Ингибиторы АПФ в терапии больных сахарным диабетом
- Аннотация
- Статья
- Ссылки
- English
В статье проанализирована роль ингибиторов ангиотензинпревращающего фермента, в частности фозиноприла, в лечении больных сахарным диабетом с артериальной гипертензией и нефропатией. Представлены новые данные о механизмах кардио- и нефропротективного действия указанных препаратов.
В статье проанализирована роль ингибиторов ангиотензинпревращающего фермента, в частности фозиноприла, в лечении больных сахарным диабетом с артериальной гипертензией и нефропатией. Представлены новые данные о механизмах кардио- и нефропротективного действия указанных препаратов.

Сахарный диабет (СД) и ассоциированные с ним заболевания остаются одной из важнейших проблем здравоохранения, что прежде всего связано с постоянно увеличивающимся, опережающим все прогнозы темпом роста числа больных. Так, по данным экспертов Международной федерации диабета (International Diabetes Federation – IDF), каждый 11-й житель Земли страдает СД, что составляет 425 млн человек. К 2045 г. количество пациентов может увеличиться до 629 млн [1]. Приводятся данные о 9 млн взрослых больных СД в Российской Федерации (IDF, 2017). Однако реальная распространенность патологии может быть значительно выше. Так, по оценкам экспертов, более чем у 4,5 млн человек СД не диагностирован [1].
Для решения указанной проблемы требуются значительные социальные, интеллектуальные и экономические ресурсы. Так, финансовые расходы здравоохранения на оказание помощи больным СД в 2017 г. превысили 650 млрд долл. США, 20 из них – расходы Российской Федерации [1].
Как было отмечено ранее, при СД поражаются разные системы и органы. Однако основной причиной смерти пациентов с СД являются сердечно-сосудистые заболевания – 65–80% случаев [2–5]. Причиной тому служит комбинация сразу нескольких заболеваний и состояний, таких как ишемическая болезнь сердца, артериальная гипертензия (АГ), альбуминурия/хроническая болезнь почек (ХБП), дислипидемия, ожирение.
При СД одной из наиболее часто встречающихся и значимых нозологий в отношении риска развития сердечно-сосудистых заболеваний признана артериальная гипертензия [6, 7]. Так, в масштабном проспективном исследовании [8] по оценке факторов сердечно-сосудистого риска установлено, что самая высокая частота развития сердечно-сосудистых заболеваний была у пациентов с СД и артериальной гипертензией. Необходимо отметить, что в исследование было включено 1211 (11,0%) пациентов с СД и артериальной гипертензией, 628 (5,7%) – только с СД, 4,022 (36,4%) – только с АГ, 5189 (47,2%) лиц без указанных патологий. Группы не различались по уровню общего холестерина. Процент курящих оказался самым высоким в группе без диабета и артериальной гипертензии. Пациенты с СД и/или АГ, как правило, были старше, страдали ожирением и сердечно-сосудистыми заболеваниями. В группе диабета и артериальной гипертензии зафиксированы самые высокие уровни систолического артериального давления, средние уровни глюкозы в крови натощак и высокая распространенность сердечно-сосудистых заболеваний. За период наблюдения (в среднем – 11,7 года) зарегистрировано 2758 случаев развития сердечно-сосудистых заболеваний и событий, из них 1672 – ишемической болезни сердца, 558 – инсульта, 1375 – сердечной недостаточности.
Риск развития сердечно-сосудистых заболеваний у взрослых с СД в два – четыре раза выше, чем у лиц без диабета [6].
Аналогичным образом риск развития сердечно-сосудистых заболеваний соотносится с повышением систолического артериального давления на 20 мм рт. ст. [9].
Сердечно-сосудистые заболевания признаны одним из наиболее значимых осложнений ХБП [10–13].
Установлено, что в отсутствие диабета артериальная гипертензия чаще встречается у мужчин, чем у женщин. Однако после 64 лет показатели заболеваемости в этих группах сравниваются [14]. В то же время среди женщин с нарушенной толерантностью к глюкозе и СД распространенность АГ выше, чем среди мужчин с такими же нарушениями [15]. У женщин с СД также более высокий относительный риск смерти от сердечно-сосудистых заболеваний [16].
