–ë–Ψ–Μ–Β–Ζ–Ϋ―¨ –Λ–Α–±―Ä–Η –≤¬†–Ω―Ä–Α–Κ―²–Η–Κ–Β ―Ä–Β–≤–Φ–Α―²–Ψ–Μ–Ψ–≥–Α
- –ê–Ϋ–Ϋ–Ψ―²–Α―Ü–Η―è
- –Γ―²–Α―²―¨―è
- –Γ―¹―΄–Μ–Κ–Η
- English
–ë–Ψ–Μ–Β–Ζ–Ϋ―¨ –Λ–Α–±―Ä–Η¬†βÄ™ –Ω―Ä–Ψ–≥―Ä–Β―¹―¹–Η―Ä―É―é―â–Α―è, –Φ―É–Μ―¨―²–Η―¹–Η―¹―²–Β–Φ–Ϋ–Α―è, –Ξ-―¹―Ü–Β–Ω–Μ–Β–Ϋ–Ϋ–Α―è –Μ–Η–Ζ–Ψ―¹–Ψ–Φ–Ϋ–Α―è –±–Ψ–Μ–Β–Ζ–Ϋ―¨ –Ϋ–Α–Κ–Ψ–Ω–Μ–Β–Ϋ–Η―è –≤―¹–Μ–Β–¥―¹―²–≤–Η–Β –¥–Β―³–Β–Κ―²–Α –≤¬†–≥–Β–Ϋ–Β, –Κ–Ψ–¥–Η―Ä―É―é―â–Β–Φ ―³–Β―Ä–Φ–Β–Ϋ―² –Α–Μ―¨―³–Α-–≥–Α–Μ–Α–Κ―²–Ψ–Ζ–Η–¥–Α–Ζ―É. –î–Α–Ϋ–Ϋ–Α―è –Ω–Α―²–Ψ–Μ–Ψ–≥–Η―è ―¹–Ψ–Ω―Ä–Ψ–≤–Ψ–Ε–¥–Α–Β―²―¹―è –≤―΄―¹–Ψ–Κ–Η–Φ ―Ä–Η―¹–Κ–Ψ–Φ –Ψ―¹–Μ–Ψ–Ε–Ϋ–Β–Ϋ–Η–Ι [1βÄ™3]. –ö―Ä–Ψ–Φ–Β ―²–Ψ–≥–Ψ, –Ψ–Ϋ–Α –Α―¹―¹–Ψ―Ü–Η–Η―Ä―É–Β―²―¹―è ―¹¬†–Ω―Ä–Β–Ε–¥–Β–≤―Ä–Β–Φ–Β–Ϋ–Ϋ–Ψ–Ι ―¹–Φ–Β―Ä―²―¨―é –Ψ―²¬†–Ω–Ψ―΅–Β―΅–Ϋ–Ψ–Ι –Ϋ–Β–¥–Ψ―¹―²–Α―²–Ψ―΅–Ϋ–Ψ―¹―²–Η, –Κ–Α―Ä–¥–Η–Ψ- –Η¬†―Ü–Β―Ä–Β–±―Ä–Ψ–≤–Α―¹–Κ―É–Μ―è―Ä–Ϋ–Ψ–Ι –Ω–Α―²–Ψ–Μ–Ψ–≥–Η–Ι [4].
–û―¹–Ϋ–Ψ–≤–Ϋ–Α―è –Ω―Ä–Η―΅–Η–Ϋ–Η –±–Ψ–Μ–Β–Ζ–Ϋ–Η¬†βÄ™ –Ϋ–Β―¹–Ω–Ψ―¹–Ψ–±–Ϋ–Ψ―¹―²―¨ ―Ä–Α―¹―â–Β–Ω–Μ―è―²―¨ –Ψ–Ω―Ä–Β–¥–Β–Μ–Β–Ϋ–Ϋ―΄–Β –≥–Μ–Η–Κ–Ψ―¹―³–Η–Ϋ–≥–Ψ–Μ–Η–Ω–Η–¥―΄. –≠―²–Ψ –Ω―Ä–Η–≤–Ψ–¥–Η―² –Κ¬†–Ω―Ä–Ψ–≥―Ä–Β―¹―¹–Η―Ä―É―é―â–Β–Φ―É, –≤–Ϋ―É―²―Ä–Η–Μ–Η–Ζ–Ψ―¹–Ψ–Φ–Α–Μ―¨–Ϋ–Ψ–Φ―É –Ϋ–Α–Κ–Ψ–Ω–Μ–Β–Ϋ–Η―é ―¹―É–±―¹―²―Ä–Α―²–Α –≤¬†―Ä–Α–Ζ–Ϋ―΄―Ö ―²–Η–Ω–Α―Ö –Κ–Μ–Β―²–Ψ–Κ [3, 4].
–†–Α―¹–Ω―Ä–Ψ―¹―²―Ä–Α–Ϋ–Β–Ϋ–Ϋ–Ψ―¹―²―¨ –±–Ψ–Μ–Β–Ζ–Ϋ–Η –Λ–Α–±―Ä–Η –≤–Α―Ä―¨–Η―Ä―É–Β―²―¹―è –Ψ―²¬†–Ψ–¥–Ϋ–Ψ–≥–Ψ ―¹–Μ―É―΅–Α―è –Ϋ–Α¬†40 ―²―΄―¹. ―ɬ†–Φ―É–Ε―΅–Η–Ϋ –¥–Ψ –Ψ–¥–Ϋ–Ψ–≥–Ψ ―¹–Μ―É―΅–Α―è –Ϋ–Α¬†117 ―²―΄―¹. –≤¬†–Ψ–±―â–Β–Ι –Ω–Ψ–Ω―É–Μ―è―Ü–Η–Η. –†–Β–Ζ―É–Μ―¨―²–Α―²―΄ –Ϋ–Β–¥–Α–≤–Ϋ–Ψ –Ω―Ä–Ψ–≤–Β–¥–Β–Ϋ–Ϋ–Ψ–≥–Ψ ―Ä–Β–≥–Η–Ψ–Ϋ–Α–Μ―¨–Ϋ–Ψ–≥–Ψ –Ω–Η–Μ–Ψ―²–Ϋ–Ψ–≥–Ψ ―¹–Κ―Ä–Η–Ϋ–Η–Ϋ–≥–Ψ–≤–Ψ–≥–Ψ –Η―¹―¹–Μ–Β–¥–Ψ–≤–Α–Ϋ–Η―è –Ϋ–Ψ–≤–Ψ―Ä–Ψ–Ε–¥–Β–Ϋ–Ϋ―΄―Ö ―¹–≤–Η–¥–Β―²–Β–Μ―¨―¹―²–≤―É―é―², ―΅―²–Ψ –Β–Β –≤―¹―²―Ä–Β―΅–Α–Β–Φ–Ψ―¹―²―¨ ―¹―Ä–Β–¥–Η –Ε–Η–≤–Ψ―Ä–Ψ–Ε–¥–Β–Ϋ–Ϋ―΄―Ö –¥–Ψ―¹―²–Η–≥–Α–Β―² –Ψ–¥–Ϋ–Ψ–≥–Ψ ―¹–Μ―É―΅–Α―è –Ϋ–Α¬†3,1 ―²―΄―¹. –ù–Β―¹–Φ–Ψ―²―Ä―è –Ϋ–Α¬†–Ξ-―¹―Ü–Β–Ω–Μ–Β–Ϋ–Ϋ―΄–Ι ―²–Η–Ω –Ϋ–Α―¹–Μ–Β–¥–Ψ–≤–Α–Ϋ–Η―è –±–Ψ–Μ–Β–Ζ–Ϋ–Η –Λ–Α–±―Ä–Η, –±–Ψ–Μ―¨―à–Α―è –¥–Ψ–Μ―è –≥–Β―²–Β―Ä–Ψ–Ζ–Η–≥–Ψ―²–Ϋ―΄―Ö –Ε–Β–Ϋ―â–Η–Ϋ ―²–Α–Κ–Ε–Β ―¹―²―Ä–Α–¥–Α–Β―² –Ψ―²¬†–≤―΄―Ä–Α–Ε–Β–Ϋ–Ϋ―΄―Ö ―¹–Η–Φ–Ω―²–Ψ–Φ–Ψ–≤ –±–Ψ–Μ–Β–Ζ–Ϋ–Η [5, 6].
–ë–Ψ–Μ–Β–Ζ–Ϋ―¨ –Λ–Α–±―Ä–Η –Φ–Ψ–Ε–Β―² –Φ–Α–Ϋ–Η―³–Β―¹―²–Η―Ä–Ψ–≤–Α―²―¨ –≤¬†―Ä–Α–Ζ–Ϋ–Ψ–Φ –≤–Ψ–Ζ―Ä–Α―¹―²–Β, –Η–Φ–Β―²―¨ ―Ä–Α–Ζ–Ϋ―΄–Ι –Ϋ–Α–±–Ψ―Ä ―¹–Η–Φ–Ω―²–Ψ–Φ–Ψ–≤ –Η¬†―Ö–Α―Ä–Α–Κ―²–Β―Ä –Ω―Ä–Ψ–≥―Ä–Β―¹―¹–Η―Ä–Ψ–≤–Α–Ϋ–Η―è. –û–¥–Ϋ–Α–Κ–Ψ ―΅–Α―â–Β –Ω–Α―²–Ψ–Μ–Ψ–≥–Η―è –Ω―Ä–Ψ―è–≤–Μ―è–Β―²―¹―è –≤¬†–¥–Β―²―¹–Κ–Ψ–Φ –≤–Ψ–Ζ―Ä–Α―¹―²–Β –Η¬†―É –Ω–Ψ–¥―Ä–Ψ―¹―²–Κ–Ψ–≤ [1, 5].
–ö–Μ–Η–Ϋ–Η―΅–Β―¹–Κ–Η–Β –Ω―Ä–Ψ―è–≤–Μ–Β–Ϋ–Η―è
–û–¥–Ϋ–Η–Φ –Η–Ζ¬†–Ϋ–Α–Η–±–Ψ–Μ–Β–Β –Η–Ϋ–≤–Α–Μ–Η–¥–Η–Ζ–Η―Ä―É―é―â–Η―Ö ―¹–Η–Φ–Ω―²–Ψ–Φ–Ψ–≤ ―ɬ†–¥–Β―²–Β–Ι –Ω―Ä–Η–Ζ–Ϋ–Α–Ϋ–Α –Ω–Β―Ä–Η―³–Β―Ä–Η―΅–Β―¹–Κ–Α―è –Ϋ–Β–Ι―Ä–Ψ–Ω–Α―²–Η―΅–Β―¹–Κ–Α―è –±–Ψ–Μ―¨. –û–Ϋ–Α –Ψ–Ω–Η―¹―΄–≤–Α–Β―²―¹―è –Κ–Α–Κ ―Ö―Ä–Ψ–Ϋ–Η―΅–Β―¹–Κ–Α―è, –Ϋ–Ψ―é―â–Α―è, –Ω–Ψ–Κ–Α–Μ―΄–≤–Α―é―â–Α―è, –Ε–≥―É―΅–Α―è –≤¬†–Μ–Α–¥–Ψ–Ϋ―è―Ö –Η¬†–Ω–Ψ–¥–Ψ―à–≤–Α―Ö. –û―¹―²―Ä–Α―è, –Φ―É―΅–Η―²–Β–Μ―¨–Ϋ–Α―è –±–Ψ–Μ―¨ –≤¬†–Κ–Ψ–Ϋ–Β―΅–Ϋ–Ψ―¹―²―è―Ö –Φ–Ψ–Ε–Β―² –±―΄―²―¨ –≤―΄–Ζ–≤–Α–Ϋ–Α –±–Ψ–Μ–Β–Ζ–Ϋ―¨―é, ―³–Η–Ζ–Η―΅–Β―¹–Κ–Η–Φ–Η ―É–Ω―Ä–Α–Ε–Ϋ–Β–Ϋ–Η―è–Φ–Η, –Μ–Η―Ö–Ψ―Ä–Α–¥–Κ–Ψ–Ι, ―¹―²―Ä–Β―¹―¹–Ψ–Φ –Η–Μ–Η –Η–Ζ–Φ–Β–Ϋ–Β–Ϋ–Η―è–Φ–Η –Ω–Ψ–≥–Ψ–¥―΄. –≠―²–Η ―ç–Ω–Η–Ζ–Ψ–¥―΄ –Ω―Ä–Ψ–¥–Ψ–Μ–Ε–Α―é―²―¹―è –Ψ―²¬†–Ϋ–Β―¹–Κ–Ψ–Μ―¨–Κ–Η―Ö –Φ–Η–Ϋ―É―² –¥–Ψ –Ϋ–Β―¹–Κ–Ψ–Μ―¨–Κ–Η―Ö –Ϋ–Β–¥–Β–Μ―¨ –Η, –Κ–Α–Κ –Ω―Ä–Α–≤–Η–Μ–Ψ, ―¹–Ψ–Ω―Ä–Ψ–≤–Ψ–Ε–¥–Α―é―²―¹―è –Μ–Η―Ö–Ψ―Ä–Α–¥–Κ–Ψ–Ι, –±–Ψ–Μ―è–Φ–Η –≤¬†―¹―É―¹―²–Α–≤–Α―Ö, –Η–Ϋ–Ψ–≥–¥–Α –Ω–Ψ–≤―΄―à–Β–Ϋ–Η–Β–Φ ―¹–Κ–Ψ―Ä–Ψ―¹―²–Η –Ψ―¹–Β–¥–Α–Ϋ–Η―è ―ç―Ä–Η―²―Ä–Ψ―Ü–Η―²–Ψ–≤ (–Γ–û–≠). –ß–Α―¹―²–Ψ –Ψ–Ϋ–Η –Ψ―à–Η–±–Ψ―΅–Ϋ–Ψ –Ω―Ä–Η–Ϋ–Η–Φ–Α―é―²―¹―è –Ζ–Α ―¹―É―¹―²–Α–≤–Ϋ―΄–Β –±–Ψ–Μ–Η [7, 8].
