Эпигенетика в неврологии нейродегенеративных заболеваний. Терапевтический потенциал факторов Яманаки
- Аннотация
- Статья
- Ссылки
- English

Введение
Нейродегенеративные заболевания представляют собой обширную гетерогенную группу патологических состояний нервной системы, которую отличает прогрессирующая и необратимая потеря специфических популяций нейронов, приводящая к клиническому дефициту соответствующих неврологических функций. Процесс дегенерации, нередко ассоциированный с накоплением аномальных белковых агрегатов в клетках головного и спинного мозга, лежит в основе инвалидизирующих расстройств, например болезней Альцгеймера и Паркинсона, бокового амиотрофического склероза, хореи Гентингтона.
Эпигенетика изучает наследуемые модификации в экспрессии генов, не затрагивающие последовательность ДНК, и регуляторные элементы генома. Развитие нейродегенеративных болезней зависит от эпигенетической регуляции. Знание данных механизмов позволяет разработать потенциальные методы лечения нейродегенеративных заболеваний.
Цель – обобщить данные научной литературы за последние пять лет, касающиеся эпигенетики нейродегенеративных заболеваний, и рассмотреть варианты потенциальной терапии.
Эпигенетические факторы развития нейродегенеративных заболеваний
Болезнь Альцгеймера (БА) характеризуется накоплением бета-амилоидных пептидов в виде внеклеточных бляшек и формированием внутриклеточных нейрофибриллярных клубков из гиперфосфорилированного тау-белка, что сопровождается синаптической дисфункцией, гибелью нейронов и кортикальной атрофией. Наряду с известными генетическими факторами риска (например, аллель APOE ε4 [1]), все больше данных указывает на важную роль эпигенетических механизмов, таких как метилирование ДНК, модификация гистонов, РНК-опосредованные механизмы регуляции.
Метилирование ДНК
Как показывают результаты исследований, БА свойственна значительная дизрегуляция метилирования ДНК. Преимущественно наблюдается гипометилирование (недостаточное метилирование) ДНК в гиппокампе и коре головного мозга [2–4]. Ряд авторов указывают на увеличение экспрессии других генов при сниженном метилировании ДНК. Так, ген APP кодирует белок-предшественник бета-амилоида. Ген APP заглушается метилированием своей премоторной области, однако в процессе старения этот ген деметилируется, способствуя собственной экспрессии и, как следствие, накоплению бета-амилоида в мозге [5–8].
Модификация гистонов
Посттрансляционные модификации, происходящие на гистоновых белках (ацетилирование, метилирование, фосфорилирование и убиквитинирование), регулируют структуру хроматина и паттерны экспрессии генов [9, 10].
Инвазия некодирующей РНК
Существует две группы некодирующих молекул РНК: микроРНК и длинные некодирующие РНК. Последние регулируют экспрессию генов в нервной системе и активно участвуют в нейродегенеративных процессах [11]. МикроРНК функционируют как короткие молекулы РНК, связываясь с комплементарными участками на целевых матричных РНК (мРНК), что приводит к деградации или подавлению трансляции этих молекул [12, 13].
Формирование эпигенетической блокады
Данные механизмы эпигенетической перестройки формируют устойчивые изменения в больных клетках, препятствуя активации генов, связанных с плюрипотентностью клетки. Таким образом, клетка становится сенесцентной. В аспекте центральной нервной системы подобные изменения приводят к снижению когнитивных способностей, поскольку долговременные формы памяти и доступ к ним требуют стабильных изменений экспрессии генов [14], которые частично регулируются эпигенетическими процессами. Среди эпигенетических модификаций, выявленных в нервной системе, ацетилирование гистонов в большей степени связано с обучением и памятью [15].
Семейство гистоновых деацетилаз и их влияние на нервную систему
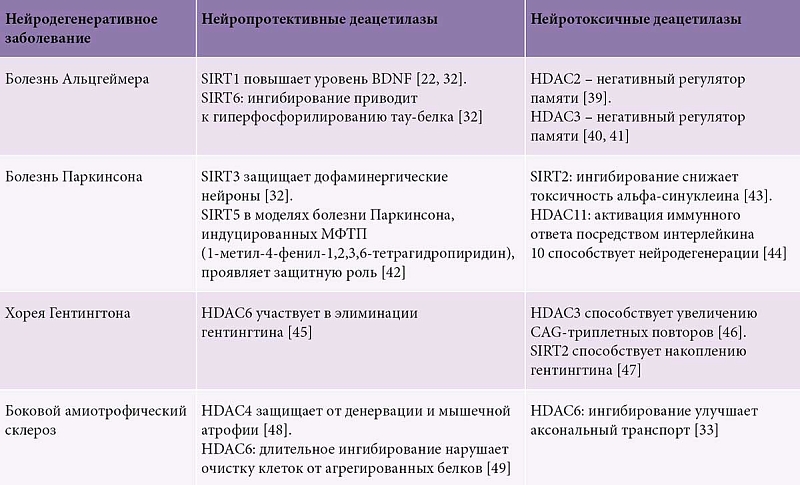
Семейство гистоновых деацетилаз (histone deacetylases, HDAC) представляет собой группу ферментов, критически важных для эпигенетической регуляции работы генома (рисунок) [16–26]. Их основная функция заключается в удалении ацетильных групп с остатков лизина в гистоновых белках, что приводит к конденсации хроматина и репрессии транскрипции [27, 28]. Нарушение баланса ацетилирования гистоновых и негистоновых белков лежит в основе патогенеза нейродегенеративных заболеваний [29–32].
