–≠—Д—Д–µ–Ї—В –Њ–Ї—Б–Є–≥–Є–і—А–Њ–Ї—Б–Є–і–∞ –ґ–µ–ї–µ–Ј–∞ –љ–∞ —Н–љ–і–Њ–≥–µ–љ–љ—Л–µ –Ї–∞–ї—М—Ж–Є–є-—Д–Њ—Б—Д–∞—В–љ—Л–µ –њ—А–Њ—В–µ–Є–љ–Њ–≤—Л–µ —З–∞—Б—В–Є—Ж—Л –њ—А–Є —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–є –±–Њ–ї–µ–Ј–љ–Є –њ–Њ—З–µ–Ї: –Њ—В –љ–Њ–≤–Њ–≥–Њ –њ–Њ–љ–Є–Љ–∞–љ–Є—П –Љ–Њ–ї–µ–Ї—Г–ї—П—А–љ—Л—Е –Љ–µ—Е–∞–љ–Є–Ј–Љ–Њ–≤ –і–Њ –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Њ–є –Ј–љ–∞—З–Є–Љ–Њ—Б—В–Є
- –Р–љ–љ–Њ—В–∞—Ж–Є—П
- –°—В–∞—В—М—П
- –°—Б—Л–ї–Ї–Є
- English
–Т–≤–µ–і–µ–љ–Є–µ
–°–µ—А–і–µ—З–љ–Њ-—Б–Њ—Б—Г–і–Є—Б—В—Л–µ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П (–°–°–Ч) —П–≤–ї—П—О—В—Б—П –Њ—Б–љ–Њ–≤–љ–Њ–є –њ—А–Є—З–Є–љ–Њ–є —Б–Љ–µ—А—В–Є –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†—Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–є –±–Њ–ї–µ–Ј–љ—М—О –њ–Њ—З–µ–Ї (–•–С–Я). –£¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–•–С–Я –њ—А–Є—Б—Г—В—Б—В–≤—Г—О—В —В—А–∞–і–Є—Ж–Є–Њ–љ–љ—Л–µ —Д–∞–Ї—В–Њ—А—Л —А–Є—Б–Ї–∞ —А–∞–Ј–≤–Є—В–Є—П –°–°–Ч, —В–∞–Ї–Є–µ –Ї–∞–Ї –њ–Њ–ґ–Є–ї–Њ–є –≤–Њ–Ј—А–∞—Б—В, –∞—А—В–µ—А–Є–∞–ї—М–љ–∞—П¬†–≥–Є–њ–µ—А—В–µ–љ–Ј–Є—П, –і–Є—Б–ї–Є–њ–Є–і–µ–Љ–Є—П, —Б–∞—Е–∞—А–љ—Л–є –і–Є–∞–±–µ—В, –Њ–ґ–Є—А–µ–љ–Є–µ, –Ї—Г—А–µ–љ–Є–µ. –Ъ—А–Њ–Љ–µ —В–Њ–≥–Њ, –≤—Б–µ –±–Њ–ї—М—И–µ —Д–∞–Ї—В–Є—З–µ—Б–Ї–Є—Е –і–∞–љ–љ—Л—Е —Б–≤–Є–і–µ—В–µ–ї—М—Б—В–≤—Г—О—В –Њ¬†—В–Њ–Љ, —З—В–Њ –љ–µ—В—А–∞–і–Є—Ж–Є–Њ–љ–љ—Л–µ —Д–∞–Ї—В–Њ—А—Л —А–Є—Б–Ї–∞, —Б–њ–µ—Ж–Є—Д–Є—З–љ—Л–µ –і–ї—П –•–С–Я, –Є–≥—А–∞—О—В –Ї–ї—О—З–µ–≤—Г—О —А–Њ–ї—М –≤¬†–њ–∞—В–Њ–≥–µ–љ–µ–Ј–µ –°–°–Ч. –Ъ¬†—В–∞–Ї–Є–Љ —Д–∞–Ї—В–Њ—А–∞–Љ –Њ—В–љ–Њ—Б—П—В—Б—П –њ–Њ—З–µ—З–љ–∞—П –∞–љ–µ–Љ–Є—П, —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–µ –≤–Њ—Б–њ–∞–ї–µ–љ–Є–µ, –њ–Њ–≤—Л—И–µ–љ–љ—Л–є –Њ–Ї–Є—Б–ї–Є—В–µ–ї—М–љ—Л–є —Б—В—А–µ—Б—Б, —Г—А–µ–Љ–Є—З–µ—Б–Ї–Є–µ —В–Њ–Ї—Б–Є–љ—Л –Є¬†–Љ–Є–љ–µ—А–∞–ї—М–љ–Њ-–Ї–Њ—Б—В–љ—Л–µ –љ–∞—А—Г—И–µ–љ–Є—П (–Ь–Ъ–Э), –Ї–Њ—В–Њ—А—Л–µ —Б—А–µ–і–Є –љ–µ—В—А–∞–і–Є—Ж–Є–Њ–љ–љ—Л—Е —Д–∞–Ї—В–Њ—А–Њ–≤ —А–Є—Б–Ї–∞ —Б—В–∞–ї–Є –≤–∞–ґ–љ—Л–Љ –Є–≥—А–Њ–Ї–Њ–Љ –≤¬†–њ–∞—В–Њ–≥–µ–љ–µ–Ј–µ –°–°–Ч [1]. –°–Њ¬†–≤—А–µ–Љ–µ–љ–Є –њ—А–Њ–≤–µ–і–µ–љ–Є—П –њ–µ—А–≤—Л—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є –љ–∞—И–µ –њ–Њ–љ–Є–Љ–∞–љ–Є–µ –Ь–Ъ–Э –њ—А–Є –•–С–Я –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ —А–∞—Б—И–Є—А–Є–ї–Њ—Б—М, –Є¬†–њ–Њ—Б—В–µ–њ–µ–љ–љ–Њ —А–∞—Б–Ї—А—Л–≤–∞—О—В—Б—П –љ–Њ–≤—Л–µ —Б–ї–Њ–ґ–љ—Л–µ –Љ–µ—Е–∞–љ–Є–Ј–Љ—Л, –ї–µ–ґ–∞—Й–Є–µ –≤¬†–µ–≥–Њ –Њ—Б–љ–Њ–≤–µ. –Ю–і–љ–∞–Ї–Њ –њ–Њ-–њ—А–µ–ґ–љ–µ–Љ—Г —Б—Г—Й–µ—Б—В–≤—Г–µ—В –Љ–љ–Њ–ґ–µ—Б—В–≤–Њ –љ–µ—А–µ—И–µ–љ–љ—Л—Е –≤–Њ–њ—А–Њ—Б–Њ–≤ –≤¬†–њ–∞—В–Њ–≥–µ–љ–µ–Ј–µ —Б–Є–љ–і—А–Њ–Љ–∞ –Ь–Ъ–Э. –Ю–і–љ–Є–Љ –Є–Ј¬†–љ–Є—Е —П–≤–ї—П–µ—В—Б—П –Є–Ј—Г—З–µ–љ–Є–µ –Љ–µ—Е–∞–љ–Є–Ј–Љ–∞ –љ–∞—А—Г—И–µ–љ–Є—П¬†–≥–Њ–Љ–µ–Њ—Б—В–∞–Ј–∞ —Д–Њ—Б—Д–∞—В–Њ–≤ —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–•–С–Я, —Б–≤—П–Ј–∞–љ–љ–Њ–≥–Њ —Б¬†—А–∞–Ј–ї–Є—З–љ–Њ–є –њ–∞—В–Њ–ї–Њ–≥–Є–µ–є —Б–µ—А–і–µ—З–љ–Њ-—Б–Њ—Б—Г–і–Є—Б—В–Њ–є —Б–Є—Б—В–µ–Љ—Л, –≤¬†–њ–µ—А–≤—Г—О –Њ—З–µ—А–µ–і—М —Н–Ї—В–Њ–њ–Є—З–µ—Б–Ї–Њ–є –Ї–∞–ї—М—Ж–Є—Д–Є–Ї–∞—Ж–Є–µ–є,¬†–≥–Є–њ–µ—А—В—А–Њ—Д–Є–µ–є –ї–µ–≤–Њ–≥–Њ –ґ–µ–ї—Г–і–Њ—З–Ї–∞, —Г–≤–µ–ї–Є—З–µ–љ–Є–µ–Љ —З–Є—Б–ї–∞ —Б–µ—А–і–µ—З–љ–Њ-—Б–Њ—Б—Г–і–Є—Б—В—Л—Е —Б–Њ–±—Л—В–Є–є, –∞¬†—В–∞–Ї–ґ–µ —Б¬†–њ–Њ–≤—А–µ–ґ–і–µ–љ–Є–µ–Љ –њ–Њ—З–µ–Ї, –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–µ–Љ –Ї–Њ—Б—В–µ–є, —А–∞–Ј–≤–Є—В–Є–µ–Љ –≤—В–Њ—А–Є—З–љ–Њ–≥–Њ¬†–≥–Є–њ–µ—А–њ–∞—А–∞—В–Є—А–µ–Њ–Ј–∞, –Ї–Њ—В–Њ—А—Л–µ –µ—Й–µ –±–Њ–ї—М—И–µ —Г—Б–Є–ї–Є–≤–∞—О—В –љ–∞—А—Г—И–µ–љ–Є–µ —Д–Њ—Б—Д–∞—В–љ–Њ–є —А–µ–≥—Г–ї—П—Ж–Є–Є, –Є¬†–њ–Њ–≤—Л—И–µ–љ–Є–µ–Љ –ї–µ—В–∞–ї—М–љ–Њ—Б—В–Є. –Ґ–µ–Љ –љ–µ¬†–Љ–µ–љ–µ–µ —Д–∞—А–Љ–∞–Ї–Њ—В–µ—А–∞–њ–Є—П, –љ–∞–њ—А–∞–≤–ї–µ–љ–љ–∞—П –љ–µ–њ–Њ—Б—А–µ–і—Б—В–≤–µ–љ–љ–Њ –љ–∞¬†–Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є–µ –њ–Њ—Б–ї–µ–і—Б—В–≤–Є—П¬†–≥–Є–њ–µ—А—Д–Њ—Б—Д–∞—В–µ–Љ–Є–Є, –њ–Њ-–њ—А–µ–ґ–љ–µ–Љ—Г –Њ—Б—В–∞–µ—В—Б—П –Њ–±–ї–∞—Б—В—М—О –±–Њ–ї—М—И–Њ–є –љ–µ—Г–і–Њ–≤–ї–µ—В–≤–Њ—А–µ–љ–љ–Њ–є –Љ–µ–і–Є—Ж–Є–љ—Б–Ї–Њ–є –њ–Њ—В—А–µ–±–љ–Њ—Б—В–Є.
–§–Њ—Б—Д–∞—В-—Ж–µ–љ—В—А–Є—З–µ—Б–Ї–∞—П –њ–∞—А–∞–і–Є–≥–Љ–∞ —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–є –±–Њ–ї–µ–Ј–љ–Є –њ–Њ—З–µ–Ї
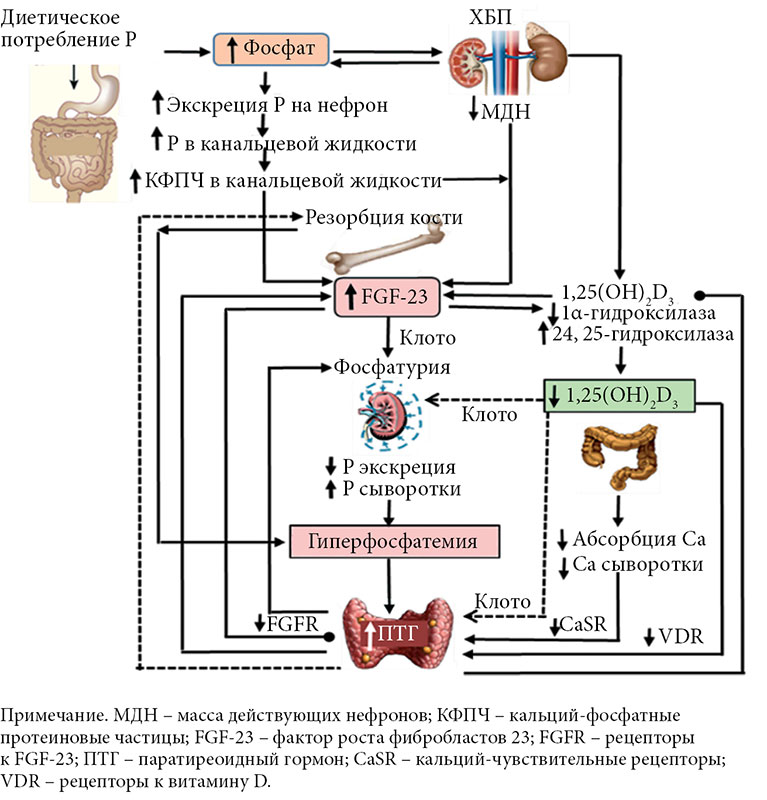
–§–Њ—Б—Д–Њ—А (P) —П–≤–ї—П–µ—В—Б—П –≤–∞–ґ–љ—Л–Љ –Љ–Є–Ї—А–Њ–Љ–Є–љ–µ—А–∞–ї–Њ–Љ, –Є–≥—А–∞—О—Й–Є–Љ –Ї–ї—О—З–µ–≤—Г—О —А–Њ–ї—М –≤¬†–Ї–ї–µ—В–Њ—З–љ–Њ–Љ –Љ–µ—В–∞–±–Њ–ї–Є–Ј–Љ–µ –Є¬†—Б—В—А—Г–Ї—В—Г—А–µ —В–Ї–∞–љ–µ–є. –£—А–Њ–≤–љ–Є –Ї–∞–ї—М—Ж–Є—П –Є¬†—Д–Њ—Б—Д–∞—В–Њ–≤ –≤¬†—Б—Л–≤–Њ—А–Њ—В–Ї–µ –Ї—А–Њ–≤–Є —Б—В—А–Њ–≥–Њ —А–µ–≥—Г–ї–Є—А—Г—О—В—Б—П –≤¬†–Њ—А–≥–∞–љ–Є–Ј–Љ–µ —З–µ–ї–Њ–≤–µ–Ї–∞. –£—А–Њ–≤–µ–љ—М –† –≤¬†—Б—Л–≤–Њ—А–Њ—В–Ї–µ –Ї—А–Њ–≤–Є –њ–Њ–і–і–µ—А–ґ–Є–≤–∞–µ—В—Б—П –≤¬†–≥–Њ–Љ–µ–Њ—Б—В–∞—В–Є—З–µ—Б–Ї–Њ–Љ –і–Є–∞–њ–∞–Ј–Њ–љ–µ –Ї–Є—И–µ—З–љ–Є–Ї–Њ–Љ, –Ї–Њ—Б—В—П–Љ–Є –Є¬†–њ–Њ—З–Ї–∞–Љ–Є. –Я–Њ—Б–Ї–Њ–ї—М–Ї—Г P —Г—З–∞—Б—В–≤—Г–µ—В –≤¬†—А—П–і–µ —Д–Є–Ј–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є—Е –њ—А–Њ—Ж–µ—Б—Б–Њ–≤, –њ–Њ–і–і–µ—А–ґ–∞–љ–Є–µ –µ–≥–Њ¬†–≥–Њ–Љ–µ–Њ—Б—В–∞–Ј–∞ –Њ—З–µ–љ—М –≤–∞–ґ–љ–Њ. –≠—В–Њ—В –њ—А–Њ—Ж–µ—Б—Б –Ї–Њ–Њ—А–і–Є–љ–Є—А—Г–µ—В—Б—П —Н–љ–і–Њ–Ї—А–Є–љ–љ–Њ–є —Б–Є—Б—В–µ–Љ–Њ–є –±–ї–∞–≥–Њ–і–∞—А—П –≤—Л—Б–Њ–Ї–Њ–Є–љ—В–µ–≥—А–Є—А–Њ–≤–∞–љ–љ–Њ–Љ—Г –і–µ–є—Б—В–≤–Є—О –љ–µ—Б–Ї–Њ–ї—М–Ї–Є—Е –Ї–ї—О—З–µ–≤—Л—Е¬†–≥–Њ—А–Љ–Њ–љ–Њ–≤, –≤–Ї–ї—О—З–∞—П —Д–∞–Ї—В–Њ—А —А–Њ—Б—В–∞ —Д–Є–±—А–Њ–±–ї–∞—Б—В–Њ–≤ (FGF-23), –Ї–Њ—А–µ—Ж–µ–њ—В–Њ—А –Ъ–ї–Њ—В–Њ, –њ–∞—А–∞—В–Є—А–µ–Њ–Є–і–љ—Л–є¬†–≥–Њ—А–Љ–Њ–љ (–Я–Ґ–У) –Є¬†1,25-–і–Є–≥–Є–і—А–Њ–Ї—Б–Є–≤–Є—В–∞–Љ–Є–љ D (1,25(OH)2D), —Г—З–∞—Б—В–≤—Г—О—Й–Є—Е –≤¬†—А–µ–≥—Г–ї—П—Ж–Є–Є¬†–≥–Њ–Љ–µ–Њ—Б—В–∞–Ј–∞ —Д–Њ—Б—Д–∞—В–Њ–≤ (—А–Є—Б—Г–љ–Њ–Ї).
–У–Њ–Љ–µ–Њ—Б—В–∞–Ј —Д–Њ—Б—Д–∞—В–Њ–≤ –њ–Њ–і–і–µ—А–ґ–Є–≤–∞–µ—В—Б—П –±–ї–∞–≥–Њ–і–∞—А—П —Б–±–∞–ї–∞–љ—Б–Є—А–Њ–≤–∞–љ–љ–Њ–Љ—Г –њ–Њ—Б—В—Г–њ–ї–µ–љ–Є—О –Є¬†–≤—Л–≤–µ–і–µ–љ–Є—О –Є–Ј¬†–Њ—А–≥–∞–љ–Є–Ј–Љ–∞ –Є¬†–Љ–Њ–ґ–µ—В –±—Л—В—М –ї–µ–≥–Ї–Њ –љ–∞—А—Г—И–µ–љ –њ—А–Є —Б–Њ—Б—В–Њ—П–љ–Є—П—Е, –њ—А–µ–≤—Л—И–∞—О—Й–Є—Е —Д–Є–Ј–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є –љ–µ–Њ–±—Е–Њ–і–Є–Љ–Њ–µ –њ–Њ—Б—В—Г–њ–ї–µ–љ–Є–µ –† –≤¬†–Њ—А–≥–∞–љ–Є–Ј–Љ. –Я–µ—А–µ–≥—А—Г–Ј–Ї–∞ –† –Љ–Њ–ґ–µ—В –±—Л—В—М –≤—Л–Ј–≤–∞–љ–∞ –њ—А–µ–≤—Л—И–µ–љ–Є–µ–Љ –њ–Њ—В—А–µ–±–ї–µ–љ–Є—П —Д–Њ—Б—Д–∞—В–∞ —Б¬†–њ–Є—Й–µ–є. –Я—А–Є–≤–µ—А–ґ–µ–љ–љ–Њ—Б—В—М –і–Є–µ—В–µ —Б¬†–≤—Л—Б–Њ–Ї–Є–Љ —Б–Њ–і–µ—А–ґ–∞–љ–Є–µ–Љ –±–µ–ї–Ї–∞, –њ–Њ–≤—Л—И–µ–љ–љ–Њ–µ –њ–Њ—В—А–µ–±–ї–µ–љ–Є–µ –Њ–±—А–∞–±–Њ—В–∞–љ–љ—Л—Е –њ–Є—Й–µ–≤—Л—Е –њ—А–Њ–і—Г–Ї—В–Њ–≤ –Є¬†–Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–љ–Є–µ –њ–Є—Й–µ–≤—Л—Е –і–Њ–±–∞–≤–Њ–Ї —Б¬†–≤—Л—Б–Њ–Ї–Є–Љ —Б–Њ–і–µ—А–ґ–∞–љ–Є–µ–Љ –љ–µ–Њ—А–≥–∞–љ–Є—З–µ—Б–Ї–Њ–≥–Њ –†¬†—Б–њ–Њ—Б–Њ–±—Б—В–≤–Њ–≤–∞–ї–Є –≤–Њ–Ј–љ–Є–Ї–љ–Њ–≤–µ–љ–Є—О –Њ—Б—В—А—Л—Е —Б–Ї–∞—З–Ї–Њ–≤ –Є–ї–Є –њ—А–µ—Е–Њ–і—П—Й–µ–Љ—Г –њ–Њ–≤—Л—И–µ–љ–Є—О —Г—А–Њ–≤–љ—П –† –≤¬†—Б—Л–≤–Њ—А–Њ—В–Ї–µ –Ї—А–Њ–≤–Є. –≠—В–Њ –Љ–Њ–ґ–µ—В –±—Л—В—М —З–∞—Б—В–Є—З–љ–Њ –Њ–±—К—П—Б–љ–µ–љ–Њ –≤—Л—Б–Њ–Ї–Њ–є —Н—Д—Д–µ–Ї—В–Є–≤–љ–Њ—Б—В—М—О –∞–±—Б–Њ—А–±—Ж–Є–Є, –Њ—Б–Њ–±–µ–љ–љ–Њ –љ–µ–Њ—А–≥–∞–љ–Є—З–µ—Б–Ї–Њ–≥–Њ –†,¬†–њ—А–Є—Б—Г—В—Б—В–≤—Г—О—Й–µ–≥–Њ –Ї–∞–Ї –≤¬†–њ–Є—Й–µ–≤—Л—Е –і–Њ–±–∞–≤–Ї–∞—Е, —В–∞–Ї –Є¬†–≤ –ї–µ–Ї–∞—А—Б—В–≤–µ–љ–љ—Л—Е –њ—А–µ–њ–∞—А–∞—В–∞—Е, –∞¬†—В–∞–Ї–ґ–µ —Ж–Є—А–Ї–∞–і–љ—Л–Љ–Є –Ї–Њ–ї–µ–±–∞–љ–Є—П–Љ–Є —Б—Л–≤–Њ—А–Њ—В–Њ—З–љ–Њ–≥–Њ –†.¬†–С—Л–ї–Њ –њ–Њ–Ї–∞–Ј–∞–љ–Њ, —З—В–Њ –і–∞–ґ–µ –Ї—А–∞—В–Ї–Њ—Б—А–Њ—З–љ—Л–µ –і–Є–µ—В—Л —Б¬†–≤—Л—Б–Њ–Ї–Є–Љ —Б–Њ–і–µ—А–ґ–∞–љ–Є–µ–Љ –† –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ —Г–≤–µ–ї–Є—З–Є–≤–∞—О—В —Ж–Є—А–Ї—Г–ї–Є—А—Г—О—Й–Є–µ —Г—А–Њ–≤–љ–Є FGF-23. –Ъ—А–Њ–Љ–µ —В–Њ–≥–Њ, –њ–µ—А–µ–≥—А—Г–Ј–Ї–∞ –† –Њ–±—Г—Б–ї–Њ–≤–ї–µ–љ–∞ —Б–љ–Є–ґ–µ–љ–Є–µ–Љ –Ї–Њ–ї–Є—З–µ—Б—В–≤–∞ —Д—Г–љ–Ї—Ж–Є–Њ–љ–Є—А—Г—О—Й–Є—Е –љ–µ—Д—А–Њ–љ–Њ–≤ –Ї–∞–Ї –≤—Б–ї–µ–і—Б—В–≤–Є–µ –њ—А–Њ—Ж–µ—Б—Б–∞ —Д–Є–Ј–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–≥–Њ —Б—В–∞—А–µ–љ–Є—П, —В–∞–Ї –Є¬†–њ—А–Њ–≥—А–µ—Б—Б–Є—А—Г—О—Й–µ–є –•–С–Я, –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П–Љ–Є –Ї–Њ—Б—В–µ–є, –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ—Л–Љ –і–Є–∞–ї–Є–Ј–Њ–Љ, –љ–Њ¬†–љ–µ –Њ–≥—А–∞–љ–Є—З–Є–≤–∞–µ—В—Б—П –Є–Љ–Є [2вАУ4].
–Ю–і–љ–∞–Ї–Њ –≤–Њ–њ—А–Њ—Б—Л –Њ¬†—В–Њ–Љ, –Ї–∞–Ї–Є–µ —А–µ–≥—Г–ї—П—В–Њ—А–љ—Л–µ –њ—А–Њ—Ж–µ—Б—Б—Л –≤¬†–њ–µ—А–≤—Г—О –Њ—З–µ—А–µ–і—М —Б–≤—П–Ј–∞–љ—Л —Б¬†–≤—Л—Б–Њ–Ї–Є–Љ–Є –љ–∞–≥—А—Г–Ј–Ї–∞–Љ–Є –† –Є¬†–Ї–∞–Ї –Њ–њ—А–µ–і–µ–ї—П–µ—В—Б—П —Б—В–µ–њ–µ–љ—М —А–µ–∞–Ї—Ж–Є–Є, –њ–Њ-–њ—А–µ–ґ–љ–µ–Љ—Г —П–≤–ї—П—О—В—Б—П –њ—А–µ–і–Љ–µ—В–Њ–Љ –Њ–±—Б—Г–ґ–і–µ–љ–Є—П. –Ю—В–Ї—А—Л—В–Є–µ —Б–Є—Б—В–µ–Љ—Л ¬ЂFGF-23¬†вАУ –Ъ–ї–Њ—В–Њ¬ї —А–∞—Б—И–Є—А–Є–ї–Њ –њ—А–µ–і—Б—В–∞–≤–ї–µ–љ–Є–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞—В–µ–ї–µ–є –Њ¬†–љ–∞—А—Г—И–µ–љ–Є—П—Е, —Б–≤—П–Ј–∞–љ–љ—Л—Е —Б¬†–њ–µ—А–µ–≥—А—Г–Ј–Ї–Њ–є —Д–Њ—Б—Д–∞—В–∞–Љ–Є.¬† –Ъ–Њ–ї–Є—З–µ—Б—В–≤–Њ —Н–Ї—Б–Ї—А–µ—Ж–Є–Є —Д–Њ—Б—Д–∞—В–Њ–≤ —Б¬†–Љ–Њ—З–Њ–є –≤¬†–њ–µ—А–≤—Г—О –Њ—З–µ—А–µ–і—М —А–µ–≥—Г–ї–Є—А—Г–µ—В—Б—П —Н–љ–і–Њ–Ї—А–Є–љ–љ–Њ–є —Б–Є—Б—В–µ–Љ–Њ–є, —Б–Њ—Б—В–Њ—П—Й–µ–є –Є–Ј¬†FGF-23 –Є¬†–µ–≥–Њ –Њ–±–ї–Є–≥–∞—В–љ–Њ–≥–Њ –Ї–Њ—А–µ—Ж–µ–њ—В–Њ—А–∞ –Ъ–ї–Њ—В–Њ. –Э–∞¬†—А–∞–љ–љ–Є—Е —Б—В–∞–і–Є—П—Е –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є—П –њ–Њ—З–µ–Ї —Б–љ–Є–ґ–∞–µ—В—Б—П —Н–Ї—Б–њ—А–µ—Б—Б–Є—П –Ъ–ї–Њ—В–Њ¬†вАУ –Љ–µ–Љ–±—А–∞–љ–љ–Њ–≥–Њ ќ≤-–≥–ї–Є–Ї–Њ–Ј–Є–і–∞–Ј–Њ–њ–Њ–і–Њ–±–љ–Њ–≥–Њ –±–µ–ї–Ї–∞ —В–Є–њ–∞ I, –Ї–Њ—В–Њ—А—Л–є –њ—А–Є–і–∞–µ—В —В–Ї–∞–љ–µ–≤—Г—О —Б–њ–µ—Ж–Є—Д–Є—З–љ–Њ—Б—В—М FGF-23. –Ф–µ—Д–Є—Ж–Є—В –Ъ–ї–Њ—В–Њ –њ–Њ–≤—Л—И–∞–µ—В —Г—А–Њ–≤–µ–љ—М Na+-–Ј–∞–≤–Є—Б–Є–Љ–Њ–≥–Њ —Д–Њ—Б—Д–∞—В–љ–Њ–≥–Њ —В—А–∞–љ—Б–њ–Њ—А—В–µ—А–∞ 2a (NaPi-2a), —Н–Ї—Б–њ—А–µ—Б—Б–Є—О NaPi-2c –≤¬†–њ–Њ—З–Ї–∞—Е –Є¬†—Г—А–Њ–≤–µ–љ—М NaPi-2b –≤¬†–Ї–Є—И–µ—З–љ–Є–Ї–µ, –Ї–Њ—В–Њ—А—Л–µ –Љ–Њ–≥—Г—В –Є–љ–Є—Ж–Є–Є—А–Њ–≤–∞—В—М –љ–∞–≥—А—Г–Ј–Ї—Г –† –њ—А–Є –љ–∞—А—Г—И–µ–љ–Є–Є —Д—Г–љ–Ї—Ж–Є–Є –њ–Њ—З–µ–Ї [5].
–£¬†—Н–Ї—Б–њ–µ—А–Є–Љ–µ–љ—В–∞–ї—М–љ—Л—Е –Љ—Л—И–µ–є –і–µ—Д–Є—Ж–Є—В –Ъ–ї–Њ—В–Њ –њ—А–Є–≤–Њ–і–Є—В –Ї¬†—Д–µ–љ–Њ—В–Є–њ—Г, —Е–∞—А–∞–Ї—В–µ—А–Є–Ј—Г—О—Й–µ–Љ—Г—Б—П –Є–Ј–Љ–µ–љ–µ–љ–љ—Л–Љ —Д–Њ—Б—Д–Њ—А–љ–Њ-–Ї–∞–ї—М—Ж–Є–µ–≤—Л–Љ –Њ–±–Љ–µ–љ–Њ–Љ —Б¬†–≥–Є–њ–µ—А—Д–Њ—Б—Д–∞—В–µ–Љ–Є–µ–є, –≤—В–Њ—А–Є—З–љ—Л–Љ¬†–≥–Є–њ–µ—А–њ–∞—А–∞—В–Є—А–µ–Њ–Ј–Њ–Љ, –Ї–∞–ї—М—Ж–Є—Д–Є–Ї–∞—Ж–Є–µ–є —Б–Њ—Б—Г–і–Њ–≤ (–Ъ–°),¬†–≥–Є–њ–µ—А—В—А–Њ—Д–Є–µ–є —Б–µ—А–і—Ж–∞, –њ—А–µ–ґ–і–µ–≤—А–µ–Љ–µ–љ–љ—Л–Љ —Б—В–∞—А–µ–љ–Є–µ–Љ –Є¬†—Б–Њ–Ї—А–∞—Й–µ–љ–Є–µ–Љ –њ—А–Њ–і–Њ–ї–ґ–Є—В–µ–ї—М–љ–Њ—Б—В–Є –ґ–Є–Ј–љ–Є [6]. –Ґ–∞–Ї–ґ–µ —Б–Њ–Њ–±—Й–∞–ї–Њ—Б—М, —З—В–Њ –Ъ–ї–Њ—В–Њ –Є–≥—А–∞–µ—В –Ї–∞–Ї FGF-23-–љ–µ–Ј–∞–≤–Є—Б–Є–Љ—Г—О, —В–∞–Ї –Є¬†FGF-23-–Ј–∞–≤–Є—Б–Є–Љ—Г—О —А–Њ–ї—М –≤¬†–≥–Њ–Љ–µ–Њ—Б—В–∞–Ј–µ –† [7, 8]. FGF-23¬†вАУ¬†–≥–Њ—А–Љ–Њ–љ, –Ї–Њ—В–Њ—А—Л–є —Б–µ–Ї—А–µ—В–Є—А—Г–µ—В—Б—П –Њ—Б—В–µ–Њ—Ж–Є—В–∞–Љ–Є –Є¬†–Њ—Б—В–µ–Њ–±–ї–∞—Б—В–∞–Љ–Є –Є¬†–і–Њ—Б—В–Є–≥–∞–µ—В —Б–≤–Њ–µ–є –Ї–ї–µ—В–Њ—З–љ–Њ–є —Б–њ–µ—Ж–Є—Д–Є—З–љ–Њ—Б—В–Є –≤¬†–њ–Њ—З–Ї–∞—Е –Є¬†–њ–∞—А–∞—Й–Є—В–Њ–≤–Є–і–љ—Л—Е –ґ–µ–ї–µ–Ј–∞—Е –±–ї–∞–≥–Њ–і–∞—А—П —Б–≤—П–Ј—Л–≤–∞–љ–Є—О –≤¬†–њ—А–Є—Б—Г—В—Б—В–≤–Є–Є —Б–≤–Њ–µ–≥–Њ –Њ–±—П–Ј–∞—В–µ–ї—М–љ–Њ–≥–Њ —В—А–∞–љ—Б–Љ–µ–Љ–±—А–∞–љ–љ–Њ–≥–Њ –±–µ–ї–Ї–∞ –Ї–Њ—А–µ—Ж–µ–њ—В–Њ—А–∞ –Ъ–ї–Њ—В–Њ, —З—В–Њ —Г–≤–µ–ї–Є—З–Є–≤–∞–µ—В —Б—А–Њ–і—Б—В–≤–Њ FGF-23 –Ї¬†–њ–Њ–≤—Б–µ–Љ–µ—Б—В–љ–Њ —Н–Ї—Б–њ—А–µ—Б—Б–Є—А—Г–µ–Љ—Л–Љ —А–µ—Ж–µ–њ—В–Њ—А–∞–Љ –§–†–§. –С–Є–љ–∞—А–љ—Л–є –Ї–Њ–Љ–њ–ї–µ–Ї—Б —А–µ—Ж–µ–њ—В–Њ—А–∞ –§–†–§ –Є¬†K–ї–Њ—В–Њ, —Н–Ї—Б–њ—А–µ—Б—Б–Є—А—Г—О—Й–Є–є—Б—П –≤¬†–њ–Њ—З–µ—З–љ—Л—Е –Ї–∞–љ–∞–ї—М—Ж–∞—Е, –њ–Њ–і–∞–≤–ї—П–µ—В —А–µ–∞–±—Б–Њ—А–±—Ж–Є—О –† –≤—Б–ї–µ–і—Б—В–≤–Є–µ –Є–љ–≥–Є–±–Є—А–Њ–≤–∞–љ–Є—П –љ–∞—В—А–Є–є-—Д–Њ—Б—Д–∞—В–љ—Л—Е –Ї–Њ—В—А–∞–љ—Б–њ–Њ—А—В–µ—А–Њ–≤ 2-–≥–Њ¬†—В–Є–њ–∞, —В–µ–Љ —Б–∞–Љ—Л–Љ —Б–њ–Њ—Б–Њ–±—Б—В–≤—Г—П –Є—Е –≤—Л–≤–µ–і–µ–љ–Є—О —Б¬†–Љ–Њ—З–Њ–є. –Я–Њ–Љ–Є–Љ–Њ —Д—Г–љ–Ї—Ж–Є–Є —Д–Њ—Б—Д–∞—В—Г—А–Є—З–µ—Б–Ї–Њ–≥–Њ¬†–≥–Њ—А–Љ–Њ–љ–∞, FGF-23 –і–µ–є—Б—В–≤—Г–µ—В –Ї–∞–Ї –Ї–Њ–љ—В—А—А–µ–≥—Г–ї—П—В–Њ—А–љ—Л–є¬†–≥–Њ—А–Љ–Њ–љ¬†вАУ –Є–љ–≥–Є–±–Є—А—Г–µ—В —Б–µ–Ї—А–µ—Ж–Є—О –Я–Ґ–У, —Б–љ–Є–ґ–∞–µ—В —Б–Є–љ—В–µ–Ј –∞–Ї—В–Є–≤–љ–Њ–≥–Њ –≤–Є—В–∞–Љ–Є–љ–∞ D (1,25-–і–Є–≥–Є–і—А–Њ–Ї—Б–Є–≤–Є—В–∞–Љ–Є–љ D3) —Б¬†–њ–Њ–Љ–Њ—Й—М—О –њ–Њ–і–∞–≤–ї–µ–љ–Є—П —Н–Ї—Б–њ—А–µ—Б—Б–Є–Є 1ќ±-–≥–Є–і—А–Њ–Ї—Б–Є–ї–∞–Ј—Л, –љ–µ–Њ–±—Е–Њ–і–Є–Љ–Њ–є –і–ї—П —Б–Є–љ—В–µ–Ј–∞, –Є¬†–њ–Њ–≤—Л—И–µ–љ–Є—П —Н–Ї—Б–њ—А–µ—Б—Б–Є–Є 24-–≥–Є–і—А–Њ–Ї—Б–Є–ї–∞–Ј—Л, –љ–µ–Њ–±—Е–Њ–і–Є–Љ–Њ–є –і–ї—П –µ–≥–Њ –Є–љ–∞–Ї—В–Є–≤–∞—Ж–Є–Є –≤¬†–Ї–ї–µ—В–Ї–∞—Е –њ—А–Њ–Ї—Б–Є–Љ–∞–ї—М–љ—Л—Е –Ї–∞–љ–∞–ї—М—Ж–µ–≤ –њ–Њ—З–µ–Ї. FGF-23, –Я–Ґ–У –Є¬†1,25(OH)2D3 –≤–Ј–∞–Є–Љ–Њ–і–µ–є—Б—В–≤—Г—О—В –і—А—Г–≥ —Б¬†–і—А—Г–≥–Њ–Љ —З–µ—А–µ–Ј –Ї–ї–∞—Б—Б–Є—З–µ—Б–Ї–Є–µ –њ–µ—В–ї–Є –Њ—В—А–Є—Ж–∞—В–µ–ї—М–љ–Њ–є —Н–љ–і–Њ–Ї—А–Є–љ–љ–Њ–є –Њ–±—А–∞—В–љ–Њ–є —Б–≤—П–Ј–Є, –Ї–Њ—В–Њ—А—Л–µ –≤–ї–Є—П—О—В –љ–∞¬†—В—А–∞–љ—Б–њ–Њ—А—В –°–∞¬†–Є¬†–† –≤¬†–њ–Њ—З–Ї–∞—Е, –Ї–Њ—Б—В—П—Е –Є¬†–Ї–Є—И–µ—З–љ–Є–Ї–µ [9]. –Ґ–∞–Ї–Є–Љ –Њ–±—А–∞–Ј–Њ–Љ, –і–≤–∞ —Д–Њ—Б—Д–∞—В—Г—А–Є—З–µ—Б–Ї–Є—Е¬†–≥–Њ—А–Љ–Њ–љ–∞¬†вАУ –Я–Ґ–У –Є¬†FGF-23 —Б–њ–Њ—Б–Њ–±—Б—В–≤—Г—О—В –≤—Л–≤–µ–і–µ–љ–Є—О –† —Б¬†–Љ–Њ—З–Њ–є –≤¬†–Њ—В–≤–µ—В –љ–∞¬†–љ–∞–≥—А—Г–Ј–Ї—Г –† –і–ї—П –њ–Њ–і–і–µ—А–ґ–∞–љ–Є—П —Д–Њ—Б—Д–∞—В–љ–Њ–≥–Њ¬†–≥–Њ–Љ–µ–Њ—Б—В–∞–Ј–∞ –≤¬†–Ї—А–∞—В–Ї–Њ—Б—А–Њ—З–љ–Њ–є (—З–∞—Б—Л) –Є¬†–≤ –і–Њ–ї–≥–Њ—Б—А–Њ—З–љ–Њ–є (–і–љ–Є) –њ–µ—А—Б–њ–µ–Ї—В–Є–≤–µ —Б–Њ–Њ—В–≤–µ—В—Б—В–≤–µ–љ–љ–Њ [4, 10, 11].
