Фенотипическая вариабельность синдрома Швахмана – Даймонда: клиническое наблюдение
- Аннотация
- Статья
- Ссылки
- English
Введение
Синдром Швахмана – Даймонда (СШД) представляет собой редкое мультисистемное генетическое заболевание, наследуемое по аутосомно-рецессивному типу, характеризующееся изменениями костного скелета, дисфункцией костного мозга, липоматозом поджелудочной железы (ПЖ) и, как следствие, ее внешнесекреторной недостаточностью [1, 2].
Как правило, синдром клинически проявляется с самого рождения или в раннем детстве [3]. Проведенные исследования показали, что причиной заболевания служит мутация гена Shwachman-Bodian-Diamond syndrome (SBDS), расположенного на длинном плече хромосомы 7, в цитогенетической позиции 7q11 [4]. Ген SBDS кодирует белок, который участвует в созревании рибосом, стабилизации микротрубочек и полимеризации актина, а также играет роль в пролиферации клеток, митозе и стромальном микроокружении костного мозга [5, 6].
В 1961 г. французскими врачами К. Незелофом и М. Уотчи впервые была описана врожденная липоматозная гипоплазия ПЖ у двоих детей [1, 4]. В 1964 г. М. Бодиан и соавт. (1964) опубликовали статью «Врожденная гипоплазия экзокринной части поджелудочной железы» [7]. В том же году Г. Швахман и соавт. описали синдром недостаточности ПЖ и дисфункции костного мозга у троих детей из одной семьи, наблюдавшихся в клинике муковисцидоза в Бостоне [8, 9]. Этот синдром позже был назван синдромом Швахмана – Даймонда.
В 2001 г. канадскими исследователями при полногеномном сканировании семей с синдромом Швахмана – Даймонда был идентифицирован ген SBDS, расположенный в хромосоме 7 и показавший связь с заболеванием [7]. Ген SBDS кодирует белок, который участвует в созревании рибосом, стабилизации микротрубочек и полимеризации актина, а также играет роль в пролиферации клеток, митозе и стромальном микроокружении костного мозга [5, 6]. У большинства (90%) пациентов с клинической картиной СШД наблюдаются мутации в гене SBDS [10]. У 10–20% пациентов с клиническим диагнозом СШД известные мутации могут быть не выявлены, однако было обнаружено, что мутации генов DNAJ21 и EFL1 вызывают СШД-подобный синдром. Эти два гена кодируют белки, участвующие в пути биогенеза рибосом [9–11].
Белок SBDS в небольшом количестве содержится практически во всех органах и тканях человеческого организма (печень, легкие, почки, головной мозг, ткани глаза), а в наибольшем количестве обнаруживается в клетках ПЖ, костного мозга, костной ткани. Этим обусловлена мультисистемность поражения [4, 12]. Наиболее характерными признаками синдрома являются нарушенная экзокринная функция ПЖ и гематологические аномалии. Экзокринная недостаточность ПЖ обычно проявляется в младенчестве, однако с возрастом может наблюдаться некоторое улучшение. Гематологические аномалии рассматриваются как генетический синдром недостаточности костного мозга. Это проявляется постоянной или периодической нейтропенией и повышенной восприимчивостью к инфекциям [7].
Высокое содержание белка SBDS в ПЖ обусловливает одно из основных проявлений заболевания – экзокринную недостаточность ПЖ вследствие жировой инфильтрации [5]. Тем не менее клетки островков Лангерганса и структура протоков остаются сохраненными [13, 14]. Вследствие экзокринной недостаточности ПЖ развиваются синдром мальабсорбции и дефицит жирорастворимых витаминов (A, D, E, K), что ведет к снижению нутритивного статуса и задержке физического развития [12]. При классическом варианте СШД эти симптомы появляются с первого года жизни [17].
Другим классическим и самым частым проявлением СШД являются гематологические нарушения, связанные с дисфункцией костного мозга и проявляющиеся прежде всего нейтропенией, которая встречается практически у 100% пациентов и диагностируется еще в неонатальном периоде [4, 13]. Нейтропения может быть как персистирующей, так и рецидивирующей, когда содержание нейтрофилов колеблется от крайне низкого до нормального уровня [12, 13, 15]. Поэтому пациенты с СШД чаще остальных подвержены различным рецидивирующим бактериальным, вирусным и грибковым инфекциям, включая средний отит, синусит, бронхопневмонию, остеомиелит, сепсис и кожные инфекции. Обширный сепсис является хорошо известным фатальным осложнением этого заболевания, особенно в раннем возрасте [15]. Другие гематологические проявления включают апластическую анемию, тромбоцитопению и повышение уровня фетального гемоглобина. Отмечается высокий риск развития миелодиспластического синдрома и острого миелоидного лейкоза [4].
