–Ъ–ї–Є–љ–Є–Ї–Њ-—Д–∞—А–Љ–∞–Ї–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–µ –Њ–±–Њ—Б–љ–Њ–≤–∞–љ–Є–µ –љ–Њ–≤—Л—Е –љ–∞–њ—А–∞–≤–ї–µ–љ–Є–є –њ–∞—В–Њ–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Њ–є —В–µ—А–∞–њ–Є–Є –і–Є–∞–±–µ—В–Є—З–µ—Б–Ї–Њ–є –њ–Њ–ї–Є–љ–µ–є—А–Њ–њ–∞—В–Є–Є
- –Р–љ–љ–Њ—В–∞—Ж–Є—П
- –°—В–∞—В—М—П
- –°—Б—Л–ї–Ї–Є
- English
–Ф–Є–∞–±–µ—В–Є—З–µ—Б–Ї–∞—П –њ–Њ–ї–Є–љ–µ–є—А–Њ–њ–∞—В–Є—П (–Ф–Я–Э)¬†вАУ —З–∞—Б—В–Њ–µ –Њ—Б–ї–Њ–ґ–љ–µ–љ–Є–µ —Б–∞—Е–∞—А–љ–Њ–≥–Њ –і–Є–∞–±–µ—В–∞ (–°–Ф) 1 –Є¬†2 —В–Є–њ–Њ–≤. –Ю–і–љ–∞–Ї–Њ –Њ–љ–Њ —А–∞–Ј–≤–Є–≤–∞–µ—В—Б—П –љ–µ¬†—Г –≤—Б–µ—Е –њ–∞—Ж–Є–µ–љ—В–Њ–≤. –§–∞–Ї—В–Њ—А–∞–Љ–Є —А–Є—Б–Ї–∞ —А–∞–Ј–≤–Є—В–Є—П –Ф–Я–Э –њ—А–Є –°–Ф¬†—П–≤–ї—П—О—В—Б—П –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ—Л–є¬†–≥–ї–Є–Ї–µ–Љ–Є—З–µ—Б–Ї–Є–є –Ї–Њ–љ—В—А–Њ–ї—М [1], —Б–µ—А–і–µ—З–љ–Њ-—Б–Њ—Б—Г–і–Є—Б—В–∞—П –Ї–Њ–Љ–Њ—А–±–Є–і–љ–Њ—Б—В—М, –≤¬†—З–∞—Б—В–љ–Њ—Б—В–Є –∞—А—В–µ—А–Є–∞–ї—М–љ–∞—П¬†–≥–Є–њ–µ—А—В–µ–љ–Ј–Є—П, –њ–Њ–≤—Л—И–µ–љ–љ—Л–є —Г—А–Њ–≤–µ–љ—М —В—А–Є–≥–ї–Є—Ж–µ—А–Є–і–Њ–≤, –Њ–ґ–Є—А–µ–љ–Є–µ, –Ї—Г—А–µ–љ–Є–µ [2], –∞¬†—В–∞–Ї–ґ–µ –≤—А–Њ–ґ–і–µ–љ–љ–∞—П (–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–∞—П) –њ—А–µ–і—А–∞—Б–њ–Њ–ї–Њ–ґ–µ–љ–љ–Њ—Б—В—М, –Ј–∞–Ї–ї—О—З–∞—О—Й–∞—П—Б—П –≤¬†–±–Њ–ї–µ–µ –≤—Л—Б–Њ–Ї–Њ–є —З—Г–≤—Б—В–≤–Є—В–µ–ї—М–љ–Њ—Б—В–Є –њ–µ—А–Є—Д–µ—А–Є—З–µ—Б–Ї–Є—Е —В–Ї–∞–љ–µ–є –Ї¬†–≥–Є–њ–µ—А–≥–ї–Є–Ї–µ–Љ–Є–Є [3].
–Я–Њ–љ–Є–Љ–∞–љ–Є–µ –њ—А–Є—З–Є–љ –≤–Њ–Ј–љ–Є–Ї–љ–Њ–≤–µ–љ–Є—П –Ф–Я–Э —В–µ–Љ –љ–µ¬†–Љ–µ–љ–µ–µ –љ–µ¬†–і–∞–µ—В –Њ—В–≤–µ—В–∞ –љ–∞¬†–≤–Њ–њ—А–Њ—Б: –њ–Њ—З–µ–Љ—Г —Г¬†–Њ–і–љ–Є—Е –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —А–∞–Ј–≤–Є–≤–∞–µ—В—Б—П —Н—В–Њ –Њ—Б–ї–Њ–ґ–љ–µ–љ–Є–µ, –∞¬†—Г –і—А—Г–≥–Є—Е –љ–µ—В?
–Ф—А—Г–≥–Њ–є –љ–µ¬†–Љ–µ–љ–µ–µ –≤–∞–ґ–љ—Л–є –≤–Њ–њ—А–Њ—Б, –љ–µ¬†—А–µ—И–µ–љ–љ—Л–є –і–Њ –љ–∞—Б—В–Њ—П—Й–µ–≥–Њ –≤—А–µ–Љ–µ–љ–Є,¬†вАУ –њ—А–Є—З–Є–љ—Л, –њ–Њ¬†–Ї–Њ—В–Њ—А—Л–Љ –љ–µ–Ї–Њ—В–Њ—А—Л–µ –њ–∞—Ж–Є–µ–љ—В—Л –Є—Б–њ—Л—В—Л–≤–∞—О—В –љ–µ–є—А–Њ–њ–∞—В–Є—З–µ—Б–Ї—Г—О –±–Њ–ї—М. –°–Њ–≥–ї–∞—Б–љ–Њ —Н–њ–Є–і–µ–Љ–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–Љ –і–∞–љ–љ—Л–Љ, –љ–µ–є—А–Њ–њ–∞—В–Є—З–µ—Б–Ї–∞—П –±–Њ–ї—М –Њ—В–Љ–µ—З–∞–µ—В—Б—П —Г¬†20вАУ50% –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–°–Ф¬†–Є¬†–њ—А–Є–Љ–µ—А–љ–Њ —Г¬†60% –±–Њ–ї—М–љ—Л—Е –Ф–Я–Э [4].
–Ъ–ї—О—З–µ–≤—Л–Љ–Є —Д–∞–Ї—В–Њ—А–∞–Љ–Є, –њ—А–Є–≤–Њ–і—П—Й–Є–Љ–Є –Ї¬†—А–∞–Ј–≤–Є—В–Є—О –Ф–Я–Э, —П–≤–ї—П—О—В—Б—П¬†–≥–Є–њ–µ—А–≥–ї–Є–Ї–µ–Љ–Є—П –Є¬†–і–Є—Б–ї–Є–њ–Є–і–µ–Љ–Є—П. –Т–ї–Є—П–љ–Є–µ —Н—В–Є—Е —Д–∞–Ї—В–Њ—А–Њ–≤ –Њ–њ–Њ—Б—А–µ–і–Њ–≤–∞–љ–Њ –Љ–µ—В–∞–±–Њ–ї–Є—З–µ—Б–Ї–Є–Љ –Є¬†–Њ–Ї–Є—Б–ї–Є—В–µ–ї—М–љ—Л–Љ —Б—В—А–µ—Б—Б–Њ–Љ, –њ—А–Є–≤–Њ–і—П—Й–Є–Љ –Ї¬†–і–µ–≥–µ–љ–µ—А–∞—Ж–Є–Є –∞–Ї—Б–Њ–љ–Њ–≤ [5, 6]. –Я–Њ–≤—Л—И–µ–љ–љ—Л–є —Г—А–Њ–≤–µ–љ—М¬†–≥–ї—О–Ї–Њ–Ј—Л —Б–њ–Њ—Б–Њ–±—Б—В–≤—Г–µ—В –∞–Ї—В–Є–≤–∞—Ж–Є–Є –њ–Њ–ї–Є–Њ–ї–Њ–≤–Њ–≥–Њ –Є¬†–≥–µ–Ї—Б–Њ–Ј–∞–Љ–Є–љ–Њ–≤–Њ–≥–Њ –њ—Г—В–µ–є, —З—В–Њ –≤–µ–і–µ—В –Ї¬†–Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є—О –∞–Ї—В–Є–≤–љ—Л—Е —Д–Њ—А–Љ –Ї–Є—Б–ї–Њ—А–Њ–і–∞ –Є¬†—А–∞–Ј–≤–Є—В–Є—О —Б–Є—Б—В–µ–Љ–љ–Њ–≥–Њ –≤–Њ—Б–њ–∞–ї–µ–љ–Є—П. –≠—В–Є –ґ–µ –њ—А–Њ—Ж–µ—Б—Б—Л –Ј–∞–њ—Г—Б–Ї–∞–µ—В –і–Є—Б–ї–Є–њ–Є–і–µ–Љ–Є—П, —З—В–Њ –≤¬†—Б–Њ–≤–Њ–Ї—Г–њ–љ–Њ—Б—В–Є –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ —Г—Б—Г–≥—Г–±–ї—П–µ—В –µ—Й–µ –Њ–і–Є–љ –Ї—А–∞–є–љ–µ –≤–∞–ґ–љ—Л–є –Љ–µ—Е–∞–љ–Є–Ј–Љ –њ–Њ—А–∞–ґ–µ–љ–Є—П –њ–µ—А–Є—Д–µ—А–Є—З–µ—Б–Ї–Њ–є –љ–µ—А–≤–љ–Њ–є —Б–Є—Б—В–µ–Љ—Л¬†вАУ –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є–µ –Љ–Є—В–Њ—Е–Њ–љ–і—А–Є–є [7]. –Ъ—А–∞–є–љ–µ –≤–∞–ґ–љ–Њ, —З—В–Њ –і–Њ–Љ–Є–љ–Є—А—Г—О—Й—Г—О —А–Њ–ї—М –≤¬†–і–µ–±—О—В–µ –Ф–Я–Э –Є–≥—А–∞–µ—В –Є–Љ–µ–љ–љ–Њ —Б–Є—Б—В–µ–Љ–љ–Њ–µ –≤–Њ—Б–њ–∞–ї–µ–љ–Є–µ [8, 9].
–Я—А–Є¬†–≥–Є–њ–µ—А–≥–ї–Є–Ї–µ–Љ–Є–Є —В–∞–Ї–ґ–µ –љ–∞–±–ї—О–і–∞–µ—В—Б—П —Г—Б–Є–ї–µ–љ–љ–Њ–µ¬†–≥–ї–Є–Ї–Є—А–Њ–≤–∞–љ–Є–µ –Љ–љ–Њ–≥–Њ—З–Є—Б–ї–µ–љ–љ—Л—Е —Б—В—А—Г–Ї—В—Г—А–љ—Л—Е –Є¬†—Д—Г–љ–Ї—Ж–Є–Њ–љ–∞–ї—М–љ—Л—Е –±–µ–ї–Ї–Њ–≤ –љ–µ—А–≤–љ–Њ–є —В–Ї–∞–љ–Є —Б¬†–Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є–µ–Љ –Ї–Њ–љ–µ—З–љ—Л—Е –њ—А–Њ–і—Г–Ї—В–Њ–≤, –Ї–Њ—В–Њ—А—Л–µ —В–Њ–ґ–µ —Б—В–Є–Љ—Г–ї–Є—А—Г—О—В –≤—Л—Б–≤–Њ–±–Њ–ґ–і–µ–љ–Є–µ –њ—А–Њ–≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ—Л—Е –Љ–Њ–ї–µ–Ї—Г–ї –Є¬†—Б–≤–Њ–±–Њ–і–љ—Л—Е —А–∞–і–Є–Ї–∞–ї–Њ–≤. –Ш–Ј–Љ–µ–љ–µ–љ–Є–µ —Д—Г–љ–Ї—Ж–Є–Є –±–µ–ї–Ї–Њ–≤ –љ–∞–±–ї—О–і–∞–µ—В—Б—П –Є¬†–њ—А–Є –µ—Б—В–µ—Б—В–≤–µ–љ–љ–Њ–Љ —Б—В–∞—А–µ–љ–Є–Є, –њ–Њ—Н—В–Њ–Љ—Г —Г¬†–≤–Њ–Ј—А–∞—Б—В–љ—Л—Е –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–і–Є–∞–±–µ—В–Њ–Љ –Љ–Њ–і–Є—Д–Є–Ї–∞—Ж–Є—П —Н–Ї—Б–њ—А–µ—Б—Б–Є–Є¬†–≥–µ–љ–Њ–≤ –Є¬†–≤–љ—Г—В—А–Є–Ї–ї–µ—В–Њ—З–љ–Њ–≥–Њ (–Є–љ—Б—Г–ї–Є–љ–Њ–≤–Њ–≥–Њ) —Б–Є–≥–љ–∞–ї–Є–љ–≥–∞ –≤—Л—А–∞–ґ–µ–љ–∞ –Њ—Б–Њ–±–µ–љ–љ–Њ —П—А–Ї–Њ [10]. –Р–љ–∞–ї–Њ–≥–Є—З–љ—Л–µ –њ—А–Њ—Ж–µ—Б—Б—Л –њ—А–Њ–Є—Б—Е–Њ–і—П—В –Є¬†–≤ –Љ–∞–ї—Л—Е —Б–Њ—Б—Г–і–∞—Е, –Є–Ј–Љ–µ–љ–µ–љ–Є—П –≤¬†–Ї–Њ—В–Њ—А—Л—Е –≤—Л–Ј—Л–≤–∞—О—В –љ–∞—А—Г—И–µ–љ–Є–µ –њ–µ—А—Д—Г–Ј–Є–Є –љ–µ—А–≤–Њ–≤, –њ—А–Њ–≤–Њ—Ж–Є—А—Г—П –Є—Е¬†–≥–Є–њ–Њ–Ї—Б–Є—О –Є¬†–і–Є—Б—Д—Г–љ–Ї—Ж–Є—О [11].
–†–∞–Ј–≤–Є—В–Є–µ –Ф–Я–Э —Б–≤—П–Ј–∞–љ–Њ —В–∞–Ї–ґ–µ —Б–Њ¬†—Б–љ–Є–ґ–µ–љ–Є–µ–Љ –±–Є–Њ–і–Њ—Б—В—Г–њ–љ–Њ—Б—В–Є —В–Ї–∞–љ–µ–є (—З—Г–≤—Б—В–≤–Є—В–µ–ї—М–љ–Њ—Б—В–Є —В–Ї–∞–љ–µ–є –Є¬†–Є—Е —А–µ—Ж–µ–њ—В–Њ—А–Њ–≤) –Ї¬†–і–µ–є—Б—В–≤–Є—О —А–∞–Ј–ї–Є—З–љ—Л—Е –∞—Г—В–Њ–Ї–Њ–Є–і–Њ–≤¬†вАУ –љ–µ–є—А–Њ–Љ–µ–і–Є–∞—В–Њ—А–Њ–≤, —Д–∞–Ї—В–Њ—А–Њ–≤ —А–Њ—Б—В–∞, –≤–Ї–ї—О—З–∞—П –Є–љ—Б—Г–ї–Є–љ–Њ–њ–Њ–і–Њ–±–љ—Л–є —Д–∞–Ї—В–Њ—А —А–Њ—Б—В–∞, —Д–∞–Ї—В–Њ—А —А–Њ—Б—В–∞ —Н–љ–і–Њ—В–µ–ї–Є—П —Б–Њ—Б—Г–і–Њ–≤, –∞¬†—В–∞–Ї–ґ–µ¬†–≥–∞–Ј–Њ—В—А–∞–љ—Б–Љ–Є—В—В–µ—А–Њ–≤ (–Њ–Ї—Б–Є–і–∞ –∞–Ј–Њ—В–∞ –Є¬†—Г–≥–ї–µ—А–Њ–і–∞), –Њ–±–µ—Б–њ–µ—З–Є–≤–∞—О—Й–Є—Е —Г—Б–ї–Њ–≤–Є—П –і–ї—П –љ–Њ—А–Љ–∞–ї—М–љ–Њ–≥–Њ —Д—Г–љ–Ї—Ж–Є–Њ–љ–Є—А–Њ–≤–∞–љ–Є—П —Б–Њ—Б—Г–і–Њ–≤ –Є¬†–љ–µ—А–≤–Њ–≤ [12, 13].
–Т–∞–ґ–љ–Њ –Њ—В–Љ–µ—В–Є—В—М, —З—В–Њ —Б—В—А–Њ–≥–Є–є –Ї–Њ–љ—В—А–Њ–ї—М —Г—А–Њ–≤–љ—П¬†–≥–ї—О–Ї–Њ–Ј—Л —Б–њ–Њ—Б–Њ–±–µ–љ —Б–љ–Є–Ј–Є—В—М —З–∞—Б—В–Њ—В—Г —А–∞–Ј–≤–Є—В–Є—П –Ф–Я–Э —В–Њ–ї—М–Ї–Њ –њ—А–Є –°–Ф¬†1 —В–Є–њ–∞. –Я—А–Є –°–Ф¬†2 —В–Є–њ–∞ –і–∞–љ–љ–Њ–µ –љ–∞–њ—А–∞–≤–ї–µ–љ–Є–µ –ї–µ—З–µ–љ–Є—П –љ–µ¬†—Б–Њ–њ—А–Њ–≤–Њ–ґ–і–∞–µ—В—Б—П –і–Њ—Б—В–Њ–≤–µ—А–љ—Л–Љ–Є –њ–Њ–ї–Њ–ґ–Є—В–µ–ї—М–љ—Л–Љ–Є —А–µ–Ј—Г–ї—М—В–∞—В–∞–Љ–Є [14]. –Я—А–Є –°–Ф¬†2 —В–Є–њ–∞ –љ–µ–≤–µ–ї–Є–Ї–∞ –Ј–љ–∞—З–Є–Љ–Њ—Б—В—М –Є¬†–Ї–Њ—А—А–µ–Ї—Ж–Є–Є¬†–≥–Є–њ–µ—А–≥–ї–Є–Ї–µ–Љ–Є–Є –Є–љ—Б—Г–ї–Є–љ–Њ–Љ –≤¬†–Њ—В–ї–Є—З–Є–µ –Њ—В¬†–°–Ф¬†1 —В–Є–њ–∞, –њ—А–Є –Ї–Њ—В–Њ—А–Њ–Љ –њ–Њ–і–Њ–±–љ–∞—П —В–µ—А–∞–њ–Є—П –і–Њ—Б—В–∞—В–Њ—З–љ–Њ —Н—Д—Д–µ–Ї—В–Є–≤–љ–∞ –≤¬†–Њ—В–љ–Њ—И–µ–љ–Є–Є –Ф–Я–Э [15].
