–Ш–љ—В–µ—А—Б—В–Є—Ж–Є–∞–ї—М–љ–Њ–µ –њ–Њ—А–∞–ґ–µ–љ–Є–µ –ї–µ–≥–Ї–Є—Е –њ—А–Є —А–µ–≤–Љ–∞—В–Є—З–µ—Б–Ї–Є—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П—Е: –љ–Њ–≤—Л–µ –≤–Њ–Ј–Љ–Њ–ґ–љ–Њ—Б—В–Є –∞–љ—В–Є–њ—А–Њ–ї–Є—Д–µ—А–∞—В–Є–≤–љ–Њ–є/–∞–љ—В–Є—Д–Є–±—А–Њ—В–Є—З–µ—Б–Ї–Њ–є —В–µ—А–∞–њ–Є–Є
- –Р–љ–љ–Њ—В–∞—Ж–Є—П
- –°—В–∞—В—М—П
- –°—Б—Л–ї–Ї–Є
- English
–Ф–ї–Є—В–µ–ї—М–љ–Њ–µ –≤—А–µ–Љ—П –∞—А—Б–µ–љ–∞–ї —А–µ–≤–Љ–∞—В–Њ–ї–Њ–≥–Њ–≤ –њ—А–Є –Ш–Ч–Ы –±—Л–ї –Њ–≥—А–∞–љ–Є—З–µ–љ –љ–µ—Б–Ї–Њ–ї—М–Ї–Є–Љ–Є –њ—А–µ–њ–∞—А–∞—В–∞–Љ–Є, –≤ —З–∞—Б—В–љ–Њ—Б—В–Є —Ж–Є–Ї–ї–Њ—Д–Њ—Б—Д–∞–Љ–Є–і–Њ–Љ –Є –Љ–Є–Ї–Њ—Д–µ–љ–Њ–ї–∞—В–∞ –Љ–Њ—Д–µ—В–Є–ї–Њ–Љ. –Ю–і–љ–∞–Ї–Њ –Њ–љ–Є –љ–µ –њ–Њ–Ј–≤–Њ–ї—П—О—В –Ї–Њ–љ—В—А–Њ–ї–Є—А–Њ–≤–∞—В—М –≤—Б–µ –≤–Њ–Ј–Љ–Њ–ґ–љ—Л–µ —Б–Є—В—Г–∞—Ж–Є–Є. –Т –љ–∞—Б—В–Њ—П—Й–µ–µ –≤—А–µ–Љ—П, –њ–Њ—Б–ї–µ –њ—Г–±–ї–Є–Ї–∞—Ж–Є–Є —А–µ–Ј—Г–ї—М—В–∞—В–Њ–≤ –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є SENCSIS –Є INBUILD, –і–ї—П –ї–µ—З–µ–љ–Є—П –Ш–Ч–Ы –њ—А–Є —Б–Є—Б—В–µ–Љ–љ–Њ–є —Б–Ї–ї–µ—А–Њ–і–µ—А–Љ–Є–Є –Є –і—А—Г–≥–Є—Е —А–µ–≤–Љ–∞—В–Є—З–µ—Б–Ї–Є—Е –±–Њ–ї–µ–Ј–љ—П—Е –Ј–∞—А–µ–≥–Є—Б—В—А–Є—А–Њ–≤–∞–љ –∞–љ—В–Є–њ—А–Њ–ї–Є—Д–µ—А–∞—В–Є–≤–љ—Л–є –Є –∞–љ—В–Є—Д–Є–±—А–Њ–Ј–љ—Л–є –њ—А–µ–њ–∞—А–∞—В –љ–Є–љ—В–µ–і–∞–љ–Є–±, —З—В–Њ –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ —А–∞—Б—И–Є—А–Є–ї–Њ —В–µ—А–∞–њ–µ–≤—В–Є—З–µ—Б–Ї–Є–µ –≤–Њ–Ј–Љ–Њ–ґ–љ–Њ—Б—В–Є.