HDAC-мишени при нейродегенеративных заболеваниях
Факторы Яманаки – набор транскрипционных факторов (Oct4, Sox2, Klf4, c-Myc, далее – OSKM), способных перепрограммировать зрелые соматические клетки в состояние плюрипотентности [33], то есть возвращать их в эмбриональное, недифференцированное состояние. Изначально технология, за разработку которой в 2012 г. Синья Яманака и Джон Гердон получили Нобелевскую премию, использовалась для получения индуцированных плюрипотентных стволовых клеток. Однако последние исследования показали, что кратковременное и контролируемое воздействие этих факторов может привести не к полной передифференцировке клетки, а к ее эпигенетическому омоложению, при котором все накопленные факторы экспрессии генов, формирующие предыдущие эпигенетические блокады, стираются [34].
Связь между OSKM и HDAC до конца не установлена. Тем не менее на данный момент достоверно известно, что SIRT1 (silent information regulator 1), деацетилаза гистонов III класса, регулирует и модулирует активность Oct4 и Sox2 [35]. Дальнейший поиск общих молекулярных механизмов регуляции может быть обусловлен общими мишенями нейрогенеза [35, 36], а также влиянием на эпигенетические часы, напрямую контролируемые деацетилазами, в частности сиртуинами [37].
Способность OSKM обращать вспять некоторые особенности старения тканей млекопитающих продемонстрирована методом эпигенетического программирования на 5ХFAD-трансгенных мышах, склонных к накоплению бета-амилоида (модель болезни Альцгеймера у людей). В исследовании интермиттирующая индукция OSKM у мышей 5ХFAD снижала эпигенетический возраст клеток зубчатой извилины гиппокампа [38]. Транзитная экспрессия OSKM в эмбриональном мозге привела к резкому увеличению пролиферации нейрональных предшественников (Pax6+, Sox2+), что в итоге вызвало увеличение неокортекса за счет роста числа нейронов и глиальных клеток. У трансгенных мышей эпигенетическое репрограммирование привело к уменьшению бета-амилоидных бляшек в гиппокампе. При этом экспрессия гена PSEN1 (ген, кодирующий пресенилин 1) не изменилась (таблица) [22, 32, 33, 39–49]. Таким образом, факторы Яманаки катализировали существующие механизмы элиминации бета-амилоида. Протеомный анализ показал, что индукция OSKM у мышей 5XFAD нормализовала патологические изменения в белках, связанных с клеточным метаболизмом.
Проблема онкогенности классических факторов Яманаки
Метод интермиттирующей индукции OSKM основан на результатах предыдущих экспериментов, показавших, что постоянная экспрессия факторов Яманаки приводит к формированию тератом [50–53]. Онкогенность прежде всего обусловлена их фундаментальной ролью в нарушении контроля клеточного роста и дифференцировки. Два фактора, c-Myc и Klf4, являются установленными онкогенами, а все четыре фактора активны в различных типах рака [52, 53]. Общий биологический принцип возникновения новообразований базируется на экспрессии и неконтролируемом делении плюрипотентных клеток. Сигнальные пути, например Wnt/бета-катенин, регулируемые OSKM, участвуют как в поддержании плюрипотентности, так и в формировании раковых клеток [53, 54].
Помимо интермиттирующей индукции разработан модифицированный вариант «коктейля Яманаки», в котором отсутствует главный онкоген – c-Myc. Данная модификация совмещает три классических фактора Яманаки и TERT-ген, кодирующий теломеразную обратную транскриптазу [55].
