NB
![Рис.1. Алгоритм лечения диабетической нейропатии* Адаптировано по [27].](/upload/iblock/c7e/c7e1150e112737199a7c2f9d27449bd4.JPG)
Диабетическая нейропатия (ДН) – патология нервной системы, проявляющаяся клинически или субклинически у больных сахарным диабетом (СД) при отсутствии других причин ее развития (конференция по проблемам диабетической нейропатии, Сан-Антонио, 1988) [1]. При данной патологии поражается как соматический отдел нервной системы, так и автономный. Клинические проявления ДН имеют примерно 50% больных СД. Субклинические формы, выявляемые дополнительными методами исследования, имеют около 90% больных СД с длительностью течения заболевания более года [2].
Дистальная периферическая сенсомоторная диабетическая полинейропатия (ДСПН) является наиболее часто встречающейся формой ДН. По данным литературы, частота данной формы сильно варьирует в зависимости от выбранных диагностических критериев. Однозначно частота выявляемости ДСПН возрастает у больных с большой давностью диабета и при недостаточной компенсации углеводного обмена. Длительная и тяжелая гипергликемия является основной причиной развития ДСПН. Самые важные этиологические факторы, связанные с развитием ДСПН, – это плохой контроль гликемии, абдоминальное ожирение, продолжительность диабета, гипертония, возраст, курение, дислипидемия [3].
Предложенная в 1970-х гг. профессором П.К. Томасом (P.K. Thomas) классификация диабетической нейропатии [4] в конце XX в. была изменена [5]. В настоящее время рекомендовано проводить дифференциальную диагностику между симметричными множественными нейропатиями и центральными или многофокальными нейропатиями. Вместе с тем у некоторых пациентов с длительным течением сахарного диабета ДCПН может отсутствовать, тогда как у части пациентов с небольшой длительностью заболевания и относительно хорошим контролем уровня гликемии ДCПН, наоборот, может присутствовать, что свидетельствует о возможном участии генетических факторов, кодирующих различные патогенетические звенья ее развития. Считается, что улучшение контроля гликемии предотвращает прогрессирование ДCПН, хотя не приводит к ее обратному развитию [6].
Эпидемиологические исследования показали, что при СД 1 типа нарушения функции периферической нервной системы могут быть выявлены в течение первых 2–8 лет после манифестации заболевания. Распространенность ДCПН незначительна в момент диагностики СД и прогрессивно нарастает по мере увеличения длительности и тяжести заболевания [7]. Было установлено, что, несмотря на выраженные метаболические нарушения, ДСПН развивается у 5–50% пациентов с СД 1 типа. В среднем периферической нейропатией страдают 25% больных СД [8, 9].
Одни авторы отмечают, что субклиническая (ЭМГ-верифицированная) патология эфферентной иннервации обнаруживается почти у всех больных СД [9, 6], другие – в 70–90% случаев [10, 11]. Различные данные статистики по распространенности ДСПН связаны с отсутствием единых диагностических подходов и критериев. Частота выявления ДСПН зависит от метода диагностики поражений нервной системы. При оценке S. Shalitin и соавт. (2002) показателей нейропатии только по шкале нейропатических жалоб/симптомов (TSS; онемение, парестезии, боль, жжение) и шкале неврологических нарушений (NDS по Янгу; вибрационная, тактильная и температурная чувствительность, снижение рефлексов) распространенность полинейропатии среди молодых пациентов с СД 1 типа составила 17,1% [12].
В полиоловом пути фермент альдозоредуктаза в норме восстанавливает токсичные альдегиды до неактивных спиртов. При повышении внутриклеточной концентрации глюкозы ее избыток восстанавливается альдозоредуктазой до сорбитола, при этом избыточно расходуется НАДФ, который необходим для регенерации восстановленного глутатиона. Глутатион, в свою очередь, является незаменимым внутриклеточным антиоксидантом, при снижении его количества клетки становятся особо чувствительными к окислению [15].