Поражения скелета встречаются у 45% пациентов с СШД и связаны со снижением минеральной плотности костной ткани. Наиболее часто наблюдаются остеопения, остеопороз, метафизарная дисплазия, дистрофия и аномалии развития грудной клетки и позвоночника, вальгусная или варусная деформация стоп, низкорослость [6].
По клиническим данным, у 50–75% пациентов встречается патология печени, проявляющаяся в гепатомегалии и повышении уровня печеночных аминотрансфераз в биохимическом анализе крови. Чаще всего выявляется у детей и, как правило, проходит с возрастом [4, 15].
Проявления пищевой аллергии встречаются у 30–35% больных, что серьезно затрудняет коррекцию нутритивного статуса пациента [4, 12].
Описаны тяжелые кардиологические нарушения при СШД, проявляющиеся гипотонией, некрозом или фиброзом миокарда, развитием сердечной недостаточности. Тяжелые формы кардиологических проявлений приводят к летальному исходу в младенчестве [5, 16].
Пациенты с СШД могут испытывать значительную задержку в физическом, психомоторном и интеллектуальном развитии [14].
Диагноз СШД ставится на основании клинических данных и результатов лабораторно-инструментального обследования, позволяющих выявить внешнесекреторную недостаточность ПЖ, гематологические отклонения, костные аномалии и другие нарушения, характерные для этого синдрома [13]. Заболевание обычно диагностируется в течение первых лет жизни, однако известны случаи, когда диагноз может быть установлен в подростковом возрасте или даже у взрослых [13].
На сегодняшний день генетическое исследование ДНК с определением мутаций в гене SBDS является признанным золотым стандартом для подтверждения диагноза СШД [1].
По предварительным данным, распространенность заболевания составляет 1:50 000 – 1:76 000 населения. Распространенность СШД в Российской Федерации на данное время неизвестна в связи с его фенотипической вариабельностью и редкостью [4, 12]. Радикального лечения таких пациентов на данный момент не существует, однако важно проводить соответствующую симптоматическую медикаментозную терапию для улучшения и облегчения их состояния [1].
Основным методом профилактики и лечения гематологических нарушений в настоящее время является применение гранулоцитарного колониестимулирующего фактора (G-CSF/Г-КСФ), что способствует улучшению качества жизни и прогноза заболевания. В редких случаях пациентам с СШД проводят трансплантацию костного мозга [17].
Коррекция экзокринной недостаточности является важным этапом в лечении данного заболевания. Она осуществляется с использованием ферментных препаратов, которые подбираются в зависимости от степени тяжести заболевания; лечение проводится пожизненно. Для восстановления трофологического статуса применяются нутритивная поддержка, витаминотерапия [12].
Вариации клинического течения заболевания весьма многообразны, а его признаки часто появляются асинхронно, что значительно затрудняет процесс диагностики [17] и, соответственно, своевременное назначение лечения.
Далее приведены два клинических наблюдения пациентов с СШД с наличием как сходств, так и различий проявлений.
Клиническое наблюдение 1
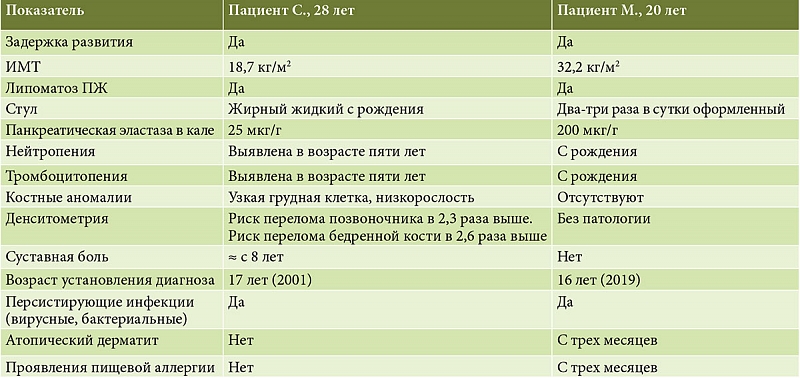
Пациент С., 28 лет, проходил стационарное лечение в отделении патологии поджелудочной железы Центрального научно-исследовательского института гастроэнтерологии (ЦНИИГ) (в настоящее время – Московский клинический научный центр им. А.С. Логинова) в 2012 г. Был госпитализирован с жалобами на метеоризм, урчание в животе, возникающие после приема пищи, кашицеобразный стул 1–2 раза в сутки и быструю утомляемость. Также пациента беспокоила боль в коленных, локтевых суставах и в шейном отделе позвоночника. При выходе на улицу, поездках в общественном транспорте пациент отмечал тревогу и страх, снижение настроения и апатию.