–Т —Б—В–∞—В—М–µ —А–∞—Б—Б–Љ–Њ—В—А–µ–љ—Л –њ–∞—В–Њ–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Є–µ –Њ—Б–љ–Њ–≤—Л –њ—А–Є–Љ–µ–љ–µ–љ–Є—П –љ–Є–љ—В–µ–і–∞–љ–Є–±–∞. –Я—А–µ–і—Б—В–∞–≤–ї–µ–љ—Л —А–µ–Ј—Г–ї—М—В–∞—В—Л –ї–µ—З–µ–љ–Є—П —Б —Д–Њ–Ї—Г—Б–Њ–Љ –љ–∞ –Ш–Ч–Ы –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —А–µ–≤–Љ–∞—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–≥–Њ –њ—А–Њ—Д–Є–ї—П.
–Ф–ї–Є—В–µ–ї—М–љ–Њ–µ –≤—А–µ–Љ—П –∞—А—Б–µ–љ–∞–ї —А–µ–≤–Љ–∞—В–Њ–ї–Њ–≥–Њ–≤ –њ—А–Є –Ш–Ч–Ы –±—Л–ї –Њ–≥—А–∞–љ–Є—З–µ–љ –љ–µ—Б–Ї–Њ–ї—М–Ї–Є–Љ–Є –њ—А–µ–њ–∞—А–∞—В–∞–Љ–Є, –≤ —З–∞—Б—В–љ–Њ—Б—В–Є —Ж–Є–Ї–ї–Њ—Д–Њ—Б—Д–∞–Љ–Є–і–Њ–Љ –Є –Љ–Є–Ї–Њ—Д–µ–љ–Њ–ї–∞—В–∞ –Љ–Њ—Д–µ—В–Є–ї–Њ–Љ. –Ю–і–љ–∞–Ї–Њ –Њ–љ–Є –љ–µ –њ–Њ–Ј–≤–Њ–ї—П—О—В –Ї–Њ–љ—В—А–Њ–ї–Є—А–Њ–≤–∞—В—М –≤—Б–µ –≤–Њ–Ј–Љ–Њ–ґ–љ—Л–µ —Б–Є—В—Г–∞—Ж–Є–Є. –Т –љ–∞—Б—В–Њ—П—Й–µ–µ –≤—А–µ–Љ—П, –њ–Њ—Б–ї–µ –њ—Г–±–ї–Є–Ї–∞—Ж–Є–Є —А–µ–Ј—Г–ї—М—В–∞—В–Њ–≤ –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є SENCSIS –Є INBUILD, –і–ї—П –ї–µ—З–µ–љ–Є—П –Ш–Ч–Ы –њ—А–Є —Б–Є—Б—В–µ–Љ–љ–Њ–є —Б–Ї–ї–µ—А–Њ–і–µ—А–Љ–Є–Є –Є –і—А—Г–≥–Є—Е —А–µ–≤–Љ–∞—В–Є—З–µ—Б–Ї–Є—Е –±–Њ–ї–µ–Ј–љ—П—Е –Ј–∞—А–µ–≥–Є—Б—В—А–Є—А–Њ–≤–∞–љ –∞–љ—В–Є–њ—А–Њ–ї–Є—Д–µ—А–∞—В–Є–≤–љ—Л–є –Є –∞–љ—В–Є—Д–Є–±—А–Њ–Ј–љ—Л–є –њ—А–µ–њ–∞—А–∞—В –љ–Є–љ—В–µ–і–∞–љ–Є–±, —З—В–Њ –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ —А–∞—Б—И–Є—А–Є–ї–Њ —В–µ—А–∞–њ–µ–≤—В–Є—З–µ—Б–Ї–Є–µ –≤–Њ–Ј–Љ–Њ–ґ–љ–Њ—Б—В–Є.
–Т —Б—В–∞—В—М–µ —А–∞—Б—Б–Љ–Њ—В—А–µ–љ—Л –њ–∞—В–Њ–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Є–µ –Њ—Б–љ–Њ–≤—Л –њ—А–Є–Љ–µ–љ–µ–љ–Є—П –љ–Є–љ—В–µ–і–∞–љ–Є–±–∞. –Я—А–µ–і—Б—В–∞–≤–ї–µ–љ—Л —А–µ–Ј—Г–ї—М—В–∞—В—Л –ї–µ—З–µ–љ–Є—П —Б —Д–Њ–Ї—Г—Б–Њ–Љ –љ–∞ –Ш–Ч–Ы –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —А–µ–≤–Љ–∞—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–≥–Њ –њ—А–Њ—Д–Є–ї—П.