–Ъ–∞–Ї —В–Њ–ї—М–Ї–Њ –Ї–Њ–ї–Є—З–µ—Б—В–≤–Њ —Д—Г–љ–Ї—Ж–Є–Њ–љ–Є—А—Г—О—Й–Є—Е –љ–µ—Д—А–Њ–љ–Њ–≤ —Б–љ–Є–ґ–∞–µ—В—Б—П –і–Њ —Г—А–Њ–≤–љ—П, —Б–Њ–Њ—В–≤–µ—В—Б—В–≤—Г—О—Й–µ–≥–Њ –•–С–Я —З–µ—В–≤–µ—А—В–Њ–є-–њ—П—В–Њ–є —Б—В–∞–і–Є–Є (—Б–Ї–Њ—А–Њ—Б—В—М –Ї–ї—Г–±–Њ—З–Ї–Њ–≤–Њ–є —Д–Є–ї—М—В—А–∞—Ж–Є–Є (–°–Ъ–§) <¬†30 –Љ–ї/–Љ–Є–љ/1,73 –Љ2), –њ—А–Њ–Є—Б—Е–Њ–і–Є—В –љ–∞—А—Г—И–µ–љ–Є–µ —А–µ–≥—Г–ї—П—Ж–Є–Є –Њ—Б–Є ¬ЂFGF-23¬†вАУ K–ї–Њ—В–Њ¬ї. –Я–Њ–≤—Л—И–µ–љ–љ—Л–µ —Г—А–Њ–≤–љ–Є FGF-23 –Є¬†–Я–Ґ–У –±–Њ–ї—М—И–µ –љ–µ¬†—Б–њ–Њ—Б–Њ–±–љ—Л –њ–Њ–і–і–µ—А–ґ–Є–≤–∞—В—М —Д–Њ—Б—Д–∞—В–љ—Л–є –±–∞–ї–∞–љ—Б —З–µ—А–µ–Ј —Г—Б–Є–ї–µ–љ–Є–µ —Н–Ї—Б–Ї—А–µ—Ж–Є–Є –† –Њ—Б—В–∞–≤—И–Є–Љ–Є—Б—П —Д—Г–љ–Ї—Ж–Є–Њ–љ–Є—А—Г—О—Й–Є–Љ–Є –љ–µ—Д—А–Њ–љ–∞–Љ–Є, –њ–Њ–≤—Л—И–∞–µ—В—Б—П –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є—П –† –≤¬†–ґ–Є–і–Ї–Њ—Б—В–Є –Ї–∞–љ–∞–ї—М—Ж–µ–≤, —З—В–Њ –≤¬†–Ї–Њ–љ–µ—З–љ–Њ–Љ –Є—В–Њ–≥–µ –њ—А–Є–≤–Њ–і–Є—В –Ї¬†—А–∞–Ј–≤–Є—В–Є—О —П–≤–љ–Њ–є¬†–≥–Є–њ–µ—А—Д–Њ—Б—Д–∞—В–µ–Љ–Є–Є, –Ї–Њ—В–Њ—А–∞—П —Б—В–Є–Љ—Г–ї–Є—А—Г–µ—В –і–∞–ї—М–љ–µ–є—И—Г—О —Б–µ–Ї—А–µ—Ж–Є—О FGF-23 –Ї–ї–µ—В–Ї–∞–Љ–Є –Ї–Њ—Б—В–љ–Њ–є —В–Ї–∞–љ–Є [12]. –Ю–і–љ–∞–Ї–Њ FGF-23, –≤–µ—А–Њ—П—В–љ–Њ, –љ–µ¬†—П–≤–ї—П–µ—В—Б—П –љ–∞–і–µ–ґ–љ—Л–Љ –±–Є–Њ–Љ–∞—А–Ї–µ—А–Њ–Љ –љ–∞–≥—А—Г–Ј–Ї–Є¬†–†,¬†–њ–Њ—Б–Ї–Њ–ї—М–Ї—Г –≤¬†–µ–≥–Њ —А–µ–≥—Г–ї—П—Ж–Є–Є —В–∞–Ї–ґ–µ —Г—З–∞—Б—В–≤—Г—О—В –Я–Ґ–У, –Ї–∞–ї—М—Ж–Є—В—А–Є–Њ–ї, –Ї–∞–ї—М—Ж–Є–є, —Н—А–Є—В—А–Њ–њ–Њ—Н—В–Є–љ (EPO), —Д–∞–Ї—В–Њ—А—Л, –Є–љ–і—Г—Ж–Є—А—Г–µ–Љ—Л–µ¬†–≥–Є–њ–Њ–Ї—Б–Є–µ–є (HIF), —А–∞–Ј–ї–Є—З–љ—Л–µ –≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ—Л–µ —Б—В–Є–Љ—Г–ї—Л –Є¬†–њ–Њ—З–µ—З–љ—Л–є –Ї–ї–Є—А–µ–љ—Б [13].
–Ъ–∞–ї—М—Ж–Є–є-—Д–Њ—Б—Д–∞—В–љ—Л–µ –њ—А–Њ—В–µ–Є–љ–Њ–≤—Л–µ —З–∞—Б—В–Є—Ж—Л: –њ—А–Њ—В–µ–Ї—В–Є–≤–љ—Л–µ –Є¬†–њ–∞—В–Њ–≥–µ–љ–љ—Л–µ —Н—Д—Д–µ–Ї—В—Л
–Э–µ–і–∞–≤–љ–Њ –±—Л–ї –≤—Л—П—Б–љ–µ–љ –Љ–Њ–ї–µ–Ї—Г–ї—П—А–љ—Л–є –Љ–µ—Е–∞–љ–Є–Ј–Љ —А–∞–љ–љ–µ–≥–Њ —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–≥–Њ –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є—П –њ–Њ—З–µ–Ї, –≤—Л–Ј–≤–∞–љ–љ–Њ–≥–Њ –њ–µ—А–µ–≥—А—Г–Ј–Ї–Њ–є –† [14]. –Ъ–∞–ї—М—Ж–Є–є –Є¬†—Д–Њ—Б—Д–∞—В, —Б–Њ–і–µ—А–ґ–∞—Й–Є–µ—Б—П –≤¬†—А–∞—Ж–Є–Њ–љ–µ, –≤—Б–∞—Б—Л–≤–∞—О—В—Б—П –њ—Г—В–µ–Љ —В—А–∞–љ—Б–Ї–ї–µ—В–Њ—З–љ–Њ–≥–Њ –Є¬†–њ–∞—А–∞–Ї–ї–µ—В–Њ—З–љ–Њ–≥–Њ —В—А–∞–љ—Б–њ–Њ—А—В–∞ —З–µ—А–µ–Ј —Н–њ–Є—В–µ–ї–Є–є –Ї–Є—И–µ—З–љ–Є–Ї–∞, –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є—П –°–∞¬†–Є¬†–† –≤¬†–Є–љ—В–µ—А—Б—В–Є—Ж–Є–∞–ї—М–љ–Њ–Љ –њ—А–Њ—Б—В—А–∞–љ—Б—В–≤–µ —Г–≤–µ–ї–Є—З–Є–≤–∞–µ—В—Б—П –Є¬†–≤ —А–µ–Ј—Г–ї—М—В–∞—В–µ —Б–њ–Њ–љ—В–∞–љ–љ–Њ –Њ–±—А–∞–Ј—Г—О—В—Б—П –Ї–Њ–ї–ї–Њ–Є–і–љ—Л–µ –љ–∞–љ–Њ—З–∞—Б—В–Є—Ж—Л, –љ–∞–Ј—Л–≤–∞–µ–Љ—Л–µ –Ї–∞–ї—М—Ж–Є–є-—Д–Њ—Б—Д–∞—В–љ—Л–Љ–Є –њ—А–Њ—В–µ–Є–љ–Њ–≤—Л–Љ–Є –Љ–Њ–љ–Њ–Љ–µ—А–∞–Љ–Є (–Ъ–§–Я–Ь), –Ї–Њ—В–Њ—А—Л–µ –њ–Њ–њ–∞–і–∞—О—В –≤¬†–њ–Њ—А—В–∞–ї—М–љ—Л–є –Є/–Є–ї–Є —Б–Є—Б—В–µ–Љ–љ—Л–є –Ї—А–Њ–≤–Њ—В–Њ–Ї —З–µ—А–µ–Ј –ї–Є–Љ—Д–∞—В–Є—З–µ—Б–Ї—Г—О —Б–Є—Б—В–µ–Љ—Г,¬†–≥–і–µ –љ–µ–Љ–µ–і–ї–µ–љ–љ–Њ —Б–≤—П–Ј—Л–≤–∞—О—В—Б—П —Б¬†—Б—Л–≤–Њ—А–Њ—В–Њ—З–љ—Л–Љ –±–µ–ї–Ї–Њ–Љ —Д–µ—В—Г–Є–љ–Њ–Љ-–Р, —Б–µ–Ї—А–µ—В–Є—А—Г–µ–Љ—Л–Љ –њ–µ—З–µ–љ—М—О, –∞–±—Б–Њ—А–±–Є—А—Г—О—В –±–µ–ї–Ї–Є –Є–Ј¬†–Њ–Ї—А—Г–ґ–∞—О—Й–µ–є —Б—А–µ–і—Л –Є¬†–Њ–±—К–µ–і–Є–љ—П—О—В –Є—Е –≤¬†–Ї–ї–∞—Б—В–µ—А—Л –±–µ–ї–Ї–∞ –Є¬†–∞–Љ–Њ—А—Д–љ–Њ–≥–Њ —Д–Њ—Б—Д–∞—В–∞ –Ї–∞–ї—М—Ж–Є—П (–°–∞3(–†–Ю4)2). –Ъ–§–Я–Ь —Д–Є–ї—М—В—А—Г—О—В—Б—П –Ї–ї—Г–±–Њ—З–Ї–∞–Љ–Є, –њ–Њ—Б—В—Г–њ–∞—О—В –≤¬†–Ї–∞–љ–∞–ї—М—Ж–µ–≤—Г—О –ґ–Є–і–Ї–Њ—Б—В—М –Є, –Ї–∞–Ї –њ—А–µ–і–њ–Њ–ї–∞–≥–∞–µ—В—Б—П, –Ј–∞–њ—Г—Б–Ї–∞—О—В —Б–µ–Ї—А–µ—Ж–Є—О FGF-23 [11, 15, 16].
–Э–µ—Б–Љ–Њ—В—А—П –љ–∞¬†—В–Њ —З—В–Њ —Б—Г—Й–µ—Б—В–≤—Г—О—В –±–Њ–ї—М—И–Є–µ –Є–љ–і–Є–≤–Є–і—Г–∞–ї—М–љ—Л–µ –Є¬†–≤–Є–і–Њ–≤—Л–µ —А–∞–Ј–ї–Є—З–Є—П –≤¬†–Ї–Њ–ї–Є—З–µ—Б—В–≤–µ –љ–µ—Д—А–Њ–љ–Њ–≤ –Є¬†–≤–µ–ї–Є—З–Є–љ–µ —Н–Ї—Б–Ї—А–µ—Ж–Є–Є –† —Б¬†–Љ–Њ—З–Њ–є, —Г¬†–Ј–і–Њ—А–Њ–≤—Л—Е –Љ–Њ–ї–Њ–і—Л—Е –ї—О–і–µ–є –љ–∞¬†–њ–Њ—З–Ї—Г –њ—А–Є—Е–Њ–і–Є—В—Б—П –Њ–Ї–Њ–ї–Њ 1 –Љ–ї–љ –љ–µ—Д—А–Њ–љ–Њ–≤ –Є¬†–≤ —Б—А–µ–і–љ–µ–Љ –≤¬†–і–µ–љ—М —Н–Ї—Б–Ї—А–µ—В–Є—А—Г–µ—В—Б—П –Њ–Ї–Њ–ї–Њ 1,0¬†–≥¬†–†.¬†–Ґ–∞–Ї–Є–Љ –Њ–±—А–∞–Ј–Њ–Љ, —Н–Ї—Б–Ї—А–µ—Ж–Є—П –† –Є–Ј¬†—А–∞—Б—З–µ—В–∞ –љ–∞¬†–Њ–і–Є–љ –љ–µ—Д—А–Њ–љ –Њ—Ж–µ–љ–Є–≤–∞–µ—В—Б—П –њ—А–Є–Љ–µ—А–љ–Њ –≤¬†0,5 –Љ–Ї–≥ –≤¬†–і–µ–љ—М. –°¬†—Г–Љ–µ–љ—М—И–µ–љ–Є–µ–Љ –Ї–Њ–ї–Є—З–µ—Б—В–≤–∞ —Д—Г–љ–Ї—Ж–Є–Њ–љ–Є—А—Г—О—Й–Є—Е –љ–µ—Д—А–Њ–љ–Њ–≤ –≤¬†–њ—А–Њ—Ж–µ—Б—Б–µ –њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є—П –•–С–Я –і–Њ 0,5 –Љ–ї–љ —Н–Ї—Б–Ї—А–µ—Ж–Є—П –† –Є–Ј¬†—А–∞—Б—З–µ—В–∞ –љ–∞¬†–љ–µ—Д—А–Њ–љ –Љ–Њ–ґ–µ—В –і–Њ—Б—В–Є–≥–∞—В—М 1,0 –Љ–Ї–≥ –≤¬†–і–µ–љ—М [17]. –Ъ–∞–Ї —В–Њ–ї—М–Ї–Њ –≤–љ–µ–Ї–ї–µ—В–Њ—З–љ–∞—П –ґ–Є–і–Ї–Њ—Б—В—М –њ–µ—А–µ–љ–∞—Б—Л—Й–∞–µ—В—Б—П –Є–Њ–љ–∞–Љ–Є –† –Є¬†–°–∞, –њ—А–µ–≤—Л—И–∞—О—Й–Є–Љ–Є –њ—А–µ–і–µ–ї —А–∞—Б—В–≤–Њ—А–Є–Љ–Њ—Б—В–Є, –Ъ–§–Я–Ь —Б–њ–Њ–љ—В–∞–љ–љ–Њ –њ–Њ–і–≤–µ—А–≥–∞—О—В—Б—П –∞–≥—А–µ–≥–∞—Ж–Є–Є, –њ—А–µ–≤—А–∞—Й–∞—П—Б—М –≤¬†–њ–µ—А–≤–Є—З–љ—Л–µ –Ї–∞–ї—М—Ж–Є–є-—Д–Њ—Б—Д–∞—В–љ—Л–µ –њ—А–Њ—В–µ–Є–љ–Њ–≤—Л–µ —З–∞—Б—В–Є—Ж—Л (–Ъ–§–Я–І-I), –Ї–Њ—В–Њ—А—Л–µ –Є–Љ–µ—О—В —Б—Д–µ—А–Є—З–µ—Б–Ї—Г—О –њ—А–Є—А–Њ–і—Г, —Ж–Є—А–Ї—Г–ї–Є—А—Г—О—В –≤¬†–њ–ї–∞–Ј–Љ–µ –Є¬†—Д—Г–љ–Ї—Ж–Є–Њ–љ–Є—А—Г—О—В –Ї–∞–Ї —Д–Є–Ј–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–є —А–µ–≥—Г–ї—П—В–Њ—А –Љ–Є–љ–µ—А–∞–ї—М–љ–Њ–≥–Њ¬†–≥–Њ–Љ–µ–Њ—Б—В–∞–Ј–∞ –Ї—А–Њ–≤–Є, –њ—А–µ–њ—П—В—Б—В–≤—Г—П –Ї–∞–ї—М—Ж–Є—Д–Є–Ї–∞—Ж–Є–Є [2, 18]. –Я–µ—А–≤–Є—З–љ—Л–µ –Ъ–§–Я–І –њ–Њ–і–≤–µ—А–≥–∞—О—В—Б—П –і–∞–ї—М–љ–µ–є—И–µ–є –∞–≥—А–µ–≥–∞—Ж–Є–Є –Є¬†—Б–Њ–Ј—А–µ–≤–∞–љ–Є—О –њ–Њ–і –≤–ї–Є—П–љ–Є–µ–Љ –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є–Є —Д–µ—В—Г–Є–љ–∞-A, —Д–Њ—Б—Д–∞—В–∞, –Є–Њ–љ–Њ–≤ –Ї–∞–ї—М—Ж–Є—П –Є¬†–Љ–∞–≥–љ–Є—П, –∞¬†—В–∞–Ї–ґ–µ —А–Э –Њ–Ї—А—Г–ґ–∞—О—Й–µ–є –Љ–Є–Ї—А–Њ—Б—А–µ–і—Л. –Т¬†—А–µ–Ј—Г–ї—М—В–∞—В–µ –Ъ–§–Я–І-I —А–µ–Њ—А–≥–∞–љ–Є–Ј—Г—О—В—Б—П –Є–Ј¬†–Ї–Њ–ї–ї–Њ–Є–і–љ—Л—Е –љ–∞–љ–Њ—З–∞—Б—В–Є—Ж –≤–Њ –≤—В–Њ—А–Є—З–љ—Л–µ –Ъ–§–Я–І-II, –Ї–Њ—В–Њ—А—Л–µ —Б–Њ–і–µ—А–ґ–∞—В –≤¬†—Б–≤–Њ–µ–є —Б–µ—А–і—Ж–µ–≤–Є–љ–µ –Ї—А–Є—Б—В–∞–ї–ї–Є—З–µ—Б–Ї–Є–є¬†–≥–Є–і—А–Њ–Ї—Б–Є–∞–њ–∞—В–Є—В, –Њ–љ–Є –Ї—А—Г–њ–љ–µ–µ –Ъ–§–Я–І-I, –±–Њ–ї–µ–µ –њ–ї–Њ—В–љ—Л–µ, —Б¬†–±–Њ¬і–ї—М—И–Є–Љ –і–Є–∞–Љ–µ—В—А–Њ–Љ, –љ–µ—А–∞—Б—В–≤–Њ—А–Є–Љ—Л –≤¬†—Б—Л–≤–Њ—А–Њ—В–Ї–µ –Ї—А–Њ–≤–Є –Є¬†–Є–Љ–µ—О—В –Є–≥–ї–Њ–Њ–±—А–∞–Ј–љ—Г—О –Ї—А–Є—Б—В–∞–ї–ї–Є—З–µ—Б–Ї—Г—О —Б—В—А—Г–Ї—В—Г—А—Г.
–Я–µ—А–µ—Б—Л—Й–µ–љ–Є–µ —Б—Л–≤–Њ—А–Њ—В–Ї–Є —В–∞–Ї–ґ–µ –Є—Б–њ–Њ–ї—М–Ј—Г–µ—В—Б—П –і–ї—П –Є–Ј–Љ–µ—А–µ–љ–Є—П –њ–Њ–ї–Њ–≤–Є–љ—Л –Љ–∞–Ї—Б–Є–Љ–∞–ї—М–љ–Њ–≥–Њ –≤—А–µ–Љ–µ–љ–Є –њ–µ—А–µ—Е–Њ–і–∞, –љ–µ–Њ–±—Е–Њ–і–Є–Љ–Њ–≥–Њ –і–ї—П —В—А–∞–љ—Б—Д–Њ—А–Љ–∞—Ж–Є–Є –Є–Ј¬†–∞–Љ–Њ—А—Д–љ–Њ–≥–Њ —Б–Њ—Б—В–Њ—П–љ–Є—П –≤¬†–Ї—А–Є—Б—В–∞–ї–ї–Є—З–µ—Б–Ї–Њ–µ. –£—Б—В–∞–љ–Њ–≤–ї–µ–љ–Њ, —З—В–Њ –њ–Њ–ї–Њ–≤–Є–љ–∞ –≤—А–µ–Љ–µ–љ–Є, –љ–µ–Њ–±—Е–Њ–і–Є–Љ–Њ–≥–Њ –і–ї—П —Б–њ–Њ–љ—В–∞–љ–љ–Њ–≥–Њ –њ–µ—А–µ—Е–Њ–і–∞ –Њ—В¬†–Ъ–§–Я–І-I –Ї¬†–Ъ–§–Я–І-II, –Њ–±–Њ–Ј–љ–∞—З–∞–µ–Љ–∞—П –Ї–∞–Ї T50, —П–≤–ї—П–µ—В—Б—П —Б–Є–ї—М–љ—Л–Љ –њ—А–µ–і–Є–Ї—В–Њ—А–Њ–Љ –Ї–∞–ї—М—Ж–Є—Д–Є—Ж–Є—А—Г—О—Й–Є—Е —Б–≤–Њ–є—Б—В–≤ —Б—Л–≤–Њ—А–Њ—В–Ї–Є. –°—Л–≤–Њ—А–Њ—В–Ї–∞ —Б¬†–±–Њ–ї–µ–µ –≤—Л—Б–Њ–Ї–Є–Љ T50 –Љ–µ–љ–µ–µ —Б–Ї–ї–Њ–љ–љ–∞ –Ї¬†–Ї–∞–ї—М—Ж–Є—Д–Є–Ї–∞—Ж–Є–Є —В–Ї–∞–љ–µ–є –њ–Њ¬†—Б—А–∞–≤–љ–µ–љ–Є—О —Б¬†—Б—Л–≤–Њ—А–Њ—В–Ї–Њ–є —Б¬†–±–Њ–ї–µ–µ –љ–Є–Ј–Ї–Є–Љ T50. –Я–Њ–≤—Л—И–µ–љ–љ–∞—П —Б–Ї–ї–Њ–љ–љ–Њ—Б—В—М —Б—Л–≤–Њ—А–Њ—В–Ї–Є –Ї¬†–Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є—О –Ъ–§–Я–І-II –љ–∞–±–ї—О–і–∞–µ—В—Б—П –≤¬†–≤–Є–і–µ —Б–љ–Є–ґ–µ–љ–Є—П T50. –Т—В–Њ—А–Є—З–љ—Л–µ –Ъ–§–Я–І –Є–љ—В–µ—А–љ–∞–ї–Є–Ј—Г—О—В—Б—П –Ї–ї–µ—В–Ї–∞–Љ–Є —Б–Њ—Б—Г–і–Њ–≤, –≤—Л–Ј—Л–≤–∞—П –Љ–∞—Б—Б–Њ–≤—Л–є –њ—А–Є—В–Њ–Ї –Є–Њ–љ–Њ–≤ –Ї–∞–ї—М—Ж–Є—П –≤¬†—Ж–Є—В–Њ–Ј–Њ–ї—М, —З—В–Њ –њ—А–Є–≤–Њ–і–Є—В –Ї¬†–њ—А–Њ–≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ–Њ–Љ—Г –Њ—В–≤–µ—В—Г, –Ї–ї–µ—В–Њ—З–љ–Њ–є –і–Є—Б—Д—Г–љ–Ї—Ж–Є–Є –Є¬†–≥–Є–±–µ–ї–Є –Ї–ї–µ—В–Њ–Ї [19, 20].
–Ю–±—А–∞–Ј–Њ–≤–∞–љ–Є–µ –Ї–∞–ї—М—Ж–Є–є-—Д–Њ—Б—Д–∞—В–љ—Л—Е –Љ–Є–Ї—А–Њ–Ї—А–Є—Б—В–∞–ї–ї–Њ–≤ —Б–Њ–њ—А–Њ–≤–Њ–ґ–і–∞–µ—В—Б—П —Б–љ–Є–ґ–µ–љ–Є–µ–Љ —Г—А–Њ–≤–љ—П –°–∞¬†–≤¬†–Ї—А–Њ–≤–Є –Є¬†–Є–љ–∞–Ї—В–Є–≤–∞—Ж–Є–µ–є –Ї–∞–ї—М—Ж–Є–є-—З—Г–≤—Б—В–≤–Є—В–µ–ї—М–љ–Њ–≥–Њ —А–µ—Ж–µ–њ—В–Њ—А–∞ (CaSR), –Є–љ–і—Г—Ж–Є—А—Г–µ—В —Б–µ–Ї—А–µ—Ж–Є—О –Я–Ґ–У. –Я–Ґ–У –Њ–±–ї–∞–і–∞–µ—В –∞–Ї—В–Є–≤–љ–Њ—Б—В—М—О, –њ–Њ–≤—Л—И–∞—О—Й–µ–є —Г—А–Њ–≤–µ–љ—М –°–∞¬†–≤¬†–Ї—А–Њ–≤–Є –Є¬†—Н–Ї—Б–Ї—А–µ—Ж–Є—О –† —Б¬†–Љ–Њ—З–Њ–є, –±–ї–∞–≥–Њ–і–∞—А—П —З–µ–Љ—Г —Г—А–Њ–≤–љ–Є –°–∞¬†–Є¬†–† –≤¬†–Ї—А–Њ–≤–Є –≤–Њ—Б—Б—В–∞–љ–∞–≤–ї–Є–≤–∞—О—В—Б—П –і–Њ –±–∞–Ј–Њ–≤–Њ–≥–Њ –Ј–љ–∞—З–µ–љ–Є—П. –≠—В–∞ –љ–µ–≥–∞—В–Є–≤–љ–∞—П –Њ–±—А–∞—В–љ–∞—П —Б–≤—П–Ј—М –і–ї—П –њ–Њ–і–і–µ—А–ґ–∞–љ–Є—П —Д–Њ—Б—Д–∞—В–љ–Њ–≥–Њ¬†–≥–Њ–Љ–µ–Њ—Б—В–∞–Ј–∞ –≤–Њ–Ј–љ–Є–Ї–∞–µ—В –≤¬†—В–µ—З–µ–љ–Є–µ –љ–µ—Б–Ї–Њ–ї—М–Ї–Є—Е —З–∞—Б–Њ–≤ –њ–Њ—Б–ї–µ –њ—А–Є–µ–Љ–∞ –† —Б¬†–њ–Є—Й–µ–є [21].
–Ъ—А–Њ–Љ–µ —В–Њ–≥–Њ, –Ъ–§–Я–І –Љ–Њ–≥—Г—В —Д—Г–љ–Ї—Ж–Є–Њ–љ–Є—А–Њ–≤–∞—В—М –Ї–∞–Ї –њ–µ—А–µ–љ–Њ—Б—З–Є–Ї, –Ї–Њ—В–Њ—А—Л–є –і–Њ—Б—В–∞–≤–ї—П–µ—В –°–∞¬†–Є¬†–†,¬†–≤—Б–∞—Б—Л–≤–∞–µ–Љ—Л–µ –Є–Ј¬†–ґ–µ–ї—Г–і–Њ—З–љ–Њ-–Ї–Є—И–µ—З–љ–Њ–≥–Њ —В—А–∞–Ї—В–∞, –љ–µ–њ–Њ—Б—А–µ–і—Б—В–≤–µ–љ–љ–Њ –≤¬†–Ї–Њ—Б—В—М [18, 21].
