Медицинский портал для врачей
количество статей
7290
Загрузка...
Пожалуйста, авторизуйтесь:
Клинические случаи
Болезнь Ниманна – Пика тип С: молекулярные механизмы патогенеза и подходы к лечению
"ЭФФЕКТИВНАЯ ФАРМАКОТЕРАПИЯ. Педиатрия" спецвыпуск
- Аннотация
- Статья
- Ссылки
Болезнь Ниманна – Пика тип С – наследственное прогрессирующее заболевание нервной системы из группы лизосомных болезней накопления, возникающее в результате нарушения внутриклеточного распределения липидов. Клиническая картина болезни многообразна и проявляется в виде прогрессирующих неврологических расстройств, нередко в сочетании с поражением внутренних органов. В статье обсуждаются новые взгляды на патогенез заболевания и современные подходы к терапии. Авторами приведены клинические примеры и первый опыт лечения данного заболевания в России.
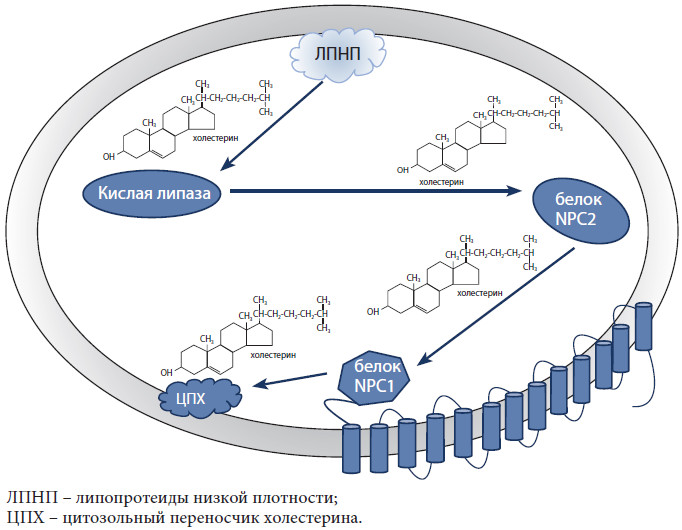
Рис. Модель переноса холестерина в поздней эндосоме/лизосоме
Этиология и клинические проявления
НПС – редкое наследственное заболевание. По разным данным, частота НПС составляет 1 : 120 000 – 1 : 150 000 живых новорожденных. В 95% случаев НПС связана c мутациями гена NPC1 (локус 18q11–q12). Около 5% случаев обусловлены мутациями гена NPC2 (локус 14q24) [2]. В зависимости от возраста появления первых клинических симптомов НПС условно подразделяют на несколько форм: неонатальная (до 3 месяцев), ранняя инфантильная (младше 2 лет), поздняя младенческая (с 2 до 6 лет), юношеская форма (с 6 до 15 лет), взрослая форма (старше 15 лет). Клинические симптомы включают различные прогрессирующие неврологические нарушения и гепатолиенальный синдром. Обычно начальными неврологическими симптомами являются неуклюжесть и прогрессирующие мозжечковые нарушения (атаксия, дисметрия), в последующем присоединяются вертикальный надъядерный офтальмопарез, пирамидные нарушения, эпилептические приступы, катаплексия, дистония и прогрессирующие когнитивные расстройства [3–6].
При поздней младенческой форме первыми клиническими симптомами являются прогрессирующие мозжечковые расстройства, когнитивные нарушения. Важный симптом болезни – развитие вертикального супрануклеарного офтальмопареза, который часто остается незамеченным на ранних стадиях болезни. Изначально возникает замедление движения глазных яблок по вертикали, постепенно прогрессирующее до полного ограничения вертикального, а иногда и горизонтального взора. Часто в данном возрастном периоде развиваются эпилептические приступы и эпизоды геластической катаплексии. Нередко выявляют дистонию, дисфагию и дизартрию [2, 7, 8]. Перечисленные симптомы наблюдаются при манифестации заболевания как в юношеском, так и во взрослом возрасте. Однако в большинстве случаев первыми и долгое время ведущими симптомами у таких пациентов являются психиатрические нарушения (расстройства поведения, шизофреноподобный синдром, маниакально-депрессивные психозы, зрительные галлюцинации, аутистические расстройства и другие), ухудшение школьной успеваемости, а также прогрессирующая деменция [9, 10].
НПС – редкое наследственное заболевание. По разным данным, частота НПС составляет 1 : 120 000 – 1 : 150 000 живых новорожденных. В 95% случаев НПС связана c мутациями гена NPC1 (локус 18q11–q12). Около 5% случаев обусловлены мутациями гена NPC2 (локус 14q24) [2]. В зависимости от возраста появления первых клинических симптомов НПС условно подразделяют на несколько форм: неонатальная (до 3 месяцев), ранняя инфантильная (младше 2 лет), поздняя младенческая (с 2 до 6 лет), юношеская форма (с 6 до 15 лет), взрослая форма (старше 15 лет). Клинические симптомы включают различные прогрессирующие неврологические нарушения и гепатолиенальный синдром. Обычно начальными неврологическими симптомами являются неуклюжесть и прогрессирующие мозжечковые нарушения (атаксия, дисметрия), в последующем присоединяются вертикальный надъядерный офтальмопарез, пирамидные нарушения, эпилептические приступы, катаплексия, дистония и прогрессирующие когнитивные расстройства [3–6].
При поздней младенческой форме первыми клиническими симптомами являются прогрессирующие мозжечковые расстройства, когнитивные нарушения. Важный симптом болезни – развитие вертикального супрануклеарного офтальмопареза, который часто остается незамеченным на ранних стадиях болезни. Изначально возникает замедление движения глазных яблок по вертикали, постепенно прогрессирующее до полного ограничения вертикального, а иногда и горизонтального взора. Часто в данном возрастном периоде развиваются эпилептические приступы и эпизоды геластической катаплексии. Нередко выявляют дистонию, дисфагию и дизартрию [2, 7, 8]. Перечисленные симптомы наблюдаются при манифестации заболевания как в юношеском, так и во взрослом возрасте. Однако в большинстве случаев первыми и долгое время ведущими симптомами у таких пациентов являются психиатрические нарушения (расстройства поведения, шизофреноподобный синдром, маниакально-депрессивные психозы, зрительные галлюцинации, аутистические расстройства и другие), ухудшение школьной успеваемости, а также прогрессирующая деменция [9, 10].
