Фиброкистозная болезнь печени: врожденный фиброз печени с комплексами фон Мейенбурга
- Аннотация
- Статья
- Ссылки
- English
![Классификация фиброкистозной болезни печени по диаметру пораженных протоков [по 1]](/upload/resize_cache/iblock/876/800_800_1/Bueverov1.jpg)
Введение
Термин «фиброкистозная болезнь печени» (ФКБП) объединяет группу генетически детерминированных заболеваний желчевыводящих путей, характеризующихся аберрантным морфогенезом желчных протоков и приводящих к формированию сегментарных расширений и фиброзу. ФКБП характеризуется уникальным патогенезом, в основе которого лежит аномальное развитие эмбриональной протоковой пластинки. Протоковые пластинчатые мальформации, в зависимости от анатомического уровня, подразделяются на комплекс фон Мейенбурга, врожденный фиброз печени, болезнь и синдром Кароли, а также кисты холедоха (таблица) [1–3]. В 1961 г. D. Kerr впервые применил термин «врожденный фиброз печени» (ВФП) для описания отличного от цирроза септального фиброза печени, диагностируемого преимущественно в детском или подростковом возрасте [4].
ФКБП относится к гораздо более широкой группе болезней развития, обозначаемых как цилиопатии. Клеточные реснички могут быть подвижны или нет. Подвижные участвуют в регуляции транспорта жидкости через эпителиальный барьер; их дисфункция ассоциирована с такими патологическими состояниями, как бронхоэктазы, situs viscerum inversus и бесплодие. Неподвижные реснички – это сенсорные органеллы, экспрессируемые поляризованными эукариотическими клетками, включая холангиоциты и эпителиальные клетки почечных канальцев. Они содержат группу белков, опосредующих межклеточные взаимодействия. Цилиопатии, обусловленные дефектами неподвижных белков ресничек, приводят, в частности, к дисгенезии протоков, что может вести к развитию кистозных образований [5, 6].
Поражение гепатобилиарной системы обусловлено мутацией гена PKHD1, локализованного на коротком плече 6-й хромосомы. Этот ген отвечает за синтез белка фиброцистина, который формирует первичные реснички эпителиальных клеток, выстилающих желчные ходы. Фиброцистин также содержится в почечном эпителии, поэтому при аномалиях гена PKHD1 болезнь Кароли нередко сопровождается поликистозом почек. Тип наследования − аутосомно-рецессивный. Помимо нарушений морфогенеза, генетический дефект обусловливает активацию передачи сигналов β-катенина, что ведет к повышению секреции цитокинов и хемокинов (CXCL1, CXCL10, CXCL12, MCP-1, CTGF), которые способны привлекать макрофаги и мезенхимальные клетки в перибилиарную область, обеспечивая в конечном итоге прогрессирующее отложение коллагена вокруг измененных протоков [1, 3].
Для ВФП характерно развитие гепатоспленомегалии и портальной гипертензии в отсутствие значимых отклонений со стороны функции печени. Частота диагностики ВФП составляет приблизительно один случай на 10–20 тысяч новорожденных [2, 7]. Описания отдельных клинических случаев в основном относятся к новорожденным и детям с комбинацией ФКБП и поликистозной болезни почек [8–11]. Выделяют четыре клинических фенотипа ФКБП [1]:
- портально-гипертензионный – с гепатоспленомегалией, цитопенией, кровотечениями из варикозно-расширенных пищевода и желудка;
- холестатический – с кожным зудом, рецидивирующим холангитом, внутрипеченочным холелитиазом;
- смешанный;
- латентный.
В русскоязычной литературе на момент подготовки статьи (декабрь 2024 г. – январь 2025 г.) наблюдений портально-гипертензионного ВФП с комплексами фон Мейенбурга мы не встретили.