–£¬†–¥–Β―²―¹–Κ–Ψ–Φ –≤–Ψ–Ζ―Ä–Α―¹―²–Β –Ψ―²–Φ–Β―΅–Α―é―²―¹―è ―Ö–Α―Ä–Α–Κ―²–Β―Ä–Ϋ―΄–Β –¥–Μ―è –±–Ψ–Μ–Β–Ζ–Ϋ–Η –Λ–Α–±―Ä–Η –Ω–Ψ―Ä–Α–Ε–Β–Ϋ–Η―è –Κ–Ψ–Ε–Η¬†βÄ™ –Α–Ϋ–≥–Η–Ψ–Κ–Β―Ä–Α―²–Ψ–Φ―΄, –Κ–Ψ―²–Ψ―Ä―΄–Β –Μ–Ψ–Κ–Α–Μ–Η–Ζ―É―é―²―¹―è ―¹–Η–Φ–Φ–Β―²―Ä–Η―΅–Ϋ–Ψ –Ϋ–Α¬†–±–Β–¥―Ä–Α―Ö, ―¹–Ω–Η–Ϋ–Β, ―è–≥–Ψ–¥–Η―Ü–Α―Ö, –≤¬†–Ψ–±–Μ–Α―¹―²–Η –Κ–Ψ–Μ–Β–Ϋ–Ϋ―΄―Ö ―¹―É―¹―²–Α–≤–Ψ–≤ –Η¬†–Ω―É–Ω–Κ–Α, –Α¬†―²–Α–Κ–Ε–Β –≥–Η–Ω–Ψ- –Η–Μ–Η –Α–Ϋ–≥–Η–¥―Ä–Ψ–Ζ.
–ü―Ä–Η –±–Ψ–Μ–Β–Ζ–Ϋ–Η –Λ–Α–±―Ä–Η –Ω―Ä–Ψ–Η―¹―Ö–Ψ–¥–Η―² –Ϋ–Α–Κ–Ψ–Ω–Μ–Β–Ϋ–Η–Β ―¹―³–Η–Ϋ–≥–Ψ–Μ–Η–Ω–Η–¥–Ψ–≤ –Η¬†–≥–Μ–Η–Κ–Ψ–Ω―Ä–Ψ―²–Β–Η–Ϋ–Α –≤¬†–Ψ―Ä–≥–Α–Ϋ–Α―Ö ―¹–Β―Ä–¥–Β―΅–Ϋ–Ψ-―¹–Ψ―¹―É–¥–Η―¹―²–Ψ–Ι ―¹–Η―¹―²–Β–Φ―΄ –Η¬†–Ω–Ψ―΅–Κ–Α―Ö. –ü–Ψ―ç―²–Ψ–Φ―É –Ψ―²–Φ–Β―΅–Α―é―²―¹―è ―¹–Η–Φ–Ω―²–Ψ–Φ―΄ ―¹―²–Β–Ϋ–Ψ–Κ–Α―Ä–¥–Η–Η, –Ω–Ψ―Ä–Α–Ε–Β–Ϋ–Η–Β –Κ–Μ–Α–Ω–Α–Ϋ–Ψ–≤ ―¹–Β―Ä–¥―Ü–Α (–Φ–Η―²―Ä–Α–Μ―¨–Ϋ–Α―è –Ϋ–Β–¥–Ψ―¹―²–Α―²–Ψ―΅–Ϋ–Ψ―¹―²―¨ –Η¬†–Α–Ψ―Ä―²–Α–Μ―¨–Ϋ―΄–Ι ―¹―²–Β–Ϋ–Ψ–Ζ), –≥–Η–Ω–Β―Ä―²―Ä–Ψ―³–Η―è –Μ–Β–≤–Ψ–≥–Ψ –Ε–Β–Μ―É–¥–Ψ―΅–Κ–Α, –Ω―Ä–Ψ―²–Β–Η–Ϋ―É―Ä–Η―è, ―¹–Ϋ–Η–Ε–Β–Ϋ–Η–Β ―³–Η–Μ―¨―²―Ä–Α―Ü–Η–Ψ–Ϋ–Ϋ–Ψ–Ι ―¹–Ω–Ψ―¹–Ψ–±–Ϋ–Ψ―¹―²–Η –Ω–Ψ―΅–Β–Κ, –Ω–Ψ―΅–Β―΅–Ϋ–Α―è –Ϋ–Β–¥–Ψ―¹―²–Α―²–Ψ―΅–Ϋ–Ψ―¹―²―¨.
–ê―Ä―²–Β―Ä–Η–Α–Μ―¨–Ϋ–Α―è –≥–Η–Ω–Β―Ä―²–Β–Ϋ–Ζ–Η―è (―¹–Κ–Ψ―Ä–Β–Β –≤―¹–Β–≥–Ψ, –Ω–Ψ―΅–Β―΅–Ϋ–Ψ–≥–Ψ –Ω―Ä–Ψ–Η―¹―Ö–Ψ–Ε–¥–Β–Ϋ–Η―è) ―É―¹–Η–Μ–Η–≤–Α–Β―² –Ϋ–Α―Ä―É―à–Β–Ϋ–Η―è ―¹–Ψ¬†―¹―²–Ψ―Ä–Ψ–Ϋ―΄ ―¹–Β―Ä–¥―Ü–Α –Η¬†―¹–Ω–Ψ―¹–Ψ–±―¹―²–≤―É–Β―² –Ω–Ψ―Ä–Α–Ε–Β–Ϋ–Η―é ―¹–Ψ―¹―É–¥–Ψ–≤ –Φ–Ψ–Ζ–≥–Α.
–ö¬†–Η–Ζ–Φ–Β–Ϋ–Β–Ϋ–Η―è–Φ ―¹–Ψ¬†―¹―²–Ψ―Ä–Ψ–Ϋ―΄ –Ε–Β–Μ―É–¥–Ψ―΅–Ϋ–Ψ-–Κ–Η―à–Β―΅–Ϋ–Ψ–≥–Ψ ―²―Ä–Α–Κ―²–Α –Ψ―²–Ϋ–Ψ―¹―è―²―¹―è ―²–Ψ―à–Ϋ–Ψ―²–Α, ―Ä–≤–Ψ―²–Α, –¥–Η–Α―Ä–Β―è, –±–Ψ–Μ–Η –≤¬†–Ε–Η–≤–Ψ―²–Β [8].
–ù–Β―Ä–Β–¥–Κ–Ψ –Ψ–±–Ϋ–Α―Ä―É–Ε–Η–≤–Α―é―²―¹―è –¥–Β―³–Ψ―Ä–Φ–Α―Ü–Η–Η –¥–Η―¹―²–Α–Μ―¨–Ϋ―΄―Ö –Φ–Β–Ε―³–Α–Μ–Α–Ϋ–≥–Ψ–≤―΄―Ö ―¹―É―¹―²–Α–≤–Ψ–≤, –Ψ―¹―²–Β–Ψ–Ω–Ψ―Ä–Ψ–Ζ –Ω–Ψ–Ζ–≤–Ψ–Ϋ–Κ–Ψ–≤ –Η¬†–Α―¹–Β–Ω―²–Η―΅–Β―¹–Κ–Η–Ι –Ϋ–Β–Κ―Ä–Ψ–Ζ –≥–Ψ–Μ–Ψ–≤–Κ–Η –±–Β–¥―Ä–Β–Ϋ–Ϋ–Ψ–Ι –Η¬†―²–Α―Ä–Α–Ϋ–Ϋ―΄―Ö –Κ–Ψ―¹―²–Β–Ι, ―΅―²–Ψ –Φ–Ψ–Ε–Β―² ―¹―²–Α―²―¨ –Ω―Ä–Η―΅–Η–Ϋ–Ψ–Ι –Ω–Ψ―¹―²–Α–Ϋ–Ψ–≤–Κ–Η –Ϋ–Β–≤–Β―Ä–Ϋ–Ψ–≥–Ψ –¥–Η–Α–≥–Ϋ–Ψ–Ζ–Α.
–£–Ψ–Ζ–Φ–Ψ–Ε–Ϋ–Α ―²–Α–Κ–Ε–Β –Ζ–Α–¥–Β―Ä–Ε–Κ–Α ―Ä–Ψ―¹―²–Α –Η¬†–Ω–Ψ–Μ–Ψ–≤–Ψ–≥–Ψ ―Ä–Α–Ζ–≤–Η―²–Η―è.
–î–Η–Α–≥–Ϋ–Ψ―¹―²–Η–Κ–Α
–ù–Β―¹–Φ–Ψ―²―Ä―è –Ϋ–Α¬†―²–Ψ ―΅―²–Ψ –±–Ψ–Μ–Β–Ζ–Ϋ―¨ –Λ–Α–±―Ä–Η, –Κ–Α–Κ –Ω―Ä–Α–≤–Η–Μ–Ψ, –Φ–Α–Ϋ–Η―³–Β―¹―²–Η―Ä―É–Β―² –≤¬†–¥–Β―²―¹–Κ–Ψ–Φ –Η–Μ–Η –Ω–Ψ–¥―Ä–Ψ―¹―²–Κ–Ψ–≤–Ψ–Φ –≤–Ψ–Ζ―Ä–Α―¹―²–Β, –Ψ–Ϋ–Α ―΅–Α―¹―²–Ψ –Ψ―¹―²–Α–Β―²―¹―è –Ϋ–Β–Ζ–Α–Φ–Β―΅–Β–Ϋ–Ϋ–Ψ–Ι –¥–Ψ ―¹–Ψ–≤–Β―Ä―à–Β–Ϋ–Ϋ–Ψ–Μ–Β―²–Η―è. –ù–Β―Ä–Β–¥–Κ–Ψ –Ω–Α―Ü–Η–Β–Ϋ―²―΄ –Ε–Η–≤―É―² ―¹¬†–Ϋ–Β–≤–Β―Ä–Ϋ―΄–Φ –¥–Η–Α–≥–Ϋ–Ψ–Ζ–Ψ–Φ –¥–Ψ 30-–Μ–Β―²–Ϋ–Β–≥–Ψ –≤–Ψ–Ζ―Ä–Α―¹―²–Α [1]. –î–Α–Ϋ–Ϋ―΄–Β ―Ä–Β–≥–Η―¹―²―Ä–Α –Ω–Α―Ü–Η–Β–Ϋ―²–Ψ–≤ ―¹¬†–±–Ψ–Μ–Β–Ζ–Ϋ―¨―é –Λ–Α–±―Ä–Η ―¹–≤–Η–¥–Β―²–Β–Μ―¨―¹―²–≤―É―é―², ―΅―²–Ψ –Ζ–Α–¥–Β―Ä–Ε–Κ–Α –≤¬†–Ω–Ψ―¹―²–Α–Ϋ–Ψ–≤–Κ–Β –¥–Η–Α–≥–Ϋ–Ψ–Ζ–Α ―¹–Ψ―¹―²–Α–≤–Μ―è–Β―² 14 –Μ–Β―² –¥–Μ―è –Φ―É–Ε―΅–Η–Ϋ –Η¬†19 –Μ–Β―² –¥–Μ―è –Ε–Β–Ϋ―â–Η–Ϋ [1]. –≠―²–Ψ –¥–Β–Φ–Ψ–Ϋ―¹―²―Ä–Η―Ä―É–Β―² –Κ–Α–Κ –Ϋ–Β―¹–Ω–Β―Ü–Η―³–Η―΅–Β―¹–Κ–Η–Ι ―Ö–Α―Ä–Α–Κ―²–Β―Ä ―Ä–Α–Ϋ–Ϋ–Η―Ö –Ε–Α–Μ–Ψ–±, ―²–Α–Κ –Η¬†–Ψ―²―¹―É―²―¹―²–≤–Η–Β ―Ä–Α―¹–Ω–Ψ–Ζ–Ϋ–Α–≤–Α–Ϋ–Η―è –≤―Ä–Α―΅–Α–Φ–Η ―Ä–Α–Ζ–Ϋ―΄―Ö ―¹–Ω–Β―Ü–Η–Α–Μ―¨–Ϋ–Ψ―¹―²–Β–Ι ―Ä–Α–Ϋ–Ϋ–Η―Ö ―¹–Η–Φ–Ω―²–Ψ–Φ–Ψ–≤ –Ω–Α―²–Ψ–Μ–Ψ–≥–Η–Η.