Общие механизмы нарушения регуляторных систем при СД, АГ и ХБП
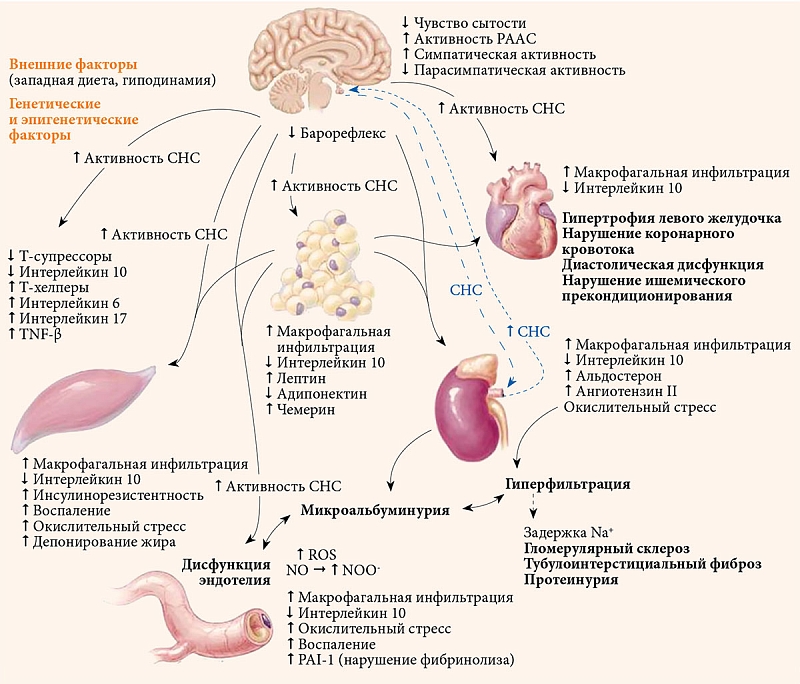
Сахарный диабет и артериальная гипертензия имеют общие патофизиологические механизмы. Речь, в частности, идет о неадекватной активации ренин-ангиотензин-альдостероновой системы (РААС), окислительном стрессе, вызванном усиленной продукцией активных форм кислорода (ROS), воспалении, нарушении опосредованной инсулином вазодилатации, активации симпатической нервной системы (СНС) и нарушении экскреции натрия [17, 18].
Ожирение в целом и висцеральное в частности являются ключевыми факторами развития как СД, так и АГ [18]. Вследствие хронического воспаления и окислительного стресса в жировой ткани увеличивается продукция ангиотензиногена и ангиотензина II, что приводит к активации РАСС [19, 20]. Кроме того, избыточная экспрессия ангиотензиногена в белой жировой ткани обусловливает повышение артериального давления [19]. Следовательно, ангиотензиноген и ангиотензин II оказывают как локальное, так и системное влияние на регуляцию артериального давления [19, 20]. Эффекты ангиотензина II реализуются через активацию рецептора 1-го типа [21]. Активация рецептора 1-го типа ангиотензина II вне ткани надпочечников связана с целым рядом внутриклеточных изменений, включая усиление продукции ROS, снижение метаболической активности инсулина, а также пролиферативные и воспалительные сосудистые реакции, приводящие к эндотелиальной дисфункции, инсулинорезистентности и артериальной гипертензии [21]. Таким образом, активация РААС играет значительную роль в патогенезе СД и АГ.
Ключевыми событиями в развитии артериальной гипертензии также являются повышение продукции альдостерона и усиление передачи сигналов через рецепторы минералокортикоидов [22]. Посредством активации последних кортикостероиды могут способствовать развитию сердечно-сосудистых заболеваний у пациентов с СД [18]. Известно, что жировая ткань продуцирует жирорастворимый фактор, который стимулирует выработку альдостерона надпочечниками [23, 24]. Комплемент C1q фактор некроза опухоли (TNF)-связанный белок 1 – адипокин, который усиливает продукцию альдостерона при ожирении и инсулинорезистентности [25]. Активация рецепторов минералокортикоидов альдостероном в почечных дистальных канальцах и собирающих протоках обусловливает задержку натрия (Na), что приводит к увеличению объема плазмы и повышению артериального давления. Через активацию рецепторов минералокортикоидов альдостерон также изменяет окислительно-восстановительные процессы в клетках и эндотелиальную вазодилатацию [22, 24]. Таким образом, жировая ткань способствует системному повышению артериального давления за счет локального производства компонентов РААС.
