–Ф–Є—Д—Д–µ—А–µ–љ—Ж–Є–∞–ї—М–љ—Л–є –і–Є–∞–≥–љ–Њ–Ј —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–є –і–Є–∞—А–µ–Є —Г –і–µ—В–µ–є
- –Р–љ–љ–Њ—В–∞—Ж–Є—П
- –°—В–∞—В—М—П
- –°—Б—Л–ї–Ї–Є

–•—А–Њ–љ–Є—З–µ—Б–Ї–Њ–є —Б—З–Є—В–∞–µ—В—Б—П –і–Є–∞—А–µ—П –њ—А–Њ–і–Њ–ї–ґ–Є—В–µ–ї—М–љ–Њ—Б—В—М—О –±–Њ–ї–µ–µ 3¬†–љ–µ–і–µ–ї—М —Б¬†—З–∞—Б—В–Њ—В–Њ–є —Б—В—Г–ї–∞ —В—А–Є –Є¬†–±–Њ–ї–µ–µ —А–∞–Ј –≤¬†–і–µ–љ—М. –£—З–Є—В—Л–≤–∞—П, —З—В–Њ —В–∞–Ї–∞—П —З–∞—Б—В–Њ—В–∞ —Б—В—Г–ї–∞ –Љ–Њ–ґ–µ—В –±—Л—В—М –љ–Њ—А–Љ–Њ–є –і–ї—П –і–µ—В–µ–є –њ–µ—А–≤—Л—Е –Љ–µ—Б—П—Ж–µ–≤ –ґ–Є–Ј–љ–Є, –љ–µ–Њ–±—Е–Њ–і–Є–Љ–Њ —В–∞–Ї–ґ–µ –Њ–±—А–∞—Й–∞—В—М –≤–љ–Є–Љ–∞–љ–Є–µ –љ–∞¬†—Е–∞—А–∞–Ї—В–µ—А —Б—В—Г–ї–∞ –Є¬†–µ–≥–Њ –Њ–±—К–µ–Љ. –Т¬†–љ–Њ—А–Љ–µ —Г¬†–Ј–і–Њ—А–Њ–≤–Њ–≥–Њ —А–µ–±–µ–љ–Ї–∞ –Њ–±—К–µ–Љ —Б—В—Г–ї–∞ –Њ–±—Л—З–љ–Њ –љ–µ¬†–њ—А–µ–≤—Л—И–∞–µ—В 10¬†–≥/–Ї–≥ –Љ–∞—Б—Б—Л —В–µ–ї–∞ –≤¬†—Б—Г—В–Ї–Є. –Т–Њ–і—П–љ–Є—Б—В—Л–є –Є–ї–Є –ґ–Є—А–љ—Л–є —Е–∞—А–∞–Ї—В–µ—А —Б—В—Г–ї–∞, –љ–∞–ї–Є—З–Є–µ –Ј–µ–ї–µ–љ–Є, —Б–ї–Є–Ј–Є, –Ї—А–Њ–≤–Є, –љ–µ–њ–µ—А–µ–≤–∞—А–µ–љ–љ—Л—Е –Њ—Б—В–∞—В–Ї–Њ–≤ –њ–Є—Й–Є —П–≤–ї—П—О—В—Б—П –њ—А–Є–Ј–љ–∞–Ї–∞–Љ–Є –њ–∞—В–Њ–ї–Њ–≥–Є–Є.
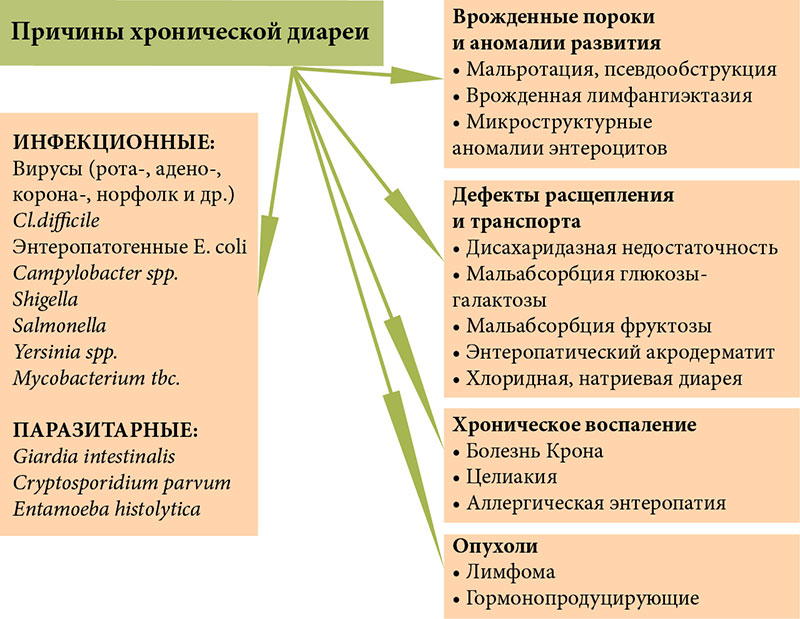
–Я—А–Є—З–Є–љ—Л —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–є –і–Є–∞—А–µ–Є (–•–Ф) –≤–µ—Б—М–Љ–∞ —А–∞–Ј–љ–Њ–Њ–±—А–∞–Ј–љ—Л (—А–Є—Б.¬†1), –љ–∞–Є–±–Њ–ї–µ–µ —З–∞—Б—В—Л–µ –Є–Ј¬†–љ–Є—Е: –Ї–Є—И–µ—З–љ—Л–µ –Є–љ—Д–µ–Ї—Ж–Є–Є (—З–∞—Й–µ –≤–Є—А—Г—Б–љ—Л–µ, —А–µ–ґ–µ –±–∞–Ї—В–µ—А–Є–∞–ї—М–љ—Л–µ, –≤—Л–Ј–≤–∞–љ–љ—Л–µ —Г—Б–ї–Њ–≤–љ–Њ-–њ–∞—В–Њ–≥–µ–љ–љ–Њ–є –Љ–Є–Ї—А–Њ—Д–ї–Њ—А–Њ–є), –∞¬†—В–∞–Ї–ґ–µ –њ–∞—А–∞–Ј–Є—В–∞—А–љ—Л–µ –Є–љ–≤–∞–Ј–Є–Є (–ї—П–Љ–±–ї–Є–Њ–Ј, –Є–љ–Њ–≥–і–∞ –Ї—А–Є–њ—В–Њ—Б–њ–Њ—А–Є–і–Є–Њ–Ј, –∞–Љ–µ–±–Є–∞–Ј –Є¬†–і—А.). –Ґ—А–∞–љ—Б—Д–Њ—А–Љ–∞—Ж–Є—П –Њ—Б—В—А–Њ–є –і–Є–∞—А–µ–Є –≤¬†—Е—А–Њ–љ–Є—З–µ—Б–Ї—Г—О –Љ–Њ–ґ–µ—В –±—Л—В—М –Њ–±—Г—Б–ї–Њ–≤–ї–µ–љ–∞ –љ–µ–±–ї–∞–≥–Њ–њ—А–Є—П—В–љ—Л–Љ —Д–Њ–љ–Њ–Љ, –љ–∞¬†–Ї–Њ—В–Њ—А–Њ–Љ –њ—А–Њ—В–µ–Ї–∞–µ—В –Є–љ—Д–µ–Ї—Ж–Є—П (–≥–Є–њ–Њ—В—А–Њ—Д–Є—П, –і–µ—Д–Є—Ж–Є—В –Љ–Є–Ї—А–Њ–љ—Г—В—А–Є–µ–љ—В–Њ–≤, –Є–Љ–Љ—Г–љ–Њ–і–µ—Д–Є—Ж–Є—В), –∞¬†—В–∞–Ї–ґ–µ –љ–µ–∞–і–µ–Ї–≤–∞—В–љ–Њ–є —В–µ—А–∞–њ–Є–µ–є. –Ъ–∞–Ї —Б–ї–µ–і—Б—В–≤–Є–µ –і–ї–Є—В–µ–ї—М–љ–Њ–≥–Њ –≤–Њ—Б–њ–∞–ї–µ–љ–Є—П, —А–∞–Ј–≤–Є–≤–∞–µ—В—Б—П –∞—В—А–Њ—Д–Є—П —Б–ї–Є–Ј–Є—Б—В–Њ–є –Њ–±–Њ–ї–Њ—З–Ї–Є —В–Њ–љ–Ї–Њ–є –Ї–Є—И–Ї–Є (–°–Ю–Ґ–Ъ) —Б¬†–≤—В–Њ—А–Є—З–љ—Л–Љ –љ–∞—А—Г—И–µ–љ–Є–µ–Љ –µ–µ —Д—Г–љ–Ї—Ж–Є–є.
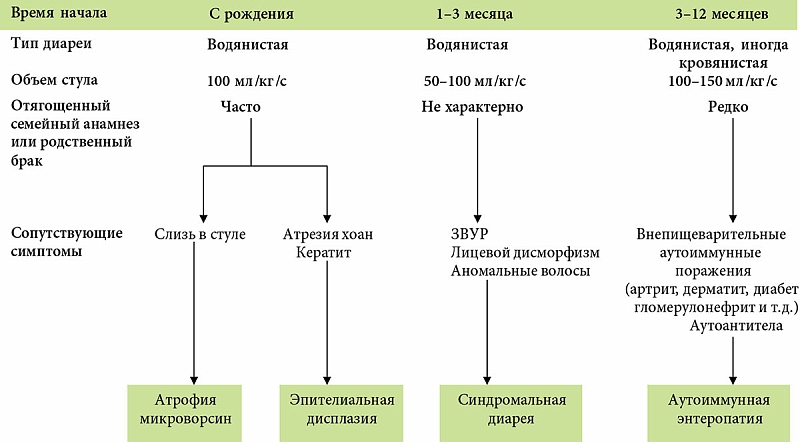
–Э–∞—А—П–і—Г —Б¬†–Є–љ—Д–µ–Ї—Ж–Є–Њ–љ–љ—Л–Љ–Є –Є¬†–њ–∞—А–∞–Ј–Є—В–∞—А–љ—Л–Љ–Є –њ—А–Є—З–Є–љ–∞–Љ–Є, –•–Ф –Љ–Њ–ґ–µ—В –±—Л—В—М –Њ–±—Г—Б–ї–Њ–≤–ї–µ–љ–∞ –≤—А–Њ–ґ–і–µ–љ–љ—Л–Љ–Є¬†–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Є –і–µ—В–µ—А–Љ–Є–љ–Є—А–Њ–≤–∞–љ–љ—Л–Љ–Є –і–µ—Д–µ–Ї—В–∞–Љ–Є –њ–Є—Й–µ–≤–∞—А–µ–љ–Є—П (–ї–∞–Ї—В–∞–Ј–љ–∞—П –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В—М, —Б–∞—Е–∞—А–∞–Ј–Њ-–Є–Ј–Њ–Љ–∞–ї—М-—В–∞–Ј–љ–∞—П –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В—М) –Є¬†–≤—Б–∞—Б—Л–≤–∞–љ–Є—П (–Љ–∞–ї—М–∞–±—Б–Њ—А–±—Ж–Є—П¬†–≥–ї—О–Ї–Њ-–Ј—Л-–≥–∞–ї–∞–Ї—В–Њ–Ј—Л, —Е–ї–Њ—А–Є–і–љ–∞—П, –љ–∞—В—А–Є–µ–≤–∞—П –і–Є–∞—А–µ—П, –Љ–∞–ї—М–∞–±—Б–Њ—А–±—Ж–Є—П –ґ–µ–ї—З–љ—Л—Е –Ї–Є—Б–ї–Њ—В, —Н–љ—В–µ—А–Њ–њ–∞—В–Є—З–µ—Б–Ї–Є–є –∞–Ї—А–Њ–і–µ—А–Љ–∞—В–Є—В –Є¬†–і—А.), –≤—А–Њ–ґ–і–µ–љ–љ—Л–Љ–Є –∞–љ–Њ–Љ–∞–ї–Є—П–Љ–Є —Б—В—А–Њ–µ–љ–Є—П —Н–љ—В–µ—А–Њ—Ж–Є—В–Њ–≤ (–∞—В—А–Њ—Д–Є—П –Љ–Є–Ї—А–Њ–≤–Њ—А—Б–Є–љ, –Є–љ—В–µ—Б—В–Є–љ–∞–ї—М–љ–∞—П —Н–њ–Є—В–µ–ї–Є–∞–ї—М–љ–∞—П –і–Є—Б–њ–ї–∞–Ј–Є—П) –Є¬†–Є–Љ–Љ—Г–љ–Њ–њ–∞—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–Љ–Є –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П–Љ–Є, —Б–Њ–њ—А–Њ–≤–Њ–ґ–і–∞—О—Й–Є–Љ–Є—Б—П —Е—А–Њ–љ–Є—З–µ—Б–Ї–Є–Љ –≤–Њ—Б–њ–∞–ї–µ–љ–Є–µ–Љ –°–Ю–Ґ–Ъ (–∞–ї–ї–µ—А–≥–Є—З–µ—Б–Ї–∞—П —Н–љ—В–µ—А–Њ–њ–∞—В–Є—П, —Ж–µ–ї–Є–∞–Ї–Є—П, –∞—Г—В–Њ–Є–Љ–Љ—Г–љ–љ–∞—П —Н–љ—В–µ—А–Њ–њ–∞—В–Є—П, –±–Њ–ї–µ–Ј–љ—М –Ъ—А–Њ–љ–∞). –Э–∞–Є–±–Њ–ї–µ–µ —В—П–ґ–µ–ї–Њ–є —Д–Њ—А–Љ–Њ–є –•–Ф —П–≤–ї—П–µ—В—Б—П —В–∞–Ї –љ–∞–Ј—Л–≤–∞–µ–Љ–∞—П —В—А—Г–і–љ–Њ–Є–Ј–ї–µ—З–Є–Љ–∞—П –Љ–ї–∞–і–µ–љ—З–µ—Б–Ї–∞—П –і–Є–∞—А–µ—П, –Њ–±—Г—Б–ї–Њ–≤–ї–µ–љ–љ–∞—П –∞–љ–Њ–Љ–∞–ї–Є—П–Љ–Є —Н–љ—В–µ—А–Њ—Ж–Є—В–Њ–≤ –Є–ї–Є –∞—Г—В–Њ–Є–Љ–Љ—Г–љ–љ–Њ–є —Н–љ—В–µ—А–Њ–њ–∞—В–Є–µ–є.
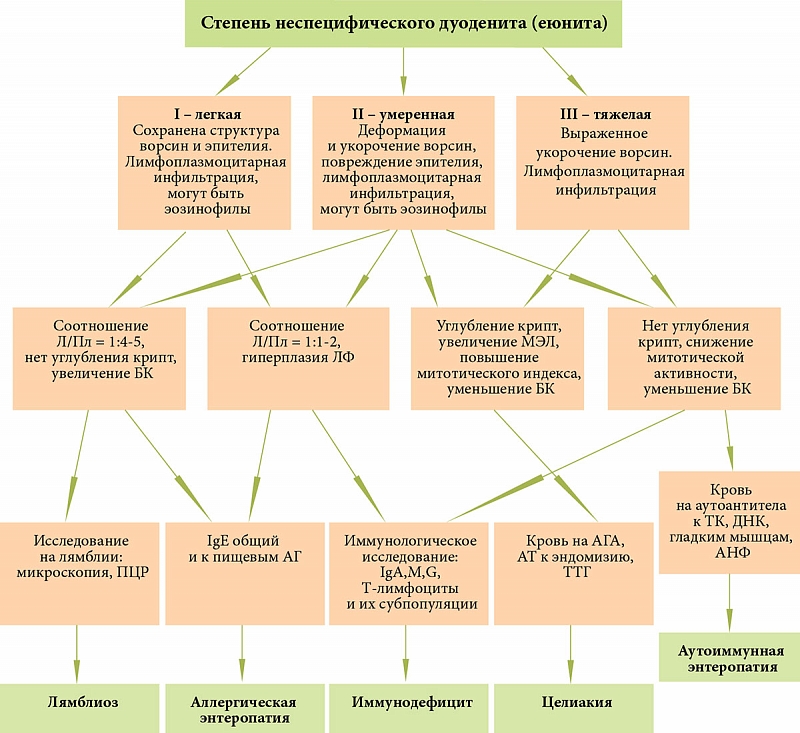
–Ф–Є–∞–≥–љ–Њ—Б—В–Є–Ї–∞ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є, –њ—А–Њ—П–≤–ї—П—О—Й–Є—Е—Б—П –•–Ф, –і–Њ–ї–ґ–љ–∞ –Њ—Б–љ–Њ–≤—Л–≤–∞—В—М—Б—П –љ–∞¬†—В—Й–∞—В–µ–ї—М–љ–Њ–є –Њ—Ж–µ–љ–Ї–µ —Е–∞—А–∞–Ї—В–µ—А–∞ –Ї–Є—И–µ—З–љ–Њ–≥–Њ —Б–Є–љ–і—А–Њ–Љ–∞, —Б–Є–Љ–њ—В–Њ–Љ–Њ–≤ —Б–Њ¬†—Б—В–Њ—А–Њ–љ—Л –і—А—Г–≥–Є—Е –Њ—А–≥–∞–љ–Њ–≤ –Є¬†—Б–Є—Б—В–µ–Љ, –і–∞–љ–љ—Л—Е –∞–љ–∞–Љ–љ–µ–Ј–∞ –Є¬†–і–Њ–њ–Њ–ї–љ–Є—В–µ–ї—М–љ—Л—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є. –Т¬†—Б–≤—П–Ј–Є —Б¬†—И–Є—А–Њ–Ї–Њ–є —А–∞—Б–њ—А–Њ—Б—В—А–∞–љ–µ–љ–љ–Њ—Б—В—М—О –Ї–Є—И–µ—З–љ—Л—Е –Є–љ—Д–µ–Ї—Ж–Є–є –Є¬†–њ–∞—А–∞–Ј–Є—В–Њ–Ј–Њ–≤ —Г¬†–і–µ—В–µ–є –њ–µ—А–≤—Л–Љ —Н—В–∞–њ–Њ–Љ –і–Є–∞–≥–љ–Њ—Б—В–Є–Ї–Є —П–≤–ї—П–µ—В—Б—П —Г—Б—В–∞–љ–Њ–≤–ї–µ–љ–Є–µ –Є–ї–Є –Є—Б–Ї–ї—О—З–µ–љ–Є–µ –і–∞–љ–љ—Л—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є. –Ф–ї—П —Н—В–Њ–≥–Њ –Ї–∞–ґ–і–Њ–Љ—Г —А–µ–±–µ–љ–Ї—Г —Б¬†–•–Ф –љ–µ–Њ–±—Е–Њ–і–Є–Љ–Њ –њ—А–Њ–≤–µ—Б—В–Є:
- –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є–є –∞–љ–∞–ї–Є–Ј –Ї—А–Њ–≤–Є;
- –њ–Њ—Б–µ–≤—Л –Ї–∞–ї–∞;
- –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ –Ї–∞–ї–∞ –љ–∞¬†—П–є—Ж–∞¬†–≥–ї–Є—Б—В–Њ–≤ –Є¬†—Ж–Є—Б—В—Л –ї—П–Љ–±–ї–Є–є –Љ–µ—В–Њ–і–∞–Љ–Є –Љ–Є–Ї—А–Њ—Б–Ї–Њ–њ–Є–Є, –Ш–§–Р –Є¬†–Я–¶–†;
- –Љ–Є–Ї—А–Њ—Б–Ї–Њ–њ–Є—О –Љ–∞–Ј–Ї–∞ –Ї–∞–ї–∞ –љ–∞¬†–Ї—А–Є–њ—В–Њ—Б–њ–Њ—А–Є–і–Є–Њ–Ј (—Б¬†–Њ–Ї—А–∞—Б–Ї–Њ–є –њ–Њ¬†–¶–Є–ї—О-–Э–Є–ї—М—Б–µ–љ—Г –Є–ї–Є –Њ–±—А–∞–±–Њ—В–Ї–Њ–є 1% —А–∞—Б—В–≤–Њ—А–Њ–Љ HCl);
- –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ –Ї–∞–ї–∞ –љ–∞¬†—В–Њ–Ї—Б–Є–љ –Р¬†–Є¬†–Т¬†Clostridium difficile –Љ–µ—В–Њ–і–Њ–Љ –Ш–§–Р;
- –Ї–Њ–њ—А–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ (–љ–µ¬†–Љ–µ–љ–µ–µ 3¬†–Ї–Њ–њ—А–Њ–≥—А–∞–Љ–Љ);
- –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ –Ї–∞–ї–∞ –љ–∞¬†—Б–Ї—А—Л—В—Г—О –Ї—А–Њ–≤—М.