–î–Μ―è ―Ä–Α–Ϋ–Ϋ–Β–≥–Ψ –≤―΄―è–≤–Μ–Β–Ϋ–Η―è –Ζ–Α–±–Ψ–Μ–Β–≤–Α–Ϋ–Η―è ―²―Ä–Β–±―É–Β―²―¹―è –≤–Ζ–Α–Η–Φ–Ψ–¥–Β–Ι―¹―²–≤–Η–Β –≥–Β–Ϋ–Β―²–Η–Κ–Ψ–≤, –Ϋ–Β―³―Ä–Ψ–Μ–Ψ–≥–Ψ–≤, –Ϋ–Β–≤―Ä–Ψ–Μ–Ψ–≥–Ψ–≤, –Ω–Β–¥–Η–Α―²―Ä–Ψ–≤, –¥–Β―Ä–Φ–Α―²–Ψ–Μ–Ψ–≥–Ψ–≤, –Κ–Α―Ä–¥–Η–Ψ–Μ–Ψ–≥–Ψ–≤, –Ψ―³―²–Α–Μ―¨–Φ–Ψ–Μ–Ψ–≥–Ψ–≤, ―Ä–Β–≤–Φ–Α―²–Ψ–Μ–Ψ–≥–Ψ–≤, –≥–Α―¹―²―Ä–Ψ―ç–Ϋ―²–Β―Ä–Ψ–Μ–Ψ–≥–Ψ–≤ –Η¬†―¹–Ω–Β―Ü–Η–Α–Μ–Η―¹―²–Ψ–≤ –Ω–Β―Ä–≤–Η―΅–Ϋ–Ψ–≥–Ψ –Ζ–≤–Β–Ϋ–Α. –≠―²–Ψ ―΅―Ä–Β–Ζ–≤―΄―΅–Α–Ι–Ϋ–Ψ –≤–Α–Ε–Ϋ–Ψ, –Ω–Ψ―¹–Κ–Ψ–Μ―¨–Κ―É ―ç―³―³–Β–Κ―²–Η–≤–Ϋ–Α―è –Ω–Α―²–Ψ–≥–Β–Ϋ–Β―²–Η―΅–Β―¹–Κ–Α―è ―²–Β―Ä–Α–Ω–Η―è –Φ–Ψ–Ε–Β―² –Ω―Ä–Β–¥–Ψ―²–≤―Ä–Α―²–Η―²―¨ –Η–Μ–Η –Ζ–Α–Φ–Β–¥–Μ–Η―²―¨ –Β–≥–Ψ –Ω―Ä–Ψ–≥―Ä–Β―¹―¹–Η―Ä–Ψ–≤–Α–Ϋ–Η–Β.
–‰―¹–Κ–Μ―é―΅–Η―²―¨ ―¹–Η―¹―²–Β–Φ–Ϋ–Ψ–Β ―Ä–Β–≤–Φ–Α―²–Η―΅–Β―¹–Κ–Ψ–Β –Ζ–Α–±–Ψ–Μ–Β–≤–Α–Ϋ–Η–Β –≤¬†–Ϋ–Α―΅–Α–Μ–Β –¥–Η–Α–≥–Ϋ–Ψ―¹―²–Η–Κ–Η –Ω–Ψ–Ζ–≤–Ψ–Μ―è–Β―² –Ϋ–Α–Μ–Η―΅–Η–Β ―²–Α–Κ–Η―Ö –Ω―Ä–Η–Ζ–Ϋ–Α–Κ–Ψ–≤, –Κ–Α–Κ –Ω–Ψ―Ä–Α–Ε–Β–Ϋ–Η–Β ―¹―É―¹―²–Α–≤–Ψ–≤, –Κ–Ψ–Ε–Η, –Ω–Ψ―΅–Β–Κ –Η¬†–Μ–Η―Ö–Ψ―Ä–Α–¥–Κ–Α.
–ü―Ä–Η –Ω–Ψ–¥–Ψ–Ζ―Ä–Β–Ϋ–Η–Η –Ϋ–Α¬†–±–Ψ–Μ–Β–Ζ–Ϋ―¨ –Λ–Α–±―Ä–Η –Ω―Ä–Ψ–≤–Ψ–¥―è―² –Ψ–±―¹–Μ–Β–¥–Ψ–≤–Α–Ϋ–Η–Β –Ϋ–Α¬†–Ϋ–Β–¥–Ψ―¹―²–Α―²–Ψ―΅–Ϋ–Ψ―¹―²―¨ ―³–Β―Ä–Φ–Β–Ϋ―²–Α –Α–Μ―¨―³–Α-–≥–Α–Μ–Α–Κ―²–Ψ–Ζ–Η–¥–Α–Ζ―΄.
–¦–Β―΅–Β–Ϋ–Η–Β
–£¬†–Ϋ–Α―¹―²–Ψ―è―â–Β–Β –≤―Ä–Β–Φ―è –Ω―Ä–Η –±–Ψ–Μ–Β–Ζ–Ϋ–Η –Λ–Α–±―Ä–Η –Η―¹–Ω–Ψ–Μ―¨–Ζ―É–Β―²―¹―è ―³–Β―Ä–Φ–Β–Ϋ―²-–Ζ–Α–Φ–Β―¹―²–Η―²–Β–Μ―¨–Ϋ–Α―è ―²–Β―Ä–Α–Ω–Η―è. –û–Ϋ–Α ―¹–Ω–Ψ―¹–Ψ–±―¹―²–≤―É–Β―² –≤–Ψ―¹―¹―²–Α–Ϋ–Ψ–≤–Μ–Β–Ϋ–Η―é ―³–Η–Ζ–Η–Ψ–Μ–Ψ–≥–Η―΅–Β―¹–Κ–Ψ–≥–Ψ ―É―Ä–Ψ–≤–Ϋ―è ―³–Β―Ä–Φ–Β–Ϋ―²–Α, ―É–Φ–Β–Ϋ―¨―à–Β–Ϋ–Η―é –Ϋ–Α–Κ–Ψ–Ω–Μ–Β–Ϋ–Η―è ―¹―É–±―¹―²―Ä–Α―²–Α (Gb3) –≤¬†―²–Κ–Α–Ϋ―è―Ö –Η¬†–Ϋ–Ψ―Ä–Φ–Α–Μ–Η–Ζ–Α―Ü–Η–Η ―³―É–Ϋ–Κ―Ü–Η–Η –≤–Ϋ―É―²―Ä–Β–Ϋ–Ϋ–Η―Ö –Ψ―Ä–≥–Α–Ϋ–Ψ–≤.
–Γ–≤–Ψ–Β–≤―Ä–Β–Φ–Β–Ϋ–Ϋ–Ψ –Ϋ–Α―΅–Α―²–Α―è –Ω–Α―²–Ψ–≥–Β–Ϋ–Β―²–Η―΅–Β―¹–Κ–Α―è ―²–Β―Ä–Α–Ω–Η―è –Φ–Ψ–Ε–Β―² –Ζ–Ϋ–Α―΅–Η―²–Β–Μ―¨–Ϋ–Ψ ―¹–Ϋ–Η–Ζ–Η―²―¨ –Η–Ϋ―²–Β–Ϋ―¹–Η–≤–Ϋ–Ψ―¹―²―¨ –Ϋ–Β–≤―Ä–Ψ–Ω–Α―²–Η―΅–Β―¹–Κ–Ψ–Ι –±–Ψ–Μ–Η –Η–Μ–Η –Ω–Ψ–Μ–Ϋ–Ψ―¹―²―¨―é –Κ―É–Ω–Η―Ä–Ψ–≤–Α―²―¨ –Β–Β, ―É–Μ―É―΅―à–Η―²―¨ ―¹–Μ―É―Ö, –Ω–Ψ–Μ–Ψ–Ε–Η―²–Β–Μ―¨–Ϋ–Ψ –Ω–Ψ–≤–Μ–Η―è―²―¨ –Ϋ–Α¬†―¹–Ψ―¹―²–Ψ―è–Ϋ–Η–Β –Ω–Ψ―΅–Β–Κ, ―¹–Β―Ä–¥–Β―΅–Ϋ–Ψ-―¹–Ψ―¹―É–¥–Η―¹―²–Ψ–Ι ―¹–Η―¹―²–Β–Φ―΄, ―É―¹―²―Ä–Α–Ϋ–Η―²―¨ –≥–Α―¹―²―Ä–Ψ–Η–Ϋ―²–Β―¹―²–Η–Ϋ–Α–Μ―¨–Ϋ―΄–Β –Ω―Ä–Ψ―è–≤–Μ–Β–Ϋ–Η―è –±–Ψ–Μ–Β–Ζ–Ϋ–Η [9].
–£¬†–Ϋ–Α―¹―²–Ψ―è―â–Β–Β –≤―Ä–Β–Φ―è –≤¬†–†–Ψ―¹―¹–Η–Η –Ζ–Α―Ä–Β–≥–Η―¹―²―Ä–Η―Ä–Ψ–≤–Α–Ϋ―΄ –¥–≤–Α –Ω―Ä–Β–Ω–Α―Ä–Α―²–Α –¥–Μ―è –Μ–Β―΅–Β–Ϋ–Η―è –±–Ψ–Μ–Β–Ζ–Ϋ–Η –Λ–Α–±―Ä–Η: –Α–≥–Α–Μ―¹–Η–¥–Α–Ζ–Α –Α–Μ―¨―³–Α 0,2 –Φ–≥/–Κ–≥ –Η¬†–Α–≥–Α–Μ―¹–Η–¥–Α–Ζ–Α –±–Β―²–Α 1 –Φ–≥/–Κ–≥. –ü―Ä–Β–Ω–Α―Ä–Α―²―΄ –≤–≤–Ψ–¥―è―²―¹―è –≤–Ϋ―É―²―Ä–Η–≤–Β–Ϋ–Ϋ–Ψ –Ψ–¥–Η–Ϋ ―Ä–Α–Ζ –≤¬†–¥–≤–Β –Ϋ–Β–¥–Β–Μ–Η [9, 10].
–ö–Μ–Η–Ϋ–Η―΅–Β―¹–Κ–Η–Ι ―¹–Μ―É―΅–Α–Ι
–ü–Α―Ü–Η–Β–Ϋ―² –ö. 29 –Μ–Β―² –¥–Ψ―¹―²–Α–≤–Μ–Β–Ϋ –±―Ä–Η–≥–Α–¥–Ψ–Ι ―¹–Κ–Ψ―Ä–Ψ–Ι –Ω–Ψ–Φ–Ψ―â–Η –≤¬†–Ψ―²–¥–Β–Μ–Β–Ϋ–Η–Β ―Ä–Β–≤–Φ–Α―²–Ψ–Μ–Ψ–≥–Η–Η –ü–Β–Ϋ–Ζ–Β–Ϋ―¹–Κ–Ψ–Ι –Ψ–±–Μ–Α―¹―²–Ϋ–Ψ–Ι –Κ–Μ–Η–Ϋ–Η―΅–Β―¹–Κ–Ψ–Ι –±–Ψ–Μ―¨–Ϋ–Η―Ü―΄ ―¹¬†–¥–Η–Α–≥–Ϋ–Ψ–Ζ–Ψ–Φ ¬Ϊ–≥–Β–Φ–Ψ―Ä―Ä–Α–≥–Η―΅–Β―¹–Κ–Η–Ι –≤–Α―¹–Κ―É–Μ–Η―²¬Μ.
