Обоснование дифференцированного подхода к назначению препаратов дефицитных факторов свертывания крови больным гемофилией А
- Аннотация
- Статья
- Ссылки
Введение
Современное обеспечение пациентов с коагулопатиями препаратами факторов свертывания крови позволило увеличить продолжительность жизни больных гемофилией и улучшить ее качество [1, 2, 3], что подтверждает международный опыт [4]. В России заместительная терапия пациентов с гемофилией осуществляется согласно национальным протоколам [5]. Тем не менее не утратила своей актуальности проблема эффективности лечения и профилактики кровотечений. При одинаковых уровнях дефицитного фактора свертывания крови интенсивность и характер кровопотерь имеет индивидуальные отличия у разных пациентов с гемофилией. Поражения суставов у больных гемофилией остаются в центре внимания специалистов, профилактическое введение препаратов дефицитных факторов свертывания крови во взрослом возрасте не устраняет нарушения, а только уменьшает частоту рецидивов гемартрозов [6, 7]. Таким образом, актуальным вопросом остается разработка подходов к индивидуализации терапии при гемофилии [8].
Цель исследования
Обосновать дифференцированный подход к проведению заместительной терапии препаратами дефицитных факторов свертывания крови у больных гемофилией с учетом гемостатического потенциала при разной AB0-групповой принадлежности крови пациентов.
Материал и методы исследования
Проведено исследование 185 госпитализаций 30 больных с тяжелой формой гемофилии А (фактор свертывания крови VIII менее 1%) в возрасте от 18 до 40 лет, проживающих в Самарской области. Заместительная терапия препаратами фактора свертывания крови VIII (Иммунат®) проводилась в режиме «по требованию» согласно моделям 4.3 и 4.4 протокола ведения больных с гемофилией от 01.10.2009, включенного в национальный стандарт Российской Федерации. 50% больных с тяжелой гемофилией А получали профилактическое лечение в средней дозе 25 МЕ/кг 3 раза в неделю, согласно модели 4.2 [5]. Комплексное исследование системы гемостаза у больных гемофилией проводили в первые сутки после возникновения кровотечения, до введения препарата дефицитного фактора свертывания крови. При этом у пациентов, находившихся на профилактической заместительной терапии, интервал времени между последним введением препарата фактора и моментом взятия крови на исследование составил минимум 3 дня. Уровень фактора Виллебранда оценивался как во время кровотечения, так и вне периода кровотечений.
Оценка коагуляционного гемостаза проводилась с помощью автоматического коагулометра STA-Compact (“Roche”, Франция), тромбоцитарного гемостаза – с помощью гематологического анализатора Sysmex КХ-21 (“Roche”, Япония) и определения агрегационной способности тромбоцитов методом визуальной детекции времени начала агрегации с индукторами: аденозиндифосфатом (АДФ); коллагеном, универсальным индуктором агрегации (УИА). Для определения групп крови по системе AB0 использовали перекрестный способ с помощью моноклональных антител.
Контрольную группу составили 185 практически здоровых мужчин в возрасте от 18 до 45 лет (28 ± 1,5 лет).
Статистическая обработка результатов исследований проведена с помощью статистического пакета SPSS 12.0 и STATISTICA 7.0 с использованием параметрических и непараметрических методов.
Результаты и обсуждение
У больных гемофилией А тяжелой степени был проведен сравнительный анализ коагуляционного и сосудисто-тромбоцитарного потенциала системы гемостаза при 0 (I) – АВ (IV) группах крови (табл. 1). Данные литературы свидетельствуют об особенностях фактора свертывания крови VIII и фактора Виллебранда при 0 (I) группе крови [9–14]. В некоторых популяционных исследованиях было показано, что у лиц с I группой крови уровень фактора свертывания VIII и уровень фактора Виллебранда на 25% ниже, чем при других группах крови [10]. В исследовании, проведенном у здоровых женщин и у женщин-носительниц гена гемофилии, был выявлен значимо более низкий уровень обоих факторов при I группе крови [14].