В развитии СД и АГ важную роль также играет резистентность к инсулину. Об этом свидетельствует тот факт, что инсулинорезистентность выявляется примерно у 50% пациентов с артериальной гипертензией [18, 26, 27]. Связывание инсулина с его рецептором приводит к активации не только транспорта глюкозы в чувствительных к инсулину тканях, но и эндотелиальной синтазы оксида азота (NO), что вызывает продукцию NO. Как следствие, возникает эндотелий-зависимая вазодилатация [28, 29]. Инсулин также влияет на тонус сосудов, опосредуемый митоген-активируемой протеинкиназой (MAPK) [28, 30]. Активируя MAPK-зависимые сигнальные пути, инсулин стимулирует секрецию вазоконстрикторных медиаторов, в частности эндотелина 1 [31, 32], а также экспрессию ингибитора активатора плазминогена 1 (PAI-1), молекулы адгезии сосудистых клеток 1 [33]. При нормальной чувствительности к инсулину баланс между вазоконстрикторными и вазодилатирующими действиями благоприятствует вазодилатации. В случае инсулинорезистентности равновесие смещается в сторону вазоконстрикторных механизмов [17, 18, 34].
Гиперинсулинемия и инсулинорезистентность приводят к нарушению сосудистой функции, жесткости сосудов, гипертрофии, фиброзу и ремоделированию [35]. Гиперинсулинемия также усиливает симпатический тонус сосудов через вентромедиальные ядра гипоталамуса [36, 37].
Кроме того, лептин – адипокин, вырабатываемый жировой тканью у лиц с ожирением, увеличивает активацию симпатической нервной системы. Возможно, это достигается за счет активации его рецептора в центральной нервной системе [38]. По всей вероятности, этот механизм опосредуется также через нефрогенную индукцию артериальной гипертензии, связанной с ожирением и инсулинорезистентностью [18].
Важно отметить, что инсулин усиливает реабсорбцию натрия, что приводит к снижению экскреции последнего [39]. Задержка натрия, вызванная гиперинсулинемией, потенциально может способствовать возникновению артериальной гипертензии. В экспериментальном исследовании установлено, что снижение активности инсулина приводит к нарушению натрийуретического ответа и артериальной гипертензии, вероятно, вследствие снижения продукции NO [40]. Наряду со снижением экспрессии Na ухудшается экскреция мочевой кислоты. Это способствует развитию гиперурикемии, которая часто наблюдается у пациентов с артериальной гипертензией [41]. Взаимосвязь механизмов патогенеза сахарного диабета, артериальной гипертензии, сердечно-сосудистых заболеваний и хронической болезни почек представлена на рисунке [18].
В то же время СД и АГ являются факторами риска развития ХБП [42–44]. Так, 40% случаев терминальной стадии почечной недостаточности приходится на больных диабетом. Около 30% случаев терминальной стадии почечной недостаточности обусловлены артериальной гипертензией [45]. Вклад диабета и артериальной гипертензии в развитие патологии почек отмечен в клинических рекомендациях по скринингу хронической болезни почек [46–48]. Сахарный диабет 2 типа является основной причиной развития терминальной стадии почечной недостаточности в Нидерландах [49], Германии [50], а также в Соединенных Штатах [51]. Диабетическая нефропатия, первоначально проявляющаяся микро- и макроальбуминурией, приводит к постепенному снижению функции почек. Данная патология может прогрессировать до терминальной стадии почечной недостаточности, которая обусловливает необходимость проведения диализа или трансплантации почки [52]. Например, в Нидерландах за последние 15 лет применение заместительной почечной терапии возросло в два раза [53]. За последние пять лет доля пациентов, которым была пересажена почка, увеличилась до 57% [53].
Антигипертензивная терапия при СД
Рациональную комплексную терапию для пациентов с СД и сопутствующими заболеваниями возможно разработать только зная о взаимосвязи указанных нозологий. Например, результат лечения ишемической болезни сердца зависит от эффективности сахароснижающей и антигипертензивной терапии. Артериальная гипертензия определяет прогноз нефропатии и хронической болезни почек, которые в свою очередь влияют на риск развития ишемической болезни сердца.
Для коррекции артериального давления у пациентов с умеренной артериальной гипертензией и нормальной функцией почек может быть достаточно модифицировать образ жизни [54–56]. Однако большинству больных требуется дополнительное медикаментозное лечение.