Из анамнеза известно, что родился в срок, масса при рождении – 3300 г, рост – 54 см, однако с рождения отмечался жирный стул. В массе тела и росте прибавлял плохо, наблюдались часто рецидивирующие тяжелые бактериальные и вирусные инфекции с необходимостью стационарного лечения, несколько отставал в психомоторном развитии. В возрасте двух лет на основании повышения уровня натрия в кале и ногтях был установлен диагноз «муковисцидоз». В возрасте трех лет пациент был обследован в научно-исследовательском институте педиатрии, диагноз муковисцидоза был отвергнут в связи с отрицательной потовой пробой и отсутствием изменений со стороны органов дыхания. Состояние трактовалось как первичная недостаточность панкреатической липазы. Тогда же при рентгенологическом исследовании кистей был диагностирован остеопороз. В возрасте пяти лет впервые были выявлены нейтропения (16%), тромбоцитопения (менее 180 × 103/л) и снижение уровня гемоглобина до 112 г/л. В последующем наблюдалась персистенция хламидийной, цитомегаловирусной, герпетической инфекции, вируса Эпштейна – Барр. Пациенту проводили терапию макролидами, Полиоксидонием, противовирусными препаратами с положительным эффектом. Уровень лейкоцитов составлял 2–3,1 × 109/л. В возрасте 17 лет в Детской городской клинической больнице им. Н.Ф. Филатова был диагностирован СШД. Клинический диагноз был подтвержден молекулярно-генетическим исследованием: выявлена мутация в гене SBDS с.183–184 ТА–СТ, с.258+2Т–С. Уровень фекальной эластазы на момент диагностики составлял 27 мкг/г, что свидетельствует о тяжелой степени экзокринной недостаточности ПЖ. Была назначена ферментозаместительная терапия: панкреатин до 100 тыс. ед. в сутки. К 19 годам пациент имел рост 156 см, весил 44 кг. В январе 2011 г. (26 лет) отметил ухудшение состояния в виде нарастания слабости, головокружения, снижения артериального давления. По данным магнитно-резонансной томографии головного мозга патологии не выявлено. По результатам эхокардиографии были выявлены малые аномалии развития сердца: пролапс трикуспидального клапана первой степени, пролапс митрального клапана с регургитацией первой степени, дополнительная трабекула. Рентгенография шейного отдела позвоночника: межпозвонковый хондроз, нестабильность сегментов. В июне 2011 г. находился на лечении в Клинике неврозов с диагнозом «органическое тревожное расстройство в связи со смешанным заболеванием». В возрасте 27 лет пациент обследован в Гематологическом научном центре. По данным миелограммы картина соответствует тяжелой гипоплазии костного мозга. Назначена терапия Г-КСФ (филграстим) 150 мкг.
Для уточнения состояния ПЖ и коррекции дозы ферментозаместительной терапии пациент был госпитализирован в ЦНИИГ.
Объективно: рост – 162 см, вес – 49 кг, индекс массы тела (ИМТ) – 18,7 кг/м2. Телосложение астеническое, узкая грудная клетка. Перкуторный звук ясный легочный. В легких везикулярное дыхание, хрипов нет. Частота дыхания (ЧД) – 17/мин. Тоны сердца ясные, ритм правильный, частота сердечных сокращений (ЧСС) – 76/мин. Артериальное давление (АД) – 90/60 мм рт. ст. Живот обычной конфигурации, в акте дыхания участвует всеми отделами. При пальпации мягкий, умеренно болезненный в эпигастральной и околопупочной областях. Печень и селезенка не увеличены. Стул кашицеобразный, 3–4 раза в день.