–Ю–њ–Є—Б–∞–љ–љ—Л–µ –Љ–µ—Е–∞–љ–Є–Ј–Љ—Л –њ—А–Є–≤–Њ–і—П—В –Ї¬†–≤–Њ–Ј–љ–Є–Ї–љ–Њ–≤–µ–љ–Є—О —А–∞–Ј–ї–Є—З–љ—Л—Е —Б–Њ–Љ–∞—В–Њ—Б–µ–љ—Б–Њ—А–љ—Л—Е —Д–µ–љ–Њ—В–Є–њ–Њ–≤ –Ф–Я–Э. –Ъ–ї–Є–љ–Є—З–µ—Б–Ї–∞—П —Б–Є–Љ–њ—В–Њ–Љ–∞—В–Є–Ї–∞ –њ—А–Є —Н—В–Њ–Љ –Њ–њ—А–µ–і–µ–ї—П–µ—В—Б—П –њ—А–µ–Є–Љ—Г—Й–µ—Б—В–≤–µ–љ–љ—Л–Љ –њ–Њ—А–∞–ґ–µ–љ–Є–µ–Љ —В–Њ–ї—Б—В—Л—Е –Є–ї–Є —В–Њ–љ–Ї–Є—Е –≤–Њ–ї–Њ–Ї–Њ–љ. –Я—А–Є –њ–Њ—А–∞–ґ–µ–љ–Є–Є —В–Њ–ї—Б—В—Л—Е –≤–Њ–ї–Њ–Ї–Њ–љ —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ –≤—Л—П–≤–ї—П—О—В—Б—П —А–∞—Б—Б—В—А–Њ–є—Б—В–≤–∞ —В–∞–Ї—В–Є–ї—М–љ–Њ–є (–Њ–љ–µ–Љ–µ–љ–Є–µ) –Є¬†–≤–Є–±—А–∞—Ж–Є–Њ–љ–љ–Њ–є —З—Г–≤—Б—В–≤–Є—В–µ–ї—М–љ–Њ—Б—В–Є, –љ–∞—А—Г—И–∞–µ—В—Б—П –≤–Њ—Б–њ—А–Є—П—В–Є–µ —В–µ–ї–∞ –≤¬†–њ—А–Њ—Б—В—А–∞–љ—Б—В–≤–µ (–њ—А–Њ–њ—А–Є–Њ—Ж–µ–њ—Ж–Є—П), –≤¬†—Б–≤—П–Ј–Є —Б¬†—З–µ–Љ –≤–Њ–Ј—А–∞—Б—В–∞–µ—В —А–Є—Б–Ї¬†–њ–∞–і–µ–љ–Є–є –Є¬†–њ–µ—А–µ–ї–Њ–Љ–Њ–≤, —Б–љ–Є–ґ–∞—О—В—Б—П –Љ–Є–Њ—В–∞—В–Є—З–µ—Б–Ї–Є–µ —А–µ—Д–ї–µ–Ї—Б—Л, –њ—А–µ–ґ–і–µ –≤—Б–µ–≥–Њ –∞—Е–Є–ї–ї–Њ–≤, –Љ–Њ–≥—Г—В –љ–∞–±–ї—О–і–∞—В—М—Б—П —Г–Љ–µ–љ—М—И–µ–љ–Є–µ –Љ—Л—И–µ—З–љ–Њ–є —Б–Є–ї—Л –Є¬†–∞—В—А–Њ—Д–Є—П –Љ—Л—И—Ж —Б—В–Њ–њ—Л. –Я–Њ—А–∞–ґ–µ–љ–Є–µ —В–Њ–љ–Ї–Є—Е –≤–Њ–ї–Њ–Ї–Њ–љ —Б–Њ–њ—А–Њ–≤–Њ–ґ–і–∞–µ—В—Б—П –њ–Њ—П–≤–ї–µ–љ–Є–µ–Љ —А–∞—Б—Б—В—А–Њ–є—Б—В–≤ —В–µ–Љ–њ–µ—А–∞—В—Г—А–љ–Њ–є —З—Г–≤—Б—В–≤–Є—В–µ–ї—М–љ–Њ—Б—В–Є. –Я–∞—Ж–Є–µ–љ—В—Л –њ—А–µ–і—К—П–≤–ї—П—О—В –ґ–∞–ї–Њ–±—Л –љ–∞¬†—З—Г–≤—Б—В–≤–Њ –њ—А–Њ—Е–Њ–ґ–і–µ–љ–Є—П —Н–ї–µ–Ї—В—А–Є—З–µ—Б–Ї–Њ–≥–Њ —В–Њ–Ї–∞, –ґ–ґ–µ–љ–Є–µ. –Ъ—А–Њ–Љ–µ —В–Њ–≥–Њ, —Г¬†–љ–Є—Е –Љ–Њ–ґ–µ—В –љ–∞–±–ї—О–і–∞—В—М—Б—П –≤–µ–≥–µ—В–∞—В–Є–≤–љ–∞—П –і–Є—Б—Д—Г–љ–Ї—Ж–Є—П.
–С–Њ–ї—М –њ—А–Є –Ф–Я–Э –≤—Б—В—А–µ—З–∞–µ—В—Б—П –Ї–∞–Ї –њ—А–Є –њ–Њ—А–∞–ґ–µ–љ–Є–Є —В–Њ–ї—Б—В—Л—Е –≤–Њ–ї–Њ–Ї–Њ–љ (–Рќ≤), —В–∞–Ї –Є¬†–њ—А–Є –њ–Њ—А–∞–ґ–µ–љ–Є–Є —В–Њ–љ–Ї–Є—Е (–°). –Я—А–Є –њ–Њ—А–∞–ґ–µ–љ–Є–Є –њ–µ—А–≤—Л—Е –±–Њ–ї—М –Є–Љ–µ–µ—В –±–Њ–ї–µ–µ¬†–≥–ї—Г–±–Њ–Ї–Њ–µ —А–∞—Б–њ—А–µ–і–µ–ї–µ–љ–Є–µ, –∞¬†–њ—А–Є –њ–Њ—А–∞–ґ–µ–љ–Є–Є –≤—В–Њ—А—Л—Е¬†вАУ –±–Њ–ї–µ–µ –њ–Њ–≤–µ—А—Е–љ–Њ—Б—В–љ–Њ–µ.
–Ґ–∞–Ї–Є–Љ –Њ–±—А–∞–Ј–Њ–Љ, –≤¬†–Ї–ї–Є–љ–Є—З–µ—Б–Ї–Њ–є –Ї–∞—А—В–Є–љ–µ –Ф–Я–Э –Љ–Њ–≥—Г—В –љ–∞–±–ї—О–і–∞—В—М—Б—П –Ї–∞–Ї —Б–Є–Љ–њ—В–Њ–Љ—Л –≤—Л–њ–∞–і–µ–љ–Є—П (–Њ–љ–µ–Љ–µ–љ–Є–µ,¬†–≥–Є–њ–Њ—А–µ—Д–ї–µ–Ї—Б–Є—П), —В–∞–Ї –Є¬†—Б–Є–Љ–њ—В–Њ–Љ—Л —А–∞–Ј–і—А–∞–ґ–µ–љ–Є—П.
–С–Њ–ї–µ–≤—Л–µ —Д–µ–љ–Њ–Љ–µ–љ—Л –Љ–Њ–≥—Г—В –±—Л—В—М —Б–њ–Њ–љ—В–∞–љ–љ—Л–Љ–Є. –Я—А–Є –њ–Њ—А–∞–ґ–µ–љ–Є–Є —В–Њ–ї—Б—В—Л—Е –≤–Њ–ї–Њ–Ї–Њ–љ —Н—В–Њ —З–∞—Й–µ –љ–Њ—О—Й–∞—П –±–Њ–ї—М –Є–ї–Є –њ–Њ–Ї–∞–ї—Л–≤–∞–љ–Є–µ, –∞¬†–њ—А–Є –њ–Њ—А–∞–ґ–µ–љ–Є–Є —В–Њ–љ–Ї–Є—Е¬†вАУ —З–∞—Й–µ –ґ–ґ–µ–љ–Є–µ, –Њ—Б—В—А–∞—П –Є–ї–Є –Ї–Њ–ї—О—Й–∞—П –±–Њ–ї—М [16]. –Ь–Њ–≥—Г—В –Њ–њ—А–µ–і–µ–ї—П—В—М—Б—П –њ–Њ–≤—Л—И–µ–љ–љ–∞—П —З—Г–≤—Б—В–≤–Є—В–µ–ї—М–љ–Њ—Б—В—М –Ї¬†–±–Њ–ї–µ–≤—Л–Љ —Б—В–Є–Љ—Г–ї–∞–Љ (–≥–Є–њ–µ—А–∞–ї–≥–µ–Ј–Є—П) –Є–ї–Є –±–Њ–ї–µ–≤—Л–µ –Њ—Й—Г—Й–µ–љ–Є—П –Ї¬†—Б—В–Є–Љ—Г–ї–∞–Љ —А–∞–Ј–ї–Є—З–љ—Л—Е –љ–µ–±–Њ–ї–µ–≤—Л—Е –Љ–Њ–і–∞–ї—М–љ–Њ—Б—В–µ–є (–∞–ї–ї–Њ–і–Є–љ–Є—П¬†вАУ —З–∞—Й–µ –њ—А–Є –њ–Њ—А–∞–ґ–µ–љ–Є–Є —В–Њ–љ–Ї–Є—Е –≤–Њ–ї–Њ–Ї–Њ–љ). –Ь–µ—Е–∞–љ–Є—З–µ—Б–Ї–∞—П¬†–≥–Є–њ–µ—А–∞–ї–≥–µ–Ј–Є—П –њ—А–Є –Ф–Я–Э –Є–Љ–µ–µ—В –і–Њ—Б—В–∞—В–Њ—З–љ–Њ –≤—Л—Б–Њ–Ї—Г—О —А–∞—Б–њ—А–Њ—Б—В—А–∞–љ–µ–љ–љ–Њ—Б—В—М [17вАУ19]. –Ґ–µ–њ–ї–Њ–≤–∞—П¬†–≥–Є–њ–µ—А–∞–ї–≥–µ–Ј–Є—П (–Њ–±—Л—З–љ–Њ –≤¬†—Б–Њ—З–µ—В–∞–љ–Є–Є —Б¬†—Б–Њ—Е—А–∞–љ–µ–љ–љ–Њ–є —Д—Г–љ–Ї—Ж–Є–µ–є —В–Њ–љ–Ї–Є—Е –≤–Њ–ї–Њ–Ї–Њ–љ¬†вАУ —Д–µ–љ–Њ—В–Є–њ —А–∞–Ј–і—А–∞–ґ–µ–љ–љ–Њ–≥–Њ –љ–Њ—Ж–Є—Ж–µ–њ—В–Њ—А–∞) –≤—Б—В—А–µ—З–∞–µ—В—Б—П —Б—Г—Й–µ—Б—В–≤–µ–љ–љ–Њ —А–µ–ґ–µ. –£¬†–±–Њ–ї—М—И–Є–љ—Б—В–≤–∞ –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–Ф–Я–Э –љ–∞–±–ї—О–і–∞–µ—В—Б—П —Б–Њ—З–µ—В–∞–љ–љ–Њ–µ –њ–Њ—А–∞–ґ–µ–љ–Є–µ —В–Њ–ї—Б—В—Л—Е –Є¬†—В–Њ–љ–Ї–Є—Е –≤–Њ–ї–Њ–Ї–Њ–љ (—Д–µ–љ–Њ—В–Є–њ –і–µ–∞—Д—Д–µ—А–µ–љ—В–∞—Ж–Є–Є), –њ—А–Є —Н—В–Њ–Љ –Љ–µ—Е–∞–љ–Є—З–µ—Б–Ї–∞—П –±–Њ–ї—М –≤¬†–≥–ї—Г–±–Њ–Ї–Є—Е —В–Ї–∞–љ—П—Е (–Њ—Ж–µ–љ–Є–≤–∞–µ—В—Б—П –њ–Њ¬†–њ–Њ—А–Њ–≥—Г –±–Њ–ї–Є –њ—А–Є –і–∞–≤–ї–µ–љ–Є–Є) –Њ—В–Љ–µ—З–∞–µ—В—Б—П —А–µ–і–Ї–Њ [20].
–Т¬†–љ–∞—Б—В–Њ—П—Й–µ–µ –≤—А–µ–Љ—П –љ–µ¬†–Є–Ј—Г—З–µ–љ—Л –Ї–ї–Є–љ–Є–Ї–Њ-–њ–∞—В–Њ–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Є–µ –њ–∞—А–∞–ї–ї–µ–ї–Є, –њ–Њ–Ј–≤–Њ–ї—П—О—Й–Є–µ –Њ–њ—А–µ–і–µ–ї–Є—В—М –≤–µ–і—Г—Й–Є–є –Љ–µ—Е–∞–љ–Є–Ј–Љ —А–∞–Ј–≤–Є—В–Є—П —В–Њ–≥–Њ –Є–ї–Є –Є–љ–Њ–≥–Њ —Д–µ–љ–Њ—В–Є–њ–∞ –Ф–Я–Э. –Ф–ї—П –±–Њ–ї–µ–≤—Л—Е —Д–Њ—А–Љ –Ф–Я–Э –Њ–њ–Є—Б–∞–љ—Л –Љ–Њ–і–Є—Д–Є—Ж–Є—А—Г–µ–Љ—Л–µ –Є¬†–љ–µ–Љ–Њ–і–Є—Д–Є—Ж–Є—А—Г–µ–Љ—Л–µ —Д–∞–Ї—В–Њ—А—Л —А–Є—Б–Ї–∞. –Э–µ–є—А–Њ–њ–∞—В–Є—З–µ—Б–Ї–∞—П –±–Њ–ї—М –њ—А–Є –Ф–Я–Э —З–∞—Й–µ –≤—Б—В—А–µ—З–∞–µ—В—Б—П —Г¬†–ґ–µ–љ—Й–Є–љ [21], –≤¬†—Б—В–∞—А—И–Є—Е –≤–Њ–Ј—А–∞—Б—В–љ—Л—Е¬†–≥—А—Г–њ–њ–∞—Е [22], —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–Њ–ґ–Є—А–µ–љ–Є–µ–Љ [23], –њ–Њ–≤—Л—И–µ–љ–љ—Л–Љ —Г—А–Њ–≤–љ–µ–Љ¬†–≥–ї–Є–Ї–Є—А–Њ–≤–∞–љ–љ–Њ–≥–Њ¬†–≥–µ–Љ–Њ–≥–ї–Њ–±–Є–љ–∞ [17], –Ј–љ–∞—З–Є—В–µ–ї—М–љ—Л–Љ –њ–Њ—В—А–µ–±–ї–µ–љ–Є–µ–Љ –∞–ї–Ї–Њ–≥–Њ–ї—П, –і–ї–Є—В–µ–ї—М–љ—Л–Љ —В–µ—З–µ–љ–Є–µ–Љ –°–Ф¬†–Є¬†–≤—Л—А–∞–ґ–µ–љ–љ—Л–Љ–Є —Б–µ–љ—Б–Њ—А–љ—Л–Љ–Є —А–∞—Б—Б—В—А–Њ–є—Б—В–≤–∞–Љ–Є [24], –љ–∞—Б–ї–µ–і—Б—В–≤–µ–љ–љ–Њ–є –њ—А–µ–і—А–∞—Б–њ–Њ–ї–Њ–ґ–µ–љ–љ–Њ—Б—В—М—О [25].
–Т¬†–њ–∞—В–Њ–≥–µ–љ–µ–Ј–µ –±–Њ–ї–Є –њ—А–Є –Ф–Я–Э –љ–µ–Љ–∞–ї–Њ–≤–∞–ґ–љ—Г—О —А–Њ–ї—М –Є–≥—А–∞—О—В —Б—В—А—Г–Ї—В—Г—А–љ–Њ-—Д—Г–љ–Ї—Ж–Є–Њ–љ–∞–ї—М–љ—Л–µ –њ–Њ—А–∞–ґ–µ–љ–Є—П —Б–Њ—Б—Г–і–Њ–≤ –љ–µ—А–≤–љ–Њ–є —Б–Є—Б—В–µ–Љ—Л [26] –Є¬†–љ–∞—А—Г—И–µ–љ–Є—П —А–µ–≥—Г–ї—П—Ж–Є–Є –њ–µ—А–Є—Д–µ—А–Є—З–µ—Б–Ї–Њ–≥–Њ –Ї—А–Њ–≤–Њ—В–Њ–Ї–∞ [27]. –Ю–±–љ–∞—А—Г–ґ–µ–љ–Њ, —З—В–Њ –њ—А–Є¬†–≥–Є–њ–Њ–Ї—Б–Є–Є, —Б–≤—П–Ј–∞–љ–љ–Њ–є —Б¬†–љ–∞—А—Г—И–µ–љ–Є–µ–Љ —А–µ–≥—Г–ї—П—Ж–Є–Є –Љ–µ—Б—В–љ–Њ–≥–Њ –Ї—А–Њ–≤–Њ—В–Њ–Ї–∞ –≤¬†–Ї–Њ–ґ–µ, –≤—Л—А–∞–±–∞—В—Л–≤–∞—О—В—Б—П —Б–њ–µ—Ж–Є—Д–Є—З–µ—Б–Ї–Є–µ –±–µ–ї–Ї–Є¬†вАУ —Д–∞–Ї—В–Њ—А, –Є–љ–і—Г—Ж–Є—А—Г–µ–Љ—Л–є¬†–≥–Є–њ–Њ–Ї—Б–Є–µ–є, 1ќ± [28], –Є¬†—Д–∞–Ї—В–Њ—А —Д–Њ–љ –Т–Є–ї–ї–µ–±—А–∞–љ–і–∞ [29], –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є—П –Ї–Њ—В–Њ—А—Л—Е –њ—А—П–Љ–Њ –Ї–Њ—А—А–µ–ї–Є—А—Г–µ—В —Б¬†–≤—Л—А–∞–ґ–µ–љ–љ–Њ—Б—В—М—О –±–Њ–ї–Є. –С—Л–ї–Њ —В–∞–Ї–ґ–µ —Г—Б—В–∞–љ–Њ–≤–ї–µ–љ–Њ, —З—В–Њ —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–±–Њ–ї–µ–≤–Њ–є —Д–Њ—А–Љ–Њ–є –Ф–Я–Э —Б–љ–Є–ґ–µ–љ–∞ –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є—П –≤–Є—В–∞–Љ–Є–љ–∞ D –≤¬†–њ–ї–∞–Ј–Љ–µ –Ї—А–Њ–≤–Є [30]. –Э–∞–Ї–Њ–љ–µ—Ж, —А–µ–Ј—Г–ї—М—В–∞—В—Л —А—П–і–∞ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є —Б–≤–Є–і–µ—В–µ–ї—М—Б—В–≤—Г—О—В –Њ¬†—В–Њ–Љ, —З—В–Њ –≤¬†–њ–∞—В–Њ–≥–µ–љ–µ–Ј–µ –љ–µ–є—А–Њ–њ–∞—В–Є—З–µ—Б–Ї–Њ–є –±–Њ–ї–Є –њ—А–Є –°–Ф¬†—Г—З–∞—Б—В–≤—Г–µ—В –Љ–µ—В–Є–ї¬≠–≥–ї–Є–Њ–Ї—Б–∞–ї—М (–∞–ї—М–і–µ–≥–Є–і –њ–Є—А–Њ–≤–Є–љ–Њ–≥—А–∞–і–љ–Њ–є –Ї–Є—Б–ї–Њ—В—Л)¬†вАУ –њ–Њ–±–Њ—З–љ—Л–є —Ж–Є—В–Њ—В–Њ–Ї—Б–Є—З–µ—Б–Ї–Є–є –њ—А–Њ–і—Г–Ї—В¬†–≥–ї–Є–Ї–Њ–ї–Є–Ј–∞, –Ї–Њ—В–Њ—А—Л–є —А–µ–∞–ї–Є–Ј—Г–µ—В —Б–≤–Њ–Є —Н—Д—Д–µ–Ї—В—Л —З–µ—А–µ–Ј –∞–Ї—В–Є–≤–∞—Ж–Є—О –°-–љ–Њ—Ж–Є—Ж–µ–њ—В–Њ—А–Њ–≤ –Є¬†—В–µ–њ–ї–Њ–≤—Г—О¬†–≥–Є–њ–µ—А–∞–ї–≥–µ–Ј–Є—О [31].