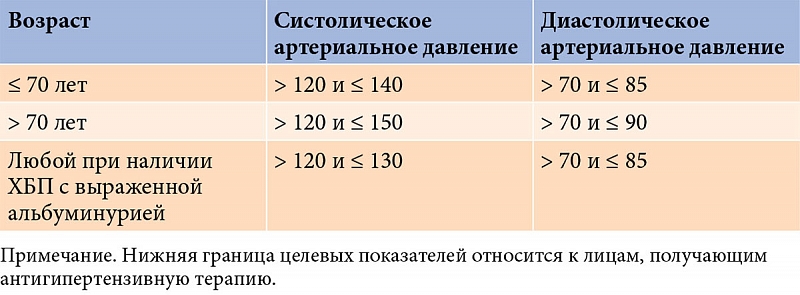
Современные отечественные рекомендации по коррекции артериальной гипертензии и целевые уровни артериального давления представлены в табл. 1 и 2 [57].
Установлено, что широкий спектр препаратов, снижающих артериальное давление, оказывают протективный эффект в отношении риска развития сердечно-сосудистых событий. К таким препаратам относятся ингибиторы ангиотензинпревращающего фермента (иАПФ), блокаторы рецепторов ангиотензина II (БРА), β-блокаторы, диуретики и блокаторы кальциевых каналов.
Учитывая вышеизложенное, предпочтение при выборе антигипертензивной терапии следует отдавать тем препаратам, которые оказывают не только патогенетическое, но и протективное действие в отношении сердечно-сосудистой системы, а также почек. Этим требованиям в большей степени отвечают блокаторы РААС, в частности иАПФ и БРА. К преимуществам данных препаратов также следует отнести потенциальную возможность влиять на инсулинорезистентность, воспаление, окислительный стресс и эндотелиальную функцию [58, 59].
Сравнение эффектов иАПФ и БРА
Если БРА блокируют рецепторы ангиотензина II, ингибиторы конвертирующего фермента ангиотензина II снижают его активность. Уменьшение активности конвертирующего фермента ангиотензина II приводит к вазодилатации и, как следствие, снижению артериального давления, а также улучшению микроциркуляции в органах и тканях [60, 61]. В исследовании HOPE сравнивали влияние иАПФ рамиприла и плацебо на сердечно-сосудистые осложнения. Средний период наблюдения – 4,5 года. Рамиприл снижал риск инфаркта миокарда, инсульта и сердечно-сосудистой смерти на 25% [62]. При субанализе результатов исследования MICRO-HOPE в подгруппе пациентов с диабетом (n = 3577) отмечен аналогичный положительный эффект иАПФ на показатели сердечно-сосудистой и общей смертности [63].
В исследовании LIFE, включавшем 1195 пациентов с СД 2 типа, продемонстрировано значительное снижение сердечно-сосудистой заболеваемости и смертности у пациентов, получавших лозартан, по сравнению с принимавшими β-блокатор атенолол. Относительный риск таковых снизился на 24%. При этом на фоне проводившегося лечения снижение артериального давления в обеих группах было сопоставимым [64].
Справедливости ради следует отметить, что в отдельных проспективных исследованиях, оценивавших долгосрочный прогноз терапии в отношении сердечно-сосудистого риска, установлено, что заболеваемость и смертность у пациентов, принимавших БРА (кандесартан в исследовании CASE-J и валсартан в исследовании VALUE), не отличались от таковых у получавших длительно действующий блокатор кальциевых каналов амлодипин. В то же время на фоне лечения БРА значительно снизилась частота возникновения СД [65, 66].
Протективный эффект в отношении сердечно-сосудистой заболеваемости и смертности телмисартана (БРА) и рамиприла (иАПФ) продемонстрирован в исследовании с участием 6391 пациента с СД [67]. Однако комбинированная терапия иАПФ и БРА ассоциировалась с большей частотой побочных эффектов, таких как артериальная гипотензия, обмороки и гиперкалиемия. Так, относительный риск гипотензии в этой группе составил 2,75 (р < 0,001). Количество пациентов, у которых уровень калия повышался более чем на 5,5 ммоль/л, достигло 480 (p < 0,001), в то время как в группе рамиприла и телмисартана таковых насчитывалось 283 и 287 соответственно. Кроме того, в группе «рамиприл + телмисартан» значительно увеличился относительный риск нарушения функции почек – 1,33 (р < 0,001). Поэтому авторы исследования сделали вывод о нецелесообразности комбинированного назначения БРА и иАПФ.
Ингибиторы РААС также оказывают защитное воздействие на почки. Согласно результатам исследований BENEDICT и ROADMAP, в которых оценивался эффект терапии иАПФ и БРА соответственно, у пациентов с СД, АГ и нормоальбуминурией (< 30 мг/сут) блокада РААС препятствовала развитию микроальбуминурии (30–300 мг/сут) [68, 69]. Необходимо отметить, что данный эффект БРА не был подтвержден у больных СД с ретинопатией и нормотензивных больных СД [70].