В клиническом анализе крови выявлена лейкопения – 2,2 × 109/л, тромбоцитопения – 125 × 109/л, эритропения – 3,87 × 1012/л, уровень гемоглобина – 119 г/л.
В биохимическом анализе крови отмечено снижение уровня альфа-амилазы до 14 Ед/л. Нарушение углеводного обмена не выявлено.
При определении уровня панкреатической эластазы в кале подтверждена тяжелая экзокринная недостаточность ПЖ: уровень 25 мкг/г при норме более 200 мкг/г.
По данным ультразвукового исследования брюшной полости: билиарный сладж в желчном пузыре, диффузные изменения ПЖ, утолщение стенок главного протока ПЖ. Конкременты почек.
При эзофагогастродуоденоскопии – признаки гастрита, дуоденита.
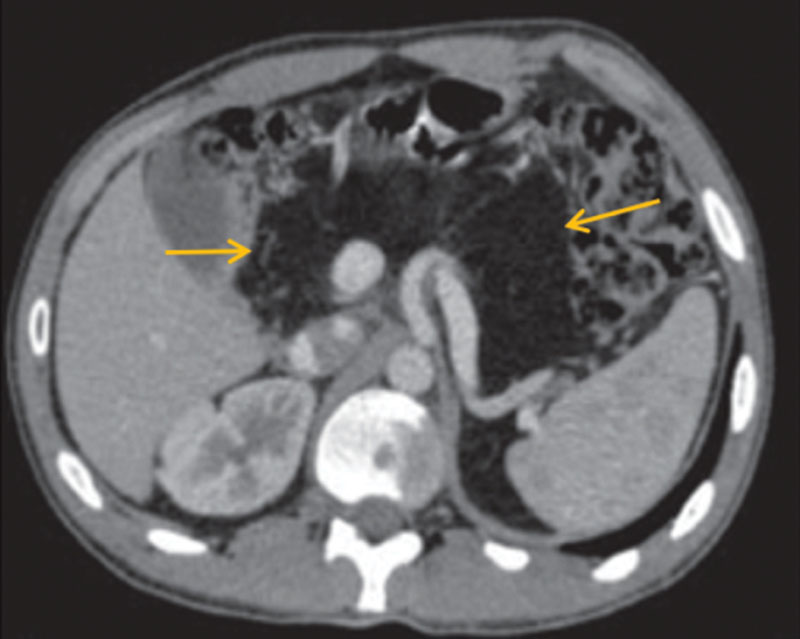
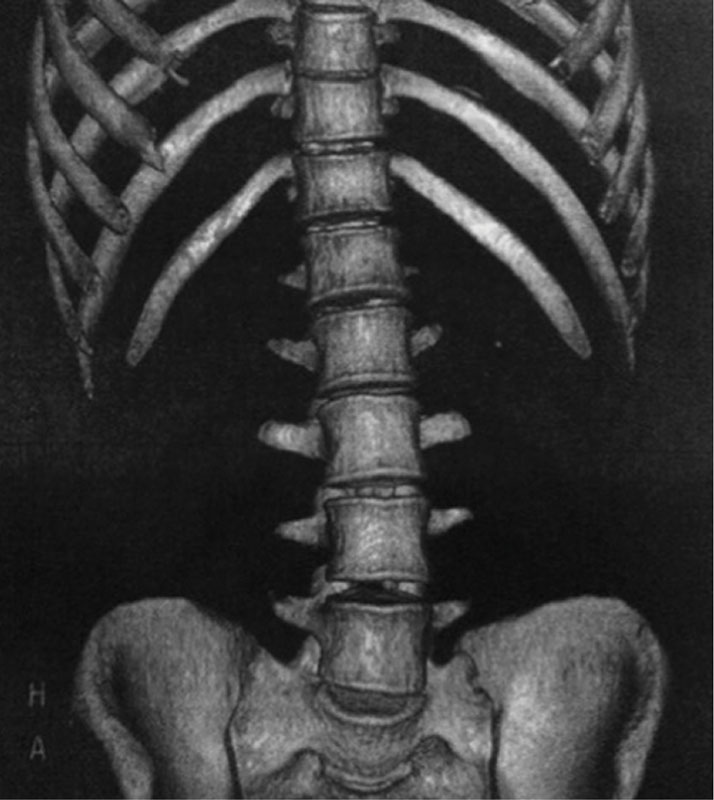
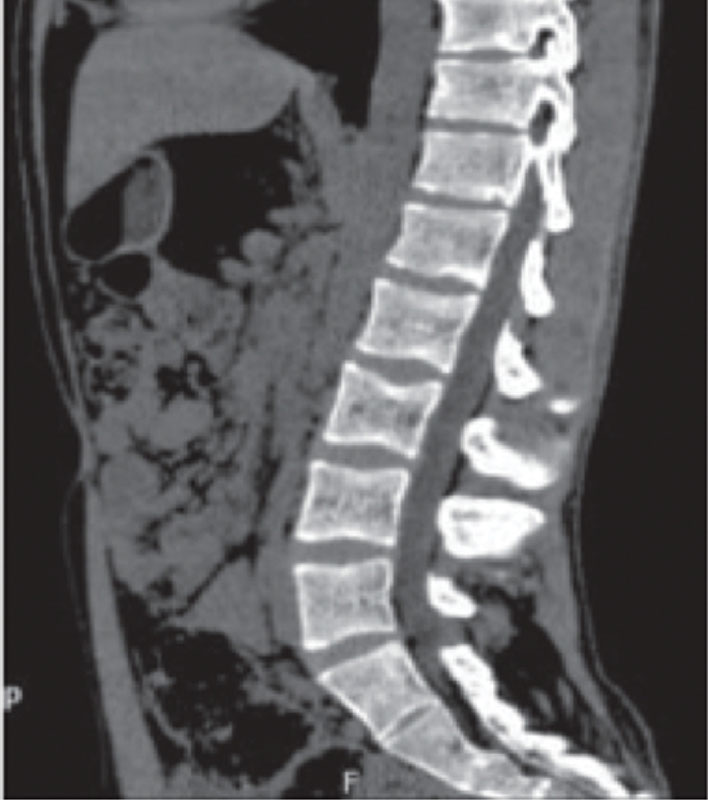
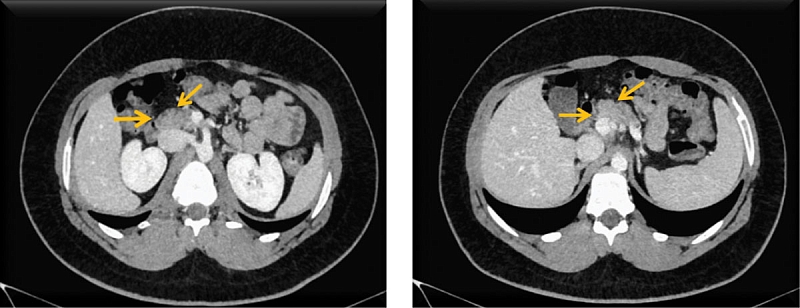
Компьютерная томография органов брюшной полости: признаки жировой инфильтрации ПЖ (рис. 1), лимфаденопатия брыжейки тонкой кишки (рис. 2). Сколиотическая деформация позвоночного столба (рис. 3) и локальный остеопороз отдельных позвонков (рис. 4).
Денситометрия – признаки остеопении: масса костной ткани поясничного отдела позвоночника (L1–L4) на 11% ниже нормы (Т-критерий – 1,1). Риск перелома позвоночника в 2,3 раза выше. Масса костной ткани бедренной кости на 11% ниже нормы (Т-критерий – 1,1). Риск перелома бедренной кости в 2,6 раза выше.
Пациент был выписан с диагнозом: «СШД (мутация в гене SBDS с.183–184 ТА–СТ, с.258+2 Т–С). Липоматоз ПЖ. Внешнесекреторная недостаточность ПЖ тяжелой степени. Вторичная остеопения. Наследственная нейтропения с гипоплазией костного мозга. Малые аномалии развития сердца – пролапс митрального клапана первой степени, пролапс трикуспидального клапана первой степени».
При выписке были даны рекомендации по увеличению объема заместительной полиферментной терапии до 200 тыс. ед. в сутки, увеличению суточного калоража и включению в питание жирорастворимых витаминов и микро- и макроэлементов, своевременному антибактериальному лечению инфекций, а также по применению рекомбинантного Г-КСФ, согласно рекомендации гематолога, для стимуляции лейкопоэза.