Клиническое наблюдение
Пациентка Л., 36 лет, домохозяйка. Ранее диагностированных хронических заболеваний и отягощенного по заболеваниям печени семейного анамнеза не имела, алкоголь в токсических дозах не употребляла. В 2023 г. при плановом обследовании впервые было обращено внимание на снижение уровня тромбоцитов до 74 тыс/мкл по Фонио при нормальных уровнях гемоглобина (129 г/л) и лейкоцитов (4,32 тыс/мкл) периферической крови. Жалоб на самочувствие пациентка не предъявляла, при физикальном исследовании были выявлены умеренно выраженные гепатомегалия и спленомегалия. При последующем обследовании в сентябре 2024 г. сохранялась тромбоцитопения, при биохимическом анализе крови отмечали нормальную активность аланинаминотрансферазы, аспартатаминотрансферазы, щелочной фосфатазы и увеличение активности гамма-глутамилтранспептидазы (ГГТП) до 2,5 норм. Поверхностный антиген вируса гепатита В и антитела к вирусу гепатита С не обнаруживались. Сывороточные уровни общего и прямого билирубина, общего белка, альбумина, протромбина, глюкозы, общего холестерина, триглицеридов, ферритина, церулоплазмина, альфа1-антитрипсина, иммуноглобулинов А, М и G, а также суточная экскреция меди с мочой находились в пределах референсных значений. При компьютерной томографии органов брюшной полости с внутривенным контрастированием в октябре 2024 г. были отмечены гепатоспленомегалия и неровность контуров печени, а также диспропорция сегментов печени, проявляющаяся гипертрофией 1-го и 4-го сегментов печени и гипотрофией 5-го и 6-го сегментов. Размеры печени составляли 236 × 120 × 120 мм, селезенки – 182 × 17 × 60 мм, воротной вены – 10 мм, селезеночной вены – 9 мм. Других патологических изменений органов брюшной полости, в т.