–£¬†–Η―é–Ϋ–Β 2015 –≥.¬†–Ω―Ä–Β–¥―ä―è–≤–Μ―è–Μ –Ε–Α–Μ–Ψ–±―΄ –Ϋ–Α¬†–Ε–≥―É―΅–Η–Β –Ω―Ä–Η―¹―²―É–Ω–Ψ–Ψ–±―Ä–Α–Ζ–Ϋ―΄–Β –±–Ψ–Μ–Η –≤¬†―¹―É―¹―²–Α–≤–Α―Ö –Κ–Η―¹―²–Β–Ι, –Μ–Ψ–Κ―²–Β–≤―΄―Ö –Η¬†–Κ–Ψ–Μ–Β–Ϋ–Ϋ―΄―Ö ―¹―É―¹―²–Α–≤–Α―Ö, –Ψ–Ϋ–Β–Φ–Β–Ϋ–Η–Β –≤¬†―Ä―É–Κ–Α―Ö –Η¬†–Ϋ–Ψ–≥–Α―Ö, ―¹–Ϋ–Η–Ε–Β–Ϋ–Η–Β ―΅―É–≤―¹―²–≤–Η―²–Β–Μ―¨–Ϋ–Ψ―¹―²–Η –≤¬†–Ϋ–Ψ–≥–Α―Ö, –≤―΄―¹―΄–Ω–Α–Ϋ–Η―è –±–Β–Ζ –Ζ―É–¥–Α –Ϋ–Α¬†–Κ–Ψ–Ε–Β –Ϋ–Ψ–≥ –Η¬†–Ε–Η–≤–Ψ―²–Α, –Ω–Μ–Ψ―Ö―É―é –Ω–Β―Ä–Β–Ϋ–Ψ―¹–Η–Φ–Ψ―¹―²―¨ –Ε–Α―Ä―΄, ―à―É–Φ –≤¬†―É―à–Α―Ö.
–‰–Ζ¬†–Α–Ϋ–Α–Φ–Ϋ–Β–Ζ–Α: ―¹¬†–¥–Β―¹―è―²–Η –Μ–Β―² ―¹―²–Α–Μ –Ψ―²–Φ–Β―΅–Α―²―¨ ―²–Β–Φ–Ϋ–Ψ-–±–Ψ―Ä–¥–Ψ–≤―΄–Β –Φ–Β–Μ–Κ–Ψ―²–Ψ―΅–Β―΅–Ϋ―΄–Β –≤―΄―¹―΄–Ω–Α–Ϋ–Η―è –≤¬†–Ψ–±–Μ–Α―¹―²–Η –Ω―É–Ω–Κ–Α, –Ζ–Α―²–Β–Φ –≤¬†–Ψ–±–Μ–Α―¹―²–Η –Μ–Ψ–Κ―²–Β–Ι, –±–Β–¥–Β―Ä –Η¬†–Ω–Ψ―è―¹–Ϋ–Η―Ü―΄, –Ϋ–Α¬†–≥―É–±–Α―Ö, –≤–Β–Κ–Α―Ö. –ü–Ψ–Μ―É―΅–Η–Μ –Κ–Ψ–Ϋ―¹―É–Μ―¨―²–Α―Ü–Η―é –¥–Β―Ä–Φ–Α―²–Ψ–Μ–Ψ–≥–Α, –Ω–Ψ―¹―²–Α–≤–Μ–Β–Ϋ –¥–Η–Α–≥–Ϋ–Ψ–Ζ ¬Ϊ–≥–Β–Φ–Α–Ϋ–≥–Η–Ψ–Φ–Α―²–Ψ–Ζ¬Μ. –Γ¬†12 –Μ–Β―²¬†βÄ™ –Ω―Ä–Η―¹―²―É–Ω–Ψ–Ψ–±―Ä–Α–Ζ–Ϋ―΄–Β –Ε–≥―É―΅–Η–Β –±–Ψ–Μ–Η –≤¬†–Κ―Ä―É–Ω–Ϋ―΄―Ö –Η¬†–Φ–Β–Μ–Κ–Η―Ö ―¹―É―¹―²–Α–≤–Α―Ö, –±–Β–Ζ –Ψ―²–Β―΅–Ϋ–Ψ―¹―²–Η, –Ϋ–Ψ¬†―¹ –Ω–Ψ–≤―΄―à–Β–Ϋ–Η–Β–Φ ―²–Β–Φ–Ω–Β―Ä–Α―²―É―Ä―΄ –¥–Ψ ―¹―É–±―³–Β–±―Ä–Η–Μ―¨–Ϋ–Ψ–Ι. –ü―Ä–Η―¹―²―É–Ω―΄ –±–Ψ–Μ–Η –Ω―Ä–Ψ–≤–Ψ―Ü–Η―Ä–Ψ–≤–Α–Μ–Η―¹―¨ –Ω―Ä–Β–±―΄–≤–Α–Ϋ–Η–Β–Φ –Ϋ–Α¬†―¹–Ψ–Μ–Ϋ―Ü–Β. –¦–Β―΅–Β–Ϋ–Η–Β –Ω―Ä–Β–¥–Ϋ–Η–Ζ–Ψ–Μ–Ψ–Ϋ–Ψ–Φ –Η¬†–Ϋ–Β―¹―²–Β―Ä–Ψ–Η–¥–Ϋ―΄–Φ–Η –Ω―Ä–Ψ―²–Η–≤–Ψ–≤–Ψ―¹–Ω–Α–Μ–Η―²–Β–Μ―¨–Ϋ―΄–Φ–Η –Ω―Ä–Β–Ω–Α―Ä–Α―²–Α–Φ–Η –Ϋ–Β¬†–±―΄–Μ–Ψ ―ç―³―³–Β–Κ―²–Η–≤–Ϋ―΄–Φ.
–Γ¬†20 –Μ–Β―² –Ω–Α―Ü–Η–Β–Ϋ―² –Ψ―²–Φ–Β―΅–Α–Μ –Ψ–Ϋ–Β–Φ–Β–Ϋ–Η–Β –Η¬†―¹–Ϋ–Η–Ε–Β–Ϋ–Η–Β ―΅―É–≤―¹―²–≤–Η―²–Β–Μ―¨–Ϋ–Ψ―¹―²–Η –≤¬†–≤–Β―Ä―Ö–Ϋ–Η―Ö –Η¬†–Ϋ–Η–Ε–Ϋ–Η―Ö –Κ–Ψ–Ϋ–Β―΅–Ϋ–Ψ―¹―²―è―Ö, –Η―Ö –Ω–Ψ―Ö–Ψ–Μ–Ψ–¥–Α–Ϋ–Η–Β.
–Γ¬†28 –Μ–Β―² –Ω―Ä–Η―¹–Ψ–Β–¥–Η–Ϋ–Η–Μ–Η―¹―¨ –≥–Ψ–Μ–Ψ–≤–Ϋ―΄–Β –±–Ψ–Μ–Η, ―à―É–Φ –≤¬†―É―à–Α―Ö, ―¹¬†29 –Μ–Β―²¬†βÄ™ –±–Ψ–Μ–Η –≤¬†–Ψ–±–Μ–Α―¹―²–Η ―¹–Β―Ä–¥―Ü–Α.
–½–Α ―²―Ä–Η –¥–Ϋ―è –¥–Ψ –≥–Ψ―¹–Ω–Η―²–Α–Μ–Η–Ζ–Α―Ü–Η–Η –Ε–Α–Μ–Ψ–≤–Α–Μ―¹―è –Ϋ–Α¬†―¹–Η–Μ―¨–Ϋ―΄–Β –Ε–≥―É―΅–Η–Β –±–Ψ–Μ–Η –≤¬†―¹―É―¹―²–Α–≤–Α―Ö.
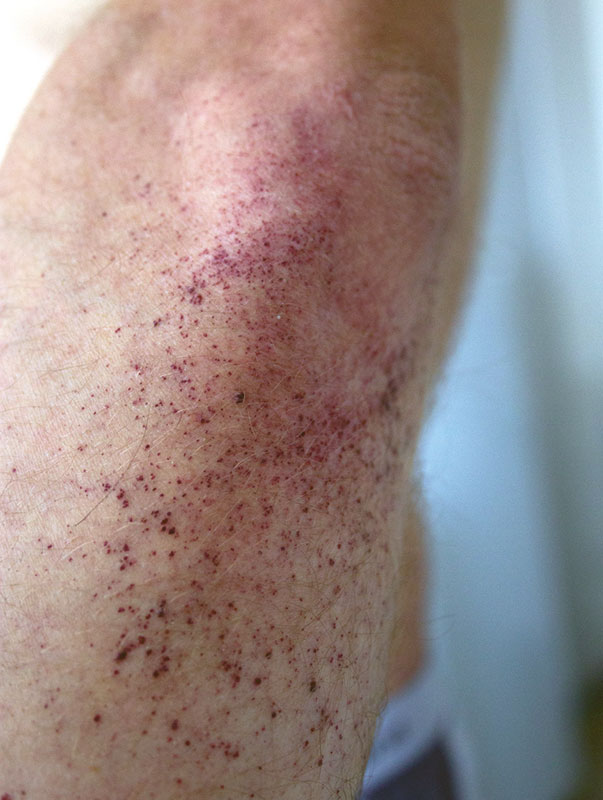
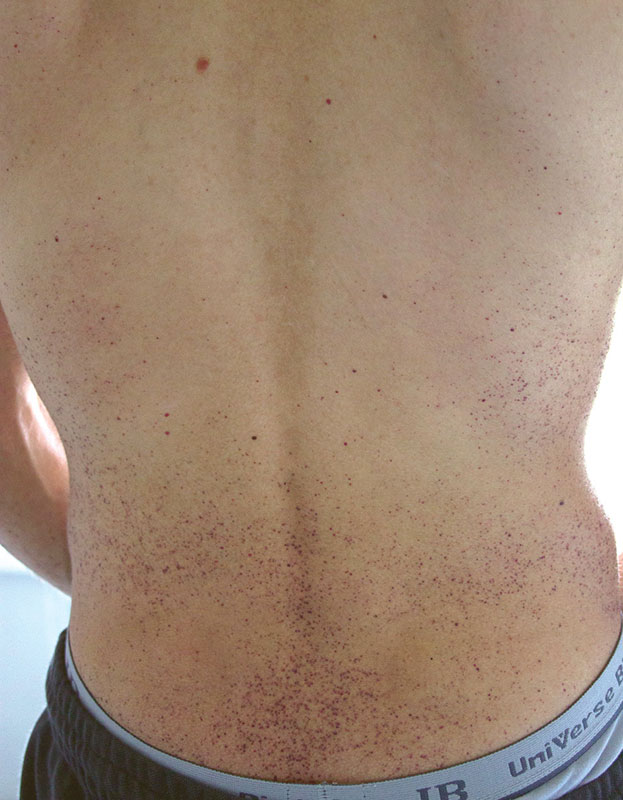
–û–±―ä–Β–Κ―²–Η–≤–Ϋ–Ψ: –Α―¹―²–Β–Ϋ–Η―΅–Β―¹–Κ–Ψ–Β ―²–Β–Μ–Ψ¬≠―¹–Μ–Ψ–Ε–Β–Ϋ–Η–Β, –Κ–Ψ–Ε–Α ―¹―É―Ö–Α―è, –≥–Η–Ω–Ψ–≥–Η–¥―Ä–Ψ–Ζ, –Ϋ–Α¬†–Κ–Ψ–Ε–Β –Ψ–±–Ψ–Η―Ö –Μ–Ψ–Κ―²–Β–Ι, –Ω–Ψ―è―¹–Ϋ–Η―Ü―΄, –Κ–Ψ–Μ–Β–Ϋ–Ϋ―΄―Ö ―¹―É―¹―²–Α–≤–Ψ–≤, ―è–≥–Ψ–¥–Η―Ü, –±–Β–¥―Ä–Α―Ö, –Ϋ–Η–Ε–Ϋ–Η―Ö –Ψ―²–¥–Β–Μ–Α―Ö –Ε–Η–≤–Ψ―²–Α –Φ–Ϋ–Ψ–Ε–Β―¹―²–≤–Β–Ϋ–Ϋ―΄–Β –Φ–Β–Μ–Κ–Ψ―²–Ψ―΅–Β―΅–Ϋ―΄–Β ―²–Β–Φ–Ϋ–Ψ-–±–Ψ―Ä–¥–Ψ–≤–Ψ–≥–Ψ ―Ü–≤–Β―²–Α –≤―΄―¹―΄–Ω–Α–Ϋ–Η―è (―Ä–Η―¹.¬†1 –Η¬†2), –Ϋ–Β¬†–Η―¹―΅–Β–Ζ–Α―é―â–Η–Β –Ω―Ä–Η –Ϋ–Α–¥–Α–≤–Μ–Η–≤–Α–Ϋ–Η–Η, ―²–Β–Μ–Β–Α–Ϋ–≥–Η―ç–Κ―²–Α–Ζ–Η–Η –Ϋ–Α¬†–≤–Β―Ä―Ö–Ϋ–Η―Ö –≤–Β–Κ–Α―Ö –Η¬†―¹–Μ–Η–Ζ–Η―¹―²–Ψ–Ι –Ψ–±–Ψ–Μ–Ψ―΅–Κ–Β –≥―É–±. –ë–Ψ–Μ–Β–Ζ–Ϋ–Β–Ϋ–Ϋ–Ψ―¹―²―¨ –Ω―Ä–Η –Ω–Α–Μ―¨–Ω–Α―Ü–Η–Η –Η¬†―É–Φ–Β―Ä–Β–Ϋ–Ϋ–Ψ–Β –Ψ–≥―Ä–Α–Ϋ–Η―΅–Β–Ϋ–Η–Β –≤¬†–Ψ–±―ä–Β–Φ–Β –¥–≤–Η–Ε–Β–Ϋ–Η–Ι –≤¬†–Κ–Ψ–Μ–Β–Ϋ–Ϋ―΄―Ö –Η¬†–Ω–Μ–Β―΅–Β–≤―΄―Ö ―¹―É―¹―²–Α–≤–Α―Ö, –Ϋ–Β–±–Ψ–Μ―¨―à–Α―è –¥–Β―³–Η–≥―É―Ä–Α―Ü–Η―è –Ω―Ä–Ψ–Κ―¹–Η–Φ–Α–Μ―¨–Ϋ―΄―Ö ―¹―É―¹―²–Α–≤–Ψ–≤ –Ψ–±–Β–Η―Ö –Κ–Η―¹―²–Β–Ι. –£¬†–Μ–Β–≥–Κ–Η―Ö –≤–Β–Ζ–Η–Κ―É–Μ―è―Ä–Ϋ–Ψ–Β –¥―΄―Ö–Α–Ϋ–Η–Β, ―΅–Α―¹―²–Ψ―²–Α –¥―΄―Ö–Α–Ϋ–Η―è¬†βÄ™ 18 –≤¬†–Φ–Η–Ϋ―É―²―É, ―²–Ψ–Ϋ―΄ ―¹–Β―Ä–¥―Ü–Α ―è―¹–Ϋ―΄–Β, ―Ä–Η―²–Φ –Ω―Ä–Α–≤–Η–Μ―¨–Ϋ―΄–Ι, ―΅–Α―¹―²–Ψ―²–Α ―¹–Β―Ä–¥–Β―΅–Ϋ―΄―Ö ―¹–Ψ–Κ―Ä–Α―â–Β–Ϋ–Η–Ι¬†βÄ™ 80 –≤¬†–Φ–Η–Ϋ―É―²―É, –Α―Ä―²–Β―Ä–Η–Α–Μ―¨–Ϋ–Ψ–Β –¥–Α–≤–Μ–Β–Ϋ–Η–Β¬†βÄ™ 120/80 –Φ–Φ ―Ä―². ―¹―². –•–Η–≤–Ψ―² –±–Β–Ζ–±–Ψ–Μ–Β–Ζ–Ϋ–Β–Ϋ–Ϋ―΄–Ι.