–Ґ–µ–Љ –љ–µ¬†–Љ–µ–љ–µ–µ –љ–µ—П—Б–љ–Њ, –Ї–∞–Ї –Њ—Б—В–µ–Њ—Ж–Є—В—Л/–Њ—Б—В–µ–Њ–±–ї–∞—Б—В—Л –≤–Њ—Б–њ—А–Є–љ–Є–Љ–∞—О—В –њ–Њ—Б—В—Г–њ–ї–µ–љ–Є–µ –†,¬†–Є–љ–і—Г—Ж–Є—А—Г—П —Н–Ї—Б–њ—А–µ—Б—Б–Є—О –Є¬†—Б–µ–Ї—А–µ—Ж–Є—О FGF-23. –Т–Њ–Ј–Љ–Њ–ґ–љ—Л–є –Љ–µ—Е–∞–љ–Є–Ј–Љ –Ј–∞–Ї–ї—О—З–∞–µ—В—Б—П –≤¬†—В–Њ–Љ, —З—В–Њ –Њ—Б—В–µ–Њ–±–ї–∞—Б—В—Л/–Њ—Б—В–µ–Њ—Ж–Є—В—Л —Б–µ–Ї—А–µ—В–Є—А—Г—О—В FGF-23, –≤–Њ—Б–њ—А–Є–љ–Є–Љ–∞—П –њ–Њ—Б—В–њ—А–∞–љ–і–Є–∞–ї—М–љ–Њ–µ –њ–Њ–≤—Л—И–µ–љ–Є–µ —Г—А–Њ–≤–љ—П –† –≤¬†–Ї—А–Њ–≤–Є —З–µ—А–µ–Ј –њ—А–µ–і–њ–Њ–ї–∞–≥–∞–µ–Љ—Л–є ¬Ђ—Д–Њ—Б—Д–∞—В-—З—Г–≤—Б—В–≤–Є—В–µ–ї—М–љ—Л–є —А–µ—Ж–µ–њ—В–Њ—А¬ї. –Ю–і–љ–∞–Ї–Њ —Д–Њ—Б—Д–∞—В-—З—Г–≤—Б—В–≤–Є—В–µ–ї—М–љ—Л–є —А–µ—Ж–µ–њ—В–Њ—А –љ–µ¬†–±—Л–ї –Є–і–µ–љ—В–Є—Д–Є—Ж–Є—А–Њ–≤–∞–љ. –≠—В–∞¬†–≥–Є–њ–Њ—В–µ–Ј–∞ –∞–љ–∞–ї–Њ–≥–Є—З–љ–∞ —В–Њ–Љ—Г —Д–∞–Ї—В—Г, —З—В–Њ –Ї–ї–µ—В–Ї–Є –њ–∞—А–∞—Й–Є—В–Њ–≤–Є–і–љ–Њ–є –ґ–µ–ї–µ–Ј—Л —Б–µ–Ї—А–µ—В–Є—А—Г—О—В –Я–Ґ–У –њ—А–Є —Б–љ–Є–ґ–µ–љ–Є–Є –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є–Є –°–∞¬†–≤¬†–Ї—А–Њ–≤–Є –±–ї–∞–≥–Њ–і–∞—А—П CaSR, —Г–ї–∞–≤–ї–Є–≤–∞—П –Є–Ј–Љ–µ–љ–µ–љ–Є—П –µ–≥–Њ —Г—А–Њ–≤–љ—П –≤¬†–Ї—А–Њ–≤–Є –Є¬†—А–µ–≥—Г–ї–Є—А—Г—П —Б–µ–Ї—А–µ—Ж–Є—О –Я–Ґ–У.¬† –Ю–і–љ–∞–Ї–Њ –љ–µ–Ї–Њ—В–Њ—А—Л–µ –і–∞–љ–љ—Л–µ –Њ–њ—А–Њ–≤–µ—А–≥–∞—О—В —Н—В—Г¬†–≥–Є–њ–Њ—В–µ–Ј—Г. –Т–Њ-–њ–µ—А–≤—Л—Е, —Г—А–Њ–≤–љ–Є FGF-23 –≤¬†—Б—Л–≤–Њ—А–Њ—В–Ї–µ –Ї—А–Њ–≤–Є –Ї–Њ—А—А–µ–ї–Є—А–Њ–≤–∞–ї–Є –љ–µ¬†—В–Њ–ї—М–Ї–Њ —Б¬†—Г—А–Њ–≤–љ–µ–Љ –† –≤¬†—Б—Л–≤–Њ—А–Њ—В–Ї–µ, –љ–Њ¬†–Є —Б¬†—Г—А–Њ–≤–љ–µ–Љ –°–∞. –Т–Њ-–≤—В–Њ—А—Л—Е, —Г–≤–µ–ї–Є—З–µ–љ–Є–µ —Б—Л–≤–Њ—А–Њ—В–Њ—З–љ–Њ–≥–Њ –† –љ–µ¬†–њ—А–Є–≤–µ–ї–Њ –Ї¬†–њ–Њ–≤—Л—И–µ–љ–Є—О —Г—А–Њ–≤–љ—П —Б—Л–≤–Њ—А–Њ—В–Њ—З–љ–Њ–≥–Њ FGF-23, –Ї–Њ–≥–і–∞ —Г—А–Њ–≤–µ–љ—М –°–∞¬†–≤¬†—Б—Л–≤–Њ—А–Њ—В–Ї–µ –±—Л–ї –љ–Є–Ј–Ї–Є–Љ, –Є¬†–љ–∞–Њ–±–Њ—А–Њ—В.¬† –Р¬†–Є–Љ–µ–љ–љ–Њ, –њ—А–Є –љ–∞–ї–Є—З–Є–Є¬†–≥–Є–њ–Њ—Д–Њ—Б—Д–∞—В–µ–Љ–Є–Є —Г—А–Њ–≤–µ–љ—М FGF-23 –љ–µ¬†–њ–Њ–≤—Л—И–∞–ї—Б—П –≤¬†–Њ—В–≤–µ—В –љ–∞¬†–њ–Њ–≤—Л—И–µ–љ–Є–µ —Г—А–Њ–≤–љ—П –°–∞¬†–≤¬†—Б—Л–≤–Њ—А–Њ—В–Ї–µ –Ї—А–Њ–≤–Є. –≠—В–Є –љ–∞–±–ї—О–і–µ–љ–Є—П —Г–Ї–∞–Ј—Л–≤–∞—О—В –љ–∞¬†—В–Њ, —З—В–Њ –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є—П –°–∞¬†–Є¬†–† –≤¬†—Б—Л–≤–Њ—А–Њ—В–Ї–µ –Ї—А–Њ–≤–Є –і–Њ–ї–ґ–љ–∞ –±—Л—В—М –≤—Л—И–µ –Њ–њ—А–µ–і–µ–ї–µ–љ–љ–Њ–≥–Њ —Г—А–Њ–≤–љ—П, —З—В–Њ–±—Л –Є–љ–і—Г—Ж–Є—А–Њ–≤–∞—В—М —Б–µ–Ї—А–µ—Ж–Є—О FGF-23 [22, 23].
–Т¬†–Ї—Г–ї—М—В–Є–≤–Є—А—Г–µ–Љ—Л—Е –Њ—Б—В–µ–Њ–±–ї–∞—Б—В–љ—Л—Е –Ї–ї–µ—В–Ї–∞—Е —Г–≤–µ–ї–Є—З–µ–љ–Є–µ –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є–Є –°–∞¬†–Є–ї–Є –† –≤¬†—Б—А–µ–і–µ –Є–љ–і—Г—Ж–Є—А–Њ–≤–∞–ї–Њ —Н–Ї—Б–њ—А–µ—Б—Б–Є—О FGF-23, –Ї–Њ—В–Њ—А–∞—П –Ј–∞–≤–Є—Б–µ–ї–∞ –Њ—В¬†–Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є—П –Ъ–§–Я–І. –Ъ–Њ–≥–і–∞ –±–Є—Б—Д–Њ—Б—Д–Њ–љ–∞—В –±–ї–Њ–Ї–Є—А–Њ–≤–∞–ї –њ–µ—А–µ—Е–Њ–і –∞–Љ–Њ—А—Д–љ–Њ–є —Д–∞–Ј—Л –Ъ–§–Я–Ь –≤¬†–Ї—А–Є—Б—В–∞–ї–ї–Є—З–µ—Б–Ї—Г—О —Д–Њ—А–Љ—Г –Ъ–§–Я–І, —Н–Ї—Б–њ—А–µ—Б—Б–Є—П FGF-23 —Г–≤–µ–ї–Є—З–Є–≤–∞–ї–∞—Б—М. –≠—В–Њ –њ–Њ–Ј–≤–Њ–ї—П–µ—В –њ—А–µ–і–њ–Њ–ї–Њ–ґ–Є—В—М, —З—В–Њ –Љ–µ–ї–Ї–Є–µ –Ъ–§–Я–І, —Б–Њ–і–µ—А–ґ–∞—Й–Є–µ –∞–Љ–Њ—А—Д–љ—Л–µ –Ї–∞–ї—М—Ж–Є–є-—Д–Њ—Б—Д–∞—В–љ—Л–µ –Њ—Б–∞–і–Ї–Є, –і–µ–є—Б—В–≤—Г—О—В –Ї–∞–Ї –±–Њ–ї–µ–µ –Љ–Њ—Й–љ—Л–є –Є–љ–і—Г–Ї—В–Њ—А FGF-23, —З–µ–Љ –±–Њ–ї–µ–µ –Ї—А—Г–њ–љ—Л–µ –Ъ–§–Я–І, —Б–Њ–і–µ—А–ґ–∞—Й–Є–µ –Ї—А–Є—Б—В–∞–ї–ї–Є—З–µ—Б–Ї–Є–µ –Ї–∞–ї—М—Ж–Є–є-—Д–Њ—Б—Д–∞—В–љ—Л–µ –Њ—Б–∞–і–Ї–Є. –£¬†–Љ—Л—И–µ–є –±–Њ–ї—О—Б–љ–Њ–µ –≤–≤–µ–і–µ–љ–Є–µ –† —З–µ—А–µ–Ј –њ–µ—А–Њ—А–∞–ї—М–љ—Л–є –Ј–Њ–љ–і –≤—А–µ–Љ–µ–љ–љ–Њ —Г–≤–µ–ї–Є—З–Є–≤–∞–ї–Њ —Г—А–Њ–≤–љ–Є —Ж–Є—А–Ї—Г–ї–Є—А—Г—О—Й–Є—Е –Ъ–§–Я–І —Б¬†–њ–Њ—Б–ї–µ–і—Г—О—Й–Є–Љ —Г–Љ–µ—А–µ–љ–љ—Л–Љ –њ–Њ–≤—Л—И–µ–љ–Є–µ–Љ —Н–Ї—Б–њ—А–µ—Б—Б–Є–Є –§–†–§ –Є¬†—Г—А–Њ–≤–љ–µ–є FGF-23 –≤¬†—Б—Л–≤–Њ—А–Њ—В–Ї–µ –Ї—А–Њ–≤–Є. –Ю–і–љ–∞–Ї–Њ –њ–Њ—Б—В–Њ—П–љ–љ–∞—П –љ–∞–≥—А—Г–Ј–Ї–∞ –† —Б¬†–њ–Є—Й–µ–є –≤—Л–Ј—Л–≤–∞–ї–∞ –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ–µ –Є¬†—Б—В–Њ–є–Ї–Њ–µ —Г–≤–µ–ї–Є—З–µ–љ–Є–µ —Ж–Є—А–Ї—Г–ї–Є—А—Г—О—Й–Є—Е –Ъ–§–Я–І –Є¬†—Г—А–Њ–≤–љ–µ–є FGF-23. –Т–Є–Ј—Г–∞–ї–Є–Ј–∞—Ж–Є—П in¬†vivo –њ–Њ–і—В–≤–µ—А–і–Є–ї–∞, —З—В–Њ –Ъ–§–Я–І, –≤–≤–µ–і–µ–љ–љ—Л–µ –≤–љ—Г—В—А–Є–≤–µ–љ–љ–Њ, –њ—А–Њ–љ–Є–Ї–∞–ї–Є –≤¬†–Ї–Њ—Б—В–љ—Л–є –Љ–Њ–Ј–≥ –Є¬†–Њ—Б–∞–ґ–і–∞–ї–Є—Б—М –љ–∞¬†–≤–љ—Г—В—А–µ–љ–љ–µ–є –њ–Њ–≤–µ—А—Е–љ–Њ—Б—В–Є –Ї–Њ—Б—В–Є, —З—В–Њ —Г–Ї–∞–Ј—Л–≤–∞–µ—В –љ–∞¬†—В–Њ, —З—В–Њ —Н—В–Є —З–∞—Б—В–Є—Ж—Л –Є–Љ–µ—О—В –њ—А—П–Љ–Њ–є –і–Њ—Б—В—Г–њ –Ї¬†–Њ—Б—В–µ–Њ–±–ї–∞—Б—В–∞–Љ. K.-I. Akiyama¬† –Є¬†—Б–Њ–∞–≤—В. –њ—А–µ–і–њ–Њ–ї–Њ–ґ–Є–ї–Є [21], —З—В–Њ –Њ—Б—В–µ–Њ–±–ї–∞—Б—В—Л –Є–љ–і—Г—Ж–Є—А—Г—О—В —Н–Ї—Б–њ—А–µ—Б—Б–Є—О –Є¬†—Б–µ–Ї—А–µ—Ж–Є—О FGF-23, –Ї–Њ–≥–і–∞ –Њ–љ–Є –Њ—Й—Г—Й–∞—О—В —Г–≤–µ–ї–Є—З–µ–љ–Є–µ –≤–љ–µ–Ї–ї–µ—В–Њ—З–љ—Л—Е –Ъ–§–Я–І –њ–Њ—Б–ї–µ –њ—А–Є–µ–Љ–∞ –†.¬†–Э–∞¬†—А–∞–љ–љ–Є—Е —Б—В–∞–і–Є—П—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П –њ–Њ—З–µ–Ї –њ–Є—Й–µ–≤–∞—П –љ–∞–≥—А—Г–Ј–Ї–∞ –†,¬†—Б¬†–Њ–і–љ–Њ–є —Б—В–Њ—А–Њ–љ—Л, –Є¬†—Г–Љ–µ–љ—М—И–µ–љ–Є–µ —З–Є—Б–ї–∞ —Д—Г–љ–Ї—Ж–Є–Њ–љ–Є—А—Г—О—Й–Є—Е –љ–µ—Д—А–Њ–љ–Њ–≤ —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–•–С–Я, —Б¬†–і—А—Г–≥–Њ–є, —Б–Њ–њ—А–Њ–≤–Њ–ґ–і–∞—О—В—Б—П —А–Њ—Б—В–Њ–Љ —Г—А–Њ–≤–љ—П FGF-23 –≤¬†—Б—Л–≤–Њ—А–Њ—В–Ї–µ –Ї—А–Њ–≤–Є. FGF-23 —Б–љ–Є–ґ–∞–µ—В –≤—Б–∞—Б—Л–≤–∞–љ–Є–µ –†,¬†–њ–Њ—Б—В—Г–њ–∞—О—Й–µ–≥–Њ —Б¬†–њ–Є—Й–µ–є, –Є–љ–≥–Є–±–Є—А—Г—П —А–µ–∞–±—Б–Њ—А–±—Ж–Є—О –† –≤¬†–њ—А–Њ–Ї—Б–Є–Љ–∞–ї—М–љ—Л—Е –Ї–∞–љ–∞–ї—М—Ж–∞—Е –њ–Њ—З–µ–Ї, —З—В–Њ –њ—А–Є–≤–Њ–і–Є—В –Ї¬†—Г–≤–µ–ї–Є—З–µ–љ–Є—О —Н–Ї—Б–Ї—А–µ—Ж–Є–Є –† —Б¬†–Љ–Њ—З–Њ–є –і–ї—П –њ–Њ–і–і–µ—А–ґ–∞–љ–Є—П —Д–Њ—Б—Д–∞—В–љ–Њ–≥–Њ –±–∞–ї–∞–љ—Б–∞ –њ—А–µ–ґ–і–µ, —З–µ–Љ –њ–Њ—П–≤—П—В—Б—П –Є–Ј–Љ–µ–љ–µ–љ–Є—П –≤¬†—Б—Л–≤–Њ—А–Њ—В–Њ—З–љ–Њ–Љ —Г—А–Њ–≤–љ–µ –†,¬†–Є¬†—А–∞—Б—Б–Љ–∞—В—А–Є–≤–∞–µ—В—Б—П –Ї–∞–Ї —Д–Є–Ј–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–∞—П —А–µ–∞–Ї—Ж–Є—П.
–≠—В–∞ —Б–µ—А–Є—П —Б–Њ–±—Л—В–Є–є —Г–Ї–∞–Ј—Л–≤–∞–µ—В –љ–∞¬†—В–Њ, —З—В–Њ –Ї–∞–Ї –°–∞, —В–∞–Ї –Є¬†–† –љ–µ–Њ–±—Е–Њ–і–Є–Љ—Л –і–ї—П –Є–љ–і—Г–Ї—Ж–Є–Є —Б–µ–Ї—А–µ—Ж–Є–Є FGF-23 –Є, –≤–Њ–Ј–Љ–Њ–ґ–љ–Њ, –љ–µ¬†—Д–Њ—Б—Д–∞—В, –∞¬†–Є–Љ–µ–љ–љ–Њ –Ъ–§–Я–І –Љ–Њ–≥—Г—В –Є–љ–і—Г—Ж–Є—А–Њ–≤–∞—В—М —Б–µ–Ї—А–µ—Ж–Є—О –Є¬†–≤—Л—А–∞–±–Њ—В–Ї—Г FGF-23, –Ї–Њ—В–Њ—А—Л–µ —П–≤–ї—П—О—В—Б—П –љ–µ¬†–±–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–Љ –њ—А–Њ—Ж–µ—Б—Б–Њ–Љ, –∞¬†—Д–Є–Ј–Є–Ї–Њ-—Е–Є–Љ–Є—З–µ—Б–Ї–Є–Љ —П–≤–ї–µ–љ–Є–µ–Љ, —А–∞–Ј–≤–Є–≤–∞—О—Й–Є–Љ—Б—П —Б–њ–Њ–љ—В–∞–љ–љ–Њ —Б¬†—В–µ—З–µ–љ–Є–µ–Љ –≤—А–µ–Љ–µ–љ–Є. –§–Є–Ј–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–є —Б–Љ—Л—Б–ї —Д–Њ—А–Љ–Є—А–Њ–≤–∞–љ–Є—П –Ъ–§–Я–І –Ј–∞–Ї–ї—О—З–∞–µ—В—Б—П –≤¬†–∞–≥—А–µ–≥–∞—Ж–Є–Є –Є–Ј–±—Л—В–Њ—З–љ—Л—Е –Є–Њ–љ–Њ–≤ –°–∞¬†–Є¬†–†,¬†–Є—Е —Б–≤–Њ–µ–≤—А–µ–Љ–µ–љ–љ–Њ–Љ –≤—Л–≤–µ–і–µ–љ–Є–Є –Љ–∞–Ї—А–Њ—Д–∞–≥–∞–Љ–Є, –Њ—Б–Њ–±–µ–љ–љ–Њ –Ї–ї–µ—В–Ї–∞–Љ–Є –Ъ—Г–њ—Д–µ—А–∞ –≤¬†–њ–µ—З–µ–љ–Є –Є –њ—А–µ–і–Њ—В–≤—А–∞—Й–µ–љ–Є–Є —Н–Ї—В–Њ–њ–Є—З–µ—Б–Ї–Њ–є –Ї–∞–ї—М—Ж–Є—Д–Є–Ї–∞—Ж–Є–Є. –Ґ–∞–Ї–Є–Љ –Њ–±—А–∞–Ј–Њ–Љ, –Ъ–§–Я–І —П–≤–ї—П—О—В—Б—П —Д–Є–Ј–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–Љ –Љ–Њ–і—Г–ї—П—В–Њ—А–Њ–Љ —Н–љ–і–Њ–Ї—А–Є–љ–љ–Њ–є –Њ—Б–Є ¬ЂFGF-23¬†вАУ K–ї–Њ—В–Њ¬ї [24вАУ26].
–Ъ–ї–Є–љ–Є—З–µ—Б–Ї–∞—П –Ј–љ–∞—З–Є–Љ–Њ—Б—В—М —Д–Њ—А–Љ–Є—А–Њ–≤–∞–љ–Є—П –Ї–∞–ї—М—Ж–Є–є-—Д–Њ—Б—Д–∞—В–љ—Л—Е –њ—А–Њ—В–µ–Є–љ–Њ–≤—Л—Е —З–∞—Б—В–Є—Ж
–Т¬†–њ–Њ—Б–ї–µ–і–љ–µ–µ –і–µ—Б—П—В–Є–ї–µ—В–Є–µ –∞–Ї—В–Є–≤–љ–Њ –њ—А–Њ–≤–Њ–і—П—В—Б—П —А–∞–±–Њ—В—Л –њ–Њ¬†–Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—О —Д–Є–Ј–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–є –Є¬†–њ–∞—В–Њ—Д–Є–Ј–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–є –Ј–љ–∞—З–Є–Љ–Њ—Б—В–Є –Ъ–§–Я–І, –њ–Њ—Б–Ї–Њ–ї—М–Ї—Г, —Б¬†–Њ–і–љ–Њ–є —Б—В–Њ—А–Њ–љ—Л, –і–∞–љ–љ—Л–µ —З–∞—Б—В–Є—Ж—Л —П–≤–ї—П—О—В—Б—П –≤–∞–ґ–љ—Л–Љ –Ј–≤–µ–љ–Њ–Љ –Љ–Є–љ–µ—А–∞–ї—М–љ–Њ–≥–Њ¬†–≥–Њ–Љ–µ–Њ—Б—В–∞–Ј–∞, –∞–Ї–Ї—Г–Љ—Г–ї–Є—А—Г—П –Є–Ј–±—Л—В–Њ—З–љ—Л–µ –Є–Њ–љ—Л –Ї–∞–ї—М—Ж–Є—П –Є¬†—Д–Њ—Б—Д–Њ—А–∞ –Є¬†–≤—Л–≤–Њ–і—П –Є—Е –Є–Ј¬†–Њ—А–≥–∞–љ–Є–Ј–Љ–∞, –∞¬†—Б –і—А—Г–≥–Њ–є¬†вАУ –њ–Њ–≤—Л—И–µ–љ–љ—Л–µ —Г—А–Њ–≤–љ–Є —Ж–Є—А–Ї—Г–ї–Є—А—Г—О—Й–Є—Е –Ъ–§–Я–І –∞—Б—Б–Њ—Ж–Є–Є—А–Њ–≤–∞–љ—Л —Б¬†–≤—Л—Б–Њ–Ї–Є–Љ —А–Є—Б–Ї–Њ–Љ –љ–µ–±–ї–∞–≥–Њ–њ—А–Є—П—В–љ—Л—Е –Є—Б—Е–Њ–і–Њ–≤, –≤–Ї–ї—О—З–∞—П¬† –Њ—Б–љ–Њ–≤–љ—Л–µ —Б–µ—А–і–µ—З–љ–Њ-—Б–Њ—Б—Г–і–Є—Б—В—Л–µ —Б–Њ–±—Л—В–Є—П –Є¬†—Б–Љ–µ—А—В–љ–Њ—Б—В—М —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–і–Њ–і–Є–∞–ї–Є–Ј–љ–Њ–є –Є¬†—В–µ—А–Љ–Є–љ–∞–ї—М–љ–Њ–є —Б—В–∞–і–Є–µ–є –њ–Њ—З–µ—З–љ–Њ–є –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В–Є (–Ґ–Я–Э), —А–µ—Ж–Є–њ–Є–µ–љ—В–Њ–≤ –њ–Њ—З–µ—З–љ–Њ–≥–Њ —В—А–∞–љ—Б–њ–ї–∞–љ—В–∞—В–∞ [27вАУ30].
–Я–µ—А–µ–≥—А—Г–Ј–Ї–∞ –† –Њ–Ї–∞–Ј—Л–≤–∞–µ—В —В–Њ–Ї—Б–Є—З–µ—Б–Ї–Њ–µ –≤–Њ–Ј–і–µ–є—Б—В–≤–Є–µ —А–∞–Ј–ї–Є—З–љ—Л–Љ–Є –њ—Г—В—П–Љ–Є, –≤–Ї–ї—О—З–∞—П –њ—А—П–Љ–Њ–µ –≤–Њ–Ј–і–µ–є—Б—В–≤–Є–µ –Є¬†–Ї–Њ—Б–≤–µ–љ–љ—Л–µ —Н—Д—Д–µ–Ї—В—Л, —Б–≤—П–Ј–∞–љ–љ—Л–µ —Б¬†–Ї–Њ–Љ–њ–µ–љ—Б–∞—В–Њ—А–љ—Л–Љ–Є —А–µ–∞–Ї—Ж–Є—П–Љ–Є, —В–∞–Ї–Є–Љ–Є –Ї–∞–Ї —Д–Њ—А–Љ–Є—А–Њ–≤–∞–љ–Є–µ –Ъ–§–Я–І, –њ–Њ–≤—Л—И–µ–љ–Є–µ —Г—А–Њ–≤–љ—П FGF-23 –Є¬†–Я–Ґ–У. –Ф–∞–љ–љ—Л–µ —Н–њ–Є–і–µ–Љ–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є —Г–Ї–∞–Ј—Л–≤–∞—О—В –љ–∞¬†—В–Њ, —З—В–Њ –њ–µ—А–µ–≥—А—Г–Ј–Ї–∞ –† —Б–≤—П–Ј–∞–љ–∞ —Б¬†–љ–µ–±–ї–∞–≥–Њ–њ—А–Є—П—В–љ—Л–Љ–Є –Є—Б—Е–Њ–і–∞–Љ–Є, –і–∞–ґ–µ –Ї–Њ–≥–і–∞ —Г—А–Њ–≤–љ–Є –† –љ–∞—Е–Њ–і—П—В—Б—П –≤¬†–њ—А–µ–і–µ–ї–∞—Е –љ–Њ—А–Љ—Л, —Г–≤–µ–ї–Є—З–Є–≤–∞—П —А–Є—Б–Ї¬†—Б–Љ–µ—А—В–Є, —Б–µ—А–і–µ—З–љ–Њ-—Б–Њ—Б—Г–і–Є—Б—В—Л—Е –Њ—Б–ї–Њ–ґ–љ–µ–љ–Є–є –Є¬†–Ъ–° –Ї–∞–Ї —Г¬†–Ј–і–Њ—А–Њ–≤—Л—Е –≤–Ј—А–Њ—Б–ї—Л—Е, —В–∞–Ї –Є¬†—Г –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–•–С–Я. –£—А–Њ–≤–љ–Є —Д–Њ—Б—Д–∞—В–Њ–≤ 3,9вАУ4,7 –Љ–≥/–і–ї –±—Л–ї–Є —Б–≤—П–Ј–∞–љ—Л —Б¬†–ї–Є–љ–µ–є–љ—Л–Љ —Г–≤–µ–ї–Є—З–µ–љ–Є–µ–Љ —З–Є—Б–ї–∞ —Б–µ—А–і–µ—З–љ–Њ-—Б–Њ—Б—Г–і–Є—Б—В—Л—Е —Б–Њ–±—Л—В–Є–є —Г¬†–ї—О–і–µ–є —Б¬†–љ–Њ—А–Љ–∞–ї—М–љ–Њ–є —Д—Г–љ–Ї—Ж–Є–µ–є –њ–Њ—З–µ–Ї –Є¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–•–С–Я –њ–µ—А–≤–Њ–є –Є¬†–≤—В–Њ—А–Њ–є —Б—В–∞–і–Є–Є [31, 32].
–Ъ–ї–Є–љ–Є—З–µ—Б–Ї–∞—П –Ј–љ–∞—З–Є–Љ–Њ—Б—В—М –Ъ–§–Я–І –Њ–±—Г—Б–ї–Њ–≤–ї–µ–љ–∞ –љ–∞–±–ї—О–і–µ–љ–Є–µ–Љ –њ–Њ–≤—Л—И–µ–љ–Є—П —Ж–Є—А–Ї—Г–ї–Є—А—Г—О—Й–Є—Е —Г—А–Њ–≤–љ–µ–є —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–•–С–Я, –Ї–Њ—А—А–µ–ї—П—Ж–Є–µ–є —Б¬†–њ–∞—А–∞–Љ–µ—В—А–∞–Љ–Є –≤–Њ—Б–њ–∞–ї–µ–љ–Є—П (–≤—Л—Б–Њ–Ї–Њ—З—Г–≤—Б—В–≤–Є—В–µ–ї—М–љ—Л–є —Г—А–Њ–≤–µ–љ—М –°-—А–µ–∞–Ї—В–Є–≤–љ–Њ–≥–Њ –±–µ–ї–Ї–∞), –ґ–µ—Б—В–Ї–Њ—Б—В—М—О —Б–Њ—Б—Г–і–Њ–≤ (—Б–Ї–Њ—А–Њ—Б—В—М –њ—Г–ї—М—Б–Њ–≤–Њ–є –≤–Њ–ї–љ—Л –≤¬†–∞–Њ—А—В–µ), –Ъ–° (–Њ—Ж–µ–љ–Ї–∞ –Ї–∞–ї—М—Ж–Є—Д–Є–Ї–∞—Ж–Є–Є –∞—А—В–µ—А–Є–є) –Є¬†—В–Њ–ї—Й–Є–љ–Њ–є –±–ї—П—И–µ–Ї –≤¬†–Ї–Њ—А–Њ–љ–∞—А–љ—Л—Е –∞—А—В–µ—А–Є—П—Е, —А–∞–Ј–≤–Є—В–Є–µ–Љ –Є¬†–њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є–µ–Љ —Г—А–µ–Љ–Є—З–µ—Б–Ї–Њ–є –∞—А—В–µ—А–Є–Њ–ї–Њ–њ–∞—В–Є–Є, –∞—В–µ—А–Њ—Б–Ї–ї–µ—А–Њ–Ј–∞, —В—П–ґ–µ—Б—В—М—О –Є¬†–њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є–µ–Љ –Ъ–°, –∞¬†—В–∞–Ї–ґ–µ —А–∞–Ј–≤–Є—В–Є–µ–Љ –Њ—Б—В—А—Л—Е —Б–µ—А–і–µ—З–љ–Њ-—Б–Њ—Б—Г–і–Є—Б—В—Л—Е —Б–Њ–±—Л—В–Є–є –Є¬†—Б–Љ–µ—А—В–љ–Њ—Б—В—М—О [30, 33вАУ37].
–Ъ–∞–Ї —В–Њ–ї—М–Ї–Њ –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є—П –† –≤¬†–ґ–Є–і–Ї–Њ—Б—В–Є –Ї–∞–љ–∞–ї—М—Ж–µ–≤ –њ—А–µ–≤—Л—И–∞–µ—В –њ–Њ—А–Њ–≥–Њ–≤–Њ–µ –Ј–љ–∞—З–µ–љ–Є–µ, –њ–Њ–≤—Л—И–∞—О—В—Б—П —Г—А–Њ–≤–µ–љ—М FGF-23 –Є¬†–Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є—П –† –≤¬†–ґ–Є–і–Ї–Њ—Б—В–Є –њ—А–Њ–Ї—Б–Є–Љ–∞–ї—М–љ—Л—Е –Ї–∞–љ–∞–ї—М—Ж–µ–≤, —Г–≤–µ–ї–Є—З–Є–≤–∞–µ—В—Б—П —А–Є—Б–Ї¬†–Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є—П –Ъ–§–Я–І –≤¬†–ґ–Є–і–Ї–Њ—Б—В–Є –Ї–∞–љ–∞–ї—М—Ж–µ–≤, –Ї–Њ—В–Њ—А—Л–µ –≤—Л–Ј—Л–≤–∞—О—В –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є–µ –Ї–∞–љ–∞–ї—М—Ж–µ–≤ —Б¬†–њ–Њ–Љ–Њ—Й—М—О —Б–≤—П–Ј—Л–≤–∞–љ–Є—П —Б¬†—Н–Ї—Б–њ—А–µ—Б—Б–Є—А—Г–µ–Љ—Л–Љ –љ–∞¬†–њ–Њ–≤–µ—А—Е–љ–Њ—Б—В–Є –Ї–ї–µ—В–Њ–Ї –Ї–∞–љ–∞–ї—М—Ж–µ–≤ Toll-–њ–Њ–і–Њ–±–љ—Л–Љ —А–µ—Ж–µ–њ—В–Њ—А–Њ–Љ 4 (TLR4). –Ъ. Shiizaki –Є¬†—Б–Њ–∞–≤—В. [14] –њ—А–Є—И–ї–Є –Ї¬†–≤—Л–≤–Њ–і—Г, —З—В–Њ TLR4-–Ј–∞–≤–Є—Б–Є–Љ–Њ–µ –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є–µ –њ–Њ—З–µ—З–љ—Л—Е –Ї–∞–љ–∞–ї—М—Ж–µ–≤ in¬†vivo –±—Л–ї–Њ –≤—Л–Ј–≤–∞–љ–Њ –љ–µ¬†–∞–Ї—В–Є–≤–∞—Ж–Є–µ–є –њ–µ—А–µ–і–∞—З–Є —Б–Є–≥–љ–∞–ї–Њ–≤ TLR4 –Ъ–§–Я–І, –∞¬†—Б–Ї–Њ—А–µ–µ –≤–Ј–∞–Є–Љ–Њ–і–µ–є—Б—В–≤–Є–µ–Љ —Б¬†–Ъ–§–Я–І –љ–∞¬†–њ–Њ–≤–µ—А—Е–љ–Њ—Б—В–Є –Ї–ї–µ—В–Њ–Ї –њ—А–Њ–Ї—Б–Є–Љ–∞–ї—М–љ—Л—Е –Ї–∞–љ–∞–ї—М—Ж–µ–≤ –≤¬†—В–µ—З–µ–љ–Є–µ —И–µ—Б—В–Є —З–∞—Б–Њ–≤ –Є–ї–Є –і–Њ–ї—М—И–µ –њ—А–Њ—В–Є–≤ –њ–Њ—В–Њ–Ї–∞ –Ї–∞–љ–∞–ї—М—Ж–µ–≤–Њ–є –ґ–Є–і–Ї–Њ—Б—В–Є, —З—В–Њ —Б–њ–Њ—Б–Њ–±—Б—В–≤–Њ–≤–∞–ї–Њ —Н–љ–і–Њ—Ж–Є—В–Њ–Ј—Г, –≤—Л–Ј—Л–≤–∞—П –љ–∞—А—Г—И–µ–љ–Є–µ —Н–љ–і–Њ—Б–Њ–Љ–∞–ї—М–љ–Њ–≥–Њ —В—А–∞–љ—Б–њ–Њ—А—В–∞ –Є¬†–њ–Њ–≤—А–µ–ґ–і–µ–љ–Є–µ –Ї–ї–µ—В–Њ–Ї –Ї–∞–љ–∞–ї—М—Ж–µ–≤. –°—В–Њ–є–Ї–Њ–µ –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є–µ –Ї–∞–љ–∞–ї—М—Ж–µ–≤ –≤—Л–Ј—Л–≤–∞–µ—В –Є–љ—В–µ—А—Б—В–Є—Ж–Є–∞–ї—М–љ—Л–є —Д–Є–±—А–Њ–Ј, —Г–Љ–µ–љ—М—И–∞–µ—В –Ї–Њ–ї–Є—З–µ—Б—В–≤–Њ –љ–µ—Д—А–Њ–љ–Њ–≤ –Є¬†–і–Њ–њ–Њ–ї–љ–Є—В–µ–ї—М–љ–Њ –њ–Њ–≤—Л—И–∞–µ—В —Г—А–Њ–≤–µ–љ—М FGF-23, –≤—Л–Ј—Л–≤–∞—П —Б–њ–Є—А–∞–ї—М —Г—Е—Г–і—И–µ–љ–Є—П, –њ—А–Є–≤–Њ–і—П—Й—Г—О –Ї¬†–њ—А–Њ–≥—А–µ—Б—Б–Є—А—Г—О—Й–µ–є –њ–Њ—В–µ—А–µ –љ–µ—Д—А–Њ–љ–Њ–≤. –£¬†–ї—О–і–µ–є –њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є–µ –•–С–Я –љ–∞—З–Є–љ–∞–ї–Њ—Б—М, –Ї–Њ–≥–і–∞ —Г—А–Њ–≤–µ–љ—М FGF-23 –≤¬†—Б—Л–≤–Њ—А–Њ—В–Ї–µ –Ї—А–Њ–≤–Є –њ—А–µ–≤—Л—И–∞–ї 53 –њ–≥/–Љ–ї [14].