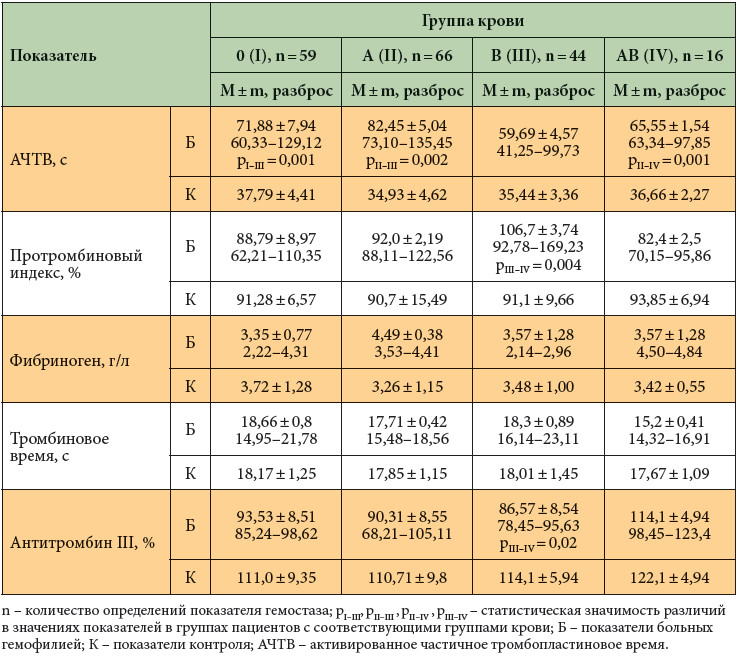
В нашем исследовании госпитализированные больные распределились по группам крови следующим образом: 0 (I) группа крови – у 32%, A (II) – у 36%, B (III) – у 24%, AB (IV) – у 8% пациентов; Rh(+) – в 85%, Rh(-) – в 15% случаев. Группоспецифические особенности коагуляционного гемостаза у обследуемых пациентов представлены в табл. 1.
У обследованных пациентов с A (II) группой крови активированное частичное тромбопластиновое время (АЧТВ) составило 82,45 ± 5,04 с и было значимо выше по сравнению со значениями у больных с другими группами крови (рII–III = 0,002, рII–IV = 0,001). Это может свидетельствовать о более слабом внутреннем механизме коагуляции у больных гемофилией с A (II) группой крови. Как показано в табл. 1, наибольшее среднее значение протромбинового индекса (ПТИ) и наименьшее значение активности антитромбина III выявлены у больных тяжелой гемофилией А c B (III) группой крови. Предположительно, у этой категории пациентов существует компенсаторная реакция внешнего пути свертывания крови и противосвертывающей системы, направленная на предотвращение чрезмерной кровопотери.
Выявленные нами группоспецифические особенности коагуляционного гемостаза у больных гемофилией А тяжелой степени имеют отличия от особенностей гемостаза в здоровой популяции. Так, в здоровой популяции мужчин отмечена тенденция к гипокоагуляции при 0 (I) группе крови (максимальные значения АЧТВ, более высокие значения тромбопластинового времени). При A (II) группе крови, в отличие от данных обследованных нами пациентов с гемофилией, выявлена склонность к гиперкоагуляции (наиболее низкие значения АЧТВ, тромбопластинового времени, антитромбина III). Как у здоровых мужчин с В (III) группой крови, так и у больных гемофилией значения АЧТВ минимальны, но значение ПТИ не имеет тенденции к увеличению. Как у здоровых мужчин, так и у больных гемофилией при АВ (IV) группе крови отмечено достаточно стабильное состояние системы гемостаза за счет сбалансированности систем про- и антикоагуляции [9, 15].