–£¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–±–Њ–ї–µ–≤–Њ–є —Д–Њ—А–Љ–Њ–є –Ф–Я–Э –њ–Њ–Љ–Є–Љ–Њ –њ–µ—А–Є—Д–µ—А–Є—З–µ—Б–Ї–Є—Е –Њ–±–љ–∞—А—Г–ґ–Є–≤–∞—О—В—Б—П —Ж–µ–љ—В—А–∞–ї—М–љ—Л–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П –љ–µ–≤—А–∞–ї—М–љ—Л—Е —Б—В—А—Г–Ї—В—Г—А. –°—В—А—Г–Ї—В—Г—А–љ—Л–µ –Є¬†—Д—Г–љ–Ї—Ж–Є–Њ–љ–∞–ї—М–љ—Л–µ –Љ–Њ–і–Є—Д–Є–Ї–∞—Ж–Є–Є –Њ–њ—А–µ–і–µ–ї—П—О—В—Б—П –≤¬†—Б–њ–Є–љ–∞–ї—М–љ—Л—Е, —Б–Њ–Љ–∞—В–Њ–Љ–Њ—В–Њ—А–љ—Л—Е, –ї–Є–Љ–±–Є—З–µ—Б–Ї–Є—Е, —В–∞–ї–∞–Љ–Є—З–µ—Б–Ї–Є—Е, –≤–Њ—Б—Е–Њ–і—П—Й–Є—Е –Є¬†–љ–Є—Б—Е–Њ–і—П—Й–Є—Е –Љ–Њ–і—Г–ї—П—Ж–Є–Њ–љ–љ—Л—Е —Б–Є—Б—В–µ–Љ–∞—Е¬†[32]. –Т—Л—П–≤–ї–µ–љ—Л –Є–Ј–Љ–µ–љ–µ–љ–Є—П –≤¬†–≤—Л—Б—И–Є—Е –Ї–Њ—А–Ї–Њ–≤—Л—Е —Ж–µ–љ—В—А–∞—Е: –∞—В—А–Њ—Д–Є—П –Є¬†–∞–љ–Њ–Љ–∞–ї—М–љ–∞—П –∞–Ї—В–Є–≤–љ–Њ—Б—В—М —Б–Њ–Љ–∞—В–Њ–Љ–Њ—В–Њ—А–љ–Њ–є –Є¬†–Њ—Б—В—А–Њ–≤–Ї–Њ–≤–Њ–є –Ї–Њ—А—Л¬†–≥–Њ–ї–Њ–≤–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞ [29, 33], —Г—Б–Є–ї–µ–љ–Є–µ –Љ–Њ–Ј–≥–Њ–≤–Њ–≥–Њ –Ї—А–Њ–≤–Њ—В–Њ–Ї–∞ –≤¬†–њ–µ—А–µ–і–љ–µ–є –њ–Њ—П—Б–љ–Њ–є –Ї–Њ—А–µ¬†[34]. –≠—В–Є –Љ–Њ–і–Є—Д–Є–Ї–∞—Ж–Є–Є –Љ–Њ–≥—Г—В –±—Л—В—М —Б–ї–µ–і—Б—В–≤–Є–µ–Љ –њ–µ—А–≤–Є—З–љ–Њ–≥–Њ –њ–Њ—А–∞–ґ–µ–љ–Є—П, –∞¬†—В–∞–Ї–ґ–µ —Е—А–Њ–љ–Є–Ј–∞—Ж–Є–Є –љ–µ–є—А–Њ–њ–∞—В–Є—З–µ—Б–Ї–Њ–є –±–Њ–ї–Є.
–Ю—В—Б—Г—В—Б—В–≤–Є–µ –µ–і–Є–љ–Њ–є —З–µ—В–Ї–Њ–є –Ї–Њ–љ—Ж–µ–њ—Ж–Є–Є, –њ–Њ–Ј–≤–Њ–ї—П—О—Й–µ–є —Г—Б—В–∞–љ–Њ–≤–Є—В—М –≤¬†–Ї–∞–ґ–і–Њ–Љ –Ї–Њ–љ–Ї—А–µ—В–љ–Њ–Љ —Б–ї—Г—З–∞–µ –≤–µ—А–Њ—П—В–љ–Њ—Б—В—М –Ј–∞–њ—Г—Б–Ї–∞ –Є¬†—А–µ–ї–µ–≤–∞–љ—В–љ–Њ—Б—В—М –Љ–µ—Е–∞–љ–Є–Ј–Љ–Њ–≤ –≤–Њ–Ј–љ–Є–Ї–љ–Њ–≤–µ–љ–Є—П –Є¬†—А–∞–Ј–≤–Є—В–Є—П –Ф–Я–Э, –≤¬†—В–Њ–Љ —З–Є—Б–ї–µ –µ–µ –±–Њ–ї–µ–≤—Л—Е —Д–Њ—А–Љ, –њ–Њ—Б–ї—Г–ґ–Є–ї–Њ –њ—А–µ–і–њ–Њ—Б—Л–ї–Ї–Њ–є –Ї¬†–њ—А–Њ–і–Њ–ї–ґ–µ–љ–Є—О –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є, –Ј–љ–∞—З–Є—В–µ–ї—М–љ–∞—П —З–∞—Б—В—М –Ї–Њ—В–Њ—А—Л—Е –≤¬†–љ–∞—Б—В–Њ—П—Й–µ–µ –≤—А–µ–Љ—П —Б–Ї–Њ–љ—Ж–µ–љ—В—А–Є—А–Њ–≤–∞–љ–∞ –љ–∞¬†–Њ—Ж–µ–љ–Ї–µ —А–Њ–ї–Є¬†–≥–ї–Є–∞–ї—М–љ—Л—Е –Ї–ї–µ—В–Њ–Ї –≤¬†–њ–∞—В–Њ–≥–µ–љ–µ–Ј–µ –љ–µ–≤—А–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є—Е –Њ—Б–ї–Њ–ґ–љ–µ–љ–Є–є –°–Ф.
–Я—А–Є –Ф–Я–Э –≤¬†–њ–µ—А–≤—Г—О –Њ—З–µ—А–µ–і—М —Б—В—А–∞–і–∞—О—В –љ–µ–є—А–Њ–љ–∞–ї—М–љ—Л–µ —Б—В—А—Г–Ї—В—Г—А—Л, –Њ–і–љ–∞–Ї–Њ –Ї–ї–µ—В–Ї–Є¬†–≥–ї–Є–Є —П–≤–ї—П—О—В—Б—П —В–∞–Ї–Њ–є –ґ–µ –Љ–Є—И–µ–љ—М—О –°–Ф, —З—В–Њ –њ–Њ–і—В–≤–µ—А–ґ–і–∞–µ—В—Б—П –≤—Л—П–≤–ї–µ–љ–Є–µ–Љ –њ—А–Є–Ј–љ–∞–Ї–Њ–≤ –і–µ–Љ–Є–µ–ї–Є–љ–Є–Ј–∞—Ж–Є–Є —Г¬†—З–∞—Б—В–Є –њ–∞—Ж–Є–µ–љ—В–Њ–≤ [7, 35]. –Ю—З–µ–≤–Є–і–љ–Њ, —З—В–Њ¬†–≥–ї–Є–∞–ї—М–љ—Л–µ –Ї–ї–µ—В–Ї–Є –њ–µ—А–Є—Д–µ—А–Є—З–µ—Б–Ї–Њ–є –љ–µ—А–≤–љ–Њ–є —Б–Є—Б—В–µ–Љ—Л —В–∞–Ї–ґ–µ —А–µ–∞–≥–Є—А—Г—О—В –љ–∞¬†–і–Є–∞–±–µ—В–Є—З–µ—Б–Ї–Є–µ —Б–Њ—Б—В–Њ—П–љ–Є—П, —З—В–Њ –≤¬†—Б–≤–Њ—О –Њ—З–µ—А–µ–і—М –≤–ї–Є—П–µ—В –љ–∞¬†–Є—Е –≤–∞–ґ–љ—Г—О —Д—Г–љ–Ї—Ж–Є—О¬†вАУ –њ–Є—В–∞–љ–Є–µ –Є¬†—Б–Њ–і–µ–є—Б—В–≤–Є–µ –≤—Л–ґ–Є–≤–∞–љ–Є—О –љ–µ–є—А–Њ–љ–Њ–≤.
–Т¬†—Н–Ї—Б–њ–µ—А–Є–Љ–µ–љ—В–∞—Е –љ–∞¬†–ґ–Є–≤–Њ—В–љ—Л—Е —Б¬†–њ–Њ–Љ–Њ—Й—М—О —Б—В—А–µ–њ—В–Њ–Ј–Њ—В–Њ—Ж–Є–љ–∞, —Б–њ–µ—Ж–Є—Д–Є—З–µ—Б–Ї–Њ–є –і–Є–µ—В—Л –Є¬†–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Њ–є –Є–љ–і—Г–Ї—Ж–Є–Є –±—Л–ї–Є —Б–Њ–Ј–і–∞–љ—Л –Љ–Њ–і–µ–ї–Є –°–Ф¬†1 –Є¬†2 —В–Є–њ–Њ–≤ [36, 37]. –Т¬†—А–∞–Љ–Ї–∞—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є –Њ–њ—А–µ–і–µ–ї—П–ї–Є—Б—М –љ–µ¬†—В–Њ–ї—М–Ї–Њ –њ–Њ—А–∞–ґ–µ–љ–Є—П –і–Є—Б—В–∞–ї—М–љ—Л—Е –∞–Ї—Б–Њ–љ–Њ–≤, –љ–Њ¬†–Є –Є–Ј–Љ–µ–љ–µ–љ–Є–µ –≤–Ј–∞–Є–Љ–Њ–і–µ–є—Б—В–≤–Є—П –љ–µ–є—А–Њ–љ–Њ–≤ —Б¬†–≥–ї–Є–∞–ї—М–љ—Л–Љ–Є –Ї–ї–µ—В–Ї–∞–Љ–Є. –Я–Њ–ї—Г—З–µ–љ–љ—Л–µ —А–µ–Ј—Г–ї—М—В–∞—В—Л —Б–≤–Є–і–µ—В–µ–ї—М—Б—В–≤—Г—О—В –Њ¬†—В–Њ–Љ, —З—В–Њ –њ—А–Є –°–Ф¬†–њ–Њ—А–∞–ґ–∞—О—В—Б—П –≤—Б–µ —Б—В—А—Г–Ї—В—Г—А—Л –њ–µ—А–≤–Є—З–љ–Њ–≥–Њ —Б–µ–љ—Б–Њ—А–љ–Њ–≥–Њ –љ–µ–є—А–Њ–љ–∞. –Ю—Б—В–∞–µ—В—Б—П –Њ—В–Ї—А—Л—В—Л–Љ –≤–Њ–њ—А–Њ—Б –Њ¬†—В–Њ–Љ, –њ–Њ–≤—А–µ–ґ–і–∞—О—В—Б—П –ї–Є –≤¬†–њ–µ—А–≤—Г—О –Њ—З–µ—А–µ–і—М –њ–µ—А–Є—Д–µ—А–Є—З–µ—Б–Ї–Є–µ –∞–Ї—Б–Њ–љ—Л –Є¬†—Б–≤—П–Ј–∞–љ–љ—Л–µ —Б¬†–љ–Є–Љ–Є —И–≤–∞–љ–љ–Њ–≤—Б–Ї–Є–µ –Ї–ї–µ—В–Ї–Є –Є–ї–Є –і–Њ–Љ–Є–љ–Є—А—Г–µ—В –њ–Њ—А–∞–ґ–µ–љ–Є–µ –њ–µ—А–Є–Ї–∞—А–Є–Њ–љ–∞ –љ–µ–є—А–Њ–љ–Њ–≤ —Б¬†—Б–∞—В–µ–ї–ї–Є—В–љ—Л–Љ–Є¬†–≥–ї–Є–∞–ї—М–љ—Л–Љ–Є –Ї–ї–µ—В–Ї–∞–Љ–Є, –љ–∞—Е–Њ–і—П—Й–Є–Љ–Є—Б—П –≤¬†–і–Њ—А—Б–∞–ї—М–љ—Л—Е —Б–њ–Є–љ–љ–Њ–Љ–Њ–Ј–≥–Њ–≤—Л—Е¬†–≥–∞–љ–≥–ї–Є—П—Е.
–°–∞—В–µ–ї–ї–Є—В–љ—Л–µ¬†–≥–ї–Є–∞–ї—М–љ—Л–µ –Ї–ї–µ—В–Ї–Є –ї–Њ–Ї–∞–ї–Є–Ј—Г—О—В—Б—П –≤¬†—Б–µ–љ—Б–Њ—А–љ—Л—Е –Є¬†–∞–≤—В–Њ–љ–Њ–Љ–љ—Л—Е¬†–≥–∞–љ–≥–ї–Є—П—Е –њ–µ—А–Є—Д–µ—А–Є—З–µ—Б–Ї–Њ–є –љ–µ—А–≤–љ–Њ–є —Б–Є—Б—В–µ–Љ—Л, –Њ–±—А–∞–Ј—Г—П —В–Њ–љ–Ї—Г—О –Є¬†–њ–ї–Њ—В–љ—Г—О –Њ–±–Њ–ї–Њ—З–Ї—Г –≤–Њ–Ї—А—Г–≥ –Ї–∞–ґ–і–Њ–є –Њ—В–і–µ–ї—М–љ–Њ–є –љ–µ–є—А–Њ–љ–∞–ї—М–љ–Њ–є —Б–Њ–Љ—Л. –Ш—Е¬†–Ї–Њ–ї–Є—З–µ—Б—В–≤–Њ –њ—А–Њ–њ–Њ—А—Ж–Є–Њ–љ–∞–ї—М–љ–Њ —А–∞–Ј–Љ–µ—А—Г –љ–µ–є—А–Њ–љ–∞. –Ф–≤—Г–љ–∞–њ—А–∞–≤–ї–µ–љ–љ–∞—П —Б–≤—П–Ј—М –Љ–µ–ґ–і—Г –љ–µ–є—А–Њ–љ–∞–Љ–Є –Є¬†—Б–∞—В–µ–ї–ї–Є—В–љ—Л–Љ–Є¬†–≥–ї–Є–∞–ї—М–љ—Л–Љ–Є –Ї–ї–µ—В–Ї–∞–Љ–Є –Њ–±–љ–∞—А—Г–ґ–µ–љ–∞ –≤¬†—Б–њ–Є–љ–љ–Њ–Љ–Њ–Ј–≥–Њ–≤—Л—Е¬†–≥–∞–љ–≥–ї–Є—П—Е –Є¬†–≤ –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ–є —Б—В–µ–њ–µ–љ–Є –Њ–њ–Њ¬≠—Б—А–µ–і–Њ–≤–∞–љ–∞ –њ—Г—А–Є–љ–µ—А–≥–Є—З–µ—Б–Ї–Є–Љ–Є —Б–Є—Б—В–µ–Љ–∞–Љ–Є, –Њ—Б–Њ–±–µ–љ–љ–Њ –њ—А–µ–і—Б—В–∞–≤–ї–µ–љ–љ—Л–Љ–Є –Ї–ї–∞—Б—Б–Њ–Љ P2¬†[38]. –°–∞—В–µ–ї–ї–Є—В–љ—Л–µ¬†–≥–ї–Є–∞–ї—М–љ—Л–µ –Ї–ї–µ—В–Ї–Є —Н–Ї—Б–њ—А–µ—Б—Б–Є—А—Г—О—В —А–µ—Ж–µ–њ—В–Њ—А—Л –љ–µ–є—А–Њ—В—А–∞–љ—Б–Љ–Є—В—В–µ—А–Њ–≤, —В—А–∞–љ—Б–њ–Њ—А—В–µ—А—Л –Є¬†–Є–Њ–љ–љ—Л–µ –Ї–∞–љ–∞–ї—Л, —З—В–Њ –њ–Њ–Ј–≤–Њ–ї—П–µ—В –Ї–Њ–љ—В—А–Њ–ї–Є—А–Њ–≤–∞—В—М –Љ–µ—В–∞–±–Њ–ї–Є—З–µ—Б–Ї–Є–є –Є¬†–љ–µ–є—А–Њ–љ–∞–ї—М–љ—Л–є¬†–≥–Њ–Љ–µ–Њ—Б—В–∞–Ј. –Ъ—А–Њ–Љ–µ —В–Њ–≥–Њ, –Њ–љ–Є –≤—Л—Б–≤–Њ–±–Њ–ґ–і–∞—О—В –љ–µ–є—А–Њ–∞–Ї—В–Є–≤–љ—Л–µ –≤–µ—Й–µ—Б—В–≤–∞, —В–∞–Ї–Є–µ –Ї–∞–Ї –∞–і–µ–љ–Њ–Ј–Є–љ—В—А–Є—Д–Њ—Б—Д–∞—В –Є¬†—Ж–Є—В–Њ–Ї–Є–љ—Л, –Ї–Њ—В–Њ—А—Л–µ —Б–њ–Њ—Б–Њ–±–љ—Л —Б–∞–Љ–Њ—Б—В–Њ—П—В–µ–ї—М–љ–Њ –њ–µ—А–µ–і–∞–≤–∞—В—М —Б–Є–≥–љ–∞–ї—Л –Њ—В¬†–≥–ї–Є–Є –Ї¬†–љ–µ–є—А–Њ–љ—Г [39].