Другим патологическим путем повреждения тканей является активация протеинкиназы С. Конечный продукт метаболизма ацетилглюкозамин вызывает изменение факторов транскрипции, что приводит к гиперэкспрессии PAI-1 и TGF-бета, а также изменению структуры белковых рецепторов, вследствие чего возникает резистентность к инсулину [16, 17]. В условиях гипергликемии идет повышенное образование конечных продуктов гликирования (КПГ) и их предшественников (например, метилглиоксаля). Метилглиоксаль – важный фактор гликирования, который реагирует со свободными аминогруппами лизина и аргинина в составе белков, образуя внутриклеточные КПГ [17, 18]. Внутриклеточная продукция КПГ, изменяя транскрипцию генов, структуру белков внеклеточного матрикса и циркулирующих белков крови, приводит к нарушению функции периферических нервных волокон. Систематические исследования выяснили, что все эти патогенетические механизмы объединяет общий процесс – повышение продукции свободнорадикального супероксид-аниона и образующихся из него активных форм кислорода (АФК) [19, 20, 21].
Супероксид-анион активирует основные пути гипергликемического повреждения, снижая активность ключевого фермента гликолиза глицеральдегид-3-фосфатдегидрогеназы (GAPDH). Супероксид-анион образуется в качестве побочного продукта окисления глюкозы в цепи электронного транспорта митохондрий. Внутриклеточная гипергликемия увеличивает количество доноров электронов в митохондриях, что повышает продукцию супероксид-аниона. Разрыв цепочек ДНК под действием этого супероксид-аниона приводит к подавлению GAPDH. Уровни всех предшествующих промежуточных продуктов этого метаболического пути гликолиза становятся повышенными, что «вытесняет» их в альтернативные пути. Глицеральдегид-3-фосфат и его производные глицерол и метилглиоксаль поступают в пути PKC и КПГ, фруктозо-6-фосфат – в гексозаминовый путь, а сама глюкоза поступает в полиольный путь. Все вышеперечисленные патологические пути утилизации метаболитов глюкозы и самой глюкозы являются причиной развития диабетических осложнений, поражения нервной ткани и сосудистой стенки (нейропатия и ангиопатия) [13, 14]. Главной причиной диабетической нейропатии, как, впрочем, и всех других осложнений сахарного диабета, является хроническая гипергликемия и активация описанных выше патологических путей метаболизма глюкозы.
В лечении диабетической нейропатии можно выделить два направления – патогенетический и симптоматический способы лечения. Целью патогенетической терапии является замедление, стабилизация или обратное развитие нейропатии [22]. Симптоматическое лечение дистальной симметричной полинейропатии направлено в основном на ослабление боли. Как симптоматические, так и патогенетические методы лечения должны сопровождаться общими мерами. Например, с целью предупреждения образования язв на стопах пациенты должны быть информированы о значении сниженной чувствительности стоп, правилах ухода за ними и выбора удобной обуви [23]. Каждому больному, страдающему диабетом, следует проходить обследование стоп не реже одного раза в год; больные со сниженной чувствительностью стоп нуждаются в регулярном наблюдении ортопеда [24].
Поскольку гипергликемия признана фактором повреждения нервной ткани при СД, оптимальный контроль гликемии является основным компонентом этиотропной терапии. В исследовании Diabetes Control and Complications Trial (DCCT, 1993) замедление прогрессирования диабетической нейропатии было достигнуто через 6,5 лет интенсивной инсулинотерапии. Этот благоприятный эффект сохранялся на протяжении не менее 8 лет наблюдения после завершения исследования Epidemiology of Diabetes Interventions and Complications (EDIC, 2002). Таким образом, в исследовании DCCT/EDIC было подтверждено положительное влияние строгого контроля уровня глюкозы на микро- и макрососудистые осложнения диабета. Целью лечения должно быть достижение уровня гликированного гемоглобина менее 7% [25, 26].