Ингибиторы АПФ замедляют прогрессирование диабетической нефропатии [71, 72]. Аналогичное воздействие оказывают БРА [73]. Однако БРА в отличие от иАПФ не повышают уровень брадикинина и, следовательно, реже вызывают сухой кашель [74].
Сравнительный анализ разных методов блокады РААС свидетельствует, что иАПФ и БРА одинаково влияют на основные сердечно-сосудистые и почечные исходы у пациентов с сахарным диабетом [75]. В частности, не было отмечено существенных различий между иАПФ и БРА в отношении показателей смертности от всех причин, сердечно-сосудистой смертности, инфаркта миокарда, инсульта, частоты стенокардии, госпитализаций по поводу сердечной недостаточности, терминальной стадии почечной недостаточности или повышения уровня креатинина в сыворотке крови.
Необходимо отметить, что профилактика терминальной стадии почечной недостаточности важна не только с медицинской, но и с экономической точки зрения. Например, на лечение ХБП в европейских странах в среднем тратится от 1 до 2% национального бюджета, терминальной стадии почечной недостаточности в Нидерландах – 42 000 евро на одного пациента в год [76–79].
В этой связи немаловажно, что терапия БРА более дорогостоящая, чем иАПФ.
Согласно большинству национальных и международных руководств, больным СД терапию иАПФ следует назначать при выявлении микро- или альбуминурии [80–82]. Однако в реальной клинической практике эти рекомендации не выполняются даже в странах с высокими бюджетами здравоохранения [83].
Исходя из моделей экономической эффективности [84–86], наилучший результат можно получить при назначении иАПФ сразу после выявления СД.
Фозиноприл
Одним из наиболее хорошо изученных иАПФ является фозиноприл.
В исследовании TRAIN (n = 290) сравнили эффект фозиноприла и плацебо у 290 пациентов с артериальной гипертензией. В среднем исходные значения артериального давления составляли 137,4/80,8 мм рт. ст. Большинство (89,8%) участников получали фозиноприл в максимальной дозе – 40 мг [87]. Прием препарата по сравнению с применением плацебо способствовал снижению как систолического (128,5 против 133,4 мм рт. ст. (p < 0,001)), так и диастолического (77,6 против 80,6 мм рт. ст. (р < 0,001)) артериального давления. Кроме того, в группе фозиноприла были зарегистрированы более низкие уровни ангиотензина II – 11,10 против 12,50 пг/мл соответственно (р < 0,001) [87].
Терапия фозиноприлом также ассоциируется с положительной динамикой маркера активации коагуляции D-димера. Так, у большинства получавших фозиноприл в максимальной дозе выявлено статистически значимое повышение (около 10%) концентрации D-димера. Высокие уровни D-димера у больных атеросклерозом свидетельствуют об активации фибринолиза [88–90]. Ингибирование АПФ запускает профибринолитический механизм, вследствие которого восстанавливается баланс между фибринолизом и коагуляцией, возможно, за счет повышения уровня брадикинина [91]. Таким образом, повышенные уровни D-димера могут свидетельствовать о деградации ранее существовавшего фибрина [88]. У пациентов с атеросклерозом периферических сосудов к значительному увеличению уровня D-димера также могут приводить субмаксимальные физические нагрузки [88].
Под действием иАПФ в сыворотке крови увеличивается концентрация инсулиноподобного фактора роста (IGF) 1 [92] и белка, связывающего IGF (IGFBP-3) [93]. Установлено, что инфузия ангиотензина II способна снижать циркулирующие концентрации IGF-1 независимо от изменений артериального давления [94]. Сам по себе IGF-1 обладает вазопротективным эффектом, а его снижение может быть связано с началом и прогрессированием атеросклероза [95, 96]. У пожилых пациентов уровень IGF-1 обратно пропорционален атерогенности липидного профиля [95] и степени стабильности артериальных бляшек [96].
IGF-1 преимущественно синтезируется печенью в ответ на гормон роста. В кровотоке от 90 до 95% IGF-1 связывается со специфическими высокоаффинными белками, при этом наиболее активным в сыворотке крови является IGFBP-3. Он не только модулирует биологическую активность IGF-1, но и вызывает дополнительные, IGF-независимые эффекты, например ингибирует рост клеток [97] и индуцирует апоптоз [98]. Согласно результатам проспективных исследований, повышение концентрации IGF-1 связано с уменьшением риска развития ишемической болезни сердца [99]. Обратная зависимость также наблюдается между сывороточными концентрациями IGF-1 и IGFBP-3 и риском развития ишемического инсульта [100]. Влияние иАПФ на IGF-1 было подтверждено в ряде клинических исследований [101, 102]. В частности, прием фозиноприла в течение шести месяцев пожилыми пациентами с высоким сердечно-сосудистым риском привел к значительному увеличению сывороточных концентраций IGF-1 и IGFBP-3 [103]. Возможно, это является одним из механизмов кардиопротективного воздействия иАПФ.