–Э–∞–Ї–∞–њ–ї–Є–≤–∞—О—В—Б—П –і–∞–љ–љ—Л–µ, —Б–≤–Є–і–µ—В–µ–ї—М—Б—В–≤—Г—О—Й–Є–µ –Њ¬†—В–Њ–Љ, —З—В–Њ –Ъ–§–Я–І, —Ж–Є—А–Ї—Г–ї–Є—А—Г—О—Й–Є–µ –љ–∞–љ–Њ—А–∞–Ј–Љ–µ—А–љ—Л–µ –∞–≥—А–µ–≥–∞—В—Л, –Њ–њ–Њ—Б—А–µ–і—Г—О—В –љ–µ–Ї–Њ—В–Њ—А—Л–µ —В–Њ–Ї—Б–Є—З–µ—Б–Ї–Є–µ —Н—Д—Д–µ–Ї—В—Л –љ–∞¬†–Љ–љ–Њ–≥–Њ—З–Є—Б–ї–µ–љ–љ—Л–µ —В–Є–њ—Л –Ї–ї–µ—В–Њ–Ї —Б–Њ—Б—Г–і–Њ–≤ –Є¬†–Ї–ї–∞–њ–∞–љ–Њ–≤, –≤–Ї–ї—О—З–∞—П —Н–љ–і–Њ—В–µ–ї–Є–∞–ї—М–љ—Л–µ –Ї–ї–µ—В–Ї–Є —Б–Њ—Б—Г–і–Њ–≤,¬†–≥–ї–∞–і–Ї–Њ–Љ—Л—И–µ—З–љ—Л–µ –Ї–ї–µ—В–Ї–Є —Б–Њ—Б—Г–і–Њ–≤ (–У–Ь–Ъ–°), –∞–і–≤–µ–љ—В–Є—Ж–Є–∞–ї—М–љ—Л–µ —Д–Є–±—А–Њ–±–ї–∞—Б—В—Л, –Є–љ—В–µ—А—Б—В–Є—Ж–Є–∞–ї—М–љ—Л–µ –Є¬†—Н–љ–і–Њ—В–µ–ї–Є–∞–ї—М–љ—Л–µ –Ї–ї–µ—В–Ї–Є –Ї–ї–∞–њ–∞–љ–Њ–≤ —Б–µ—А–і—Ж–∞, –Є–љ–і—Г—Ж–Є—А—Г—О—В —Н–Ї—Б–њ—А–µ—Б—Б–Є—О –Є¬†—Б–µ–Ї—А–µ—Ж–Є—О –њ—А–Њ–≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ—Л—Е —Ж–Є—В–Њ–Ї–Є–љ–Њ–≤, –≤–Ї–ї—О—З–∞—П –Є–љ—В–µ—А–ї–µ–є–Ї–Є–љ-1ќ≤ (–Ш–Ы-1ќ≤), –Ш–Ы-6, –Ш–Ы-8 –Є¬†—Д–∞–Ї—В–Њ—А –љ–µ–Ї—А–Њ–Ј–∞ –Њ–њ—Г—Е–Њ–ї–Є –∞–ї—М—Д–∞ (–§–Э–Ю-ќ±) [38, 39].
–†–µ–Ј—Г–ї—М—В–∞—В—Л –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П, –њ—А–Њ–≤–µ–і–µ–љ–љ–Њ–≥–Њ –Ъ. Shiizaki –Є¬†—Б–Њ–∞–≤—В. [14], –њ–Њ–Ї–∞–Ј–∞–ї–Є, —З—В–Њ –Ъ–§–Я–І –Њ—В–≤–µ—В—Б—В–≤–µ–љ–љ—Л –Ј–∞¬†–≥–Є–±–µ–ї—М –Ї–ї–µ—В–Њ–Ї, –≤—Л–Ј–≤–∞–љ–љ—Г—О –≤—Л—Б–Њ–Ї–Є–Љ —Б–Њ–і–µ—А–ґ–∞–љ–Є–µ–Љ –≤–љ–µ–Ї–ї–µ—В–Њ—З–љ–Њ–≥–Њ –†,¬†–Є¬†–њ–Њ–і—В–≤–µ—А–і–Є–ї–Є, —З—В–Њ –і–Њ–±–∞–≤–ї–µ–љ–Є–µ –∞–ї–µ–љ–і—А–Њ–љ–∞—В–∞, –Ї–Њ—В–Њ—А—Л–є –Є–љ–≥–Є–±–Є—А—Г–µ—В –њ–µ—А–µ—Е–Њ–і —Д–Њ—Б—Д–∞—В–∞ –Ї–∞–ї—М—Ж–Є—П –Є–Ј¬†–∞–Љ–Њ—А—Д–љ–Њ–є —Д–∞–Ј—Л –≤¬†–Ї—А–Є—Б—В–∞–ї–ї–Є—З–µ—Б–Ї—Г—О, –і–Њ–Ј–Њ–Ј–∞–≤–Є—Б–Є–Љ—Л–Љ –Њ–±—А–∞–Ј–Њ–Љ –њ–Њ–і–∞–≤–ї—П–µ—В –Є–љ–і—Г—Ж–Є—А—Г–µ–Љ—Г—О –†¬†–≥–Є–±–µ–ї—М –Ї–ї–µ—В–Њ–Ї –Є, –љ–∞–Њ–±–Њ—А–Њ—В, –і–Њ–±–∞–≤–ї–µ–љ–Є–µ —Б–Є–љ—В–µ–Ј–Є—А–Њ–≤–∞–љ–љ—Л—Е –Ъ–§–Я–І –≤¬†–Њ–±—Л—З–љ—Г—О —Б—А–µ–і—Г —Б–љ–Є–ґ–∞–ї–Њ –ґ–Є–Ј–љ–µ—Б–њ–Њ—Б–Њ–±–љ–Њ—Б—В—М –Ї–ї–µ—В–Њ–Ї –і–Њ–Ј–Њ–Ј–∞–≤–Є—Б–Є–Љ—Л–Љ –Њ–±—А–∞–Ј–Њ–Љ. –≠—В–Є –љ–∞–±–ї—О–і–µ–љ–Є—П —Г–Ї–∞–Ј—Л–≤–∞—О—В –љ–∞¬†—В–Њ, —З—В–Њ –Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є–µ –Ї—А–Є—Б—В–∞–ї–ї–Њ–≤ —Д–Њ—Б—Д–∞—В–∞ –Ї–∞–ї—М—Ж–Є—П –љ–µ–Њ–±—Е–Њ–і–Є–Љ–Њ –Є¬†–і–Њ—Б—В–∞—В–Њ—З–љ–Њ –і–ї—П —В–Њ–≥–Њ, —З—В–Њ–±—Л –≤—Л—Б–Њ–Ї–Њ–µ —Б–Њ–і–µ—А–ґ–∞–љ–Є–µ –≤–љ–µ–Ї–ї–µ—В–Њ—З–љ–Њ–≥–Њ –† –њ–Њ–≤—А–µ–ґ–і–∞–ї–Њ –Ї–ї–µ—В–Ї–Є –њ—А–Њ–Ї—Б–Є–Љ–∞–ї—М–љ—Л—Е –Ї–∞–љ–∞–ї—М—Ж–µ–≤. –•–Њ—В—П –Њ–±–µ —Д–Њ—А–Љ—Л –Ъ–§–Я–І –Њ–±–ї–∞–і–∞—О—В –њ—А–Њ–≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ—Л–Љ –і–µ–є—Б—В–≤–Є–µ–Љ –Є¬†–Є–љ–і—Г—Ж–Є—А—Г—О—В¬†–≥–Є–њ–µ—А–њ–ї–∞–Ј–Є—О –Є–љ—В–Є–Љ—Л –У–Ь–Ъ–°, –Ї–Њ—В–Њ—А—Л–µ –±—Л–ї–Є –±–Њ–ї–µ–µ –≤—Л—А–∞–ґ–µ–љ—Л —Г¬†–Ъ–§–Я–І-II. –≠—В–Њ –Љ–Њ–ґ–љ–Њ –Њ–±—К—П—Б–љ–Є—В—М —Б–Њ–і–µ—А–ґ–∞–љ–Є–µ–Љ –≤¬†–љ–Є—Е¬†–≥–Є–і—А–Њ–Ї—Б–Є–∞–њ–∞—В–Є—В–∞ –≤¬†–Ї—А–Є—Б—В–∞–ї–ї–Є—З–µ—Б–Ї–Њ–є —Д–Њ—А–Љ–µ –Є¬†—А–∞–Ј–ї–Є—З–љ—Л–Љ —Б—А–Њ–і—Б—В–≤–Њ–Љ –Ї¬†—Б–≤—П–Ј—Л–≤–∞–љ–Є—О —Б¬†—А–µ—Ж–µ–њ—В–Њ—А–∞–Љ–Є, —З—В–Њ –Ј–∞–њ—Г—Б–Ї–∞–µ—В —А–∞–Ј–ї–Є—З–љ—Л–µ —Б–Є–≥–љ–∞–ї—М–љ—Л–µ –Ї–∞—Б–Ї–∞–і—Л [39вАУ43].
–Ъ–∞–ї—М—Ж–Є–є-—Д–Њ—Б—Д–∞—В–љ—Л–µ –њ—А–Њ—В–µ–Є–љ–Њ–≤—Л–µ —З–∞—Б—В–Є—Ж—Л¬†вАУ –Љ–µ–і–Є–∞—В–Њ—А—Л –Ї–∞–ї—М—Ж–Є—Д–Є–Ї–∞—Ж–Є–Є —Б–Њ—Б—Г–і–Њ–≤
–Ъ–° –±—Л–ї–∞ –њ—А–Є–Ј–љ–∞–љ–∞ –Њ–і–љ–Њ–є –Є–Ј¬†–Њ—Б–љ–Њ–≤–љ—Л—Е –њ—А–Є—З–Є–љ –њ–Њ–≤—Л—И–µ–љ–Є—П –ґ–µ—Б—В–Ї–Њ—Б—В–Є –∞—А—В–µ—А–Є–∞–ї—М–љ–Њ–є —Б—В–µ–љ–Ї–Є, –њ—А–Є–≤–Њ–і—П—Й–µ–є –Ї¬†–Ј–љ–∞—З–Є—В–µ–ї—М–љ—Л–Љ –Љ–µ—Е–∞–љ–Є—З–µ—Б–Ї–Є–Љ –Є–Ј–Љ–µ–љ–µ–љ–Є—П–Љ, –Ї–Њ—В–Њ—А—Л–µ –Є–Ј–Љ–µ–љ—П—О—В —А–∞—Б—В—П–ґ–Є–Љ–Њ—Б—В—М —Б–Њ—Б—Г–і–Њ–≤, –≤—Л–Ј—Л–≤–∞—П —Г–≤–µ–ї–Є—З–µ–љ–Є–µ —Б–Ї–Њ—А–Њ—Б—В–Є –њ—Г–ї—М—Б–Њ–≤–Њ–є –≤–Њ–ї–љ—Л –Є¬†–њ–Њ–≤—Л—И–µ–љ–Є–µ –∞—А—В–µ—А–Є–∞–ї—М–љ–Њ–≥–Њ –і–∞–≤–ї–µ–љ–Є—П. –°—В—А—Г–Ї—В—Г—А–љ—Л–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П, –≤—Л–Ј–≤–∞–љ–љ—Л–µ –Ъ–°, –≤¬†—Б–≤–Њ—О –Њ—З–µ—А–µ–і—М, —Б–Њ–њ—А–Њ–≤–Њ–ґ–і–∞—О—В—Б—П –∞—А—В–µ—А–Є–∞–ї—М–љ–Њ–є¬†–≥–Є–њ–µ—А—В–µ–љ–Ј–Є–µ–є,¬†–≥–Є–њ–µ—А—В—А–Њ—Д–Є–µ–є –ї–µ–≤–Њ–≥–Њ –ґ–µ–ї—Г–і–Њ—З–Ї–∞ –Є¬†—Б–µ—А–і–µ—З–љ–Њ–є –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В—М—О, —З—В–Њ –њ–Њ–Ј–≤–Њ–ї—П–µ—В —Б—З–Є—В–∞—В—М –Ъ–° –Њ—Б–љ–Њ–≤–љ—Л–Љ –љ–µ–Ј–∞–≤–Є—Б–Є–Љ—Л–Љ —Д–∞–Ї—В–Њ—А–Њ–Љ —А–Є—Б–Ї–∞ —А–∞–Ј–≤–Є—В–Є—П –°–°–Ч –Є¬†–њ–Њ–≤—Л—И–µ–љ–Є—П –Њ–±—Й–µ–є —Б–Љ–µ—А—В–љ–Њ—Б—В–Є —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–•–С–Я [44]. –ЪC –±—Л–ї–∞ –Њ–њ—А–µ–і–µ–ї–µ–љ–∞ –Ї–∞–Ї –њ–∞—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–µ –Њ—В–ї–Њ–ґ–µ–љ–Є–µ –Ї—А–Є—Б—В–∞–ї–ї–Њ–≤¬†–°–∞¬†–≤¬†—Б–Њ—Б—Г–і–Є—Б—В–Њ–є —Б–µ—В–Є. –£¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–Ґ–Я–Э —Г—А–µ–Љ–Є—З–µ—Б–Ї–∞—П —Б—А–µ–і–∞ —Е–∞—А–∞–Ї—В–µ—А–Є–Ј—Г–µ—В—Б—П –љ–∞–ї–Є—З–Є–µ–Љ –±–Њ–ї—М—И–Њ–≥–Њ –Ї–Њ–ї–Є—З–µ—Б—В–≤–∞ —Г—А–µ–Љ–Є—З–µ—Б–Ї–Є—Е —В–Њ–Ї—Б–Є–љ–Њ–≤ –Є¬†–Ъ–§–Я–І-II, –Ї–Њ—В–Њ—А—Л–µ —П–≤–ї—П—О—В—Б—П –Є–љ–і—Г–Ї—В–Њ—А–∞–Љ–Є –Ъ–°, –Є¬†—А–∞–Ј–≤–Є–≤–∞–µ—В—Б—П —А–∞–љ—М—И–µ –њ–Њ¬†—Б—А–∞–≤–љ–µ–љ–Є—О —Б–Њ¬†–Ј–і–Њ—А–Њ–≤—Л–Љ–Є —Б—Г–±—К–µ–Ї—В–∞–Љ–Є. –Ъ–° –њ—А–Є –•–С–Я –њ—А–µ–і—Б—В–∞–≤–ї–µ–љ–∞ –і–≤—Г–Љ—П –Њ—Б–љ–Њ–≤–љ—Л–Љ–Є —В–Є–њ–∞–Љ–Є –Ї–∞–ї—М—Ж–Є—Д–Є–Ї–∞—Ж–Є–Є: –Є–љ—В–Є–Љ–∞–ї—М–љ–Њ–є –Є¬†–Љ–µ–і–Є–∞–ї—М–љ–Њ–є. –Ъ–∞–ї—М—Ж–Є—Д–Є–Ї–∞—Ж–Є—П –Є–љ—В–Є–Љ—Л —Б–≤—П–Ј–∞–љ–∞ —Б¬†–∞—В–µ—А–Њ—Б–Ї–ї–µ—А–Њ–Ј–Њ–Љ, –њ—А–Є –Ї–Њ—В–Њ—А–Њ–Љ Ca2+ –Њ—В–Ї–ї–∞–і—Л–≤–∞–µ—В—Б—П –≤–Љ–µ—Б—В–µ —Б¬†–ї–Є–њ–Њ–њ—А–Њ—В–µ–Є–љ–∞–Љ–Є, –∞¬†—В–∞–Ї–ґ–µ —Д–Њ—Б—Д–Њ–ї–Є–њ–Є–і–∞–Љ–Є –≤¬†–∞—В–µ—А–Њ—Б–Ї–ї–µ—А–Њ—В–Є—З–µ—Б–Ї–Є—Е –±–ї—П—И–Ї–∞—Е, –≤¬†—В–Њ –≤—А–µ–Љ—П –Ї–∞–Ї –Љ–µ–і–Є–∞–ї—М–љ–∞—П –Ї–∞–ї—М—Ж–Є—Д–Є–Ї–∞—Ж–Є—П, –±–Њ–ї–µ–µ —А–∞—Б–њ—А–Њ—Б—В—А–∞–љ–µ–љ–љ–∞—П –њ—А–Є –•–С–Я, —П–≤–ї—П–µ—В—Б—П —А–µ–Ј—Г–ї—М—В–∞—В–Њ–Љ –Њ—Б—В–µ–Њ–≥–µ–љ–љ–Њ–≥–Њ –њ—А–Њ—Ж–µ—Б—Б–∞, —Б—Е–Њ–і–љ–Њ–≥–Њ —Б¬†–≤–љ—Г—В—А–Є–Љ–µ–Љ–±—А–∞–љ–Њ–Ј–љ–Њ–є –Њ—Б—Б–Є—Д–Є–Ї–∞—Ж–Є–µ–є, –Ї–Њ—В–Њ—А–∞—П –љ–µ¬†–Ј–∞–≤–Є—Б–Є—В –Њ—В¬†–∞—В–µ—А–Њ—Б–Ї–ї–µ—А–Њ–Ј–∞ –Є¬†–≤—Л–Ј—Л–≤–∞–µ—В —Б–љ–Є–ґ–µ–љ–Є–µ –њ–Њ–і–∞—В–ї–Є–≤–Њ—Б—В–Є —Б–Њ—Б—Г–і–Є—Б—В–Њ–є —Б—В–µ–љ–Ї–Є [45вАУ47].
–Ъ–° –њ—А–µ–і—Б—В–∞–≤–ї—П–µ—В —Б–Њ–±–Њ–є —Б–ї–Њ–ґ–љ—Л–є –Љ–љ–Њ–≥–Њ—Д–∞–Ї—В–Њ—А–љ—Л–є –њ—А–Њ—Ж–µ—Б—Б, —Ж–µ–љ—В—А–∞–ї—М–љ—Г—О —А–Њ–ї—М –≤¬†–Ї–Њ—В–Њ—А–Њ–Љ –Є–≥—А–∞—О—В —В—А–∞–љ—Б–і–Є—Д—Д–µ—А–µ–љ—Ж–Є—А–Њ–≤–Ї–∞ –У–Ь–Ъ–° –Є¬†–∞–њ–Њ–њ—В–Њ–Ј. –§–Њ—Б—Д–∞—В –љ–∞–њ—А—П–Љ—Г—О –≤–Њ–Ј–і–µ–є—Б—В–≤—Г–µ—В –љ–∞¬†—А–µ—Ж–µ–њ—В–Њ—А PiT1, —Б—В–Є–Љ—Г–ї–Є—А—Г—П –У–Ь–Ъ–° –њ–Њ—Б—В–Њ—П–љ–љ–Њ —В—А–∞–љ—Б—Д–Њ—А–Љ–Є—А–Њ–≤–∞—В—М—Б—П –Є–Ј¬†—Б–Њ–Ї—А–∞—В–Є—В–µ–ї—М–љ–Њ–≥–Њ –≤¬†–Њ—Б—В–µ–Њ—Е–Њ–љ–і—А–Њ–≥–µ–љ–љ—Л–є —Д–µ–љ–Њ—В–Є–њ, –Њ–±–ї–∞–і–∞—О—Й–Є–є –Ї–∞–ї—М—Ж–Є—Д–Є—Ж–Є—А—Г—О—Й–µ–є —Б–њ–Њ—Б–Њ–±–љ–Њ—Б—В—М—О. –Ъ–∞–Ї —В–Њ–ї—М–Ї–Њ –њ—А–Њ–Є—Б—Е–Њ–і–Є—В —Н—В–∞ —В—А–∞–љ—Б—Д–Њ—А–Љ–∞—Ж–Є—П, —Н—В–Є –Њ—Б—В–µ–Њ–±–ї–∞—Б—В–Њ–њ–Њ–і–Њ–±–љ—Л–µ –Ї–ї–µ—В–Ї–Є –љ–∞—З–Є–љ–∞—О—В –≤—Л—А–∞–±–∞—В—Л–≤–∞—В—М –Њ—В–ї–Њ–ґ–µ–љ–Є—П¬†–≥–Є–і—А–Њ–Ї—Б–Є–∞–њ–∞—В–Є—В–∞, –Ї–Њ—В–Њ—А—Л–µ –Њ–±—Л—З–љ–Њ –љ–∞—Е–Њ–і—П—В—Б—П –≤¬†–Ї–Њ—Б—В–Є. –Я—А–Є —А–∞–Ј–≤–Є—В–Є–Є –Ґ–Я–Э –У–Ь–Ъ–° —А–µ–∞–≥–Є—А—Г—О—В –љ–∞¬†—В–Њ–Ї—Б–Є—З–љ—Г—О —Г—А–µ–Љ–Є—З–µ—Б–Ї—Г—О —Б—А–µ–і—Г, —Г—Б–Є–ї–Є–≤–∞—П –∞–љ–∞–ї–Њ–≥–Є—З–љ—Г—О —В—А–∞–љ—Б—Д–Њ—А–Љ–∞—Ж–Є—О, –Љ–Њ–≥—Г—В —Б—В–∞–љ–Њ–≤–Є—В—М—Б—П –∞–њ–Њ–њ—В–Њ—В–Є—З–µ—Б–Ї–Є–Љ–Є –Є¬†–і–µ–є—Б—В–≤–Њ–≤–∞—В—М –Ї–∞–Ї –Њ—З–∞–≥ –Є–љ–Є—Ж–Є–∞—Ж–Є–Є –Ї–∞–ї—М—Ж–Є—Д–Є–Ї–∞—Ж–Є–Є [48].
–Т¬†–Њ—В–ї–Є—З–Є–µ –Њ—В¬†–Ъ–§–Я–Ь, –Ї–Њ—В–Њ—А—Л–µ –Ї–∞–ґ—Г—В—Б—П –≤¬†–Њ—Б–љ–Њ–≤–љ–Њ–Љ –Є–љ–µ—А—В–љ—Л–Љ–Є –њ–Њ¬†–Њ—В–љ–Њ—И–µ–љ–Є—О –Ї¬†–Ї–ї–µ—В–Ї–∞–Љ —Б–Њ—Б—Г–і–Њ–≤ in¬†vitro¬†[16], —Б—Д–Њ—А–Љ–Є—А–Њ–≤–∞–љ–љ—Л–µ –Ъ–§–Я–І –±—Л–ї–Є –≤–Њ–≤–ї–µ—З–µ–љ—Л –≤¬†–Ї–∞—З–µ—Б—В–≤–µ –њ–Њ—В–µ–љ—Ж–Є–∞–ї—М–љ—Л—Е –Љ–µ–і–Є–∞—В–Њ—А–Њ–≤ –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є—П —Б–Њ—Б—Г–і–Њ–≤, –≤—Л–Ј—Л–≤–∞—О—Й–Є—Е –≤–Њ—Б–њ–∞–ї–µ–љ–Є–µ, —Н–љ–і–Њ—В–µ–ї–Є–∞–ї—М–љ—Г—О –і–Є—Б—Д—Г–љ–Ї—Ж–Є—О –Є¬†–Ъ–° [35, 38, 49]. –≠–љ–і–Њ—В–µ–ї–Є–є –њ—А–µ–і—Б—В–∞–≤–ї—П–µ—В —Б–Њ–±–Њ–є –±–∞—А—М–µ—А –Љ–µ–ґ–і—Г —Ж–Є—А–Ї—Г–ї–Є—А—Г—О—Й–Є–Љ–Є –Ъ–§–Я–І –Є¬†–њ–Њ–і–ї–µ–ґ–∞—Й–µ–є —Б–Њ—Б—Г–і–Є—Б—В–Њ–є —В–Ї–∞–љ—М—О –Є¬†—П–≤–ї—П–µ—В—Б—П –њ–µ—А–≤–Њ–є –њ–Њ–њ—Г–ї—П—Ж–Є–µ–є –Ї–ї–µ—В–Њ–Ї, –њ–Њ–і–≤–µ—А–≥–∞—О—Й–Є—Е—Б—П –≤–Њ–Ј–і–µ–є—Б—В–≤–Є—О –Ъ–§–Я–І –њ–Њ—Б–ї–µ –Є—Е –Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є—П. –≠–љ–і–Њ—В–µ–ї–Є–∞–ї—М–љ–∞—П –і–Є—Б—Д—Г–љ–Ї—Ж–Є—П –Њ–њ—А–µ–і–µ–ї—П–µ—В—Б—П –Ї–∞–Ї –њ–∞—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–µ —Б–Њ—Б—В–Њ—П–љ–Є–µ, –њ—А–Є –Ї–Њ—В–Њ—А–Њ–Љ –њ—А–Њ–Є—Б—Е–Њ–і–Є—В —Б—Г–ґ–µ–љ–Є–µ —Б–Њ—Б—Г–і–Њ–≤ –≤—Б–ї–µ–і—Б—В–≤–Є–µ –і–Є—Б–±–∞–ї–∞–љ—Б–∞ –Њ—В–љ–Њ—Б–Є—В–µ–ї—М–љ–Њ–≥–Њ –≤–Ї–ї–∞–і–∞ —Н–љ–і–Њ—В–µ–ї–Є–∞–ї—М–љ—Л—Е —А–∞—Б—Б–ї–∞–±–ї—П—О—Й–Є—Е –Є¬†—Б–Њ–Ї—А–∞—Й–∞—О—Й–Є—Е —Д–∞–Ї—В–Њ—А–Њ–≤. –Т–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ–∞—П –∞–Ї—В–Є–≤–∞—Ж–Є—П —Н–љ–і–Њ—В–µ–ї–Є—П –Є¬†—Н–љ–і–Њ—В–µ–ї–Є–∞–ї—М–љ–∞—П –і–Є—Б—Д—Г–љ–Ї—Ж–Є—П –Ј–∞–њ—Г—Б–Ї–∞—О—В—Б—П –њ—А–Њ–∞—В–µ—А–Њ–≥–µ–љ–љ—Л–Љ–Є –Є¬†–њ—А–Њ–≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ—Л–Љ–Є —Б–Є–≥–љ–∞–ї—М–љ—Л–Љ–Є –Љ–Њ–ї–µ–Ї—Г–ї–∞–Љ–Є –Є¬†–Є–≥—А–∞—О—В –Ї–ї—О—З–µ–≤—Г—О —А–Њ–ї—М –≤¬†—А–∞–Ј–≤–Є—В–Є–Є –∞—В–µ—А–Њ—Б–Ї–ї–µ—А–Њ–Ј–∞ –Є¬†–Ъ–°.
–Э–µ–і–∞–≤–љ–Є–µ –і–∞–љ–љ—Л–µ —Б–≤–Є–і–µ—В–µ–ї—М—Б—В–≤—Г—О—В –Њ¬†—В–Њ–Љ, —З—В–Њ –Ъ–§–Я–І —П–≤–ї—П—О—В—Б—П –њ—А—П–Љ—Л–Љ–Є –Љ–µ–і–Є–∞—В–Њ—А–∞–Љ–Є —Н–љ–і–Њ—В–µ–ї–Є–∞–ї—М–љ–Њ–є –і–Є—Б—Д—Г–љ–Ї—Ж–Є–Є [50, 51]. –£—Б—В–∞–љ–Њ–≤–ї–µ–љ–Њ, —З—В–Њ –њ–Њ—Б–ї–µ–і—Б—В–≤–Є–µ–Љ –Є–љ—В–µ—А–љ–∞–ї–Є–Ј–∞—Ж–Є–Є –Ъ–§–Я–І —П–≤–ї—П–µ—В—Б—П –њ–∞—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–∞—П –∞–Ї—В–Є–≤–∞—Ж–Є—П —Н–љ–і–Њ—В–µ–ї–Є—П, —Б–Њ–њ—А–Њ–≤–Њ–ґ–і–∞–µ–Љ–∞—П –≤—Л–і–µ–ї–µ–љ–Є–µ–Љ –њ—А–Њ–≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ—Л—Е —Ж–Є—В–Њ–Ї–Є–љ–Њ–≤ –≤¬†–Љ–Є–Ї—А–Њ–Њ–Ї—А—Г–ґ–µ–љ–Є–µ –Є¬†—Б–Є—Б—В–µ–Љ–љ—Л–є –Ї—А–Њ–≤–Њ—В–Њ–Ї, –∞–і–≥–µ–Ј–Є–µ–є –ї–µ–є–Ї–Њ—Ж–Є—В–Њ–≤ –Ї¬†—Н–љ–і–Њ—В–µ–ї–Є—О, —А–∞–Ј–≤–Є—В–Є–µ–Љ —Н–љ–і–Њ—В–µ–ї–Є–∞–ї—М–љ–Њ-–Љ–µ–Ј–µ–љ—Е–Є–Љ–∞–ї—М–љ–Њ–≥–Њ –њ–µ—А–µ—Е–Њ–і–∞ —Б¬†–њ–Њ—Б—В–µ–њ–µ–љ–љ–Њ–є –њ–Њ—В–µ—А–µ–є –Ї–ї–µ—В–Ї–∞–Љ–Є —Н–љ–і–Њ—В–µ–ї–Є–∞–ї—М–љ–Њ–≥–Њ —Д–µ–љ–Њ—В–Є–њ–∞ –≤¬†—Б–Њ—З–µ—В–∞–љ–Є–Є —Б¬†–њ—А–Є–Њ–±—А–µ—В–µ–љ–Є–µ–Љ —Д–µ–љ–Њ—В–Є–њ–∞ –Љ–µ–Ј–µ–љ—Е–Є–Љ–∞–ї—М–љ–Њ–≥–Њ [52]. –Ю–і–љ–Є–Љ –Є–Ј¬†–≤–Њ–Ј–Љ–Њ–ґ–љ—Л—Е –Љ–µ—Е–∞–љ–Є–Ј–Љ–Њ–≤, —Б¬†–њ–Њ–Љ–Њ—Й—М—О –Ї–Њ—В–Њ—А–Њ–≥–Њ –Ъ–§–Я–І –Љ–Њ–≥—Г—В –Є–љ–і—Г—Ж–Є—А–Њ–≤–∞—В—М —Н–љ–і–Њ—В–µ–ї–Є–∞–ї—М–љ—Г—О –і–Є—Б—Д—Г–љ–Ї—Ж–Є—О, —П–≤–ї—П–µ—В—Б—П —Б–љ–Є–ґ–µ–љ–Є–µ —Н–Ї—Б–њ—А–µ—Б—Б–Є–Є –Љ–†–Э–Ъ —Н–љ–і–Њ—В–µ–ї–Є–∞–ї—М–љ–Њ–є —Б–Є–љ—В–∞–Ј—Л –Њ–Ї—Б–Є–і–∞ –∞–Ј–Њ—В–∞ (eNOS) –Є¬†–≤—Л—А–∞–±–Њ—В–Ї–Є –љ–Є—В—А–Є—В–Њ–≤, —З—В–Њ —Г–Ї–∞–Ј—Л–≤–∞–µ—В –љ–∞¬†—Б–љ–Є–ґ–µ–љ–Є–µ –±–Є–Њ–і–Њ—Б—В—Г–њ–љ–Њ—Б—В–Є –Њ–Ї—Б–Є–і–∞ –∞–Ј–Њ—В–∞ (NO) –њ—Г—В–µ–Љ –њ–Њ–і–∞–≤–ї–µ–љ–Є—П —Н–Ї—Б–њ—А–µ—Б—Б–Є–Є –Є–ї–Є –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є —Н–љ–і–Њ—В–µ–ї–Є–∞–ї—М–љ–Њ–є eNOS –ї–Є–±–Њ –њ—Г—В–µ–Љ –Њ–њ–Њ—Б—А–µ–і–Њ–≤–∞–љ–љ–Њ–≥–Њ –∞–Ї—В–Є–≤–љ—Л–Љ–Є —Д–Њ—А–Љ–∞–Љ–Є –Ї–Є—Б–ї–Њ—А–Њ–і–∞ –њ–Њ–≥–ї–Њ—Й–µ–љ–Є—П NO [52].