–Ф–∞–ґ–µ –≤¬†—Б–ї—Г—З–∞–µ –Њ—В—А–Є—Ж–∞—В–µ–ї—М–љ–Њ–≥–Њ —А–µ–Ј—Г–ї—М—В–∞—В–∞ –±–∞–Ї—В–µ—А–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–≥–Њ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П –њ—А–Є –њ–Њ–і–Њ–Ј—А–µ–љ–Є–Є –љ–∞¬†–Є–љ—Д–µ–Ї—Ж–Є–Њ–љ–љ—Л–є¬†–≥–µ–љ–µ–Ј –і–Є–∞—А–µ–Є –њ–Њ–Ї–∞–Ј–∞–љ –Ї—Г—А—Б –∞–љ—В–Є–±–∞–Ї—В–µ—А–Є–∞–ї—М–љ–Њ–є —В–µ—А–∞–њ–Є–Є ex juvantibus. –Ю–±—Л—З–љ–Њ –≤¬†—Н—В–Є—Е —Б–ї—Г—З–∞—П—Е –љ–∞–Ј–љ–∞—З–∞—О—В –љ–Є—В—А–Њ—Д—Г—А–∞–љ—Л (–љ–Є—Д—Г—А–∞—В–µ–ї—М 15¬†–Љ–≥/–Ї–≥ –Є–ї–Є –љ–Є—Д—Г—А–Њ–Ї—Б–∞–Ј–Є–і 100¬†–Љ–≥ 2¬†—А–∞–Ј–∞ –≤¬†–і–µ–љ—М), –∞–Љ–Є–љ–Њ–≥–ї–Є–Ї–Њ–Ј–Є–і—Л (–≥–µ–љ—В–∞–Љ–Є—Ж–Є–љ 3вАУ5¬†–Љ–≥/–Ї–≥, –∞–Љ–Є–Ї–∞—Ж–Є–љ 10¬†–Љ–≥/–Ї–≥) –Є–ї–Є —Ж–µ—Д–∞–ї–Њ—Б–њ–Њ—А–Є–љ—Л¬†III –њ–Њ–Ї–Њ–ї–µ–љ–Є—П (—Ж–µ—Д–Њ—В–∞–Ї—Б–Є–Љ 50¬†–Љ–≥/–Ї–≥, —Ж–µ—Д—В—А–Є–∞–Ї—Б–Њ–љ 50¬†–Љ–≥/–Ї–≥). –Я–Њ—Б–Ї–Њ–ї—М–Ї—Г –і–Є–∞—А–µ—П –≤¬†–і–µ—В—Б–Ї–Њ–Љ –≤–Њ–Ј—А–∞—Б—В–µ –Є–Љ–µ–µ—В –њ—А–µ–Є–Љ—Г—Й–µ—Б—В–≤–µ–љ–љ–Њ –≤–Є—А—Г—Б–љ—Г—О —Н—В–Є–Њ–ї–Њ–≥–Є—О –Є¬†—Е–∞—А–∞–Ї—В–µ—А–Є–Ј—Г–µ—В—Б—П –≤–Њ–і—П–љ–Є—Б—В—Л–Љ —Б—В—Г–ї–Њ–Љ —Б–Њ¬†–Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ–є –њ–Њ—В–µ—А–µ–є –ґ–Є–і–Ї–Њ—Б—В–Є –Є¬†—Б–Њ–ї–µ–є, –≤–∞–ґ–љ–Њ —Б¬†–њ–µ—А–≤—Л—Е¬†–ґ–µ –і–љ–µ–є –±–Њ–ї–µ–Ј–љ–Є –њ—А–Њ–≤–Њ–і–Є—В—М –∞–і–µ–Ї–≤–∞—В–љ—Г—О —А–µ–≥–Є–і—А–∞—В–∞—Ж–Є—О –Є¬†–Ї–Њ—А—А–µ–Ї—Ж–Є—О —Н–ї–µ–Ї—В—А–Њ–ї–Є—В–љ–Њ–≥–Њ –Њ–±–Љ–µ–љ–∞. –Э–µ¬†–Љ–µ–љ–µ–µ –≤–∞–ґ–љ–Њ –Њ–±–µ—Б–њ–µ—З–Є—В—М –њ–Њ–ї–љ–Њ—Ж–µ–љ–љ–Њ–µ –њ–Є—В–∞–љ–Є–µ —А–µ–±–µ–љ–Ї–∞ —Б¬†—Г—З–µ—В–Њ–Љ —Б–љ–Є–ґ–µ–љ–љ—Л—Е –≤–Њ–Ј–Љ–Њ–ґ–љ–Њ—Б—В–µ–є –њ–Є—Й–µ–≤–∞—А–µ–љ–Є—П.
–Ф–ї—П –Ї–Њ—А—А–µ–Ї—Ж–Є–Є –Љ–Є–Ї—А–Њ–±–Є–Њ—Ж–µ–љ–Њ–Ј–∞ –Ї–Є—И–µ—З–љ–Є–Ї–∞ –њ—А–Є –њ–Њ—Б—В–Є–љ—Д–µ–Ї—Ж–Є–Њ–љ–љ—Л—Е –і–Є–∞—А–µ—П—Е –њ—А–Є–Љ–µ–љ—П—О—В –њ—А–µ–њ–∞—А–∞—В—Л-–њ—А–Њ–±–Є–Њ—В–Є–Ї–Є. –Я—А–Є –љ–∞–Ј–љ–∞—З–µ–љ–Є–Є –њ—А–Њ–±–Є–Њ—В–Є—З–µ—Б–Ї–Њ–≥–Њ –њ—А–µ–њ–∞—А–∞—В–∞ –љ–µ–Њ–±—Е–Њ–і–Є–Љ–Њ —Г–±–µ–і–Є—В—М—Б—П –≤¬†–Њ—В—Б—Г—В—Б—В–≤–Є–Є –ї–∞–Ї—В–Њ–Ј—Л –≤¬†–µ–≥–Њ —Б–Њ—Б—В–∞–≤–µ. –Ф–ї—П –і–µ—В–µ–є —А–∞–љ–љ–µ–≥–Њ –љ–µ–Њ–љ–∞—В–∞–ї—М–љ–Њ–≥–Њ –њ–µ—А–Є–Њ–і–∞ –≤—Л–њ—Г—Б–Ї–∞–µ—В—Б—П –±–µ–Ј–ї–∞–Ї—В–Њ–Ј–љ—Л–є –њ—А–Њ–±–Є–Њ—В–Є–Ї –Я—А–Є–Љ–∞–і–Њ—Д–Є–ї—Г—Б –Ф–µ—В—Б–Ї–Є–є, –Ї–Њ—В–Њ—А—Л–є —Б–Њ–і–µ—А–ґ–Є—В –њ–Њ–і–Њ–±—А–∞–љ–љ—Л–µ –≤¬†—Б–Њ–Њ—В–≤–µ—В—Б—В–≤–Є–Є —Б¬†–≤–Њ–Ј—А–∞—Б—В–Њ–Љ —И—В–∞–Љ–Љ—Л –ї–∞–Ї—В–Њ- –Є¬†–±–Є—Д–Є–і–Њ–±–∞–Ї—В–µ—А–Є–є –≤¬†–≤–Њ–Ј—А–∞—Б—В–љ–Њ–є –і–Њ–Ј–Є—А–Њ–≤–Ї–µ 2 —Е 10 9 –Ъ–Ю–Х –≤¬†—Б—Г—В–Њ—З–љ–Њ–є –і–Њ–Ј–µ.
–Х—Б–ї–Є —А–µ–±–µ–љ–Њ–Ї –љ–∞—Е–Њ–і–Є—В—Б—П –љ–∞¬†–µ—Б—В–µ—Б—В–≤–µ–љ–љ–Њ–Љ –≤—Б–Ї–∞—А–Љ–ї–Є–≤–∞–љ–Є–Є, –µ–≥–Њ –Љ–Њ–ґ–љ–Њ –њ—А–Њ–і–Њ–ї–ґ–Є—В—М, –љ–Њ¬†–≤¬†–Ї–∞–ґ–і–Њ–µ –Ї–Њ—А–Љ–ї–µ–љ–Є–µ —А–µ–Ї–Њ–Љ–µ–љ–і—Г–µ—В—Б—П –і–Њ–±–∞–≤–ї—П—В—М —Д–µ—А–Љ–µ–љ—В –Ы–∞–Ї—В–∞–Ј–∞ –С—Н–±–Є.
–Я—А–Є –Є—Б–Ї—Г—Б—Б—В–≤–µ–љ–љ–Њ–Љ –≤—Б–Ї–∞—А–Љ–ї–Є–≤–∞–љ–Є–Є —Б¬†—Г—З–µ—В–Њ–Љ –≤–µ—А–Њ—П—В–љ–Њ–≥–Њ —А–∞–Ј–≤–Є—В–Є—П –≤—В–Њ—А–Є—З–љ–Њ–є –ї–∞–Ї—В–∞–Ј–љ–Њ–є –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В–Є –љ–∞¬†—Д–Њ–љ–µ –Ї–Є—И–µ—З–љ–Њ–є –Є–љ—Д–µ–Ї—Ж–Є–Є —Ж–µ–ї–µ—Б–Њ–Њ–±—А–∞–Ј–љ–Њ –Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞—В—М –±–µ–Ј–ї–∞–Ї—В–Њ–Ј–љ—Л–µ –Є–ї–Є –Ї–Є—Б–ї–Њ–Љ–Њ–ї–Њ—З–љ—Л–µ —Б–Љ–µ—Б–Є: –Э–Р–Э –Ї–Є—Б–ї–Њ–Љ–Њ–ї–Њ—З–љ—Л–є, –Э–Р–Э –±–µ–Ј–ї–∞–Ї—В–Њ–Ј–љ—Л–є, –≠–љ—Д–∞–Љ–Є–ї –Ы–∞–Ї—В–Њ—Д—А–Є.
–Ы–∞–Ї—В–∞–Ј–љ–∞—П –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В—М —Г¬†–і–µ—В–µ–є —А–∞–љ–љ–µ–≥–Њ –≤–Њ–Ј—А–∞—Б—В–∞ –Љ–Њ–ґ–µ—В –±—Л—В—М –љ–µ¬†—В–Њ–ї—М–Ї–Њ –≤—В–Њ—А–Є—З–љ–Њ–є, –љ–Њ¬†–Є¬†–њ–µ—А–≤–Є—З–љ–Њ–є: –≤—А–Њ–ґ–і–µ–љ–љ–Њ–є –Є–ї–Є —В—А–∞–љ–Ј–Є—В–Њ—А–љ–Њ–є. –Т—А–Њ–ґ–і–µ–љ–љ–∞—П –ї–∞–Ї—В–∞–Ј–љ–∞—П –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В—М¬†вАУ –Ї—А–∞–є–љ–µ —А–µ–і–Ї–∞—П –њ–∞—В–Њ–ї–Њ–≥–Є—П, –њ—А–Є –Ї–Њ—В–Њ—А–Њ–є –≤¬†—А–µ–Ј—Г–ї—М—В–∞—В–µ¬†–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Є—Е –Љ—Г—В–∞—Ж–Є–є –ї–∞–Ї—В–∞–Ј–∞ –љ–µ¬†—В—А–∞–љ—Б–њ–Њ—А—В–Є—А—Г–µ—В—Б—П –Є–Ј¬†–∞–њ–њ–∞—А–∞—В–∞ –У–Њ–ї—М–і–ґ–Є –љ–∞¬†–њ–Њ–≤–µ—А—Е–љ–Њ—Б—В—М –Љ–µ–Љ–±—А–∞–љ—Л —Н–љ—В–µ—А–Њ—Ж–Є—В–∞. –°–Є–Љ–њ—В–Њ–Љ–∞—В–Є–Ї–∞ –≤—А–Њ–ґ–і–µ–љ–љ–Њ–є –ї–∞–Ї—В–∞–Ј–љ–Њ–є –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В–Є –њ—А–Њ—П–≤–ї—П–µ—В—Б—П —Б¬†–њ–µ—А–≤—Л—Е –і–љ–µ–є –ґ–Є–Ј–љ–Є –Њ—З–µ–љ—М —П—А–Ї–Њ –≤¬†–≤–Є–і–µ –Љ–љ–Њ–≥–Њ–Ї—А–∞—В–љ–Њ–є –≤–Њ–і—П–љ–Є—Б—В–Њ–є –і–Є–∞—А–µ–Є, —Б–Њ–њ—А–Њ–≤–Њ–ґ–і–∞—О—Й–µ–є—Б—П –≤–Ј–і—Г—В–Є–µ–Љ –ґ–Є–≤–Њ—В–∞, –њ–Њ—В–µ—А–µ–є –Љ–∞—Б—Б—Л —В–µ–ї–∞ –Є¬†–і–µ–≥–Є–і—А–∞—В–∞—Ж–Є–µ–є. –Т¬†–∞–±—Б–Њ–ї—О—В–љ–Њ–Љ –±–Њ–ї—М—И–Є–љ—Б—В–≤–µ —Б–ї—Г—З–∞–µ–≤ –ї–∞–Ї—В–∞–Ј–љ–∞—П –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В—М —Г¬†–љ–Њ–≤–Њ—А–Њ–ґ–і–µ–љ–љ–Њ–≥–Њ –Є–ї–Є –љ–µ–і–Њ–љ–Њ—И–µ–љ–љ–Њ–≥–Њ —А–µ–±–µ–љ–Ї–∞ –љ–Њ—Б–Є—В —В—А–∞–љ–Ј–Є—В–Њ—А–љ—Л–є —Е–∞—А–∞–Ї—В–µ—А –Є¬†–Њ–±—Г—Б–ї–Њ–≤–ї–µ–љ–∞ –љ–µ–Ј—А–µ–ї–Њ—Б—В—М—О –Ї–Є—И–µ—З–љ—Л—Е –≤–Њ—А—Б–Є–љ–Њ–Ї –Є¬†–љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ–є –∞–Ї—В–Є–≤–љ–Њ—Б—В—М—О —Д–µ—А–Љ–µ–љ—В–∞ –ї–∞–Ї—В–∞–Ј—Л –љ–∞¬†–њ–Њ–≤–µ—А—Е–љ–Њ—Б—В–Є —Н–њ–Є—В–µ–ї–Є—П (–≥–Є–њ–Њ–ї–∞–Ї—В–∞–Ј–Є–µ–є).
–Ґ—А–∞–љ–Ј–Є—В–Њ—А–љ–∞—П –ї–∞–Ї—В–∞–Ј–љ–∞—П –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В—М –Љ–Њ–ґ–µ—В –±—Л—В—М –њ—А–Є—З–Є–љ–Њ–є –љ–µ—В—П–ґ–µ–ї–Њ–є ¬Ђ–Ї–Є—Б–ї–Њ–є¬ї –≤–Њ–і—П–љ–Є—Б—В–Њ–є –і–Є–∞—А–µ–Є –≤¬†–њ–µ—А–≤—Л–µ –Љ–µ—Б—П—Ж—Л –ґ–Є–Ј–љ–Є, –Ї–Њ—В–Њ—А–∞—П –љ–µ—А–µ–і–Ї–Њ —Б–Њ–њ—А–Њ–≤–Њ–ґ–і–∞–µ—В—Б—П –Ї–Є—И–µ—З–љ—Л–Љ–Є –Ї–Њ–ї–Є–Ї–∞–Љ–Є –Є¬†–±–µ—Б–њ–Њ–Ї–Њ–є—Б—В–≤–Њ–Љ —А–µ–±–µ–љ–Ї–∞, –љ–Њ¬†–љ–µ –≤–ї–Є—П–µ—В –љ–∞¬†–µ–≥–Њ —Д–Є–Ј–Є—З–µ—Б–Ї–Њ–µ —А–∞–Ј–≤–Є—В–Є–µ. –°–Є–Љ–њ—В–Њ–Љ—Л –њ–Њ—Б—В–µ–њ–µ–љ–љ–Њ —Б–Љ—П–≥—З–∞—О—В—Б—П –љ–∞¬†—Д–Њ–љ–µ –њ—А–Њ–і–Њ–ї–ґ–µ–љ–Є—П¬†–≥—А—Г–і–љ–Њ–≥–Њ –≤—Б–Ї–∞—А–Љ–ї–Є–≤–∞–љ–Є—П. –≠—В–Њ –Њ–±—К—П—Б–љ—П–µ—В—Б—П —В–µ–Љ, —З—В–Њ –ґ–µ–љ—Б–Ї–Њ–µ –Љ–Њ–ї–Њ–Ї–Њ –Њ–±–ї–∞–і–∞–µ—В –Љ–љ–Њ–≥–Њ—З–Є—Б–ї–µ–љ–љ—Л–Љ–Є —Д–∞–Ї—В–Њ—А–∞–Љ–Є —А–Њ—Б—В–∞ (EGF, IGF-1, TGF-ќ≤, —Б–њ–µ—А–Љ–Є–љ, —Б–њ–µ—А–Љ–Є–і–Є–љ, –љ—Г–Ї–ї–µ–Њ—В–Є–і—Л, –Ї–Њ—А–Њ—В–Ї–Њ—Ж–µ–њ–Њ—З–µ—З–љ—Л–µ –ґ–Є—А–љ—Л–µ –Ї–Є—Б–ї–Њ—В—Л –Є¬†—В.–і.), –Ї–Њ—В–Њ—А—Л–µ —Б–њ–Њ—Б–Њ–±—Б—В–≤—Г—О—В —А–Њ—Б—В—Г –Є¬†—Б–Њ–Ј—А–µ–≤–∞–љ–Є—О –Ї–Є—И–µ—З–љ–Њ–≥–Њ —Н–њ–Є—В–µ–ї–Є—П –Є¬†–њ–Њ–≤—Л—И–µ–љ–Є—О –ї–∞–Ї—В–∞–Ј–љ–Њ–є –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є.