–î–Α–Ϋ–Ϋ―΄–Β –Μ–Α–±–Ψ―Ä–Α―²–Ψ―Ä–Ϋ–Ψ–≥–Ψ –Ψ–±―¹–Μ–Β–¥–Ψ–≤–Α–Ϋ–Η―è: –≤¬†–Ψ–±―â–Β–Φ –Α–Ϋ–Α–Μ–Η–Ζ–Β –Κ―Ä–Ψ–≤–Η –≥–Β–Φ–Ψ–≥–Μ–Ψ–±–Η–Ϋ¬†βÄ™ 133 –≥/–Μ (–Ϋ–Ψ―Ä–Φ–Α¬†βÄ™ 120βÄ™140 –≥/–Μ), ―ç―Ä–Η―²―Ä–Ψ―Ü–Η―²―΄¬†βÄ™ 4,4 Ο½ 1012/–Μ, –Μ–Β–Ι–Κ–Ψ―Ü–Η―²―΄¬†βÄ™ 8,8 Ο½ 109/–Μ, ―²―Ä–Ψ–Φ–±–Ψ―Ü–Η―²―΄¬†βÄ™ 224 Ο½ 109/–Μ, –Ω–Α–Μ–Ψ―΅–Κ–Ψ―è–¥–Β―Ä–Ϋ―΄–Β –Ϋ–Β–Ι―²―Ä–Ψ―³–Η–Μ―΄¬†βÄ™ 6%, ―¹–Β–≥–Φ–Β–Ϋ―²–Ψ―è–¥–Β―Ä–Ϋ―΄–Β –Ϋ–Β–Ι―²―Ä–Ψ―³–Η–Μ―΄¬†βÄ™ 59%, –Μ–Η–Φ―³–Ψ―Ü–Η―²―΄¬†βÄ™ 32%, –Φ–Ψ–Ϋ–Ψ―Ü–Η―²―΄¬†βÄ™ 7%, –±–Α–Ζ–Ψ―³–Η–Μ―΄¬†βÄ™ 2%, –Γ–û–≠¬†βÄ™ 28 –Φ–Φ/―΅, –≤¬†–±–Η–Ψ―Ö–Η–Φ–Η―΅–Β―¹–Κ–Ψ–Φ: –Γ-―Ä–Β–Α–Κ―²–Η–≤–Ϋ―΄–Ι –±–Β–Μ–Ψ–Κ –Ψ―²―Ä–Η―Ü–Α―²–Β–Μ―¨–Ϋ―΄–Ι, –Α–Μ–Α–Ϋ–Η–Ϋ–Α–Φ–Η–Ϋ–Ψ―²―Ä–Α–Ϋ―¹―³–Β―Ä–Α–Ζ–Α¬†βÄ™ 8 –ï–¥/–Μ, –Α―¹–Ω–Α―Ä―²–Α―²–Α–Φ–Η–Ϋ–Ψ―²―Ä–Α–Ϋ―¹―³–Β―Ä–Α–Ζ–Α¬†βÄ™ 15 –ï–¥/–Μ, –Κ―Ä–Β–Α―²–Η–Ϋ–Η–Ϋ¬†βÄ™ 87 –Φ–Κ–Φ–Ψ–Μ―¨/–Μ, –±–Η–Μ–Η―Ä―É–±–Η–Ϋ¬†βÄ™ 12 –Φ–Κ–Φ–Ψ–Μ―¨/–Μ, ―³–Η–±―Ä–Η–Ϋ–Ψ–≥–Β–Ϋ¬†βÄ™ 3,2 –≥/–Μ, –Η–Φ–Φ―É–Ϋ–Ψ–Μ–Ψ–≥–Η―΅–Β―¹–Κ–Ψ–Φ: ―Ü–Η―Ä–Κ―É–Μ–Η―Ä―É―é―â–Η―Ö –Η–Φ–Φ―É–Ϋ–Ϋ―΄―Ö –Κ–Ψ–Φ–Ω–Μ–Β–Κ―¹–Ψ–≤¬†βÄ™ 50 –Ψ–Ω―². –Β–¥., ―Ä–Β–≤–Φ–Α―²–Ψ–Η–¥–Ϋ―΄–Ι ―³–Α–Κ―²–Ψ―Ä –Ψ―²―Ä–Η―Ü–Α―²–Β–Μ―¨–Ϋ―΄–Ι. –£¬†–Ψ–±―â–Β–Φ –Α–Ϋ–Α–Μ–Η–Ζ–Β –Φ–Ψ―΅–Η: –Ω–Μ–Ψ―²–Ϋ–Ψ―¹―²―¨¬†βÄ™ 1021 –≥/–Μ, ―Ä–ù¬†βÄ™ 6,0, –±–Β–Μ–Ψ–Κ¬†βÄ™ 0,5 –≥/–Μ, –Μ–Β–Ι–Κ–Ψ―Ü–Η―²―΄¬†βÄ™ 2βÄ™4 –≤¬†–Ω/–Ζ―Ä, ―¹―É―²–Ψ―΅–Ϋ–Α―è –Ω―Ä–Ψ―²–Β–Η–Ϋ―É―Ä–Η―è¬†βÄ™ 0,4 –≥/–Μ.
–ù–Α¬†―Ä–Β–Ϋ―²–≥–Β–Ϋ–Ψ–≥―Ä–Α–Φ–Φ–Β –Κ–Η―¹―²–Β–Ι –Η¬†―¹―²–Ψ–Ω¬†βÄ™ –Ψ―¹―²–Β–Ψ–Ω–Ψ―Ä–Ψ–Ζ, –≤―΄―¹–Ψ―²–Α ―¹―É―¹―²–Α–≤–Ϋ―΄―Ö ―â–Β–Μ–Β–Ι –Ϋ–Β¬†–Η–Ζ–Φ–Β–Ϋ–Β–Ϋ–Α, –Κ–Ψ―¹―²–Β–Ι ―²–Α–Ζ–Α¬†βÄ™ –≤―΄―¹–Ψ―²–Α ―¹―É―¹―²–Α–≤–Ϋ―΄―Ö ―â–Β–Μ–Β–Ι ―²–Α–Ζ–Ψ–±–Β–¥―Ä–Β–Ϋ–Ϋ―΄―Ö ―¹―É―¹―²–Α–≤–Ψ–≤ –Ϋ–Β¬†–Η–Ζ–Φ–Β–Ϋ–Β–Ϋ–Α, ―à–Η―Ä–Η–Ϋ–Α ―¹–Α–Κ―Ä–Ψ–Η–Μ–Β–Α–Μ―¨–Ϋ―΄―Ö ―¹–Ψ―΅–Μ–Β–Ϋ–Β–Ϋ–Η–Ι ―¹–Ψ―Ö―Ä–Α–Ϋ–Β–Ϋ–Α, –Ψ―Ä–≥–Α–Ϋ–Ψ–≤ –≥―Ä―É–¥–Ϋ–Ψ–Ι –Κ–Μ–Β―²–Κ–Η¬†βÄ™ –Μ–Β–≥–Κ–Η–Β –±–Β–Ζ –Ψ―΅–Α–≥–Ψ–≤―΄―Ö –Η¬†–Η–Ϋ―³–Η–Μ―¨―²―Ä–Α―²–Η–≤–Ϋ―΄―Ö ―²–Β–Ϋ–Β–Ι, –Ω–Μ–Β–≤―Ä–Α–Μ―¨–Ϋ―΄–Β ―¹–Η–Ϋ―É―¹―΄ ―¹–≤–Ψ–±–Ψ–¥–Ϋ―΄–Β, ―¹–Β―Ä–¥―Ü–Β –≤¬†―Ä–Α–Ζ–Φ–Β―Ä–Α―Ö –Ϋ–Β¬†―É–≤–Β–Μ–Η―΅–Β–Ϋ–Ψ. –‰―¹―Ö–Ψ–¥―è –Η–Ζ¬†–Α–Ϋ–Α–Φ–Ϋ–Β–Ζ–Α, –Κ–Μ–Η–Ϋ–Η–Κ–Η, –Ω–Ψ–Κ–Α–Ζ–Α―²–Β–Μ–Β–Ι –Μ–Α–±–Ψ―Ä–Α―²–Ψ―Ä–Ϋ―΄―Ö –Α–Ϋ–Α–Μ–Η–Ζ–Ψ–≤, –Ϋ–Β¬†–Ω–Ψ–¥―²–≤–Β―Ä–Ε–¥–Α―é―â–Η―Ö ―Ä–Β–≤–Φ–Α―²–Ψ–Μ–Ψ–≥–Η―΅–Β―¹–Κ―É―é –Ω―Ä–Η―Ä–Ψ–¥―É –Ω–Ψ―Ä–Α–Ε–Β–Ϋ–Η―è ―¹―É―¹―²–Α–≤–Ψ–≤, ―¹–¥–Β–Μ–Α–Ϋ–Ψ –Ω―Ä–Β–¥–Ω–Ψ–Μ–Ψ–Ε–Β–Ϋ–Η–Β –Ψ¬†–Ϋ–Α–Μ–Η―΅–Η–Η –±–Ψ–Μ–Β–Ζ–Ϋ–Η –Λ–Α–±―Ä–Η.
–£–Ζ―è―² –Ψ–±―Ä–Α–Ζ–Β―Ü ―¹―É―Ö–Ψ–≥–Ψ –Ω―è―²–Ϋ–Α –Κ―Ä–Ψ–≤–Η –Ϋ–Α¬†–Α–Κ―²–Η–≤–Ϋ–Ψ―¹―²―¨ ―³–Β―Ä–Φ–Β–Ϋ―²–Α –Α–Μ―¨―³–Α-–≥–Α–Μ–Α–Κ―²–Ψ–Ζ–Η–¥–Α–Ζ―΄ (–Ω―Ä–Ψ–≤–Ψ–¥–Η―²―¹―è –≤¬†–†–Ψ―¹―¹–Η–Η –±–Β―¹–Ω–Μ–Α―²–Ϋ–Ψ). –£―΄―è–≤–Μ–Β–Ϋ–Ψ ―¹–Ϋ–Η–Ε–Β–Ϋ–Η–Β ―É―Ä–Ψ–≤–Ϋ―è –Α–Κ―²–Η–≤–Ϋ–Ψ―¹―²–Η ―³–Β―Ä–Φ–Β–Ϋ―²–Α –Α–Μ―¨―³–Α-–≥–Α–Μ–Α–Κ―²–Ψ–Ζ–Η–¥–Α–Ζ―΄ –≤¬†–Ω–Μ–Α–Ζ–Φ–Β –Κ―Ä–Ψ–≤–Η –¥–Ψ 8,46 –Ω–Φ–Ψ–Μ―¨/–¥–Η―¹–Κ*20 ―΅ (–Ϋ–Ψ―Ä–Φ–Α¬†βÄ™ 160βÄ™2000 –Ω–Φ–Ψ–Μ―¨/–¥–Η―¹–Κ*20 ―΅), ―΅―²–Ψ ―Ö–Α―Ä–Α–Κ―²–Β―Ä–Ϋ–Ψ –¥–Μ―è –¥–Α–Ϋ–Ϋ–Ψ–Ι –Ω–Α―²–Ψ–Μ–Ψ–≥–Η–Η.