–Ь–Є—В–Њ—Е–Њ–љ–і—А–Є–Є —П–≤–ї—П—О—В—Б—П –Њ—Б–љ–Њ–≤–љ—Л–Љ –Є—Б—В–Њ—З–љ–Є–Ї–Њ–Љ –Њ–Ї–Є—Б–ї–Є—В–µ–ї—М–љ–Њ–≥–Њ —Б—В—А–µ—Б—Б–∞ –≤¬†–Ї–ї–µ—В–Ї–∞—Е, –∞¬†–≤—Л—Б–Њ–Ї–Є–µ —Г—А–Њ–≤–љ–Є P,¬†–≤–Њ–Ј–і–µ–є—Б—В–≤—Г—О—Й–Є–µ –љ–∞¬†–Љ–Є—В–Њ—Е–Њ–љ–і—А–Є–Є, —Г–≤–µ–ї–Є—З–Є–≤–∞—О—В –≤—Л—Б–≤–Њ–±–Њ–ґ–і–µ–љ–Є–µ —Б–≤–Њ–±–Њ–і–љ—Л—Е —А–∞–і–Є–Ї–∞–ї–Њ–≤. –Ю–і–љ–Є–Љ –Є–Ј¬†–Њ—Б–љ–Њ–≤–љ—Л—Е —Н—Д—Д–µ–Ї—В–Њ—А–Њ–≤, –Є–љ–і—Г—Ж–Є—А—Г–µ–Љ—Л—Е P,¬†–≤–µ—А–Њ—П—В–љ–Њ, —П–≤–ї—П–µ—В—Б—П –≤–љ—Г—В—А–Є–Ї–ї–µ—В–Њ—З–љ–Њ–µ –њ–Њ–≤—Л—И–µ–љ–Є–µ —Г—А–Њ–≤–љ—П –°–∞, –Ї–Њ—В–Њ—А–Њ–µ –≤—Л–Ј—Л–≤–∞–µ—В –≤–љ—Г—В—А–Є–Ї–ї–µ—В–Њ—З–љ–Њ–µ –Њ–Ї–Є—Б–ї–Є—В–µ–ї—М–љ–Њ–µ —Д–Њ—Б—Д–Њ—А–Є–ї–Є—А–Њ–≤–∞–љ–Є–µ. –°–≤—П–Ј—М –Љ–µ–ґ–і—Г –Њ–Ї–Є—Б–ї–Є—В–µ–ї—М–љ—Л–Љ —Б—В—А–µ—Б—Б–Њ–Љ –Є¬†–Ъ–° –±—Л–ї–∞ –і–Њ–Ї–∞–Ј–∞–љ–∞, –Ї–Њ–≥–і–∞ –±—Л–ї–Њ –Њ–±–љ–∞—А—Г–ґ–µ–љ–Њ, —З—В–Њ –љ–∞–Ї–Њ–њ–ї–µ–љ–Є–µ –∞–Ї—В–Є–≤–љ—Л—Е —Д–Њ—А–Љ –Ї–Є—Б–ї–Њ—А–Њ–і–∞ —Б–њ–Њ—Б–Њ–±–љ–Њ –Є–љ–і—Г—Ж–Є—А–Њ–≤–∞—В—М —Н–Ї—Б–њ—А–µ—Б—Б–Є—О RUNX2, –њ–Њ–і–і–µ—А–ґ–Є–≤–∞—О—Й—Г—О –Њ—Б—В–µ–Њ–±–ї–∞—Б—В–љ—Г—О —В—А–∞–љ—Б–і–Є—Д—Д–µ—А–µ–љ—Ж–Є—А–Њ–≤–Ї—Г –У–Ь–Ъ–°. –Я–µ—А–µ–≥—А—Г–Ј–Ї–∞ –† –≤—Л–Ј—Л–≤–∞–µ—В –і–Є—Б–±–∞–ї–∞–љ—Б –Љ–µ–ґ–і—Г –∞–љ—В–Є–Њ–Ї—Б–Є–і–∞–љ—В–∞–Љ–Є –Є¬†–∞–Ї—В–Є–≤–љ—Л–Љ–Є —Д–Њ—А–Љ–∞–Љ–Є –Ї–Є—Б–ї–Њ—А–Њ–і–∞ –≤¬†–У–Ь–Ъ–°, –Є¬†–µ—Й–µ –Њ–і–љ–Є–Љ —Б–≤–Є–і–µ—В–µ–ї—М—Б—В–≤–Њ–Љ —Г—З–∞—Б—В–Є—П –Њ–Ї–Є—Б–ї–Є—В–µ–ї—М–љ–Њ–≥–Њ —Б—В—А–µ—Б—Б–∞ —П–≤–ї—П–µ—В—Б—П —Н—Д—Д–µ–Ї—В –∞–љ—В–Є–Њ–Ї—Б–Є–і–∞–љ—В–Њ–≤, –Ї–Њ—В–Њ—А—Л–µ —П–≤–ї—П—О—В—Б—П –Ј–∞—Й–Є—В–љ—Л–Љ–Є –њ—А–Є –Ї–∞–ї—М—Ж–Є—Д–Є–Ї–∞—Ж–Є–Є –У–Ь–Ъ–° [53]. –Р–ї—М—В–µ—А–љ–∞—В–Є–≤–љ–Њ –Ъ–§–Я–І –Љ–Њ–≥—Г—В –њ–Њ–≤—Л—И–∞—В—М —Г—А–Њ–≤–љ–Є –∞—Б–Є–Љ–Љ–µ—В—А–Є—З–љ–Њ–≥–Њ –і–Є–Љ–µ—В–Є–ї–∞—А–≥–Є–љ–Є–љ–∞, —Н–љ–і–Њ–≥–µ–љ–љ–Њ–≥–Њ –Є–љ–≥–Є–±–Є—В–Њ—А–∞ NO [54]. –Ґ–Њ—З–љ—Л–є –Љ–µ—Е–∞–љ–Є–Ј–Љ, —Б¬†–њ–Њ–Љ–Њ—Й—М—О –Ї–Њ—В–Њ—А–Њ–≥–Њ –Ъ–§–Я–І –Є–љ–і—Г—Ж–Є—А—Г—О—В —Н–љ–і–Њ—В–µ–ї–Є–∞–ї—М–љ—Г—О –і–Є—Б—Д—Г–љ–Ї—Ж–Є—О, –љ—Г–ґ–і–∞–µ—В—Б—П –≤¬†–і–∞–ї—М–љ–µ–є—И–µ–Љ –Є–Ј—Г—З–µ–љ–Є–Є.
–Т—В–Њ—А–Є—З–љ—Л–µ –Ъ–§–Я–І –Љ–Њ–≥—Г—В –Є–љ–і—Г—Ж–Є—А–Њ–≤–∞—В—М –Ї–∞–ї—М—Ж–Є—Д–Є–Ї–∞—Ж–Є—О –≤¬†–Ї—Г–ї—М—В–Є–≤–Є—А—Г–µ–Љ—Л—Е –У–Ь–Ъ–° –Є¬†–≤—А–Њ–ґ–і–µ–љ–љ—Л–µ –Є–Љ–Љ—Г–љ–љ—Л–µ —А–µ–∞–Ї—Ж–Є–Є –≤¬†–Ї—Г–ї—М—В–Є–≤–Є—А—Г–µ–Љ—Л—Е –Љ–∞–Ї—А–Њ—Д–∞–≥–∞—Е, –Ї–∞–Ї –µ—Б–ї–Є –±—Л –Њ–љ–Є –±—Л–ї–Є –њ–∞—В–Њ–≥–µ–љ–Њ–Љ.¬† –Ъ–§–Я–І-II (–≤¬†–Њ—В–ї–Є—З–Є–µ –Њ—В¬†–Ъ–§–Я–І-I) –љ–µ–њ–Њ—Б—А–µ–і—Б—В–≤–µ–љ–љ–Њ –Є–љ–і—Г—Ж–Є—А—Г—О—В –≤–Њ—Б–њ–∞–ї–µ–љ–Є–µ –Є¬†—Г—Б–Ї–Њ—А—П—О—В —Д–µ–љ–Њ—В–Є–њ–Є—З–µ—Б–Ї–Є–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П –≤¬†–У–Ь–Ъ–°, —З—В–Њ –њ—А–Є–≤–Њ–і–Є—В –Ї¬†–Ї–∞–ї—М—Ж–Є—Д–Є–Ї–∞—Ж–Є–Є —Б–Њ—Б—Г–і–Є—Б—В–Њ–є —Б—В–µ–љ–Ї–Є –Є¬†—Б–µ—А–і–µ—З–љ—Л—Е –Ї–ї–∞–њ–∞–љ–Њ–≤, –Є–љ–і—Г—Ж–Є—А—Г–µ—В —Б–Є–љ—В–µ–Ј –≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ—Л—Е —Ж–Є—В–Њ–Ї–Є–љ–Њ–≤ –≤¬†–ї–µ–є–Ї–Њ—Ж–Є—В–∞—Е, –Љ–∞–Ї—А–Њ—Д–∞–≥–∞—Е, –У–Ь–Ъ–° –Є¬†—Б–Њ—Б—Г–і–Є—Б—В—Л—Е –Є–љ—В–µ—А—Б—В–Є—Ж–Є–∞–ї—М–љ—Л—Е –Ї–ї–µ—В–Ї–∞—Е. –Р–Ї—В–Є–≤–∞—Ж–Є—П –Є–љ—Д–ї–∞–Љ–Љ–∞—Б–Њ–Љ –≤¬†–Њ—В–≤–µ—В –љ–∞¬†–Ъ–§–Я–І-II –≤¬†–љ–∞—Б—В–Њ—П—Й–µ–µ –≤—А–µ–Љ—П —А–∞—Б—Б–Љ–∞—В—А–Є–≤–∞–µ—В—Б—П –Ї–∞–Ї –Њ–і–Є–љ –Є–Ј¬†–≤–∞–ґ–љ–µ–є—И–Є—Е —Н—В–∞–њ–Њ–≤ —Б–µ—А–і–µ—З–љ–Њ-—Б–Њ—Б—Г–і–Є—Б—В–Њ–є –Ї–∞–ї—М—Ж–Є—Д–Є–Ї–∞—Ж–Є–Є [55, 56].
–Ш—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П –њ–Њ–Ї–∞–Ј–∞–ї–Є, —З—В–Њ –Ъ–§–Я–І-II –Є–љ–і—Г—Ж–Є—А—Г—О—В –Ї–∞–ї—М—Ж–Є—Д–Є–Ї–∞—Ж–Є—О –У–Ь–Ъ–° in¬†vitro, –∞¬†—В–∞–Ї–ґ–µ —Б–µ–Ї—А–µ—Ж–Є—О –§–Э–Ю-ќ± –≤¬†–Љ–∞–Ї—А–Њ—Д–∞–≥–∞—Е, —Г–≤–µ–ї–Є—З–Є–≤–∞—О—В —Н–Ї—Б–њ—А–µ—Б—Б–Є—О –Ї–Њ—Б—В–љ–Њ–≥–Њ –Љ–Њ—А—Д–Њ–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Њ–≥–Њ –±–µ–ї–Ї–∞-2, –∞¬†—В–∞–Ї–ґ–µ —П–і–µ—А–љ–Њ–≥–Њ —Д–∞–Ї—В–Њ—А–∞ –Ї–∞–њ–њ–∞-–Т¬†–≤ –У–Ь–Ъ–°. –С—Л–ї–Њ —В–∞–Ї–ґ–µ –њ–Њ–Ї–∞–Ј–∞–љ–Њ, —З—В–Њ –Ї–∞–ї—М—Ж–Є—Д–Є–Ї–∞—Ж–Є—П –У–Ь–Ъ–° —П–≤–ї—П–µ—В—Б—П —А–µ–Ј—Г–ї—М—В–∞—В–Њ–Љ –њ–Њ–≥–ї–Њ—Й–µ–љ–Є—П –Ъ–§–Я–І-II –Ї–ї–µ—В–Ї–∞–Љ–Є, –њ—А–Є—З–µ–Љ –Ъ–§–Я–І-II –Њ–±–љ–∞—А—Г–ґ–Є–≤–∞—О—В—Б—П –≤–љ—Г—В—А–Є–Ї–ї–µ—В–Њ—З–љ–Њ –≤¬†–Ї–∞–ї—М—Ж–Є–љ–Є—А–Њ–≤–∞–љ–љ—Л—Е –У–Ь–Ъ–° [39, 55]. –Ю–і–љ–∞–Ї–Њ —Б—Г—Й–µ—Б—В–≤—Г—О—В —А–∞–Ј–љ–Њ–≥–ї–∞—Б–Є—П –Њ—В–љ–Њ—Б–Є—В–µ–ї—М–љ–Њ –Є–љ–і—Г–Ї—Ж–Є–Є –Њ—Б—В–µ–Њ—Е–Њ–љ–і—А–Њ–≥–µ–љ–љ–Њ–є –і–Є—Д—Д–µ—А–µ–љ—Ж–Є—А–Њ–≤–Ї–Є —Б¬†–њ–Њ–Љ–Њ—Й—М—О –Ъ–§–Я–І. –Ф–ї—П –Є–ї–ї—О—Б—В—А–∞—Ж–Є–Є –≤¬†–љ–µ–Ї–Њ—В–Њ—А—Л—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П—Е —Б–Њ–Њ–±—Й–∞–µ—В—Б—П –Њ¬†—Б–љ–Є–ґ–µ–љ–Є–Є –Њ—Б—В–µ–Њ—Е–Њ–љ–і—А–Њ–≥–µ–љ–љ–Њ–є –і–µ–і–Є—Д—Д–µ—А–µ–љ—Ж–Є—А–Њ–≤–Ї–Є, –Ї–Њ–≥–і–∞ –±–ї–Њ–Ї–Є—А—Г–µ—В—Б—П –Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є–µ –≤—В–Њ—А–Є—З–љ—Л—Е –Ъ–§–Я–І [57], —В–Њ–≥–і–∞ –Ї–∞–Ї –≤¬†–і—А—Г–≥–Є—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П—Е –љ–µ¬†—Г–і–∞–ї–Њ—Б—М –Є–і–µ–љ—В–Є—Д–Є—Ж–Є—А–Њ–≤–∞—В—М –њ—А–Є–Ј–љ–∞–Ї–Є –Њ—Б—В–µ–Њ—Е–Њ–љ–і—А–Њ–≥–µ–љ–љ—Л—Е¬†–≥–µ–љ–Њ–≤ –≤¬†–Ї–∞–ї—М—Ж–Є–љ–Є—А–Њ–≤–∞–љ–љ—Л—Е –њ–Њ—А–∞–ґ–µ–љ–Є—П—Е [58]. –Ь–µ—Е–∞–љ–Є—Б—В–Є—З–µ—Б–Ї–Њ–µ –њ–Њ–љ–Є–Љ–∞–љ–Є–µ –≤–ї–Є—П–љ–Є—П –Ъ–§–Я–І –љ–∞¬†–Њ—Б—В–µ–Њ—Е–Њ–љ–і—А–Њ–≥–µ–љ–љ—Г—О –і–µ–і–Є—Д—Д–µ—А–µ–љ—Ж–Є—А–Њ–≤–Ї—Г –У–Ь–Ъ–° –Њ–≥—А–∞–љ–Є—З–µ–љ–Њ, –Њ–і–љ–∞–Ї–Њ —Н–ї–Є–Љ–Є–љ–∞—Ж–Є—П –Ъ–§–Я–І –Є–Ј¬†—Б—Л–≤–Њ—А–Њ—В–Ї–Є –Ї—А–Њ–≤–Є –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–Ґ–Я–Э —Б–љ–Є–ґ–∞–µ—В —Б–њ–Њ—Б–Њ–±–љ–Њ—Б—В—М —Б—Л–≤–Њ—А–Њ—В–Ї–Є –Є–љ–і—Г—Ж–Є—А–Њ–≤–∞—В—М –Њ—Б—В–µ–Њ—Е–Њ–љ–і—А–Њ–≥–µ–љ–љ—Г—О –і–µ–і–Є—Д—Д–µ—А–µ–љ—Ж–Є—А–Њ–≤–Ї—Г –Є¬†—Б–≤–Њ–і–Є—В –љ–∞¬†–љ–µ—В –µ–µ –њ—А–Њ–Ї–∞–ї—М—Ж–Є—Д–Є—Ж–Є—А—Г—О—Й—Г—О —Б–њ–Њ—Б–Њ–±–љ–Њ—Б—В—М. –Р–љ–∞–ї–Њ–≥–Є—З–љ—Л–Љ –Њ–±—А–∞–Ј–Њ–Љ –і–Њ–±–∞–≤–ї–µ–љ–Є–µ –Ъ–§–Я–І, –њ–Њ–ї—Г—З–µ–љ–љ—Л—Е –Њ—В¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–Ґ–Я–Э, –≤¬†—Б—Л–≤–Њ—А–Њ—В–Ї—Г –Ї—А–Њ–≤–Є –Ј–і–Њ—А–Њ–≤—Л—Е –і–Њ–љ–Њ—А–Њ–≤ —Б–њ–Њ—Б–Њ–±—Б—В–≤—Г–µ—В –Њ—Б—В–µ–Њ—Е–Њ–љ–і—А–Њ–≥–µ–љ–љ–Њ–є –і–µ–і–Є—Д—Д–µ—А–µ–љ—Ж–Є—А–Њ–≤–Ї–µ –У–Ь–Ъ–° [59]. –Ъ–§–Я–І-–Є–љ–і—Г—Ж–Є—А–Њ–≤–∞–љ–љ–∞—П –Њ—Б—В–µ–Њ—Е–Њ–љ–і—А–Њ–≥–µ–љ–љ–∞—П –і–µ–і–Є—Д—Д–µ—А–µ–љ—Ж–Є—А–Њ–≤–Ї–∞, –њ–Њ-–≤–Є–і–Є–Љ–Њ–Љ—Г, –Њ–≥—А–∞–љ–Є—З–Є–≤–∞–µ—В—Б—П –Ъ–§–Я–І-II, –њ–Њ—Б–Ї–Њ–ї—М–Ї—Г –Є–љ–≥–Є–±–Є—А–Њ–≤–∞–љ–Є–µ –њ–µ—А–µ—Е–Њ–і–∞ –Є–Ј¬†–∞–Љ–Њ—А—Д–љ–Њ–≥–Њ —Б–Њ—Б—В–Њ—П–љ–Є—П –≤¬†–Ї—А–Є—Б—В–∞–ї–ї–Є—З–µ—Б–Ї–Њ–µ –њ—А–µ–і–Њ—В–≤—А–∞—Й–∞–µ—В –Ї–∞–ї—М—Ж–Є—Д–Є–Ї–∞—Ж–Є—О [57].
–Ъ–° –њ—А–Њ–Є—Б—Е–Њ–і–Є—В –≤–Њ –≤–љ–µ–Ї–ї–µ—В–Њ—З–љ–Њ–Љ –њ—А–Њ—Б—В—А–∞–љ—Б—В–≤–µ –Є¬†–Є–љ–Є—Ж–Є–Є—А—Г–µ—В—Б—П —Б–µ–Ї—А–µ—Ж–Є–µ–є –Ї–∞–ї—М—Ж–Є—Д–Є—Ж–Є—А—Г—О—Й–Є—Е –Љ–Є–Ї—А–Њ–≤–µ–Ј–Є–Ї—Г–ї (–Ъ–Ь–Т) –Є–Ј¬†–У–Ь–Ъ–° –Є¬†–Љ–∞–Ї—А–Њ—Д–∞–≥–Њ–≤ –±–ї—П—И–µ–Ї, –Ї–Њ—В–Њ—А—Л–µ –њ—А–µ–і—Б—В–∞–≤–ї—П—О—В —Б–Њ–±–Њ–є –Љ–µ—Б—В–∞ –Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є—П —П–і–µ—А –і–ї—П –Ї–∞–ї—М—Ж–Є—Д–Є–Ї–∞—Ж–Є–Є –Љ–∞—В—А–Є–Ї—Б–∞ [60вАУ62]. –Ъ–Ь–Т –Ї–ї–µ—В–Њ—З–љ–Њ–≥–Њ –њ—А–Њ–Є—Б—Е–Њ–ґ–і–µ–љ–Є—П –Њ—В–ї–Є—З–∞—О—В—Б—П –Њ—В¬†–Ъ–Ь–Т, –њ–µ—А–µ–љ–Њ—Б–Є–Љ—Л—Е —З–µ—А–µ–Ј –Ї—А–Њ–≤—М. –Ъ–Ь–Т –Є¬†–Ъ–§–Я–І —А–∞–Ј–ї–Є—З–∞—О—В—Б—П –њ–Њ¬†–њ—А–Њ–Є—Б—Е–Њ–ґ–і–µ–љ–Є—О, —А–∞–Ј–Љ–µ—А—Г, –љ–∞–ї–Є—З–Є—О –Љ–µ–Љ–±—А–∞–љ–љ—Л—Е –±–µ–ї–Ї–Њ–≤ –Є¬†–ї–Є–њ–Є–і–Њ–≤ –Є¬†–Ї—А–Є—Б—В–∞–ї–ї–Є—З–љ–Њ—Б—В–Є –Є¬†–њ—А–µ–і—Б—В–∞–≤–ї—П—О—В —Б–Њ–±–Њ–є¬†–≥–µ—В–µ—А–Њ–≥–µ–љ–љ—Г—О¬†–≥—А—Г–њ–њ—Г —Б–µ–Ї—А–µ—В–Є—А—Г–µ–Љ—Л—Е –≤–µ–Ј–Є–Ї—Г–ї, –≤–Ї–ї—О—З–∞—П –Љ–∞—В—А–Є–Ї—Б–љ—Л–µ –≤–µ–Ј–Є–Ї—Г–ї—Л –Є¬†—Н–Ї–Ј–Њ—Б–Њ–Љ—Л, –Ї–Њ—В–Њ—А—Л–µ —Д—Г–љ–Ї—Ж–Є–Њ–љ–Є—А—Г—О—В –і–ї—П –њ–Њ–і–і–µ—А–ґ–∞–љ–Є—П –Љ–Є–љ–µ—А–∞–ї—М–љ–Њ–≥–Њ¬†–≥–Њ–Љ–µ–Њ—Б—В–∞–Ј–∞ [60, 63]. –Т¬†—Д–Є–Ј–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є—Е —Г—Б–ї–Њ–≤–Є—П—Е –Ъ–Ь–Т —Б–Њ–і–µ—А–ґ–∞—В –Є–љ–≥–Є–±–Є—В–Њ—А—Л –Ї–∞–ї—М—Ж–Є—Д–Є–Ї–∞—Ж–Є–Є, —В–Њ–≥–і–∞ –Ї–∞–Ї –≤¬†–њ–∞—В–Њ–≥–µ–љ–љ—Л—Е —Г—Б–ї–Њ–≤–Є—П—Е –њ—А–Є—Б—Г—В—Б—В–≤—Г—О—В –њ—А–Њ–Љ–Њ—В–Њ—А—Л –Ї–∞–ї—М—Ж–Є—Д–Є–Ї–∞—Ж–Є–Є. –Я–Њ—Б–ї–µ –≤—Л—Б–≤–Њ–±–Њ–ґ–і–µ–љ–Є—П –≤–Њ –≤–љ–µ–Ї–ї–µ—В–Њ—З–љ–Њ–µ –њ—А–Њ—Б—В—А–∞–љ—Б—В–≤–Њ –Ъ–Ь–Т –∞–≥—А–µ–≥–Є—А—Г—О—В—Б—П –Є¬†—Б–≤—П–Ј—Л–≤–∞—О—В—Б—П —Б¬†–Љ–∞—В—А–Є–Ї—Б–љ—Л–Љ–Є –Ї–Њ–ї–ї–∞–≥–µ–љ–∞–Љ–Є —Б¬†–Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є–µ–Љ —Ж–µ–љ—В—А–Њ–≤ –Ј–∞—А–Њ–ґ–і–µ–љ–Є—П –Ї–∞–ї—М—Ж–Є—Д–Є–Ї–∞—Ж–Є–Є, –Ї—Г–ї—М–Љ–Є–љ–∞—Ж–Є–µ–є –Ї–Њ—В–Њ—А—Л—Е —П–≤–ї—П—О—В—Б—П –Љ–Є–Ї—А–Њ–Ї–∞–ї—М—Ж–Є—Д–Є–Ї–∞—Ж–Є–Є, –Ї–Њ—В–Њ—А—Л–µ –Љ–Њ–≥—Г—В —Б–ї–Є–≤–∞—В—М—Б—П —Б¬†–Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є–µ–Љ –Љ–∞–Ї—А–Њ–Ї–∞–ї—М—Ж–Є—Д–Є–Ї–∞—Ж–Є–Є –≤¬†—Б—В–µ–љ–Ї–µ —Б–Њ—Б—Г–і–∞ [64вАУ66].
–Ъ–§–Я–І –Љ–Њ–≥—Г—В –≤–ї–Є—П—В—М –љ–∞¬†–Ї–∞–ї—М—Ж–Є—Д–Є–Ї–∞—Ж–Є—О –њ–Њ—Б—А–µ–і—Б—В–≤–Њ–Љ –Ъ–Ь–Т –љ–µ—Б–Ї–Њ–ї—М–Ї–Є–Љ–Є —Б–њ–Њ—Б–Њ–±–∞–Љ–Є. –Т–Њ-–њ–µ—А–≤—Л—Е, –Ъ–§–Я–І –Є–љ–і—Г—Ж–Є—А—Г—О—В –∞–њ–Њ–њ—В–Њ–Ј –У–Ь–Ъ–°, –Є¬†–∞–њ–Њ–њ—В–Њ—В–Є—З–µ—Б–Ї–Є–µ —В–µ–ї—М—Ж–∞ –Њ–±—А–∞–Ј—Г—О—В –Њ—З–∞–≥ –Ї–∞–ї—М—Ж–Є—Д–Є–Ї–∞—Ж–Є–Є. –Т–Њ-–≤—В–Њ—А—Л—Е, –Ъ–§–Я–І –≤—Л–Ј—Л–≤–∞—О—В –њ–Њ–≤—Л—И–µ–љ–Є–µ —Ж–Є—В–Њ–њ–ї–∞–Ј–Љ–∞—В–Є—З–µ—Б–Ї–Њ–≥–Њ Ca2+, –∞¬†–≤—Л—Б–Њ–Ї–Є–µ —Ж–Є—В–Њ–Ј–Њ–ї—М–љ—Л–µ —Г—А–Њ–≤–љ–Є Ca2+ –≤¬†–У–Ь–Ъ–° –њ—А–Є–≤–Њ–і—П—В –Ї¬†–Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є—О –њ—А–Њ–Ї–∞–ї—М—Ж–Є—Д–Є—Ж–Є—А—Г—О—Й–Є—Е –Ъ–Ь–Т. –Т-—В—А–µ—В—М–Є—Е, –Ъ–§–Я–І –Љ–Њ–≥—Г—В –±—Л—В—М –≤—Л–і–µ–ї–µ–љ—Л –Є–Ј¬†–Ї–∞–ї—М—Ж–Є–љ–Є—А–Њ–≤–∞–љ–љ—Л—Е –∞—В–µ—А–Њ–≥–µ–љ–љ—Л—Е –њ–Њ—А–∞–ґ–µ–љ–Є–є,¬†–≥–і–µ –Ъ–§–Я–І –Љ–Њ–≥—Г—В —Б–ї–Є–≤–∞—В—М—Б—П —Б¬†—А–∞–Ј–≤–Є–≤–∞—О—Й–Є–Љ–Є—Б—П –Љ–Є–Ї—А–Њ–Ї–∞–ї—М—Ж–Є—Д–Є–Ї–∞—В–∞–Љ–Є –Є¬†–Є–љ—В–µ–≥—А–Є—А–Њ–≤–∞—В—М—Б—П –≤¬†–љ–Є—Е. –Ь–µ—Е–∞–љ–Є–Ј–Љ –≤–ї–Є—П–љ–Є—П –Ъ–§–Я–І –љ–∞¬†–Ї–∞–ї—М—Ж–Є—Д–Є–Ї–∞—Ж–Є—О, –Њ–њ–Њ—Б—А–µ–і–Њ–≤–∞–љ–љ—Г—О –Ъ–Ь–Т, –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ –Є–Ј—Г—З–µ–љ, –Є¬†–њ–Њ–ї–љ–∞—П –Ї–∞—А—В–Є–љ–∞ –Њ—В—Б—Г—В—Б—В–≤—Г–µ—В. –Ґ–µ–Љ –љ–µ¬†–Љ–µ–љ–µ–µ —Б–Ї–ї–Њ–љ–љ–Њ—Б—В—М –Ї¬†–Ї–∞–ї—М—Ж–Є—Д–Є–Ї–∞—Ж–Є–Є —Б—Л–≤–Њ—А–Њ—В–Ї–Є –Є¬†–Ј—А–µ–ї–Њ—Б—В—М –Ъ–§–Я–І —Б–≤—П–Ј–∞–љ—Л —Б¬†—А–∞–Ј–Љ–µ—А–Њ–Љ –Ї–∞–ї—М—Ж–Є—Д–Є—Ж–Є—А–Њ–≤–∞–љ–љ–Њ–≥–Њ –Њ—З–∞–≥–∞, —З—В–Њ —Г–Ї–∞–Ј—Л–≤–∞–µ—В –љ–∞¬†–≤–Ј–∞–Є–Љ–Њ—Б–≤—П–Ј—М, –Ї–Њ—В–Њ—А–∞—П –Ј–∞—Б–ї—Г–ґ–Є–≤–∞–µ—В –і–∞–ї—М–љ–µ–є—И–µ–є –Њ—Ж–µ–љ–Ї–Є [37, 67]. –Ш—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П –њ–Њ–Ї–∞–Ј—Л–≤–∞—О—В, —З—В–Њ –≤¬†–і–Є–∞–ї–Є–Ј–∞—В–µ –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–Ґ–Я–Э —Б–Њ–і–µ—А–ґ–Є—В—Б—П –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ–µ –Ї–Њ–ї–Є—З–µ—Б—В–≤–Њ –Ъ–§–Я–І, –Ї–Њ—В–Њ—А–Њ–µ —В–∞–Ї–ґ–µ –±—Л–ї–Њ –њ—А—П–Љ–Њ –њ—А–Њ–њ–Њ—А—Ж–Є–Њ–љ–∞–ї—М–љ–Њ —Б–Њ–і–µ—А–ґ–∞–љ–Є—О Ca2+ –≤¬†–і–Є–∞–ї–Є–Ј–∞—В–µ. –≠—В–Њ –њ–Њ–Ј–≤–Њ–ї—П–µ—В –њ—А–µ–і–њ–Њ–ї–Њ–ґ–Є—В—М, —З—В–Њ –Ъ–§–Я–І –Љ–Њ–ґ–µ—В –±—Л—В—М –≤—Л–≤–µ–і–µ–љ –Є–Ј¬†–њ–ї–∞–Ј–Љ—Л –њ–∞—Ж–Є–µ–љ—В–Њ–≤ –њ–Њ—Б—А–µ–і—Б—В–≤–Њ–Љ¬†–≥–µ–Љ–Њ–і–Є–∞–ї–Є–Ј–∞ (–У–Ф) [68]. –Т¬†—В–Њ –ґ–µ –≤—А–µ–Љ—П –±—Л–ї–Њ –Њ–±–љ–∞—А—Г–ґ–µ–љ–Њ, —З—В–Њ –У–Ф —Г–≤–µ–ї–Є—З–Є–≤–∞–µ—В T50, —Б–љ–Є–ґ–∞—П —Б–Ї–ї–Њ–љ–љ–Њ—Б—В—М –Ї¬†–Ї–∞–ї—М—Ж–Є—Д–Є–Ї–∞—Ж–Є–Є –њ–ї–∞–Ј–Љ—Л –њ–∞—Ж–Є–µ–љ—В–∞ [69, 70]. –Я–Њ-–≤–Є–і–Є–Љ–Њ–Љ—Г, —Н—В–Њ —Б–≤–Є–і–µ—В–µ–ї—М—Б—В–≤—Г–µ—В –Њ¬†—В–Њ–Љ, —З—В–Њ —Г—А–Њ–≤–љ–Є –Ъ–§–Я–І- I –Є¬†–Ъ–§–Я–І-II –≤¬†—Б—Л–≤–Њ—А–Њ—В–Ї–µ –љ–µ¬†–њ–Њ–і–≤–µ—А–ґ–µ–љ—Л –≤–ї–Є—П–љ–Є—О —Б—В–∞–љ–і–∞—А—В–љ–Њ–≥–Њ –У–Ф [71]. –Т–Њ-–њ–µ—А–≤—Л—Е, —Н—В–Њ –њ–Њ–Ј–≤–Њ–ї—П–µ—В –њ—А–µ–і–њ–Њ–ї–Њ–ґ–Є—В—М, —З—В–Њ —Г–≤–µ–ї–Є—З–µ–љ–Є–µ T50 –њ–Њ—Б–ї–µ –љ–∞—З–∞–ї–∞ –У–Ф —Б–≤—П–Ј–∞–љ–Њ –љ–µ¬†—Б –Ї–ї–Є—А–µ–љ—Б–Њ–Љ –Ъ–§–Я–І –Ї–∞–Ї —В–∞–Ї–Њ–≤—Л–Љ, –∞¬†—Б —Г–Љ–µ–љ—М—И–µ–љ–Є–µ–Љ —Д–∞–Ї—В–Њ—А–Њ–≤, —Г—Б–Ї–Њ—А—П—О—Й–Є—Е –њ—А–Њ—Ж–µ—Б—Б –Є—Е —Б–Њ–Ј—А–µ–≤–∞–љ–Є—П, –љ–∞–Є–±–Њ–ї–µ–µ –≤–µ—А–Њ—П—В–љ–Њ —Б–≤—П–Ј–∞–љ–љ—Л–є —Б¬†—Г–Љ–µ–љ—М—И–µ–љ–Є–µ–Љ Ca2+–Є¬†P.¬†–Т–Њ-–≤—В–Њ—А—Л—Е, –Ъ–§–Я–І, —Е–Њ—В—П –Є¬†–љ–µ –≤—Л–≤–Њ–і—П—В—Б—П –Є–Ј¬†—Б—Л–≤–Њ—А–Њ—В–Ї–Є –њ—А–Є —Б—В–∞–љ–і–∞—А—В–љ—Л—Е —Г—Б–ї–Њ–≤–Є—П—Е –У–Ф, –≤¬†–љ–µ–Ї–Њ—В–Њ—А–Њ–є —Б—В–µ–њ–µ–љ–Є –≤—Л–≤–Њ–і—П—В—Б—П –њ—А–Є –њ–µ—А–Є—В–Њ–љ–µ–∞–ї—М–љ–Њ–Љ –і–Є–∞–ї–Є–Ј–µ (–Я–Ф). –Ю–і–љ–∞–Ї–Њ, –µ—Б–ї–Є –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є—П –Є–Њ–љ–Њ–≤ –Ьg –≤¬†—А–∞—Б—В–≤–Њ—А–µ –і–ї—П –У–Ф –њ–Њ–≤—Л—И–µ–љ–∞, –Ъ–§–Я–І, –њ–Њ-–≤–Є–і–Є–Љ–Њ–Љ—Г, –њ—А–Њ—Е–Њ–і—П—В —З–µ—А–µ–Ј –і–Є–∞–ї–Є–Ј–љ—Г—О –Љ–µ–Љ–±—А–∞–љ—Г –Є¬†–≤—Л–≤–Њ–і—П—В—Б—П –Є–Ј¬†—Б—Л–≤–Њ—А–Њ—В–Ї–Є –њ–∞—Ж–Є–µ–љ—В–∞ [71]. –≠—В–Њ –Њ–±—К—П—Б–љ—П–µ—В –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ–µ –њ–Њ–≤—Л—И–µ–љ–Є–µ T50 —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤, –њ–Њ–ї—Г—З–∞–≤—И–Є—Е –і–Є–∞–ї–Є–Ј–љ—Л–є —А–∞—Б—В–≤–Њ—А, —Б–Њ–і–µ—А–ґ–∞—Й–Є–є –±–Њ–ї—М—И—Г—О –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є—О Mg2+ –њ–Њ¬†—Б—А–∞–≤–љ–µ–љ–Є—О —Б–Њ¬†—Б—В–∞–љ–і–∞—А—В–љ—Л–Љ —А–∞—Б—В–≤–Њ—А–Њ–Љ [72].