Лабораторная диагностика
Основным методом лабораторной диагностики НПС является нагрузочный тест с филипином: при окрашивании культуры клеток фибробластов наблюдаются интенсивные флюоресцирующие области, сконцентрированные вокруг ядра клетки, которые соответствуют накоплению неэтерифицированного холестерина. Это исследование проводят только в небольшом числе зарубежных лабораторий. Дополнительными биохимическими маркерами могут быть: снижение активности сфингомиелиназы в лейкоцитах крови (20–30% от нормы), повышение активности маркерного фермента лизосом – хитотриозидазы в плазме крови. ДНК-диагностика является наиболее предпочтительным методом верификации диагноза. Описано несколько относительно распространенных мутаций в гене NPC1, которые локализованы в 20–22 экзонах гена NPC1, но в большинстве случаев требуется полное секвенирование генов NPC1 и NPC2 для выявления мутаций [11]. Информативность такого тестирования составляет около 94%.
Перспективным методом биохимической диагностики является определение производных холестерина. В ряде работ было показано, что в клетках и плазме крови пациентов с НПС происходит накопление холестан-3,5,6-триола и 7-кетостерола [12]. При этом у пациентов с другими нейродегенеративными заболеваниями и ЛБН уровень этих соединений в крови не повышен [13]. Данный показатель коррелирует с возрастом начала заболевания и тяжестью клинических проявлений. Также на животных моделях НПС было показано, что концентрация этих соединений снижается на фоне лечения. Концентрацию холестан-3,5,6-триола и 7-кетостерола можно определить с помощью метода газовой хроматографии масс-спектрометрии (ГХ-МС) или высокоэффективной жидкостной хроматографии тандемной масс-спектрометрии (ВЭЖХ-МС/МС). Последний метод является более предпочтительным, поскольку позволяет определять эти соединения в небольшом количестве биологического материала, длительная пробоподготовка, как при ГХ-МС, не требуется. Возможно, что уже в ближайшее время данный методический подход будет применяться на первом этапе диагностики этого редкого заболевания.
Основным методом лабораторной диагностики НПС является нагрузочный тест с филипином: при окрашивании культуры клеток фибробластов наблюдаются интенсивные флюоресцирующие области, сконцентрированные вокруг ядра клетки, которые соответствуют накоплению неэтерифицированного холестерина. Это исследование проводят только в небольшом числе зарубежных лабораторий. Дополнительными биохимическими маркерами могут быть: снижение активности сфингомиелиназы в лейкоцитах крови (20–30% от нормы), повышение активности маркерного фермента лизосом – хитотриозидазы в плазме крови. ДНК-диагностика является наиболее предпочтительным методом верификации диагноза. Описано несколько относительно распространенных мутаций в гене NPC1, которые локализованы в 20–22 экзонах гена NPC1, но в большинстве случаев требуется полное секвенирование генов NPC1 и NPC2 для выявления мутаций [11]. Информативность такого тестирования составляет около 94%.
Перспективным методом биохимической диагностики является определение производных холестерина. В ряде работ было показано, что в клетках и плазме крови пациентов с НПС происходит накопление холестан-3,5,6-триола и 7-кетостерола [12]. При этом у пациентов с другими нейродегенеративными заболеваниями и ЛБН уровень этих соединений в крови не повышен [13]. Данный показатель коррелирует с возрастом начала заболевания и тяжестью клинических проявлений. Также на животных моделях НПС было показано, что концентрация этих соединений снижается на фоне лечения. Концентрацию холестан-3,5,6-триола и 7-кетостерола можно определить с помощью метода газовой хроматографии масс-спектрометрии (ГХ-МС) или высокоэффективной жидкостной хроматографии тандемной масс-спектрометрии (ВЭЖХ-МС/МС). Последний метод является более предпочтительным, поскольку позволяет определять эти соединения в небольшом количестве биологического материала, длительная пробоподготовка, как при ГХ-МС, не требуется. Возможно, что уже в ближайшее время данный методический подход будет применяться на первом этапе диагностики этого редкого заболевания.
Патогенез болезни
Молекулярные механизмы развития НПС пока еще полностью не расшифрованы. В отличие от других ЛБН, при НПС накапливаются метаболиты липидов различных классов – холестерин, сфингомиелин, гликосфинголипиды, сфингозин. Другой отличительной чертой НПС является нарушение эндоцитоза и внутрилизосомного метаболизма Ca2+ [14, 15]. За последние 20 лет было предложено несколько теорий, объясняющих патогенез этого заболевания, в каждой из которых ключевая роль отведена определенному метаболиту неэстерифицированному холестерину, сфингомиелину, сфингозину.
Молекулярные механизмы развития НПС пока еще полностью не расшифрованы. В отличие от других ЛБН, при НПС накапливаются метаболиты липидов различных классов – холестерин, сфингомиелин, гликосфинголипиды, сфингозин. Другой отличительной чертой НПС является нарушение эндоцитоза и внутрилизосомного метаболизма Ca2+ [14, 15]. За последние 20 лет было предложено несколько теорий, объясняющих патогенез этого заболевания, в каждой из которых ключевая роль отведена определенному метаболиту неэстерифицированному холестерину, сфингомиелину, сфингозину.
Холестерин Холестерин является одним из важнейших компонентов клеточной мембраны млекопитающих. Его уникальные физико-химические свойства позволяют стабилизировать мембрану клетки, придавая ей определенную «жесткость». Холестерин также является предшественником стероидных гормонов, оксистеролов, витамина D и участвует в регулировании многочисленных процессов в клетке. Около 80% холестерина синтезируется в клетках, 20% поступает с пищей. Холестерин в крови образует комплексы с особыми белками-транспортерами – аполипопротеинами. Эти комплексы называют липопротеидами и в зависимости от размеров и физико-химических свойств их разделяют на несколько групп. В центральной нервной системе единственным источником холестерина является его синтез, поскольку липопротеиды низкой плотности не могут проникать через гематоэнцефалический барьер. Свободный холестерин синтезируется в клетках глии и, соединяясь с аполипопротеном Е, проникает в нейроны [16]. Гены NPC1 и NPC2 кодируют белки, отвечающие за транспорт холестерина и липидов внутри клетки. NPC1 относится к семейству генов, кодирующих мембранно-связанные стеролчувствительные белки. Белок NPC2 – внутрилизосомный переносчик/транспортер холестерина. В периферических тканях после освобождения липопротеида в поздних эндосомах/лизосомах эфиры холестерина гидролизуются при участии лизосомной кислой липазы. Затем свободный холестерин переносится на белок NPC2 и далее на белок NPC1. После этого свободный холестерин отщепляется от белка NPC1, переносится на мембрану поздней эндосомы/лизосомы и может спонтанно проникать в цитозоль клетки, после чего транспортируется в различные части клетки специальными цитоплазматическими переносчиками (рис.).