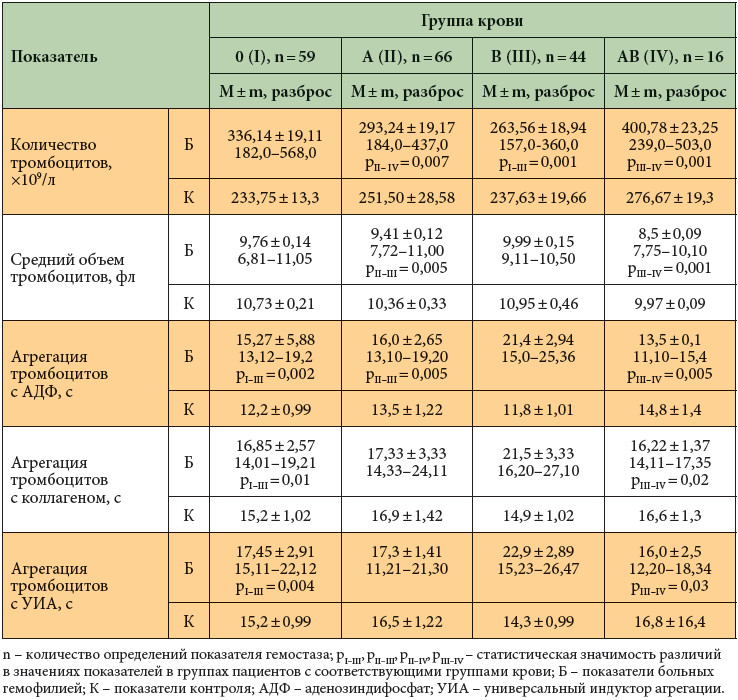
При исследовании тромбоцитарного звена гемостаза у больных тяжелой гемофилией А в период кровотечений также выявлены группоспецифические особенности (табл. 2).
У всех исследуемых больных определено более высокое количество тромбоцитов по сравнению со значениями контрольной группы, возможно, это обусловлено реакцией тромбоцитарного гемостаза на кровопотерю. Однако наибольшее количество тромбоцитов определено у пациентов с AB (IV) группой крови (табл. 2). Установлено, что при B (III) группе крови у больных тяжелой гемофилией А тромбоцитарное звено гемостаза развито хуже, чем при других группах крови: количество тромбоцитов наименьшее, средний объем тромбоцитов максимален, нарушена стимулированная агрегация тромбоцитов. Полученные нами данные существенно не отличаются от параметров тромбоцитарного гемостаза, определенных в ряде исследований у здоровой популяции мужчин [9, 15].
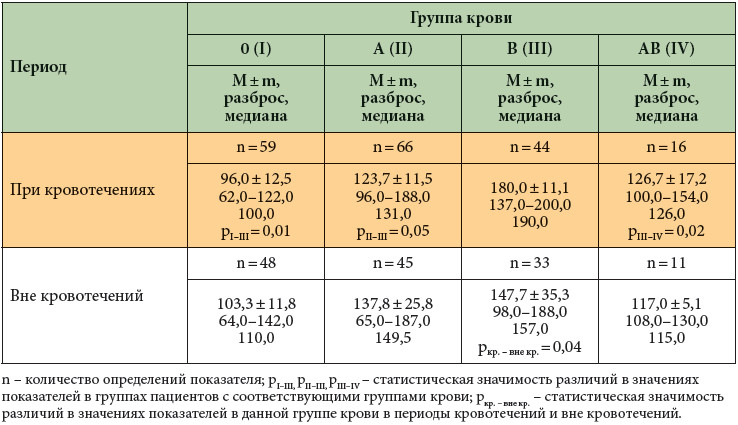
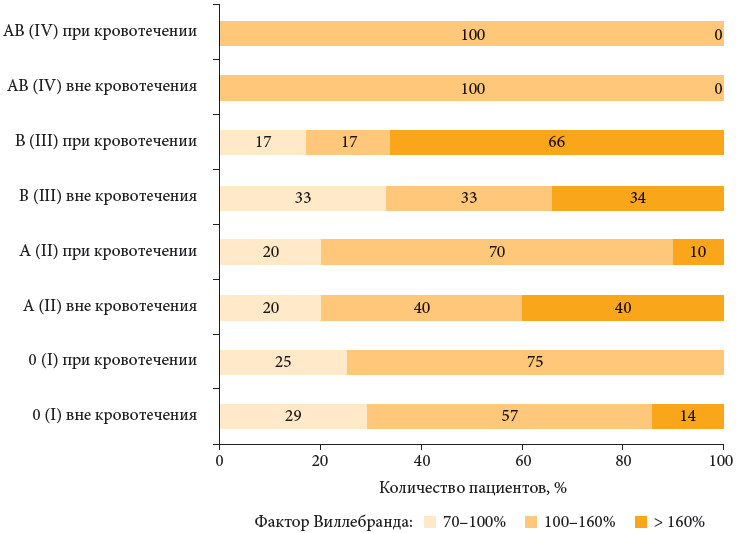
Для характеристики гемостатического потенциала эндотелия сосудов у больных гемофилией исследован фактор Виллебранда, который, как известно, поступает в кровоток преимущественно из эндотелиальных клеток (табл. 3, рисунок).