Клиническое наблюдение 2
Пациент М., 20 лет, проходил обследование в МКНЦ им. А.С. Логинова в 2023 г. Был госпитализирован с жалобами на умеренную слабость, вздутие в животе. Из анамнеза известно, что на первом году жизни отмечались частые вирусные заболевания, по поводу которых неоднократно проходил стационарное лечение, была выявлена хроническая лейконейтропения. Тогда же был выявлен атопический дерматит. В возрасте семи месяцев был обследован аллергологом, выявлена пищевая непереносимость белка коровьего молока, глютена, мяса курицы, яиц. По рекомендации аллерголога пациент находился на элиминационной диете в течение нескольких лет. В дальнейшем явления атопического дерматита регрессировали. Кроме того, отмечались жирный стул со слизью, периодически «овечий», и недостаточная прибавка массы тела. При обследовании в возрасте четырех лет в клиническом анализе крови отмечался агранулоцитоз, уровень гемоглобина составлял 128 г/л, лейкоциты – 4,7 × 109/л, нейтрофилы – 3%, лимфоциты – 71%, моноциты – 14%. В биохимическом анализе крови выявлены отклонения уровней щелочной фосфатазы – 620 Ед/л, аланинаминотрансферазы – 108 Ед/л, аспартатаминотрансферазы – 52 Ед/л, лактатдегидрогеназы – 377 Ед/л. В анализе крови на оппортунистические инфекции – повышение IgG к вирусу Эпштейна – Барр. Пациенту был назначен курс Кипферона и Энгистола. В дальнейшем пациент периодически проходил стационарное лечение и обследование в связи с нейтропенией, тромбоцитопенией и персистирующими инфекциями.
В 14 лет (2017 г.) в миелограмме впервые было выявлено минимальное сужение нейтрофильного ростка, эритроидный росток раздражен, тромбоцитарный – в пределах нормы. В 16 лет (2019 г.) было проведено цитогенетическое исследование костного мозга, обнаружена делеция хромосомы 20 (20q). Установлен диагноз миелодиспластического синдрома. В миелограмме – сужение нейтрофильного ростка. В клиническом анализе крови сохранялись нейтропения, тромбоцитопения, лейкопения. В Национальном медицинском исследовательском центре детской гематологии, онкологии и иммунологии им. Дмитрия Рогачева пациенту было проведено генетическое исследование. Выявлены две мутации в гене SBDS (c.258+2T>C и c.184A>T, в компаунд-гетерозиготном состоянии, подтверждены методом прямого секвенирования по Сенгеру), характерные для СШД. Установлен диагноз: «Синдром Швахмана – Даймонда (мутации в гене SBDS c.258+2T>C и c.184A>T), протекающий с первичным иммунодефицитом, хронической нейтропенией, экзокринной недостаточностью ПЖ. Миелодиспластический синдром (делеция 20q)». В лабораторных исследованиях отмечалась нейтропения – 38%, амилаза сыворотки крови составляла 20 Ед/л, щелочная фосфатаза – 190 Ед/л. Уровень фекальной эластазы – 376 мкг/г. В дальнейшем при цитогенетическом исследовании костного мозга выявлена имевшаяся ранее делеция 20q (20/100 клеток), наличие миелодиспластического синдрома на момент исследования не подтвердилось. К 17 годам пациент имел рост 166 см и вес 83 кг.
Объективно: рост – 168 см, вес – 91 кг, ИМТ – 32,2 кг/м2. Кожные покровы и видимые слизистые обычной окраски. Телосложение гиперстеническое. Костно-мышечная система развита правильно. Гинекомастия. Перкуторный звук ясный легочный. В легких везикулярное дыхание, хрипов нет. ЧД – 16/мин. Тоны сердца ясные, ритм правильный, ЧСС – 76/мин. АД – 130/85 мм рт. ст. Живот обычной конфигурации, в акте дыхания участвует всеми отделами, при пальпации мягкий, безболезненный, увеличен за счет жировой клетчатки. Печень пальпируется по краю реберной дуги. Селезенка не пальпируется. Стул 2–3 раза в сутки, оформленный, без патологических примесей.
В клиническом анализе крови: тромбоцитопения – 179 × 109/л, лейкопения – 3,73 × 109 /л и нейтропения: абсолютное количество – 1,06 × 109/л, относительное количество – 28,3%.
В биохимическом анализе крови: снижение уровня альфа-амилазы – 20,2 МЕ/л, холестерина – 2,70 ммоль/л, повышение щелочной фосфатазы – 131,4 МЕ/л, остальные показатели в пределах референсных значений. Панкреатическая эластаза в кале – 200 мкг/г, что свидетельствует о сохранной внешнесекреторной функции ПЖ.