Гликосфинголипиды В клетках нервной системы при НПС основным накапливаемым материалом является не только холестерин, но и разнообразные гликосфинголипиды: глюкозилцерамид, сфингозин, лактоцерамид и ганглиозиды GM2 и GM3 [17]. Предполагается, что к их накоплению приводят нарушения везикулярного транспорта или вторичные нарушения процессов расщепления гликосфинголипидов. Так, в ряде работ было показано, что накопление сфингомиелина связано со снижением активности сфингомиелиназы в результате нарушения ее посттрансляционных модификаций, которые вызваны накоплением холестерина.
Подходы к лечению
Первоначально казалось, что подход к лечению НПС очень прост: снизить уровень холестерина и таким образом остановить прогрессирование заболевания. Эта уверенность подкреплялась тем, что существует большое разнообразие высокоэффективных лекарственных препаратов, снижающих холестерин. В 1993 г. было показано, что применение подобных препаратов приводит к снижению уровня холестерина в печени и сыворотке крови пациентов с НПС, но, к сожалению, не оказывает никакого влияния на неврологическую симптоматику
Клинические наблюдения
[20]. В дальнейших исследованиях были предприняты попытки снизить уровень других токсичных метаболитов, таких как сфингомиелин, сфингозин, с помощью субстрат-редуцирующей терапии, и этот подход оказался более эффективным.
Субстрат-редуцирующая терапия Миглустат (N-бутил-деоксино-жиримицин; NB-DNJ; OGT-918) – небольшая молекула аминосахарида, которая обратимо ингибирует синтез глюкоцерамидсинтазы – первого фермента, участвующего в синтезе гликосфинголипидов [18]. На животных моделях c НПС было убедительно продемонстрировано, что на фоне лечения снижается накопление ганглиозидов, останавливается нейрональная дисфункция и значительно (25%) увеличивается продолжительность жизни у леченных животных [19]. Способность миглустата проникать через гематоэнцефалический барьер обусловливает возможность применения препарата при ЛБН с преимущественным вовлечением в патологический процесс нервной системы. На основании результатов проведенных рандомизированных плацебоконтролируемых клинических испытаний и динамического наблюдения пациентов с НПС в январе 2009 г. миглустат был одобрен как препарат для лечения прогрессирующих неврологических нарушений при НПС как у взрослых, так и у детей. Применение данного препарата является единственным видом терапии НПС, которая позволяет замедлить прогрессирование болезни и не имеет выраженных побочных эффектов [20, 21]. Препарат назначают перорально. Он разрешен к применению с 4-летнего возраста. Рекомендуемая доза для взрослых и детей старше 12 лет составляет 200 мг 3 раза в день. Детям от 4 до 11 лет доза рассчитывается исходя из площади поверхности тела. Поскольку при приеме препарата часто наблюдается диарея, связанная с блокированием фермента, расщепляющего ди- и олигосахариды, рекомендовано соблюдение диеты с пониженным содержанием этих углеводов в первые три недели терапии с постепенным включением их в рацион в дальнейшем.
Первоначально казалось, что подход к лечению НПС очень прост: снизить уровень холестерина и таким образом остановить прогрессирование заболевания. Эта уверенность подкреплялась тем, что существует большое разнообразие высокоэффективных лекарственных препаратов, снижающих холестерин. В 1993 г. было показано, что применение подобных препаратов приводит к снижению уровня холестерина в печени и сыворотке крови пациентов с НПС, но, к сожалению, не оказывает никакого влияния на неврологическую симптоматику
Клинические наблюдения
[20]. В дальнейших исследованиях были предприняты попытки снизить уровень других токсичных метаболитов, таких как сфингомиелин, сфингозин, с помощью субстрат-редуцирующей терапии, и этот подход оказался более эффективным.
Субстрат-редуцирующая терапия Миглустат (N-бутил-деоксино-жиримицин; NB-DNJ; OGT-918) – небольшая молекула аминосахарида, которая обратимо ингибирует синтез глюкоцерамидсинтазы – первого фермента, участвующего в синтезе гликосфинголипидов [18]. На животных моделях c НПС было убедительно продемонстрировано, что на фоне лечения снижается накопление ганглиозидов, останавливается нейрональная дисфункция и значительно (25%) увеличивается продолжительность жизни у леченных животных [19]. Способность миглустата проникать через гематоэнцефалический барьер обусловливает возможность применения препарата при ЛБН с преимущественным вовлечением в патологический процесс нервной системы. На основании результатов проведенных рандомизированных плацебоконтролируемых клинических испытаний и динамического наблюдения пациентов с НПС в январе 2009 г. миглустат был одобрен как препарат для лечения прогрессирующих неврологических нарушений при НПС как у взрослых, так и у детей. Применение данного препарата является единственным видом терапии НПС, которая позволяет замедлить прогрессирование болезни и не имеет выраженных побочных эффектов [20, 21]. Препарат назначают перорально. Он разрешен к применению с 4-летнего возраста. Рекомендуемая доза для взрослых и детей старше 12 лет составляет 200 мг 3 раза в день. Детям от 4 до 11 лет доза рассчитывается исходя из площади поверхности тела. Поскольку при приеме препарата часто наблюдается диарея, связанная с блокированием фермента, расщепляющего ди- и олигосахариды, рекомендовано соблюдение диеты с пониженным содержанием этих углеводов в первые три недели терапии с постепенным включением их в рацион в дальнейшем.
Циклодекстрины Циклодекстрины – углеводы, в составе которых остатки D-(+)-глюкопиранозы объединены в макроциклы α-D-1,4-гликозидными связями. Несмотря на то что циклодекстрины не способны свободно проникать через мембрану клетки, они могут переноситься внутрь путем пиноцитоза. Гидроксипропил-β-циклодекстрин (HPBCD) состоит из семи β-1,4- глюкопиранозных единиц. В ряде работ было показано, что введение этого соединения внутрибрюшинно или интратекально «нокаутным» мышам (NPC -|-) приводит к замедлению прогрессирования неврологических нарушений и увеличению продолжительности жизни у животных [22]. Считается, что циклодекстрины могут выполнять функцию измененных в результате мутаций белков NPC1 и NPC2 в эндосомах и лизосомах. Возможно, что создание лекарственных форм на основе циклодекстринов станет одним из методов терапии НПС.