–Я–Њ—Н—В–Њ–Љ—Г –њ—А–Є —Б–Є–Љ–њ—В–Њ–Љ–∞—В–Є–Ї–µ¬†–≥–Є–њ–Њ–ї–∞–Ї—В–∞–Ј–Є–Є¬†–≥—А—Г–і–љ–Њ–µ –≤—Б–Ї–∞—А–Љ–ї–Є–≤–∞–љ–Є–µ —Б–ї–µ–і—Г–µ—В –њ—А–Њ–і–Њ–ї–ґ–Є—В—М, –љ–Њ¬†–і–ї—П —Г—Б—В—А–∞–љ–µ–љ–Є—П —Б–Є–Љ–њ—В–Њ–Љ–Њ–≤ —Ж–µ–ї–µ—Б–Њ-–Њ–±—А–∞–Ј–љ–Њ –њ—А–Є–Љ–µ–љ—П—В—М —Д–µ—А–Љ–µ–љ—В –Ы–∞–Ї—В–∞–Ј–∞ –С—Н–±–Є –њ–Њ¬†700¬†–µ–і. –≤¬†–љ–∞—З–∞–ї–µ –Ї–∞–ґ–і–Њ–≥–Њ –Ї–Њ—А–Љ–ї–µ–љ–Є—П. –Ф–µ—В—П–Љ —Б¬†—А–Њ–ґ–і–µ–љ–Є—П –љ–∞–Ј–љ–∞—З–∞–µ—В—Б—П —Б–Њ–і–µ—А–ґ–Є–Љ–Њ–µ 1¬†–Ї–∞–њ—Б—Г–ї—Л –Ы–∞–Ї—В–∞–Ј—Л –С—Н–±–Є –љ–∞¬†100¬†–Љ–ї –Љ–Њ–ї–Њ–Ї–∞. –Я–µ—А–µ–і –Ї–Њ—А–Љ–ї–µ–љ–Є–µ–Љ —Б–ї–µ–і—Г–µ—В —Б—Ж–µ–і–Є—В—М 20¬†–Љ–ї –Љ–Њ–ї–Њ–Ї–∞ –Є¬†–≤—Л—Б—Л–њ–∞—В—М —Б–Њ–і–µ—А–ґ–Є–Љ–Њ–µ –Ї–∞–њ—Б—Г–ї. –Ъ–Њ—А–Љ–ї–µ–љ–Є–µ –љ–∞—З–Є–љ–∞—В—М —З–µ—А–µ–Ј 10¬†–Љ–Є–љ—Г—В —Б¬†—Н—В–Њ–є –њ–Њ—А—Ж–Є–Є. –Т¬†—Б–ї—Г—З–∞–µ –µ—Б–ї–Є —А–µ–±–µ–љ–Њ–Ї –њ–Њ–ї—Г—З–∞–µ—В —В–Њ–ї—М–Ї–Њ —Б—Ж–µ–ґ–µ–љ–љ–Њ–µ¬†–≥—А—Г–і–љ–Њ–µ –Љ–Њ–ї–Њ–Ї–Њ, –Ы–∞–Ї—В–∞–Ј—Г –С—Н–±–Є –і–Њ–±–∞–≤–ї—П—О—В –≤¬†–њ–Њ–ї–љ—Л–є –Њ–±—К–µ–Љ –њ–Є—В–∞–љ–Є—П. –Я—А–Є –Є—Б–Ї—Г—Б—Б—В–≤–µ–љ–љ–Њ–Љ –≤—Б–Ї–∞—А–Љ–ї–Є–≤–∞–љ–Є–Є –љ–µ–Њ–±—Е–Њ–і–Є–Љ–Њ–µ –Ї–Њ–ї–Є—З–µ—Б—В–≤–Њ —Д–µ—А–Љ–µ–љ—В–∞ —В–∞–Ї–ґ–µ –і–Њ–±–∞–≤–ї—П–µ—В—Б—П –≤¬†–њ–Њ–ї–љ—Л–є –Њ–±—К–µ–Љ –Љ–Њ–ї–Њ—З–љ–Њ–≥–Њ –њ–Є—В–∞–љ–Є—П. –Т—А–µ–Љ—П –і–ї—П —Д–µ—А–Љ–µ–љ—В–∞—Ж–Є–Є —Б–Њ—Б—В–∞–≤–ї—П–µ—В 10¬†–Љ–Є–љ—Г—В.
–Ф–ї—П –њ–Њ–і—В–≤–µ—А–ґ–і–µ–љ–Є—П –ї–∞–Ї—В–∞–Ј–љ–Њ–є –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В–Є –ї—О–±–Њ–≥–Њ¬†–≥–µ–љ–µ–Ј–∞ —Г¬†—А–µ–±–µ–љ–Ї–∞ –Љ–Њ–ґ–љ–Њ –Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞—В—М —А–∞–Ј–ї–Є—З–љ—Л–µ —В–µ—Б—В—Л: –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ —А–Э –Ї–∞–ї–∞ (–љ–Є–ґ–µ 5,5),¬†–≥–ї–Є–Ї–µ–Љ–Є—З–µ—Б–Ї—Г—О –Ї—А–Є–≤—Г—О —Б¬†–ї–∞–Ї—В–Њ–Ј–љ–Њ–є –љ–∞–≥—А—Г–Ј–Ї–Њ–є –Є–Ј¬†—А–∞—Б—З–µ—В–∞ 2¬†–≥/–Ї–≥ (–њ—А–Є—А–Њ—Б—В —Г—А–Њ–≤–љ—П¬†–≥–ї—О–Ї–Њ–Ј—Л –љ–µ¬†–±–Њ–ї–µ–µ 25% –Њ—В¬†–Є—Б—Е–Њ–і–љ–Њ–≥–Њ), –≤–Њ–і–Њ—А–Њ–і–љ—Л–є —В–µ—Б—В (–њ—А–Є—А–Њ—Б—В —Г—А–Њ–≤–љ—П –≤–Њ–і–Њ—А–Њ–і–∞ –≤¬†–≤—Л–і—Л—Е–∞–µ–Љ–Њ–Љ –≤–Њ–Ј–і—Г—Е–µ –≤—Л—И–µ 20¬†ppm).
–Х—Б–ї–Є –Ї–Є—И–µ—З–љ–∞—П –Є–љ—Д–µ–Ї—Ж–Є—П –Є¬†–ї–∞–Ї—В–∞–Ј–љ–∞—П –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В—М (–љ–∞–Є–±–Њ–ї–µ–µ —З–∞—Б—В—Л–µ –њ—А–Є—З–Є–љ—Л —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–є –і–Є–∞—А–µ–Є) –Є—Б–Ї–ї—О—З–µ–љ—Л, –∞¬†–і–Є–∞—А–µ—П, –љ–µ—Б–Љ–Њ—В—А—П –љ–∞¬†–∞–љ—В–Є–±–∞–Ї—В–µ—А–Є–∞–ї—М–љ—Г—О —В–µ—А–∞–њ–Є—О –Є¬†–±–µ–Ј–ї–∞–Ї—В–Њ–Ј–љ—Г—О –і–Є–µ—В—Г, –њ—А–Њ–і–Њ–ї–ґ–∞–µ—В—Б—П, –љ–µ–Њ–±—Е–Њ–і–Є–Љ–Њ –њ—А–Њ–і–Њ–ї–ґ–Є—В—М –Њ–±—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ, –Њ–±–µ—Б–њ–µ—З–Є–≤–∞—П –∞–і–µ–Ї–≤–∞—В–љ—Г—О –љ—Г—В—А–Є—Ж–Є–Њ–љ–љ—Г—О –њ–Њ–і–і–µ—А–ґ–Ї—Г –њ—Г—В–µ–Љ —Б–Њ—З–µ—В–∞–љ–Є—П –њ–∞—А–µ–љ—В–µ—А–∞–ї—М–љ–Њ–≥–Њ –Є¬†—Н–љ—В–µ—А–∞–ї—М–љ–Њ–≥–Њ –њ–Є—В–∞–љ–Є—П —Б–Љ–µ—Б—П–Љ–Є –±–µ–ї–Ї–Њ–≤—Л—Е¬†–≥–Є–і—А–Њ–ї–Є–Ј–∞—В–Њ–≤ –±–µ–Ј –ї–∞–Ї—В–Њ–Ј—Л: –Р–ї—М—Д–∞—А–µ, –Э—Г—В—А–Є–ї–Њ–љ –Я–µ–њ—В–Є –Ґ–°–¶, –Э—Г—В—А–∞–Љ–Є–≥–µ–љ, –§—А–Є—Б–Њ–њ–µ–њ –Р–°, –Я—А–µ–≥–µ—Б—В–Є–Љ–Є–ї.
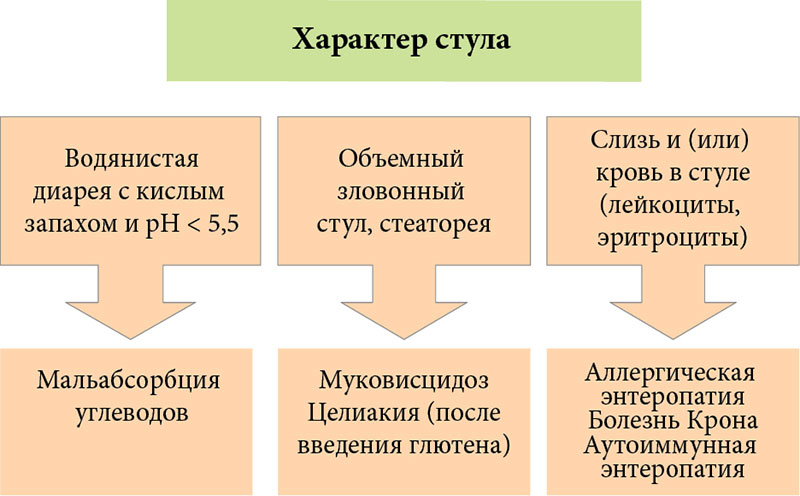
–Ф–∞–ї—М–љ–µ–є—И–Є–є –∞–ї–≥–Њ—А–Є—В–Љ –і–Є–∞–≥–љ–Њ—Б—В–Є–Ї–Є –•–Ф –Њ—Б–љ–Њ–≤—Л–≤–∞–µ—В—Б—П –љ–∞¬†–Њ—Ж–µ–љ–Ї–µ —Е–∞—А–∞–Ї—В–µ—А–∞ —Б—В—Г–ї–∞ (—А–Є—Б.¬†2). –°–ї–µ–і—Г–µ—В —А–∞–Ј–ї–Є—З–∞—В—М –≤–Њ–і—П–љ–Є—Б—В—Г—О –і–Є–∞—А–µ—О, –і–Є–∞—А–µ—О —Б–Њ¬†—Б—В–µ–∞—В–Њ—А–µ–µ–є –Є¬†–і–Є–∞—А–µ—О —Б¬†–Ї–Њ–ї–Є—В–Є—З–µ—Б–Ї–Є–Љ —Б–Є–љ–і—А–Њ–Љ–Њ–Љ (–Ї—А–Њ–≤—М—О –Є¬†—Б–ї–Є–Ј—М—О –≤¬†—Б—В—Г–ї–µ).
–Т–Њ–і—П–љ–Є—Б—В–∞—П –і–Є–∞—А–µ—П
–Я—А–Є —Б–Њ—Е—А–∞–љ—П—О—Й–µ–є—Б—П, –љ–µ—Б–Љ–Њ—В—А—П –љ–∞¬†–Њ—В–Љ–µ–љ—Г –ї–∞–Ї—В–Њ–Ј—Л, –≤–Њ–і—П–љ–Є—Б—В–Њ–є ¬Ђ–Ї–Є—Б–ї–Њ–є¬ї –і–Є–∞—А–µ–µ (—А–Э –Ї–∞–ї–∞¬†<¬†5,5) —Г¬†–љ–Њ–≤–Њ—А–Њ–ґ–і–µ–љ–љ–Њ–≥–Њ —А–µ–±–µ–љ–Ї–∞ –≤–µ—А–Њ—П—В–љ—Л–Љ –і–Є–∞–≥–љ–Њ–Ј–Њ–Љ –Љ–Њ–ґ–µ—В –±—Л—В—М –Љ–∞–ї—М–∞–±—Б–Њ—А–±—Ж–Є—П¬†–≥–ї—О–Ї–Њ–Ј—Л-–≥–∞–ї–∞–Ї—В–Њ–Ј—Л, –і–ї—П –њ–Њ–і—В–≤–µ—А–ґ–і–µ–љ–Є—П –Ї–Њ—В–Њ—А–Њ–≥–Њ –њ—А–Њ–≤–Њ–і–Є—В—Б—П –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ —Н–Ї—Б–Ї—А–µ—Ж–Є–Є —Б–∞—Е–∞—А–Њ–≤ —Б¬†–Ї–∞–ї–Њ–Љ –Є¬†–њ—А–Њ–±—Л –љ–∞¬†—В–Њ–ї–µ—А–∞–љ—В–љ–Њ—Б—В—М –Ї¬†–≥–ї—О–Ї–Њ–Ј–µ –Є¬†—Д—А—Г–Ї—В–Њ–Ј–µ. –Я–Њ—Б–Ї–Њ–ї—М–Ї—Г –њ—А–Є —Н—В–Њ–Љ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–Є –љ–∞—А—Г—И–µ–љ–∞ —А–∞–±–Њ—В–∞¬†–≥–ї—О–Ї–Њ–Ј–Њ-–≥–∞–ї–∞–Ї—В–Њ–Ј–љ–Њ–≥–Њ —В—А–∞–љ—Б–њ–Њ—А—В–µ—А–∞, –≤¬†—В–Њ¬†–≤—А–µ–Љ—П –Ї–∞–Ї —Д—Г–љ–Ї—Ж–Є–Є —Д—А—Г–Ї—В–Њ–Ј–љ–Њ–≥–Њ —В—А–∞–љ—Б–њ–Њ—А—В–µ—А–∞ –≤¬†–°–Ю–Ґ–Ъ —Б–Њ—Е—А–∞–љ—П—О—В—Б—П, –љ–∞–≥—А—Г–Ј–Ї—Г —Н—В–Є–Љ–Є –Љ–Њ–љ–Њ—Б–∞—Е–∞—А–Є–і–∞–Љ–Є –Њ—А–≥–∞–љ–Є–Ј–Љ —А–µ–±–µ–љ–Ї–∞ –≤–Њ—Б–њ—А–Є–љ–Є–Љ–∞–µ—В –њ–Њ-—А–∞–Ј–љ–Њ–Љ—Г. –Я–Њ—Б–ї–µ –њ—А–Є–µ–Љ–∞¬†–≥–ї—О–Ї–Њ–Ј—Л –і–Є–∞—А–µ—П —Г—Б–Є–ї–Є–≤–∞–µ—В—Б—П, –∞¬†–≥–ї–Є–Ї–µ–Љ–Є—З–µ—Б–Ї–∞—П –Ї—А–Є–≤–∞—П –Є–Љ–µ–µ—В –њ–ї–Њ—Б–Ї–Є–є –≤–Є–і. –§—А—Г–Ї—В–Њ–Ј–∞¬†–ґ–µ —П–≤–ї—П–µ—В—Б—П –µ–і–Є–љ—Б—В–≤–µ–љ–љ—Л–Љ —Г–≥–ї–µ–≤–Њ–і–Њ–Љ, –Ї–Њ—В–Њ—А—Л–є —Е–Њ—А–Њ—И–Њ –њ–µ—А–µ–љ–Њ—Б–Є—В—Б—П –і–µ—В—М–Љ–Є —Б¬†–Љ–∞–ї—М–∞–±—Б–Њ—А–±—Ж–Є–µ–є¬†–≥–ї—О–Ї–Њ–Ј—Л-–≥–∞–ї–∞–Ї—В–Њ–Ј—Л, –њ–Њ—Н—В–Њ–Љ—Г –њ—А–Є –њ–Њ–і—В–≤–µ—А–ґ–і–µ–љ–Є–Є –і–Є–∞–≥–љ–Њ–Ј–∞ –≤—Б–Ї–∞—А–Љ–ї–Є–≤–∞–љ–Є–µ —Б–ї–µ–і—Г–µ—В –њ—А–Њ–≤–Њ–і–Є—В—М –∞—А—В–Є—Д–Є—Ж–Є–∞–ї—М–љ–Њ–є —Б–Љ–µ—Б—М—О, –њ—А–Є–≥–Њ—В–Њ–≤–ї–µ–љ–љ–Њ–є –Є–Ј¬†–Ї–∞–Ј–µ–Є–љ–∞—В–∞ –Ї–∞–ї—М—Ж–Є—П, —А–∞—Б—В–Є—В–µ–ї—М–љ–Њ–≥–Њ –Љ–∞—Б–ї–∞ –Є¬†—Д—А—Г–Ї—В–Њ–Ј—Л.
–Т—Б–µ –љ–∞—А—Г—И–µ–љ–Є—П —А–∞—Б—Й–µ–њ–ї–µ–љ–Є—П –Є¬†–≤—Б–∞—Б—Л–≤–∞–љ–Є—П —Г–≥–ї–µ–≤–Њ–і–Њ–≤¬†вАУ –ї–∞–Ї—В–∞–Ј–љ–∞—П, —Б–∞—Е–∞—А–∞–Ј–Њ-–Є–Ј–Њ–Љ–∞–ї—М—В–∞–Ј–љ–∞—П –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В—М, –Љ–∞–ї—М–∞–±—Б–Њ—А–±—Ж–Є—П¬†–≥–ї—О–Ї–Њ–Ј—Л-–≥–∞–ї–∞–Ї—В–Њ–Ј—Л, –Љ–∞–ї—М–∞–±—Б–Њ—А–±—Ж–Є—П —Д—А—Г–Ї—В–Њ–Ј—Л¬†вАУ –њ—А–Њ—П–≤–ї—П—О—В—Б—П –Њ—Б–Љ–Њ—В–Є—З–µ—Б–Ї–Њ–є –і–Є–∞—А–µ–µ–є, –Ї–Њ—В–Њ—А–∞—П —А–∞–Ј–≤–Є–≤–∞–µ—В—Б—П –≤¬†—А–µ–Ј—Г–ї—М—В–∞—В–µ –Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є—П –Є–Ј¬†–љ–µ–≤—Б–Њ—Б–∞–≤—И–Є—Е—Б—П —Г–≥–ї–µ–≤–Њ–і–Њ–≤ –Њ—Б–Љ–Њ—В–Є—З–µ—Б–Ї–Є –∞–Ї—В–Є–≤–љ—Л—Е –њ—А–Њ–і—Г–Ї—В–Њ–≤ –±–∞–Ї—В–µ—А–Є–∞–ї—М–љ–Њ–≥–Њ –±—А–Њ–ґ–µ–љ–Є—П. –Т—А–µ–Љ–µ–љ–љ—Л–є –њ–µ—А–µ–≤–Њ–і —А–µ–±–µ–љ–Ї–∞ –љ–∞¬†–њ–∞—А–µ–љ—В–µ—А–∞–ї—М–љ–Њ–µ –њ–Є—В–∞–љ–Є–µ –Є–ї–Є –Є—Б–Ї–ї—О—З–µ–љ–Є–µ –Є–Ј¬†—А–∞—Ж–Є–Њ–љ–∞ —Г–≥–ї–µ–≤–Њ–і–∞, –≤—Л–Ј–≤–∞–≤—И–µ–≥–Њ –і–Є–∞—А–µ—О, —Б–Њ–њ—А–Њ–≤–Њ–ґ–і–∞—О—В—Б—П –Ї—Г–њ–Є—А–Њ–≤–∞–љ–Є–µ–Љ —Б–Є–Љ–њ—В–Њ–Љ–Њ–≤.