По данным ультразвукового исследования брюшной полости: признаки диффузных изменений паренхимы печени и ПЖ.
Эзофагогастродуоденоскопия: недостаточность кардии. Дуоденогастральный рефлюкс.
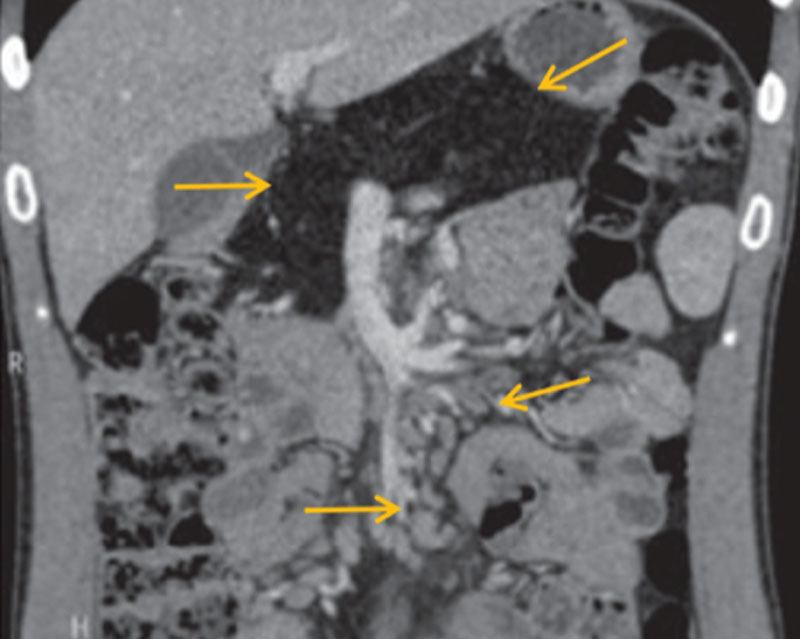
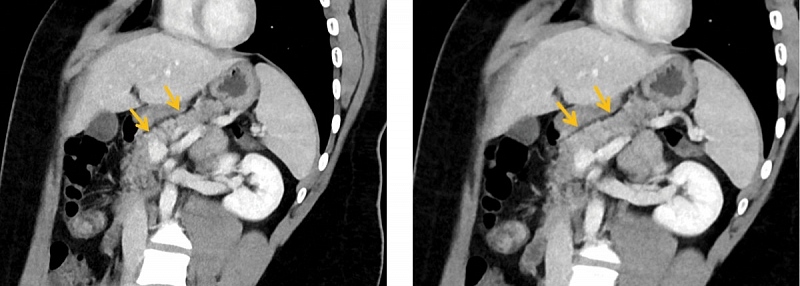
По данным компьютерной томографии признаки жировой дегенерации ПЖ. Поджелудочная железа обычно расположена, не увеличена, размерами: головка – 29 мм, тело – 19 мм, хвост – 17 мм. Контуры ПЖ четкие и ровные. Плотность паренхимы составляет 28 ед.Н. Структура дольчатая, контрастируется равномерно, имеются жировые прослойки в головке и хвосте. Главный панкреатический проток не расширен. Парапанкреатическая клетчатка не изменена (рис. 5, 6).
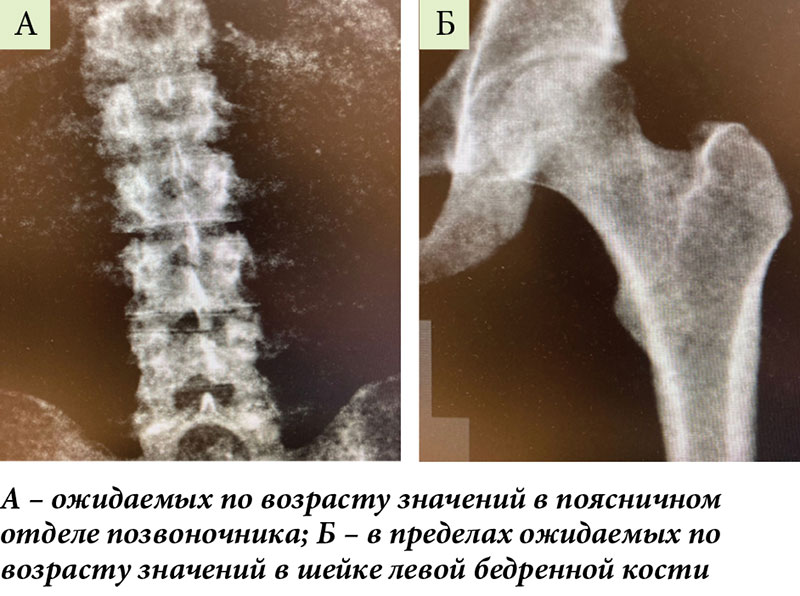
По данным денситометрии, значения минеральной плотности костной ткани в поясничном отделе позвоночника и в шейке левой бедренной кости находятся в пределах ожидаемых по возрасту значений (рис. 7).