ч. аномалий развития и тромбозов сосудов портальной венозной системы и патологических изменений паренхимы почек не было выявлено. Медиана жесткости печени при эластографии составила 14,6 кПа, что соответствовало стадии фиброза F4 по METAVIR. При эзофагогастродуоденоскопии варикозно расширенные вены пищевода или желудка отсутствовали. Была также проведена трепанобиопсия, которая позволила исключить гематологическое заболевание.
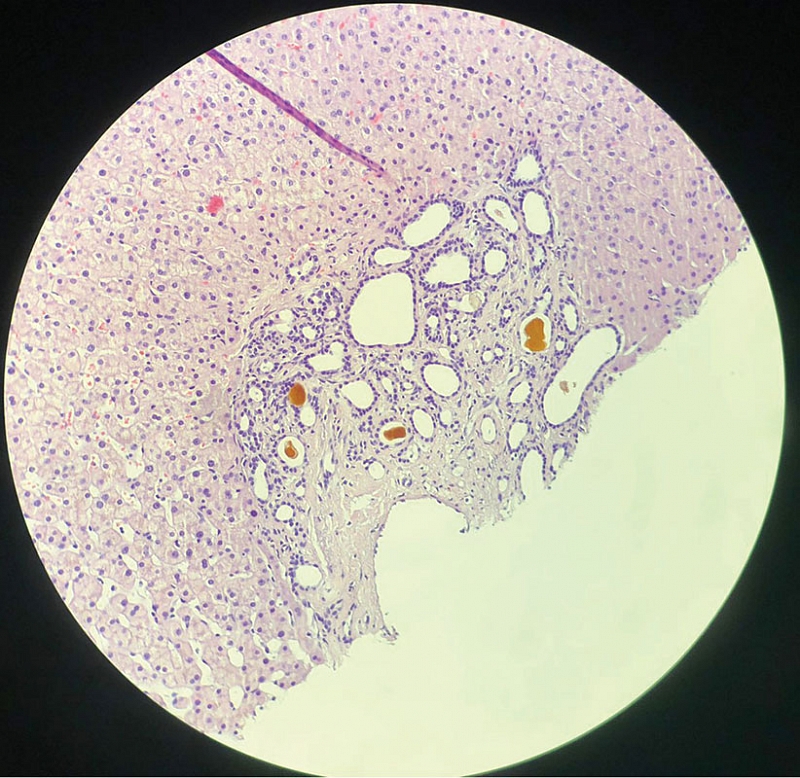
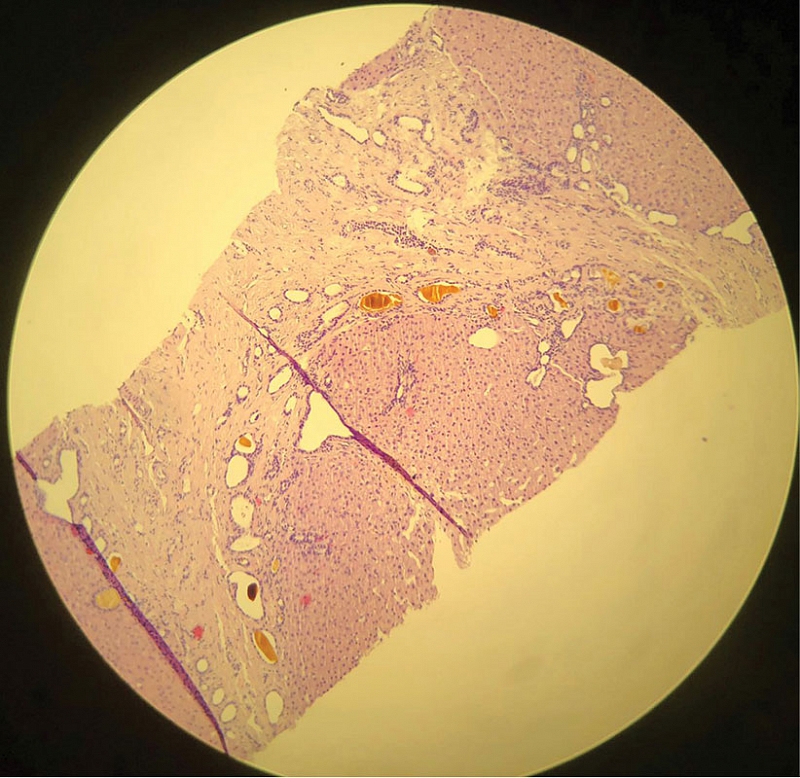
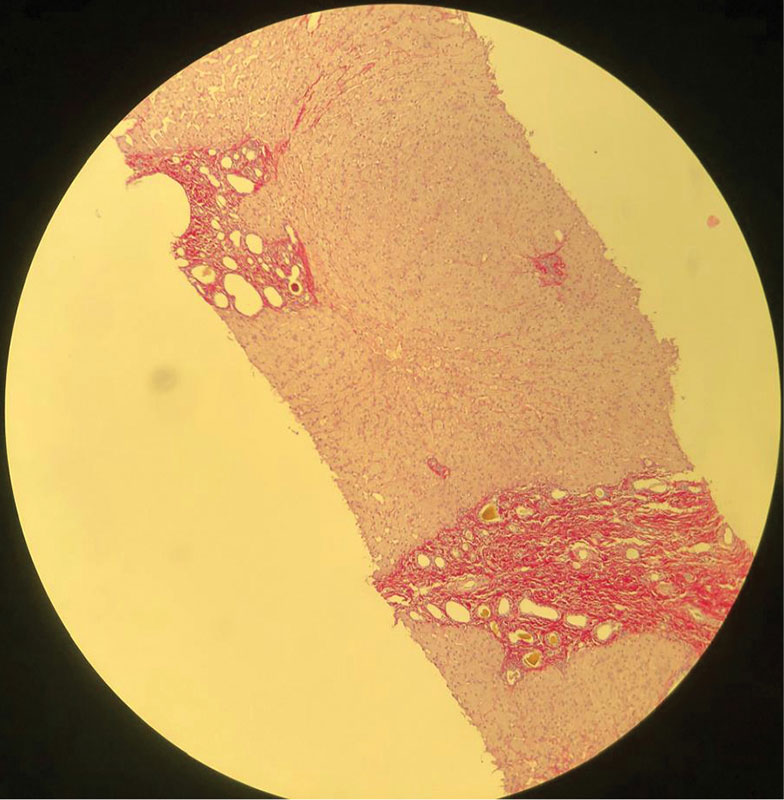
Таким образом, результаты проведенного обследования позволили предположить наличие хронического заболевания печени, осложненного портальной гипертензией. Вместе с тем не было выявлено специфических изменений лабораторных показателей, которые могли бы указывать на причину развития заболевания. С целью установления характера поражения печени в октябре 2024 г. выполнена пункционная биопсия печени. При гистологическом исследовании биоптата наблюдали большое количество мелких сливающихся островков малоклеточной фиброзной стромы с многочисленными тубулярными структурами, выстланными кубическим эпителием без признаков атипии, с просветами различного диаметра, формы и заполненными желчью. Гистологическая картина соответствовала множественным микрогамартомам (комплексы фон Мейенбурга) с кистозно расширенными протоками и интраканаликулярным холестазом (рис. 1–3).