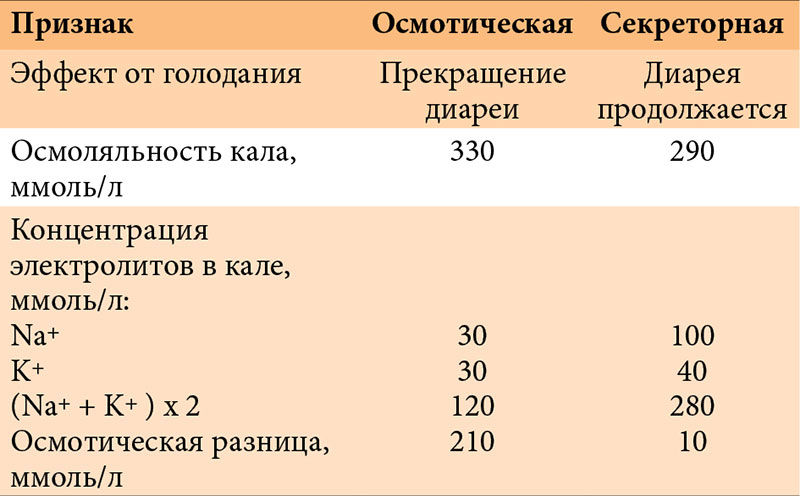
–Ф–Є–∞—А–µ—П, —Б–Њ—Е—А–∞–љ—П—О—Й–∞—П—Б—П –њ–Њ—Б–ї–µ –њ–µ—А–µ–≤–Њ–і–∞ —А–µ–±–µ–љ–Ї–∞ –љ–∞¬†–њ–Њ–ї–љ–Њ–µ –њ–∞—А–µ–љ—В–µ—А–∞–ї—М–љ–Њ–µ –њ–Є—В–∞–љ–Є–µ, —Б–≤–Є–і–µ—В–µ–ї—М—Б—В–≤—Г–µ—В –Њ¬†–µ–µ —Б–µ–Ї—А–µ—В–Њ—А–љ–Њ–Љ —Е–∞—А–∞–Ї—В–µ—А–µ. –Ю—Б–љ–Њ–≤–љ—Л–Љ –Њ—В–ї–Є—З–Є–µ–Љ —Б–µ–Ї—А–µ—В–Њ—А–љ–Њ–є –і–Є–∞—А–µ–Є –Њ—В¬†–Њ—Б–Љ–Њ—В–Є—З–µ—Б–Ї–Њ–є —П–≤–ї—П–µ—В—Б—П –Њ—В—Б—Г—В—Б—В–≤–Є–µ —Н—Д—Д–µ–Ї—В–∞ –Њ—В¬†¬Ђ–≥–Њ–ї–Њ–і–∞–љ–Є—П¬ї, —В–Њ –µ—Б—В—М –Њ—В¬†–Њ—В–Љ–µ–љ—Л –Њ–±—Л—З–љ–Њ–≥–Њ –њ–Є—В–∞–љ–Є—П –Є¬†–њ–µ—А–µ–≤–Њ–і–∞ —А–µ–±–µ–љ–Ї–∞ –љ–∞¬†–њ–∞—А–µ–љ—В–µ—А–∞–ї—М–љ–Њ–µ (—В–∞–±–ї.¬†1). –Ф–ї—П –њ–Њ–і—В–≤–µ—А–ґ–і–µ–љ–Є—П —Б–µ–Ї—А–µ—В–Њ—А–љ–Њ–≥–Њ —Е–∞—А–∞–Ї—В–µ—А–∞ –і–Є–∞—А–µ–Є –љ–µ–Њ–±—Е–Њ–і–Є–Љ–Њ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ —Г—А–Њ–≤–љ—П —Н–ї–µ–Ї—В—А–Њ–ї–Є—В–Њ–≤ –≤¬†–Ї—А–Њ–≤–Є –Є¬†—Н–Ї—Б–Ї—А–µ—Ж–Є–Є –Є—Е —Б¬†–Ї–∞–ї–Њ–Љ. –Я—А–Є —Б–µ–Ї—А–µ—В–Њ—А–љ–Њ–є –і–Є–∞—А–µ–µ –Њ—Б–Љ–Њ–ї—П–ї—М–љ–Њ—Б—В—М –Ї–∞–ї–∞ –Ј–∞–≤–Є—Б–Є—В¬†–≥–ї–∞–≤–љ—Л–Љ –Њ–±—А–∞–Ј–Њ–Љ –Њ—В¬†—Б–Њ–і–µ—А–ґ–∞–љ–Є—П –≤¬†–љ–µ–Љ —Н–ї–µ–Ї—В—А–Њ–ї–Є—В–Њ–≤ –Є¬†–њ—А–µ–і—Б—В–∞–≤–ї—П–µ—В —Б–Њ–±–Њ–є —Б—Г–Љ–Љ—Г –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є–є –Є–Њ–љ–Њ–≤ Na+ –Є¬†K+ (–Є–ї–Є Na+, K+ –Є¬†Cl-), —Г–Љ–љ–Њ–ґ–µ–љ–љ—Г—О –љ–∞¬†–і–≤–∞. –Я—А–Є –Њ—Б–Љ–Њ—В–Є—З–µ—Б–Ї–Њ–є –і–Є–∞—А–µ–µ –Њ—Б–Љ–Њ–ї—П–ї—М–љ–Њ—Б—В—М –Ї–∞–ї–∞ –Њ–±—Г—Б–ї–Њ–≤–ї–µ–љ–∞ –љ–µ—А–∞—Б—Й–µ–њ–ї–µ–љ–љ—Л–Љ–Є –Є–ї–Є –љ–µ–≤—Б–Њ—Б–∞–≤—И–Є–Љ–Є—Б—П —З–∞—Б—В–Є—З–Ї–∞–Љ–Є –њ–Є—Й–Є, –њ—А–Є —Н—В–Њ–Љ –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є—П —Н–ї–µ–Ї—В—А–Њ–ї–Є—В–Њ–≤ –≤¬†–љ–µ–Љ –Њ—Б—В–∞–µ—В—Б—П –љ–Є–Ј–Ї–Њ–є, —З—В–Њ –Њ–±—К—П—Б–љ—П–µ—В –±–Њ–ї—М—И—Г—О –Њ—Б–Љ–Њ—В–Є—З–µ—Б–Ї—Г—О —А–∞–Ј–љ–Є—Ж—Г, –њ–Њ—Н—В–Њ–Љ—Г —Б—Г–Љ–Љ–∞ —Н–ї–µ–Ї—В—А–Њ–ї–Є—В–Њ–≤ –Ї–∞–ї–∞ (Na++K++Cl-) –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ –Љ–µ–љ—М—И–µ –Њ—Б–Љ–Њ–ї—П–ї—М–љ–Њ—Б—В–Є. –°–µ–Ї—А–µ—В–Њ—А–љ–∞—П –і–Є–∞—А–µ—П —З–∞—Й–µ –љ–∞–±–ї—О–і–∞–µ—В—Б—П –њ—А–Є –Њ—Б—В—А—Л—Е (—В–∞–Ї –љ–∞–Ј—Л–≤–∞–µ–Љ—Л—Е –≤–Њ–і—П–љ–Є—Б—В—Л—Е) –і–Є–∞—А–µ—П—Е, –Њ–і–љ–∞–Ї–Њ –і–ї–Є—В–µ–ї—М–љ–Њ–µ —Г–њ–Њ—А–љ–Њ–µ –µ–µ —В–µ—З–µ–љ–Є–µ –Љ–Њ–ґ–µ—В –љ–∞–±–ї—О–і–∞—В—М—Б—П –њ—А–Є —В–∞–Ї–Є—Е —А–µ–і–Ї–Є—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П—Е, –Ї–∞–Ї –≤—А–Њ–ґ–і–µ–љ–љ–∞—П —Е–ї–Њ—А–Є–і–љ–∞—П –Є–ї–Є –љ–∞—В—А–Є–µ–≤–∞—П –і–Є–∞—А–µ—П, –∞¬†—В–∞–Ї–ґ–µ –њ—А–Є –Љ–∞–ї—М–∞–±—Б–Њ—А–±—Ж–Є–Є –ґ–µ–ї—З–љ—Л—Е –Ї–Є—Б–ї–Њ—В. –Ф–ї—П —Е–ї–Њ—А–Є–і–љ–Њ–є –і–Є–∞—А–µ–Є —Е–∞—А–∞–Ї—В–µ—А–љ–Њ –њ—А–µ–Њ–±–ї–∞–і–∞–љ–Є–µ –≤¬†–Ї–∞–ї–µ –Є–Њ–љ–Њ–≤ —Е–ї–Њ—А–∞ –љ–∞–і –і—А—Г–≥–Є–Љ–Є —Н–ї–µ–Ї—В—А–Њ–ї–Є—В–∞–Љ–Є, –і–ї—П –љ–∞—В—А–Є–µ–≤–Њ–є –і–Є–∞—А–µ–Є¬†вАУ –Є–Њ–љ–Њ–≤ –љ–∞—В—А–Є—П. –Я—А–Є –њ–Њ–і—В–≤–µ—А–ґ–і–µ–љ–Є–Є —Б–µ–ї–µ–Ї—В–Є–≤–љ–Њ–є –њ–Њ—В–µ—А–Є —Б–Њ–Њ—В–≤–µ—В—Б—В–≤—Г—О—Й–µ–≥–Њ —Н–ї–µ–Ї—В—А–Њ–ї–Є—В–∞ –њ–Њ–Ї–∞–Ј–∞–љ–∞ –Ј–∞–Љ–µ—Б—В–Є—В–µ–ї—М–љ–∞—П —В–µ—А–∞–њ–Є—П –≤¬†–≤–Є–і–µ —Б–Њ–ї–µ–≤—Л—Е —А–∞—Б—В–≤–Њ—А–Њ–≤, —Б–љ–∞—З–∞–ї–∞ –≤–љ—Г—В—А–Є–≤–µ–љ–љ–Њ, –Ј–∞—В–µ–Љ –њ–µ—А–Њ—А–∞–ї—М–љ–Њ.
–Э–∞–±–ї—О–і–∞–µ–Љ–∞—П —Б¬†—А–Њ–ґ–і–µ–љ–Є—П —В—П–ґ–µ–ї–∞—П —Б–Љ–µ—И–∞–љ–љ–∞—П –і–Є–∞—А–µ—П —Б¬†—Б–µ–Ї—А–µ—В–Њ—А–љ—Л–Љ –Ї–Њ–Љ–њ–Њ–љ–µ–љ—В–Њ–Љ, –њ–Њ—В–µ—А–µ–є –±–Њ–ї—М—И–Њ–≥–Њ –Ї–Њ–ї–Є—З–µ—Б—В–≤–∞ –ґ–Є–і–Ї–Њ—Б—В–Є (–±–Њ–ї–µ–µ 300¬†–Љ–ї –≤¬†—Б—Г—В–Ї–Є) –≤¬†—Б–Њ—З–µ—В–∞–љ–Є–Є —Б¬†–њ–Њ—В–µ—А–µ–є –њ–ї–∞–Ј–Љ–µ–љ–љ–Њ–≥–Њ –±–µ–ї–Ї–∞ —Е–∞—А–∞–Ї—В–µ—А–љ–∞ –і–ї—П –≤—А–Њ–ґ–і–µ–љ–љ—Л—Е –∞–љ–Њ–Љ–∞–ї–Є–є —Н–љ—В–µ—А–Њ—Ж–Є—В–Њ–≤ (—В–∞–±–ї.¬†2). –≠—В–Њ–Љ—Г —В–Є–њ—Г –і–Є–∞—А–µ–Є —Б–≤–Њ–є—Б—В–≤–µ–љ–љ–Њ –Њ—В—Б—Г—В—Б—В–≤–Є–µ —Н—Д—Д–µ–Ї—В–∞ –Њ—В¬†–њ–µ—А–µ–≤–Њ–і–∞ —А–µ–±–µ–љ–Ї–∞ –љ–∞¬†–њ–∞—А–µ–љ—В–µ—А–∞–ї—М–љ–Њ–µ –њ–Є—В–∞–љ–Є–µ –Є¬†—Г—Б–Є–ї–µ–љ–Є–µ –њ–Њ–љ–Њ—Б–∞ –њ–Њ—Б–ї–µ –µ–і—Л. –Ю—Б–Љ–Њ—В–Є—З–µ—Б–Ї–∞—П —А–∞–Ј–љ–Є—Ж–∞ –≤¬†–Ї–∞–ї–µ –Њ—В—Б—Г—В—Б—В–≤—Г–µ—В, –Ї–∞–Ї –Є¬†–њ—А–Є —Б–µ–Ї—А–µ—В–Њ—А–љ–Њ–є –і–Є–∞—А–µ–µ. –Ґ–∞–Ї–∞—П –і–Є–∞—А–µ—П —П–≤–ї—П–µ—В—Б—П –њ–Њ–Ї–∞–Ј–∞–љ–Є–µ–Љ –Ї¬†–њ—А–Њ–≤–µ–і–µ–љ–Є—О –±–Є–Њ–њ—Б–Є–Є –°–Ю–Ґ–Ъ –Є¬†—Н–ї–µ–Ї—В—А–Њ–љ–љ–Њ-–Љ–Є–Ї—А–Њ—Б–Ї–Њ–њ–Є—З–µ—Б–Ї–Њ–≥–Њ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П –±–Є–Њ–њ—В–∞—В–Њ–≤. –Ю–±–љ–∞—А—Г–ґ–µ–љ–Є–µ —Д–µ–љ–Њ–Љ–µ–љ–∞ ¬Ђ–≤–Ї–ї—О—З–µ–љ–љ—Л—Е –Љ–Є–Ї—А–Њ–≤–Њ—А—Б–Є–љ¬ї –Є–ї–Є ¬Ђ–њ—Г—З–Ї–Њ–≤–Њ–є –і–Є—Б–њ–ї–∞–Ј–Є–Є¬ї —Н–љ—В–µ—А–Њ—Ж–Є—В–Њ–≤ –њ–Њ–і—В–≤–µ—А–ґ–і–∞–µ—В —Д–∞–Ї—В –≤—А–Њ–ґ–і–µ–љ–љ–Њ–є –∞–љ–Њ–Љ–∞–ї–Є–Є –Є—Е —Б—В—А–Њ–µ–љ–Є—П. –Х–і–Є–љ—Б—В–≤–µ–љ–љ—Л–Љ —Б–њ–Њ—Б–Њ–±–Њ–Љ –ї–µ—З–µ–љ–Є—П —Н—В–Є—Е –≤—А–Њ–ґ–і–µ–љ–љ—Л—Е –∞–љ–Њ–Љ–∞–ї–Є–є —П–≤–ї—П–µ—В—Б—П –њ–µ—А–µ—Б–∞–і–Ї–∞ —В–Њ–љ–Ї–Њ–є –Ї–Є—И–Ї–Є.
–Ф–Є–∞—А–µ—П —Б–Њ¬†—Б—В–µ–∞—В–Њ—А–µ–µ–є
–°—В–µ–∞—В–Њ—А–µ—П —Б–Њ–њ—А–Њ–≤–Њ–ґ–і–∞–µ—В—Б—П –љ–µ¬†—Б—В–Њ–ї—М–Ї–Њ —Г—З–∞—Й–µ–љ–љ—Л–Љ, —Б–Ї–Њ–ї—М–Ї–Њ –ґ–Є—А–љ—Л–Љ –љ–∞¬†–≤–Є–і, –Њ–±—К–µ–Љ–љ—Л–Љ –Є¬†–Ј–ї–Њ–≤–Њ–љ–љ—Л–Љ —Б—В—Г–ї–Њ–Љ, –Ї–Њ—В–Њ—А—Л–є –Њ–±—Л—З–љ–Њ –Є–Љ–µ–µ—В –Ї–∞—И–Є—Ж–µ–Њ–±—А–∞–Ј–љ—Л–є —Е–∞—А–∞–Ї—В–µ—А, –њ–ї–Њ—Е–Њ –Њ—В–Љ—Л–≤–∞–µ—В—Б—П –Њ—В¬†–≥–Њ—А—И–Ї–∞. –Я—А–Є –Ї–Њ–њ—А–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–Љ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–Є –≤–∞–ґ–љ–Њ —А–∞–Ј–ї–Є—З–∞—В—М –і–≤–∞ —В–Є–њ–∞ —Б—В–µ–∞—В–Њ—А–µ–Є: —Б—В–µ–∞—В–Њ—А–µ—О –љ–µ–є—В—А–∞–ї—М–љ—Л–Љ –ґ–Є—А–Њ–Љ, –Ї–Њ—В–Њ—А–∞—П —П–≤–ї—П–µ—В—Б—П —Б–Є–Љ–њ—В–Њ–Љ–Њ–Љ –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В–Є –њ–Њ–і–ґ–µ–ї—Г–і–Њ—З–љ–Њ–є –ґ–µ–ї–µ–Ј—Л, –Є¬†—Б—В–µ–∞—В–Њ—А–µ—О –ґ–Є—А–љ—Л–Љ–Є –Ї–Є—Б–ї–Њ—В–∞–Љ–Є, –Ї–Њ—В–Њ—А–∞—П —Е–∞—А–∞–Ї—В–µ—А–љ–∞ –і–ї—П –њ–∞—В–Њ–ї–Њ–≥–Є–Є —В–Њ–љ–Ї–Њ–є –Ї–Є—И–Ї–Є.