–°–∞—В–µ–ї–ї–Є—В–љ—Л–µ¬†–≥–ї–Є–∞–ї—М–љ—Л–µ –Ї–ї–µ—В–Ї–Є, –Њ–Ї—А—Г–ґ–∞—О—Й–Є–µ –љ–µ–є—А–Њ–љ—Л —Б–њ–Є–љ–љ–Њ–Љ–Њ–Ј–≥–Њ–≤–Њ–≥–Њ¬†–≥–∞–љ–≥–ї–Є—П, —А–µ–∞–≥–Є—А—Г—О—В –љ–∞¬†–њ–Њ–≤—А–µ–ґ–і–µ–љ–Є–µ –љ–µ—А–≤–∞, –≤—Л–Ј—Л–≤–∞—П —Б–Њ–њ—А—П–ґ–µ–љ–љ—Г—О –∞–Ї—В–Є–≤–љ–Њ—Б—В—М –љ–µ–є—А–Њ–љ–Њ–≤, –∞¬†–±–ї–Њ–Ї–Є—А–Њ–≤–∞–љ–Є–µ —Н—В–Є—Е –Ї–Њ–љ—В–∞–Ї—В–Њ–≤ –Њ—Б–ї–∞–±–ї—П–µ—В¬†–≥–Є–њ–µ—А–≤–Њ–Ј–±—Г–і–Є–Љ–Њ—Б—В—М –љ–µ—А–≤–љ—Л—Е –Ї–ї–µ—В–Њ–Ї –Є¬†–Љ–µ—Е–∞–љ–Є—З–µ—Б–Ї—Г—О¬†–≥–Є–њ–µ—А–∞–ї–≥–µ–Ј–Є—О [40]. –Т–Љ–µ—Б—В–µ —Б¬†—В–µ–Љ —А–µ–∞–Ї—Ж–Є—П —Б–∞—В–µ–ї–ї–Є—В–љ—Л—Е¬†–≥–ї–Є–∞–ї—М–љ—Л—Е –Ї–ї–µ—В–Њ–Ї –љ–∞¬†–°–Ф¬†–Љ–Њ–ґ–µ—В –±—Л—В—М –њ–µ—А–≤–Є—З–љ–Њ–є, –Є¬†—В–Њ–≥–і–∞ –Є—Е –≤–Ј–∞–Є–Љ–Њ—Б–≤—П–Ј—М —Б¬†—Б–Њ–Љ–Њ–є –љ–µ–є—А–Њ–љ–Њ–≤, –Њ–њ–Њ—Б—А–µ–і–Њ–≤–∞–љ–љ–∞—П –њ—Г—А–Є–љ–µ—А–≥–Є—З–µ—Б–Ї–Є–Љ–Є —А–µ—Ж–µ–њ—В–Њ—А–∞–Љ–Є, –Њ–њ—А–µ–і–µ–ї—П–µ—В —Д–µ–љ–Њ–Љ–µ–љ –љ–µ–є—А–Њ–љ–∞–ї—М–љ–Њ–≥–Њ —А–∞–Ј–і—А–∞–ґ–µ–љ–Є—П, –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є–Љ–Є —Н–Ї–≤–Є–≤–∞–ї–µ–љ—В–∞–Љ–Є –Ї–Њ—В–Њ—А–Њ–≥–Њ –≤¬†—В–Њ–Љ —З–Є—Б–ї–µ —П–≤–ї—П—О—В—Б—П –±–Њ–ї–µ–≤—Л–µ —Д–µ–љ–Њ–Љ–µ–љ—Л. –Я–Њ–і—В–≤–µ—А–ґ–і–µ–љ–Є–µ–Љ —Н—В–Њ–≥–Њ —В–µ–Ј–Є—Б–∞ —Б–ї—Г–ґ–∞—В —А–µ–Ј—Г–ї—М—В–∞—В—Л –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є, –≤¬†–Ї–Њ—В–Њ—А—Л—Е —Г¬†–Ї—А—Л—Б —Б¬†–Љ–Њ–і–µ–ї—М—О –і–Є–∞–±–µ—В–∞ –Є–љ–≥–Є–±–Є—А–Њ–≤–∞–ї–Є –њ—Г—А–Є–љ–µ—А–≥–Є—З–µ—Б–Ї—Г—О —Б–≤—П–Ј—М –њ–Њ—А–∞–ґ–µ–љ–љ—Л—Е —Б–∞—В–µ–ї–ї–Є—В–љ—Л—Е¬†–≥–ї–Є–∞–ї—М–љ—Л—Е –Ї–ї–µ—В–Њ–Ї —Б¬†–љ–µ–є—А–Њ–љ–∞–Љ–Є –Є¬†–њ–Њ–ї—Г—З–∞–ї–Є –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є–є —Н—Д—Д–µ–Ї—В –≤¬†–≤–Є–і–µ —Б–љ–Є–ґ–µ–љ–Є—П –≤—Л—А–∞–ґ–µ–љ–љ–Њ—Б—В–Є –Љ–µ—Е–∞–љ–Є—З–µ—Б–Ї–Њ–є –Є¬†—В–µ–њ–ї–Њ–≤–Њ–є¬†–≥–Є–њ–µ—А–∞–ї–≥–µ–Ј–Є–Є [41].
–Я—А–Є –°–Ф¬†—В–∞–Ї–ґ–µ –љ–∞—А—Г—И–∞–µ—В—Б—П —Д—Г–љ–Ї—Ж–Є—П —И–≤–∞–љ–љ–Њ–≤—Б–Ї–Є—Е –Ї–ї–µ—В–Њ–Ї, –њ—А–Є—З–µ–Љ –ї–µ–≥–Ї–∞—П —Б–µ–≥–Љ–µ–љ—В–∞—А–љ–∞—П –і–µ–Љ–Є–µ–ї–Є–љ–Є–Ј–∞—Ж–Є—П –љ–∞–±–ї—О–і–∞–µ—В—Б—П —Г–ґ–µ –≤¬†–і–µ–±—О—В–µ –Ф–Я–Э –њ—А–Є —Б–Њ—Е—А–∞–љ–љ–Њ–Љ –∞–Ї—Б–Њ–љ–µ. –≠—В–Њ –њ–Њ–Ј–≤–Њ–ї—П–µ—В –њ—А–µ–і–њ–Њ–ї–Њ–ґ–Є—В—М, —З—В–Њ –Љ–Є–µ–ї–Є–љ–Њ–њ–∞—В–Є—П –Љ–Њ–ґ–µ—В –ї–µ–ґ–∞—В—М –≤¬†–Њ—Б–љ–Њ–≤–µ –њ–µ—А–≤–Є—З–љ–Њ–≥–Њ –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є—П –љ–µ—А–≤–љ—Л—Е –≤–Њ–ї–Њ–Ї–Њ–љ –Є¬†–±—Л—В—М –њ–µ—А–≤—Л–Љ –Ј–≤–µ–љ–Њ–Љ –≤¬†–њ–∞—В–Њ–≥–µ–љ–µ–Ј–µ –Ф–Я–Э [42]. –Ь–µ—Е–∞–љ–Є–Ј–Љ—Л –њ–Њ—А–∞–ґ–µ–љ–Є—П –љ–µ—А–≤–љ—Л—Е –≤–Њ–ї–Њ–Ї–Њ–љ –њ—А–Є –°–Ф¬†—В–∞–Ї–ґ–µ –∞–Ї—В—Г–∞–ї—М–љ—Л –і–ї—П —И–≤–∞–љ–љ–Њ–≤—Б–Ї–Є—Е –Ї–ї–µ—В–Њ–Ї, –њ—А–Є—З–µ–Љ –љ–µ–Ї–Њ—В–Њ—А—Л–µ –Є–Ј¬†–љ–Є—Е –њ—А–µ–Њ–±–ї–∞–і–∞—О—В –Є–Љ–µ–љ–љ–Њ –≤¬†–і–∞–љ–љ—Л—Е¬†–≥–ї–Є–∞–ї—М–љ—Л—Е —Н–ї–µ–Љ–µ–љ—В–∞—Е. –Т¬†—З–∞—Б—В–љ–Њ—Б—В–Є, —Н—В–Њ –Ї–∞—Б–∞–µ—В—Б—П –њ–Њ–ї–Є–Њ–ї–Њ–≤–Њ–≥–Њ –њ—Г—В–Є –Ї–∞–Ї –љ–∞–Є–±–Њ–ї–µ–µ –Є–Ј—Г—З–µ–љ–љ–Њ–≥–Њ –Љ–µ—Е–∞–љ–Є–Ј–Љ–∞ —А–∞–Ј–≤–Є—В–Є—П –Ф–Я–Э. –Ф–Њ–Ї–∞–Ј–∞–љ–Њ, —З—В–Њ –≤–∞–ґ–љ–µ–є—И–Є–є —Д–µ—А–Љ–µ–љ—В —Н—В–Њ–≥–Њ –Љ–µ—В–∞–±–Њ–ї–Є—З–µ—Б–Ї–Њ–≥–Њ –њ—А–Њ—Ж–µ—Б—Б–∞¬†вАУ –∞–ї—М–і–Њ–Ј–Њ—А–µ–і—Г–Ї—В–∞–Ј–∞ —Н–Ї—Б–њ—А–µ—Б—Б–Є—А—Г–µ—В—Б—П –Є–Љ–µ–љ–љ–Њ –≤¬†—И–≤–∞–љ–љ–Њ–≤—Б–Ї–Є—Е –Ї–ї–µ—В–Ї–∞—Е, —З—В–Њ –њ—А–Є–≤–Њ–і–Є—В –Ї¬†—Б–љ–Є–ґ–µ–љ–Є—О –≤—Л—А–∞–±–Њ—В–Ї–Є –±–µ–ї–Ї–Њ–≤ –Љ–Є–µ–ї–Є–љ–∞ –Є¬†–љ–µ–є—А–Њ—В—А–Њ—Д–Є—З–µ—Б–Ї–Є—Е —Д–∞–Ї—В–Њ—А–Њ–≤ [43, 44]. –Я—А–Є —Н—В–Њ–Љ —В–∞–Ї–ґ–µ —Б–љ–Є–ґ–∞–µ—В—Б—П —Н–Ї—Б–њ—А–µ—Б—Б–Є—П —Б–њ–µ—Ж–Є—Д–Є—З–µ—Б–Ї–Є—Е –±–µ–ї–Ї–Њ–≤ –Ї–ї–µ—В–Њ—З–љ–Њ–≥–Њ —Б–Є–≥–љ–∞–ї–Є–љ–≥–∞, —З—В–Њ –і–Њ–њ–Њ–ї–љ–Є—В–µ–ї—М–љ–Њ —Б–њ–Њ—Б–Њ–±—Б—В–≤—Г–µ—В –њ–Њ—А–∞–ґ–µ–љ–Є—О –љ–µ—А–≤–љ—Л—Е –≤–Њ–ї–Њ–Ї–Њ–љ, –љ–∞—А—Г—И–µ–љ–Є—О —Б–Ї–Њ—А–Њ—Б—В–Є –њ—А–Њ–≤–µ–і–µ–љ–Є—П –≤–Њ–Ј–±—Г–ґ–і–µ–љ–Є—П, –Є–љ–і—Г—Ж–Є—А—Г–µ—В —Н–љ–і–Њ—В–µ–ї–Є–∞–ї—М–љ—Г—О –і–Є—Б—Д—Г–љ–Ї—Ж–Є—О –Є¬†–≤ –Ї–Њ–љ–µ—З–љ–Њ–Љ —Б—З–µ—В–µ —Б–њ–Њ—Б–Њ–±—Б—В–≤—Г–µ—В —А–∞–Ј–≤–Є—В–Є—О –Ф–Я–Э [45, 46].
–Т¬†—И–≤–∞–љ–љ–Њ–≤—Б–Ї–Є—Е –Ї–ї–µ—В–Ї–∞—Е, —В–∞–Ї –ґ–µ –Ї–∞–Ї –≤¬†–љ–µ—А–≤–љ—Л—Е –≤–Њ–ї–Њ–Ї–љ–∞—Е, –њ—А–Є –і–Є–∞–±–µ—В–µ –љ–∞–±–ї—О–і–∞—О—В—Б—П —Г–ї—М—В—А–∞—Б—В—А—Г–Ї—В—Г—А–љ—Л–µ –∞–љ–Њ–Љ–∞–ї–Є–Є –≤¬†–Љ–Є—В–Њ—Е–Њ–љ–і—А–Є—П—Е [47]. –Ъ—А–Њ–Љ–µ —В–Њ–≥–Њ, —Б–њ–µ—Ж–Є—Д–Є—З–µ—Б–Ї–Є–µ —А–µ—Ж–µ–њ—В–Њ—А—Л, –љ–∞¬†–Ї–Њ—В–Њ—А—Л–µ –і–µ–є—Б—В–≤—Г—О—В –Ї–Њ–љ–µ—З–љ—Л–µ –њ—А–Њ–і—Г–Ї—В—Л —Г—Б–Є–ї–µ–љ–љ–Њ–≥–Њ¬†–≥–ї–Є–Ї–Є—А–Њ–≤–∞–љ–Є—П, —А–∞—Б–њ–Њ–ї–Њ–ґ–µ–љ—Л –љ–µ¬†—В–Њ–ї—М–Ї–Њ –љ–∞¬†–∞–Ї—Б–Њ–љ–∞—Е, –љ–Њ¬†–Є –љ–∞¬†—И–≤–∞–љ–љ–Њ–≤—Б–Ї–Є—Е –Ї–ї–µ—В–Ї–∞—Е. –≠—В–Њ –њ–Њ–Ј–≤–Њ–ї—П–µ—В –њ—А–µ–і–њ–Њ–ї–∞–≥–∞—В—М –≤–Њ–Ј–Љ–Њ–ґ–љ—Л–є –Ј–∞–њ—Г—Б–Ї –Њ–Ї–Є—Б–ї–Є—В–µ–ї—М–љ–Њ–≥–Њ —Б—В—А–µ—Б—Б–∞ –Є¬†–≤–Њ—Б–њ–∞–ї–µ–љ–Є—П –≤¬†—Н—В–Є—Е —Б—В—А—Г–Ї—В—Г—А–∞—Е –µ—Й–µ –і–Њ –Љ–∞–љ–Є—Д–µ—Б—В–∞—Ж–Є–Є –Ф–Я–Э [48].
–Я—А–Є –і–Є–∞–±–µ—В–µ —Г—Б–Є–ї–Є–≤–∞—О—В—Б—П –Є–Љ–Љ—Г–љ–љ—Л–µ —А–µ–∞–Ї—Ж–Є–Є. –®–≤–∞–љ–љ–Њ–≤—Б–Ї–Є–µ –Ї–ї–µ—В–Ї–Є —Б—З–Є—В–∞—О—В—Б—П –Є–Љ–Љ—Г–љ–Њ–Ї–Њ–Љ–њ–µ—В–µ–љ—В–љ—Л–Љ–Є, –њ–Њ—Б–Ї–Њ–ї—М–Ї—Г —Н–Ї—Б–њ—А–µ—Б—Б–Є—А—Г—О—В –Њ—Б–љ–Њ–≤–љ—Л–µ –Љ–Њ–ї–µ–Ї—Г–ї—Л –Ї–Њ–Љ–њ–ї–µ–Ї—Б–∞¬†–≥–Є—Б—В–Њ—Б–Њ–≤–Љ–µ—Б—В–Є–Љ–Њ—Б—В–Є II –Є¬†–љ–µ—Б–Ї–Њ–ї—М–Ї–Њ Toll-–њ–Њ–і–Њ–±–љ—Л—Е –Є¬†–≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ—Л—Е —А–µ—Ж–µ–њ—В–Њ—А–Њ–≤, –∞¬†—В–∞–Ї–ґ–µ –њ—А–Њ–і—Г—Ж–Є—А—Г—О—В —А—П–і –њ—А–Њ–≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ—Л—Е —Ж–Є—В–Њ–Ї–Є–љ–Њ–≤, –Ї–Њ—В–Њ—А—Л–µ —Г—З–∞—Б—В–≤—Г—О—В –≤¬†–њ–∞—В–Њ–≥–µ–љ–µ–Ј–µ –Ф–Я–Э, –Љ–Њ–≥—Г—В —Б–µ–љ—Б–Є–±–Є–ї–Є–Ј–Є—А–Њ–≤–∞—В—М Aќ≤- –Є¬†–°-–≤–Њ–ї–Њ–Ї–љ–∞ –Є¬†—Б–њ–Њ—Б–Њ–±—Б—В–≤–Њ–≤–∞—В—М —А–∞–Ј–≤–Є—В–Є—О –±–Њ–ї–Є [49].
–Ґ–∞–Ї–Є–Љ –Њ–±—А–∞–Ј–Њ–Љ, —Г—З–∞—Б—В–Є–µ¬†–≥–ї–Є–∞–ї—М–љ—Л—Е –Ї–ї–µ—В–Њ–Ї –≤¬†–њ–∞—В–Њ–≥–µ–љ–µ–Ј–µ –Ф–Я–Э –Њ—З–µ–≤–Є–і–љ–Њ. –Я—А–Є —Н—В–Њ–Љ –Љ–Њ—А—Д–Њ—Д—Г–љ–Ї—Ж–Є–Њ–љ–∞–ї—М–љ—Л–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П –≤¬†–љ–Є—Е –Є–Љ–µ—О—В –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є–µ —Н–Ї–≤–Є–≤–∞–ї–µ–љ—В—Л –ї–Є—И—М –љ–∞¬†–њ–Њ–Ј–і–љ–Є—Е —Б—В–∞–і–Є—П—Е –Ф–Я–Э, –Њ–і–љ–∞–Ї–Њ –Љ–Њ–≥—Г—В —Б—В–∞—В—М –Ј–љ–∞—З–Є–Љ—Л–Љ –Є–ї–Є –і–∞–ґ–µ –њ—Г—Б–Ї–Њ–≤—Л–Љ –Љ–µ—Е–∞–љ–Є–Ј–Љ–Њ–Љ –њ–Њ—А–∞–ґ–µ–љ–Є—П –љ–µ—А–≤–љ—Л—Е –≤–Њ–ї–Њ–Ї–Њ–љ –Є¬†–њ–Њ—П–≤–ї–µ–љ–Є—П –±–Њ–ї–Є —Г–ґ–µ –≤¬†–і–µ–±—О—В–µ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П.