Таким образом, результаты проведенного обследования позволили диагностировать ВФП с комплексами фон Мейенбурга. Назначена урсодезоксихолевая кислота (УДХК, Урдокса®) 750 мг/сут с целью возможного замедления пролиферации желчных протоков. С учетом риска прогрессирования портальной гипертензии и развития злокачественных опухолей печени пациентке рекомендовано наблюдение у гепатолога и трансплантолога.
Обсуждение
В нашем клиническом наблюдении мы представили редкий вариант ВФП, характеризовавшийся сочетанием аномалий развития фиброзной стромы печени и системы желчевыводящих протоков (комплексы фон Мейенбурга). Диагноз был установлен на основании специфической морфологической картины в биоптате печени. Заболевание протекало бессимптомно, значимых изменений лабораторных показателей, кроме секвестрационной тромбоцитопении умеренной степени выраженности и изолированного умеренного повышения активности ГГТП, не наблюдалось. Единственным клиническим проявлением заболевания стала гепатоспленомегалия, развившаяся вследствие пресинусоидальной портальной гипертензии, обусловленной прогрессирующим фиброзом печени. К особенностям данного клинического случая относится также отсутствие поликистозных изменений почек.
Эффективные стратегии замедления печеночного фиброгенеза на сегодняшний день отсутствуют. Ввиду невозможности проведения этиотропной терапии, лечение ВФП направлено на профилактику осложнений портальной гипертензии: лигирование варикозно расширенных вен пищевода и желудка, трансъюгулярное внутрипеченочное портосистемное шунтирование и т.д. Назначение УДХК было обусловлено данными в отношении замедления пролиферации билиарного эпителия. Так, мальформации протоковой пластинки в процессе эмбриогенеза и аномальный рост холангиоцитов представляют собой ключевые патогенетические механизмы поликистозной болезни печени. УДХК и ее конъюгаты рассматриваются в качестве потенциально эффективного средства для лечения поликистоза печени, способствующего ингибированию роста кистозных холангиоцитов, что продемонстрировано как в экспериментальных, так и в клинических исследованиях [12, 13].
Поскольку у этих больных, помимо осложнений портальной гипертензии, повышен риск гепатоцеллюлярного и холангиоцеллюлярного рака [1, 14, 15], при прогрессировании ВФП в качестве метода выбора рассматривается трансплантация печени. Показатели выживаемости после трансплантации по поводу ВФП составляют 89, 86 и 76% через год, пять и 10 лет соответственно [1, 14].
Авторы выражают благодарность к.м.н. Т.В. Павловой за подготовку рисунков.
A.O. Bueverov, PhD, Prof., M.V. Kalashnikov, S.V. Koblov, PhD, P.O. Bogomolov, PhD
I.M. Sechenov First Moscow State Medical University
Moscow Regional Research Clinical Institute named after M.F. Vladimirsky
Contact person: Mikhail V. Kalashnikov, mk1408@mail.ru
The report is devoted to the description of a clinical case of portal-hypertensive form of fibrocystic liver disease – congenital liver fibrosis with von Meyenburg complexes in a 36-year-old woman. The reason for the examination was accidentally detected thrombocytopenia. Viral, toxic, autoimmune liver diseases, storage diseases, and hematologic pathology were excluded. Renal involvement was absent in the patient. The diagnosis was confirmed by pathognomonic histopathological picture. Due to the lack of possibility to influence the process of fibrogenesis, a patient was consulted by a transplant surgeon and dynamic follow-up was recommended.
Уважаемый посетитель uMEDp!
Уведомляем Вас о том, что здесь содержится информация, предназначенная исключительно для специалистов здравоохранения.
Если Вы не являетесь специалистом здравоохранения, администрация не несет ответственности за возможные отрицательные последствия, возникшие в результате самостоятельного использования Вами информации с портала без предварительной консультации с врачом.
Нажимая на кнопку «Войти», Вы подтверждаете, что являетесь врачом или студентом медицинского вуза.