Потенциал к нейрорегенерации и новый молекулярный каскад экспрессии продемонстрированы на примере регенерации зрительного нерва декоративной рыбки Данио-рерио [56]. Ключевым моментом работы является демонстрация быстрой и последовательной активации каскада, включающего HSF1 (heat shock factor protein 1) и OSKM, в сетчатке Данио-рерио после повреждения зрительного нерва. Показано, что экспрессия мРНК HSF1 является наиболее ранним откликом и значительно возрастает через 30 минут после травмы, достигая пика к шести часам и оставаясь повышенной в течение 24 часов. Иммуногистохимический анализ подтвердил, что белок HSF1 присутствует во всех ядерных слоях сетчатки. За активацией HSF1 следует быстрая индукция генов трех факторов Яманаки – Klf4, Oct4 и Sox2. Экспрессия их мРНК достоверно увеличивается в течение 1–3 часов после повреждения зрительного нерва. При этом отмечаются различия в кинетике и локализации: Klf4 активируется быстрее и транзиторно. В то же время Sox2 демонстрирует более длительную экспрессию, распространяясь из ганглионарного слоя на все ядерные слои сетчатки.
Влияние кетоновых тел на нейродегенерацию
Во время голодания или ограничения углеводов печень катаболизирует мобилизованный жир из жировой ткани в кетоновые тела, которые попадают в кровоток, достигают мозга и поддерживают его биоэнергетику. Однако механическое влияние кетоновых тел выходит за рамки генерации аденозинтрифосфата. Бета-гидроксибутират, основное кетоновое тело по концентрации в крови, повышает митохондриальное дыхание и снижает фосфорилирование Akt в сигнальном пути PI3K – Akt – mTOR в нейронах (первичные нейроны крысы и клеточная линия SH-SY5Y), но не в астроцитах. В астроцитах бета-гидроксибутират, напротив, ремоделирует биоэнергетику, увеличивая максимальную дыхательную способность [57]. Бета-гидроксибутират специфически повышает экспрессию ключевых факторов репрограммирования Яманаки в нейрональных клетках (линия SH-SY5Y), но не в астроцитах. В клетках SH-SY5Y хроническое воздействие бета-гидроксибутирата достоверно увеличивает уровень мРНК Oct4. Индукция OSKM в данном контексте не направлена на приобретение плюрипотентности, это их классическая функция. Повышенная экспрессия OSKM ассоциируется с вхождением клетки в состояние обратимого покоя. Это состояние характеризуется замедлением клеточного цикла (показано на клетках SH-SY5Y через накопление в фазе G0–G1), снижением общей метаболической активности и подавлением пролиферативного сигнального пути PI3K – Akt – mTOR.
Заключение
Эпигенетические механизмы возникновения нейродегенеративных заболеваний сложны и до конца не изучены. На данный момент на эпигенетической карте биологических реакций много белых пятен. Дальнейшие исследования должны быть направлены на оценку роли факторов Яманаки в различных каскадах экспрессии, связи с другими эпигенетическими механизмами регуляции. Терапия OSKM обладает огромным потенциалом, поскольку использует механизмы саморегуляции макроорганизма. Необходима экспериментальная оценка потенциальной комбинированной эпигенетической терапии OSKM и ингибиторами деацетилаз гистонов. Перспективным направлением считается также метаболическая эпигенетика. Такие соединения, как бета-гидроксибутират, выступают в роли эндогенных ингибиторов HDAC, что объясняет нейропротекторный потенциал кетогенной диеты.
Несмотря на значительный прогресс, остаются ключевые проблемы – обеспечение селективности и специфичности эпигенетических препаратов и достижение долговременного и контролируемого эффекта без побочных последствий. Будущие исследования должны быть направлены на создание методов комбинированной терапии, интегрирующих эпигенетическую модуляцию с традиционными подходами, а также на разработку персонализированных схем лечения на основе эпигенетических биомаркеров.
Таким образом, эпигенетика не только углубляет наше понимание фундаментальных основ нейродегенерации, но и открывает путь к разработке принципиально новых, патогенетически обоснованных и потенциально обратимых методов лечения, которые позволят решить одну из самых серьезных проблем современной медицины.
E.T. Guseynov
I.M. Sechenov First Moscow State Medical University
Contact person: Eldar T. Guseynov, eldartar@outlook.com
Neurodegenerative diseases are associated with profound epigenetic dysregulation that disrupts the expression of genes critical to the functioning of neurons. Based on the analysis of modern literature data, it has been established that epigenetic blockade mediated by HDAC2 overexpression is a key pathogenetic link. Controlled reprogramming by Yamanaka factors and metabolic modulation with beta-hydroxybutyrate are promising areas of therapy.



Уважаемый посетитель uMEDp!
Уведомляем Вас о том, что здесь содержится информация, предназначенная исключительно для специалистов здравоохранения.
Если Вы не являетесь специалистом здравоохранения, администрация не несет ответственности за возможные отрицательные последствия, возникшие в результате самостоятельного использования Вами информации с портала без предварительной консультации с врачом.
Нажимая на кнопку «Войти», Вы подтверждаете, что являетесь врачом или студентом медицинского вуза.