–°—В–µ–∞—В–Њ—А–µ—П –њ–∞–љ–Ї—А–µ–∞—В–Є—З–µ—Б–Ї–Њ–≥–Њ —В–Є–њ–∞ (–љ–µ–є—В—А–∞–ї—М–љ—Л–Љ –ґ–Є—А–Њ–Љ) –≤–Њ–Ј–Љ–Њ–ґ–љ–∞ –њ—А–Є –Љ—Г–Ї–Њ–≤–Є—Б—Ж–Є–і–Њ–Ј–µ, —Б–Є–љ–і—А–Њ–Љ–µ –®–≤–∞—Е–Љ–∞–љ–∞-–Ф–∞–є–Љ–Њ–љ–і–∞, —Б–Є–љ–і—А–Њ–Љ–µ –Я–Є—А—Б–Њ–љ–∞, –Є–Ј–Њ–ї–Є—А–Њ–≤–∞–љ–љ–Њ–Љ –і–µ—Д–Є—Ж–Є—В–µ –ї–Є–њ–∞–Ј—Л (—Б–Є–љ–і—А–Њ–Љ–µ –®–µ–ї–і–Њ–љ–∞-–†–µ—П). –Я—А–Є –µ–µ –Њ–±–љ–∞—А—Г–ґ–µ–љ–Є–Є –≤¬†—Б–µ—А–Є–Є –Ї–Њ–њ—А–Њ–≥—А–∞–Љ–Љ –њ–Њ–Ї–∞–Ј–∞–љ–∞ –±–Њ–ї–µ–µ —В–Њ—З–љ–∞—П –Њ—Ж–µ–љ–Ї–∞ —Н–Ї–Ј–Њ–Ї—А–Є–љ–љ–Њ–є —Д—Г–љ–Ї—Ж–Є–Є –њ–Њ–і–ґ–µ–ї—Г–і–Њ—З–љ–Њ–є –ґ–µ–ї–µ–Ј—Л —Б¬†–њ–Њ–Љ–Њ—Й—М—О —В–µ—Б—В–∞ –љ–∞¬†—Н–ї–∞—Б—В–∞–Ј—Г-1¬†–≤¬†–Ї–∞–ї–µ. –°–љ–Є–ґ–µ–љ–Є–µ —Г—А–Њ–≤–љ—П —Н–ї–∞—Б—В–∞–Ј—Л-1¬†–љ–Є–ґ–µ 200¬†–Љ–≥/–≥¬†–њ–Њ–і—В–≤–µ—А–ґ–і–∞–µ—В –≤–µ—А–Њ—П—В–љ–Њ—Б—В—М –њ–∞–љ–Ї—А–µ–∞—В–Є—З–µ—Б–Ї–Њ–є –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В–Є. –Я–Њ—Б–ї–µ–і—Г—О—Й–Є–є –і–Є–∞–≥–љ–Њ—Б—В–Є—З–µ—Б–Ї–Є–є –∞–ї–≥–Њ—А–Є—В–Љ –љ–∞–њ—А–∞–≤–ї–µ–љ –љ–∞¬†–≤—Л—П–≤–ї–µ–љ–Є–µ –Ї–Њ–љ–Ї—А–µ—В–љ–Њ–≥–Њ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П –њ–Њ–і–ґ–µ–ї—Г–і–Њ—З–љ–Њ–є –ґ–µ–ї–µ–Ј—Л. –Ф–Є–∞–≥–љ–Њ–Ј –Љ—Г–Ї–Њ–≤–Є—Б—Ж–Є–і–Њ–Ј–∞ –њ–Њ–і—В–≤–µ—А–ґ–і–∞–µ—В—Б—П –љ–∞¬†–Њ—Б–љ–Њ–≤–∞–љ–Є–Є –њ–Њ–≤—Л—И–µ–љ–Є—П —Г—А–Њ–≤–љ—П —Е–ї–Њ—А–Є–і–Њ–≤ –≤¬†–њ–Њ—В–µ –Є¬†–Њ–±–љ–∞—А—Г–ґ–µ–љ–Є—П –Љ—Г—В–∞—Ж–Є–є CFTR-–≥–µ–љ–∞.
–Ф–ї—П —Б–Є–љ–і—А–Њ–Љ–∞ –®–≤–∞—Е–Љ–∞–љ–∞-–Ф–∞–є–Љ–Њ–љ–і–∞ —Е–∞—А–∞–Ї—В–µ—А–љ–Њ —Б–Њ—З–µ—В–∞–љ–Є–µ –њ–∞–љ–Ї—А–µ–∞—В–Є—З–µ—Б–Ї–Њ–є –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В–Є –Є¬†–≥–µ–Љ–∞—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є—Е –љ–∞—А—Г—И–µ–љ–Є–є (–љ–µ–є—В—А–Њ–њ–µ–љ–Є–Є, —А–µ–ґ–µ —В—А–Њ–Љ–±–Њ—Ж–Є—В–Њ–њ–µ–љ–Є–Є –Є¬†–∞–љ–µ–Љ–Є–Є), –∞¬†—В–∞–Ї–ґ–µ –Њ—В—Б—В–∞–≤–∞–љ–Є–µ –≤¬†—А–Њ—Б—В–µ –Є¬†–љ–∞–ї–Є—З–Є–µ –љ–µ–Ї–Њ—В–Њ—А—Л—Е –Ї–Њ—Б—В–љ—Л—Е –∞–љ–Њ–Љ–∞–ї–Є–є (–і–Є—Б—Е–Њ–љ–і—А–Њ–њ–ї–∞–Ј–Є–Є —В–∞–Ј–Њ–±–µ–і—А–µ–љ–љ—Л—Е —Б—Г—Б—В–∞–≤–Њ–≤, –љ–µ—Б—А–∞—Й–µ–љ–Є–µ —А–µ–±–µ—А, –Ї–ї–Є–љ–Њ–≤–Є–і–љ—Л–µ –њ–∞–ї—М—Ж—Л –Є¬†—В.–і.).
–°–Є–љ–і—А–Њ–Љ –Я–Є—А—Б–Њ–љ–∞ —В–Њ–ґ–µ –њ—А–Њ—П–≤–ї—П–µ—В—Б—П¬†–≥–µ–Љ–∞—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–Љ–Є –љ–∞—А—Г—И–µ–љ–Є—П–Љ–Є, –љ–Њ¬†–Њ–љ–Є, –Ї–∞–Ї –њ—А–∞–≤–Є–ї–Њ, –±–Њ–ї–µ–µ —В—П–ґ–µ–ї—Л–µ, —Е–∞—А–∞–Ї—В–µ—А–љ–Њ —Г–њ–Њ—А–љ–Њ–µ —В–µ—З–µ–љ–Є–µ –∞–љ–µ–Љ–Є–Є –Є¬†—В—А–Њ–Љ–±–Њ—Ж–Є—В–Њ–њ–µ–љ–Є–Є. –Ъ–Њ—Б—В–љ—Л–µ –∞–љ–Њ–Љ–∞–ї–Є–Є –њ—А–Є —Б–Є–љ–і—А–Њ–Љ–µ –Я–Є—А—Б–Њ–љ–∞ –Њ—В—Б—Г—В—Б—В–≤—Г—О—В.
–°–Є–љ–і—А–Њ–Љ –®–µ–ї–і–Њ–љ–∞-–†–µ—П —Е–∞—А–∞–Ї—В–µ—А–Є–Ј—Г–µ—В—Б—П –Њ—В—Б—Г—В—Б—В–≤–Є–µ–Љ –ї–Є–њ–∞–Ј—Л –≤¬†–Ї—А–Њ–≤–Є –Є¬†–Љ–Њ—З–µ, –≤—Л—А–∞–ґ–µ–љ–љ–Њ–є —Б—В–µ–∞—В–Њ—А–µ–є; –љ–∞—А—Г—И–µ–љ–Є–є —А–Њ—Б—В–∞ –љ–µ—В.
–Ю—Б–љ–Њ–≤–Њ–є –ї–µ—З–µ–љ–Є—П –≤—Б–µ—Е —Н—В–Є—Е¬†–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Є –і–µ—В–µ—А–Љ–Є–љ–Є—А–Њ–≤–∞–љ–љ—Л—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є —П–≤–ї—П–µ—В—Б—П –Ј–∞–Љ–µ—Б—В–Є—В–µ–ї—М–љ–∞—П —В–µ—А–∞–њ–Є—П –њ–∞–љ–Ї—А–µ–∞—В–Є—З–µ—Б–Ї–Є–Љ–Є —Д–µ—А–Љ–µ–љ—В–∞–Љ–Є. –Ф–Њ–Ј–∞ —Д–µ—А–Љ–µ–љ—В–Њ–≤ –њ–Њ–і–±–Є—А–∞–µ—В—Б—П –Є–љ–і–Є–≤–Є–і—Г–∞–ї—М–љ–Њ, –Њ–±—Л—З–љ–Њ –љ–∞—З–Є–љ–∞—О—В —Б¬†2 000¬†–Х–Ф/–Ї–≥ –ї–Є–њ–∞–Ј—Л –≤¬†–і–µ–љ—М, –њ—А–Є –љ–µ–Њ–±—Е–Њ–і–Є–Љ–Њ—Б—В–Є –і–Њ–Ј—Г –њ–Њ–≤—Л—И–∞—О—В. –Я—А–µ–і–њ–Њ—З—В–Є—В–µ–ї—М–љ—Л –њ—А–µ–њ–∞—А–∞—В—Л —Б¬†–Љ–Є–Ї—А–Њ—Б—Д–µ—А–Є—З–µ—Б–Ї–Є–Љ–Є —Д–µ—А–Љ–µ–љ—В–∞–Љ–Є (–Ъ—А–µ–Њ–љ, –Я–∞–љ—Ж–Є—В—А–∞—В), –Ї–Њ—В–Њ—А—Л–µ —Б–ї–µ–і—Г–µ—В –і–∞–≤–∞—В—М —А–µ–±–µ–љ–Ї—Г –≤¬†–љ–∞—З–∞–ї–µ –Ї–∞–ґ–і–Њ–≥–Њ –њ—А–Є–µ–Љ–∞ –њ–Є—Й–Є –Є¬†—А–∞—Б–њ—А–µ–і–µ–ї—П—В—М –≤¬†—Б–Њ–Њ—В–≤–µ—В—Б—В–≤–Є–Є —Б¬†–µ–µ –Ї–Њ–ї–Є—З–µ—Б—В–≤–Њ–Љ. –Я—А–Є —В—П–ґ–µ–ї–Њ–є¬†–≥–Є–њ–Њ—В—А–Њ—Д–Є–Є –≤¬†–њ–Є—В–∞–љ–Є–Є –±–Њ–ї—М–љ—Л—Е –Є—Б–њ–Њ–ї—М–Ј—Г—О—В —Б–Љ–µ—Б–Є —Б–Њ¬†—Б—А–µ–і–љ–µ—Ж–µ–њ–Њ—З–µ—З–љ—Л–Љ–Є —В—А–Є–≥–ї–Є—Ж–µ—А–Є–і–∞–Љ–Є (–°–¶–Ґ): –Р–ї—М—Д–∞—А–µ, –Э—Г—В—А–Є–ї–Њ–љ –Я–µ–њ—В–Є –Ґ–°–¶, –Я—А–µ–≥–µ—Б—В–Є–Љ–Є–ї, –Я–µ–њ—В–∞–Љ–µ–љ, –Я–Њ—А—В–∞–≥–µ–љ. –Ф–Њ–њ–Њ–ї–љ–Є—В–µ–ї—М–љ–Њ –љ–∞–Ј–љ–∞—З–∞—О—В –ґ–Є—А–Њ—А–∞—Б—В–≤–Њ—А–Є–Љ—Л–µ –≤–Є—В–∞–Љ–Є–љ—Л: –Р, –Х, –Ф, –Ъ.
–°—В–µ–∞—В–Њ—А–µ—П –Ї–Є—И–µ—З–љ–Њ–≥–Њ —В–Є–њ–∞ (–ґ–Є—А–љ—Л–Љ–Є –Ї–Є—Б–ї–Њ—В–∞–Љ–Є) –љ–µ¬†—П–≤–ї—П–µ—В—Б—П —Б–њ–µ—Ж–Є—Д–Є—З–µ—Б–Ї–Є–Љ –њ—А–Є–Ј–љ–∞–Ї–Њ–Љ –Њ–њ—А–µ–і–µ–ї–µ–љ–љ—Л—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є, –љ–Њ¬†–і–Њ–≤–Њ–ї—М–љ–Њ —З–∞—Б—В–Њ –Њ—В–Љ–µ—З–∞–µ—В—Б—П –њ—А–Є –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П—Е —В–Њ–љ–Ї–Њ–є –Ї–Є—И–Ї–Є. –Ф–ї—П —Г—В–Њ—З–љ–µ–љ–Є—П —Е–∞—А–∞–Ї—В–µ—А–∞ –Є–љ—В–µ—Б—В–Є–љ–∞–ї—М–љ–Њ–є –њ–∞—В–Њ–ї–Њ–≥–Є–Є –љ–µ–Њ–±—Е–Њ–і–Є–Љ–Њ –Љ–Њ—А—Д–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ –±–Є–Њ–њ—В–∞—В–Њ–≤ –°–Ю–Ґ–Ъ, –≤–Ј—П—В—Л—Е –≤¬†—Е–Њ–і–µ —Н–љ–і–Њ—Б–Ї–Њ–њ–Є—З–µ—Б–Ї–Њ–≥–Њ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П –Є–Ј¬†–і–Є—Б—В–∞–ї—М–љ–Њ–≥–Њ –Њ—В–і–µ–ї–∞ –і–≤–µ–љ–∞–і—Ж–∞—В–Є–њ–µ—А—Б—В–љ–Њ–є –Є–ї–Є –њ—А–Њ–Ї—Б–Є–Љ–∞–ї—М–љ–Њ–≥–Њ –Њ—В–і–µ–ї–∞ —В–Њ—Й–µ–є –Ї–Є—И–Ї–Є.
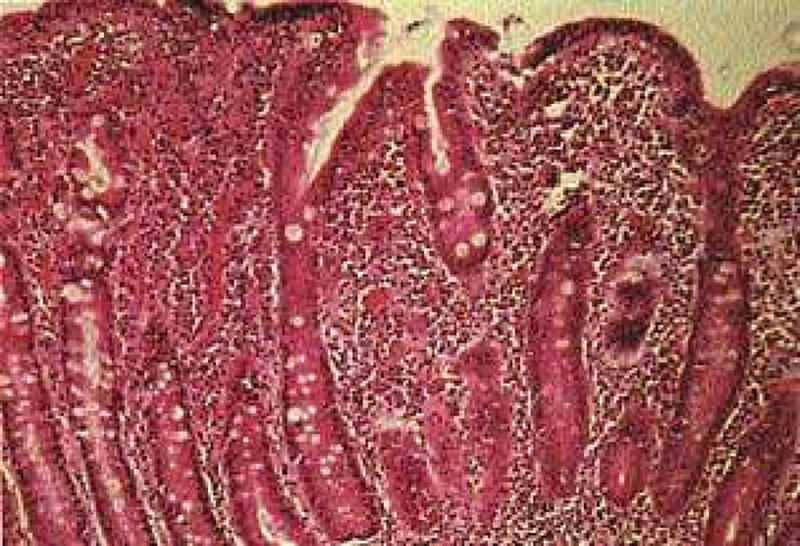
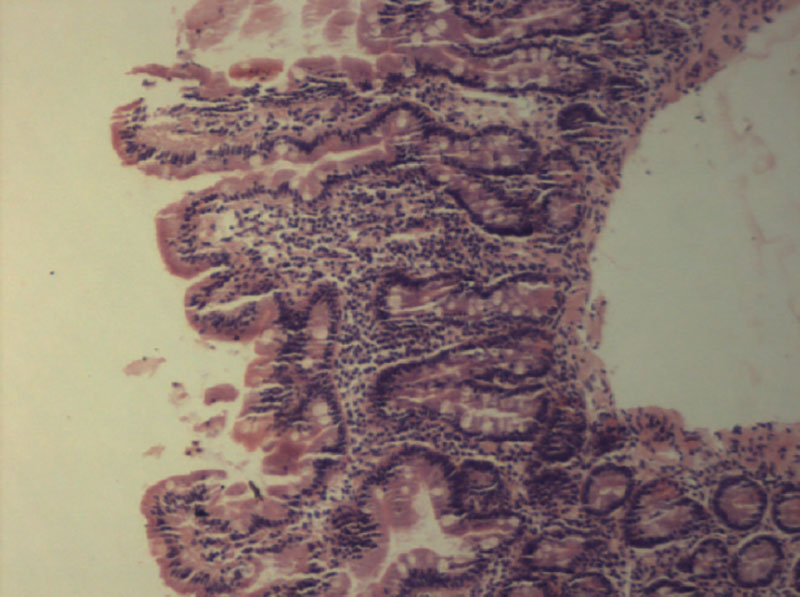
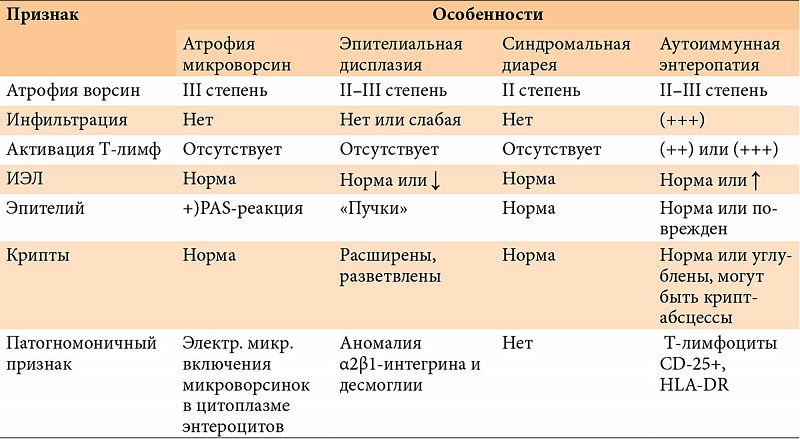
–Э–∞–ї–Є—З–Є–µ –ї–Є–Љ—Д–Њ–њ–ї–∞–Ј–Љ–Њ—Ж–Є—В–∞—А–љ–Њ–є –Є–љ—Д–Є–ї—М—В—А–∞—Ж–Є–Є —Б–Њ–±—Б—В–≤–µ–љ–љ–Њ–є –њ–ї–∞—Б—В–Є–љ–Ї–Є –°–Ю–Ґ–Ъ —Б–≤–Є–і–µ—В–µ–ї—М—Б—В–≤—Г–µ—В –Њ¬†—Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–Љ –≤–Њ—Б–њ–∞–ї–µ–љ–Є–Є, –Ї–Њ—В–Њ—А–Њ–µ –≤¬†–±–Њ–ї—М—И–Є–љ—Б—В–≤–µ —Б–ї—Г—З–∞–µ–≤ —Б–Њ–њ—А–Њ–≤–Њ–ґ–і–∞–µ—В—Б—П –∞—В—А–Њ—Д–Є–µ–є —Б–ї–Є–Ј–Є—Б—В–Њ–є, –њ—А–Њ—П–≤–ї—П—О—Й–µ–є—Б—П —Г–Ї–Њ—А–Њ—З–µ–љ–Є–µ–Љ –≤–Њ—А—Б–Є–љ –Є¬†–Є—Б—В–Њ–љ—З–µ–љ–Є–µ–Љ —Н–њ–Є—В–µ–ї–Є—П. –Ю—Ж–µ–љ–Ї–∞ —Б—В–µ–њ–µ–љ–Є –∞—В—А–Њ—Д–Є–Є –°–Ю–Ґ–Ъ –њ—А–Њ–≤–Њ–і–Є—В—Б—П –≤¬†—Б–Њ–Њ—В–≤–µ—В—Б—В–≤–Є–Є —Б¬†–Ї–ї–∞—Б—Б–Є—Д–Є–Ї–∞—Ж–Є–µ–є Marsh, —А–∞–Ј–ї–Є—З–∞—О—В 3¬†—Б—В–µ–њ–µ–љ–Є. –Т¬†—Б–ї—Г—З–∞–µ –Њ–±–љ–∞—А—Г–ґ–µ–љ–Є—П –∞—В—А–Њ—Д–Є–Є 2вАС–є¬†–Є–ї–Є 3вАС–є¬†—Б—В–µ–њ–µ–љ–Є (—Г–Ї–Њ—А–Њ—З–µ–љ–Є–µ –≤–Њ—А—Б–Є–љ, —Г–≥–ї—Г–±–ї–µ–љ–Є–µ –Ї—А–Є–њ—В,¬†–≥—Г—Б—В–∞—П —А–∞–≤–љ–Њ–Љ–µ—А–љ–∞—П –ї–Є–Љ—Д–Њ–њ–ї–∞–Ј–Љ–Њ—Ж–Є—В–∞—А–љ–∞—П –Є–љ—Д–Є–ї—М—В—А–∞—Ж–Є—П —Б–Њ–±—Б—В–≤–µ–љ–љ–Њ–є –њ–ї–∞—Б—В–Є–љ–Ї–Є, —Г–≤–µ–ї–Є—З–µ–љ–Є–µ –Љ–µ–ґ—Н–њ–Є—В–µ–ї–Є–∞–ї—М–љ—Л—Е –ї–Є–Љ—Д–Њ—Ж–Є—В–Њ–≤¬†вАУ –Ь–≠–Ы) (—А–Є—Б.¬†3) —Г¬†—А–µ–±–µ–љ–Ї–∞, –Ї–Њ—В–Њ—А—Л–є —Г–ґ–µ –њ–Њ–ї—Г—З–∞–ї¬†–≥–ї—О—В–µ–љ, –љ–µ–Њ–±—Е–Њ–і–Є–Љ–Њ –њ—А–Њ–≤–µ—Б—В–Є –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ —Г—А–Њ–≤–љ—П –∞–љ—В–Є–≥–ї–Є–∞–і–Є–љ–Њ–≤—Л—Е –∞–љ—В–Є—В–µ–ї (IgA, IgG), –∞–љ—В–Є—В–µ–ї –Ї¬†—В–Ї–∞–љ–µ–≤–Њ–є —В—А–∞–љ—Б–≥–ї—О—В–∞–Љ–Є–љ–∞–Ј–µ (IgA, IgG) –Є¬†—Н–љ–і–Њ–Љ–Є–Ј–Є—О. –Я—А–Є –њ–Њ–≤—Л—И–µ–љ–љ–Њ–Љ —Г—А–Њ–≤–љ–µ —Н—В–Є—Е –Є–Љ–Љ—Г–љ–Њ–≥–ї–Њ–±—Г–ї–Є–љ–Њ–≤ —Г—Б—В–∞–љ–∞–≤–ї–Є–≤–∞–µ—В—Б—П –і–Є–∞–≥–љ–Њ–Ј —Ж–µ–ї–Є–∞–Ї–Є–Є, —А–µ–±–µ–љ–Ї—Г –љ–∞–Ј–љ–∞—З–∞–µ—В—Б—П –њ–Њ–ґ–Є–Ј–љ–µ–љ–љ–∞—П —Б—В—А–Њ–≥–∞—П –±–µ–Ј–≥–ї—О—В–µ–љ–Њ–≤–∞—П –і–Є–µ—В–∞. –Ъ–ї–∞—Б—Б–Є—З–µ—Б–Ї–Є–Љ–Є –њ—А–Є–Ј–љ–∞–Ї–∞–Љ–Є —Ж–µ–ї–Є–∞–Ї–Є–Є –≤¬†—А–∞–љ–љ–µ–Љ –≤–Њ–Ј—А–∞—Б—В–µ —П–≤–ї—П—О—В—Б—П –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–∞—П –њ—А–Є–±–∞–≤–Ї–∞ –≤–µ—Б–∞ —Б¬†—А–∞–Ј–≤–Є—В–Є–µ–Љ¬†–≥–Є–њ–Њ—В—А–Њ—Д–Є–Є –Є¬†—Г–≤–µ–ї–Є—З–µ–љ–Є–µ –ґ–Є–≤–Њ—В–∞.