–£¬†–Η―é–Μ–Β 2015 –≥.¬†–Ω–Α―Ü–Η–Β–Ϋ―² –Ϋ–Α–Ω―Ä–Α–≤–Μ–Β–Ϋ –≤¬†–Ψ―²–¥–Β–Μ–Β–Ϋ–Η–Β ―Ä–Β–≤–Φ–Α―²–Ψ–Μ–Ψ–≥–Η–Η –ö–Μ–Η–Ϋ–Η–Κ–Η –Ϋ–Β―³―Ä–Ψ–Μ–Ψ–≥–Η–Η, –≤–Ϋ―É―²―Ä–Β–Ϋ–Ϋ–Η―Ö –Η¬†–Ω―Ä–Ψ―³–Β―¹―¹–Η–Ψ–Ϋ–Α–Μ―¨–Ϋ―΄―Ö –Ζ–Α–±–Ψ–Μ–Β–≤–Α–Ϋ–Η–Ι –Η–Φ. –ï.–€. –Δ–Α―Ä–Β–Β–≤–Α –ü–Β―Ä–≤–Ψ–≥–Ψ –€–Ψ―¹–Κ–Ψ–≤―¹–Κ–Ψ–≥–Ψ –≥–Ψ―¹―É–¥–Α―Ä―¹―²–≤–Β–Ϋ–Ϋ–Ψ–≥–Ψ –Φ–Β–¥–Η―Ü–Η–Ϋ―¹–Κ–Ψ–≥–Ψ ―É–Ϋ–Η–≤–Β―Ä―¹–Η―²–Β―²–Α –Η–Φ. –‰.–€. –Γ–Β―΅–Β–Ϋ–Ψ–≤–Α.
–î–Ψ–Ω–Ψ–Μ–Ϋ–Η―²–Β–Μ―¨–Ϋ–Ψ –Ω―Ä–Ψ–≤–Β–¥–Β–Ϋ–Ψ ―Ö–Ψ–Μ―²–Β―Ä–Ψ–≤―¹–Κ–Ψ–Β –Φ–Ψ–Ϋ–Η―²–Ψ―Ä–Η―Ä–Ψ–≤–Α–Ϋ–Η–Β (―¹―É―²–Ψ―΅–Ϋ–Ψ–Β –Φ–Ψ–Ϋ–Η―²–Ψ―Ä–Η―Ä–Ψ–≤–Α–Ϋ–Η–Β ―ç–Μ–Β–Κ―²―Ä–Ψ–Κ–Α―Ä–¥–Η–Ψ–≥―Ä–Α–Φ–Φ―΄ (–≠–ö–™)). –û–±–Ϋ–Α―Ä―É–Ε–Β–Ϋ―΄ ―¹―É–Ω―Ä–Α–≤–Β–Ϋ―²―Ä–Η–Κ―É–Μ―è―Ä–Ϋ―΄–Β ―ç–Κ―¹―²―Ä–Α―¹–Η―¹―²–Ψ–Μ―΄, ―É–Κ–Ψ―Ä–Ψ―΅–Β–Ϋ–Η–Β –Η–Ϋ―²–Β―Ä–≤–Α–Μ–Α PQ. –ù–Α¬†―ç―Ö–Ψ–Κ–Α―Ä–¥–Η–Ψ–≥―Ä–Α―³–Η–Η¬†βÄ™ –±–Β–Ζ –≤―΄―Ä–Α–Ε–Β–Ϋ–Ϋ–Ψ–Ι –Ω–Α―²–Ψ–Μ–Ψ–≥–Η–Η, –Ω―Ä–Η –Φ–Α–≥–Ϋ–Η―²–Ϋ–Ψ-―Ä–Β–Ζ–Ψ–Ϋ–Α–Ϋ―¹–Ϋ–Ψ–Ι ―²–Ψ–Φ–Ψ–≥―Ä–Α―³–Η–Η (–€–†–Δ) ―¹–Β―Ä–¥―Ü–Α ―¹¬†–Κ–Ψ–Ϋ―²―Ä–Α―¹―²–Η―Ä–Ψ–≤–Α–Ϋ–Η–Β–Φ¬†βÄ™ –Φ–Η―²―Ä–Α–Μ―¨–Ϋ–Α―è ―Ä–Β–≥―É―Ä–≥–Η―²–Α―Ü–Η―è –Ω–Β―Ä–≤–Ψ–Ι ―¹―²–Β–Ω–Β–Ϋ–Η.
–€–Η–Κ―Ä–Ψ–Α–Μ―¨–±―É–Φ–Η–Ϋ―É―Ä–Η―è¬†βÄ™ 349 –Φ–≥/–Μ (–Ϋ–Ψ―Ä–Φ–Α¬†βÄ™ 30 –Φ–≥/–Μ).
–ö–Ψ–Ϋ―¹―É–Μ―¨―²–Α―Ü–Η―è –Ψ―³―²–Α–Μ―¨–Φ–Ψ–Μ–Ψ–≥–Α: –Ω―Ä–Η–Ζ–Ϋ–Α–Κ–Η –≤–Ψ―Ä–Ψ–Ϋ–Κ–Ψ–≤–Η–¥–Ϋ–Ψ–Ι –Κ–Β―Ä–Α―²–Ψ–Ω–Α―²–Η–Η.
–ù–Α¬†–€–†–Δ –≥–Ψ–Μ–Ψ–≤–Ϋ–Ψ–≥–Ψ –Φ–Ψ–Ζ–≥–Α¬†βÄ™ –≤¬†–Ω―Ä–Α–≤–Ψ–Ι –Ζ–Α–¥–Ϋ–Β–≤–Η―¹–Ψ―΅–Ϋ–Ψ–Ι –Ψ–±–Μ–Α―¹―²–Η –Ψ―΅–Α–≥–Ψ–≤―΄–Β –Η–Ζ–Φ–Β–Ϋ–Β–Ϋ–Η―è, –Κ–Ψ―²–Ψ―Ä―΄–Β –Φ–Ψ–≥―É―² –±―΄―²―¨ ―Ä–Α―¹―Ü–Β–Ϋ–Β–Ϋ―΄ –Κ–Α–Κ ―Ö–Η–Φ–Η―΅–Β―¹–Κ–Η–Ι ―¹–¥–≤–Η–≥ –Η–Ζ-–Ζ–Α –Φ–Η–Κ―Ä–Ψ–Κ–Α–Μ―¨―Ü–Η–Ϋ–Α―²–Ψ–≤.
–™–Β–Ϋ–Β―²–Η―΅–Β―¹–Κ–Ψ–Β –Η―¹―¹–Μ–Β–¥–Ψ–≤–Α–Ϋ–Η–Β. –ü―Ä―è–Φ–Ψ–Β ―¹–Β–Κ–≤–Β–Ϋ–Η―Ä–Ψ–≤–Α–Ϋ–Η–Β –≥–Β–Ϋ–Α –≤―΄―è–≤–Η–Μ–Ψ –Φ―É―²–Α―Ü–Η―é ―¹.550–Δ>–Γ¬†–≤¬†–≥–Β–Φ–Η–Ζ–Η–≥–Ψ―²–Ϋ–Ψ–Φ ―¹–Ψ―¹―²–Ψ―è–Ϋ–Η–Η (–Φ–Ψ–Μ–Β–Κ―É–Μ―è―Ä–Ϋ–Ψ-–≥–Β–Ϋ–Β―²–Η―΅–Β―¹–Κ–Ψ–Β –Η―¹―¹–Μ–Β–¥–Ψ–≤–Α–Ϋ–Η–Β –Ϋ–Α¬†–±–Ψ–Μ–Β–Ζ–Ϋ―¨ –Λ–Α–±―Ä–Η –Ω―Ä–Ψ–≤–Ψ–¥–Η―²―¹―è –≤¬†–†–Ψ―¹―¹–Η–Η –±–Β―¹–Ω–Μ–Α―²–Ϋ–Ψ).
–î–Η–Α–≥–Ϋ–Ψ–Ζ –Ω–Ψ¬†―Ä–Β–Ζ―É–Μ―¨―²–Α―²–Α–Φ –Κ–Ψ–Ϋ―¹–Η–Μ–Η―É–Φ–Α: –±–Ψ–Μ–Β–Ζ–Ϋ―¨ –Λ–Α–±―Ä–Η (–Φ―É―²–Α―Ü–Η―è –≤¬†–≥–Β–Ϋ–Β GLA) ―¹¬†–Ω–Ψ―Ä–Α–Ε–Β–Ϋ–Η–Β–Φ –Κ–Ψ–Ε–Η (–Α–Ϋ–≥–Η–Ψ–Κ–Β―Ä–Α―²–Ψ–Φ―΄), ―Ü–Β–Ϋ―²―Ä–Α–Μ―¨–Ϋ–Ψ–Ι (–Ψ―΅–Α–≥–Ψ–≤―΄–Β –Η–Ζ–Φ–Β–Ϋ–Β–Ϋ–Η―è –≤¬†―²–Κ–Α–Ϋ―è―Ö –≥–Ψ–Μ–Ψ–≤–Ϋ–Ψ–≥–Ψ –Φ–Ψ–Ζ–≥–Α) –Η¬†–Ω–Β―Ä–Η―³–Β―Ä–Η―΅–Β―¹–Κ–Ψ–Ι (–Α–Κ―Ä–Ψ–Ω–Α―Ä–Β―¹―²–Β–Ζ–Η–Η, ―¹–Β–Ϋ―¹–Ψ―Ä–Ϋ―΄–Β –Ϋ–Α―Ä―É―à–Β–Ϋ–Η―è) –Ϋ–Β―Ä–≤–Ϋ–Ψ–Ι ―¹–Η―¹―²–Β–Φ―΄, –Φ―΄―à―Ü –Η¬†―¹―É―¹―²–Α–≤–Ψ–≤ (–Φ–Η–Α–Μ–≥–Η–Η, –Α―Ä―²―Ä–Α–Μ–≥–Η–Η), –Ω–Ψ―²–Ψ–≤―΄―Ö –Ε–Β–Μ–Β–Ζ (–≥–Η–Ω–Ψ–≥–Η–¥―Ä–Ψ–Ζ), –Ω–Ψ―΅–Β–Κ (–Ω―Ä–Ψ―²–Β–Η–Ϋ―É―Ä–Η―è –¥–Ψ 0,5 –≥/―¹―É―²), –Ψ―Ä–≥–Α–Ϋ–Α –Ζ―Ä–Β–Ϋ–Η―è (–≤–Ψ―Ä–Ψ–Ϋ–Κ–Ψ–≤–Η–¥–Ϋ–Α―è –Κ–Β―Ä–Α―²–Ψ–Ω–Α―²–Η―è), –Μ–Η―Ö–Ψ―Ä–Α–¥–Ψ―΅–Ϋ―΄–Ι ―¹–Η–Ϋ–¥―Ä–Ψ–Φ.
–Δ–Α–Κ–Η–Φ –Ψ–±―Ä–Α–Ζ–Ψ–Φ, ―ɬ†–Ω–Α―Ü–Η–Β–Ϋ―²–Α –Ψ―΅–Β–≤–Η–¥–Ϋ―΄–Β ―Ä–Β–≤–Φ–Α―²–Ψ–Μ–Ψ–≥–Η―΅–Β―¹–Κ–Η–Β –Ω―Ä–Η―΅–Η–Ϋ―΄ –¥–Α–Ϋ–Ϋ–Ψ–Ι –Κ–Μ–Η–Ϋ–Η―΅–Β―¹–Κ–Ψ–Ι –Κ–Α―Ä―²–Η–Ϋ―΄ –Η―¹–Κ–Μ―é―΅–Β–Ϋ―΄. –Γ–Η–Φ–Ω―²–Ψ–Φ–Α―²–Η–Κ–Α ―Ä–Α―¹―Ü–Β–Ϋ–Β–Ϋ–Α –≤¬†―Ä–Α–Φ–Κ–Α―Ö –±–Ψ–Μ–Β–Ζ–Ϋ–Η –Λ–Α–±―Ä–Η, –Ω–Ψ–¥―²–≤–Β―Ä–Ε–¥–Β–Ϋ–Α ―Ä–Β–Ζ―É–Μ―¨―²–Α―²–Α–Φ–Η –≥–Β–Ϋ–Β―²–Η―΅–Β―¹–Κ–Η―Ö –Η¬†–±–Η–Ψ―Ö–Η–Φ–Η―΅–Β―¹–Κ–Η―Ö ―²–Β―¹―²–Ψ–≤.