–°¬†—Н—В–Њ–є —В–Њ—З–Ї–Є –Ј—А–µ–љ–Є—П –Њ–њ—А–µ–і–µ–ї–µ–љ–љ—Л–Љ –њ—А–Њ—А—Л–≤–Њ–Љ –≤¬†–ї–µ—З–µ–љ–Є–Є –Ф–Я–Э –Љ–Њ–≥–ї–Њ –±—Л —Б—В–∞—В—М –њ—А–Є–Љ–µ–љ–µ–љ–Є–µ –њ—А–µ–њ–∞—А–∞—В–Њ–≤, –≤–ї–Є—П—О—Й–Є—Е –љ–∞¬†–њ—А–Њ—Ж–µ—Б—Б—Л —А–µ–≥–µ–љ–µ—А–∞—Ж–Є–Є –љ–µ—А–≤–љ–Њ–є —В–Ї–∞–љ–Є, —Б–Ї–Њ—А–Њ—Б—В—М —Е–Є–Љ–Є—З–µ—Б–Ї–Њ–≥–Њ —Б–Є–љ—В–µ–Ј–∞ –Љ–Є–µ–ї–Є–љ–∞ –Є¬†–Љ–µ—В–∞–±–Њ–ї–Є–Ј–Љ¬†–≥–ї–Є–∞–ї—М–љ—Л—Е —Н–ї–µ–Љ–µ–љ—В–Њ–≤. –Ш—Е¬†—В—А–∞–і–Є—Ж–Є–Њ–љ–љ–Њ –Њ—В–љ–Њ—Б—П—В –Ї¬†–љ–µ—Б—В–µ—А–Њ–Є–і–љ—Л–Љ –∞–љ–∞–±–Њ–ї–Є—З–µ—Б–Ї–Є–Љ —Б—А–µ–і—Б—В–≤–∞–Љ. –Я–µ—А—Б–њ–µ–Ї—В–Є–≤–љ—Л–Љ–Є —Б–Њ–µ–і–Є–љ–µ–љ–Є—П–Љ–Є —Н—В–Њ–є¬†–≥—А—Г–њ–њ—Л —П–≤–ї—П—О—В—Б—П –≤–µ—Й–µ—Б—В–≤–∞, –Њ–±–µ—Б–њ–µ—З–Є–≤–∞—О—Й–Є–µ –Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є–µ –њ–Є—А–Є–Љ–Є–і–Є–љ–Њ–≤ –Є¬†–њ—Г—А–Є–љ–Њ–≤, —Г—З–∞—Б—В–≤—Г—О—Й–Є–µ –≤¬†—Б–Є–љ—В–µ–Ј–∞—Е –Ф–Э–Ъ –Є¬†–†–Э–Ъ. –°—А–µ–і–Є –љ–Є—Е –≤¬†–±–Њ–ї—М—И–µ–є —Б—В–µ–њ–µ–љ–Є –Є–Ј—Г—З–µ–љ–Њ –і–µ–є—Б—В–≤–Є–µ –њ—А–Њ–Є–Ј–≤–Њ–і–љ—Л—Е —Г—А–Є–і–Є–љ–∞, –∞–≥–Њ–љ–Є—Б—В–∞ –†2Y-–Љ–µ–і–Є–∞—В–Њ—А–љ—Л—Е –њ—Г—А–Є–љ–µ—А–≥–Є—З–µ—Б–Ї–Є—Е —Б–Є—Б—В–µ–Љ, –≤¬†–Ї–Њ—В–Њ—А—Л—Е –Њ–љ –≤—Л–њ–Њ–ї–љ—П–µ—В —В—А–∞–љ—Б–Љ–Є—В—В–µ—А–љ—Г—О —Д—Г–љ–Ї—Ж–Є—О, —Б—В–Њ–ї—М –≤–∞–ґ–љ—Г—О, —З—В–Њ –Ј–∞ —Н—В–Њ –Њ—В–Ї—А—Л—В–Є–µ –≤¬†1972¬†–≥.¬†–±—Л–ї–∞ –њ—А–Є—Б—Г–ґ–і–µ–љ–∞ –Э–Њ–±–µ–ї–µ–≤—Б–Ї–∞—П –њ—А–µ–Љ–Є—П.
–Я—Г—А–Є–љ–µ—А–≥–Є—З–µ—Б–Ї–Є–µ —А–µ—Ж–µ–њ—В–Њ—А—Л –Њ—В–љ–Њ—Б—П—В –Ї¬†–Ї–ї–∞—Б—Б—Г —В–Є–њ–Є—З–љ—Л—Е 7–Ґ–Ь-—А–µ—Ж–µ–њ—В–Њ—А–Њ–≤. –≠—В–Њ —В—А–∞–љ—Б–Љ–µ–Љ–±—А–∞–љ–љ—Л–µ —А–µ—Ж–µ–њ—В–Њ—А—Л —Б¬†—Б–µ–Љ—М—О –≤–љ–µ—И–љ–Є–Љ–Є –њ–µ—В–ї—П–Љ–Є —Г–Ј–љ–∞–≤–∞–љ–Є—П –ї–Є–≥–∞–љ–і–∞ —Г—А–Є–і–Є–љ–∞, –Є–Љ–µ—О—Й–Є–µ —Б–≤–Њ—О —В–Њ–њ–Є–Ї—Г –Є¬†—З–µ—В–Ї–Њ –Њ—З–µ—А—З–µ–љ–љ—Г—О —Д—Г–љ–Ї—Ж–Є—О. –£—А–Є–і–Є–љ —Г—З–∞—Б—В–≤—Г–µ—В –≤¬†—Б–Є—Б—В–µ–Љ–љ–Њ–є –љ–µ–є—А–Њ—В—А–∞–љ—Б–Љ–Є—В—В–µ—А–љ–Њ–є —А–µ–≥—Г–ї—П—Ж–Є–Є –ґ–Є–Ј–љ–µ–љ–љ–Њ –≤–∞–ґ–љ—Л—Е —Д—Г–љ–Ї—Ж–Є–є –Њ—А–≥–∞–љ–Є–Ј–Љ–∞, —В–∞–Ї–Є—Е –Ї–∞–Ї –њ—А–Њ–Љ–µ–ґ—Г—В–Њ—З–љ—Л–є –Њ–±–Љ–µ–љ, —В–µ—А–Љ–Њ—А–µ–≥—Г–ї—П—Ж–Є—П, –Ї—А–Њ–≤–Њ—В–Њ–Ї, —Б–Њ–Ї—А–∞—В–Є–Љ–Њ—Б—В—М. –Ю–љ –Њ–Ї–∞–Ј—Л–≤–∞–µ—В –Ї–Њ-—В—А–∞–љ—Б–Љ–Є—В—В–µ—А–љ–Њ–µ –і–µ–є—Б—В–≤–Є–µ –њ—А–µ–ґ–і–µ –≤—Б–µ–≥–Њ –≤¬†–Њ—В–љ–Њ—И–µ–љ–Є–Є –Ї¬†–У–Р–Ь–Ъ-, —Е–Њ–ї–Є–љ- –Є¬†–і–Њ—Д–∞–Љ–Є–љ–µ—А–≥–Є—З–µ—Б–Ї–Є—Е —Б–Є—Б—В–µ–Љ, –Ї–∞–ґ–і–∞—П –Є–Ј¬†–Ї–Њ—В–Њ—А—Л—Е –љ–µ—Б–µ—В –∞–љ—В–Є–љ–Њ—Ж–Є—Ж–µ–њ—В–Є–≤–љ—Л–є –њ–Њ—В–µ–љ—Ж–Є–∞–ї. –Э–µ–є—А–Њ–љ–∞–ї—М–љ–∞—П —Б–µ—В—М –њ—Г—А–Є–љ–µ—А–≥–Є—З–µ—Б–Ї–Є—Е —Б–Є—Б—В–µ–Љ –Њ–±—А–∞–Ј—Г–µ—В —А–µ–≥—Г–ї–Є—А—Г—О—Й–Є–є –љ–Њ—Ж–Є—Ж–µ–њ—Ж–Є—О Hub-—А–µ—Ж–µ–њ—В–Њ—А. –Ф–Њ–Ї–∞–Ј–∞–љ–Њ, —З—В–Њ –љ–∞¬†—А–∞–љ–љ–Є—Е —Б—В–∞–і–Є—П—Е –њ–Њ—А–∞–ґ–µ–љ–Є—П –љ–µ—А–≤–љ—Л—Е —Б—В–≤–Њ–ї–Њ–≤ –њ—А–Є —Б–±–Њ–µ –њ—А–Њ—Ж–µ—Б—Б–Є–љ–≥–∞ (—Б–Њ–Ј—А–µ–≤–∞–љ–Є–µ pre-mRNA) –Є¬†—Б–њ–ї–∞–є—Б–Є–љ–≥–∞ (–≤—Л—А–µ–Ј–∞–љ–Є–µ –Є¬†—Б–Њ–µ–і–Є–љ–µ–љ–Є–µ –љ—Г–Ї–ї–µ–Њ—В–Є–і–Њ–≤) —Г—Б–Є–ї–Є–≤–∞–µ—В—Б—П –њ–Њ–≥–ї–Њ—Й–µ–љ–Є–µ —Г—А–Є–і–Є–љ–∞ –Є¬†—Ж–Є—В–Є–і–Є–љ–∞, –∞¬†–Є—Е –љ–∞–Ј–љ–∞—З–µ–љ–Є–µ —Б–Њ–њ—А–Њ–≤–Њ–ґ–і–∞–µ—В—Б—П –љ–Њ—А–Љ–∞–ї—М–љ—Л–Љ —Б–Є–љ—В–µ–Ј–Њ–Љ –Є¬†—А–µ–≥–µ–љ–µ—А–∞—Ж–Є–µ–є –Ї–Њ–Љ–њ–Њ–љ–µ–љ—В–Њ–≤ –љ–µ—А–≤–љ–Њ–≥–Њ –Ї–ї–∞—Б—В–µ—А–∞, –њ—А–Є —Н—В–Њ–Љ –њ–Њ–≤—Л—И–∞—О—В—Б—П –±–Њ–ї–µ–≤–Њ–є –њ–Њ—А–Њ–≥ –Є¬†–њ—А–Њ–≤–Њ–і–Є–Љ–Њ—Б—В—М –≤–Њ–Ј–±—Г–ґ–і–µ–љ–Є—П –њ–Њ¬†–љ–µ—А–≤—Г [50].
–Т¬†–Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П—Е –љ–∞¬†–Ї–ї–µ—В–Њ—З–љ—Л—Е –Ї—Г–ї—М—В—Г—А–∞—Е —Г—Б—В–∞–љ–Њ–≤–ї–µ–љ–Њ, —З—В–Њ –≤–љ–µ–Ї–ї–µ—В–Њ—З–љ—Л–є —Г—А–Є–і–Є–љ—В—А–Є—Д–Њ—Б—Д–∞—В (–£–Ґ–§) –≤–Ј–∞–Є–Љ–Њ–і–µ–є—Б—В–≤—Г–µ—В —Б¬†—А–µ—Ж–µ–њ—В–Њ—А–∞–Љ–Є P2Y —И–≤–∞–љ–љ–Њ–≤—Б–Ї–Є—Е –Ї–ї–µ—В–Њ–Ї –Є¬†–∞–Ї—В–Є–≤–Є—А—Г–µ—В –Љ–Њ–ї–µ–Ї—Г–ї—П—А–љ—Л–µ –Љ–µ—Е–∞–љ–Є–Ј–Љ—Л, –Ї–Њ—В–Њ—А—Л–µ –≤—Л–Ј—Л–≤–∞—О—В –Є–Ј–Љ–µ–љ–µ–љ–Є—П –≤¬†–Є—Е —Ж–Є—В–Њ—Б–Ї–µ–ї–µ—В–µ, —Г–ї—Г—З—И–∞—О—В –Љ–µ–ґ–Ї–ї–µ—В–Њ—З–љ—Л–µ –Ї–Њ–љ—В–∞–Ї—В—Л –Є¬†—Б—В–Є–Љ—Г–ї–Є—А—Г—О—В –Є—Е –Љ–Є–≥—А–∞—Ж–Є—О¬†[51, 52]. –Э–∞–Ї–Њ–њ–ї–µ–љ–љ—Л–µ –Ї¬†–љ–∞—Б—В–Њ—П—Й–µ–Љ—Г –≤—А–µ–Љ–µ–љ–Є –і–∞–љ–љ—Л–µ –њ–Њ–Ј–≤–Њ–ї—П—О—В –Ї–Њ–љ—Б—В–∞—В–Є—А–Њ–≤–∞—В—М, —З—В–Њ —Г—А–Є–і–Є–љ, –≤–Ї–ї—О—З–∞—П—Б—М –≤¬†–Љ–µ—Е–∞–љ–Є–Ј–Љ—Л –њ—А–Њ—Ж–µ—Б—Б–Є–љ–≥–∞, –њ–Њ¬†—Б—Г—В–Є –њ–µ—А–µ–њ—А–Њ–≥—А–∞–Љ–Љ–Є—А—Г–µ—В —И–≤–∞–љ–љ–Њ–≤—Б–Ї–Є–µ –Ї–ї–µ—В–Ї–Є, —А–µ–Ј—Г–ї—М—В–∞—В–Њ–Љ —З–µ–≥–Њ —П–≤–ї—П–µ—В—Б—П –∞–Ї—В–Є–≤–∞—Ж–Є—П –≤–Њ—Б—Б—В–∞–љ–Њ–≤–ї–µ–љ–Є—П –љ–µ—А–≤–љ—Л—Е –≤–Њ–ї–Њ–Ї–Њ–љ [53]. –Я–Њ–ї—Г—З–µ–љ–љ—Л–µ –љ–∞¬†–Ї–ї–µ—В–Њ—З–љ—Л—Е –Ї—Г–ї—М—В—Г—А–∞—Е —А–µ–Ј—Г–ї—М—В–∞—В—Л –±—Л–ї–Є –њ–Њ–і—В–≤–µ—А–ґ–і–µ–љ—Л –і–∞–љ–љ—Л–Љ–Є —Н–Ї—Б–њ–µ—А–Є–Љ–µ–љ—В–Њ–≤ –љ–∞¬†–ґ–Є–≤–Њ—В–љ—Л—Е. –Т¬†—Е–Њ–і–µ –Є—Е –њ—А–Њ–≤–µ–і–µ–љ–Є—П –Ј–∞—Д–Є–Ї—Б–Є—А–Њ–≤–∞–љ–∞ –Ј–љ–∞—З–Є—В–µ–ї—М–љ–∞—П –∞–Ї—В–Є–≤–∞—Ж–Є—П –њ—А–Њ—Ж–µ—Б—Б–Њ–≤ —А–µ–Љ–Є–µ–ї–Є–љ–Є–Ј–∞—Ж–Є–Є –Є¬†—А–µ–≥–µ–љ–µ—А–∞—Ж–Є–Є –∞–Ї—Б–Њ–љ–Њ–≤ —В—А–∞–≤–Љ–∞—В–Є—З–µ—Б–Ї–Є –њ–Њ–≤—А–µ–ґ–і–µ–љ–љ–Њ–≥–Њ –љ–µ—А–≤–∞ —Б¬†–≤–Њ—Б—Б—В–∞–љ–Њ–≤–ї–µ–љ–Є–µ–Љ —Б–Ї–Њ—А–Њ—Б—В–Є –њ—А–Њ–≤–µ–і–µ–љ–Є—П –≤–Њ–Ј–±—Г–ґ–і–µ–љ–Є—П –њ—А–Є –Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–љ–Є–Є —Г—А–Є–і–Є–љ–Љ–Њ–љ–Њ—Д–Њ—Б—Д–∞—В–∞ (–£–Ь–§) –Є¬†—Ж–Є—В–Є–і–Є–љ–Љ–Њ–љ–Њ—Д–Њ—Б—Д–∞—В–∞ (–¶–Ь–§) –≤¬†—В–µ—З–µ–љ–Є–µ 60 –і–љ–µ–є [54].
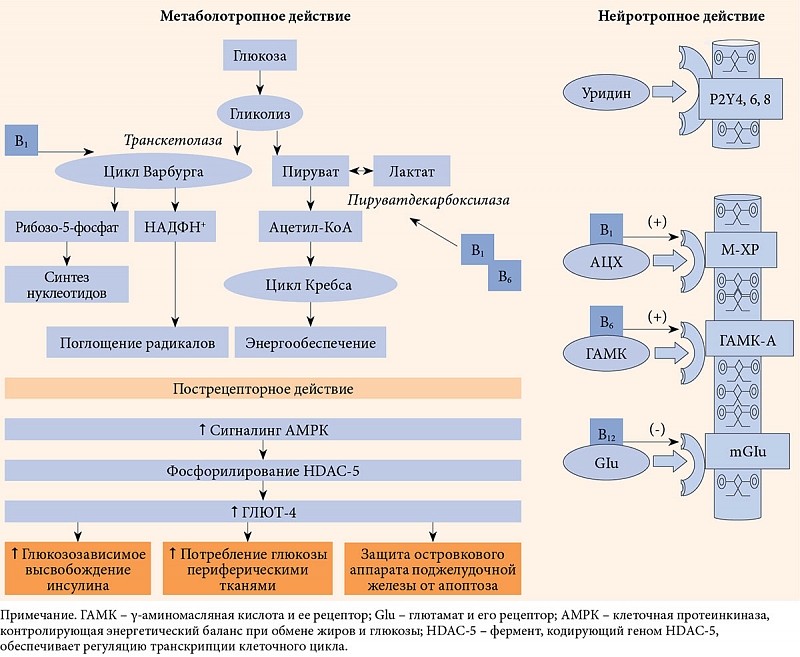
–°–ї–µ–і—Г–µ—В —А–∞–Ј–ї–Є—З–∞—В—М –і–µ–є—Б—В–≤–Є–µ —Г—А–Є–і–Є–љ–∞-–Љ–µ–і–Є–∞—В–Њ—А–∞ –Є¬†—Г—А–Є–і–Є–љ–∞-–Љ–µ—В–∞–±–Њ–ї–Є—В–∞. –Ь–µ—В–∞–±–Њ–ї–Њ—В—А–Њ–њ–љ—Л–µ —А–µ–∞–Ї—Ж–Є–Є –њ—А–Њ–Љ–µ–ґ—Г—В–Њ—З–љ–Њ–≥–Њ –Є¬†–ї–Є–њ–Є–і–љ–Њ–≥–Њ –Њ–±–Љ–µ–љ–Њ–≤ —А–µ–≥—Г–ї–Є—А—Г–µ—В —Г—А–Є–і–Є–љ-–Љe—В–∞–±–Њ–ї–Є—В, –Њ–і–љ–Њ–є –Є–Ј¬†–Љ–µ—В–∞–±–Њ–ї–Є—З–µ—Б–Ї–Є—Е –≤–µ—В–≤–µ–є –Ї–Њ—В–Њ—А–Њ–≥–Њ —П–≤–ї—П–µ—В—Б—П –њ–Њ–і–і–µ—А–ґ–∞–љ–Є–µ —Б–Є–љ—В–µ–Ј–∞ –љ—Г–Ї–ї–µ–Њ—В–Є–і–Њ–≤, –Њ–±–µ—Б–њ–µ—З–Є–≤–∞—О—Й–Є—Е —Б–Є–љ—В–µ–Ј –Є¬†–Ї—А—Г–≥–Њ–Њ–±–Њ—А–Њ—В –Љ–Є–µ–ї–Є–љ–∞. –Я–Њ–Љ–Є–Љ–Њ —Н—В–Њ–≥–Њ —Г—А–Є–і–Є–љ-–Љ–µ—В–∞–±–Њ–ї–Є—В (–≤¬†–≤–Є–і–µ –£–Ь–§) –Ї–Њ–Њ–њ–µ—А–∞—В–Є–≤–љ–Њ —Б–≤—П–Ј–∞–љ —Б¬†—Ж–Є—В–Є–і–Є–љ–Њ–Љ –Є¬†—Д–Њ—Б—Д–Њ—Е–Њ–ї–Є–љ–Њ–Љ. –Т¬†—Н—В–Њ–є —Б–≤—П–Ј–Ї–µ –Њ–љ —А–µ–≥—Г–ї–Є—А—Г–µ—В —Б–Є–љ—В–µ–Ј —Д–Њ—Б—Д–∞—В–Є–і–Є–ї—Е–Њ–ї–Є–љ–∞, —Б—В—А—Г–Ї—В—Г—А–Њ–Њ–±—А–∞–Ј—Г—О—Й–µ–≥–Њ —Д–Њ—Б—Д–Њ–ї–Є–њ–Є–і–∞ –Ї–ї–µ—В–Њ—З–љ—Л—Е –Љ–µ–Љ–±—А–∞–љ –ї—О–±–Њ–≥–Њ —В–Є–њ–∞. –§–Њ—Б—Д–∞—В–Є–і–Є–ї—Е–Њ–ї–Є–љ –Њ–±–µ—Б–њ–µ—З–Є–≤–∞–µ—В –њ–Њ—Б—В–Њ—П–љ—Б—В–≤–Њ –Ї–∞—А–Ї–∞—Б–љ–Њ–є –Є¬†–Љ–∞—В—А–Є—З–љ–Њ–є —Д—Г–љ–Ї—Ж–Є–є –≤¬†–Љ–µ–Љ–±—А–∞–љ–љ–Њ–Љ —Ж–Є–Ї–ї–µ –Ъ–µ–љ–љ–µ–і–Є. –Т¬†–і–∞–љ–љ–Њ–Љ —Ж–Є–Ї–ї–µ —Д–Є–љ–∞–ї—М–љ—Л–Љ–Є –Љ–µ—В–∞–±–Њ–ї–Є—В–∞–Љ–Є —Д–Њ—Б—Д–∞—В–Є–і–Є–ї—Е–Њ–ї–Є–љ–∞ —П–≤–ї—П—О—В—Б—П –Љ–Є–µ–ї–Є–љ –Є¬†—Б—Г—А—Д–∞–Ї—В–∞–љ—В¬†вАУ¬†–≥–ї–∞–≤–љ—Л–µ –Љ–Є—И–µ–љ–Є –і–Є–∞–±–µ—В–Є—З–µ—Б–Ї–Њ–є –љ–µ–є—А–Њ–њ–∞—В–Є–Є –Є¬†—А–∞–Ј–ї–Є—З–љ—Л—Е –±–Њ–ї–µ–Ј–љ–µ–є –ї–µ–≥–Ї–Є—Е. –°–ї–µ–і–Њ–≤–∞—В–µ–ї—М–љ–Њ, —Б–љ–Є–ґ–µ–љ–Є–µ –њ—Г–ї–∞ –£–Ь–§ –Љ–Њ–ґ–µ—В —Б–Њ–њ—А–Њ–≤–Њ–ґ–і–∞—В—М—Б—П –љ–µ¬†—В–Њ–ї—М–Ї–Њ –љ–∞—А—Г—И–µ–љ–Є–µ–Љ —Ж–µ–ї–Њ—Б—В–љ–Њ—Б—В–Є –Љ–µ–Љ–±—А–∞–љ, –љ–Њ¬†–Є —Б–љ–Є–ґ–µ–љ–Є–µ–Љ –Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є—П –Љ–Є–µ–ї–Є–љ–∞ –Є¬†—Б—Г—А—Д–∞–Ї—В–∞–љ—В–∞ [55].