–Т¬†–і–Њ–њ–Њ–ї–љ–µ–љ–Є–µ –Ї¬†–±–ї–∞–≥–Њ–њ—А–Є—П—В–љ–Њ–Љ—Г –≤–ї–Є—П–љ–Є—О –њ–Њ–≤—Л—И–µ–љ–љ–Њ–≥–Њ —Б–Њ–і–µ—А–ґ–∞–љ–Є—П Mg2+ –≤¬†–і–Є–∞–ї–Є–Ј–љ–Њ–Љ —А–∞—Б—В–≤–Њ—А–µ –љ–∞¬†—Б–Ї–ї–Њ–љ–љ–Њ—Б—В—М –Ї¬†–Ї–∞–ї—М—Ж–Є—Д–Є–Ї–∞—Ж–Є–Є —Б—Л–≤–Њ—А–Њ—В–Ї–Є —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–•–С–Я –±—Л–ї–Њ –Њ–±–љ–∞—А—Г–ґ–µ–љ–Њ, —З—В–Њ –Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–љ–Є–µ –љ–µ¬†—Б–Њ–і–µ—А–ґ–∞—Й–µ–≥–Њ –∞—Ж–µ—В–∞—В–∞, –њ–Њ–і–Ї–Є—Б–ї–µ–љ–љ–Њ–≥–Њ —Ж–Є—В—А–∞—В–Њ–Љ –і–Є–∞–ї–Є–Ј–љ–Њ–≥–Њ —А–∞—Б—В–≤–Њ—А–∞ —В–∞–Ї–ґ–µ —Г–≤–µ–ї–Є—З–Є–≤–∞–µ—В T50, —Б–љ–Є–ґ–∞—П —Б–Ї–ї–Њ–љ–љ–Њ—Б—В—М –Ї¬†–Ї–∞–ї—М—Ж–Є—Д–Є–Ї–∞—Ж–Є–Є [73, 74]. –Э–∞–њ—А–Њ—В–Є–≤, –Ъ–§–Я–І –≤¬†—Б—Л–≤–Њ—А–Њ—В–Ї–µ –Ї—А–Њ–≤–Є –њ–Њ–≤—Л—И–∞—О—В—Б—П —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–•–С–Я —В—А–µ—В—М–µ–є-—З–µ—В–≤–µ—А—В–Њ–є —Б—В–∞–і–Є–Є, –њ—А–Є—З–µ–Љ –Њ–љ —Б–∞–Љ—Л–є –≤—Л—Б–Њ–Ї–Є–є —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ –љ–∞¬†–У–Ф [75], –љ–Њ¬†—Б –Љ–µ–љ—М—И–Є–Љ —Б–Њ–і–µ—А–ґ–∞–љ–Є–µ–Љ —Д–µ—В—Г–Є–љ–∞-–Р¬†–њ–Њ –Љ–µ—А–µ –њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є—П –•–С–Я. –Т–µ—А–Њ—П—В–љ–Њ, —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–•–С–Я –Љ–µ—В–∞–±–Њ–ї–Є–Ј–Љ –Ъ–§–Я–І —Г—Б–Ї–Њ—А—П–µ—В—Б—П, –љ–Њ¬†—Д–µ—В—Г–Є–љ-–Р –њ–Њ—В—А–µ–±–ї—П–µ—В—Б—П, –Њ–Ї–∞–Ј—Л–≤–∞—П —Б–Є—Б—В–µ–Љ–љ–Њ–µ –њ—А–Њ—В–Є–≤–Њ–Ї–∞–ї—М—Ж–Є—Д–Є—Ж–Є—А—Г—О—Й–µ–µ –і–µ–є—Б—В–≤–Є–µ, –љ–µ–Њ–±—Е–Њ–і–Є–Љ–Њ–µ –і–ї—П –њ—А–Њ—В–Є–≤–Њ–і–µ–є—Б—В–≤–Є—П –Ъ–° –њ—А–Є —Г—Е—Г–і—И–µ–љ–Є–Є —Д—Г–љ–Ї—Ж–Є–Є –њ–Њ—З–µ–Ї [59].
–І—В–Њ –Ї–∞—Б–∞–µ—В—Б—П –ЪC, —В–Њ –Ъ–§–Я–І –≤¬†—Б—Л–≤–Њ—А–Њ—В–Ї–µ –Ї—А–Њ–≤–Є, –њ–Њ-–≤–Є–і–Є–Љ–Њ–Љ—Г, –≤–µ–і—Г—В —Б–µ–±—П –њ–Њ-—А–∞–Ј–љ–Њ–Љ—Г –≤¬†–Њ—В–љ–Њ—И–µ–љ–Є–Є –і–Є–љ–∞–Љ–Є–Ї–Є —Д–µ—В—Г–Є–љ–∞-A —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–•–С–Я: –±–Њ–ї–µ–µ –≤—Л—Б–Њ–Ї–Є–µ —Г—А–Њ–≤–љ–Є –Ъ–§–Я–І —Б–≤—П–Ј–∞–љ—Л —Б¬†–њ–Њ–≤—Л—И–µ–љ–љ–Њ–є –ґ–µ—Б—В–Ї–Њ—Б—В—М—О –∞–Њ—А—В—Л [19], –∞¬†—Г –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–ЪC –±—Л–ї –Њ–±–љ–∞—А—Г–ґ–µ–љ –±–Њ–ї—М—И–Є–є –і–Є–∞–Љ–µ—В—А –Ъ–§–Я–І-II [67]. –Ъ–∞–Ї –Є¬†—Б–ї–µ–і–Њ–≤–∞–ї–Њ –Њ–ґ–Є–і–∞—В—М, —Г—А–Њ–≤–µ–љ—М –Ґ50 –±—Л–ї –Њ–±—А–∞—В–љ–Њ –њ—А–Њ–њ–Њ—А—Ж–Є–Њ–љ–∞–ї–µ–љ —В—П–ґ–µ—Б—В–Є –Ї–∞–ї—М—Ж–Є—Д–Є–Ї–∞—Ж–Є–Є –Ї–Њ—А–Њ–љ–∞—А–љ—Л—Е –∞—А—В–µ—А–Є–є —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–•–С–Я [37], —В–∞–Ї–Є–Љ –Њ–±—А–∞–Ј–Њ–Љ, —В–µ—Б—В –љ–∞¬†–Ґ50, –њ–Њ-–≤–Є–і–Є–Љ–Њ–Љ—Г, –Є–Љ–Є—В–Є—А—Г–µ—В –Є–Ј–Љ–µ–љ–µ–љ–Є—П —Г—А–Њ–≤–љ—П —Д–µ—В—Г–Є–љ–∞-–Р¬†–≤ —Б—Л–≤–Њ—А–Њ—В–Ї–µ –Ї—А–Њ–≤–Є –≤¬†–Њ—В–љ–Њ—И–µ–љ–Є–Є –ЪC, –њ–Њ—Б–Ї–Њ–ї—М–Ї—Г –±—Л–ї–Њ –Њ–±–љ–∞—А—Г–ґ–µ–љ–Њ, —З—В–Њ –Њ–љ–Є —Б–≤—П–Ј–∞–љ—Л. –£—А–Њ–≤–љ–Є –Ъ–§–Я–І –Њ–±—А–∞—В–љ–Њ –Ї–Њ—А—А–µ–ї–Є—А—Г—О—В —Б¬†–°–Ъ–§¬†[24], –∞¬†–љ–µ —Б¬†T50, –Ї–Њ—В–Њ—А—Л–є –љ–µ¬†–Ј–∞–≤–Є—Б–µ–ї –Њ—В¬†–°–Ъ–§¬†[76]. –І—В–Њ –Ї–∞—Б–∞–µ—В—Б—П –њ–Њ—В–µ—А–Є –Љ–Є–љ–µ—А–∞–ї–Њ–≤ —Б–Ї–µ–ї–µ—В–∞, —В–Њ C.N. Silaghi –Є¬†—Б–Њ–∞–≤—В. –љ–µ¬†–љ–∞—И–ї–Є —Г–±–µ–і–Є—В–µ–ї—М–љ—Л—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є —Б–≤—П–Ј–Є T50 –Ъ–§–Я–І —Б¬†–Љ–Є–љ–µ—А–∞–ї—М–љ–Њ–є –њ–ї–Њ—В–љ–Њ—Б—В—М—О –Ї–Њ—Б—В–љ–Њ–є —В–Ї–∞–љ–Є¬†[77]. –Ю–і–љ–∞–Ї–Њ –њ–Њ–њ—Л—В–Ї–∞ –Њ–±—К—П—Б–љ–Є—В—М –њ–∞—А–∞–і–Њ–Ї—Б –Ї–∞–ї—М—Ж–Є—Д–Є–Ї–∞—Ж–Є–Є –љ–∞¬†–Њ—Б–Є ¬Ђ—Б–Њ—Б—Г–і–Є—Б—В–∞—П —Б–µ—В—М¬†вАУ –Ї–Њ—Б—В—М¬†вАУ –њ–Њ—З–Ї–∞¬ї —В–Њ–ї—М–Ї–Њ —Б¬†—В–Њ—З–Ї–Є –Ј—А–µ–љ–Є—П —Б–Њ–і–µ—А–ґ–∞–љ–Є—П –Ъ–§–Я–І –≤¬†—Д–µ—В—Г–Є–љ–µ-–Р —П–≤–ї—П–µ—В—Б—П –њ–Њ–њ—Л—В–Ї–Њ–є —Г–њ—А–Њ—Й–µ–љ–Є—П. –°¬†—Г—З–µ—В–Њ–Љ —Н—В–Њ–є —В–Њ—З–Ї–Є –Ј—А–µ–љ–Є—П –љ–µ–Њ–±—Е–Њ–і–Є–Љ—Л –±–Њ–ї–µ–µ —Ж–µ–ї–µ–љ–∞–њ—А–∞–≤–ї–µ–љ–љ—Л–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П, —З—В–Њ–±—Л –њ—А–Њ–і–µ–Љ–Њ–љ—Б—В—А–Є—А–Њ–≤–∞—В—М, —З—В–Њ –Ъ–§–Я–І, —Б–Ї–Њ—А–µ–µ –≤—Б–µ–≥–Њ, —П–≤–ї—П—О—В—Б—П –Ї–ї—О—З–Њ–Љ –Ї¬†–њ–Њ–љ–Є–Љ–∞–љ–Є—О —В–Њ–≥–Њ, –Ї–∞–Ї —Д–Є–Ј–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–µ –Њ–Ї–Њ—Б—В–µ–љ–µ–љ–Є–µ —Б–Њ–Њ—В–љ–Њ—Б–Є—В—Б—П —Б¬†–њ–∞—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–Љ –Ї–∞–ї—М—Ж–Є–љ–Њ–Ј–Њ–Љ.
–Т–ї–Є—П–љ–Є–µ –Њ–Ї—Б–Є–≥–Є–і—А–Њ–Ї—Б–Є–і–∞ –ґ–µ–ї–µ–Ј–∞ –љ–∞¬†—Н–љ–і–Њ–≥–µ–љ–љ—Л–µ —З–∞—Б—В–Є—Ж—Л –Ї–∞–ї—М—Ж–Є–њ—А–Њ—В–µ–Є–љ–∞, –≤–Њ—Б–њ–∞–ї–µ–љ–Є–µ –Є¬†—Б–Њ—Б—Г–і–Є—Б—В—Л–µ –Ї–ї–µ—В–Ї–Є —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤, –љ–∞—Е–Њ–і—П—Й–Є—Е—Б—П –љ–∞¬†–і–Є–∞–ї–Є–Ј–µ
–Ы–µ—З–µ–љ–Є–µ –Ь–Ъ–Э –њ—А–Є –•–С–Я —Б¬†–Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–љ–Є–µ–Љ —В—А–∞–і–Є—Ж–Є–Њ–љ–љ—Л—Е —В–µ—А–∞–њ–µ–≤—В–Є—З–µ—Б–Ї–Є—Е –њ–Њ–і—Е–Њ–і–Њ–≤, –Ї–Њ—В–Њ—А—Л–µ –≤–Ї–ї—О—З–∞—О—В –Њ–≥—А–∞–љ–Є—З–µ–љ–Є–µ –њ–Њ—В—А–µ–±–ї–µ–љ–Є—П –† —Б¬†–њ–Є—Й–µ–є, –љ–∞–Ј–љ–∞—З–µ–љ–Є–µ —Д–Њ—Б—Д–∞—В-—Б–≤—П–Ј—Л–≤–∞—О—Й–Є—Е –њ—А–µ–њ–∞—А–∞—В–Њ–≤ (–§–°–Я), –Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–љ–Є–µ –∞–Ї—В–Є–≤–љ–Њ–≥–Њ –≤–Є—В–∞–Љ–Є–љ–∞ D –Є¬†–Ї–∞–ї—М—Ж–Є–Љ–Є–Љ–µ—В–Є–Ї–Њ–≤ (–≤–ї–Є—П–љ–Є–µ –љ–∞¬†–Ї–Њ—Б—В–љ—Л–є –Љ–µ—В–∞–±–Њ–ї–Є–Ј–Љ), –∞–і–µ–Ї–≤–∞—В–љ—Л–є –і–Є–∞–ї–Є–Ј —Г –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б –Ґ–Я–Э –Љ–Њ–≥—Г—В –Ј–∞–Љ–µ–і–ї–Є—В—М –њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є–µ –≤–љ–µ–Ї–Њ—Б—В–љ–Њ–є –Ї–∞–ї—М—Ж–Є—Д–Є–Ї–∞—Ж–Є–Є, –љ–Њ¬†—Н—В–Є –њ–Њ–і—Е–Њ–і—Л –Њ—Б—В–∞—О—В—Б—П —Б–њ–Њ—А–љ—Л–Љ–Є, –њ–Њ—Б–Ї–Њ–ї—М–Ї—Г —А–µ–Ї–Њ–Љ–µ–љ–і—Г–µ–Љ—Л–µ –±–Є–Њ—Е–Є–Љ–Є—З–µ—Б–Ї–Є–µ –њ–Њ–Ї–∞–Ј–∞—В–µ–ї–Є —В—А—Г–і–љ–Њ–і–Њ—Б—В–Є–ґ–Є–Љ—Л. –С–Њ–ї–µ–µ –њ–Њ–ї–Њ–≤–Є–љ—Л –њ–∞—Ж–Є–µ–љ—В–Њ–≤, –љ–∞—Е–Њ–і—П—Й–Є—Е—Б—П –љ–∞¬†–і–Є–∞–ї–Є–Ј–µ, –Є–Љ–µ—О—В –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є—О –† –≤¬†—Б—Л–≤–Њ—А–Њ—В–Ї–µ –Ї—А–Њ–≤–Є –≤—Л—И–µ —Ж–µ–ї–µ–≤—Л—Е –Ј–љ–∞—З–µ–љ–Є–є –Є¬†–љ—Г–ґ–і–∞—О—В—Б—П –≤¬†–ї–µ—З–µ–љ–Є–Є –§–°–Я –і–ї—П —Б–љ–Є–ґ–µ–љ–Є—П –≤—Б–∞—Б—Л–≤–∞–љ–Є—П –† –≤¬†–ґ–µ–ї—Г–і–Њ—З–љ–Њ-–Ї–Є—И–µ—З–љ–Њ–Љ —В—А–∞–Ї—В–µ –Є¬†–і–Њ—Б—В–Є–ґ–µ–љ–Є—П –Ї–Њ–љ—В—А–Њ–ї—П —Г—А–Њ–≤–љ—П –† –≤¬†—Б—Л–≤–Њ—А–Њ—В–Ї–µ –Ї—А–Њ–≤–Є. –Ф–∞–љ–љ—Л–µ –њ–µ—А–µ–Ї—А–µ—Б—В–љ—Л—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є –њ–Њ–Ї–∞–Ј—Л–≤–∞—О—В, —З—В–Њ —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤, –љ–∞—Е–Њ–і—П—Й–Є—Е—Б—П –љ–∞¬†–і–Є–∞–ї–Є–Ј–µ, –њ–Њ–ї—Г—З–∞—О—Й–Є—Е —В–µ—А–∞–њ–Є—О –§–°–Я, –њ–Њ–Ї–∞–Ј–∞—В–µ–ї–Є —Б–Љ–µ—А—В–љ–Њ—Б—В–Є –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ –љ–Є–ґ–µ –њ–Њ¬†—Б—А–∞–≤–љ–µ–љ–Є—О —Б¬†—В–µ–Љ–Є, –Ї—В–Њ –љ–µ¬†–њ–Њ–ї—Г—З–∞–µ—В –§–°–Я, –і–∞–ґ–µ –њ–Њ—Б–ї–µ —Б–Њ–њ–Њ—Б—В–∞–≤–ї–µ–љ–Є—П –њ–Њ–Ї–∞–Ј–∞—В–µ–ї–µ–є —Б–Ї–ї–Њ–љ–љ–Њ—Б—В–Є –Є¬†–Ї–Њ—А—А–µ–Ї—В–Є—А–Њ–≤–Ї–Є —Б—В–∞—В—Г—Б–∞ –њ–Є—В–∞–љ–Є—П [78, 79].
–Ъ—А—Г–њ–љ—Л–µ —Н–њ–Є–і–µ–Љ–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П –њ–Њ–і—В–≤–µ—А–ґ–і–∞—О—В, —З—В–Њ –њ–Њ–≤—Л—И–µ–љ–љ—Л–µ –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є–Є –† –≤¬†—Б—Л–≤–Њ—А–Њ—В–Ї–µ –Ї—А–Њ–≤–Є –Є–і–µ–љ—В–Є—Д–Є—Ж–Є—А–Њ–≤–∞–љ—Л –Ї–∞–Ї –Њ—Б–љ–Њ–≤–љ–Њ–є –љ–µ—В—А–∞–і–Є—Ж–Є–Њ–љ–љ—Л–є —Д–∞–Ї—В–Њ—А —А–Є—Б–Ї–∞ –°–°–Ч –Є¬†—Б–Љ–µ—А—В–љ–Њ—Б—В–Є –Њ—В¬†–≤—Б–µ—Е –њ—А–Є—З–Є–љ —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–•–С–Я, –Њ—Б–Њ–±–µ–љ–љ–Њ –љ–∞—Е–Њ–і—П—Й–Є—Е—Б—П –љ–∞¬†–і–Є–∞–ї–Є–Ј–µ. –Т¬†—Н—В–Њ–Љ –Ї–Њ–љ—В–µ–Ї—Б—В–µ –Љ–Њ–ґ–љ–Њ –њ—А–µ–і–њ–Њ–ї–Њ–ґ–Є—В—М, —З—В–Њ –Ї–Њ–љ—В—А–Њ–ї—М —Г—А–Њ–≤–љ—П –† –≤¬†—Б—Л–≤–Њ—А–Њ—В–Ї–µ –Ї—А–Њ–≤–Є –Љ–Њ–ґ–µ—В –њ—А–Є–≤–µ—Б—В–Є –Ї¬†–Љ–µ–љ—М—И–µ–є –≤–љ–µ–Ї–Њ—Б—В–љ–Њ–є –Ї–∞–ї—М—Ж–Є—Д–Є–Ї–∞—Ж–Є–Є –Є¬†–љ–µ–±–ї–∞–≥–Њ–њ—А–Є—П—В–љ—Л–Љ –Є—Б—Е–Њ–і–∞–Љ, –Є¬†—Н—В–Њ –±—Л–ї–Њ –Њ—В–ї–Є—З–Є—В–µ–ї—М–љ–Њ–є —З–µ—А—В–Њ–є —В–µ—А–∞–њ–µ–≤—В–Є—З–µ—Б–Ї–Њ–є —Б—В—А–∞—В–µ–≥–Є–Є —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–•–С–Я –љ–∞¬†–њ—А–Њ—В—П–ґ–µ–љ–Є–Є –Љ–љ–Њ–≥–Є—Е –ї–µ—В. –Э–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В—М —Б—Г—Й–µ—Б—В–≤—Г—О—Й–Є—Е —Б—В—А–∞—В–µ–≥–Є–є —А–µ–≥—Г–ї–Є—А–Њ–≤–∞–љ–Є—П —Г—А–Њ–≤–љ—П –† –і–ї—П –њ–Њ—Б–ї–µ–і–Њ–≤–∞—В–µ–ї—М–љ–Њ–≥–Њ –і–Њ—Б—В–Є–ґ–µ–љ–Є—П –Є¬†–њ–Њ–і–і–µ—А–ґ–∞–љ–Є—П —Ж–µ–ї–µ–≤—Л—Е —Г—А–Њ–≤–љ–µ–є —Е–Њ—А–Њ—И–Њ –Ј–∞–і–Њ–Ї—Г–Љ–µ–љ—В–Є—А–Њ–≤–∞–љ–∞ [80].
–Ю–Ї—Б–Є–≥–Є–і—А–Њ–Ї—Б–Є–і –ґ–µ–ї–µ–Ј–∞ (–Ю–Ц) (Velphoro¬Ѓ)¬†вАУ —Б–Є–ї—М–љ–Њ–і–µ–є—Б—В–≤—Г—О—Й–Є–є –§–°–Я –љ–∞¬†–Њ—Б–љ–Њ–≤–µ –ґ–µ–ї–µ–Ј–∞, —Б–Њ—Б—В–Њ–Є—В –Є–Ј¬†—Б–Љ–µ—Б–Є —Б–∞—Е–∞—А–Њ–Ј—Л, –Ї—А–∞—Е–Љ–∞–ї–Њ–≤ –Є¬†–∞–Ї—В–Є–≤–љ–Њ–≥–Њ –Ї–Њ–Љ–њ–Њ–љ–µ–љ—В–∞, –Љ–љ–Њ–≥–Њ—П–і–µ—А–љ–Њ–≥–Њ –Ю–Ц(III) —Б¬†–љ–Є–Ј–Ї–Њ–є —Б—Г—В–Њ—З–љ–Њ–є –і–Њ–Ј–Њ–є —В–∞–±–ї–µ—В–Њ–Ї, –Њ–і–Њ–±—А–µ–љ–љ—Л–є –і–ї—П –Ї–Њ–љ—В—А–Њ–ї—П –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є–Є –† –≤¬†—Б—Л–≤–Њ—А–Њ—В–Ї–µ –Ї—А–Њ–≤–Є —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–•–С–Я, –љ–∞—Е–Њ–і—П—Й–Є—Е—Б—П –љ–∞¬†–і–Є–∞–ї–Є–Ј–µ [81].
–≠—Д—Д–µ–Ї—В–Є–≤–љ–Њ—Б—В—М —В–µ—А–∞–њ–Є–Є –Ю–Ц –≤¬†—А–µ–∞–ї—М–љ—Л—Е –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є—Е —Г—Б–ї–Њ–≤–Є—П—Е –±—Л–ї–∞ —В—Й–∞—В–µ–ї—М–љ–Њ –Њ—Ж–µ–љ–µ–љ–∞ –≤¬†—А—П–і–µ –љ–∞–±–ї—О–і–∞—В–µ–ї—М–љ—Л—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є, –њ—А–Њ–≤–µ–і–µ–љ–љ—Л—Е —Б¬†—Г—З–∞—Б—В–Є–µ–Љ –±–Њ–ї—М—И–Њ–≥–Њ —З–Є—Б–ї–∞ –њ–∞—Ж–Є–µ–љ—В–Њ–≤, –љ–∞—Е–Њ–і—П—Й–Є—Е—Б—П –љ–∞¬†–У–Ф (> 6400) –Є¬†–Я–Ф (~ 500) –≤¬†–°–®–Р –Є¬†–Х–≤—А–Њ–њ–µ, –Ї–∞–Ї –≤¬†—А–µ—В—А–Њ—Б–њ–µ–Ї—В–Є–≤–љ—Л—Е, —В–∞–Ї –Є¬†–≤ –њ—А–Њ—Б–њ–µ–Ї—В–Є–≤–љ—Л—Е –Ї–Њ–≥–Њ—А—В–∞—Е –Є¬†—А–∞–љ–і–Њ–Љ–Є–Ј–Є—А–Њ–≤–∞–љ–љ—Л—Е –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П—Е (–†–Ъ–Ш). –Т¬†—Б–Њ–Њ—В–≤–µ—В—Б—В–≤–Є–Є —Б¬†–і–∞–љ–љ—Л–Љ–Є –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є, —А–µ–∞–ї—М–љ—Л–µ –љ–∞–±–ї—О–і–∞—В–µ–ї—М–љ—Л–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П –њ—А–Њ–і–µ–Љ–Њ–љ—Б—В—А–Є—А–Њ–≤–∞–ї–Є, —З—В–Њ –Ю–Ц –Љ–Њ–ґ–µ—В —Н—Д—Д–µ–Ї—В–Є–≤–љ–Њ —Б–љ–Є–ґ–∞—В—М —Г—А–Њ–≤–µ–љ—М –† –≤¬†—Б—Л–≤–Њ—А–Њ—В–Ї–µ –Ї—А–Њ–≤–Є –њ—А–Є –Љ–µ–љ—М—И–µ–Љ –µ–ґ–µ–і–љ–µ–≤–љ–Њ–Љ –њ—А–Є–µ–Љ–µ —В–∞–±–ї–µ—В–Њ–Ї, —З–µ–Љ –±–Њ–ї—М—И–Є–љ—Б—В–≤–Њ –і—А—Г–≥–Є—Е –§–°–Я. –≠—В–Є –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П —В–∞–Ї–ґ–µ –њ–Њ–Ї–∞–Ј–∞–ї–Є, —З—В–Њ –Ю–Ц –Њ–±–µ—Б–њ–µ—З–Є–≤–∞–µ—В —Н—Д—Д–µ–Ї—В–Є–≤–љ—Л–є –Ї–Њ–љ—В—А–Њ–ї—М —Г—А–Њ–≤–љ—П –† –≤¬†—Б—Л–≤–Њ—А–Њ—В–Ї–µ –Ї—А–Њ–≤–Є –њ—А–Є —А–∞–Ј–ї–Є—З–љ—Л—Е —Г—Б–ї–Њ–≤–Є—П—Е –ї–µ—З–µ–љ–Є—П, –≤¬†—В–Њ–Љ —З–Є—Б–ї–µ –≤¬†–Ї–∞—З–µ—Б—В–≤–µ –Љ–Њ–љ–Њ—В–µ—А–∞–њ–Є–Є —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤, –љ–µ¬†–њ—А–Є–љ–Є–Љ–∞—О—Й–Є—Е –§–°–Я –Є–ї–Є –њ–µ—А–µ—Е–Њ–і—П—Й–Є—Е –љ–∞¬†–Ю–Ц —Б¬†–і—А—Г–≥–Є—Е –§–°–Я, –Є–ї–Є –њ—А–Є –Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–љ–Є–Є –≤¬†–Ї–Њ–Љ–±–Є–љ–∞—Ж–Є–Є —Б¬†–і—А—Г–≥–Є–Љ–Є –§–°–Я, –Њ–±–ї–∞–і–∞–µ—В –≤—Л—Б–Њ–Ї–Њ–є —Д–Њ—Б—Д–∞—В-—Б–≤—П–Ј—Л–≤–∞—О—Й–µ–є —Б–њ–Њ—Б–Њ–±–љ–Њ—Б—В—М—О. –≠—В–Є –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П —Г–Ї–∞–Ј—Л–≤–∞—О—В –љ–∞¬†–±–ї–∞–≥–Њ–њ—А–Є—П—В–љ—Л–є –њ—А–Њ—Д–Є–ї—М –±–µ–Ј–Њ–њ–∞—Б–љ–Њ—Б—В–Є –Є¬†–њ–µ—А–µ–љ–Њ—Б–Є–Љ–Њ—Б—В–Є, –∞¬†—В–∞–Ї–ґ–µ –љ–∞¬†–Љ–Є–љ–Є–Љ–∞–ї—М–љ—Г—О, –µ—Б–ї–Є —В–∞–Ї–Њ–≤–∞—П –Є–Љ–µ–µ—В—Б—П, —Б–Є—Б—В–µ–Љ–љ—Г—О –∞–±—Б–Њ—А–±—Ж–Є—О –ґ–µ–ї–µ–Ј–∞ [82].
–Ы–µ—З–µ–љ–Є–µ –Ю–Ц –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ —Б–љ–Є–ґ–∞–ї–Њ —Г—А–Њ–≤–µ–љ—М FGF-23 –≤¬†—Б—Л–≤–Њ—А–Њ—В–Ї–µ –Ї—А–Њ–≤–Є –≤¬†–±–Њ–ї—М—И–µ–є —Б—В–µ–њ–µ–љ–Є, —З–µ–Љ –Ї–∞—А–±–Њ–љ–∞—В –Ї–∞–ї—М—Ж–Є—П, –Є¬†–њ—А–µ–≤–Њ—Б—Е–Њ–і–Є–ї–Њ –Ї–∞—А–±–Њ–љ–∞—В –Ї–∞–ї—М—Ж–Є—П –≤¬†–њ—А–µ–і–Њ—В–≤—А–∞—Й–µ–љ–Є–Є —А–∞–Ј–≤–Є—В–Є—П –Ъ–°, –∞¬†—В–∞–Ї–ґ–µ –Њ–Ї–∞–Ј—Л–≤–∞–ї–Њ –±–ї–∞–≥–Њ–њ—А–Є—П—В–љ–Њ–µ –≤–ї–Є—П–љ–Є–µ –љ–∞¬†–Љ–µ—В–∞–±–Њ–ї–Є–Ј–Љ –Ї–Њ—Б—В–љ–Њ–є —В–Ї–∞–љ–Є [83вАУ87]. –Ф—А—Г–≥–Є–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П, –њ—А–Њ–≤–µ–і–µ–љ–љ—Л–µ —Б¬†–њ—А–Є–Љ–µ–љ–µ–љ–Є–µ–Љ –Ю–Ц, –њ–Њ–Ї–∞–Ј–∞–ї–Є –µ–≥–Њ —Н—Д—Д–µ–Ї—В–Є–≤–љ–Њ—Б—В—М –≤¬†—Б–љ–Є–ґ–µ–љ–Є–Є –ЪC –≤¬†–∞–і–µ–љ–Є–љ–Њ–≤–Њ–є –Љ–Њ–і–µ–ї–Є –•–Я–Э. –°—А–∞–≤–љ–µ–љ–Є–µ O–Ц –Є¬†–§–°–Я –љ–∞¬†–Њ—Б–љ–Њ–≤–µ –Ї–∞–ї—М—Ж–Є—П (CaCO3) –њ–Њ–Ї–∞–Ј–∞–ї–Њ, —З—В–Њ –Њ–±–∞ –Љ–µ—В–Њ–і–∞ –ї–µ—З–µ–љ–Є—П –Є–Љ–µ–ї–Є —Б—Е–Њ–і–љ—Л–є —Н—Д—Д–µ–Ї—В –≤¬†—Б–љ–Є–ґ–µ–љ–Є–Є P –≤¬†—Б—Л–≤–Њ—А–Њ—В–Ї–µ –Ї—А–Њ–≤–Є, –љ–Њ¬†–Ю–Ц –Њ–±–ї–∞–і–∞–ї –±–Њ–ї—М—И–µ–є —Н—Д—Д–µ–Ї—В–Є–≤–љ–Њ—Б—В—М—О –≤¬†—Б–љ–Є–ґ–µ–љ–Є–Є FGF-23 –Є¬†–њ—А–µ–і–Њ—В–≤—А–∞—Й–µ–љ–Є–Є —А–∞–Ј–≤–Є—В–Є—П –≤–љ–µ–Ї–Њ—Б—В–љ–Њ–є –Ї–∞–ї—М—Ж–Є—Д–Є–Ї–∞—Ж–Є–Є [83].
–°–Є—Б—В–µ–Љ–∞—В–Є—З–µ—Б–Ї–Є–є –Њ–±–Ј–Њ—А –Є¬†–Љ–µ—В–∞–∞–љ–∞–ї–Є–Ј, –≤–Ї–ї—О—З–∞—О—Й–Є–µ 36 –†–Ъ–Ш –Є¬†–њ—П—В—М –њ—А–Њ—Б–њ–µ–Ї—В–Є–≤–љ—Л—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є (7590¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤), –њ—А–Њ–і–µ–Љ–Њ–љ—Б—В—А–Є—А–Њ–≤–∞–ї–Є, —З—В–Њ –Њ–≥—А–∞–љ–Є—З–µ–љ–Є–µ¬†–† —Б¬†–њ–Є—Й–µ–є –Љ–µ–љ–µ–µ 800 –Љ–≥ –≤¬†—Б—Г—В–Ї–Є –і–∞–ї–Њ –љ–µ–Ј–љ–∞—З–Є—В–µ–ї—М–љ—Л–є —Н—Д—Д–µ–Ї—В –љ–∞¬†—Б–љ–Є–ґ–µ–љ–Є–µ FGF-23, –≤¬†—В–Њ –≤—А–µ–Љ—П –Ї–∞–Ї —Б—А–µ–і–Є –§–°–Я, –љ–µ¬†—Б–Њ–і–µ—А–ґ–∞—Й–Є—Е –Ї–∞–ї—М—Ж–Є–є, –≤–Ї–ї—О—З–∞—О—Й–Є—Е –Є¬†–Ю–Ц, –≤—Л—П–≤–ї–µ–љ–Њ —Н—Д—Д–µ–Ї—В–Є–≤–љ–Њ–µ —Б–љ–Є–ґ–µ–љ–Є–µ FGF-23 —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–Ґ–Я–Э [88].