Диагноз: «Синдром Швахмана – Даймонда (мутации в гене SBDS c.258+2T>C и c.184A>T). Первичный иммунодефицит. Хроническая нейтропения». Сопутствующий диагноз: «Экзогенно-конституциональное ожирение первой степени (ИМТ 32,2 кг/м2). Поливалентная сенсибилизация: пищевая, пыльцевая и бытовая».
С учетом наличия липоматоза ПЖ, подтвержденного результатами компьютерной томографии, при удовлетворительной внешнесекреторной функции ПЖ пациенту рекомендован прием полиферментных препаратов в капсулах с минисубстанциями в минимальной дозе: по 10 000 Ед два раза во время завтрака и ужина и по 25 000 Ед во время обеда. Контроль уровня фекальной эластазы один раз в год с целью своевременного выявления и коррекции возможного нарушения внешнесекреторной функции ПЖ.
Обсуждение
В представленных клинических наблюдениях отмечена разница в фенотипических проявлениях синдрома Швахмана – Даймонда у пациентов с идентичным генотипом (таблица). Основное различие наблюдается в функциональных возможностях ПЖ: если в первом клиническом случае у пациента (С., 28 лет) имеется явная внешнесекреторная недостаточность, то во втором (М., 20 лет) – явных признаков внешнесекреторной недостаточности нет. При этом липоматоз ПЖ как характерное проявление СШД [18] выявлен у обоих пациентов. Нельзя исключить, что именно этот факт явился определяющим и в других проявлениях, таких как остеопороз, скелетные аномалии у первого и их отсутствие у второго пациента. Из анамнеза видно, что синдром мальабсорбции имел место у пациента С. с раннего детства и отсутствовал у пациента М. Этим можно объяснить и большее отставание в физическом развитии первого пациента, и выраженные различия в ИМТ (см. таблицу) на момент обследования. В связи с наличием внешнесекреторной недостаточности ПЖ тяжелой степени пациенту С. дополнительно проводили высокодозовую ферментозаместительную терапию.
У обоих пациентов с момента рождения наблюдались частые вирусные инфекции вследствие нейтропении и нарушений иммунного статуса, что также является патогномоничным признаком СШД [6]. Основным методом лечения гематологических нарушений у обоих пациентов явилось применение Г-КСФ.
Следует заметить, что обоим пациентам диагноз был установлен в подростковом возрасте, хотя заболевание проявлялось с младенчества. Проблема диагностики и лечения пациентов с клиническим диагнозом СШД обусловлена широкой вариабельностью фенотипа этого заболевания, которая может изменяться не только у разных пациентов, но даже у одного и того же человека с течением времени [10].
N.A. Neyasova, L.V. Vinokurova, PhD, E.A. Dubtsova, PhD, Yu.V. Embutnieks, PhD, I.V. Savina, K.A. Lesko, PhD, D.S. Bordin, PhD, Prof.
Contact person: Natalia A. Neyasova, n.neyasova@mknc.ru
Schwachman – Diamond syndrome is a rare genetic disease manifested mainly by extrinsic pancreatic insufficiency and neutropenia, as well as changes in the bone skeleton. The disease is characterized by phenotypic variability. The presented observations consider similar and different manifestations of the Schwachman – Diamond syndrome.
Уважаемый посетитель uMEDp!
Уведомляем Вас о том, что здесь содержится информация, предназначенная исключительно для специалистов здравоохранения.
Если Вы не являетесь специалистом здравоохранения, администрация не несет ответственности за возможные отрицательные последствия, возникшие в результате самостоятельного использования Вами информации с портала без предварительной консультации с врачом.
Нажимая на кнопку «Войти», Вы подтверждаете, что являетесь врачом или студентом медицинского вуза.