В нашем исследовании определено, что у больных тяжелой формой гемофилии с B (III) группой крови наблюдается наилучший гемостатический потенциал эндотелия. В данной группе больных как вне кровотечений, так и в период кровотечений определялся максимальный уровень фактора Виллебранда (рисунок), при этом в период кровотечений фактор Виллебранда достоверно повышался и в 66% случаев был более 160%. У больных с AB (IV) группой крови данный показатель находился всегда в диапазоне 100–160%. Наиболее слабые резервные возможности эндотелия сосудов наблюдались при 0 (I) и A (II) группах крови; при кровотечениях у данных больных определена тенденция к снижению фактора Виллебранда.
Заключение
В нашем исследовании были выявлены AB0-группоспецифические особенности показателей коагуляционного и сосудисто-тромбоцитарного гемостаза при тяжелой гемофилии А. Полученные нами данные свидетельствуют о возможности проведения лечения этих пациентов с учетом групповой принадлежности крови. Наиболее стабильное состояние системы коагуляционного и сосудисто-тромбоцитарного гемостаза выявлено у больных тяжелой гемофилией А с АВ (IV) группой крови. У пациентов с группой крови B (III) определена более высокая активность коагуляционного гемостаза, в том числе внешних механизмов свертывания крови, и снижение активности антитромбина III по сравнению с другими группами крови, что, предположительно, имеет компенсаторное значение. У этих пациентов также определен достаточно высокий гемостатический потенциал эндотелия, о чем косвенно свидетельствует высокая активность фактора Виллебранда. В то же время у данных больных выявлены неполноценность тромбоцитарного звена, нарушение АДФ-, коллаген-, УИА-индуцированной агрегации тромбоцитов. Следовательно, у больных гемофилией с группой крови B (III) при использовании для профилактического лечения препаратов дефицитных факторов свертывания крови, содержащих фактор Виллебранда, целесообразно проводить лабораторный мониторинг этого показателя и контролировать агрегационные свойства тромбоцитов. У больных с гемофилией А тяжелой степени при 0 (I) и при A (II) группах крови наблюдались низкие, по сравнению с другими группами, коагуляционная активность внутреннего пути свертывания крови и гемостатический потенциал эндотелия, обусловленный снижением фактора Виллебранда при кровопотерях. У больных гемофилией с данными группами крови не отмечалось компенсаторной активации внешнего пути свертывания крови.
У больных гемофилией А тяжелой степени с 0 (I) и A (II) группами крови возможно патогенетически обоснованное использование в качестве заместительной терапии препаратов, содержащих фактор Виллебранда (Иммунат и др.). У этой категории пациентов при развитии тяжелых кровотечений, не купируемых введением больших доз концентрата фактора VIII, по жизненным показаниям возможно применение препаратов с шунтирующим механизмом действия, например, антиингибиторного коагулянтного комплекса. Таким образом, у больных тяжелой формой гемофилии А выявлены AB0-группоспецифичные отличия в коагуляционном и тромбоцитарном звеньях гемостаза, что может быть учтено при назначении заместительной терапии концентратами факторов крови.
Уважаемый посетитель uMEDp!
Уведомляем Вас о том, что здесь содержится информация, предназначенная исключительно для специалистов здравоохранения.
Если Вы не являетесь специалистом здравоохранения, администрация не несет ответственности за возможные отрицательные последствия, возникшие в результате самостоятельного использования Вами информации с портала без предварительной консультации с врачом.
Нажимая на кнопку «Войти», Вы подтверждаете, что являетесь врачом или студентом медицинского вуза.