Химические шапероны Ряд мутаций в генах приводит к нарушениям фолдинга (сворачивания) белка и вызывает либо его накопление, либо быстрое расщепление. Как правило, это миссенс-мутации и небольшие делеции без сдвига рамки считывания, которые не затрагивают функционально значимые домены белка (такие как активные центры, рецептор-связывающие сайты). Такие белки сохраняют свою функциональную активность, если достигают места своего назначения. Известно, что некоторые соединения могут служить стабилизаторами белков, помогают им образовывать более устойчивую конформацию. Эти вещества получили название химических шаперонов по аналогии с белками- шаперонами, которые принимают участие в поддержании третичной структуры и доставке синтезированных в клетке белков. Мутация Ile1061Thr в гене NPC1 относится к числу мутаций, приводящих к нарушению фолдинга белка [23]. Эти данные позволяют надеяться, что высокоспецифичные фармакологические шапероны могут быть синтезированы и для лечения этого заболевания наряду с другими ЛБН.
Мы приводим собственные клинические данные и первый опыт лечения НПС в России.
Клинический пример 1 Пациентка Г.П., 2002 г.р., впервые поступила в стационар в 2004 г. с жалобами на утрату ранее приобретенных психоречевых и двигательных навыков, непроизвольные насильственные движения в мимической мускулатуре и языке, ограничение движения глазных яблок во всех направлениях, неустойчивость при ходьбе, эпизоды внезапных падений.
Анамнез жизни: ребенок от беременности, протекавшей на фоне умеренного токсикоза, вторых срочных родов, масса при рождении 2990 г, рост 50 см. Раннее развитие: голову держит с 1 месяца, сидит самостоятельно с 6 месяцев, стоит с 10 месяцев, ходит с 1 года.
Анамнез болезни: со слов мамы, первые симптомы заболевания появились в возрасте 1 года 2 месяцев через 2 недели после вакцинации АКДС и полиомиелита – сначала возникло «подворачивание» правой стопы при ходьбе, затем присоединилось «подворачивание» левой стопы, и ребенок стал часто спотыкаться, падать. С 1,5 лет появились поперхивания при еде, с 1,6 года – перестала самостоятельно ходить, с 1,8 года – насильственные движения в мышцах языка, лицевой мускулатуры. Отмечалась выраженная атаксия, интенционный тремор (не могла собрать пирамидку, не могла есть из ложки жидкую пищу – проливала). При КТ и МРТ головного мозга в возрасте 1,8 года патологических изменений головного мозга, костей свода и основания черепа не выявлено. С 2,2 года ребенок неоднократно проходил обследование и лечение в отделении психоневрологии № 2 Российской детской клинической больницы (РДКБ). Впервые поступила в стационар с направляющим диагнозом «острый диссеминированный энцефаломиелополиневрит». При осмотре в неврологическом статусе отмечалась легкая асимметрия глазных щелей, OD > OS, опущение угла рта справа, расходящееся косоглазие, периодически поперхивание при еде, оживление глоточных рефлексов, положительные симптомы орального автоматизма, походка атактическая, статическая и динамическая атаксия; диффузная мышечная гипотония, анизорефлексия сухожильных рефлексов с рук, рефлексы с ног – торпидные, периодически отмечались гиперкинезы в мышцах лица (в виде гримасничанья). В связи с подозрением на острый диссеминированный энцефаломиелополиневрит пациентке была проведена терапия преднизолоном в дозе 2 мг/кг/сут в течение 1 месяца с незначительной положительной динамикой в виде уменьшения атаксии в начале курса терапии. Со слов мамы, после выписки уверенно ходила, уменьшились гиперкинезы. При снижении дозы преднизолона до 1 мг/кг/сут появились гиперкинезы в мимической мускулатуре, несколько хуже стала ходить. В последующем курсами получала нейротрофическую терапию без явного положительного эффекта. К 2 годам 9 месяцам появилось умеренное ограничение движений глазных яблок, в последующем стала ухудшаться походка, появились и усилились гиперкинезы конечностей и лицевой мускулатуры, ухудшилась речь.
В возрасте 9 лет в неврологическом статусе: общемозговых и менингеальных симптомов нет. Голова округлой формы. Умеренно выражена венозная сеть. Окружность головы 52 см, OD > OS, расходящееся косоглазие, непостоянное альтернирующее. Офтальмопарез – ограничение движения глазных яблок во всех направлениях. Ослаблена конвергенция. Периодически – миоклонии век. При фиксации взора – спонтанный нистагм с ротаторным компонентом. Слух ориентировочно не снижен. Язык беспокойный, миоклонии языка, гиперкинезы языка, периодически – девиация языка вправо. Периодически отмечаются гиперкинезы в мышцах лица (в виде гримасничанья), хореические гиперкинезы. Голос с дисфоничным оттенком. Иногда поперхивается при глотании. Глоточные рефлексы вызываются после латентной паузы. Может самостоятельно пройти несколько шагов, походка атактико-полиневропатическая. Рекурвация коленных суставов. Статическая и динамическая атаксия. Мышечный тонус: диффузная мышечная гипотония. Сухожильные рефлексы на руках снижены, D > S, на ногах – не вызываются. Умеренная гипотрофия дистальных отделов конечностей. Сила мышц снижена до 4,5 балла. Координаторные пробы – с грубой интенцией и дисметрией с двух сторон. Чувствительность (болевая, тактильная, температурная) ориентировочно сохранена. Гипергидроз ладоней, стоп. Функции тазовых органов контролирует.
Речь активная отсутствует; задержка психоречевого развития. Девочка выполняет инструкции и команды по подражанию, обслуживает себя, память и интеллект снижены, контактна, окружающим интересуется, одевается и кушает самостоятельно. Таким образом, при сопоставлении клинических, нейрорадиологических данных и течения заболевания было заподозрено заболевание из группы ЛБН – НПС, которое было подтверждено молекулярно-генетическими методами (выявлена замена IVS4+2G-A в гене NPC2 в гетерозиготном состоянии). Решением консилиума ребенку было назначено патогенетическое лечение препаратом Завеска (миглустат) (Actelion Pharmaceuticals Ltd, Швейцария) в дозе 200 мг 2 раза в день (утро, вечер) длительно, постоянно и предложена диета с пониженным содержанием ди- и олигосахаридов. Через 5 месяцев после назначения специфического лечения отмечается некоторая положительная динамика в виде улучшения походки, уменьшения выраженности гиперкинетических расстройств. Девочка стала спокойнее, больше интересуется окружающим, стала произносить три слова («мама», «папа», «баба»), приступов катаплексии не было. По данным литературы, было показано, что чем раньше манифестирует заболевание, тем хуже восстанавливаются когнитивные функции, однако в нашем случае наблюдалось их улучшение [24]. В нашем случае не назначались трициклические антидепрессанты, ингибиторы селективного захвата серотонина для купирования катаплексии [25, 29], однако через 2 месяца после назначения препарата Завеска у пациентки наблюдалось значительное уменьшение частоты эпизодов катаплексии и через 4 месяца их полное отсутствие, что совпадает с данными литературы [4].