–Т–∞–ґ–љ–Њ–є –Њ—Б–Њ–±–µ–љ–љ–Њ—Б—В—М—О –і–µ–є—Б—В–≤–Є—П —Г—А–Є–і–Є–љ–∞-–Љ–µ–і–Є–∞—В–Њ—А–∞ –Є¬†–њ—А–µ–њ–∞—А–∞—В–Њ–≤, —А–∞–Ј—А–∞–±–Њ—В–∞–љ–љ—Л—Е –љ–∞¬†–µ–≥–Њ –Њ—Б–љ–Њ–≤–µ, —П–≤–ї—П–µ—В—Б—П –њ–Њ—Б—В—А–µ—Ж–µ–њ—В–Њ—А–љ–Њ–µ –і–µ–є—Б—В–≤–Є–µ –њ—Г—А–Є–љ–µ—А–≥–Є—З–µ—Б–Ї–Є—Е —Б–Є—Б—В–µ–Љ, –Њ—Б–Њ–±–µ–љ–љ–Њ —А–µ–∞–ї–Є–Ј—Г–µ–Љ–Њ–µ —З–µ—А–µ–Ј –†2Y- –Є¬†P4Y-—А–µ—Ж–µ–њ—В–Њ—А—Л. –Ю–љ–Њ –≤–Ї–ї—О—З–∞–µ—В —Ж–µ–њ—М –Љ–Њ–ї–µ–Ї—Г–ї—П—А–љ—Л—Е —А–µ–∞–Ї—Ж–Є–є, –Њ–њ–Њ—Б—А–µ–і—Г–µ–Љ—Л—Е –Р–Ь–†–Ъ-—Б–Є—Б—В–µ–Љ–Њ–є¬†вАУ –≤–љ—Г—В—А–Є–Ї–ї–µ—В–Њ—З–љ—Л–Љ –±–µ–ї–Ї–Њ–≤—Л–Љ –Ї–Њ–Љ–њ–ї–µ–Ї—Б–Њ–Љ, –Ї–Њ—В–Њ—А—Л–є –Њ–±–µ—Б–њ–µ—З–Є–≤–∞–µ—В –Њ–Ї–Є—Б–ї–µ–љ–Є–µ¬†–≥–ї—О–Ї–Њ–Ј—Л –Є¬†–ґ–Є—А–љ—Л—Е –Ї–Є—Б–ї–Њ—В –≤¬†—Г—Б–ї–Њ–≤–Є—П—Е –љ–Є–Ј–Ї–Њ–є –≤—Л—А–∞–±–Њ—В–Ї–Є –Ї–ї–µ—В–Њ—З–љ–Њ–є —Н–љ–µ—А–≥–Є–Є, —З—В–Њ –љ–∞–±–ї—О–і–∞–µ—В—Б—П –њ—А–Є –і–Є–∞–±–µ—В–µ, —В–µ–Љ —Б–∞–Љ—Л–Љ —Б—В–Є–Љ—Г–ї–Є—А—Г–µ—В—Б—П –Љ–Є—В–Њ—Е–Њ–љ–і—А–Є–∞–ї—М–љ—Л–є –±–Є–Њ–≥–µ–љ–µ–Ј –Ј–∞ —Б—З–µ—В —Г–≤–µ–ї–Є—З–µ–љ–Є—П –њ—Г–ї–∞ –µ–≥–Њ —Д–µ—А–Љ–µ–љ—В–Њ–≤, —В–∞–Ї–Є—Е –Ї–∞–Ї —Ж–Є—В–Њ—Е—А–Њ–Љ –°,¬†—Б—Г–Ї—Ж–Є–љ–∞—В–і–µ–≥–Є–і—А–Њ–≥–µ–љ–∞–Ј–∞, –Љ–∞–ї–∞—В–і–µ–≥–Є–і—А–Њ–≥–µ–љ–∞–Ј–∞ [56].
–Я—Г—А–Є–љ–µ—А–≥–Є—З–µ—Б–Ї–Є–є —Б–Є–≥–љ–∞–ї–Є–љ–≥ —В–∞–Ї–ґ–µ –Ј–∞—В—А–∞–≥–Є–≤–∞–µ—В –≤–µ—А—Е–љ–Є–µ —Н—В–∞–ґ–Є –±–Є–Њ—В—А–∞–љ—Б—Д–Њ—А–Љ–∞—Ж–Є–Є¬†–≥–ї—О–Ї–Њ–Ј—Л, —Г–≤–µ–ї–Є—З–Є–≤–∞–µ—В –∞–Ї—В–Є–≤–љ–Њ—Б—В—М —Д–µ—А–Љ–µ–љ—В–Њ–≤ –µ–µ –Љ–∞—Б—Б–Њ–њ–µ—А–µ–љ–Њ—Б–∞, —В–∞–Ї–Є—Е –Ї–∞–Ї¬†–≥–ї—О–Ї–Њ–Ј–љ—Л–є —В—А–∞–љ—Б–њ–Њ—А—В–µ—А 4 (–У–Ы–Ѓ–Ґ-4) –Є¬†–≥–µ–Ї—Б–Њ–Ї–Є–љ–∞–Ј–∞ [57].
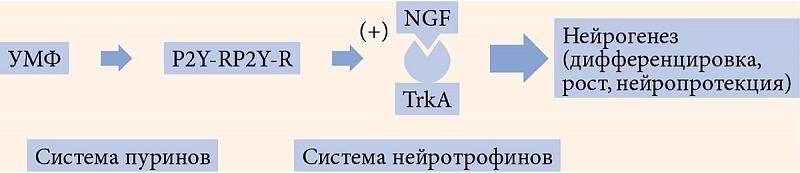
–Я–Њ–Љ–Є–Љ–Њ —Г—Б–Є–ї–µ–љ–Є—П –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є –Р–Ь–†–Ъ-—Б–Є—Б—В–µ–Љ—Л –≤–∞–ґ–љ–Њ–є –Њ—Б–Њ–±–µ–љ–љ–Њ—Б—В—М—О –њ–Њ—Б—В—А–µ—Ж–µ–њ—В–Њ—А–љ–Њ–≥–Њ –і–µ–є—Б—В–≤–Є—П —Г—А–Є–і–Є–љ–∞ —П–≤–ї—П–µ—В—Б—П –∞–Ї—В–Є–≤–∞—Ж–Є—П —В–Є—А–Њ–Ј–Є–љ–Ї–Є–љ–∞–Ј–љ—Л—Е —А–µ—Ж–µ–њ—В–Њ—А–Њ–≤, —А–∞—Б–њ–Њ–ї–Њ–ґ–µ–љ–љ—Л—Е –љ–∞¬†–Љ–µ–Љ–±—А–∞–љ–∞—Е —П–і–µ—А –Ї–ї–µ—В–Њ–Ї, –≤¬†—З–∞—Б—В–љ–Њ—Б—В–Є TrkA-—А–µ—Ж–µ–њ—В–Њ—А–Њ–≤. –≠—В–∞ –≤–Ј–∞–Є–Љ–Њ—Б–≤—П–Ј—М –і–≤—Г—Е —Б–Є—Б—В–µ–Љ –Њ—З–µ–љ—М –≤–∞–ґ–љ–∞, —В–∞–Ї –Ї–∞–Ї –ї–Є–≥–∞–љ–і–∞–Љ–Є Trk-—А–µ—Ж–µ–њ—В–Њ—А–Њ–≤ —П–≤–ї—П—О—В—Б—П –љ–µ–є—А–Њ—В—А–Њ—Д–Є–љ—Л, –Њ–і–Є–љ –Є–Ј¬†–Ї–Њ—В–Њ—А—Л—Е¬†вАУ —Д–∞–Ї—В–Њ—А —А–Њ—Б—В–∞ –љ–µ—А–≤–Њ–≤ (NGF) –њ–Њ–і–і–µ—А–ґ–Є–≤–∞–µ—В –ґ–Є–Ј–љ–µ—Б–њ–Њ—Б–Њ–±–љ–Њ—Б—В—М –љ–µ–є—А–Њ–љ–Њ–≤ –Є¬†—Б—В–Є–Љ—Г–ї–Є—А—Г–µ—В –Є—Е —А–∞–Ј–≤–Є—В–Є–µ –Є¬†–∞–Ї—В–Є–≤–љ–Њ—Б—В—М (—А–Є—Б.¬†1) [58].
–Ґ–∞–Ї–Є–Љ –Њ–±—А–∞–Ј–Њ–Љ, –њ–Њ—Б—В—А–µ—Ж–µ–њ—В–Њ—А–љ–Њ–µ –і–µ–є—Б—В–≤–Є–µ –њ—Г—А–Є–љ–µ—А–≥–Є—З–µ—Б–Ї–Є—Е —Б–Є—Б—В–µ–Љ –Њ–±–µ—Б–њ–µ—З–Є–≤–∞–µ—В [59]:
- —Г–≤–µ–ї–Є—З–µ–љ–Є–µ¬†–≥–ї—О–Ї–Њ–Ј–Њ–Ј–∞–≤–Є—Б–Є–Љ–Њ–≥–Њ –≤—Л—Б–≤–Њ–±–Њ–ґ–і–µ–љ–Є—П –Є–љ—Б—Г–ї–Є–љ–∞;
- —Г—Б–Є–ї–µ–љ–Є–µ –њ–Њ—В—А–µ–±–ї–µ–љ–Є—П¬†–≥–ї—О–Ї–Њ–Ј—Л –њ–µ—А–Є—Д–µ—А–Є—З–µ—Б–Ї–Є–Љ–Є —В–Ї–∞–љ—П–Љ–Є;
- –Ј–∞—Й–Є—В—Г –Њ—Б—В—А–Њ–≤–Ї–Њ–≤–Њ–≥–Њ –∞–њ–њ–∞—А–∞—В–∞ –њ–Њ–і–ґ–µ–ї—Г–і–Њ—З–љ–Њ–є –ґ–µ–ї–µ–Ј—Л –Њ—В¬†–∞–њ–Њ–њ—В–Њ–Ј–∞.
–Ш–љ—Л–Љ–Є —Б–ї–Њ–≤–∞–Љ–Є, –≤¬†–і–µ–є—Б—В–≤–Є–Є —Г—А–Є–і–Є–љ–∞ –Є¬†–њ—А–µ–њ–∞—А–∞—В–Њ–≤, —Б–Њ–Ј–і–∞–љ–љ—Л—Е –љ–∞¬†–µ–≥–Њ –Њ—Б–љ–Њ–≤–µ, –≤¬†—З–∞—Б—В–љ–Њ—Б—В–Є –Ї–Њ–Љ–њ–ї–µ–Ї—Б–∞ –Э–µ–є—А–Њ—Г—А–Є–і–Є–љ, –њ—А–Њ—Б–ї–µ–ґ–Є–≤–∞—О—В—Б—П —В—А–Є –≤–∞–ґ–љ—Л—Е –Ї–Њ–Љ–њ–Њ–љ–µ–љ—В–∞: –Љ–µ—В–∞–±–Њ–ї–Є—З–µ—Б–Ї–Є–є, –љ–µ–є—А–Њ—В—А–Њ–њ–љ—Л–є –Є¬†–њ–Њ—Б—В—А–µ—Ж–µ–њ—В–Њ—А–љ—Л–є, –Ї–∞–ґ–і—Л–є –Є–Ј¬†–Ї–Њ—В–Њ—А—Л—Е –≤–љ–Њ—Б–Є—В —Б–≤–Њ–є –≤–Ї–ї–∞–і –≤¬†—А–µ–њ–∞—А–∞—В–Є–≤–љ—Л–µ –њ—А–Њ—Ж–µ—Б—Б—Л, –Є–Љ–µ—О—Й–Є–µ –Љ–µ—Б—В–Њ –≤¬†—А–∞–Ј–≤–Є—В–Є–Є –і–Є–∞–±–µ—В–Є—З–µ—Б–Ї–Њ–є –љ–µ–є—А–Њ–њ–∞—В–Є–Є (—А–Є—Б.¬†2).
–Э–∞–Ї–Њ–њ–ї–µ–љ–љ–∞—П –і–Њ–Ї–∞–Ј–∞—В–µ–ї—М–љ–∞—П –±–∞–Ј–∞ –њ–Њ–Ј–≤–Њ–ї–Є–ї–∞ –њ—А–Њ–≤–µ—Б—В–Є —А—П–і –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є –њ–Њ¬†–Њ—Ж–µ–љ–Ї–µ —Ж–µ–ї–µ—Б–Њ¬≠–Њ–±—А–∞–Ј–љ–Њ—Б—В–Є –њ—А–Є–Љ–µ–љ–µ–љ–Є—П —Г—А–Є–і–Є–љ–∞ –Є¬†–µ–≥–Њ –њ—А–Њ–Є–Ј–≤–Њ–і–љ—Л—Е –њ—А–Є –Ф–Я–Э. –Т¬†–Њ–і–љ–Њ–Љ –Є–Ј¬†–љ–Є—Е —Н—Д—Д–µ–Ї—В–Є–≤–љ–Њ—Б—В—М –њ–Є—А–Є–Љ–Є–і–Є–љ–Њ–≤—Л—Е –љ—Г–Ї–ї–µ–Њ—В–Є–і–Њ–≤ –Њ—Ж–µ–љ–Є–≤–∞–ї–∞—Б—М —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–°–Ф 2 —В–Є–њ–∞ –Є¬†–Ф–Я–Э –≤—В–Њ—А–Њ–є –Є¬†—В—А–µ—В—М–µ–є —Б—В–∞–і–Є–є. –Ю–±—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ –њ–Њ–Ї–∞–Ј–∞–ї–Њ, —З—В–Њ –і–µ–Љ–Є–µ–ї–Є–љ–Є–Ј–∞—Ж–Є—П —Г¬†–±–Њ–ї—М–љ—Л—Е –Є–Љ–µ–ї–∞ –љ–µ¬†—В–Њ–ї—М–Ї–Њ –≤—В–Њ—А–Є—З–љ–Њ–µ, –љ–Њ¬†–Є –њ–µ—А–≤–Є—З–љ–Њ–µ –њ—А–Њ–Є—Б—Е–Њ–ґ–і–µ–љ–Є–µ, –Ї–Њ—В–Њ—А–Њ–µ –∞–≤—В–Њ—А—Л —Б–≤—П–Ј–∞–ї–Є —Б¬†–Є–Љ–Љ—Г–љ–љ—Л–Љ–Є –Љ–µ—Е–∞–љ–Є–Ј–Љ–∞–Љ–Є –і–Є–∞–±–µ—В–∞. –Ґ–µ—А–∞–њ–Є—П –Њ—Б—Г—Й–µ—Б—В–≤–ї—П–ї–∞—Б—М –Ї–Њ–Љ–њ–ї–µ–Ї—Б–Њ–Љ —Г—А–Є–і–Є–љ–і–Є—Д–Њ—Б—Д–∞—В–∞ (–£–Ф–§), –£–Ґ–§, –£–Ь–§, –¶–Ь–§ –≤¬†—В–µ—З–µ–љ–Є–µ 30 –і–љ–µ–є. –Я–Њ–ї—Г—З–µ–љ–љ—Л–µ —А–µ–Ј—Г–ї—М—В–∞—В—Л —Б–≤–Є–і–µ—В–µ–ї—М—Б—В–≤–Њ–≤–∞–ї–Є –Њ¬†—Б—В–∞—В–Є—Б—В–Є—З–µ—Б–Ї–Є –Ј–љ–∞—З–Є–Љ–Њ–Љ —Г–ї—Г—З—И–µ–љ–Є–Є –њ–Њ–Ї–∞–Ј–∞—В–µ–ї–µ–є —А–µ–Љ–Є–µ–ї–Є–љ–Є–Ј–∞—Ж–Є–Є –Є¬†—А–µ–Є–љ–љ–µ—А–≤–∞—Ж–Є–Є —Г¬†–њ—А–Є–љ–Є–Љ–∞–≤—И–Є—Е –њ–Є—А–Є–Љ–Є–і–Є–љ–Њ–≤—Л–µ –љ—Г–Ї–ї–µ–Њ—В–Є–і—Л. –Я—А–Є —Н—В–Њ–Љ –і–Њ—Б—В–Є–≥–љ—Г—В–∞—П –њ–Њ–ї–Њ–ґ–Є—В–µ–ї—М–љ–∞—П –і–Є–љ–∞–Љ–Є–Ї–∞ —Б–Њ—Е—А–∞–љ—П–ї–∞—Б—М –≤¬†—В–µ—З–µ–љ–Є–µ —В—А–µ—Е –Љ–µ—Б—П—Ж–µ–≤ –њ–Њ—Б–ї–µ –љ–∞—З–∞–ї–∞ —В–µ—А–∞–њ–Є–Є [60].