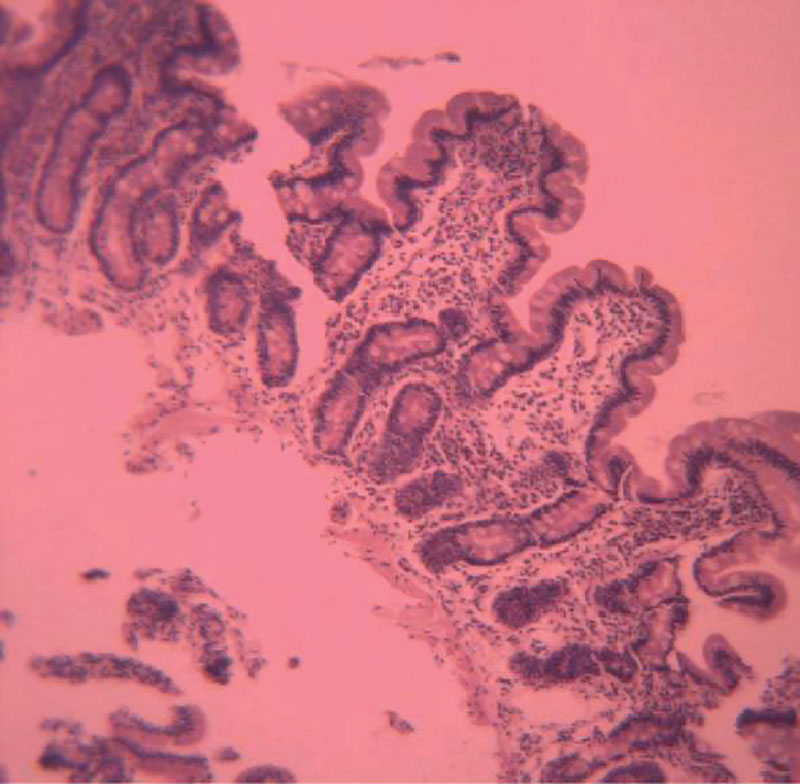
–°—Е–Њ–і–љ—Л–є —Б¬†—Ж–µ–ї–Є–∞–Ї–Є–µ–є —Н–љ—В–µ—А–∞–ї—М–љ—Л–є —Б–Є–љ–і—А–Њ–Љ –љ–µ—А–µ–і–Ї–Њ –љ–∞–±–ї—О–і–∞–µ—В—Б—П —Г¬†–і–µ—В–µ–є —Б¬†–њ–Є—Й–µ–≤–Њ–є –∞–ї–ї–µ—А–≥–Є–µ–є. –°–Є–Љ–њ—В–Њ–Љ—Л –∞–ї–ї–µ—А–≥–Є—З–µ—Б–Ї–Њ–є —Н–љ—В–µ—А–Њ–њ–∞—В–Є–Є –Љ–Њ–≥—Г—В –њ—А–Њ—П–≤–ї—П—В—М—Б—П —Г–ґ–µ –≤¬†–њ–µ—А–≤—Л–µ –Љ–µ—Б—П—Ж—Л –ґ–Є–Ј–љ–Є –≤¬†–≤–Є–і–µ –±–µ—Б–њ–Њ–Ї–Њ–є—Б—В–≤–∞, –Ї–Є—И–µ—З–љ–Њ–є –Ї–Њ–ї–Є–Ї–Є, —Б—А—Л–≥–Є–≤–∞–љ–Є–є, –њ–Њ–Ј–і–љ–µ–µ –Љ–Њ–ґ–µ—В –њ—А–Є—Б–Њ–µ–і–Є–љ—П—В—М—Б—П –ґ–µ–ї–µ–Ј–Њ–і–µ—Д–Є—Ж–Є—В–љ–∞—П –∞–љ–µ–Љ–Є—П. –Ю–і–љ–∞–Ї–Њ –≤¬†—А—П–і–µ —Б–ї—Г—З–∞–µ–≤ —Г¬†–і–µ—В–µ–є –і–Њ¬†2¬†–ї–µ—В –Љ–Њ–≥—Г—В –і–Њ–Љ–Є–љ–Є—А–Њ–≤–∞—В—М —Б–Є–Љ–њ—В–Њ–Љ—Л –∞–ї–ї–µ—А–≥–Є—З–µ—Б–Ї–Њ–≥–Њ –Ї–Њ–ї–Є—В–∞ —Б¬†–њ–Њ—П–≤–ї–µ–љ–Є–µ–Љ –Ї—А–Њ–≤–Є –Є¬†—Б–ї–Є–Ј–Є –≤¬†—Б—В—Г–ї–µ. –Ъ–Є—И–µ—З–љ—Л–µ —Б–Є–Љ–њ—В–Њ–Љ—Л —Г¬†–±–Њ–ї—М—И–Є–љ—Б—В–≤–∞ –і–µ—В–µ–є —Б–Њ—З–µ—В–∞—О—В—Б—П —Б¬†–Ї–∞—А—В–Є–љ–Њ–є –∞—В–Њ–њ–Є—З–µ—Б–Ї–Њ–≥–Њ –і–µ—А–Љ–∞—В–Є—В–∞. –Ь–Њ—А—Д–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–∞—П –Ї–∞—А—В–Є–љ–∞ –∞–ї–ї–µ—А–≥–Є—З–µ—Б–Ї–Њ–є —Н–љ—В–µ—А–Њ–њ–∞—В–Є–Є —Е–∞—А–∞–Ї—В–µ—А–Є–Ј—Г–µ—В—Б—П –њ—А–Є–Ј–љ–∞–Ї–∞–Љ–Є —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–≥–Њ –≤–Њ—Б–њ–∞–ї–µ–љ–Є—П –°–Ю–Ґ–Ъ —Б¬†–љ–Њ—А–Љ–∞–ї—М–љ–Њ–є –≤—Л—Б–Њ—В–Њ–є (–Є–ї–Є –љ–µ–Ј–љ–∞—З–Є—В–µ–ї—М–љ—Л–Љ —Г–Ї–Њ—А–Њ—З–µ–љ–Є–µ–Љ) –Є¬†–≥–ї—Г–±–Є–љ–Њ–є –≤–Њ—А—Б–Є–љ –Є¬†–љ–∞–ї–Є—З–Є–µ–Љ —Н–Њ–Ј–Є–љ–Њ—Д–Є–ї–Њ–≤ –≤¬†–Є–љ—Д–Є–ї—М—В—А–∞—В–µ (—А–Є—Б.¬†4). –Ф–Є–∞–≥–љ–Њ–Ј –Љ–Њ–ґ–µ—В –њ–Њ–і—В–≤–µ—А–ґ–і–∞—В—М—Б—П –њ–Њ–≤—Л—И–µ–љ–Є–µ–Љ –≤¬†–Ї—А–Њ–≤–Є –Њ–±—Й–µ–≥–Њ –Є¬†—Б–њ–µ—Ж–Є—Д–Є—З–µ—Б–Ї–Є—Е¬†IgE, –љ–Њ¬†–њ–Њ—Б–Ї–Њ–ї—М–Ї—Г¬†–≥–∞—Б—В—А–Њ–Є–љ—В–µ—Б—В–Є–љ–∞–ї—М–љ–∞—П –∞–ї–ї–µ—А–≥–Є—П —З–∞—Й–µ —А–µ–∞–ї–Є–Ј—Г–µ—В—Б—П —З–µ—А–µ–Ј –Ї–ї–µ—В–Њ—З–љ—Л–µ –Љ–µ—Е–∞–љ–Є–Ј–Љ—Л —Б–µ–љ—Б–Є–±–Є–ї–Є–Ј–∞—Ж–Є–Є, –љ–Њ—А–Љ–∞–ї—М–љ—Л–є —Г—А–Њ–≤–µ–љ—М¬†IgE —Г¬†—А–µ–±–µ–љ–Ї–∞ –≤–Њ–≤—Б–µ –љ–µ¬†–Є—Б–Ї–ї—О—З–∞–µ—В –≤–µ—А–Њ—П—В–љ–Њ—Б—В—М –∞–ї–ї–µ—А–≥–Є—З–µ—Б–Ї–Њ–≥–Њ –њ–Њ—А–∞–ґ–µ–љ–Є—П –Ї–Є—И–µ—З–љ–Є–Ї–∞. –°—Е–Њ–і–љ–∞—П –Љ–Њ—А—Д–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–∞—П –Ї–∞—А—В–Є–љ–∞ —Б–≤–Њ–є—Б—В–≤–µ–љ–љ–∞ —В–∞–Ї–ґ–µ –ї—П–Љ–±–ї–Є–Њ–Ј—Г. –Р–ї–≥–Њ—А–Є—В–Љ –і–Є–∞–≥–љ–Њ—Б—В–Є–Ї–Є –љ–∞¬†–Њ—Б–љ–Њ–≤–∞–љ–Є–Є –Љ–Њ—А—Д–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є—Е –њ—А–Є–Ј–љ–∞–Ї–Њ–≤ –њ—А–µ–і—Б—В–∞–≤–ї–µ–љ –љ–∞¬†—А–Є—Б.¬†5.
–°–Њ—З–µ—В–∞–љ–Є–µ —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–є –і–Є–∞—А–µ–Є —Б¬†–њ–Њ—А–∞–ґ–µ–љ–Є—П–Љ–Є –Ї–Њ–ґ–Є —Б–≤–Њ–є—Б—В–≤–µ–љ–љ–Њ —В–∞–Ї–ґ–µ —Н–љ—В–µ—А–Њ–њ–∞—В–Є—З–µ—Б–Ї–Њ–Љ—Г –∞–Ї—А–Њ–і–µ—А–Љ–∞—В–Є—В—Г, –≤—Л–Ј–≤–∞–љ–љ–Њ–Љ—Г –і–µ—Д–Є—Ж–Є—В–Њ–Љ —Ж–Є–љ–Ї–∞. –Я–Њ—Б–Ї–Њ–ї—М–Ї—Г —Ж–Є–љ–Ї –≤—Е–Њ–і–Є—В –≤¬†—Б–Њ—Б—В–∞–≤ –±–Њ–ї–µ–µ 40¬†–Љ–µ—В–∞–ї–ї–Њ—Б–Њ–і–µ—А–ґ–∞—Й–Є—Е —Д–µ—А–Љ–µ–љ—В–Њ–≤ –Є¬†—Г—З–∞—Б—В–≤—Г–µ—В –≤¬†–Њ–±–µ—Б–њ–µ—З–µ–љ–Є–Є –Є–Љ–Љ—Г–љ–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–є –Ј–∞—Й–Є—В—Л, –і–Є–∞—А–µ—П –њ—А–Є —Н—В–Њ–Љ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–Є –Љ–Њ–ґ–µ—В –њ–Њ–і–і–µ—А–ґ–Є–≤–∞—В—М—Б—П —Н–љ–і–Њ–≥–µ–љ–љ–Њ–є –Љ–Є–Ї—А–Њ—Д–ї–Њ—А–Њ–є, –≤¬†—З–∞—Б—В–љ–Њ—Б—В–Є –љ–µ—А–µ–і–Ї–Њ —А–∞–Ј–≤–Є–≤–∞–µ—В—Б—П –≤—В–Њ—А–Є—З–љ—Л–є –Ї–∞–љ–і–Є–і–Њ–Ј. –Ь–Њ—А—Д–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П –°–Ю–Ґ–Ъ —Е–∞—А–∞–Ї—В–µ—А–Є–Ј—Г—О—В—Б—П —Б–Њ—З–µ—В–∞–љ–Є–µ–Љ –≤–Њ—Б–њ–∞–ї–µ–љ–Є—П –Є¬†—Г–Љ–µ—А–µ–љ–љ–Њ–є –∞—В—А–Њ—Д–Є–Є –љ–∞¬†—Д–Њ–љ–µ —Б–љ–Є–ґ–µ–љ–љ–Њ–≥–Њ –Ї–Њ–ї–Є—З–µ—Б—В–≤–∞ –Ь–≠–Ы, –њ–ї–∞–Ј–Љ–Њ—Ж–Є—В–Њ–≤ –≤¬†—Б–Њ–±—Б—В–≤–µ–љ–љ–Њ–є –њ–ї–∞—Б—В–Є–љ–Ї–µ –Є¬†—Б–љ–Є–ґ–µ–љ–љ–Њ–є —А–µ–≥–µ–љ–µ—А–∞—В–Њ—А–љ–Њ–є —Б–њ–Њ—Б–Њ–±–љ–Њ—Б—В—М—О —Н–њ–Є—В–µ–ї–Є—П. –Ъ–Њ–ґ–љ—Л–µ –њ—А–Њ—П–≤–ї–µ–љ–Є—П –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П —Е–∞—А–∞–Ї—В–µ—А–Є–Ј—Г—О—В—Б—П —З–µ—В–Ї–Њ –Њ—В–≥—А–∞–љ–Є—З–µ–љ–љ—Л–Љ–Є –Њ—В¬†–љ–Њ—А–Љ–∞–ї—М–љ–Њ–є –Ї–Њ–ґ–Є –Ј–Њ–љ–∞–Љ–Є¬†–≥–Є–њ–µ—А–µ–Љ–Є–Є, –Є–љ—Д–Є–ї—М—В—А–∞—Ж–Є–Є –Є¬†—Н–Ї—Б–Ї–Њ—А–Є–∞—Ж–Є–Є, –ї–Њ–Ї–∞–ї–Є–Ј—Г—О—Й–Є–Љ–Є—Б—П –Њ–±—Л—З–љ–Њ –љ–∞¬†—П–≥–Њ–і–Є—Ж–∞—Е, –≤–Њ–Ї—А—Г–≥ —А—В–∞, –љ–∞¬†–њ–Њ–і–±–Њ—А–Њ–і–Ї–µ –Є¬†—Й–µ–Ї–∞—Е. –≠–љ—В–µ—А–Њ–њ–∞—В–Є—З–µ—Б–Ї–Є–є –∞–Ї—А–Њ–і–µ—А–Љ–∞—В–Є—В –њ—А–Њ—П–≤–ї—П–µ—В—Б—П –њ–Њ—Б–ї–µ –Њ—В–Љ–µ–љ—Л¬†–≥—А—Г–і–љ–Њ–≥–Њ –≤—Б–Ї–∞—А–Љ–ї–Є–≤–∞–љ–Є—П, –њ–Њ—Б–Ї–Њ–ї—М–Ї—Г –ґ–µ–љ—Б–Ї–Њ–µ –Љ–Њ–ї–Њ–Ї–Њ —Б–Њ–і–µ—А–ґ–Є—В —Ж–Є–љ–Ї-–≤—Б–∞—Б—Л–≤–∞—О—Й–Є–є –ї–Є–≥–∞–љ–і, –њ—А–µ–њ—П—В—Б—В–≤—Г—О—Й–Є–є —А–∞–Ј–≤–Є—В–Є—О –і–µ—Д–Є—Ж–Є—В–∞ —Н—В–Њ–≥–Њ –Љ–Є–Ї—А–Њ—Н–ї–µ–Љ–µ–љ—В–∞. –Ш–љ–Њ–≥–і–∞ –±–Њ–ї–µ–Ј–љ—М –љ–∞—З–Є–љ–∞–µ—В—Б—П –љ–∞¬†—Д–Њ–љ–µ –Њ—Б—В—А–Њ–є –Ї–Є—И–µ—З–љ–Њ–є –Є–љ—Д–µ–Ї—Ж–Є–Є, —З—В–Њ —Б–≤—П–Ј–∞–љ–Њ —Б¬†–Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ–є —Н–љ—В–µ—А–∞–ї—М–љ–Њ–є –њ–Њ—В–µ—А–µ–є —Ж–Є–љ–Ї–∞. –Ґ–∞–Ї–Є–Љ –Њ–±—А–∞–Ј–Њ–Љ, —Д–Њ—А–Љ–Є—А—Г–µ—В—Б—П –њ–Њ—А–Њ—З–љ—Л–є –Ї—А—Г–≥: –Њ—Б—В—А–∞—П –і–Є–∞—А–µ—П –≤—Л–Ј—Л–≤–∞–µ—В –і–µ—Д–Є—Ж–Є—В —Ж–Є–љ–Ї–∞, –Ї–Њ—В–Њ—А—Л–є –≤¬†—Б–≤–Њ—О –Њ—З–µ—А–µ–і—М –њ–Њ–і–і–µ—А–ґ–Є–≤–∞–µ—В –Є¬†—Г—Б—Г–≥—Г–±–ї—П–µ—В –і–Є–∞—А–µ—О, —Б–њ–Њ—Б–Њ–±—Б—В–≤—Г—П –µ–µ –њ–µ—А–µ—Е–Њ–і—Г –≤¬†—Е—А–Њ–љ–Є—З–µ—Б–Ї—Г—О. –£—А–Њ–≤–µ–љ—М —Ж–Є–љ–Ї–∞ –≤¬†–Ї—А–Њ–≤–Є –Њ–±—Л—З–љ–Њ –Њ–њ—Г—Б–Ї–∞–µ—В—Б—П –љ–Є–ґ–µ 8¬†–Љ–Љ–Њ–ї—М/–ї. –Я–µ—А–Њ—А–∞–ї—М–љ–Њ–µ –љ–∞–Ј–љ–∞—З–µ–љ–Є–µ –њ—А–µ–њ–∞—А–∞—В–Њ–≤ —Ж–Є–љ–Ї–∞ –≤¬†–≤—Л—Б–Њ–Ї–Є—Е –і–Њ–Ј–∞—Е (—Б—Г–ї—М—Д–∞—В —Ж–Є–љ–Ї–∞ 50вАУ100¬†–Љ–≥/—Б—Г—В) –±—Л—Б—В—А–Њ –Ї—Г–њ–Є—А—Г–µ—В –і–Є–∞—А–µ—О –Є¬†–Ї–Њ–ґ–љ—Л–µ —Б–Є–Љ–њ—В–Њ–Љ—Л.