Анамнез жизни: ребенок от беременности, протекавшей на фоне умеренного токсикоза, вторых срочных родов, масса при рождении 2990 г, рост 50 см. Раннее развитие: голову держит с 1 месяца, сидит самостоятельно с 6 месяцев, стоит с 10 месяцев, ходит с 1 года.
Анамнез болезни: со слов мамы, первые симптомы заболевания появились в возрасте 1 года 2 месяцев через 2 недели после вакцинации АКДС и полиомиелита – сначала возникло «подворачивание» правой стопы при ходьбе, затем присоединилось «подворачивание» левой стопы, и ребенок стал часто спотыкаться, падать. С 1,5 лет появились поперхивания при еде, с 1,6 года – перестала самостоятельно ходить, с 1,8 года – насильственные движения в мышцах языка, лицевой мускулатуры. Отмечалась выраженная атаксия, интенционный тремор (не могла собрать пирамидку, не могла есть из ложки жидкую пищу – проливала). При КТ и МРТ головного мозга в возрасте 1,8 года патологических изменений головного мозга, костей свода и основания черепа не выявлено. С 2,2 года ребенок неоднократно проходил обследование и лечение в отделении психоневрологии № 2 Российской детской клинической больницы (РДКБ). Впервые поступила в стационар с направляющим диагнозом «острый диссеминированный энцефаломиелополиневрит». При осмотре в неврологическом статусе отмечалась легкая асимметрия глазных щелей, OD > OS, опущение угла рта справа, расходящееся косоглазие, периодически поперхивание при еде, оживление глоточных рефлексов, положительные симптомы орального автоматизма, походка атактическая, статическая и динамическая атаксия; диффузная мышечная гипотония, анизорефлексия сухожильных рефлексов с рук, рефлексы с ног – торпидные, периодически отмечались гиперкинезы в мышцах лица (в виде гримасничанья). В связи с подозрением на острый диссеминированный энцефаломиелополиневрит пациентке была проведена терапия преднизолоном в дозе 2 мг/кг/сут в течение 1 месяца с незначительной положительной динамикой в виде уменьшения атаксии в начале курса терапии. Со слов мамы, после выписки уверенно ходила, уменьшились гиперкинезы. При снижении дозы преднизолона до 1 мг/кг/сут появились гиперкинезы в мимической мускулатуре, несколько хуже стала ходить. В последующем курсами получала нейротрофическую терапию без явного положительного эффекта. К 2 годам 9 месяцам появилось умеренное ограничение движений глазных яблок, в последующем стала ухудшаться походка, появились и усилились гиперкинезы конечностей и лицевой мускулатуры, ухудшилась речь.
В возрасте 9 лет в неврологическом статусе: общемозговых и менингеальных симптомов нет. Голова округлой формы. Умеренно выражена венозная сеть. Окружность головы 52 см, OD > OS, расходящееся косоглазие, непостоянное альтернирующее. Офтальмопарез – ограничение движения глазных яблок во всех направлениях. Ослаблена конвергенция. Периодически – миоклонии век. При фиксации взора – спонтанный нистагм с ротаторным компонентом. Слух ориентировочно не снижен. Язык беспокойный, миоклонии языка, гиперкинезы языка, периодически – девиация языка вправо. Периодически отмечаются гиперкинезы в мышцах лица (в виде гримасничанья), хореические гиперкинезы. Голос с дисфоничным оттенком. Иногда поперхивается при глотании. Глоточные рефлексы вызываются после латентной паузы. Может самостоятельно пройти несколько шагов, походка атактико-полиневропатическая. Рекурвация коленных суставов. Статическая и динамическая атаксия. Мышечный тонус: диффузная мышечная гипотония. Сухожильные рефлексы на руках снижены, D > S, на ногах – не вызываются. Умеренная гипотрофия дистальных отделов конечностей. Сила мышц снижена до 4,5 балла. Координаторные пробы – с грубой интенцией и дисметрией с двух сторон. Чувствительность (болевая, тактильная, температурная) ориентировочно сохранена. Гипергидроз ладоней, стоп. Функции тазовых органов контролирует.
Речь активная отсутствует; задержка психоречевого развития. Девочка выполняет инструкции и команды по подражанию, обслуживает себя, память и интеллект снижены, контактна, окружающим интересуется, одевается и кушает самостоятельно. Таким образом, при сопоставлении клинических, нейрорадиологических данных и течения заболевания было заподозрено заболевание из группы ЛБН – НПС, которое было подтверждено молекулярно-генетическими методами (выявлена замена IVS4+2G-A в гене NPC2 в гетерозиготном состоянии). Решением консилиума ребенку было назначено патогенетическое лечение препаратом Завеска (миглустат) (Actelion Pharmaceuticals Ltd, Швейцария) в дозе 200 мг 2 раза в день (утро, вечер) длительно, постоянно и предложена диета с пониженным содержанием ди- и олигосахаридов. Через 5 месяцев после назначения специфического лечения отмечается некоторая положительная динамика в виде улучшения походки, уменьшения выраженности гиперкинетических расстройств. Девочка стала спокойнее, больше интересуется окружающим, стала произносить три слова («мама», «папа», «баба»), приступов катаплексии не было. По данным литературы, было показано, что чем раньше манифестирует заболевание, тем хуже восстанавливаются когнитивные функции, однако в нашем случае наблюдалось их улучшение [24]. В нашем случае не назначались трициклические антидепрессанты, ингибиторы селективного захвата серотонина для купирования катаплексии [25, 29], однако через 2 месяца после назначения препарата Завеска у пациентки наблюдалось значительное уменьшение частоты эпизодов катаплексии и через 4 месяца их полное отсутствие, что совпадает с данными литературы [4].
Клинический пример 2 Пациент Б.Г., 9 лет, поступил в стационар с жалобами на шаткость походки, частые падения, прихрамывание при ходьбе, навязчивые движения в конечностях, нарушение речи, эпизоды головокружений, снижение памяти и утомляемость [2].
Анамнез жизни: ребенок от второй физиологически протекавшей беременности (от первой беременности –
здоровый мальчик), вторых родов в срок путем кесарева сечения. Физическое развитие в раннем возрасте соответствовало средним
значениям.