–ù–Α–Ζ–Ϋ–Α―΅–Β–Ϋ–Ψ –Μ–Β―΅–Β–Ϋ–Η–Β: –Α–≥–Α–Μ―¹–Η–¥–Α–Ζ–Α –±–Β―²–Α 1 –Φ–≥/–Κ–≥ –Κ–Α–Ε–¥―΄–Β –¥–≤–Β –Ϋ–Β–¥–Β–Μ–Η –Ω–Ψ–Ε–Η–Ζ–Ϋ–Β–Ϋ–Ϋ–Ψ.
–Γ¬†―³–Β–≤―Ä–Α–Μ―è 2016 –≥.¬†–Ω–Α―Ü–Η–Β–Ϋ―² –Β–Ε–Β–Ϋ–Β–¥–Β–Μ―¨–Ϋ–Ψ –≤¬†―¹―²–Α―Ü–Η–Ψ–Ϋ–Α―Ä–Β –Ω–Ψ–Μ―É―΅–Α–Β―² –Ω―Ä–Β–Ω–Α―Ä–Α―² 1 –Φ–≥/–Κ–≥ –≤–Ϋ―É―²―Ä–Η–≤–Β–Ϋ–Ϋ–Ψ –Κ–Α–Ω–Β–Μ―¨–Ϋ–Ψ.
–£¬†―Ä–Β–Ζ―É–Μ―¨―²–Α―²–Β –Μ–Β―΅–Β–Ϋ–Η―è ―¹–Α–Φ–Ψ―΅―É–≤―¹―²–≤–Η–Β –±–Ψ–Μ―¨–Ϋ–Ψ–≥–Ψ ―É–Μ―É―΅―à–Η–Μ–Ψ―¹―¨: –Ψ―²―¹―É―²―¹―²–≤–Η–Β –Φ―É―΅–Η―²–Β–Μ―¨–Ϋ―΄―Ö –±–Ψ–Μ–Β–Ι –≤¬†―¹―É―¹―²–Α–≤–Α―Ö, –Μ–Η―Ö–Ψ―Ä–Α–¥–Κ–Η, ―É–Φ–Β–Ϋ―¨―à–Β–Ϋ–Η–Β –Ψ–±―â–Β–Ι ―¹–Μ–Α–±–Ψ―¹―²–Η, –Ω–Ψ–≤―΄―à–Β–Ϋ–Η–Β –Α–Ω–Ω–Β―²–Η―²–Α.
–ë―΄–Μ–Η –≤–Ζ―è―²―΄ –Α–Ϋ–Α–Μ–Η–Ζ―΄ –Κ―Ä–Ψ–≤–Η ―ɬ†―Ä–Ψ–¥―¹―²–≤–Β–Ϋ–Ϋ–Η–Κ–Ψ–≤ –±–Ψ–Μ―¨–Ϋ–Ψ–≥–Ψ.
–Θ¬†–Φ–Α―²–Β―Ä–Η, –±–Α–±―É―à–Κ–Η –Ω–Ψ¬†–Φ–Α―²–Β―Ä–Η–Ϋ―¹–Κ–Ψ–Ι –Μ–Η–Ϋ–Η–Η, –¥–≤–Ψ―é―Ä–Ψ–¥–Ϋ―΄―Ö ―¹–Β―¹―²–Β―Ä –Ω–Ψ–¥―²–≤–Β―Ä–Ε–¥–Β–Ϋ–Ψ –Ϋ–Ψ―¹–Η―²–Β–Μ―¨―¹―²–≤–Ψ –≥–Β–Ϋ–Α –±–Ψ–Μ–Β–Ζ–Ϋ–Η –Λ–Α–±―Ä–Η.
–£¬†–Η―é–Μ–Β 2015 –≥.¬†―ɬ†–±―Ä–Α―²–Α –Ω–Α―Ü–Η–Β–Ϋ―²–Α –Η¬†–¥―è–¥–Η –¥–Η–Α–≥–Ϋ–Ψ―¹―²–Η―Ä–Ψ–≤–Α–Ϋ–Α –±–Ψ–Μ–Β–Ζ–Ϋ―¨ –Λ–Α–±―Ä–Η. –Θ¬†–¥―è–¥–Η (45 –Μ–Β―²) –Ω–Ψ¬†–Μ–Η–Ϋ–Η–Η –Φ–Α―²–Β―Ä–Η ―΅–Β―²―΄―Ä–Β –≥–Ψ–¥–Α –Ψ―²–Φ–Β―΅–Α–Μ–Η―¹―¨ –Φ―É―΅–Η―²–Β–Μ―¨–Ϋ―΄–Β –±–Ψ–Μ–Η –≤¬†–Κ–Η―¹―²―è―Ö –Η¬†―¹―²–Ψ–Ω–Α―Ö –¥–Μ–Η―²–Β–Μ―¨–Ϋ–Ψ―¹―²―¨―é –¥–Ψ –¥–≤―É―Ö –¥–Ϋ–Β–Ι, –Κ–Ψ―²–Ψ―Ä―΄–Β ―É―¹–Η–Μ–Η–≤–Α–Μ–Η―¹―¨ –Ω―Ä–Η ―³–Η–Ζ–Η―΅–Β―¹–Κ–Ψ–Ι –Ϋ–Α–≥―Ä―É–Ζ–Κ–Β –Η¬†–≤ –Ε–Α―Ä―É, –Ω–Ψ–≤―΄―à–Β–Ϋ–Η–Β ―²–Β–Φ–Ω–Β―Ä–Α―²―É―Ä―΄ ―²–Β–Μ–Α –¥–Ψ 39 ¬Κ–Γ, –≤―΄―¹―΄–Ω–Α–Ϋ–Η―è –Ϋ–Α¬†–Κ–Ψ–Ε–Β –±–Β–Ζ –Ζ―É–¥–Α (–≤¬†–Ϋ–Α―¹―²–Ψ―è―â–Β–Β –≤―Ä–Β–Φ―è –Ω–Α―Ü–Η–Β–Ϋ―² –Ψ–±―¹–Μ–Β–¥―É–Β―²―¹―è). –Θ¬†―Ä–Ψ–¥–Ϋ–Ψ–≥–Ψ –±―Ä–Α―²–Α –Ω–Α―Ü–Η–Β–Ϋ―²–Α –Ψ―²–Φ–Β―΅–Α―é―²―¹―è –Ω–Β―Ä–Β–±–Ψ–Η –≤¬†―Ä–Α–±–Ψ―²–Β ―¹–Β―Ä–¥―Ü–Α –Ω–Ψ―¹–Μ–Β ―³–Η–Ζ–Η―΅–Β―¹–Κ–Ψ–Ι –Ϋ–Α–≥―Ä―É–Ζ–Κ–Η –Η¬†–≤―΄―¹―΄–Ω–Α–Ϋ–Η―è –Ϋ–Α¬†–Κ–Ψ–Ε–Β –≤¬†–Ω–Α―Ö–Ψ–≤–Ψ–Ι –Ψ–±–Μ–Α―¹―²–Η –Η¬†–Ψ–±–Μ–Α―¹―²–Η –Ω―É–Ω–Κ–Α. –‰–Ζ¬†–Α–Ϋ–Α–Φ–Ϋ–Β–Ζ–Α: ―¹¬†18 –Μ–Β―² –≤―΄―¹―΄–Ω–Α–Ϋ–Η―è –±―É―Ä–Ψ–≤–Ψ–≥–Ψ ―Ü–≤–Β―²–Α –≤¬†–Ψ–±–Μ–Α―¹―²–Η –Ω―É–Ω–Κ–Α –Η¬†–Ϋ–Α ―¹–Μ–Η–Ζ–Η―¹―²–Ψ–Ι –Ψ–±–Ψ–Μ–Ψ―΅–Κ–Β –≤–Β―Ä―Ö–Ϋ–Β–Ι –≥―É–±―΄, –Ω–Β―Ä–Β–±–Ψ–Η –≤¬†―Ä–Α–±–Ψ―²–Β ―¹–Β―Ä–¥―Ü–Α. –ü―Ä–Η –Ψ–±―¹–Μ–Β–¥–Ψ–≤–Α–Ϋ–Η–Η –Ω–Α―Ü–Η–Β–Ϋ―²–Α –≤¬†–Ψ―²–¥–Β–Μ–Β–Ϋ–Η–Η ―Ä–Β–≤–Φ–Α―²–Ψ–Μ–Ψ–≥–Η–Η –ö–Μ–Η–Ϋ–Η–Κ–Η –Ϋ–Β―³―Ä–Ψ–Μ–Ψ–≥–Η–Η, –≤–Ϋ―É―²―Ä–Β–Ϋ–Ϋ–Η―Ö –Η¬†–Ω―Ä–Ψ―³–Β―¹―¹–Η–Ψ–Ϋ–Α–Μ―¨–Ϋ―΄―Ö –±–Ψ–Μ–Β–Ζ–Ϋ–Β–Ι –Η–Φ. –ï.–€. –Δ–Α―Ä–Β–Β–≤–Α –Ω―Ä–Ψ–≤–Β–¥–Β–Ϋ–Ψ –≥–Β–Ϋ–Β―²–Η―΅–Β―¹–Κ–Ψ–Β –Η―¹―¹–Μ–Β–¥–Ψ–≤–Α–Ϋ–Η–Β, –Ω–Ψ–¥―²–≤–Β―Ä–¥–Η–≤―à–Β–Β –Φ―É―²–Α―Ü–Η―é. –û–±―ä–Β–Κ―²–Η–≤–Ϋ–Ψ: –≤¬†–Ω–Α―Ö–Ψ–≤―΄―Ö –Ψ–±–Μ–Α―¹―²―è―Ö –Η¬†–Ψ–Κ–Ψ–Μ–Ψ–Ω―É–Ω–Ψ―΅–Ϋ–Ψ–Ι –Ψ–±–Μ–Α―¹―²–Η –Ψ–±–Ϋ–Α―Ä―É–Ε–Β–Ϋ―΄ –Α–Ϋ–≥–Η–Ψ–Κ–Β―Ä–Α―²–Ψ–Φ―΄, ―¹―É―¹―²–Α–≤―΄ –Ϋ–Β¬†–Η–Ζ–Φ–Β–Ϋ–Β–Ϋ―΄, ―²–Ψ–Ϋ―΄ ―¹–Β―Ä–¥―Ü–Α ―è―¹–Ϋ―΄–Β, ―Ä–Η―²–Φ –Ω―Ä–Α–≤–Η–Μ―¨–Ϋ―΄–Ι, ―΅–Α―¹―²–Ψ―²–Α ―¹–Β―Ä–¥–Β―΅–Ϋ―΄―Ö ―¹–Ψ–Κ―Ä–Α―â–Β–Ϋ–Η–Ι¬†βÄ™ 76 –≤¬†–Φ–Η–Ϋ―É―²―É, –Α―Ä―²–Β―Ä–Η–Α–Μ―¨–Ϋ–Ψ–Β –¥–Α–≤–Μ–Β–Ϋ–Η–Β¬†βÄ™ 110/70 –Φ–Φ ―Ä―². ―¹―². –£¬†–Ψ–±―â–Β–Φ –Α–Ϋ–Α–Μ–Η–Ζ–Β –Φ–Ψ―΅–Η: –±–Β–Μ–Ψ–Κ βÄ™ 0,09 –≥/–Μ, ―¹―É―²–Ψ―΅–Ϋ–Α―è –Ω―Ä–Ψ―²–Β–Η–Ϋ―É―Ä–Η―è¬†βÄ™ 0,04 –≥/–Μ, –Φ–Η–Κ―Ä–Ψ–Α–Μ―¨–±―É–Φ–Η–Ϋ―É―Ä–Η―è¬†βÄ™ 76 –Φ–≥/–Μ. –ù–Α¬†–≠–ö–™¬†βÄ™ ―¹–Η–Ϋ–¥―Ä–Ψ–Φ –Ω―Ä–Β–Ε–¥–Β–≤―Ä–Β–Φ–Β–Ϋ–Ϋ–Ψ–≥–Ψ –≤–Ψ–Ζ–±―É–Ε–¥–Β–Ϋ–Η―è –Ε–Β–Μ―É–¥–Ψ―΅–Κ–Ψ–≤, –€–†–Δ ―¹–Β―Ä–¥―Ü–Α ―¹¬†–Κ–Ψ–Ϋ―²―Ä–Α―¹―²–Η―Ä–Ψ–≤–Α–Ϋ–Η–Β–Φ¬†βÄ™ –Φ–Η―²―Ä–Α–Μ―¨–Ϋ–Α―è ―Ä–Β–≥―É―Ä–≥–Η―²–Α―Ü–Η―è –Ω–Β―Ä–≤–Ψ–Ι ―¹―²–Β–Ω–Β–Ϋ–Η. –ö–Ψ–Ϋ―¹―É–Μ―¨―²–Α―Ü–Η―è –Ψ―³―²–Α–Μ―¨–Φ–Ψ–Μ–Ψ–≥–Α: –Ω―Ä–Η–Ζ–Ϋ–Α–Κ–Η –≤–Ψ―Ä–Ψ–Ϋ–Κ–Ψ–≤–Η–¥–Ϋ–Ψ–Ι –Κ–Β―Ä–Α―²–Ψ–Ω–Α―²–Η–Η. –î–Η–Α–≥–Ϋ–Ψ―¹―²–Η―Ä–Ψ–≤–Α–Ϋ–Α –±–Ψ–Μ–Β–Ζ–Ϋ―¨ –Λ–Α–±―Ä–Η ―¹¬†–Ω–Ψ―Ä–Α–Ε–Β–Ϋ–Η–Β–Φ –Κ–Ψ–Ε–Η (–Α–Ϋ–≥–Η–Ψ–Κ–Β―Ä–Α―²–Ψ–Φ―΄), –Ω–Ψ―΅–Β–Κ (–Φ–Η–Κ―Ä–Ψ–Α–Μ―¨–±―É–Φ–Η–Ϋ―É―Ä–Η―è), –Ψ―Ä–≥–Α–Ϋ–Α –Ζ―Ä–Β–Ϋ–Η―è (–≤–Ψ―Ä–Ψ–Ϋ–Κ–Ψ–Ψ–±―Ä–Α–Ζ–Ϋ–Α―è –Κ–Β―Ä–Α―²–Ψ–Ω–Α―²–Η―è).