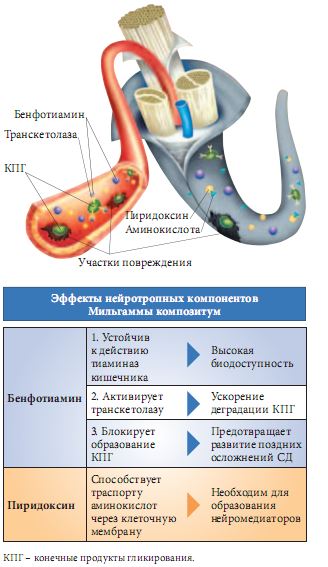
Как видно из рисунка 1, бенфотиамин относится к препаратам патогенетической терапии [27]. Тиаминдифосфат (ТДФ) – активная форма тиамина – является коферментом транскетолазы. Транскетолаза функционирует в пентозофосфатном пути, участвуя в переносе гликоальдегидного радикала между кето- и альдосахарами. В 1929 г. Х. Эйкман (C. Eijkman) был удостоен Нобелевской премии за открытие витамина В1. Водорастворимая форма витамина В1 достаточно трудно проникает через клеточные мембраны нейронов и разрушается тиаминазой кишечника, что делает эту форму фармакологически мало активной. В 1952 г. японской исследовательской группе Фудзивары удалось синтезировать новую жирорастворимую форму витамина В1 и тем самым сделать сенсационное открытие. До этого времени применение витамина В1 было эффективно только при его дефиците. Бенфотиамин обладает преимуществами по сравнению с водорастворимым тиамином: более устойчив к разрушающему действию тиаминазы, обладает липофильностью, легко проникает через гематоневральный барьер, что позволяет повышать внутриклеточную концентрацию витамина В1 и оказывать фармакологическое влияние на тиаминзависимые процессы.
Терапевтический аспект в истории развития бенфотиамина играет все более заметную роль. Тиамин в форме кофермента необходим для нормального процесса утилизации углеводов и обеспечения энергией клеток организма. Наиболее активно этот процесс идет в нервной ткани и печени. Так как нервные клетки получают энергию преимущественно в результате окисления углеводов, они особенно восприимчивы к соответствующим нарушениям. По этой причине тиамин можно назвать «витамином защиты нервов», или нейротропным фактором. В условиях нарушения углеводного обмена (у больных СД) особенно значимо влияние тиамина (входит в состав лекарственных форм Мильгамма и Мильгамма композитум).
Данные клинических плацебоконтролируемых двойных слепых исследований подтверждают эффективность влияния бенфотиамина и других витаминов группы В (В6 и В12) на развитие нейропатий. Так, 36 пациентам с диабетической нейропатией назначали бенфотиамин в дозе 320 мг, пиридоксин и цианокобаламин [28]. Результаты терапии оценивались по шкале неврологических нарушений и вибрационной чувствительности. У пациентов уже через три недели лечения отмечалось достоверное улучшение по этим показателям по сравнению с группой контроля. В плацебоконтролируемом исследовании BEDIP (BEnfotiamine in the treatment of DIabetic Polineuropathy) исследовали 40 пациентов с СД 1 и 2 типа и полинейропатией [29]. В течение 3 недель проводилось лечение бенфотиамином 400 мг в день или плацебо. Показатели нейропатии оценивались по шкале неврологических нарушений и вибрационной чувствительности. Было отмечено достоверное снижение количества баллов по шкале нейропатических нарушений. Таким образом, терапевтическая эффективность бенфотиамина при диабетической нейропатии может быть обоснована не только патогенетически, но и клинически.
Пиридоксин (витамин В6) входит в состав лекарственных форм Мильгамма и Мильгамма композитум, является коферментом множества ферментных комплексов, участвующих в аминокислотном обмене. Пиридоксин участвует в синтезе нейромедиаторов, таких как норадреналин и серотонин. Возможно, этим определяется антиноцицептивное действие пиридоксина. Активируя синтез транспортных белков в осевых цилиндрах нервных волокон, витамин В6 способствует ускорению регенерации периферических нервов, тем самым проявляя нейротропный эффект. От витамина В6 зависит внутриклеточный пул магния, играющего важную роль в метаболизме нервной клетки. Одновременное введение В1 и В6 способствует регенеративным процессам в нерве [30, 31]. Есть данные о способности В6 снижать уровень гликированного гемоглобина HbA1c в крови [32].