Анамнез болезни: в возрасте 5 лет на фоне полного здоровья у мальчика появились жалобы на приступы головокружений, в связи с чем наблюдался у невролога по месту жительства с диагнозом «синдром вегетативной дистонии, ветибулопатия». В 7 лет стала заметна неустойчивость при ходьбе, появились жалобы на выраженную усталость к вечеру, а также трудности в усвоении школьного материала (снижение памяти, внимания) и заикание. В связи с прогрессирующими неврологическими расстройствами ребенок поступил на обследование и лечение в отделение психоневрологии № 1 РДКБ с направляющим диагнозом «дегенеративное заболевание нервной системы, подострый склерозирующий панэнцефалит».
При поступлении в стационар: общемозговых и менингеальных симптомов нет. Черепные нервы: обоняние сохранено, взгляд фиксирует и следит, глазные щели с легкой асимметрией, OD > OS, нарушения движения глазных яблок в виде ограничения по вертикали, лицо симметричное, слух не снижен, горизонтальный мелкоамплитудный нистагм в крайнем левом отведении, глоточные и небные рефлексы высокие, саливация достаточная, мозжечковая дизартрия. Дисфонии, дисфагии нет. Язык в полости рта и при высовывании по средней линии. Походка с элементами динамической атаксии. Мышечный тонус изменен по пластическому типу на фоне диффузной мышечной гипотонии. Сухожильные рефлексы высокие, без четкой разницы сторон. Сила мышц снижена до 4 баллов. Рефлекс Бабинского положительный с двух сторон, больше слева. Дистоническая установка кистей рук. Редкие хореические гиперкинезы лицевой мускулатуры. В позе Ромберга неустойчив. Дисметрия и интенционный тремор при выполнении координаторных проб. Тазовые функции контролирует. Высшие корковые функции: пациент в сознании, ориентирован, несколько негативен к осмотру, плаксив, инструкции выполняет, правильно отвечает на вопросы, интеллект снижен.
Дифференциальный диагноз проводился с тромбозом поперечного синуса, митохондриальными заболеваниями, атаксией с окуломоторной апраксией, синдромом Луи – Бар. Пациент получал ноотропную, нейрометаболическую терапию без явного положительного эффекта. С течением времени отмечалось нарастание мозжечковых расстройств, снижение интеллекта и прогрессирование вертикального офтальмопареза. С учетом клинических данных, результатов МРТ головного мозга, прогрессирующего течения заболевания у ребенка была заподозрена НПС. Диагноз был подтвержден молекулярно-генетическим методом: обнаружены мутации в гене NPC1, которые являются патогенными, – с3614del C / Ser954Leu. Решением консилиума ребенку была назначена терапия препаратом Завеска (миглустат) в дозе 200 мг 2 раза в день (утро, вечер) длительно, постоянно, а также была рекомендована диета с пониженным содержанием ди- и олигосахаридов. Через 8 месяцев после назначения патогенетического лечения отмечается положительная динамика: пациент стал устойчивым, менее выражены гиперкинетические расстройства, однако сохранялся интеллектуальный дефицит, отмечались дисфагия, дизартрия, прежняя степень вертикального офтальмопареза.
По данным литературы, отмечено значительное улучшение неврологического статуса у пациента 9 лет после назначения специфического лечения миглустатом в виде улучшения походки, атаксии, дизартрии и офтальмопареза [26]. У других пациентов (9 и 14 лет) после назначения специфического лечения через 6 месяцев наблюдалось улучшение функции глотания и движения [27]. Через 4 месяца у нашего пациента после назначения терапии наблюдалось снижение веса, других побочных эффектов не отмечалось. Была скорректирована диета, и ребенок начал прибавлять в весе. По данным литературы, в 80% случаев на фоне терапии появляются эпизоды диареи, отрицательная весовая кривая, особенно на первом году применения препарата, в 30% – тремор, реже – нарушения сна, парестезии и полиневропатия [20, 26, 27].
Анамнез жизни: ребенок от второй физиологически протекавшей беременности (от первой беременности –
здоровый мальчик), вторых родов в срок путем кесарева сечения. Физическое развитие в раннем возрасте соответствовало средним
значениям.
Анамнез болезни: в возрасте 5 лет на фоне полного здоровья у мальчика появились жалобы на приступы головокружений, в связи с чем наблюдался у невролога по месту жительства с диагнозом «синдром вегетативной дистонии, ветибулопатия». В 7 лет стала заметна неустойчивость при ходьбе, появились жалобы на выраженную усталость к вечеру, а также трудности в усвоении школьного материала (снижение памяти, внимания) и заикание. В связи с прогрессирующими неврологическими расстройствами ребенок поступил на обследование и лечение в отделение психоневрологии № 1 РДКБ с направляющим диагнозом «дегенеративное заболевание нервной системы, подострый склерозирующий панэнцефалит».
При поступлении в стационар: общемозговых и менингеальных симптомов нет. Черепные нервы: обоняние сохранено, взгляд фиксирует и следит, глазные щели с легкой асимметрией, OD > OS, нарушения движения глазных яблок в виде ограничения по вертикали, лицо симметричное, слух не снижен, горизонтальный мелкоамплитудный нистагм в крайнем левом отведении, глоточные и небные рефлексы высокие, саливация достаточная, мозжечковая дизартрия. Дисфонии, дисфагии нет. Язык в полости рта и при высовывании по средней линии. Походка с элементами динамической атаксии. Мышечный тонус изменен по пластическому типу на фоне диффузной мышечной гипотонии. Сухожильные рефлексы высокие, без четкой разницы сторон. Сила мышц снижена до 4 баллов. Рефлекс Бабинского положительный с двух сторон, больше слева. Дистоническая установка кистей рук. Редкие хореические гиперкинезы лицевой мускулатуры. В позе Ромберга неустойчив. Дисметрия и интенционный тремор при выполнении координаторных проб. Тазовые функции контролирует. Высшие корковые функции: пациент в сознании, ориентирован, несколько негативен к осмотру, плаксив, инструкции выполняет, правильно отвечает на вопросы, интеллект снижен.