–Γ¬†―³–Β–≤―Ä–Α–Μ―è 2016 –≥.¬†–Ω–Ψ –Ω―Ä–Η–±―΄―²–Η–Η –Ϋ–Α¬†–Φ–Β―¹―²–Ψ –Ε–Η―²–Β–Μ―¨―¹―²–≤–Α –Ω–Α―Ü–Η–Β–Ϋ―² –Β–Ε–Β–Ϋ–Β–¥–Β–Μ―¨–Ϋ–Ψ –Ω–Ψ–Μ―É―΅–Α–Β―² –≤¬†―¹―²–Α―Ü–Η–Ψ–Ϋ–Α―Ä–Β –Α–≥–Α–Μ―¹–Η–¥–Α–Ζ―É –±–Β―²–Α 1 –Φ–≥/–Κ–≥ –≤–Ϋ―É―²―Ä–Η–≤–Β–Ϋ–Ϋ–Ψ –Κ–Α–Ω–Β–Μ―¨–Ϋ–Ψ. –≠―³―³–Β–Κ―² ―²–Β―Ä–Α–Ω–Η–Η –Ω–Ψ–Μ–Ψ–Ε–Η―²–Β–Μ―¨–Ϋ―΄–Ι.
–½–Α–Κ–Μ―é―΅–Β–Ϋ–Η–Β
–£¬†―¹–≤–Ψ–Β–Ι –Ω―Ä–Α–Κ―²–Η–Κ–Β ―Ä–Β–≤–Φ–Α―²–Ψ–Μ–Ψ–≥–Η –Φ–Ψ–≥―É―² ―¹―²–Ψ–Μ–Κ–Ϋ―É―²―¨―¹―è ―¹¬†―Ä–Β–≤–Φ–Α―²–Ψ–Μ–Ψ–≥–Η―΅–Β―¹–Κ–Η–Φ–Η –Ω―Ä–Ψ―è–≤–Μ–Β–Ϋ–Η―è–Φ–Η –±–Ψ–Μ–Β–Ζ–Ϋ–Η –Λ–Α–±―Ä–Η. –Γ–≤–Ψ–Β–≤―Ä–Β–Φ–Β–Ϋ–Ϋ–Α―è –¥–Η–Α–≥–Ϋ–Ψ―¹―²–Η–Κ–Α ―ç―²–Ψ–Ι –Ω–Α―²–Ψ–Μ–Ψ–≥–Η–Η –Ω–Ψ–Ζ–≤–Ψ–Μ–Η―² –Ϋ–Α–Ζ–Ϋ–Α―΅–Η―²―¨ –Α–¥–Β–Κ–≤–Α―²–Ϋ–Ψ–Β –Μ–Β―΅–Β–Ϋ–Η–Β,¬†―É–Μ―É―΅―à–Η―²―¨ –Ω―Ä–Ψ–≥–Ϋ–Ψ–Ζ –Η¬†–Κ–Α―΅–Β―¹―²–≤–Ψ –Ε–Η–Ζ–Ϋ–Η –Ω–Α―Ü–Η–Β–Ϋ―²–Ψ–≤.¬†
–ü―Ä–Ψ–Ζ―Ä–Α―΅–Ϋ–Ψ―¹―²―¨ –Η―¹―¹–Μ–Β–¥–Ψ–≤–Α–Ϋ–Η–Ι. –‰―¹―¹–Μ–Β–¥–Ψ–≤–Α–Ϋ–Η–Β –Ϋ–Β¬†–Η–Φ–Β–Μ–Ψ ―¹–Ω–Ψ–Ϋ―¹–Ψ―Ä―¹–Κ–Ψ–Ι –Ω–Ψ–¥–¥–Β―Ä–Ε–Κ–Η. –ê–≤―²–Ψ―Ä―΄ –Ϋ–Β―¹―É―² –Ω–Ψ–Μ–Ϋ―É―é –Ψ―²–≤–Β―²―¹―²–≤–Β–Ϋ–Ϋ–Ψ―¹―²―¨ –Ζ–Α –Ω―Ä–Β–¥–Ψ―¹―²–Α–≤–Μ–Β–Ϋ–Η–Β –Ψ–Κ–Ψ–Ϋ―΅–Α―²–Β–Μ―¨–Ϋ–Ψ–Ι –≤–Β―Ä―¹–Η–Η ―Ä―É–Κ–Ψ–Ω–Η―¹–Η –≤¬†–Ω–Β―΅–Α―²―¨.
–î–Β–Κ–Μ–Α―Ä–Α―Ü–Η―è –Ψ¬†―³–Η–Ϋ–Α–Ϋ―¹–Ψ–≤―΄―Ö –Η¬†–¥―Ä―É–≥–Η―Ö –≤–Ζ–Α–Η–Φ–Ψ–Ψ―²–Ϋ–Ψ―à–Β–Ϋ–Η―è―Ö. –£―¹–Β –Α–≤―²–Ψ―Ä―΄ –Ω―Ä–Η–Ϋ–Η–Φ–Α–Μ–Η ―É―΅–Α―¹―²–Η–Β –≤¬†―Ä–Α–Ζ―Ä–Α–±–Ψ―²–Κ–Β –Κ–Ψ–Ϋ―Ü–Β–Ω―Ü–Η–Η ―¹―²–Α―²―¨–Η –≤¬†–Ϋ–Α–Ω–Η―¹–Α–Ϋ–Η–Η ―Ä―É–Κ–Ψ–Ω–Η―¹–Η. –û–Κ–Ψ–Ϋ―΅–Α―²–Β–Μ―¨–Ϋ–Α―è –≤–Β―Ä―¹–Η―è ―Ä―É–Κ–Ψ–Ω–Η―¹–Η –±―΄–Μ–Α –Ψ–¥–Ψ–±―Ä–Β–Ϋ–Α –≤―¹–Β–Φ–Η –Α–≤―²–Ψ―Ä–Α–Φ–Η. –ê–≤―²–Ψ―Ä―΄ –Ϋ–Β¬†–Ω–Ψ–Μ―É―΅–Α–Μ–Η –≥–Ψ–Ϋ–Ψ―Ä–Α―Ä–Ψ–≤ –Ζ–Α ―¹―²–Α―²―¨―é.
V.T. Komarov, N.S. Hichina
Burdenko Regional Clinical Hospital of Penza
Contact person: Nataliya Serafimovna Hichina, nataliyahichina@rambler.ru
Pain in distal interphalangeal joints of the upper and lower limbs is usually regarded as a sign of rheumatic pathologies. However, it may be the result of diseases of other etiologies. In the article on the specific example the similar case is considered. For a long time a patient with arthralgia was observed with regard to the different rheumatic diseases. In 17 years the correct diagnosis of Fabry disease (a rare genetic disorder) was made. Its timely diagnosis will allow providing adequate treatment which as a result will improve the prognosis and quality of life of patients.
–Θ–≤–Α–Ε–Α–Β–Φ―΄–Ι –Ω–Ψ―¹–Β―²–Η―²–Β–Μ―¨ uMEDp!
–Θ–≤–Β–¥–Ψ–Φ–Μ―è–Β–Φ –£–Α―¹ –Ψ ―²–Ψ–Φ, ―΅―²–Ψ –Ζ–¥–Β―¹―¨ ―¹–Ψ–¥–Β―Ä–Ε–Η―²―¹―è –Η–Ϋ―³–Ψ―Ä–Φ–Α―Ü–Η―è, –Ω―Ä–Β–¥–Ϋ–Α–Ζ–Ϋ–Α―΅–Β–Ϋ–Ϋ–Α―è –Η―¹–Κ–Μ―é―΅–Η―²–Β–Μ―¨–Ϋ–Ψ –¥–Μ―è ―¹–Ω–Β―Ü–Η–Α–Μ–Η―¹―²–Ψ–≤ –Ζ–¥―Ä–Α–≤–Ψ–Ψ―Ö―Ä–Α–Ϋ–Β–Ϋ–Η―è.
–ï―¹–Μ–Η –£―΄ –Ϋ–Β ―è–≤–Μ―è–Β―²–Β―¹―¨ ―¹–Ω–Β―Ü–Η–Α–Μ–Η―¹―²–Ψ–Φ –Ζ–¥―Ä–Α–≤–Ψ–Ψ―Ö―Ä–Α–Ϋ–Β–Ϋ–Η―è, –Α–¥–Φ–Η–Ϋ–Η―¹―²―Ä–Α―Ü–Η―è –Ϋ–Β –Ϋ–Β―¹–Β―² –Ψ―²–≤–Β―²―¹―²–≤–Β–Ϋ–Ϋ–Ψ―¹―²–Η –Ζ–Α –≤–Ψ–Ζ–Φ–Ψ–Ε–Ϋ―΄–Β –Ψ―²―Ä–Η―Ü–Α―²–Β–Μ―¨–Ϋ―΄–Β –Ω–Ψ―¹–Μ–Β–¥―¹―²–≤–Η―è, –≤–Ψ–Ζ–Ϋ–Η–Κ―à–Η–Β –≤ ―Ä–Β–Ζ―É–Μ―¨―²–Α―²–Β ―¹–Α–Φ–Ψ―¹―²–Ψ―è―²–Β–Μ―¨–Ϋ–Ψ–≥–Ψ –Η―¹–Ω–Ψ–Μ―¨–Ζ–Ψ–≤–Α–Ϋ–Η―è –£–Α–Φ–Η –Η–Ϋ―³–Ψ―Ä–Φ–Α―Ü–Η–Η ―¹ –Ω–Ψ―Ä―²–Α–Μ–Α –±–Β–Ζ –Ω―Ä–Β–¥–≤–Α―Ä–Η―²–Β–Μ―¨–Ϋ–Ψ–Ι –Κ–Ψ–Ϋ―¹―É–Μ―¨―²–Α―Ü–Η–Η ―¹ –≤―Ä–Α―΅–Ψ–Φ.
–ù–Α–Ε–Η–Φ–Α―è –Ϋ–Α –Κ–Ϋ–Ψ–Ω–Κ―É ¬Ϊ–£–Ψ–Ι―²–Η¬Μ, –£―΄ –Ω–Ψ–¥―²–≤–Β―Ä–Ε–¥–Α–Β―²–Β, ―΅―²–Ψ ―è–≤–Μ―è–Β―²–Β―¹―¨ –≤―Ä–Α―΅–Ψ–Φ –Η–Μ–Η ―¹―²―É–¥–Β–Ϋ―²–Ψ–Φ –Φ–Β–¥–Η―Ü–Η–Ϋ―¹–Κ–Ψ–≥–Ψ –≤―É–Ζ–Α.