–Т¬†–і—А—Г–≥–Њ–Љ –і–≤–Њ–є–љ–Њ–Љ —Б–ї–µ–њ–Њ–Љ –њ–ї–∞—Ж–µ–±–Њ-–Ї–Њ–љ—В—А–Њ–ї–Є—А—Г–µ–Љ–Њ–Љ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–Є —Н—Д—Д–µ–Ї—В–Є–≤–љ–Њ—Б—В—М —Г—А–Є–і–Є–љ–∞ –Њ—Ж–µ–љ–Є–≤–∞–ї–∞—Б—М —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–°–Ф¬†1 –Є¬†2 —В–Є–њ–Њ–≤. –Я—А–µ–њ–∞—А–∞—В –љ–∞–Ј–љ–∞—З–∞–ї–Є —В—А–µ—Е–Ї—А–∞—В–љ–Њ –≤¬†–і–Њ–Ј–µ 900 –Љ–≥/—Б—Г—В –≤¬†—В–µ—З–µ–љ–Є–µ 180 –і–љ–µ–є. –Р–≤—В–Њ—А—Л –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П –Ї–Њ–љ—Б—В–∞—В–Є—А–Њ–≤–∞–ї–Є –њ–Њ–≤—Л—И–µ–љ–Є–µ —Б–Ї–Њ—А–Њ—Б—В–Є —А–∞—Б–њ—А–Њ—Б—В—А–∞–љ–µ–љ–Є—П –≤–Њ–Ј–±—Г–ґ–і–µ–љ–Є—П –њ–Њ¬†–љ–µ—А–≤—Г —Г¬†–њ—А–Є–љ–Є–Љ–∞–≤—И–Є—Е —Г—А–Є–і–Є–љ –≤¬†—В–µ—З–µ–љ–Є–µ –≤—Б–µ–≥–Њ –≤—А–µ–Љ–µ–љ–Є –љ–∞–±–ї—О–і–µ–љ–Є—П. –Я—А–Є–Љ–µ—З–∞—В–µ–ї—М–љ—Л–Љ —П–≤–ї—П–µ—В—Б—П —В–Њ—В —Д–∞–Ї—В, —З—В–Њ –њ–Њ–±–Њ—З–љ—Л—Е —Н—Д—Д–µ–Ї—В–Њ–≤ –Є¬†–Њ—Б–ї–Њ–ґ–љ–µ–љ–Є–є –љ–∞¬†—Д–Њ–љ–µ —В–µ—А–∞–њ–Є–Є –≤—Л—П–≤–ї–µ–љ–Њ –љ–µ¬†–±—Л–ї–Њ [61].
–С—Л–ї–Њ —В–∞–Ї–ґ–µ –њ—А–Њ–≤–µ–і–µ–љ–Њ –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Њ–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ —Н—Д—Д–µ–Ї—В–Є–≤–љ–Њ—Б—В–Є –њ—А–Є–Љ–µ–љ–µ–љ–Є—П —Г—А–Є–і–Є–љ–∞ –њ—А–Є –±–Њ–ї–µ–≤—Л—Е —Д–Њ—А–Љ–∞—Е –Ф–Я–Э. –Я–∞—Ж–Є–µ–љ—В–∞–Љ —Б¬†–°–Ф¬†1 –Є¬†2 —В–Є–њ–Њ–≤ –≤¬†—В–µ—З–µ–љ–Є–µ —В—А–µ—Е –Љ–µ—Б—П—Ж–µ–≤ –љ–∞–Ј–љ–∞—З–∞–ї—Б—П –Ї–Њ–Љ–њ–ї–µ–Ї—Б –њ–Є—А–Є–Љ–Є–і–Є–љ–Њ–≤—Л—Е –љ—Г–Ї–ї–µ–Њ—В–Є–і–Њ–≤ (–£–Ґ–§, –£–Ф–§, –£–Ь–§, –¶–Ь–§). –Э–∞—А—П–і—Г —Б¬†—Г–ї—Г—З—И–µ–љ–Є–µ–Љ –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є—Е –Є¬†–Є–љ—Б—В—А—Г–Љ–µ–љ—В–∞–ї—М–љ—Л—Е –њ–Њ–Ї–∞–Ј–∞—В–µ–ї–µ–є, —Б–≤–Є–і–µ—В–µ–ї—М—Б—В–≤—Г—О—Й–Є—Е –Њ–±¬†—Г—Б–Ї–Њ—А–µ–љ–Є–Є —А–µ–≥–µ–љ–µ—А–∞—Ж–Є–Є –љ–µ—А–≤–Њ–≤ –Є¬†–њ—А–Њ—Ж–µ—Б—Б–Њ–≤ —А–µ–Љ–Є–µ–ї–Є–љ–Є–Ј–∞—Ж–Є–Є, –Њ—В–Љ–µ—З–µ–љ–Њ –≤—Л—А–∞–ґ–µ–љ–љ–Њ–µ —Б–љ–Є–ґ–µ–љ–Є–µ –±–Њ–ї–µ–≤–Њ–≥–Њ —Б–Є–љ–і—А–Њ–Љ–∞¬†вАУ –њ—А–Є–Љ–µ—А–љ–Њ –љ–∞¬†30% [62].
–Р–љ–∞–ї—М–≥–µ—В–Є—З–µ—Б–Ї–Є–µ —Н—Д—Д–µ–Ї—В—Л —Г—А–Є–і–Є–љ–∞ —Б–≤—П–Ј–∞–љ—Л —Б¬†–µ–≥–Њ –њ—А—П–Љ—Л–Љ –Є¬†–Њ–њ–Њ—Б—А–µ–і–Њ–≤–∞–љ–љ—Л–Љ –і–µ–є—Б—В–≤–Є–µ–Љ –љ–∞¬†–Љ–µ—Е–∞–љ–Є–Ј–Љ—Л –±–Њ–ї–Є. –Я—А—П–Љ–Њ–µ –і–µ–є—Б—В–≤–Є–µ –њ—А–µ–ґ–і–µ –≤—Б–µ–≥–Њ –њ–Њ–і—А–∞–Ј—Г–Љ–µ–≤–∞–µ—В –≤–ї–Є—П–љ–Є–µ –љ–∞¬†—Д–∞–Ї—В–Њ—А—Л —Б–Є—Б—В–µ–Љ–љ–Њ–≥–Њ –≤–Њ—Б–њ–∞–ї–µ–љ–Є—П¬†вАУ –њ—А–Є–Њ—А–Є—В–µ—В–љ–Њ–є —Б–Њ—Б—В–∞–≤–ї—П—О—Й–µ–є —А–∞–Ј–≤–Є—В–Є—П –Ф–Я–Э, –Њ—Б–Њ–±–µ–љ–љ–Њ –љ–∞¬†–љ–∞—З–∞–ї—М–љ—Л—Е —Б—В–∞–і–Є—П—Е –њ–Њ—А–∞–ґ–µ–љ–Є—П –љ–µ–≤—А–∞–ї—М–љ—Л—Е —Б—В—А—Г–Ї—В—Г—А. –Ю–њ–Њ—Б—А–µ–і–Њ–≤–∞–љ–љ–Њ–µ –і–µ–є—Б—В–≤–Є–µ —А–µ–∞–ї–Є–Ј—Г–µ—В—Б—П —З–µ—А–µ–Ј –≤–Њ—Б—Б—В–∞–љ–Њ–≤–ї–µ–љ–Є–µ —Б—В—А—Г–Ї—В—Г—А–љ–Њ–є —Ж–µ–ї–Њ—Б—В–љ–Њ—Б—В–Є –Є¬†–љ–Њ—А–Љ–∞–ї—М–љ–Њ–≥–Њ —Д—Г–љ–Ї—Ж–Є–Њ–љ–Є—А–Њ–≤–∞–љ–Є—П –љ–µ–≤—А–∞–ї—М–љ—Л—Е –Є¬†–≥–ї–Є–∞–ї—М–љ—Л—Е —Н–ї–µ–Љ–µ–љ—В–Њ–≤.
–Ъ–∞–Ї —Г–ґ–µ –±—Л–ї–Њ —Б–Ї–∞–Ј–∞–љ–Њ –≤—Л—И–µ, –ї—О–±–Њ–µ –њ–Њ—А–∞–ґ–µ–љ–Є–µ –љ–µ—А–≤–Њ–≤ —Б–Њ–њ—А–Њ–≤–Њ–ґ–і–∞–µ—В—Б—П –і–µ—Д–Є—Ж–Є—В–Њ–Љ —Г—А–Є–і–Є–љ–∞, —П–≤–ї—П—О—Й–µ–≥–Њ—Б—П –≤–∞–ґ–љ—Л–Љ –Ї–Њ–Љ–њ–Њ–љ–µ–љ—В–Њ–Љ –Ф–Э–Ъ –Є¬†–†–Э–Ъ. –Ф–µ—Д–Є—Ж–Є—В –≤–Њ–Ј–љ–Є–Ї–∞–µ—В –Є–Ј-–Ј–∞ –≤–Њ–Ј—А–Њ—Б—И–µ–є –њ–Њ—В—А–µ–±–љ–Њ—Б—В–Є –≤¬†—Н—В–Њ–Љ –љ—Г–Ї–ї–µ–Њ—В–Є–і–µ –Є¬†–љ–µ–≤–Њ–Ј–Љ–Њ–ґ–љ–Њ—Б—В–Є –µ–≥–Њ –≤–љ—Г—В—А–Є–Ї–ї–µ—В–Њ—З–љ–Њ–≥–Њ —Б–Є–љ—В–µ–Ј–∞. –Я–Њ-–≤–Є–і–Є–Љ–Њ–Љ—Г, –њ–Њ—Н—В–Њ–Љ—Г –њ—А–Є –љ–µ–є—А–Њ–њ–∞—В–Є—З–µ—Б–Ї–Њ–є –±–Њ–ї–Є –љ–∞–±–ї—О–і–∞–µ—В—Б—П —Н–Ї—Б–њ—А–µ—Б—Б–Є—П –њ—Г—А–Є–љ–Њ–≤—Л—Е —А–µ—Ж–µ–њ—В–Њ—А–Њ–≤ –≤¬†—А–∞–Ј–ї–Є—З–љ—Л—Е —Б—В—А—Г–Ї—В—Г—А–∞—Е —Ж–µ–љ—В—А–∞–ї—М–љ–Њ–є –Є¬†–њ–µ—А–Є—Д–µ—А–Є—З–µ—Б–Ї–Њ–є –љ–µ—А–≤–љ–Њ–є —Б–Є—Б—В–µ–Љ—Л. –Э–∞–њ—А–Є–Љ–µ—А, —А–µ—Ж–µ–њ—В–Њ—А—Л P2Y2, –Ї–Њ—В–Њ—А—Л–µ —А–∞—Б–њ–Њ–ї–Њ–ґ–µ–љ—Л –≤¬†–љ–µ–є—А–Њ–љ–∞—Е —В—А–Њ–є–љ–Є—З–љ–Њ–≥–Њ¬†–≥–∞–љ–≥–ї–Є—П, –љ–∞—З–Є–љ–∞—О—В –Є–љ–Є—Ж–Є–Є—А–Њ–≤–∞—В—М –Є¬†–њ–Њ–і–і–µ—А–ґ–Є–≤–∞—В—М –∞–ї–ї–Њ–і–Є–љ–Є–Є [63]. –†–µ—Ж–µ–њ—В–Њ—А—Л P2Y6 –Є¬†P2Y11 —Н–Ї—Б–њ—А–µ—Б—Б–Є—А—Г—О—В—Б—П –≤¬†—Б–њ–Є–љ–∞–ї—М–љ–Њ–є –Љ–Є–Ї—А–Њ–≥–ї–Є–Є –Є¬†—А–µ–≥—Г–ї–Є—А—Г—О—В—Б—П –≤¬†–Њ—В–≤–µ—В –љ–∞¬†–њ–Њ–≤—А–µ–ґ–і–µ–љ–Є–µ —Б–њ–Є–љ–љ–Њ–Љ–Њ–Ј–≥–Њ–≤–Њ–≥–Њ –љ–µ—А–≤–∞, —З—В–Њ –њ—А–Њ—П–≤–ї—П–µ—В—Б—П¬†–≥–Є–њ–µ—А—З—Г–≤—Б—В–≤–Є—В–µ–ї—М–љ–Њ—Б—В—М—О –Ї¬†–Љ–µ—Е–∞–љ–Є—З–µ—Б–Ї–Њ–є –±–Њ–ї–Є –Є¬†—В–∞–Ї—В–Є–ї—М–љ–Њ–є –∞–ї–ї–Њ–і–Є–љ–Є–µ–є [64, 65].
–Ґ–∞–Ї–Є–Љ –Њ–±—А–∞–Ј–Њ–Љ, —Б–≤–Њ–µ–≤—А–µ–Љ–µ–љ–љ–Њ–µ –њ—А–Є–Љ–µ–љ–µ–љ–Є–µ —Г—А–Є–і–Є–љ–∞ –њ—А–µ–і–Њ—В–≤—А–∞—Й–∞–µ—В —Е—А–Њ–љ–Є–Ј–∞—Ж–Є—О –±–Њ–ї–µ–≤—Л—Е —Д–µ–љ–Њ–Љ–µ–љ–Њ–≤.
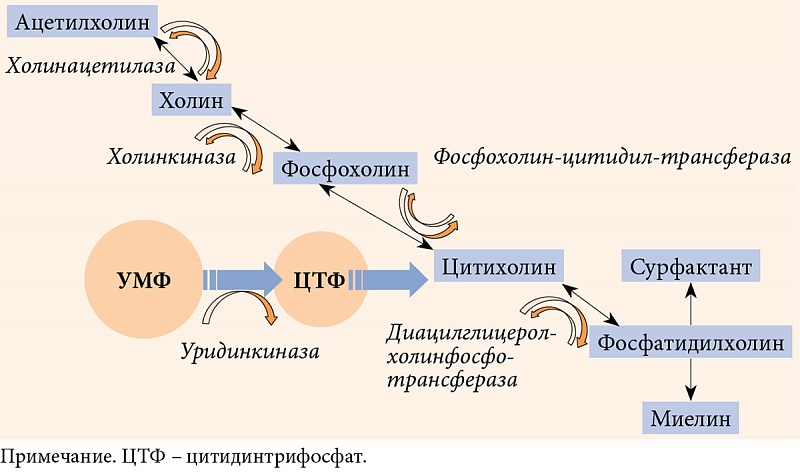
–Я—А–µ–і—Б—В–∞–≤–ї–µ–љ–љ—Л–є –љ–∞¬†–Њ—В–µ—З–µ—Б—В–≤–µ–љ–љ–Њ–Љ —А—Л–љ–Ї–µ –Ї–Њ–Љ–њ–ї–µ–Ї—Б –Э–µ–є—А–Њ—Г—А–Є–і–Є–љ —Б–Њ–і–µ—А–ґ–Є—В 150 –Љ–≥ –£–Ь–§, —Е–Њ–ї–Є–љ, —Д–Њ–ї–Є–µ–≤—Г—О –Ї–Є—Б–ї–Њ—В—Г –Є¬†–≤–Є—В–∞–Љ–Є–љ—Л¬†–≥—А—Г–њ–њ—Л –Т¬†(–Т1, –Т6 –Є¬†–Т12). –Э–µ–є—А–Њ—Г—А–Є–і–Є–љ –Њ—В–љ–Њ—Б–Є—В—Б—П –Ї¬†–±–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–Љ –∞–Ї—В–Є–≤–љ—Л–Љ –і–Њ–±–∞–≤–Ї–∞–Љ, —З—В–Њ –љ–µ¬†—Г–Љ–∞–ї—П–µ—В –µ–≥–Њ —Б–≤–Њ–є—Б—В–≤. –Ъ–Њ–Љ–њ–Њ–љ–µ–љ—В—Л –Э–µ–є—А–Њ—Г—А–Є–і–Є–љ–∞ –Њ—А–≥–∞–љ–Є—З–љ–Њ –і–Њ–њ–Њ–ї–љ—П—О—В –і–µ–є—Б—В–≤–Є–µ –£–Ь–§ –љ–∞¬†–љ–µ—А–≤–љ—Г—О —В–Ї–∞–љ—М. –Т¬†—З–∞—Б—В–љ–Њ—Б—В–Є, —Б–Є–љ—В–µ–Ј —Е–Њ–ї–Є–љ–∞, –Є–Ј¬†–Ї–Њ—В–Њ—А–Њ–≥–Њ –Њ–±—А–∞–Ј—Г–µ—В—Б—П –∞—Ж–µ—В–Є–ї—Е–Њ–ї–Є–љ-–Љ–µ–і–Є–∞—В–Њ—А (–Р–¶–•), –њ–Њ–і–і–µ—А–ґ–Є–≤–∞–µ—В—Б—П –њ—Г–ї–Њ–Љ —Г—А–Є–і–Є–љ–∞, –±–µ–Ј –Ї–Њ—В–Њ—А–Њ–≥–Њ —Г—А–Њ–≤–µ–љ—М –Р–¶–• —Б–љ–Є–ґ–∞–µ—В—Б—П. –°—Г–±—Б—В—А–∞—В —Е–Њ–ї–Є–љ–∞ –±—Л–ї –≤–≤–µ–і–µ–љ –≤¬†—А–µ—Ж–µ–њ—В—Г—А—Г –Э–µ–є—А–Њ—Г—А–Є–і–Є–љ–∞ –µ—Й–µ –њ–Њ¬†–Њ–і–љ–Њ–є –њ—А–Є—З–Є–љ–µ. –Я—А–Є —Б–Є–љ—В–µ–Ј–µ –Љ–Є–µ–ї–Є–љ–∞, –≤¬†–Ї–Њ—В–Њ—А–Њ–Љ —Г—З–∞—Б—В–≤—Г–µ—В —Г—А–Є–і–Є–љ, –≤–µ–Ї—В–Њ—А —Ж–Є–Ї–ї–∞ –Ъ–µ–љ–љ–µ–і–Є —Б–Љ–µ—Й–∞–µ—В—Б—П –≤¬†—Б—В–Њ—А–Њ–љ—Г –Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є—П —Д–Њ—Б—Д–∞—В–Є–і–Є–ї—Е–Њ–ї–Є–љ–∞, –≤—Б–ї–µ–і—Б—В–≤–Є–µ —З–µ–≥–Њ —Б–Є–љ—В–µ–Ј —Е–Њ–ї–Є–љ–∞, –Є–Ј¬†–Ї–Њ—В–Њ—А–Њ–≥–Њ –Њ–±—А–∞–Ј—Г–µ—В—Б—П –Р–¶–•, —Б–љ–Є–ґ–∞–µ—В—Б—П (—А–Є—Б.¬†3). –•–Њ–ї–Є–љ 82,5 –Љ–≥ –њ—А–Є —А–µ–Ј–Њ—А–±—Ж–Є–Є, —А–∞—Б–њ—А–µ–і–µ–ї–µ–љ–Є–Є –Є¬†–њ–Њ—Б—В—Г–њ–ї–µ–љ–Є–Є –≤¬†–Љ–µ–Љ–±—А–∞–љ—Л —Б–Њ–Ј–і–∞–µ—В —В–Њ—В –љ–µ–Њ–±—Е–Њ–і–Є–Љ—Л–є –њ—Г–ї, –њ—А–Є –Ї–Њ—В–Њ—А–Њ–Љ —Б–Є–љ—В–µ–Ј –Р–¶–• –љ–µ¬†—Б—В—А–∞–і–∞–µ—В. –Ґ–∞–Ї–Є–Љ –Њ–±—А–∞–Ј–Њ–Љ, –њ—А–Њ–і—Г–Љ–∞–љ–љ—Л–є —Б–Њ—Б—В–∞–≤ –Э–µ–є—А–Њ—Г—А–Є–і–Є–љ–∞ –њ–Њ–Ј–≤–Њ–ї—П–µ—В –Њ–±–µ—Б–њ–µ—З–Є—В—М —Б–Є–љ—В–µ–Ј –Љ–Є–µ–ї–Є–љ–∞ –Є¬†–љ–µ –і–Њ–њ—Г—Б—В–Є—В—М –њ–Њ—В–µ—А–Є –Р–¶–•.