Дифференциальный диагноз проводился с тромбозом поперечного синуса, митохондриальными заболеваниями, атаксией с окуломоторной апраксией, синдромом Луи – Бар. Пациент получал ноотропную, нейрометаболическую терапию без явного положительного эффекта. С течением времени отмечалось нарастание мозжечковых расстройств, снижение интеллекта и прогрессирование вертикального офтальмопареза. С учетом клинических данных, результатов МРТ головного мозга, прогрессирующего течения заболевания у ребенка была заподозрена НПС. Диагноз был подтвержден молекулярно-генетическим методом: обнаружены мутации в гене NPC1, которые являются патогенными, – с3614del C / Ser954Leu. Решением консилиума ребенку была назначена терапия препаратом Завеска (миглустат) в дозе 200 мг 2 раза в день (утро, вечер) длительно, постоянно, а также была рекомендована диета с пониженным содержанием ди- и олигосахаридов. Через 8 месяцев после назначения патогенетического лечения отмечается положительная динамика: пациент стал устойчивым, менее выражены гиперкинетические расстройства, однако сохранялся интеллектуальный дефицит, отмечались дисфагия, дизартрия, прежняя степень вертикального офтальмопареза.
По данным литературы, отмечено значительное улучшение неврологического статуса у пациента 9 лет после назначения специфического лечения миглустатом в виде улучшения походки, атаксии, дизартрии и офтальмопареза [26]. У других пациентов (9 и 14 лет) после назначения специфического лечения через 6 месяцев наблюдалось улучшение функции глотания и движения [27]. Через 4 месяца у нашего пациента после назначения терапии наблюдалось снижение веса, других побочных эффектов не отмечалось. Была скорректирована диета, и ребенок начал прибавлять в весе. По данным литературы, в 80% случаев на фоне терапии появляются эпизоды диареи, отрицательная весовая кривая, особенно на первом году применения препарата, в 30% – тремор, реже – нарушения сна, парестезии и полиневропатия [20, 26, 27].
Заключение
В настоящее время единственно возможным вариантом патогенетического лечения тяжелого прогрессирующего неврологического заболевания – НПС – является препарат Завеска (миглустат). Небольшое число пациентов с НПС, находящихся на лечении, и короткий период наблюдения не являются достаточным основанием для окончательного суждения об эффективности данного вида терапии. Однако с уверенностью можно сказать, что миглустат позволяет стабилизировать состояние пациентов и/или приводит к улучшению неврологических расстройств, а эффективность терапии зависит от времени установления точного диагноза. Необходимо продолжение исследований с целью изучения патогенеза этого тяжелого инвалидизирующего заболевания для создания новых высокоэффективных препаратов. Авторы благодарят Региональный общественный благотворительный фонд помощи тяжелобольным и обездоленным детям, организацию Aiutateci a Salvare i Bambini Onlus и лично Л.З. Салтыкову и Эннио Бордато за помощь, которая была оказана нашим пациентам при обследовании и лечении.
В настоящее время единственно возможным вариантом патогенетического лечения тяжелого прогрессирующего неврологического заболевания – НПС – является препарат Завеска (миглустат). Небольшое число пациентов с НПС, находящихся на лечении, и короткий период наблюдения не являются достаточным основанием для окончательного суждения об эффективности данного вида терапии. Однако с уверенностью можно сказать, что миглустат позволяет стабилизировать состояние пациентов и/или приводит к улучшению неврологических расстройств, а эффективность терапии зависит от времени установления точного диагноза. Необходимо продолжение исследований с целью изучения патогенеза этого тяжелого инвалидизирующего заболевания для создания новых высокоэффективных препаратов. Авторы благодарят Региональный общественный благотворительный фонд помощи тяжелобольным и обездоленным детям, организацию Aiutateci a Salvare i Bambini Onlus и лично Л.З. Салтыкову и Эннио Бордато за помощь, которая была оказана нашим пациентам при обследовании и лечении.
1. Carstea E.D., Morris J.A., Coleman K.G. et al. Niemann – Pick C1 disease gene: homology to mediators of cholesterol homeostasis // Science. 1997. Vol. 277. № 5323. P. 228–231.
2. Михайлова С.В., Захарова Е.Ю., Букина Т.М. Болезнь Ниманна – Пика тип С: клинические примеры // Педиатрическая фармакология. 2010. Т. 7. № 5. С. 48–53.
3. Михайлова С.В., Захарова Е.Ю., Петрухин А.С. Нейрометаболические заболевания у детей и подростков: диагностика и подходы к лечению. М.: Литтерра, 2011. 356 с.
4. Iturriaga C., Pineda M., Fernández-Valero E.M., Vanier M.T., Coll M.J. Niemann – Pick C disease in Spain: clinical spectrum and development of a disability scale // J. Neurol. Sci. 2006. Vol. 249. № 1. P. 1–6.
5. Wraith J.E., Guffon N., Rohrbach M., Hwu W.L., Korenke G.C., Bembi B., Luzy C., Giorgino R., Sedel F. Natural history of Niemann – Pick disease type C in a multicentre observational retrospective cohort study // Mol. Genet. Metab. 2009. Vol. 98. № 3. P. 250–254.
6. NP-C Guidelines Working Group, Wraith J.E., Baumgartner M.R., Bembi B., Covanis A., Levade T., Mengel E., Pineda M., Sedel F., Topçu M., Vanier M.T., Widner H., Wijburg F.A., Patterson M.C. Recommendations on the diagnosis and management of Niemann – Pick disease type C // Mol. Genet. Metab. 2009. Vol. 98. № 1–2. P. 152–165.
7. Oyama K., Takahashi T., Shoji Y., Oyamada M., Noguchi A., Tamura H., Takada G., Kanbayashi T. Niemann – Pick disease type C: cataplexy and hypocretin in cerebrospinal fluid // Tohoku J. Exp. Med. 2006. Vol. 209. № 3. P. 263–267.
8. Paciorkowski A.R., Westwell M., Ounpuu S., Bell K., Kagan J., Mazzarella C., Greenstein R.M. Motion analysis of a child with Niemann – Pick disease type C treated with miglustat // Mov. Disord. 2008. Vol. 23. № 1. P. 124–128.
9. Руденская Г.Е., Захарова Е.Ю., Букина Т.М., Волкова Э.Ю., Козлов А.С. Болезнь Ниманна – Пика тип С (ювенильный дистонический липидоз) // Журнал неврологии и психиатрии им. С.С. Корсакова. 2008. № 5. С. 76–79.
10. Руденская Г.Е., Букина Т.М., Захарова Е.Ю. Болезнь Ниманна – Пика тип С: взрослая форма с преобладанием психических расстройств // Журнал неврологии и психиатрии им. С.С. Корсакова. 2011. № 7. С. 71–75.
11. Park W.D., O'Brien J.F., Lundquist P.A., Kraft D.L., Vockley C.W., Karnes P.S., Patterson M.C., Snow K. Identification of 58 novel mutations in Niemann – Pick disease type C: correlation with biochemical phenotype and importance of PTC1-like domains in NPC1 // Hum. Mutat. 2003.Vol. 22. № 4. P. 313–325.