–Т¬†–Љ–љ–Њ–≥–Њ—Ж–µ–љ—В—А–Њ–≤–Њ–Љ –†–Ъ–Ш —Б¬†–і–≤—Г—Е—Д–∞–Ї—В–Њ—А–љ—Л–Љ –і–Є–Ј–∞–є–љ–Њ–Љ EPISODE –њ—А–Њ–≤–µ–і–µ–љ–Њ —Б—А–∞–≤–љ–µ–љ–Є–µ –≤–ї–Є—П–љ–Є—П –љ–∞¬†–њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є–µ –Ї–∞–ї—М—Ж–Є—Д–Є–Ї–∞—Ж–Є–Є –Ї–Њ—А–Њ–љ–∞—А–љ—Л—Е –∞—А—В–µ—А–Є–є (KKA) –Ю–Ц –Є–ї–Є –Ї–∞—А–±–Њ–љ–∞—В–∞ –ї–∞–љ—В–∞–љ–∞ (–Ъ–Ы) –≤¬†–і–≤—Г—Е —А–∞–Ј–ї–Є—З–љ—Л—Е —Ж–µ–ї–µ–≤—Л—Е –і–Є–∞–њ–∞–Ј–Њ–љ–∞—Е –† (3,5вАУ4,5 –Љ–≥/–і–ї –≤¬†–≥—А—Г–њ–њ–µ —Б—В—А–Њ–≥–Њ–≥–Њ –Ї–Њ–љ—В—А–Њ–ї—П –Є¬†5,0вАУ6,0 –Љ–≥/–і–ї –≤¬†—Б—В–∞–љ–і–∞—А—В–љ–Њ–є¬†–≥—А—Г–њ–њ–µ)¬†[89]. –Я–µ—А–≤–Є—З–љ–Њ–є –Ї–Њ–љ–µ—З–љ–Њ–є —В–Њ—З–Ї–Њ–є –±—Л–ї–Њ –њ—А–Њ—Ж–µ–љ—В–љ–Њ–µ (%) –Є–Ј–Љ–µ–љ–µ–љ–Є–µ –њ–Њ–Ї–∞–Ј–∞—В–µ–ї–µ–є –Ъ–Ъ–Р –њ–Њ¬†–і–∞–љ–љ—Л–Љ –Љ—Г–ї—М—В–Є–і–µ—В–µ–Ї—В–Њ—А–љ–Њ–є –Ї–Њ–Љ–њ—М—О—В–µ—А–љ–Њ–є —В–Њ–Љ–Њ–≥—А–∞—Д–Є–Є –љ–∞¬†–Њ—Б–љ–Њ–≤–µ –Љ–µ—В–Њ–і–∞ –Р. Agatston –Є¬†—Б–Њ–∞–≤—В. —З–µ—А–µ–Ј 12 –Љ–µ—Б—П—Ж–µ–≤ –њ–Њ—Б–ї–µ –ї–µ—З–µ–љ–Є—П. –Т—В–Њ—А–Є—З–љ—Л–µ –Ї–Њ–љ–µ—З–љ—Л–µ —В–Њ—З–Ї–Є –≤–Ї–ї—О—З–∞–ї–Є –∞–±—Б–Њ–ї—О—В–љ–Њ–µ –Є–Ј–Љ–µ–љ–µ–љ–Є–µ –њ–Њ–Ї–∞–Ј–∞—В–µ–ї–µ–є –Ъ–Ъ–Р –Є¬†–і–Њ–ї—О –њ–∞—Ж–Є–µ–љ—В–Њ–≤, –і–Њ—Б—В–Є–≥—И–Є—Е —Ж–µ–ї–µ–≤–Њ–≥–Њ —Г—А–Њ–≤–љ—П —Д–Њ—Б—Д–∞—В–Њ–≤ –≤¬†—Б—Л–≤–Њ—А–Њ—В–Ї–µ –Ї—А–Њ–≤–Є –Ї¬†–Ї–Њ–љ—Ж—Г –њ–µ—А–Є–Њ–і–∞ –ї–µ—З–µ–љ–Є—П. –Т¬†—Н—В–Њ–Љ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–Є –њ—А–Њ—Ж–µ–љ—В–љ–Њ–µ –Є–Ј–Љ–µ–љ–µ–љ–Є–µ –њ–Њ–Ї–∞–Ј–∞—В–µ–ї–µ–є –Ъ–Ъ–Р –≤¬†–≥—А—Г–њ–њ–µ —Б—В—А–Њ–≥–Њ–≥–Њ –Ї–Њ–љ—В—А–Њ–ї—П –† (–Љ–µ–і–Є–∞–љ–∞ (–Ь–µ) 8,52; –Љ–µ–ґ–Ї–≤–∞—А—В–Є–ї—М–љ—Л–є –і–Є–∞–њ–∞–Ј–Њ–љ (IQR) 1,0вАУ23,9) –±—Л–ї–Њ –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ –љ–Є–ґ–µ, —З–µ–Љ –≤¬†—Б—В–∞–љ–і–∞—А—В–љ–Њ–є¬†–≥—А—Г–њ–њ–µ (–Ь–µ 21,8; IQR¬†вАУ 10,0¬†вАУ 36,1; p =¬†0,006). –≠—В–Њ—В —Н—Д—Д–µ–Ї—В –±—Л–ї –±–Њ–ї–µ–µ –≤—Л—А–∞–ґ–µ–љ —Г¬†–њ–Њ–ґ–Є–ї—Л—Е –њ–∞—Ж–Є–µ–љ—В–Њ–≤ (65вАУ74 –ї–µ—В) –њ–Њ¬†—Б—А–∞–≤–љ–µ–љ–Є—О —Б¬†–±–Њ–ї–µ–µ –Љ–Њ–ї–Њ–і—Л–Љ–Є (20вАУ64¬†–ї–µ—В), —А = 0,003. –Р–љ–∞–ї–Њ–≥–Є—З–љ–Њ –њ—А–Њ—Ж–µ–љ—В–љ–Њ–Љ—Г –Є–Ј–Љ–µ–љ–µ–љ–Є—О –∞–±—Б–Њ–ї—О—В–љ–Њ–µ –Є–Ј–Љ–µ–љ–µ–љ–Є–µ –њ–Њ–Ї–∞–Ј–∞—В–µ–ї–µ–є –Ъ–Ъ–Р –±—Л–ї–Њ –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ –љ–Є–ґ–µ –≤¬†–≥—А—Г–њ–њ–µ —Б—В—А–Њ–≥–Њ–≥–Њ –Ї–Њ–љ—В—А–Њ–ї—П (–Ь–µ 66,1; IQR 3,8вАУ220,1) –≤¬†—Б—А–∞–≤–љ–µ–љ–Є–Є —Б–Њ¬†—Б—В–∞–љ–і–∞—А—В–љ–Њ–є¬†–≥—А—Г–њ–њ–Њ–є (–Ь–µ 125,9; IQR 66,6вАУ321,0; p = 0,01). –°–ї–µ–і—Г–µ—В –Њ—В–Љ–µ—В–Є—В—М, —З—В–Њ –∞–±—Б–Њ–ї—О—В–љ–Њ–µ –Є–Ј–Љ–µ–љ–µ–љ–Є–µ –њ–Њ–Ї–∞–Ј–∞—В–µ–ї–µ–є –Ъ–Ъ–Р –±—Л–ї–Њ –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ –љ–Є–ґ–µ –≤¬†–≥—А—Г–њ–њ–µ –Ю–Ц (–Ь–µ 74,4; IQR 9,8вАУ173,3), —З–µ–Љ –≤¬†–≥—А—Г–њ–њ–µ –Ъ–Ы (131,5; IQR 26,9вАУ314,5; p = 0,03). –Ф–Њ–ї—П –њ–∞—Ж–Є–µ–љ—В–Њ–≤, —Г¬†–Ї–Њ—В–Њ—А—Л—Е —З–µ—А–µ–Ј 12 –Љ–µ—Б—П—Ж–µ–≤ –њ–Њ—Б–ї–µ –ї–µ—З–µ–љ–Є—П —Б–љ–Є–Ј–Є–ї–Є—Б—М –њ–Њ–Ї–∞–Ј–∞—В–µ–ї–Є –Ъ–Ъ–Р, –±—Л–ї–∞ –±–Њ–ї—М—И–µ –≤¬†–≥—А—Г–њ–њ–µ –Ю–Ц (28,3%) –≤¬†—Б—А–∞–≤–љ–µ–љ–Є–Є —Б¬†–Ъ–Ы (14,5%),¬†–≥—А—Г–њ–њ–∞–Љ–Є —Б—В—А–Њ–≥–Њ–≥–Њ –Є¬†—Б—В–∞–љ–і–∞—А—В–љ–Њ–≥–Њ –Ї–Њ–љ—В—А–Њ–ї—П (27,6 –Є¬†14,0%) —Б–Њ–Њ—В–≤–µ—В—Б—В–≤–µ–љ–љ–Њ. –°–ї–µ–і—Г–µ—В –Њ—В–Љ–µ—В–Є—В—М, —З—В–Њ –≤¬†–Њ—Ж–µ–љ–Ї–µ –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є –Ј–љ–∞—З–Є–Љ—Л—Е —А–∞–Ј–ї–Є—З–Є–є –≤–ї–Є—П–љ–Є—П –і–≤—Г—Е —В–Є–њ–Њ–≤ –±–µ–Ј –Ї–∞–ї—М—Ж–Є–µ–≤—Л—Е –§–°–Я –љ–∞¬†–Ъ–° –љ–µ–ї—М–Ј—П –Є—Б–Ї–ї—О—З–Є—В—М –Њ–≥—А–∞–љ–Є—З–µ–љ–Є—П –њ–Њ¬†—А–∞–Ј–Љ–µ—А—Г –≤—Л–±–Њ—А–Ї–Є.
–Ф–∞–љ–љ–Њ–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ –њ—А–Њ–і–µ–Љ–Њ–љ—Б—В—А–Є—А–Њ–≤–∞–ї–Њ, —З—В–Њ —Б—В—А–Њ–≥–Є–є –Ї–Њ–љ—В—А–Њ–ї—М —Г—А–Њ–≤–љ—П –† –≤¬†–њ—А–µ–і–µ–ї–∞—Е –љ–Њ—А–Љ–∞–ї—М–љ–Њ–≥–Њ –і–Є–∞–њ–∞–Ј–Њ–љ–∞ —Б¬†–Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–љ–Є–µ–Љ –§–°–Я —Н—Д—Д–µ–Ї—В–Є–≤–µ–љ –≤¬†–Ј–∞–Љ–µ–і–ї–µ–љ–Є–Є –Ъ–° —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤, –љ–∞—Е–Њ–і—П—Й–Є—Е—Б—П –љ–∞¬†–У–Ф.
–Ю–±—К—П—Б–љ–µ–љ–Є–µ–Љ —Н—Д—Д–µ–Ї—В–Є–≤–љ–Њ—Б—В–Є –§–°–Я –Љ–Њ–ґ–µ—В –њ–Њ—Б–ї—Г–ґ–Є—В—М –Є—Е –≤–ї–Є—П–љ–Є–µ –љ–∞¬†–Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є–µ –Ъ–§–Я–І, –Ї–Њ—В–Њ—А–Њ–µ —А–∞—Б—Б–Љ–∞—В—А–Є–≤–∞–µ—В—Б—П –Ї–∞–Ї –Ј–∞—Й–Є—В–љ—Л–є –Љ–µ—Е–∞–љ–Є–Ј–Љ –њ—А–Њ—В–Є–≤ –љ–µ–ґ–µ–ї–∞—В–µ–ї—М–љ–Њ–≥–Њ —А–Њ—Б—В–∞ –Ї—А–Є—Б—В–∞–ї–ї–Њ–≤ —Д–Њ—Б—Д–∞—В–∞ –Ї–∞–ї—М—Ж–Є—П –≤¬†–Ї—А–Њ–≤–Є –Є¬†–ґ–Є–і–Ї–Њ—Б—В–Є –њ–Њ—З–µ—З–љ—Л—Е –Ї–∞–љ–∞–ї—М—Ж–µ–≤ –њ—А–Є –њ–µ—А–µ–≥—А—Г–Ј–Ї–µ –†.¬†–Ъ–∞–Ї –±—Л–ї–Њ –Њ–њ–Є—Б–∞–љ–Њ –≤—Л—И–µ, –Ъ–§–Я–І –њ—А–µ–і—Б—В–∞–≤–ї—П—О—В —Б–Њ–±–Њ–є –њ–Њ–ї–Є–і–Є—Б–њ–µ—А—Б–љ—Л–µ –Ї–Њ–ї–ї–Њ–Є–і—Л, –Ї–ї–∞—Б—Б–Є—Д–Є—Ж–Є—А—Г—О—В—Б—П –љ–∞¬†–Њ—Б–љ–Њ–≤–µ –њ–ї–Њ—В–љ–Њ—Б—В–Є –Є¬†–Ї—А–Є—Б—В–∞–ї–ї–Є—З–љ–Њ—Б—В–Є —Д–Њ—Б—Д–∞—В–∞ –Ї–∞–ї—М—Ж–Є—П. –Ъ–§–Я–І –љ–Є–Ј–Ї–Њ–є –њ–ї–Њ—В–љ–Њ—Б—В–Є, —Б–Њ–і–µ—А–ґ–∞—Й–Є–µ –∞–Љ–Њ—А—Д–љ—Л–є (–љ–µ–Ї—А–Є—Б—В–∞–ї–ї–Є—З–µ—Б–Ї–Є–є) —Д–Њ—Б—Д–∞—В –Ї–∞–ї—М—Ж–Є—П, —Д—Г–љ–Ї—Ж–Є–Њ–љ–Є—А—Г—О—В –Ї–∞–Ї –Є–љ–і—Г–Ї—В–Њ—А —Н–Ї—Б–њ—А–µ—Б—Б–Є–Є FGF-23 –≤¬†–Њ—Б—В–µ–Њ–±–ї–∞—Б—В–∞—Е –Є¬†–њ–µ—А–µ–љ–Њ—Б—З–Є–Ї —Д–Њ—Б—Д–∞—В–∞ –Ї–∞–ї—М—Ж–Є—П –≤¬†–Ї–Њ—Б—В—М. –Ю–і–љ–∞–Ї–Њ –њ–Њ—Б–ї–µ —В—А–∞–љ—Б—Д–Њ—А–Љ–∞—Ж–Є–Є –≤¬†–Ъ–§–Я–І –≤—Л—Б–Њ–Ї–Њ–є –њ–ї–Њ—В–љ–Њ—Б—В–Є, —Б–Њ–і–µ—А–ґ–∞—Й–Є–є –Ї—А–Є—Б—В–∞–ї–ї–Є—З–µ—Б–Ї–Є–є —Д–Њ—Б—Д–∞—В –Ї–∞–ї—М—Ж–Є—П, –Ъ–§–Я–І —Б—В–∞–љ–Њ–≤—П—В—Б—П —Ж–Є—В–Њ—В–Њ–Ї—Б–Є—З–љ—Л–Љ–Є, –≤—Л–Ј—Л–≤–∞—П –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є–µ –њ–Њ—З–µ—З–љ—Л—Е –Ї–∞–љ–∞–ї—М—Ж–µ–≤, —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–µ –≤–Њ—Б–њ–∞–ї–µ–љ–Є–µ, –Ї–∞–ї—М—Ж–Є—Д–Є–Ї–∞—Ж–Є—О –У–Ь–Ъ–° –Є¬†–≤—А–Њ–ґ–і–µ–љ–љ—Л–є –Є–Љ–Љ—Г–љ–љ—Л–є –Њ—В–≤–µ—В –Љ–∞–Ї—А–Њ—Д–∞–≥–Њ–≤. –Ъ–§–Я–І, —Д–Њ—А–Љ–Є—А—Г—О—Й–Є–µ—Б—П –њ—А–Є –њ–µ—А–µ–≥—А—Г–Ј–Ї–µ –†,¬†—П–≤–ї—П—О—В—Б—П –њ–Њ—В–µ–љ—Ж–Є–∞–ї—М–љ—Л–Љ —А–∞–љ–љ–Є–Љ –±–Є–Њ–Љ–∞—А–Ї–µ—А–Њ–Љ –Ь–Ъ–Э –Є¬†–њ—А–µ–і—Б—В–∞–≤–ї—П—О—В —Б–Њ–±–Њ–є –Љ–Њ–і–Є—Д–Є—Ж–Є—А—Г–µ–Љ—Л–є —Д–∞–Ї—В–Њ—А —А–Є—Б–Ї–∞. –Ъ–§–Я–І —Б—В–∞–ї–Є –Љ–љ–Њ–≥–Њ–Њ–±–µ—Й–∞—О—Й–µ–є —В–µ—А–∞–њ–µ–≤—В–Є—З–µ—Б–Ї–Њ–є –Љ–Є—И–µ–љ—М—О, –≤¬†—Б–≤—П–Ј–Є —Б¬†—З–µ–Љ –њ—А–Њ–і–Њ–ї–ґ–∞—О—В—Б—П –љ–Њ–≤–∞—В–Њ—А—Б–Ї–Є–µ –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є–µ –Є—Б–њ—Л—В–∞–љ–Є—П, –љ–∞–њ—А–∞–≤–ї–µ–љ–љ—Л–µ –љ–∞¬†—А–∞–Ј—А–∞–±–Њ—В–Ї—Г —Н—Д—Д–µ–Ї—В–Є–≤–љ—Л—Е —Б—В—А–∞—В–µ–≥–Є–є –њ—А–µ–і—Г–њ—А–µ–ґ–і–µ–љ–Є—П –Є¬†–Ј–∞–Љ–µ–і–ї–µ–љ–Є—П —Б–µ—А–і–µ—З–љ–Њ-—Б–Њ—Б—Г–і–Є—Б—В—Л—Е –Њ—Б–ї–Њ–ґ–љ–µ–љ–Є–є —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–•–С–Я.
–Ґ–∞–Ї, –≤–ї–Є—П–љ–Є–µ —В–µ—А–∞–њ–Є–Є –§–°–Я –љ–∞¬†—Н–љ–і–Њ–≥–µ–љ–љ—Л–є —Г—А–Њ–≤–µ–љ—М –Ъ–§–Я–І –њ—А–µ–ґ–і–µ –≤—Б–µ–≥–Њ –±—Л–ї–Њ –Є–Ј—Г—З–µ–љ–Њ –≤¬†—Н–Ї—Б–њ–µ—А–Є–Љ–µ–љ—В–∞–ї—М–љ—Л—Е –Љ–Њ–і–µ–ї—П—Е –Є¬†—А–∞—Б—Б–Љ–Њ—В—А–µ–љ–Њ –≤¬†–љ–µ—Б–Ї–Њ–ї—М–Ї–Є—Е –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П—Е, —З–µ—В—Л—А–µ –Є–Ј¬†–Ї–Њ—В–Њ—А—Л—Е –≤–Ї–ї—О—З–∞–ї–Є –і–Є–∞–ї–Є–Ј–љ—Л—Е –њ–∞—Ж–Є–µ–љ—В–Њ–≤ [90вАУ98]. –Э–µ–і–∞–≤–љ–µ–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ –≤–ї–Є—П–љ–Є—П O–Ц –љ–∞¬†—Н–љ–і–Њ–≥–µ–љ–љ—Л–µ –Ъ–§–Я–І –љ–∞¬†–Љ–Њ–і–µ–ї–Є –ґ–Є–≤–Њ—В–љ—Л—Е —Б¬†–Њ—Б—В–∞—В–Њ—З–љ–Њ–є –њ–Њ—З–Ї–Њ–є –њ—А–Њ–і–µ–Љ–Њ–љ—Б—В—А–Є—А–Њ–≤–∞–ї–Њ –Ј–∞—Й–Є—В–љ—Л–є —Н—Д—Д–µ–Ї—В –њ—А–Є –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є–Є –њ–Њ—З–µ–Ї, –Њ–±—Г—Б–ї–Њ–≤–ї–µ–љ–љ—Л–є —Б–љ–Є–ґ–µ–љ–Є–µ–Љ —Г—А–Њ–≤–љ—П –† –≤¬†–Ї—А–Њ–≤–Є. –°–ї–µ–і—Г–µ—В –Њ—В–Љ–µ—В–Є—В—М, —З—В–Њ –∞–≤—В–Њ—А—Л –Њ–±–љ–∞—А—Г–ґ–Є–ї–Є —Б–љ–Є–ґ–µ–љ–Є–µ —Б–Њ–і–µ—А–ґ–∞–љ–Є—П –Ъ–§–Я–І, –љ–µ—Б–Љ–Њ—В—А—П –љ–∞¬†–Њ—В—Б—Г—В—Б—В–≤–Є–µ —Н–Ї—В–Њ–њ–Є—З–µ—Б–Ї–Њ–є –Ї–∞–ї—М—Ж–Є—Д–Є–Ї–∞—Ж–Є–Є –≤¬†–њ–Њ—З–Ї–∞—Е¬†[90].¬† –°–љ–Є–ґ–µ–љ–Є–µ –Ъ–§–Я–І –Љ–Њ–ґ–µ—В –±—Л—В—М —Б–≤—П–Ј–∞–љ–Њ —Б¬†—Г–ї—Г—З—И–µ–љ–Є–µ–Љ —Д—Г–љ–Ї—Ж–Є–Є –њ–Њ—З–µ–Ї, –њ–Њ—Б–Ї–Њ–ї—М–Ї—Г –±—Л–ї–Њ –њ—А–Њ–і–µ–Љ–Њ–љ—Б—В—А–Є—А–Њ–≤–∞–љ–Њ, —З—В–Њ –љ–∞–Ї–Њ–њ–ї–µ–љ–Є–µ –Є–Љ–µ–љ–љ–Њ –Ъ–§–Я–І-II –љ–µ–њ–Њ—Б—А–µ–і—Б—В–≤–µ–љ–љ–Њ –Є–љ–і—Г—Ж–Є—А—Г–µ—В –≤–Њ—Б–њ–∞–ї–µ–љ–Є–µ –Є¬†—Г—Б–Ї–Њ—А—П–µ—В —Д–µ–љ–Њ—В–Є–њ–Є—З–µ—Б–Ї–Є–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П –≤¬†–У–Ь–ЪC, –њ—А–Є–≤–Њ–і—П—Й–Є–µ –Ї¬†–Ї–∞–ї—М—Ж–Є—Д–Є–Ї–∞—Ж–Є–Є —Б–Њ—Б—Г–і–Є—Б—В–Њ–є —Б—В–µ–љ–Ї–Є –Є¬†–Ї–ї–∞–њ–∞–љ–Њ–≤ —Б–µ—А–і—Ж–∞, –Є¬†–Є—Е —Б–љ–Є–ґ–µ–љ–Є–µ –Љ–Њ–ґ–µ—В –≤–љ–µ—Б—В–Є –њ–Њ–ї–Њ–ґ–Є—В–µ–ї—М–љ—Л–є –≤–Ї–ї–∞–і –≤¬†–њ—А–µ–і–Њ—В–≤—А–∞—Й–µ–љ–Є–µ –Ї–∞–ї—М—Ж–Є—Д–Є–Ї–∞—Ж–Є–Є [55, 59].
–°–ї–µ–і—Г–µ—В –Њ—В–Љ–µ—В–Є—В—М, —З—В–Њ –≤¬†—Н–Ї—Б–њ–µ—А–Є–Љ–µ–љ—В–∞—Е –љ–∞¬†–ґ–Є–≤–Њ—В–љ—Л—Е –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є–µ –њ–Њ—З–µ—З–љ—Л—Е –Ї–∞–љ–∞–ї—М—Ж–µ–≤ –Є¬†–Є–љ—В–µ—А—Б—В–Є—Ж–Є–∞–ї—М–љ—Л–є —Д–Є–±—А–Њ–Ј –±—Л–ї–Є –њ—А–µ–і–Њ—В–≤—А–∞—Й–µ–љ—Л –≤–≤–µ–і–µ–љ–Є–µ–Љ –±–Є—Б—Д–Њ—Б—Д–Њ–љ–∞—В–Њ–≤, –Ї–Њ—В–Њ—А—Л–µ —В–∞–Ї–ґ–µ –Є–љ–≥–Є–±–Є—А—Г—О—В —Б–Њ–Ј—А–µ–≤–∞–љ–Є–µ –Ъ–§–Я–І in¬†vitro [55].¬†
–Т¬†2023¬†–≥.¬†U. Thiem –Є¬†—Б–Њ–∞–≤—В. –Њ–њ—Г–±–ї–Є–Ї–Њ–≤–∞–ї–Є —А–µ–Ј—Г–ї—М—В–∞—В—Л –Њ—В–Ї—А—Л—В–Њ–≥–Њ —А–∞–љ–і–Њ–Љ–Є–Ј–Є—А–Њ–≤–∞–љ–љ–Њ–≥–Њ –Ї–Њ–љ—В—А–Њ–ї–Є—А—Г–µ–Љ–Њ–≥–Њ –њ–µ—А–µ–Ї—А–µ—Б—В–љ–Њ–≥–Њ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П, –Є–Ј—Г—З–∞–≤—И–µ–≥–Њ –≤–ї–Є—П–љ–Є–µ –Ю–Ц –љ–∞¬†—Г—А–Њ–≤–љ–Є —Н–љ–і–Њ–≥–µ–љ–љ–Њ–≥–Њ –Љ–Њ–љ–Њ–Љ–µ—А–∞ –Ї–∞–ї—М—Ж–Є–њ—А–Њ—В–µ–Є–љ–∞ –Є¬†–Ъ–§–Я–І, –≤–Њ—Б–њ–∞–ї–µ–љ–Є–µ –Є¬†–Ї–ї–µ—В–Ї–Є —Б–Њ—Б—Г–і–Њ–≤ —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ –љ–∞¬†–Ј–∞–Љ–µ—Б—В–Є—В–µ–ї—М–љ–Њ–є –њ–Њ—З–µ—З–љ–Њ–є —В–µ—А–∞–њ–Є–Є –і–Є–∞–ї–Є–Ј–Њ–Љ [98]. –Ф–ї—П –∞–љ–∞–ї–Є–Ј–∞ –Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–ї–Є—Б—М –Њ–±—А–∞–Ј—Ж—Л –Ї—А–Њ–≤–Є, –≤–Ј—П—В—Л–µ —Г¬†—В—А–µ—Е¬†–≥—А—Г–њ–њ —Г—З–∞—Б—В–љ–Є–Ї–Њ–≤: –њ–∞—Ж–Є–µ–љ—В–Њ–≤, –љ–∞—Е–Њ–і—П—Й–Є—Е—Б—П –љ–∞¬†–У–Ф, –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–•–С–Я —В—А–µ—В—М–µ–є-—З–µ—В–≤–µ—А—В–Њ–є —Б—В–∞–і–Є–Є –Є¬†–Ј–і–Њ—А–Њ–≤—Л—Е –і–Њ–±—А–Њ–≤–Њ–ї—М—Ж–µ–≤. –Я–∞—Ж–Є–µ–љ—В—Л –љ–∞¬†–У–Ф –њ–Њ–ї—Г—З–∞–ї–Є –њ–µ—А–Њ—А–∞–ї—М–љ–Њ –љ–Є–Ј–Ї–Є–µ –і–Њ–Ј—Л –Ю–Ц (250 –Љ–≥/—Б—Г—В), –Ј–∞—В–µ–Љ –≤—Л—Б–Њ–Ї–Є–µ –і–Њ–Ј—Л –Ю–Ц (2000 –Љ–≥/—Б—Г—В) –≤¬†—В–µ—З–µ–љ–Є–µ –і–≤—Г—Е –љ–µ–і–µ–ї—М –Ї–∞–ґ–і–∞—П –Є–ї–Є, –љ–∞–Њ–±–Њ—А–Њ—В, —Б¬†–і–≤—Г—Е–љ–µ–і–µ–ї—М–љ—Л–Љ–Є —Д–∞–Ј–∞–Љ–Є –≤—Л–Љ—Л–≤–∞–љ–Є—П (–Њ—В—Б—Г—В—Б—В–≤–Є–µ –§–°–Я) –і–Њ –Є¬†–њ–Њ—Б–ї–µ –Ї–∞–ґ–і–Њ–є —Д–∞–Ј—Л –ї–µ—З–µ–љ–Є—П. –С—Л–ї–Њ –≤—Л—П–≤–ї–µ–љ–Њ —Б–љ–Є–ґ–µ–љ–Є–µ —Г—А–Њ–≤–љ–µ–є –† –≤¬†—Б—Л–≤–Њ—А–Њ—В–Ї–µ –Ї—А–Њ–≤–Є –њ—А–Є –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ–Љ —Г–≤–µ–ї–Є—З–µ–љ–Є–Є T50 –њ—А–Є —В–µ—А–∞–њ–Є–Є –≤—Л—Б–Њ–Ї–Є–Љ–Є –і–Њ–Ј–∞–Љ–Є O–Ц. –Т¬†—В–µ–Ї—Г—Й–µ–Љ –∞–љ–∞–ї–Є–Ј–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П —Б—Л–≤–Њ—А–Њ—В–Њ—З–љ–Њ–≥–Њ –† –≤¬†–Њ—В–≤–µ—В –љ–∞¬†O–Ц —Б–Є–ї—М–љ–Њ –Ї–Њ—А—А–µ–ї–Є—А–Њ–≤–∞–ї–Є —Б¬†–Є–Ј–Љ–µ–љ–µ–љ–Є—П–Љ–Є –Ъ–§–Я–І-I (r = 0,64 (95%-–љ—Л–є –і–Њ–≤–µ—А–Є—В–µ–ї—М–љ—Л–є –Є–љ—В–µ—А–≤–∞–ї (95% –Ф–Ш) 0,35вАУ0,82), p < 0,001). –†–µ–Ј—Г–ї—М—В–∞—В—Л –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П —В–∞–Ї–ґ–µ —Г–Ї–∞–Ј—Л–≤–∞—О—В –љ–∞¬†–±–Њ–ї—М—И–Є–є –Ї–∞–ї—М—Ж–Є—Д–Є—Ж–Є—А—Г—О—Й–Є–є –њ–Њ—В–µ–љ—Ж–Є–∞–ї —Г—А–µ–Љ–Є—З–µ—Б–Ї–Є—Е –Ъ–§–Я–І-II –њ–Њ¬†—Б—А–∞–≤–љ–µ–љ–Є—О —Б¬†—Б–Є–љ—В–µ—В–Є—З–µ—Б–Ї–Є–Љ–Є –Ъ–§–Я–І-II —Н–Ї–≤–Є–≤–∞–ї–µ–љ—В–љ–Њ–≥–Њ —А–∞–Ј–Љ–µ—А–∞ –Є¬†–Ї–Њ–ї–Є—З–µ—Б—В–≤–∞, —Б–Њ–Ј–і–∞–љ–љ—Л–Љ–Є –≤¬†–љ–µ—Г—А–µ–Љ–Є—З–µ—Б–Ї–Њ–є —Б—А–µ–і–µ, —З—В–Њ –њ–Њ–і—А–∞–Ј—Г–Љ–µ–≤–∞–µ—В –і–Њ–њ–Њ–ї–љ–Є—В–µ–ї—М–љ–Њ–µ –Ї–Њ–љ–і–Є—Ж–Є–Њ–љ–Є—А–Њ–≤–∞–љ–Є–µ –Ъ–§–Я–І, —Б–њ–Њ—Б–Њ–±—Б—В–≤—Г—О—Й–µ–µ –Ї–∞–ї—М—Ж–Є—Д–Є–Ї–∞—Ж–Є–Є –У–Ь–Ъ–° –њ–Њ–і –≤–Њ–Ј–і–µ–є—Б—В–≤–Є–µ–Љ —Г—А–µ–Љ–Є—З–µ—Б–Ї–Є—Е —Д–∞–Ї—В–Њ—А–Њ–≤. –І–∞—Б—В–Є—Ж—Л —Б–Є–љ—В–µ—В–Є—З–µ—Б–Ї–Є—Е –Ъ–§–Я–І, –њ–Њ–ї—Г—З–µ–љ–љ—Л—Е –Є–Ј¬†–Њ–±—К–µ–і–Є–љ–µ–љ–љ–Њ–є —Б—Л–≤–Њ—А–Њ—В–Ї–Є –Ј–і–Њ—А–Њ–≤—Л—Е –Ї–Њ–љ—В—А–Њ–ї—М–љ—Л—Е¬†–≥—А—Г–њ–њ –Є¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤, –љ–∞—Е–Њ–і—П—Й–Є—Е—Б—П –љ–∞¬†–і–Є–∞–ї–Є–Ј–µ, –Є–љ–і—Г—Ж–Є—А—Г—О—В –Ї–∞–ї—М—Ж–Є—Д–Є–Ї–∞—Ж–Є—О –У–Ь–Ъ–° in¬†vitro. –І–∞—Б—В–Є—Ж—Л —Н–љ–і–Њ–≥–µ–љ–љ—Л—Е –Ъ–§–Я–І —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–•–С–Я –Њ–њ–Њ—Б—А–µ–і—Г—О—В –Ї–∞–ї—М—Ж–Є—Д–Є–Ї–∞—Ж–Є—О –У–Ь–Ъ–°, –Є–љ–і—Г—Ж–Є—А–Њ–≤–∞–љ–љ—Г—О —Б—Л–≤–Њ—А–Њ—В–Ї–Њ–є –Ї—А–Њ–≤–Є –њ–∞—Ж–Є–µ–љ—В–∞ in¬†vitro. –Ъ–∞–Ї –±—Л–ї–Њ –Њ–њ–Є—Б–∞–љ–Њ –≤—Л—И–µ, –њ–Њ¬†—А–µ–Ј—Г–ї—М—В–∞—В–∞–Љ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П Y.¬†Nemoto –Є¬†—Б–Њ–∞–≤—В.¬†[90], –ї–µ—З–µ–љ–Є–µ –Ю–Ц —Г¬†–Ї—А—Л—Б, –њ–Њ–і–≤–µ—А–≥—И–Є—Е—Б—П –љ–µ—Д—А—Н–Ї—В–Њ–Љ–Є–Є 5/6 –њ–Њ—З–Ї–Є, –Њ—Б–ї–∞–±–ї—П–ї–Њ –≤–Њ—Б–њ–∞–ї–µ–љ–Є–µ –њ–Њ—З–µ–Ї, –љ–Њ¬†—Н—Д—Д–µ–Ї—В—Л —Г¬†–ї—О–і–µ–є –Є¬†–љ–∞ —Б–Є—Б—В–µ–Љ–љ—Л–µ –Љ–∞—А–Ї–µ—А—Л –≤–Њ—Б–њ–∞–ї–µ–љ–Є—П –Њ—Б—В–∞—О—В—Б—П –љ–µ–Є–Ј–≤–µ—Б—В–љ—Л–Љ–Є.