–С–Њ–ї–µ–µ —В–Њ–≥–Њ, –≤¬†—Н–Ї—Б–њ–µ—А–Є–Љ–µ–љ—В–∞–ї—М–љ—Л—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П—Е –±—Л–ї–Є –њ—А–Њ–і–µ–Љ–Њ–љ—Б—В—А–Є—А–Њ–≤–∞–љ—Л –∞–љ–∞–ї—М–≥–µ–Ј–Є—А—Г—О—Й–Є–є —Н—Д—Д–µ–Ї—В —Е–Њ–ї–Є–љ–∞, –≤¬†—В–Њ–Љ —З–Є—Б–ї–µ –њ—А–Є –љ–µ–є—А–Њ–њ–∞—В–Є—З–µ—Б–Ї–Њ–є –±–Њ–ї–Є, —Г–ї—Г—З—И–µ–љ–Є–µ —Д—Г–љ–Ї—Ж–Є–Њ–љ–∞–ї—М–љ–Њ–≥–Њ –≤–Њ—Б—Б—В–∞–љ–Њ–≤–ї–µ–љ–Є—П –Є¬†—А–µ–≥–µ–љ–µ—А–∞—Ж–Є–Є –љ–µ—А–≤–∞, —Б–µ–љ—Б–Њ—А–љ—Л—Е —Б–Є–Љ–њ—В–Њ–Љ–Њ–≤ –Є¬†—Б–Ї–Њ—А–Њ—Б—В–Є –њ—А–Њ–≤–µ–і–µ–љ–Є—П –≤–Њ–Ј–±—Г–ґ–і–µ–љ–Є—П –њ–Њ¬†–Љ–Њ—В–Њ—А–љ–Њ–Љ—Г –љ–µ—А–≤—Г, —А–µ–≥–µ–љ–µ—А–∞—Ж–Є–Є –Є¬†–≤–Њ—Б—Б—В–∞–љ–Њ–≤–ї–µ–љ–Є—П –∞–Ї—Б–Њ–љ–Њ–≤ –њ—А–Є —А–∞—Б—Б–µ—З–µ–љ–Є–Є –њ–µ—А–Є—Д–µ—А–Є—З–µ—Б–Ї–Њ–≥–Њ –љ–µ—А–≤–∞ [66вАУ68]. –Я—А–µ–Є–Љ—Г—Й–µ—Б—В–≤–∞ –∞–љ–∞–ї—М–≥–µ—В–Є—З–µ—Б–Ї–Њ–≥–Њ –і–µ–є—Б—В–≤–Є—П —Е–Њ–ї–Є–љ–∞ –≤¬†–Ї–Њ–Љ–±–Є–љ–∞—Ж–Є–Є —Б¬†–њ–Є—А–Є–Љ–Є–і–Є–љ–Њ–≤—Л–Љ–Є –љ—Г–Ї–ї–µ–Њ—В–Є–і–∞–Љ–Є –њ—А–µ–і—Б—В–∞–≤–ї–µ–љ—Л –≤¬†—А—П–і–µ —Д—Г–љ–і–∞–Љ–µ–љ—В–∞–ї—М–љ—Л—Е —А–∞–±–Њ—В, –љ–∞¬†–Њ—Б–љ–Њ–≤–∞–љ–Є–Є –∞–љ–∞–ї–Є–Ј–∞ —А–µ–Ј—Г–ї—М—В–∞—В–Њ–≤ –Ї–Њ—В–Њ—А—Л—Е —Б–і–µ–ї–∞–љ –≤—Л–≤–Њ–і –Њ¬†–≤–Њ–Ј–Љ–Њ–ґ–љ–Њ–Љ —Б–Є–љ–µ—А–≥–Є–Ј–Љ–µ —Н—Д—Д–µ–Ї—В–Њ–≤ –Ї–Њ–Љ–њ–Њ–љ–µ–љ—В–Њ–≤ –і–∞–љ–љ–Њ–є –Ї–Њ–Љ–±–Є–љ–∞—Ж–Є–Є [69].
–§–Њ–ї–Є–µ–≤–∞—П –Ї–Є—Б–ї–Њ—В–∞ –Є¬†–≤–Є—В–∞–Љ–Є–љ—Л¬†–≥—А—Г–њ–њ—Л –Т, –≤—Е–Њ–і—П—Й–Є–µ –≤¬†—Б–Њ—Б—В–∞–≤ –Э–µ–є—А–Њ—Г—А–Є–і–Є–љ–∞, —Б–Њ–і–µ—А–ґ–∞—В—Б—П –≤¬†–њ—А–µ–і–µ–ї–∞—Е —Б—Г—В–Њ—З–љ–Њ–є –њ–Њ—В—А–µ–±–љ–Њ—Б—В–Є, –Ї–Њ—В–Њ—А–∞—П –Њ–±–µ—Б–њ–µ—З–Є–≤–∞–µ—В –Є—Е —Г—З–∞—Б—В–Є–µ –Ї–∞–Ї –≤¬†–Љ–µ—В–∞–±–Њ–ї–Є—З–µ—Б–Ї–Є—Е –њ—А–Њ—Ж–µ—Б—Б–∞—Е, —А–µ–≥—Г–ї–Є—А—Г–µ–Љ—Л—Е –≤–Є—В–∞–Љ–Є–љ–Њ–њ—А–µ–њ–∞—А–∞—В–∞–Љ–Є, —В–∞–Ї –Є¬†–≤ —Б–Є–љ–∞–њ—В–Є—З–µ—Б–Ї–Є—Е —Б–Є—Б—В–µ–Љ–∞—Е,¬†–≥–і–µ –Њ–љ–Є –≤—Л–њ–Њ–ї–љ—П—О—В —А–Њ–ї—М –∞–ї–ї–Њ—Б—В–µ—А–Є—З–µ—Б–Ї–Є—Е —Н—Д—Д–µ–Ї—В–Њ—А–Њ–≤ (—Б–Љ. —А–Є—Б.¬†2). –°–ї–µ–і—Г–µ—В –і–Њ–±–∞–≤–Є—В—М, —З—В–Њ —В–∞–Ї–Є–µ –і–Њ–Ј—Л –љ–µ¬†–Њ–±–µ—Б–њ–µ—З–Є–≤–∞—О—В —Б–∞–Љ–Њ—Б—В–Њ—П—В–µ–ї—М–љ–Њ–≥–Њ –∞–љ–∞–ї—М–≥–µ—В–Є—З–µ—Б–Ї–Њ–≥–Њ –і–µ–є—Б—В–≤–Є—П, –љ–Њ¬†—Б–њ–Њ—Б–Њ–±—Б—В–≤—Г—О—В —А–µ–≥–µ–љ–µ—А–∞—Ж–Є–Є –љ–µ—А–≤–Њ–≤, —Б–љ–Є–ґ–∞—О—В —А–Є—Б–Ї¬†–љ–µ–ґ–µ–ї–∞—В–µ–ї—М–љ—Л—Е –њ—А–Њ—П–≤–ї–µ–љ–Є–є —Д–∞—А–Љ–∞–Ї–Њ—В–µ—А–∞–њ–Є–Є –Є¬†–њ–Њ–і–і–µ—А–ґ–Є–≤–∞—О—В Hub-—А–µ—Ж–µ–њ—В–Њ—А –≤¬†–µ–≥–Њ –≤–Ј–∞–Є–Љ–Њ—Б–≤—П–Ј—П—Е —Б¬†–і—А—Г–≥–Є–Љ–Є —Б–Є–љ–∞–њ—В–Є—З–µ—Б–Ї–Є–Љ–Є —Б–Є—Б—В–µ–Љ–∞–Љ–Є.
–°–ї–µ–і–Њ–≤–∞—В–µ–ї—М–љ–Њ, –њ—А–Є–Љ–µ–љ–µ–љ–Є–µ –Э–µ–є—А–Њ—Г—А–Є–і–Є–љ–∞ —Г–ґ–µ –љ–∞¬†—Б–∞–Љ—Л—Е —А–∞–љ–љ–Є—Е —Н—В–∞–њ–∞—Е –Ф–Я–Э —Б–њ–Њ—Б–Њ–±–љ–Њ —Г–ї—Г—З—И–Є—В—М –Љ–µ—В–∞–±–Њ–ї–Є—З–µ—Б–Ї–Є–µ –Є¬†—А–µ–≥–µ–љ–µ—А–∞—В–Є–≤–љ—Л–µ –њ—А–Њ—Ж–µ—Б—Б—Л –≤¬†–њ–µ—А–Є—Д–µ—А–Є—З–µ—Б–Ї–Є—Е –љ–µ—А–≤–∞—Е, –∞¬†—В–∞–Ї–ґ–µ —Д—Г–љ–Ї—Ж–Є–Њ–љ–∞–ї—М–љ–Њ–µ —Б–Њ—Б—В–Њ—П–љ–Є–µ –љ–µ—А–≤–љ–Њ–є —В–Ї–∞–љ–Є.
–Я—А–Њ–≤–µ–і–µ–љ–љ—Л–є –Њ–±–Ј–Њ—А –ї–Є—В–µ—А–∞—В—Г—А—Л –љ–µ¬†–њ–Њ–і—А–∞–Ј—Г–Љ–µ–≤–∞–µ—В –њ—А–Є–Љ–µ–љ–µ–љ–Є—П —Г—А–Є–і–Є–љ–Њ–≤—Л—Е –Ї–Њ–Љ–њ–ї–µ–Ї—Б–Њ–≤ –њ—А–Є –Ф–Я–Э –≤¬†–Ї–∞—З–µ—Б—В–≤–µ –Љ–Њ–љ–Њ—В–µ—А–∞–њ–Є–Є. –Ґ–µ–Љ –љ–µ¬†–Љ–µ–љ–µ–µ —Г—А–Є–і–Є–љ–Љ–Њ–љ–Њ—Д–Њ—Б—Д–∞—В –Љ–Њ–ґ–µ—В –±—Л—В—М –Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–љ –≤¬†–Ї–∞—З–µ—Б—В–≤–µ –і–Њ–њ–Њ–ї–љ–Є—В–µ–ї—М–љ–Њ–≥–Њ —Б—А–µ–і—Б—В–≤–∞ –њ–∞—В–Њ–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Њ–≥–Њ –ї–µ—З–µ–љ–Є—П –љ–∞—А—П–і—Г —Б¬†–њ—А–µ–њ–∞—А–∞—В–∞–Љ–Є, –њ–Њ–Ј–≤–Њ–ї—П—О—Й–Є–Љ–Є –Ї–Њ—А—А–µ–Ї—В–Є—А–Њ–≤–∞—В—М¬†–≥–Є–њ–µ—А–≥–ї–Є–Ї–µ–Љ–Є—О, –і–Є—Б–ї–Є–њ–Є–і–µ–Љ–Є—О, –і—А—Г–≥–Є–µ —Д–∞–Ї—В–Њ—А—Л —А–Є—Б–Ї–∞ —А–∞–Ј–≤–Є—В–Є—П –Ф–Я–Э, ќ±-–ї–Є–њ–Њ–µ–≤–Њ–є –Ї–Є—Б–ї–Њ—В–Њ–є, –±–µ–љ—Д–Њ—В–Є–∞–Љ–Є–љ–Њ–Љ, –Є–љ–≥–Є–±–Є—В–Њ—А–∞–Љ–Є –∞–ї—М–і–Њ–Ј–Њ—А–µ–і—Г–Ї—В–∞–Ј—Л –Є¬†–њ—А–Њ—В–µ–Є–љ–Ї–Є–љ–∞–Ј—Л –°,¬†ќ≥-–ї–Є–љ–Њ–ї–µ–љ–Њ–≤–Њ–є –Ї–Є—Б–ї–Њ—В–Њ–є. –Я–µ—А—Б–њ–µ–Ї—В–Є–≤–љ—Л–Љ —В–∞–Ї–ґ–µ –≤–Є–і–Є—В—Б—П –њ—А–Є–Љ–µ–љ–µ–љ–Є–µ —Г—А–Є–і–Є–љ–Љ–Њ–љ–Њ—Д–Њ—Б—Д–∞—В–∞ –Ї–∞–Ї –Ї–Њ-–∞–љ–∞–ї—М–≥–µ—В–Є–Ї–∞ –њ—А–Є –±–Њ–ї–µ–≤—Л—Е —Д–Њ—А–Љ–∞—Е –Ф–Я–Э –≤¬†–і–Њ–њ–Њ–ї–љ–µ–љ–Є–µ –Ї¬†–∞–љ—В–Є–і–µ–њ—А–µ—Б—Б–∞–љ—В–∞–Љ –Є¬†–≥–∞–±–∞–њ–µ–љ—В–Є–љ–Њ–Є–і–∞–Љ.¬†
D.A. Iskra, MD, PhD, Prof., V.V. Afanasyev, MD, PhD, Prof., A.R. Volkova, MD, PhD, Prof.
St. Petersburg State Pediatric Medical University
North-Western State Medical University named after I.I. Mechnikov
Academician I.P. Pavlov First St. Petersburg State Medical University
Contact person: Dmitriy A. Iskra, iskradm@mail.ru
Analyzed modern conceptions about the etiopathogenesis of diabetic polyneuropathy. Thus, the main etiological factors leading to the development of the disease are hyperglycemia and dyslipidemia, which cause metabolic disorders in the neuronal cluster. The development of diabetic polyneuropathy is implemented through the mechanisms of impaired glycolysis, oxidative stress, systemic inflammation, mitochondrial dysfunction, and systemic inflammation plays a leading role in the onset of damage to the nervous system. The probability of neuropathic pain is associated with the presence of modifiable and unmodifiable risk factors: female gender, age, obesity, elevated levels of glycated hemoglobin, excessive alcohol consumption, prolonged diabetes, severity of sensory disorders, genetic predisposition. The clinical and pathogenetic relationship of pain with hypoxia of peripheral tissues, vitamin D deficiency, excessive methylglyoxal formation has been proven. The manifestation of clinical signs is caused not only by the defeat of the entire cluster, but also by its glial elements. Dysfunction of Schwann cells, sattelitic glial cells and mild segmental demyelination are observed already in the onset of diabetic polyneuropathy. These mechanisms lead to disruption of the neuroglial interactions underlying the trophic neurons and the formation of their genome. Metabotropic P2Y receptor agonists, pyrimidine nucleotides (uridine, cytidine), play an important role in the metabolism of glial elements. The prospects of using uridine-containing complexes in the treatment of diabetic polyneuropathy with the restoration of the structural integrity of the nerve fiber, regression of clinical symptoms, including the decrease in the severity of pain, have been confirmed by the results of laboratory and clinical studies.
–£–≤–∞–ґ–∞–µ–Љ—Л–є –њ–Њ—Б–µ—В–Є—В–µ–ї—М uMEDp!
–£–≤–µ–і–Њ–Љ–ї—П–µ–Љ –Т–∞—Б –Њ —В–Њ–Љ, —З—В–Њ –Ј–і–µ—Б—М —Б–Њ–і–µ—А–ґ–Є—В—Б—П –Є–љ—Д–Њ—А–Љ–∞—Ж–Є—П, –њ—А–µ–і–љ–∞–Ј–љ–∞—З–µ–љ–љ–∞—П –Є—Б–Ї–ї—О—З–Є—В–µ–ї—М–љ–Њ –і–ї—П —Б–њ–µ—Ж–Є–∞–ї–Є—Б—В–Њ–≤ –Ј–і—А–∞–≤–Њ–Њ—Е—А–∞–љ–µ–љ–Є—П.
–Х—Б–ї–Є –Т—Л –љ–µ —П–≤–ї—П–µ—В–µ—Б—М —Б–њ–µ—Ж–Є–∞–ї–Є—Б—В–Њ–Љ –Ј–і—А–∞–≤–Њ–Њ—Е—А–∞–љ–µ–љ–Є—П, –∞–і–Љ–Є–љ–Є—Б—В—А–∞—Ж–Є—П –љ–µ –љ–µ—Б–µ—В –Њ—В–≤–µ—В—Б—В–≤–µ–љ–љ–Њ—Б—В–Є –Ј–∞ –≤–Њ–Ј–Љ–Њ–ґ–љ—Л–µ –Њ—В—А–Є—Ж–∞—В–µ–ї—М–љ—Л–µ –њ–Њ—Б–ї–µ–і—Б—В–≤–Є—П, –≤–Њ–Ј–љ–Є–Ї—И–Є–µ –≤ —А–µ–Ј—Г–ї—М—В–∞—В–µ —Б–∞–Љ–Њ—Б—В–Њ—П—В–µ–ї—М–љ–Њ–≥–Њ –Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–љ–Є—П –Т–∞–Љ–Є –Є–љ—Д–Њ—А–Љ–∞—Ж–Є–Є —Б –њ–Њ—А—В–∞–ї–∞ –±–µ–Ј –њ—А–µ–і–≤–∞—А–Є—В–µ–ї—М–љ–Њ–є –Ї–Њ–љ—Б—Г–ї—М—В–∞—Ж–Є–Є —Б –≤—А–∞—З–Њ–Љ.
–Э–∞–ґ–Є–Љ–∞—П –љ–∞ –Ї–љ–Њ–њ–Ї—Г ¬Ђ–Т–Њ–є—В–Є¬ї, –Т—Л –њ–Њ–і—В–≤–µ—А–ґ–і–∞–µ—В–µ, —З—В–Њ —П–≤–ї—П–µ—В–µ—Б—М –≤—А–∞—З–Њ–Љ –Є–ї–Є —Б—В—Г–і–µ–љ—В–Њ–Љ –Љ–µ–і–Є—Ж–Є–љ—Б–Ї–Њ–≥–Њ –≤—Г–Ј–∞.