12. Porter F.D., Scherrer D.E., Lanier M.H., Langmade S.J., Molugu V., Gale S.E., Olzeski D., Sidhu R., Dietzen D.J., Fu R., Wassif C.A., Yanjanin N.M., Marso S.P., House J., Vite C., Schaffer J.E., Ory D.S. Cholesterol oxidation products are sensitive and specific blood-based biomarkers for Niemann – Pick C1 disease // Sci. Transl. Med. 2010. Vol. 3. № 2. P. 56–81.
13. Jiang X., Sidhu R., Porter F.D., Yanjanin N.M., Speak A.O., te Vruchte D.T., Platt F.M., Fujiwara H., Scherrer D.E., Zhang J., Dietzen D.J., Schaffer J.E., Ory D.S. A sensitive and specific LC-MS/MS method for rapid diagnosis of Niemann – Pick C1 disease from human plasma // J. Lipid. Res. 2011. Vol. 52. № 7. P. 1435–1445.
14. Ko D.C., Gordon M.D., Jin J.Y., Scott M.P. Dynamic movements of organelles containing Niemann – Pick C1 protein: NPC1 involvement in late endocytic events // Mol. Biol. Cell. 2001. Vol. 12. № 3. P. 601–614.
15. Lloyd-Evans E., Morgan A.J., He X., Smith D.A., Elliot-Smith E., Sillence D.J., Churchill G.C., Schuchman E.H., Galione A., Platt F.M. Niemann – Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium // Nat. Med. 2008. Vol. 14. № 11. P. 1247–1255.
16. Dietschy J.M., Turley S.D. Thematic review series: brain Lipids. Cholesterol metabolism in the central nervous system during early development and in the mature animal // J. Lipid. Res. 2004. Vol. 45. № 8. P. 1375–1397.
17. Patterson M.C., Di Bisceglie A.M., Higgins J.J., Abel R.B., Schiffmann R., Parker C.C., Argoff C.E., Grewal R.P., Yu K., Pentchev P.G. et al. The effect of cholesterol-lowering agents on hepatic and plasma cholesterol in Niemann – Pick disease type C // Neurology. 1993. Vol. 43. № 1. P. 61–64.
18. Norris-Cervetto E., Callaghan R., Platt F.M., Dwek R.A., Butters T.D. Inhibition of glucosylceramide synthase does not reverse drug resistance in cancer cells // J. Biol. Chem. 2004. Vol. 279. № 39. P. 40412–40418.
19. Zervas M., Somers K.L., Thrall M.A., Walkley S.U. Critical role for glycosphingolipids in Niemann – Pick disease type C // Curr. Biol. 2001. Vol. 11. № 16. P. 1283–1287.
20. Patterson M.C., Vecchio D., Prady H., Abel L., Wraith J.E. Miglustat for treatment of Niemann – Pick C disease: a randomised controlled study // Lancet Neurol. 2007. V. 6. P. 765–772.
21. Pineda M., Wraith J.E, Mengel E. et al. Miglustat for treatment of Niemann – Pick C disease: a randomised controlled study // Lancet Neurol. 2007. Vol. 6. № 9. P. 765–772.
22. Madra M., Sturley S.L. Niemann – Pick type C pathogenesis and treatment: from statins to sugars // Clin. Lipidol. 2010. Vol. 5. № 3. P. 387–395.
23. Gelsthorpe M.E., Baumann N., Millard E., Gale S.E., Langmade S.J., Schaffer J.E., Ory D.S. Niemann – Pick type C1 I1061T mutant encodes a functional protein that is selected for endoplasmic reticulum-associated degradation due to protein misfolding // J. Biol. Chem. 2008. Vol. 283. № 13. P. 8229–8236.
24. Pineda M., Perez-Poyato M.S., O'Callaghan M., Vilaseca M.A., Pocovi M., Domingo R., Portal L.R., Pérez A.V., Temudo T., Gaspar A., Peñas J.J., Roldán S., Fumero L.M., de la Barca O.B., Silva M.T., Macías-Vidal J., Coll M.J. Clinical experience with miglustat therapy in pediatric patients with Niemann – Pick disease type C: a case series // Mol. Genet. Metab. 2010. Vol. 99. № 4. P. 358–366.
25. Kothare S.V., Kaleyias J. Narcolepsy and other hypersomnias in children // Curr. Opin. Pediatr. 2008. Vol. 20. № 6. P. 666–675.
26. Santos M.L., Raskin S., Telles D.S., Löhr A. Jr, Liberalesso P.B., Vieira S.C., Cordeiro M.L. Treatment of a child diagnosed with Niemann – Pick disease type C with miglustat: a case report in Brazil // J. Inherit. Metab. Dis. 2008. Vol. 31. Suppl. 2. P. S357–361.
27. Chien Y.H., Lee N.C., Tsai L.K., Huang A.C., Peng S.F., Chen S.J., Hwu W.L. Treatment of Niemann – Pick disease type C in two children with miglustat: initial responses and maintenance of effects over 1 year // J. Inherit. Metab. Dis. 2007. Vol. 30. № 5. P. 826.
28. Vruchte D., Lloyd-Evans E., Veldman R.J. et al. Accumulation of glycosphingolipids in Niemann – Pick C disease disrupts endosomal transport // J. Biol. Chem. 2004. Vol. 279. P. 26167–26175.
29. Zarowski M., Steinborn B., Gurda B., Dvorakova L., Vlaskova H., Kothare S.V. Treatment of cataplexy in Niemann – Pick disease type C with the use of miglustat // Eur. J. Paediatr. Neurol. 2011. Vol. 15. № 1. P. 84–87.
Новости на тему
Отправить статью по электронной почте
Ваш адрес электронной почты:
Адрес электронной почты получателя:
Разделите несколько адресов электронной почты запятой
Сообщение(не обязательно)
Не более 1500 символов
Анти спам:
Для предотвращения спама, пожалуйста, введите в поле слово, которое видите ниже.
Обновить код
* адреса предоставленные Вами будут использоваться только для отправки электронной почты.
Уважаемый посетитель uMEDp!
Уведомляем Вас о том, что здесь содержится информация, предназначенная исключительно для специалистов здравоохранения.
Если Вы не являетесь специалистом здравоохранения, администрация не несет ответственности за возможные отрицательные последствия, возникшие в результате самостоятельного использования Вами информации с портала без предварительной консультации с врачом.
Нажимая на кнопку «Войти», Вы подтверждаете, что являетесь врачом или студентом медицинского вуза.