U. Thiem –Є¬†—Б–Њ–∞–≤—В. [98] –≤–њ–µ—А–≤—Л–µ –Ј–∞–і–Њ–Ї—Г–Љ–µ–љ—В–Є—А–Њ–≤–∞–ї–Є –≤–ї–Є—П–љ–Є–µ O–Ц –љ–∞¬†–Љ–∞—А–Ї–µ—А—Л —Б–Є—Б—В–µ–Љ–љ–Њ–≥–Њ –≤–Њ—Б–њ–∞–ї–µ–љ–Є—П —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤, –љ–∞—Е–Њ–і—П—Й–Є—Е—Б—П –љ–∞¬†–і–Є–∞–ї–Є–Ј–µ, –Є¬†—Г—Б—В–∞–љ–Њ–≤–Є–ї–Є –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ–µ —Б–љ–Є–ґ–µ–љ–Є–µ —Г—А–Њ–≤–љ–µ–є –і–µ–≤—П—В–Є –Є–Ј¬†14 —Ж–Є—В–Њ–Ї–Є–љ–Њ–≤, –Њ—Б–Њ–±–µ–љ–љ–Њ –Ш–Ы-1, –Ш–Ы-6 –Є¬†–Ш–Ы-8, –Ї–Њ—В–Њ—А—Л–µ –Љ–Њ–≥—Г—В –±—Л—В—М –≤–Њ–≤–ї–µ—З–µ–љ—Л –≤¬†—Б–Њ—Б—Г–і–Є—Б—В—Г—О –њ–∞—В–Њ–ї–Њ–≥–Є—О. –Ґ–∞–Ї, –њ—А–Є –Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–љ–Є–Є –Ї–Њ–Љ–±–Є–љ–∞—Ж–Є–Є –Є–Љ–Љ—Г–љ–Њ—Д–µ—А–Љ–µ–љ—В–љ—Л—Е –∞–љ–∞–ї–Є–Ј–Њ–≤ –Є¬†–Љ—Г–ї—М—В–Є–њ–ї–µ–Ї—Б–љ—Л—Е –Љ–∞—В—А–Є—Ж –±—Л–ї–Њ –Њ–±–љ–∞—А—Г–ґ–µ–љ–Њ, —З—В–Њ –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є–Є –Є–љ—В–µ—А–ї–µ–є–Ї–Є–љ–Њ–≤ –≤¬†–њ–ї–∞–Ј–Љ–µ –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ —Б–љ–Є–ґ–∞—О—В—Б—П –њ—А–Є –њ—А–Є–µ–Љ–µ –≤—Л—Б–Њ–Ї–Є—Е –і–Њ–Ј –Ю–Ц (2000 –Љ–≥/—Б—Г—В) –≤¬†—Б—А–∞–≤–љ–µ–љ–Є–Є —Б¬†–Њ—В—Б—Г—В—Б—В–≤–Є–µ–Љ –њ—А–Є–Љ–µ–љ–µ–љ–Є—П –§–°–Я. –Э–∞–Є–±–Њ–ї—М—И–Є–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П –Љ–∞—А–Ї–µ—А–Њ–≤ —Б–Є—Б—В–µ–Љ–љ–Њ–≥–Њ –≤–Њ—Б–њ–∞–ї–µ–љ–Є—П (–Ш–Ы-1ќ± (–Ь–µ -62% (95% –Ф–Ш –Њ—В¬†-76 –і–Њ -26), p < 0,0001), –Ш–Ы-8 (–Ь–µ -46% (95% –Ф–Ш –Њ—В¬†-73 –і–Њ -17), p < 0,0001) –Є¬†–Ш–Ы-6 (–Ь–µ -31% (95% –Ф–Ш –Њ—В¬†-51 –і–Њ -1), p < 0,001)) –љ–∞–±–ї—О–і–∞–ї–Є—Б—М –≤¬†–њ–ї–∞–Ј–Љ–µ –Ї—А–Њ–≤–Є —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤, –љ–∞—Е–Њ–і—П—Й–Є—Е—Б—П –љ–∞¬†–і–Є–∞–ї–Є–Ј–µ, –њ–Њ–ї—Г—З–∞–≤—И–Є—Е –≤—Л—Б–Њ–Ї–Є–µ –і–Њ–Ј—Л –Ю–Ц. –≠—В–Њ—В –њ—А–µ–Є–Љ—Г—Й–µ—Б—В–≤–µ–љ–љ–Њ –њ—А–Њ—В–Є–≤–Њ–≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ—Л–є —Н—Д—Д–µ–Ї—В –±—Л–ї –Њ—В—А–∞–ґ–µ–љ –≤¬†–Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ –±–Њ–ї–µ–µ –љ–Є–Ј–Ї–Є—Е —Г—А–Њ–≤–љ—П—Е –≤—Л—Б–Њ–Ї–Њ—З—Г–≤—Б—В–≤–Є—В–µ–ї—М–љ–Њ–≥–Њ –°-—А–µ–∞–Ї—В–Є–≤–љ–Њ–≥–Њ –±–µ–ї–Ї–∞ (hs CRP) –љ–∞¬†—Д–Њ–љ–µ —В–µ—А–∞–њ–Є–Є –Ю–Ц (3,90 –Љ–≥/–ї (95% –Ф–Ш 1,95вАУ7,18) –њ–Њ¬†—Б—А–∞–≤–љ–µ–љ–Є—О —Б¬†2,45 (95% –Ф–Ш 0,99вАУ5,94); p <¬†0,05) —Б—А–µ–і–Є –њ–∞—Ж–Є–µ–љ—В–Њ–≤, –љ–µ¬†–њ–Њ–ї—Г—З–∞–≤—И–Є—Е –§–°–Я (–≤—Л–Љ—Л–≤–∞–љ–Є–µ). –Т–∞–ґ–љ–Њ—Б—В—М –≤–ї–Є—П–љ–Є—П –љ–∞¬†—Ж–Є—В–Њ–Ї–Є–љ—Л –≤¬†—Н—В–Њ–Љ –Ї–Њ–љ—В–µ–Ї—Б—В–µ –Љ–Њ–ґ–µ—В –±—Л—В—М –њ–Њ–і—З–µ—А–Ї–љ—Г—В–∞ –≤¬†–љ–µ–і–∞–≤–љ–µ–Љ –†–Ъ–Ш, –њ–Њ–Ї–∞–Ј–∞–≤—И–µ–Љ –±–Њ–ї–µ–µ –љ–Є–Ј–Ї—Г—О —Б–µ—А–і–µ—З–љ–Њ-—Б–Њ—Б—Г–і–Є—Б—В—Г—О –Ј–∞–±–Њ–ї–µ–≤–∞–µ–Љ–Њ—Б—В—М —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†—Г—Б—В–∞–љ–Њ–≤–ї–µ–љ–љ—Л–Љ –°–°–Ч –Є¬†–њ–Њ–≤—Л—И–µ–љ–љ—Л–Љ–Є —Г—А–Њ–≤–љ—П–Љ–Є hs CRP, –њ–Њ–ї—Г—З–∞–≤—И–Є—Е —В–µ—А–∞–њ–Є—О –∞–љ—В–Є-–Ш–Ы-1ќ≤ –Є–ї–Є –∞–љ—В–Є-–Ш–Ы-6 [99].
–≠–љ–і–Њ–≥–µ–љ–љ—Л–µ –Ъ–§–Я–І –Њ–њ–Њ—Б—А–µ–і—Г—О—В —Н—Д—Д–µ–Ї—В –ї–µ—З–µ–љ–Є—П –Ю–Ц –љ–∞¬†–∞–Ї—В–Є–≤–∞—Ж–Є—О —Н–љ–і–Њ—В–µ–ї–Є–∞–ї—М–љ—Л—Е –Ї–ї–µ—В–Њ–Ї, –Є–љ–і—Г—Ж–Є—А—Г–µ–Љ—Г—О —Б—Л–≤–Њ—А–Њ—В–Ї–Њ–є –Ї—А–Њ–≤–Є –њ–∞—Ж–Є–µ–љ—В–∞ in¬†vitro. –Ґ–µ–Љ –љ–µ¬†–Љ–µ–љ–µ–µ —Г—А–Њ–≤–љ–Є –Љ–†–Э–Ъ –Њ—Б—В–∞–≤–∞–ї–Є—Б—М –≤—Л—И–µ, —З–µ–Љ –≤¬†–Ї–Њ–љ—В—А–Њ–ї—М–љ–Њ–є —Б—Л–≤–Њ—А–Њ—В–Ї–µ, —З—В–Њ –µ—Й–µ —А–∞–Ј –њ–Њ–і—З–µ—А–Ї–Є–≤–∞–µ—В, —З—В–Њ —В–µ—А–∞–њ–Є—П –§–°–Я –љ–µ¬†–њ–Њ–ї–љ–Њ—Б—В—М—О —Б–љ–Є–ґ–∞–µ—В —В–Њ–Ї—Б–Є—З–љ–Њ—Б—В—М —Г—А–µ–Љ–Є—З–µ—Б–Ї–Њ–є —Б—Л–≤–Њ—А–Њ—В–Ї–Є.
–Я—А–Є –≤—В–Њ—А–Є—З–љ–Њ–Љ –∞–љ–∞–ї–Є–Ј–µ —А–∞–љ–і–Њ–Љ–Є–Ј–Є—А–Њ–≤–∞–љ–љ–Њ–≥–Њ –Ї–Њ–љ—В—А–Њ–ї–Є—А—Г–µ–Љ–Њ–≥–Њ –њ–µ—А–µ–Ї—А–µ—Б—В–љ–Њ–≥–Њ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П –і–Є–∞–ї–Є–Ј–љ—Л—Е –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–≥–Є–њ–µ—А—Д–Њ—Б—Д–∞—В–µ–Љ–Є–µ–є, —Ж–µ–ї—М—О –Ї–Њ—В–Њ—А–Њ–≥–Њ —П–≤–ї—П–ї–Њ—Б—М –Є–Ј—Г—З–µ–љ–Є–µ –≤–ї–Є—П–љ–Є—П –Ю–Ц –љ–∞¬†—Б–Ї–ї–Њ–љ–љ–Њ—Б—В—М —Б—Л–≤–Њ—А–Њ—В–Ї–Є –Ї—А–Њ–≤–Є –Ї¬†–Ї–∞–ї—М—Ж–Є—Д–Є–Ї–∞—Ж–Є–Є, –Є–Ј–Љ–µ—А–µ–љ–љ—Г—О –њ–Њ¬†T50-—В–µ—Б—В—Г [97], –±—Л–ї–Њ –њ—А–Њ–і–µ–Љ–Њ–љ—Б—В—А–Є—А–Њ–≤–∞–љ–Њ, —З—В–Њ —В–µ—А–∞–њ–Є—П –≤—Л—Б–Њ–Ї–Є–Љ–Є –і–Њ–Ј–∞–Љ–Є –Ю–Ц (2000 –Љ–≥/—Б—Г—В) –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ —Б–љ–Є–ґ–∞–ї–∞ —Г—А–Њ–≤–љ–Є —Н–љ–і–Њ–≥–µ–љ–љ—Л—Е –Ї–∞–ї—М—Ж–Є–є-–њ—А–Њ—В–µ–Є–љ–Њ–≤—Л—Е –Љ–Њ–љ–Њ–Љ–µ—А–Њ–≤, –Ъ–§–Я–І-I –Є¬†–Ъ–§–Я–І-II –≤¬†—Б—Л–≤–Њ—А–Њ—В–Ї–µ –Ї—А–Њ–≤–Є –Є¬†–±—Л–ї–∞ —Б–≤—П–Ј–∞–љ–∞ —Б¬†–Є–Љ–Љ—Г–љ–Њ–Љ–Њ–і—Г–ї–Є—А—Г—О—Й–Є–Љ–Є —Н—Д—Д–µ–Ї—В–∞–Љ–Є. –Ю–њ–Њ—Б—А–µ–і–Њ–≤–∞–љ–љ—Л–µ —В–∞–Ї–Є–Љ –Њ–±—А–∞–Ј–Њ–Љ —Н—Д—Д–µ–Ї—В—Л –љ–∞¬†—Б—Л–≤–Њ—А–Њ—В–Ї—Г –Ї—А–Њ–≤–Є —Г¬†–і–Є–∞–ї–Є–Ј–љ—Л—Е –њ–∞—Ж–Є–µ–љ—В–Њ–≤ –≤—Л—А–∞–ґ–∞–ї–Є—Б—М –≤¬†—Г–≤–µ–ї–Є—З–µ–љ–Є–Є –Ґ50 (52 –Љ–Є–љ—Г—В—Л (95% –Ф–Ш 31вАУ74 –Љ–Є–љ—Г—В—Л, —А < 0,0001)), —Б–љ–Є–ґ–µ–љ–Є–Є —Г—А–Њ–≤–љ—П –† –≤¬†—Б—Л–≤–Њ—А–Њ—В–Ї–µ –Ї—А–Њ–≤–Є —Б¬†2,18 ¬± 0,5 –і–Њ 1,64 ¬± 0,46 –Љ–Љ–Њ–ї—М/–ї –Є¬†–≤ —Б–љ–Є–ґ–µ–љ–Є–Є –Ї–∞–ї—М—Ж–Є—Д–Є–Ї–∞—Ж–Є–Є –У–Ь–Ъ–° –Є¬†–∞–Ї—В–Є–≤–∞—Ж–Є–Є —Н–љ–і–Њ—В–µ–ї–Є–∞–ї—М–љ—Л—Е –Ї–ї–µ—В–Њ–Ї in¬†vitro. –Ґ–∞–Ї–ґ–µ –±—Л–ї–Њ –≤—Л—П–≤–ї–µ–љ–Њ, —З—В–Њ –Ъ–§–Я–І —П–≤–ї—П—О—В—Б—П –њ–Њ—В–µ–љ—Ж–Є–∞–ї—М–љ—Л–Љ–Є –Њ—Б–љ–Њ–≤–љ—Л–Љ–Є –і–µ—В–µ—А–Љ–Є–љ–∞–љ—В–∞–Љ–Є. –≠—В–Є —А–µ–Ј—Г–ї—М—В–∞—В—Л —Г–Ї–∞–Ј—Л–≤–∞—О—В –љ–∞¬†—В–Њ, —З—В–Њ —Б–љ–Є–ґ–µ–љ–Є–µ —Г—А–Њ–≤–љ—П –† –≤¬†—Б—Л–≤–Њ—А–Њ—В–Ї–µ –Ї—А–Њ–≤–Є —П–≤–ї—П–µ—В—Б—П –Њ—Б–љ–Њ–≤–љ—Л–Љ —Д–∞–Ї—В–Њ—А–Њ–Љ, —Б–њ–Њ—Б–Њ–±—Б—В–≤—Г—О—Й–Є–Љ –Є–Ј–Љ–µ–љ–µ–љ–Є—П–Љ —Г—А–Њ–≤–љ—П –Ґ50, –≤—Л–Ј–≤–∞–љ–љ—Л–Љ —В–µ—А–∞–њ–Є–µ–є –Ю–Ц. –Ш—Б—Е–Њ–і—П –Є–Ј¬†—Н—В–Њ–≥–Њ, —Б–љ–Є–ґ–µ–љ–Є–µ —Г—А–Њ–≤–љ—П –† –≤¬†—Б—Л–≤–Њ—А–Њ—В–Ї–µ –Ї—А–Њ–≤–Є –љ–∞¬†0,1 –Љ–Љ–Њ–ї—М/–ї –њ—А–Є–≤–Њ–і–Є—В –Ї¬†–њ–Њ–≤—Л—И–µ–љ–Є—О —Г—А–Њ–≤–љ—П –Ґ50 –њ—А–Є–±–ї–Є–Ј–Є—В–µ–ї—М–љ–Њ –љ–∞¬†10 –Љ–Є–љ—Г—В.
–Ю—Б–љ–Њ–≤–љ—Л–Љ –њ—А–µ–Є–Љ—Г—Й–µ—Б—В–≤–Њ–Љ –∞–љ–∞–ї–Є–Ј–Є—А—Г–µ–Љ—Л—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є –±—Л–ї –Ї–Њ–љ—В—А–Њ–ї–Є—А—Г–µ–Љ—Л–є –Ї—А–Њ—Б—Б–Њ–≤–µ—А —Б¬†–њ—А–µ—А—Л–≤–Є—Б—В—Л–Љ–Є —Д–∞–Ј–∞–Љ–Є –≤—Л–Љ—Л–≤–∞–љ–Є—П, —З—В–Њ –њ–Њ–Ј–≤–Њ–ї–Є–ї–Њ –Є–Ј—Г—З–Є—В—М –≤–ї–Є—П–љ–Є–µ –Љ–Њ—Й–љ–Њ–є —В–µ—А–∞–њ–Є–Є –Ю–Ц –њ–Њ¬†—Б—А–∞–≤–љ–µ–љ–Є—О —Б¬†—В–µ—А–∞–њ–Є–µ–є –±–µ–Ј –§–°–Я, —Н—Д—Д–µ–Ї—В–Є–≤–љ–Њ–µ –Є¬†–љ–µ—Н—Д—Д–µ–Ї—В–Є–≤–љ–Њ–µ (–Ю–Ц 2000 –Љ–≥/—Б—Г—В –≤¬†—Б—А–∞–≤–љ–µ–љ–Є–Є —Б¬†–Ю–Ц 250 –Љ–≥/—Б—Г—В) —Б–љ–Є–ґ–µ–љ–Є–µ –†,¬†–∞¬†—В–∞–Ї–ґ–µ –Њ–±—А–∞—В–Є–Љ–Њ—Б—В—М —Н—Д—Д–µ–Ї—В–Њ–≤ —В–µ—А–∞–њ–Є–Є –§–°–Я (–Ю–Ц 2000 –Љ–≥/—Б—Г—В –њ–Њ¬†—Б—А–∞–≤–љ–µ–љ–Є—О —Б¬†–≤—Л–Љ—Л–≤–∞–љ–Є–µ–Љ) –љ–∞¬†–Ґ50. –Ґ–∞–Ї–Є–Љ –Њ–±—А–∞–Ј–Њ–Љ, –њ–Њ–Ї–∞–Ј–∞–љ–Њ, —З—В–Њ –≤—Л—Б–Њ–Ї–Є–µ –і–Њ–Ј—Л –Ю–Ц —Б–љ–Є–ґ–∞–ї–Є –Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є–µ —Н–љ–і–Њ–≥–µ–љ–љ—Л—Е –Ъ–§–Я–І –≤¬†—Б—Л–≤–Њ—А–Њ—В–Ї–µ –Ї—А–Њ–≤–Є, –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ –њ–Њ–≤—Л—И–∞–ї–Є —Г—А–Њ–≤–µ–љ—М –Ґ50, —Б–љ–Є–ґ–∞–ї–Є —Б–њ–Њ—Б–Њ–±–љ–Њ—Б—В—М —Г—А–µ–Љ–Є—З–µ—Б–Ї–Њ–є —Б—Л–≤–Њ—А–Њ—В–Ї–Є –Є–љ–і—Г—Ж–Є—А–Њ–≤–∞—В—М –Ъ–° –Є¬†–∞–Ї—В–Є–≤–Є—А–Њ–≤–∞—В—М —Н–љ–і–Њ—В–µ–ї–Є–∞–ї—М–љ—Л–µ –Ї–ї–µ—В–Ї–Є in¬†vitro, –Њ—Б–ї–∞–±–ї—П–ї–Є —Б–Є—Б—В–µ–Љ–љ–Њ–µ –≤–Њ—Б–њ–∞–ї–µ–љ–Є–µ —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤, –љ–∞—Е–Њ–і—П—Й–Є—Е—Б—П –љ–∞¬†–У–Ф.
–Т–µ—А–Њ—П—В–љ—Л–Љ –њ—А–µ–і—Б—В–∞–≤–ї—П–µ—В—Б—П —Д–∞–Ї—В, —З—В–Њ —Б–љ–Є–ґ–µ–љ–Є–µ –Ъ–§–Я–Ь –Ю–Ц –Љ–Њ–ґ–µ—В –±—Л—В—М –±–Њ–ї–µ–µ —З—Г–≤—Б—В–≤–Є—В–µ–ї—М–љ—Л–Љ –њ–Њ–Ї–∞–Ј–∞—В–µ–ї–µ–Љ –Њ—Б—В—А—Л—Е –Є–Ј–Љ–µ–љ–µ–љ–Є–є –Љ–Є–љ–µ—А–∞–ї—М–љ–Њ–є –љ–∞–≥—А—Г–Ј–Ї–Є, —Б¬†—Г—З–µ—В–Њ–Љ –Є—Е –Ї–Є—И–µ—З–љ–Њ–≥–Њ –њ—А–Њ–Є—Б—Е–Њ–ґ–і–µ–љ–Є—П –Є¬†—Б–Є–≥–Љ–Њ–Є–і–∞–ї—М–љ–Њ–є –≤–Ј–∞–Є–Љ–Њ—Б–≤—П–Ј–Є –Љ–µ–ґ–і—Г —Б—Л–≤–Њ—А–Њ—В–Њ—З–љ—Л–Љ –† –Є¬†–Ъ–§–Я–Ь [24, 100].
–Ч–∞–Ї–ї—О—З–µ–љ–Є–µ
–У–Є–њ–µ—А—Д–Њ—Б—Д–∞—В–µ–Љ–Є—П —Б—З–Є—В–∞–µ—В—Б—П –Њ—Б–љ–Њ–≤–љ—Л–Љ –љ–µ—В—А–∞–і–Є—Ж–Є–Њ–љ–љ—Л–Љ —Д–∞–Ї—В–Њ—А–Њ–Љ —А–Є—Б–Ї–∞ —А–∞–Ј–≤–Є—В–Є—П —Б–µ—А–і–µ—З–љ–Њ-—Б–Њ—Б—Г–і–Є—Б—В—Л—Е –Њ—Б–ї–Њ–ґ–љ–µ–љ–Є–є, –Є¬†–љ–∞–Ї–∞–њ–ї–Є–≤–∞–µ—В—Б—П –≤—Б–µ –±–Њ–ї—М—И–µ –і–∞–љ–љ—Л—Е, —Б–≤–Є–і–µ—В–µ–ї—М—Б—В–≤—Г—О—Й–Є—Е –Њ¬†—В–Њ–Љ, —З—В–Њ —Ж–Є—А–Ї—Г–ї–Є—А—Г—О—Й–Є–µ –Ъ–§–Я–І –Љ–Њ–≥—Г—В –±—Л—В—М –ї—Г—З—И–Є–Љ –њ–Њ–Ї–∞–Ј–∞—В–µ–ї–µ–Љ —В–Њ–Ї—Б–Є—З–љ–Њ—Б—В–Є –†,¬†—З–µ–Љ —Б—Л–≤–Њ—А–Њ—В–Њ—З–љ—Л–є —Д–Њ—Б—Д–∞—В —Б–∞–Љ –њ–Њ¬†—Б–µ–±–µ. –Ю—В–Ї—А—Л—В–Є–µ –Ъ–§–Я–І —Б–≤–Є–і–µ—В–µ–ї—М—Б—В–≤—Г–µ—В –Њ¬†–љ–Њ–≤—Л—Е –≤–Њ–Ј–Љ–Њ–ґ–љ–Њ—Б—В—П—Е –њ—А–Њ—Д–Є–ї–∞–Ї—В–Є–Ї–Є –ЪC –Є¬†–Ї–Њ–ї–Є—З–µ—Б—В–≤–µ–љ–љ–Њ–є –Њ—Ж–µ–љ–Ї–Є —Б–Ї–ї–Њ–љ–љ–Њ—Б—В–Є –Ї¬†–Ї–∞–ї—М—Ж–Є—Д–Є–Ї–∞—Ж–Є–Є —Б—Л–≤–Њ—А–Њ—В–Ї–Є —Б¬†–њ–Њ–Љ–Њ—Й—М—О —В–µ—Б—В–∞ T50. –Э–µ—Б–Љ–Њ—В—А—П –љ–∞¬†—В–Њ —З—В–Њ —А–∞–Ј–ї–Є—З–љ—Л–µ —Д–∞–Ї—В–Њ—А—Л, –≤–ї–Є—П—О—Й–Є–µ –љ–∞¬†—Г—А–Њ–≤–љ–Є –Ъ–§–Я–І –≤¬†—Б—Л–≤–Њ—А–Њ—В–Ї–µ –Ї—А–Њ–≤–Є, –≤–Ї–ї—О—З–∞—П –њ—А–Њ—Ж–µ—Б—Б –Є—Е —Б–Њ–Ј—А–µ–≤–∞–љ–Є—П, –∞¬†—В–∞–Ї–ґ–µ –љ–∞¬†T50 –Є¬†–µ–≥–Њ –≤–∞—А–Є–∞—Ж–Є–Є –њ—А–Є —А–∞–Ј–ї–Є—З–љ—Л—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П—Е, –Є–Ј—Г—З–µ–љ—Л –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ, –њ–Њ—П–≤–ї—П–µ—В—Б—П –≤—Б–µ –±–Њ–ї—М—И–µ –і–Њ–Ї–∞–Ј–∞—В–µ–ї—М—Б—В–≤, —Б–≤–Є–і–µ—В–µ–ї—М—Б—В–≤—Г—О—Й–Є—Е –Њ¬†—В–Њ–Љ, —З—В–Њ T50 –Љ–Њ–ґ–µ—В –±—Л—В—М —Н—Д—Д–µ–Ї—В–Є–≤–љ—Л–Љ –Љ–∞—А–Ї–µ—А–Њ–Љ –њ—А–Є –Њ—Ж–µ–љ–Ї–µ –ЪC. –С–Њ–ї–µ–µ —В–Њ–≥–Њ, –Њ–њ—А–µ–і–µ–ї–µ–љ–Є–µ —В–µ—Б—В–∞ T50 –Љ–Њ–ґ–µ—В –±—Л—В—М –њ–Њ–ї–µ–Ј–љ—Л–Љ –њ—А–Є –ї–µ—З–µ–љ–Є–Є –≤–љ–µ–Ї–Њ—Б—В–љ–Њ–є –Ї–∞–ї—М—Ж–Є—Д–Є–Ї–∞—Ж–Є–Є —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–•–С–Я, –Њ—Б–Њ–±–µ–љ–љ–Њ —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ –љ–∞¬†–Ј–∞–Љ–µ—Б—В–Є—В–µ–ї—М–љ–Њ–є –њ–Њ—З–µ—З–љ–Њ–є —В–µ—А–∞–њ–Є–Є –У–Ф, —Г¬†–Ї–Њ—В–Њ—А—Л—Е –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ –њ–Њ–≤—Л—И–µ–љ —А–Є—Б–Ї¬†—А–∞–Ј–≤–Є—В–Є—П –°–°–Ч. –Т¬†—Н—В–Є—Е —Б–Є—В—Г–∞—Ж–Є—П—Е —А–∞–љ–љ–µ–µ –≤–љ–µ–і—А–µ–љ–Є–µ —Б—В—А–∞—В–µ–≥–Є–Є –ї–µ—З–µ–љ–Є—П, –њ–Њ–≤—Л—И–∞—О—Й–µ–є T50, –Љ–Њ–≥–ї–Њ –±—Л —Б–Љ—П–≥—З–Є—В—М –Њ—З–µ–≤–Є–і–љ—Л–µ –Њ—Б–ї–Њ–ґ–љ–µ–љ–Є—П, —Б–≤—П–Ј–∞–љ–љ—Л–µ —Б¬†–ЪC.
–Я–Њ–ї—Г—З–µ–љ–љ—Л–µ —А–µ–Ј—Г–ї—М—В–∞—В—Л –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є –≤–ї–Є—П–љ–Є—П –Њ–Ї—Б–Є–≥–Є–і—А–Њ–Ї—Б–Є–і–∞ –ґ–µ–ї–µ–Ј–∞ (Velphoro¬Ѓ) –љ–∞¬†–Ъ–§–Я–І —Г¬†–і–Є–∞–ї–Є–Ј–љ—Л—Е –њ–∞—Ж–Є–µ–љ—В–Њ–≤ –њ–Њ–Ї–∞–Ј–∞–ї–Є, —З—В–Њ —Д–Њ—Б—Д–∞—В-—Б–≤—П–Ј—Л–≤–∞—О—Й–∞—П —В–µ—А–∞–њ–Є—П –≤—Л—Б–Њ–Ї–Є–Љ–Є –і–Њ–Ј–∞–Љ–Є –Ю–Ц —Б–љ–Є–ґ–∞–ї–∞ —Б—Л–≤–Њ—А–Њ—В–Њ—З–љ—Л–µ —Г—А–Њ–≤–љ–Є –Ї–∞–ї—М—Ж–Є–є-—Д–Њ—Б—Д–∞—В–љ—Л—Е –Љ–Њ–љ–Њ–Љ–µ—А–Њ–≤, –Ъ–§–Я–І-I –Є¬†–Ъ–§–Я–І-II –Є¬†–±—Л–ї–∞ —Б–≤—П–Ј–∞–љ–∞ —Б¬†–Є–Љ–Љ—Г–љ–Њ–Љ–Њ–і—Г–ї–Є—А—Г—О—Й–Є–Љ–Є —Н—Д—Д–µ–Ї—В–∞–Љ–Є. –°—Л–≤–Њ—А–Њ—В–Ї–∞ –њ–∞—Ж–Є–µ–љ—В–Њ–≤, –њ–Њ–ї—Г—З–∞–≤—И–Є—Е –ї–µ—З–µ–љ–Є–µ –Ю–Ц, –Њ–±–ї–∞–і–∞–ї–∞ –±–Њ–ї–µ–µ –љ–Є–Ј–Ї–Є–Љ–Є –њ—А–Њ–Ї–∞–ї—М—Ж–Є—Д–Є—Ж–Є—А—Г—О—Й–Є–Љ–Є –Є¬†–≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ—Л–Љ–Є —Б–≤–Њ–є—Б—В–≤–∞–Љ–Є in¬†vitro, –Є¬†–±—Л–ї–Њ –Њ–±–љ–∞—А—Г–ґ–µ–љ–Њ, —З—В–Њ –Ъ–§–Я–І –Њ–њ–Њ—Б—А–µ–і—Г—О—В —Н—В–Є —Н—Д—Д–µ–Ї—В—Л. –Ґ–∞–Ї–Є–Љ –Њ–±—А–∞–Ј–Њ–Љ, –њ–Њ–ї—Г—З–µ–љ–љ—Л–µ –і–∞–љ–љ—Л–µ –њ–Њ–і—В–≤–µ—А–ґ–і–∞—О—В –њ—А–Є—З–Є–љ–љ—Г—О —А–Њ–ї—М –Ъ–§–Я–І –≤¬†—А–∞–Ј–≤–Є—В–Є–Є –Є¬†–њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є–Є —Б–Њ—Б—Г–і–Є—Б—В—Л—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є –Є¬†—Б–≤–Є–і–µ—В–µ–ї—М—Б—В–≤—Г—О—В –Њ¬†—Б–љ–Є–ґ–µ–љ–Є–Є –Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є—П —Н–љ–і–Њ–≥–µ–љ–љ—Л—Е –Ъ–§–Я–І –Є¬†—Б–њ–Њ—Б–Њ–±–љ–Њ—Б—В–Є —Г—А–µ–Љ–Є—З–µ—Б–Ї–Њ–є —Б—Л–≤–Њ—А–Њ—В–Ї–Є –≤—Л–Ј—Л–≤–∞—В—М –Ъ–°, –∞–Ї—В–Є–≤–Є—А–Њ–≤–∞—В—М —Н–љ–і–Њ—В–µ–ї–Є–∞–ї—М–љ—Л–µ –Ї–ї–µ—В–Ї–Є in¬†vitro, –∞¬†—В–∞–Ї–ґ–µ –Њ¬†–љ–µ–і–Њ–Њ—Ж–µ–љ–µ–љ–љ–Њ–є —А–Њ–ї–Є –њ—А–Њ—В–Є–≤–Њ–≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ–Њ–≥–Њ —Н—Д—Д–µ–Ї—В–∞ –Ю–Ц —Г¬†–±–Њ–ї—М—И–Є–љ—Б—В–≤–∞ –њ–∞—Ж–Є–µ–љ—В–Њ–≤, –љ–∞—Е–Њ–і—П—Й–Є—Е—Б—П –љ–∞¬†–і–Є–∞–ї–Є–Ј–µ.
G.V. Volgina, PhD, Prof., M.Yu. Dudko
Russian University of Medicine, Moscow
S.P. Botkin City Clinical Hospital
Contact person: Galina V. Volgina, volginagv@mail.ru
Phosphate overload is the primary driver of all manifestations of bone mineral disorders in chronic kidney disease (BMD-CKD) and the development of cardiovascular complications. A new understanding of cardiovascular pathology, including the formation of newly discovered calcium phosphate protein particles (CPhPP), which are a potential biomarker of the occurrence and progression of vascular calcification, modification of this risk factor for BMD and cardiovascular pathology, will undoubtedly lead to improved therapeutic interventions and possibly improve adverse outcomes in patients with CKD. The obtained data on the effect of iron hydroxide on CPHR indicate a decrease in the formation of endodogenous CPhPP, the ability of uremic serum to cause vascular calcification, activate endothelial cells and weaken systemic inflammation in most patients receiving renal replacement therapy by dialysis.
–£–≤–∞–ґ–∞–µ–Љ—Л–є –њ–Њ—Б–µ—В–Є—В–µ–ї—М uMEDp!
–£–≤–µ–і–Њ–Љ–ї—П–µ–Љ –Т–∞—Б –Њ —В–Њ–Љ, —З—В–Њ –Ј–і–µ—Б—М —Б–Њ–і–µ—А–ґ–Є—В—Б—П –Є–љ—Д–Њ—А–Љ–∞—Ж–Є—П, –њ—А–µ–і–љ–∞–Ј–љ–∞—З–µ–љ–љ–∞—П –Є—Б–Ї–ї—О—З–Є—В–µ–ї—М–љ–Њ –і–ї—П —Б–њ–µ—Ж–Є–∞–ї–Є—Б—В–Њ–≤ –Ј–і—А–∞–≤–Њ–Њ—Е—А–∞–љ–µ–љ–Є—П.
–Х—Б–ї–Є –Т—Л –љ–µ —П–≤–ї—П–µ—В–µ—Б—М —Б–њ–µ—Ж–Є–∞–ї–Є—Б—В–Њ–Љ –Ј–і—А–∞–≤–Њ–Њ—Е—А–∞–љ–µ–љ–Є—П, –∞–і–Љ–Є–љ–Є—Б—В—А–∞—Ж–Є—П –љ–µ –љ–µ—Б–µ—В –Њ—В–≤–µ—В—Б—В–≤–µ–љ–љ–Њ—Б—В–Є –Ј–∞ –≤–Њ–Ј–Љ–Њ–ґ–љ—Л–µ –Њ—В—А–Є—Ж–∞—В–µ–ї—М–љ—Л–µ –њ–Њ—Б–ї–µ–і—Б—В–≤–Є—П, –≤–Њ–Ј–љ–Є–Ї—И–Є–µ –≤ —А–µ–Ј—Г–ї—М—В–∞—В–µ —Б–∞–Љ–Њ—Б—В–Њ—П—В–µ–ї—М–љ–Њ–≥–Њ –Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–љ–Є—П –Т–∞–Љ–Є –Є–љ—Д–Њ—А–Љ–∞—Ж–Є–Є —Б –њ–Њ—А—В–∞–ї–∞ –±–µ–Ј –њ—А–µ–і–≤–∞—А–Є—В–µ–ї—М–љ–Њ–є –Ї–Њ–љ—Б—Г–ї—М—В–∞—Ж–Є–Є —Б –≤—А–∞—З–Њ–Љ.
–Э–∞–ґ–Є–Љ–∞—П –љ–∞ –Ї–љ–Њ–њ–Ї—Г ¬Ђ–Т–Њ–є—В–Є¬ї, –Т—Л –њ–Њ–і—В–≤–µ—А–ґ–і–∞–µ—В–µ, —З—В–Њ —П–≤–ї—П–µ—В–µ—Б—М –≤—А–∞—З–Њ–Љ –Є–ї–Є —Б—В—Г–і–µ–љ—В–Њ–Љ –Љ–µ–і–Є—Ж–Є–љ—Б–Ї–Њ–≥–Њ –≤—Г–Ј–∞.