–Ф–Є–∞—А–µ—П —Б¬†–Ї—А–Њ–≤—М—О –Є¬†—Б–ї–Є–Ј—М—О –≤¬†—Б—В—Г–ї–µ
–Я–Њ—П–≤–ї–µ–љ–Є–µ –Ї—А–Њ–≤–Є –Є¬†—Б–ї–Є–Ј–Є –≤¬†—Б—В—Г–ї–µ —Б–≤–Њ–є—Б—В–≤–µ–љ–љ–Њ –Ї–Њ–ї–Є—В–Є—З–µ—Б–Ї–Њ–Љ—Г —Б–Є–љ–і—А–Њ–Љ—Г. –Я–Њ—Б–ї–µ –Є—Б–Ї–ї—О—З–µ–љ–Є—П –Є–љ—Д–µ–Ї—Ж–Є–є (–і–Є–Ј–µ–љ—В–µ—А–Є–Є, —Б–∞–ї—М–Љ–Њ–љ–µ–ї–ї–µ–Ј–∞, –Є–µ—А—Б–Є–љ–Є–Њ–Ј–∞, –Ї–∞–Љ–њ–Є–ї–Њ–±–∞–Ї—В–µ—А–Є–Њ–Ј–∞) —Г—Б—В–∞–љ–Њ–≤–ї–µ–љ–Є–µ –њ—А–Є—З–Є–љ –і–Є–∞—А–µ–Є –њ—А–µ–і–њ–Њ–ї–∞–≥–∞–µ—В –њ—А–µ–ґ–і–µ –≤—Б–µ–≥–Њ –Љ–Њ—А—Д–Њ–ї–Њ–≥–Є—З–µ—Б–Ї—Г—О –Є¬†–Є–Љ–Љ—Г–љ–Њ–ї–Њ–≥–Є—З–µ—Б–Ї—Г—О –≤–µ—А–Є—Д–Є–Ї–∞—Ж–Є—О. –Т¬†–ї—О–±–Њ–Љ —Б–ї—Г—З–∞–µ –і–Є–∞—А–µ—П —Б–Њ¬†—Б–ї–Є–Ј—М—О –Є¬†–Ї—А–Њ–≤—М—О –≤¬†—Б—В—Г–ї–µ —Г–Ї–∞–Ј—Л–≤–∞–µ—В –љ–∞¬†–≤—Л—Б–Њ–Ї—Г—О –∞–Ї—В–Є–≤–љ–Њ—Б—В—М –≤–Њ—Б–њ–∞–ї–µ–љ–Є—П –≤¬†–Ї–Є—И–µ—З–љ–Є–Ї–µ –Є¬†–љ–∞–ї–Є—З–Є–µ –і–µ—Д–µ–Ї—В–Њ–≤ —Б–ї–Є–Ј–Є—Б—В–Њ–є –Њ–±–Њ–ї–Њ—З–Ї–Є. –Ъ–Њ—Б–≤–µ–љ–љ—Л–Љ –і–Њ–Ї–∞–Ј–∞—В–µ–ї—М—Б—В–≤–Њ–Љ –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є –≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ–Њ–≥–Њ –њ—А–Њ—Ж–µ—Б—Б–∞ –Љ–Њ–ґ–µ—В —Б–ї—Г–ґ–Є—В—М –Њ—Ж–µ–љ–Ї–∞ —Г—А–Њ–≤–љ—П –Ї–∞–ї—М–њ—А–Њ—В–µ–Ї—В–Є–љ–∞ –≤¬†–Ї–∞–ї–µ, —Г—А–Њ–≤–µ–љ—М –Ї–Њ—В–Њ—А–Њ–≥–Њ –≤¬†–љ–Њ—А–Љ–µ –љ–µ¬†–њ—А–µ–≤—Л—И–∞–µ—В 50, –∞¬†–њ—А–Є –≤–Њ—Б–њ–∞–ї–µ–љ–Є–Є –≤¬†–Ї–Є—И–µ—З–љ–Є–Ї–µ –≤–Њ–Ј—А–∞—Б—В–∞–µ—В –≤¬†–љ–µ—Б–Ї–Њ–ї—М–Ї–Њ —А–∞–Ј.
–Ф–ї—П –Љ–Њ—А—Д–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–є –≤–µ—А–Є—Д–Є–Ї–∞—Ж–Є–Є –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П –њ–∞—Ж–Є–µ–љ—В—Г —Б¬†—Е—А–Њ–љ–Є—З–µ—Б–Ї–Є–Љ –Ї–Њ–ї–Є—В–Є—З–µ—Б–Ї–Є–Љ —Б–Є–љ–і—А–Њ–Љ–Њ–Љ –њ–Њ–Ї–∞–Ј–∞–љ–Њ –њ—А–Њ–≤–µ–і–µ–љ–Є–µ –Ї–Њ–ї–Њ–љ–Њ—Б–Ї–Њ–њ–Є–Є —Б¬†–Є–ї–µ–Њ—Б–Ї–Њ–њ–Є–µ–є. –≠–љ–і–Њ—Б–Ї–Њ–њ–Є—З–µ—Б–Ї–Њ–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ –Љ–Њ–ґ–µ—В –≤—Л—П–≤–Є—В—М —Б–њ–µ—Ж–Є—Д–Є—З–µ—Б–Ї–Є–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П, —Б–≤–Њ–є—Б—В–≤–µ–љ–љ—Л–µ –±–Њ–ї–µ–Ј–љ–Є –Ъ—А–Њ–љ–∞ –Є–ї–Є —П–Ј–≤–µ–љ–љ–Њ–Љ—Г –Ї–Њ–ї–Є—В—Г.
–°–Њ—З–µ—В–∞–љ–Є–µ —В—П–ґ–µ–ї–Њ–є —Н–љ—В–µ—А–Њ–њ–∞—В–Є–Є —Б¬†–Ї–Њ–ї–Є—В–Є—З–µ—Б–Ї–Є–Љ —Б–Є–љ–і—А–Њ–Љ–Њ–Љ —Б–≤–Њ–є—Б—В–≤–µ–љ–љ–Њ –∞—Г—В–Њ–Є–Љ–Љ—Г–љ–љ–Њ–є —Н–љ—В–µ—А–Њ–њ–∞—В–Є–Є. –Ч–∞–±–Њ–ї–µ–≤–∞–љ–Є–µ —З–∞—Й–µ —А–∞–Ј–≤–Є–≤–∞–µ—В—Б—П —Г¬†–і–µ—В–µ–є –њ–µ—А–≤—Л—Е –і–≤—Г—Е –ї–µ—В (–Є–љ–Њ–≥–і–∞ –њ–µ—А–≤—Л—Е –Љ–µ—Б—П—Ж–µ–≤) –ґ–Є–Ј–љ–Є, –Ї–Є—И–µ—З–љ—Л–µ —Б–Є–Љ–њ—В–Њ–Љ—Л —Б–Њ—З–µ—В–∞—О—В—Б—П —Б¬†–њ—А–Є–Ј–љ–∞–Ї–∞–Љ–Є –њ–Њ—А–∞–ґ–µ–љ–Є—П –і—А—Г–≥–Є—Е –Њ—А–≥–∞–љ–Њ–≤ (–њ–Њ—З–µ–Ї, –њ–µ—З–µ–љ–Є, –ї–µ–≥–Ї–Є—Е, –Ї–Њ–ґ–Є, —Б—Г—Б—В–∞–≤–Њ–≤ –Є¬†—В.–і.). –Р—Г—В–Њ–Є–Љ–Љ—Г–љ–љ–∞—П —Н–љ—В–µ—А–Њ–њ–∞—В–Є—П —Г¬†–Љ–∞–ї—М—З–Є–Ї–Њ–≤ –Љ–Њ–ґ–µ—В —Б–Њ—З–µ—В–∞—В—М—Б—П —Б¬†–њ–Њ–ї–Є—Н–љ–і–Њ–Ї—А–Є–љ–Њ–њ–∞—В–Є–µ–є (IPEX) –Є–ї–Є –±—Л—В—М –њ—А–Њ—П–≤–ї–µ–љ–Є–µ–Љ —В—П–ґ–µ–ї—Л—Е –Є–Љ–Љ—Г–љ–Њ–і–µ—Д–Є—Ж–Є—В–љ—Л—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є.
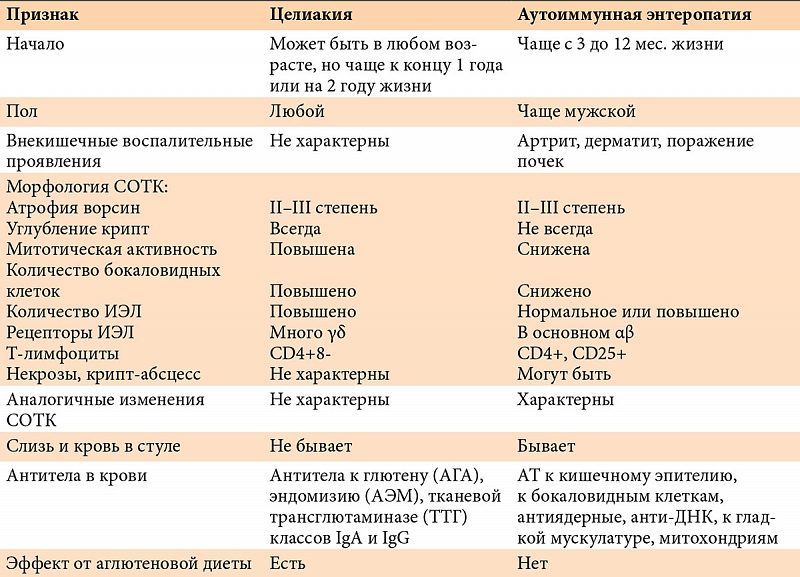
–Ь–Њ—А—Д–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П –°–Ю–Ґ–Ъ –њ—А–Є –∞—Г—В–Њ–Є–Љ–Љ—Г–љ–љ–Њ–є —Н–љ—В–µ—А–Њ–њ–∞—В–Є–Є (—А–Є—Б.¬†6) —Е–∞—А–∞–Ї—В–µ—А–Є–Ј—Г—О—В—Б—П —Г–Љ–µ—А–µ–љ–љ–Њ–є –Є–ї–Є –≤—Л—А–∞–ґ–µ–љ–љ–Њ–є –∞—В—А–Њ—Д–Є–µ–є –≤–Њ—А—Б–Є–љ, –Ї–Њ—В–Њ—А–∞—П –≤—Б–µ–≥–і–∞ –∞—Б—Б–Њ—Ж–Є–Є—А–Њ–≤–∞–љ–∞ —Б¬†–≤—Л—А–∞–ґ–µ–љ–љ–Њ–є –Љ–Њ–љ–Њ–љ—Г–Ї–ї–µ–∞—А–љ–Њ–є –Є–љ—Д–Є–ї—М—В—А–∞—Ж–Є–µ–є —Б–Њ–±—Б—В–≤–µ–љ–љ–Њ–є –њ–ї–∞—Б—В–Є–љ–Ї–Є. –У–Є–њ–µ—А–њ–ї–∞–Ј–Є—П –Ї—А–Є–њ—В –Љ–Њ–ґ–µ—В –±—Л—В—М –≤–∞—А–Є–∞–±–µ–ї—М–љ–∞. –Т¬†–±–Њ–ї—М—И–Є–љ—Б—В–≤–µ —Б–ї—Г—З–∞–µ–≤ —В—П–ґ–µ–ї–∞—П –Є–ї–Є —В–Њ—В–∞–ї—М–љ–∞—П –∞—В—А–Њ—Д–Є—П –≤–Њ—А—Б–Є–љ —Б–Њ—З–µ—В–∞–µ—В—Б—П —Б¬†—Г–≥–ї—Г–±–ї–µ–љ–Є–µ–Љ –Ї—А–Є–њ—В. –Т—Л—А–∞–ґ–µ–љ–љ–∞—П –∞—В—А–Њ—Д–Є—П –≤¬†—Б–Њ—З–µ—В–∞–љ–Є–Є —Б¬†–Љ–Њ–љ–Њ–љ—Г–Ї–ї–µ–∞—А–љ–Њ–є –Є–љ—Д–Є–ї—М—В—А–∞—Ж–Є–µ–є –Љ–Њ–≥—Г—В —А–∞—Б—Ж–µ–љ–Є–≤–∞—В—М—Б—П –њ–µ—А–≤–Њ–љ–∞—З–∞–ї—М–љ–Њ –Ї–∞–Ї –њ—А–Є–Ј–љ–∞–Ї–Є —Ж–µ–ї–Є–∞–Ї–Є–Є (—В–∞–±–ї.¬†3), –Њ–і–љ–∞–Ї–Њ –і–ї—П –љ–µ–µ —Е–∞—А–∞–Ї—В–µ—А–љ–Њ –њ–Њ–≤—Л—И–µ–љ–Є–µ –Љ–Є—В–Њ—В–Є—З–µ—Б–Ї–Њ–є –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є —Н–њ–Є—В–µ–ї–Є—П –Є¬†—Г–≤–µ–ї–Є—З–µ–љ–Є–µ –Ї–Њ–ї–Є—З–µ—Б—В–≤–∞ –Ь–≠–Ы, –≤¬†—В–Њ¬†–≤—А–µ–Љ—П –Ї–∞–Ї –њ—А–Є –∞—Г—В–Њ–Є–Љ–Љ—Г–љ–љ–Њ–є —Н–љ—В–µ—А–Њ–њ–∞—В–Є–Є –Љ–Є—В–Њ—В–Є—З–µ—Б–Ї–∞—П –∞–Ї—В–Є–≤–љ–Њ—Б—В—М —Б–љ–Є–ґ–µ–љ–∞, –∞¬†–Є–љ—Д–Є–ї—М—В—А–∞—В –ї–Њ–Ї–∞–ї–Є–Ј—Г–µ—В—Б—П –≤¬†—Б–Њ–±—Б—В–≤–µ–љ–љ–Њ–є –њ–ї–∞—Б—В–Є–љ–Ї–µ –±–µ–Ј —Г–≤–µ–ї–Є—З–µ–љ–Є—П –Є–ї–Є —Б¬†–љ–µ–±–Њ–ї—М—И–Є–Љ —Г–≤–µ–ї–Є—З–µ–љ–Є–µ–Љ –Ї–Њ–ї–Є—З–µ—Б—В–≤–∞ –Ь–≠–Ы. –Ь–≠–Ы –Є–Љ–µ—О—В –њ—А–Є —Н—В–Њ–Љ –≤¬†–Њ—Б–љ–Њ–≤–љ–Њ–Љ ќ±ќ≤-—А–µ—Ж–µ–њ—В–Њ—А—Л, –≤¬†—В–Њ¬†–≤—А–µ–Љ—П –Ї–∞–Ї –њ—А–Є —Ж–µ–ї–Є–∞–Ї–Є–Є –≤–Њ–Ј—А–∞—Б—В–∞–µ—В —З–Є—Б–ї–Њ –Ь–≠–Ы, –љ–µ—Б—Г—Й–Є—Е ќ≥ќі-—А–µ—Ж–µ–њ—В–Њ—А—Л. –£¬†–љ–µ–Ї–Њ—В–Њ—А—Л—Е –±–Њ–ї—М–љ—Л—Е —В–Њ—В–∞–ї—М–љ–∞—П –∞—В—А–Њ—Д–Є—П –≤–Њ—А—Б–Є–љ –∞—Б—Б–Њ—Ж–Є–Є—А–Њ–≤–∞–љ–∞ —Б¬†–љ–µ–Ї—А–Њ–Ј–∞–Љ–Є —Н–њ–Є—В–µ–ї–Є–∞–ї—М–љ—Л—Е –Ї–ї–µ—В–Њ–Ї –Є¬†—Д–Њ—А–Љ–Є—А–Њ–≤–∞–љ–Є–µ–Љ –Ї—А–Є–њ—В-–∞–±—Б—Ж–µ—Б—Б–Њ–≤. –Я–Њ–≤–µ—А—Е–љ–Њ—Б—В–љ—Л–є —Н–њ–Є—В–µ–ї–Є–є —Г–њ–ї–Њ—Й–µ–љ, –і–Є—Б—В—А–Њ—Д–Є—А–Њ–≤–∞–љ. –І–Є—Б–ї–Њ –±–Њ–Ї–∞–ї–Њ–≤–Є–і–љ—Л—Е –Ї–ї–µ—В–Њ–Ї —Б–љ–Є–ґ–µ–љ–Њ. –Ь–Њ–љ–Њ–љ—Г–Ї–ї–µ–∞—А—Л –њ—А–µ–і—Б—В–∞–≤–ї–µ–љ—Л –≤¬†–Њ—Б–љ–Њ–≤–љ–Њ–Љ CD4+ T-–ї–Є–Љ—Д–Њ—Ж–Є—В–∞–Љ–Є –Є¬†–Љ–∞–Ї—А–Њ—Д–∞–≥–∞–Љ–Є. –Ь–Њ—А—Д–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П –љ–∞–Є–±–Њ–ї–µ–µ –≤—Л—А–∞–ґ–µ–љ—Л –≤¬†—Б–ї–Є–Ј–Є—Б—В–Њ–є –Њ–±–Њ–ї–Њ—З–Ї–µ —В–Њ–љ–Ї–Њ–є –Ї–Є—И–Ї–Є, –љ–Њ¬†–Љ–Њ–≥—Г—В –Њ–±–љ–∞—А—Г–ґ–Є–≤–∞—В—М—Б—П –Є¬†–≤¬†–і—А—Г–≥–Є—Е –Њ—В–і–µ–ї–∞—Е –њ–Є—Й–µ–≤–∞—А–Є—В–µ–ї—М–љ–Њ–≥–Њ —В—А–∞–Ї—В–∞¬†вАУ –ґ–µ–ї—Г–і–Ї–µ –Є–ї–Є —В–Њ–ї—Б—В–Њ–є –Ї–Є—И–Ї–µ. –Ю–±—И–Є—А–љ–Њ–µ –њ–Њ—А–∞–ґ–µ–љ–Є–µ –Ц–Ъ–Ґ –Є–Љ–µ–µ—В –љ–∞–Є–±–Њ–ї–µ–µ —В—П–ґ–µ–ї—Л–µ –њ—А–Њ—П–≤–ї–µ–љ–Є—П –Є¬†–њ–ї–Њ—Е–Њ–є –њ—А–Њ–≥–љ–Њ–Ј.