Были выявлены и другие возможные механизмы влияния фозиноприла на состояние эндотелия. Так, фактор роста эндотелия сосудов (VEGF) стимулирует пролиферацию васкулярных эндотелиальных клеток и участвует в ангиогенезе. Еще одним маркером эндотелиальной дисфункции является чемерин. Этот адипокин стимулирует скорость и интенсивность ангиогенеза эндотелиальных клеток. Его уровень коррелирует с показателями инсулинорезистентности. У больных СД обнаружена повышенная экспрессия чемерина и VEGF как в сыворотке крови, так и в почечной ткани [104, 105]. Более того, выявлена положительная корреляция чемерина и VEGF со степенью альбуминурии. Это свидетельствует о важной роли чемерина и VEGF в патогенезе диабетической нефропатии. На фоне терапии фозиноприлом концентрация этих двух белков достоверно снижалась как в сыворотке крови, так и в почечной ткани, что может объяснять один из механизмов его нефропротективного действия [106]. При этом указанные механизмы нефропротекции не связаны с влиянием иАПФ на гликемию и инсулинорезистентность [107]. Положительное воздействие иАПФ на инсулинорезистентность у пациентов с АГ без СД было более выражено, чем у БРА, при сопоставимости двух препаратов в отношении эффективности контроля артериального давления [108].
Еще один из механизмов влияния фозиноприла (при добавлении к гидрохлортиазиду) на эндотелиальную функцию и провоспалительные цитокины был выявлен у больных артериальной гипертензией с избыточной массой тела и ожирением. Так, в группе комбинированной антигипертензивной терапии в отличие от группы контроля наблюдалось значительное снижение активности i-NOS, снижение TNF-α I типа и его растворимого рецептора (sTNF-αRI), а также 8-изопростана (маркера окислительного стресса). На основании полученных результатов был сделан вывод, что такая терапия позволяет улучшить функции эндотелия, уменьшить воспаление и окислительный стресс [109].
Как было отмечено выше, реализация нефропротективного эффекта иАПФ, в частности фозиноприла, происходит независимо от уровня артериального давления, даже у нормотензивных пациентов. В последнем случае суточные дозы препарата должны составлять 10–20 мг [110].
Заключение
Нарушение регуляторных систем при СД, АГ и ХБП обусловлено общими механизмами, важнейшим из которых является неадекватное функционирование (активация) РААС. Назначение иАПФ снижает негативное влияние активированной РААС на органы и ткани, включая сердце, сосуды и почки. Благодаря этому достигается кардио- и нефропротективный эффект. Одним из эффективных иАПФ признан фозиноприл. Помимо гипотензивного эффекта он оказывает метаболические эффекты, которые обусловливают снижение инсулинорезистентности, уменьшение гликемии и воспаления.
Назначение фозиноприла при выявлении СД является клинически и фармакоэкономически обоснованным.
V.I. Novikov, DM, Prof., K.Yu. Novikov
Smolensk State Medical University
Contact person: Vladimir Ivanovich Novikov, endo@smolgmu.ru
The most important problem unsolved in diabetes – therapy of hypertension, coronary heart disease, nephropathy. Violation of regulatory systems in the case of these diseases has common pathogenetic links, the most important of which is the inadequate functioning (activation) of the renin-angiotensin-aldosterone system.
The article analyzes the role of angiotensin-converting enzyme inhibitors, in particular fosinopril, in the treatment of patients with diabetes mellitus with hypertension and nephropathy. Presented new data on the mechanisms of cardio- and nephroprotective action of indicated drugs.
Уважаемый посетитель uMEDp!
Уведомляем Вас о том, что здесь содержится информация, предназначенная исключительно для специалистов здравоохранения.
Если Вы не являетесь специалистом здравоохранения, администрация не несет ответственности за возможные отрицательные последствия, возникшие в результате самостоятельного использования Вами информации с портала без предварительной консультации с врачом.
Нажимая на кнопку «Войти», Вы подтверждаете, что являетесь врачом или студентом медицинского вуза.