различались: 5,8 и 5,9 месяца (p = 0,95) [57]. Медиана времени до прогрессирования составила 3,8 и 2,9 месяца соответственно (p = 0,07), частота объективных ответов – 13 и 10%.
В двойном слепом рандомизированном исследовании III фазы AVITA, где оценивался режим терапии комбинацией гемцитабина и эрлотиниба в сочетании с бевацизумабом и без него, было выявлено увеличение времени до прогрессирования (4,6 и 3,6 месяца, p = 0,0002), но не общей выживаемости (7,1 и 6 месяцев, p = 0,2087) [58].
Ингибиторы циклооксигеназы 2 типа
Циклооксигеназа (ЦОГ) является ферментом, превращающим арахидоновую кислоту в простагландины, простациклины и тромбоксаны. ЦОГ представлена двумя изоформами: ЦОГ-1, которая постоянно экспрессируется в нормальных тканях, и ЦОГ-2, синтез которой главным образом индуцируется провоспалительными цитокинами, факторами роста и онкогенами. Повышенная экспрессия ЦОГ-2 часто наблюдается и способствует канцерогенезу при многих солидных опухолях, в том числе и РПЖ, поэтому является плохим прогностическим фактором [59, 60, 61].
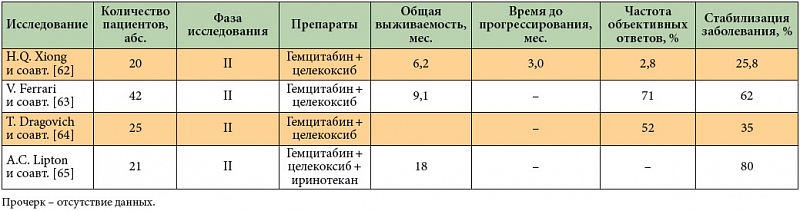
Ингибитор ЦОГ-2 целекоксиб в доклинических испытаниях проявил синергетический эффект при комбинации с цитотоксическими агентами (5-фторурацил, гемцитабин), лучевой терапией и с другими таргетными препаратами (эрлотиниб, куркумин). Однако результаты нескольких небольших исследований II фазы противоречивы [62–65], соответственно, требуются дополнительные исследования (табл. 4).
Инсулиноподобный фактор роста 1 типа
Рецептор инсулиноподобного фактора роста 1 типа (IGFR1) представляет собой трансмембранный тирозинкиназный рецептор, гиперэкспрессия которого наблюдается в 64% случаев РПЖ [66]. В настоящее время проводятся рандомизированные исследования II фазы, в которых изучается эффективность ингибиторов IGFR1, молекул AMG 479 и MK-0646 [67, 68].
Мутация гена BRCA 2
По разным данным, герминальные мутации гена BRCA 2, главным образом 6174delT и 6158insT, встречаются с частотой от 6 до 17% случаев семейного РПЖ [69–72] и в 10% спорадических случаев [73, 74]. Опухолевые клетки с мутациями в генах BRCA 1 / BRCA 2 высокочувствительны к воздействию агентов-кросслинкеров (Митомицин С,
препараты платины). Описаны случаи, когда у пациентов – носителей мутаций BRCA на фоне лечения препаратами платины наступала полная регрессия опухоли поджелудочной железы [75]. В связи с этим одним из перспективных путей развития таргетной терапии РПЖ является изучение PARP-ингибиторов (олапариб, велипариб, BSI-201).
Заключение
На сегодняшний день ни один из химиотерапевтических режимов не является эталоном в лечении диссеминированного РПЖ.
За последние годы было оценено более 33 рандомизированных исследований с участием 6026 больных. Сравнение симптоматического лечения и химиотерапии выявило преимущество последней с точки зрения продолжительности жизни: риск смерти снизился на 36%. Доказано, что продолжительность жизни увеличивается при полихимиотерапии, включающей гемцитабин, по сравнению с монотерапией гемцитабином: риск смерти снизился на 9% (14 исследований с участием 4060 больных). Наиболее активными дуплетами, влияющими на эффективность терапии и на время до прогрессирования, являются комбинации гемцитабина и препаратов платины (3 исследования, 1077 больных) или капецитабина (3 исследования, 935 больных). Наилучшие результаты по продолжительности жизни получены у пациентов, общее состояние которых оценивалось как «хорошее». При лечении ослабленных пациентов предпочтение следует отдавать монотерапии. Химиотерапевтический триплет продемонстрировал преимущества по всем показателям в сравнении с монотерапией гемцитабином, однако требуются проверочные исследования. Из препаратов целевой терапии для лечения РПЖ в настоящее время используется только эрлотиниб. Комбинация гемцитабина с эрлотинибом приводит к незначительному увеличению сроков жизни, но при этом существенно возрастает стоимость лечения и усиливается токсичность.
Отрицательные результаты приведенных выше исследований по комбинированию гемцитабина с таргетными агентами подтверждают теорию о сложности канцерогенеза РПЖ. По-видимому, для реализации противоопухолевого эффекта недостаточно подавления только одного звена патогенетического пути. Необходимо разрабатывать мультимодальный подход к лечению этого заболевания. Наиболее перспективным направлением целевой терапии РПЖ следует считать изучение эффективности PARP-ингибиторов, ингибиторов сигнального пути PI3K-Akt, выявление мишеней и антагонистов к ним в микроокружении опухоли.