Инъекционная форма препарата Мильгамма содержит витамин В12 (цианокобаламин). Свойства этого витамина не до конца изучены, но хорошо известно влияние активного метаболита В12 – метилкобаламина – на регенераторные процессы в миелиновой оболочке, способность восстанавливать миелин при повреждении нерва. Предполагают, что метилкобаламин действует непосредственно на синтез метионина в метаболизме ДНК и именно высокие концентрации регулируют транскрипцию генов, которые могут увеличить синтез белка для регенерации нерва. Эти эффекты не связаны с дефицитом витамина B12. Исследование, проведенное G.L. Mauro [33], показало положительное влияние применения больших доз витамина В12 при радикулопатиях по сравнению с мануальной терапией и терапией НПВС. Понимание механизмов развития осложнений у больных СД позволяет проводить поиск новых терапевтических воздействий, связанных с предупреждением развития осложнений. Такие крупномасштабные многоцентровые исследования, как DCCT (1993) и последующее наблюдательное исследование EDIC (2002) [25, 34], показали не только зависимость развития диабетических осложнений от гипергликемии, но также позволили увидеть новую проблему, которая позже получила название гипергликемической памяти [35].
В наблюдательном исследовании EDIC группа пациентов с плохой компенсацией сахарного диабета была переведена на интенсифицированную инсулинотерапию, в результате были достигнуты удовлетворительные показатели углеводного обмена по гликемии и НbА1с. В течение 6–10 лет проводилось наблюдение за развитием осложнений (ретинопатия, нейропатия и др.). Результатами наблюдения исследователи были расстроены, так как изменения на глазном дне и другие осложнения сахарного диабета не только не подверглись регрессии, но даже не стабилизировались. Таким образом, ткани «запомнили» предшествовавший контролю заболевания период гипергликемии, запустив патологические метаболические процессы. Даже продолжительный контроль уровня глюкозы в крови не влияет на скорость развития осложнений, начавших развиваться во время гипергликемии. Этот феномен также наблюдается у больных СД 2 типа. Причем именно в случае диабета 2 типа концепция гипергликемической памяти приобретает особое значение, поскольку в большинстве случаев заболевание диагностируют лишь через 8–10 лет после появления его первых признаков, сопровождающихся гипергликемией различной степени тяжести.
В основе явления гипергликемии лежат молекулярные механизмы, показанные М. Браунли (M. Brownlee). Выделяются четыре основных независимых механизма повреждения тканей, вызванного гипергликемией [14, 25], – полиоловый путь, гексозаминовый путь, путь протеинкиназы С и путь образования КПГ. Существование феномена гипергликемической памяти убедительно свидетельствует о срочной необходимости раннего лечения для восстановления контроля уровня глюкозы и необходимости введения препаратов, снижающих количество внутриклеточных активных форм кислорода и уровень гликирования, для уменьшения скорости развития или устранения последствий отдаленных осложнений диабета [36].
NB
Мильгамма композитум («Вёрваг Фарма», Германия) – нейротропный комплекс с высокой биодоступностью
Нейротропная комбинация 100 мг бенфотиамина и 100 мг пиридоксина в составе драже Мильгаммы композитум позволяет всесторонне улучшить метаболизм периферических нервных волокон. Компоненты не взаимодействуют между собой благодаря послойному методу нанесения. При диабетической нейропатии рекомендуется принимать Мильгамму композитум по 1 драже 3 раза в день 4–6 недель с повторными курсами 2–3 раза в год.
Уважаемый посетитель uMEDp!
Уведомляем Вас о том, что здесь содержится информация, предназначенная исключительно для специалистов здравоохранения.
Если Вы не являетесь специалистом здравоохранения, администрация не несет ответственности за возможные отрицательные последствия, возникшие в результате самостоятельного использования Вами информации с портала без предварительной консультации с врачом.
Нажимая на кнопку «Войти», Вы подтверждаете, что являетесь врачом или студентом медицинского вуза.