–Т¬†–Ї—А–Њ–≤–Є –Њ–±–љ–∞—А—Г–ґ–Є–≤–∞—О—В –∞—Г—В–Њ–∞–љ—В–Є—В–µ–ї–∞ (–њ—А–µ–Є–Љ—Г—Й–µ—Б—В–≤–µ–љ–љ–Њ –Ї–ї–∞—Б—Б–∞¬†IgG) –Ї¬†–Ї–Є—И–µ—З–љ–Њ–Љ—Г —Н–њ–Є—В–µ–ї–Є—О, –љ–∞–њ—А–∞–≤–ї–µ–љ–љ—Л–µ –њ—А–Њ—В–Є–≤ –Ї–Њ–Љ–њ–Њ–љ–µ–љ—В–Њ–≤ —Й–µ—В–Њ—З–љ–Њ–є –Ї–∞–є–Љ—Л –Є–ї–Є —Ж–Є—В–Њ–њ–ї–∞–Ј–Љ—Л —Н–љ—В–µ—А–Њ—Ж–Є—В–Њ–≤ –љ–Њ—А–Љ–∞–ї—М–љ–Њ–є –Є–љ—В–µ—Б—В–Є–љ–∞–ї—М–љ–Њ–є —Б–ї–Є–Ј–Є—Б—В–Њ–є. –Ь–Њ–≥—Г—В –њ—А–Є—Б—Г—В—Б—В–≤–Њ–≤–∞—В—М –∞–љ—В–Є—В–µ–ї–∞ –Ї¬†–±–Њ–Ї–∞–ї–Њ–≤–Є–і–љ—Л–Љ –Ї–ї–µ—В–Ї–∞–Љ. –Э–µ—Б–њ–µ—Ж–Є—Д–Є—З–µ—Б–Ї–Є–µ –∞—Г—В–Њ–∞–љ—В–Є—В–µ–ї–∞ –Љ–Њ–≥—Г—В –±—Л—В—М —В–∞–Ї–ґ–µ –љ–∞–њ—А–∞–≤–ї–µ–љ—Л –њ—А–Њ—В–Є–≤ —П–і–µ—А, –Ф–Э–Ъ,¬†–≥–ї–∞–і–Ї–Њ–є –Љ—Г—Б–Ї—Г–ї–∞—В—Г—А—Л –Є–ї–Є –Љ–Є—В–Њ—Е–Њ–љ–і—А–Є–є, —З—В–Њ –Њ–±—Л—З–љ–Њ —Б–Њ—З–µ—В–∞–µ—В—Б—П —Б¬†–≤–љ–µ–Ї–Є—И–µ—З–љ—Л–Љ–Є –њ—А–Њ—П–≤–ї–µ–љ–Є—П–Љ–Є. –Т¬†—Б–ї—Г—З–∞—П—Е –∞—Б—Б–Њ—Ж–Є–∞—Ж–Є–Є –∞—Г—В–Њ–Є–Љ–Љ—Г–љ–љ–Њ–є —Н–љ—В–µ—А–Њ–њ–∞—В–Є–Є —Б¬†–њ–Њ—А–∞–ґ–µ–љ–Є–µ–Љ –њ–Њ—З–µ–Ї –Њ–њ–Є—Б–∞–љ–∞ —Ж–Є—А–Ї—Г–ї—П—Ж–Є—П –∞—Г—В–Њ–∞–љ—В–Є—В–µ–ї –њ—А–Њ—В–Є–≤ –њ–Њ—З–µ—З–љ–Њ–є —В–Ї–∞–љ–Є, –≤¬†—З–∞—Б—В–љ–Њ—Б—В–Є –њ—А–Њ—В–Є–≤ 75¬†kDa-–∞–љ—В–Є–≥–µ–љ–∞. –Я–Њ—Б–ї–µ —Г—Б—В–∞–љ–Њ–≤–ї–µ–љ–Є—П –і–Є–∞–≥–љ–Њ–Ј–∞ –∞—Г—В–Њ–Є–Љ–Љ—Г–љ–љ–Њ–є —Н–љ—В–µ—А–Њ–њ–∞—В–Є–Є –±–Њ–ї—М–љ–Њ–Љ—Г –њ–Њ–Ї–∞–Ј–∞–љ–∞ —В–µ—А–∞–њ–Є—П¬†–≥–ї—О–Ї–Њ–Ї–Њ—А—В–Є–Ї–Њ—Б—В–µ—А–Њ–Є–і–∞–Љ–Є –≤¬†—Б–Њ—З–µ—В–∞–љ–Є–Є —Б¬†—Ж–Є—В–Њ—Б—В–∞—В–Є–Ї–∞–Љ–Є.
–Р—Г—В–Њ–Є–Љ–Љ—Г–љ–љ–∞—П –і–Є–∞—А–µ—П, –љ–∞—А—П–і—Г —Б¬†–≤—А–Њ–ґ–і–µ–љ–љ—Л–Љ–Є –∞–љ–Њ–Љ–∞–ї–Є—П–Љ–Є —Н–љ—В–µ—А–Њ—Ж–Є—В–Њ–≤, –Њ—В–љ–Њ—Б–Є—В—Б—П –Ї¬†–≥—А—Г–њ–њ–µ —В–∞–Ї –љ–∞–Ј—Л–≤–∞–µ–Љ—Л—Е —В—А—Г–і–љ–Њ–Є–Ј–ї–µ—З–Є–Љ—Л—Е –Љ–ї–∞–і–µ–љ—З–µ—Б–Ї–Є—Е –і–Є–∞—А–µ–є. –°–ї–µ–і—Г–µ—В –µ—Й–µ —А–∞–Ј –њ–Њ–і—З–µ—А–Ї–љ—Г—В—М –Њ—Б–љ–Њ–≤–љ—Л–µ –Њ—Б–Њ–±–µ–љ–љ–Њ—Б—В–Є –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є —Н—В–Њ–є¬†–≥—А—Г–њ–њ—Л:¬†–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–∞—П –њ—А–µ–і—А–∞—Б–њ–Њ–ї–Њ–ґ–µ–љ–љ–Њ—Б—В—М;
- —А–∞–љ–љ–µ–µ –љ–∞—З–∞–ї–Њ;
- –Њ—В—Б—Г—В—Б—В–≤–Є–µ —Б–≤—П–Ј–Є —Б¬†–Ї–∞–Ї–Є–Љ-–ї–Є–±–Њ –Є–љ—Д–µ–Ї—Ж–Є–Њ–љ–љ—Л–Љ –∞–≥–µ–љ—В–Њ–Љ –Є–ї–Є –њ–Є—Й–µ–≤—Л–Љ —Б—Г–±—Б—В—А–∞—В–Њ–Љ;
- —Г–њ–Њ—А–љ—Л–є —Е–∞—А–∞–Ї—В–µ—А –≤–Њ–і—П–љ–Є—Б—В–Њ–є –і–Є–∞—А–µ–Є —Б–µ–Ї—А–µ—В–Њ—А–љ–Њ–≥–Њ —В–Є–њ–∞, –љ–µ¬†–Ї—Г–њ–Є—А—Г—О—Й–µ–є—Б—П –њ—А–Є –њ–µ—А–µ–≤–Њ–і–µ —А–µ–±–µ–љ–Ї–∞ –љ–∞¬†–њ–∞—А–µ–љ—В–µ—А–∞–ї—М–љ–Њ–µ –њ–Є—В–∞–љ–Є–µ;
- –≤—Л—А–∞–ґ–µ–љ–љ–∞—П –∞—В—А–Њ—Д–Є—П –°–Ю–Ґ–Ъ.
–Р–ї–≥–Њ—А–Є—В–Љ –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Њ–є –і–Є–∞–≥–љ–Њ—Б—В–Є–Ї–Є —В—А—Г–і–љ–Њ–Є–Ј–ї–µ—З–Є–Љ—Л—Е –Љ–ї–∞–і–µ–љ—З–µ—Б–Ї–Є—Е –і–Є–∞—А–µ–є –њ—А–µ–і—Б—В–∞–≤–ї–µ–љ –љ–∞¬†—А–Є—Б—Г–љ–Ї–µ¬†7,¬†–≥–Є—Б—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–µ –Њ—Б–Њ–±–µ–љ–љ–Њ—Б—В–Є –°–Ю–Ґ–Ъ¬†вАУ –≤¬†—В–∞–±–ї–Є—Ж–µ 4.
–Ґ–∞–Ї–Є–Љ –Њ–±—А–∞–Ј–Њ–Љ, —Е—А–Њ–љ–Є—З–µ—Б–Ї–∞—П –і–Є–∞—А–µ—П —Г¬†–і–µ—В–µ–є –Љ–Њ–ґ–µ—В –±—Л—В—М –Њ–±—Г—Б–ї–Њ–≤–ї–µ–љ–∞ –Љ–љ–Њ–ґ–µ—Б—В–≤–Њ–Љ —А–∞–Ј–ї–Є—З–љ—Л—Е –њ—А–Є—З–Є–љ. –£—Б–њ–µ—Е –≤¬†–ї–µ—З–µ–љ–Є–Є —Н—В–Њ–≥–Њ –Ї—А–∞–є–љ–µ —Б–ї–Њ–ґ–љ–Њ–≥–Њ –≤¬†–њ–ї–∞–љ–µ –і–Є—Д—Д–µ—А–µ–љ—Ж–Є–∞–ї—М–љ–Њ–є –і–Є–∞–≥–љ–Њ—Б—В–Є–Ї–Є —Б–Є–љ–і—А–Њ–Љ–∞ –≤–Њ¬†–Љ–љ–Њ–≥–Њ–Љ –Ј–∞–≤–Є—Б–Є—В –Њ—В¬†–Ј–љ–∞–љ–Є–є –≤—А–∞—З–∞ –Є¬†–µ–≥–Њ —Б—В—А–µ–Љ–ї–µ–љ–Є—П –љ–∞–є—В–Є –Є—Б—В–Є–љ—Г.
–£—Б—В–∞–љ–Њ–≤–ї–µ–љ–Є–µ –њ—А–∞–≤–Є–ї—М–љ–Њ–≥–Њ –і–Є–∞–≥–љ–Њ–Ј–∞, –Ї–∞–Ї –Є–Ј–≤–µ—Б—В–љ–Њ, —П–≤–ї—П–µ—В—Б—П –Ї–ї—О—З–Њ–Љ –Ї¬†–≤—Л–±–Њ—А—Г –њ—А–∞–≤–Є–ї—М–љ–Њ–є —В–µ—А–∞–њ–Є–Є. –Ъ–∞–Ї —Г–Ї–∞–Ј—Л–≤–∞–ї–Њ—Б—М –≤—Л—И–µ, –≤¬†–ї–µ—З–µ–љ–Є–Є —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–є –і–Є–∞—А–µ–Є, –Є—Б–њ–Њ–ї—М–Ј—Г—О—В –њ—А–µ–ґ–і–µ –≤—Б–µ–≥–Њ —А–∞–Ј–ї–Є—З–љ—Л–µ –і–Є–µ—В–Є—З–µ—Б–Ї–Є–µ –њ–Њ–і—Е–Њ–і—Л. –І—В–Њ –Ї–∞—Б–∞–µ—В—Б—П –Љ–µ–і–Є–Ї–∞–Љ–µ–љ—В–Њ–Ј–љ–Њ–є —В–µ—А–∞–њ–Є–Є, —В–Њ¬†–Њ–±—Й–µ–є —Б—В—А–∞—В–µ–≥–Є–µ–є –ї–µ—З–µ–љ–Є—П –≤—Б–µ—Е –і–Є–∞—А–µ–є–љ—Л—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є —П–≤–ї—П–µ—В—Б—П –Ї–Њ—А—А–µ–Ї—Ж–Є—П –≤–Њ–і-–љ–Њ-—Н–ї–µ–Ї—В—А–Њ–ї–Є—В–љ—Л—Е –љ–∞—А—Г—И–µ–љ–Є–є –Є¬†—Г—Б–Є–ї–µ–љ–Є–µ –Ї–Є—И–µ—З–љ–Њ–≥–Њ –±–∞—А—М–µ—А–∞. –Я–Њ—Б–ї–µ–і–љ–µ–µ –і–Њ—Б—В–Є–≥–∞–µ—В—Б—П –љ–∞–Ј–љ–∞—З–µ–љ–Є–µ–Љ —Ж–Є—В–Њ–њ—А–Њ—В–µ–Ї—В–Њ—А–Њ–≤ –Є¬†–∞–і—Б–Њ—А–±–µ–љ—В–Њ–≤.
–£–≤–∞–ґ–∞–µ–Љ—Л–є –њ–Њ—Б–µ—В–Є—В–µ–ї—М uMEDp!
–£–≤–µ–і–Њ–Љ–ї—П–µ–Љ –Т–∞—Б –Њ —В–Њ–Љ, —З—В–Њ –Ј–і–µ—Б—М —Б–Њ–і–µ—А–ґ–Є—В—Б—П –Є–љ—Д–Њ—А–Љ–∞—Ж–Є—П, –њ—А–µ–і–љ–∞–Ј–љ–∞—З–µ–љ–љ–∞—П –Є—Б–Ї–ї—О—З–Є—В–µ–ї—М–љ–Њ –і–ї—П —Б–њ–µ—Ж–Є–∞–ї–Є—Б—В–Њ–≤ –Ј–і—А–∞–≤–Њ–Њ—Е—А–∞–љ–µ–љ–Є—П.
–Х—Б–ї–Є –Т—Л –љ–µ —П–≤–ї—П–µ—В–µ—Б—М —Б–њ–µ—Ж–Є–∞–ї–Є—Б—В–Њ–Љ –Ј–і—А–∞–≤–Њ–Њ—Е—А–∞–љ–µ–љ–Є—П, –∞–і–Љ–Є–љ–Є—Б—В—А–∞—Ж–Є—П –љ–µ –љ–µ—Б–µ—В –Њ—В–≤–µ—В—Б—В–≤–µ–љ–љ–Њ—Б—В–Є –Ј–∞ –≤–Њ–Ј–Љ–Њ–ґ–љ—Л–µ –Њ—В—А–Є—Ж–∞—В–µ–ї—М–љ—Л–µ –њ–Њ—Б–ї–µ–і—Б—В–≤–Є—П, –≤–Њ–Ј–љ–Є–Ї—И–Є–µ –≤ —А–µ–Ј—Г–ї—М—В–∞—В–µ —Б–∞–Љ–Њ—Б—В–Њ—П—В–µ–ї—М–љ–Њ–≥–Њ –Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–љ–Є—П –Т–∞–Љ–Є –Є–љ—Д–Њ—А–Љ–∞—Ж–Є–Є —Б –њ–Њ—А—В–∞–ї–∞ –±–µ–Ј –њ—А–µ–і–≤–∞—А–Є—В–µ–ї—М–љ–Њ–є –Ї–Њ–љ—Б—Г–ї—М—В–∞—Ж–Є–Є —Б –≤—А–∞—З–Њ–Љ.
–Э–∞–ґ–Є–Љ–∞—П –љ–∞ –Ї–љ–Њ–њ–Ї—Г ¬Ђ–Т–Њ–є—В–Є¬ї, –Т—Л –њ–Њ–і—В–≤–µ—А–ґ–і–∞–µ—В–µ, —З—В–Њ —П–≤–ї—П–µ—В–µ—Б—М –≤—А–∞—З–Њ–Љ –Є–ї–Є —Б—В—Г–і–µ–љ—В–Њ–Љ –Љ–µ–і–Є—Ж–Є–љ—Б–Ї–Њ–≥–Њ –≤—Г–Ј–∞.