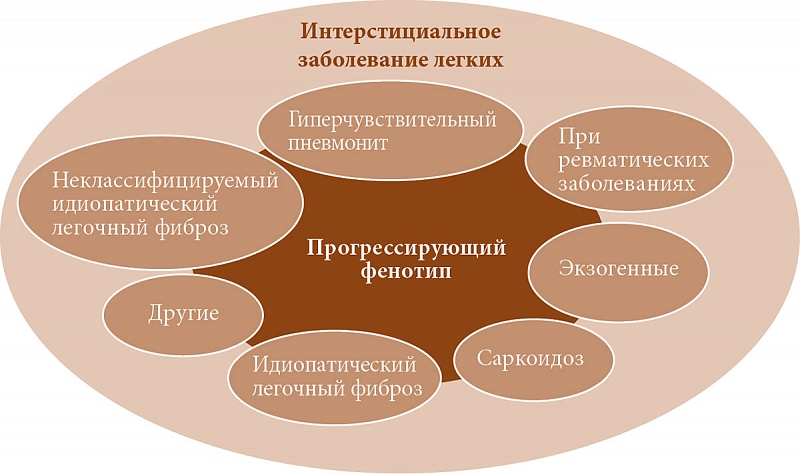


–Ш–љ—В–µ—А—Б—В–Є—Ж–Є–∞–ї—М–љ–Њ–µ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–µ –ї–µ–≥–Ї–Є—Е (–Ш–Ч–Ы)¬†вАУ —И–Є—А–Њ–Ї–Њ —А–∞—Б–њ—А–Њ—Б—В—А–∞–љ–µ–љ–љ—Л–є —Б–Є–љ–і—А–Њ–Љ, –≤–Ї–ї—О—З–∞—О—Й–Є–є –≤¬†—Б–µ–±—П –±–Њ–ї—М—И—Г—О –Є¬†—А–∞–Ј–љ–Њ—А–Њ–і–љ—Г—О –≥—А—Г–њ–њ—Г –њ–∞—А–µ–љ—Е–Є–Љ–∞—В–Њ–Ј–љ—Л—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є –ї–µ–≥–Ї–Є—Е, –Ї–Њ—В–Њ—А—Л–µ –Љ–Њ–≥—Г—В –±—Л—В—М —Б–≤—П–Ј–∞–љ—Л —Б¬†—Б–Є—Б—В–µ–Љ–љ—Л–Љ–Є —А–µ–≤–Љ–∞—В–Є—З–µ—Б–Ї–Є–Љ–Є –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П–Љ–Є, –≤–Њ–Ј–і–µ–є—Б—В–≤–Є–µ–Љ —Н–Ї–Ј–Њ–≥–µ–љ–љ—Л—Е —Д–∞–Ї—В–Њ—А–Њ–≤ –Є–ї–Є –Є–Љ–µ—В—М –љ–µ–Є–Ј–≤–µ—Б—В–љ—Г—О —Н—В–Є–Њ–ї–Њ–≥–Є—О (–Є–і–Є–Њ–њ–∞—В–Є—З–µ—Б–Ї–Њ–µ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–µ) [1]. –Т¬†–Њ—В–µ—З–µ—Б—В–≤–µ–љ–љ–Њ–є –ї–Є—В–µ—А–∞—В—Г—А–µ –і–ї—П –Њ–±–Њ–Ј–љ–∞—З–µ–љ–Є—П –і–∞–љ–љ–Њ–≥–Њ —Б–Є–љ–і—А–Њ–Љ–∞ —А–µ–≤–Љ–∞—В–Њ–ї–Њ–≥–Є —З–∞—Б—В–Њ –Є—Б–њ–Њ–ї—М–Ј—Г—О—В —В–µ—А–Љ–Є–љ ¬Ђ–Є–љ—В–µ—А—Б—В–Є—Ж–Є–∞–ї—М–љ–Њ–µ –њ–Њ—А–∞–ґ–µ–љ–Є–µ –ї–µ–≥–Ї–Є—Е¬ї [2, 3]. –Ю–і–љ–∞–Ї–Њ —Б¬†—Г—З–µ—В–Њ–Љ —В–Њ–≥–Њ, —З—В–Њ –њ—А–Є –Њ–њ–Є—Б–∞–љ–Є–Є –Њ—В–µ—З–µ—Б—В–≤–µ–љ–љ–Њ–є –њ—Г–ї—М–Љ–Њ–љ–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–є –Є¬†–Њ–±—Й–µ—В–µ—А–∞–њ–µ–≤—В–Є—З–µ—Б–Ї–Њ–є –њ—А–∞–Ї—В–Є–Ї–Є, –Ї–∞–Ї –њ—А–∞–≤–Є–ї–Њ, –њ—А–Є–Љ–µ–љ—П–µ—В—Б—П —В–µ—А–Љ–Є–љ ¬Ђ–Є–љ—В–µ—А—Б—В–Є—Ж–Є–∞–ї—М–љ–Њ–µ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–µ –ї–µ–≥–Ї–Є—Е¬ї [4], –∞¬†—В–∞–Ї–ґ–µ –Њ–±—Й–µ–њ—А–Є–љ—П—В–Њ—Б—В–Є –њ–Њ—Б–ї–µ–і–љ–µ–≥–Њ –≤¬†–Ј–∞—А—Г–±–µ–ґ–љ–Њ–є –ї–Є—В–µ—А–∞—В—Г—А–µ [5], –≤¬†–і–∞–љ–љ–Њ–є –њ—Г–±–ї–Є–Ї–∞—Ж–Є–Є –Љ—Л –±—Г–і–µ–Љ –њ—А–Є–і–µ—А–ґ–Є–≤–∞—В—М—Б—П –Є–Љ–µ–љ–љ–Њ –µ–≥–Њ.
–£ —А—П–і–∞ –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —А–∞–Ј–≤–Є–≤–∞–µ—В—Б—П –њ—А–Њ–≥—А–µ—Б—Б–Є—А—Г—О—Й–Є–є —Д–µ–љ–Њ—В–Є–њ –Ш–Ч–Ы (–Я–§-–Ш–Ч–Ы), –Ї–Њ—В–Њ—А—Л–є —Е–∞—А–∞–Ї—В–µ—А–Є–Ј—Г–µ—В—Б—П –њ—А–Њ–≥—А–µ–і–Є–µ–љ—В–љ—Л–Љ —Г—Е—Г–і—И–µ–љ–Є–µ–Љ —Д—Г–љ–Ї—Ж–Є–Є –ї–µ–≥–Ї–Є—Е, –љ–∞—А–∞—Б—В–∞–љ–Є–µ–Љ –і—Л—Е–∞—В–µ–ї—М–љ–Њ–є –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В–Є –Є¬†—Б–љ–Є–ґ–µ–љ–Є–µ–Љ –Ї–∞—З–µ—Б—В–≤–∞ –ґ–Є–Ј–љ–Є, –∞¬†—В–∞–Ї–ґ–µ —А–Є—Б–Ї–Њ–Љ —А–∞–љ–љ–µ–є —Б–Љ–µ—А—В–Є (—А–Є—Б. 1) [1, 6].
–Э–∞–Є–±–Њ–ї–µ–µ —З–∞—Б—В–Њ –Я–§-–Ш–Ч–Ы –љ–∞–±–ї—О–і–∞–µ—В—Б—П –њ—А–Є –Є–і–Є–Њ–њ–∞—В–Є—З–µ—Б–Ї–Њ–Љ –ї–µ–≥–Њ—З–љ–Њ–Љ —Д–Є–±—А–Њ–Ј–µ (–Ш–Ы–§). –Ш–і–Є–Њ–њ–∞—В–Є—З–µ—Б–Ї–Є–є –ї–µ–≥–Њ—З–љ—Л–є —Д–Є–±—А–Њ–Ј –Њ—В–љ–Њ—Б–Є—В—Б—П –Ї¬†–Ш–Ч–Ы –љ–µ–Є–Ј–≤–µ—Б—В–љ–Њ–є –њ—А–Є—З–Є–љ—Л. –Ф–∞–љ–љ–∞—П –њ–∞—В–Њ–ї–Њ–≥–Є—П –Њ—В–ї–Є—З–∞–µ—В—Б—П –љ–µ–Є–Ј–Љ–µ–љ–љ—Л–Љ –њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є–µ–Љ –Є¬†–∞—Б—Б–Њ—Ж–Є–Є—А—Г–µ—В—Б—П —Б–Њ —Б—А–µ–і–љ–µ–є –≤—Л–ґ–Є–≤–∞–µ–Љ–Њ—Б—В—М—О —В—А–Є-—З–µ—В—Л—А–µ –≥–Њ–і–∞ [6вАУ8]. –Я—А–Њ–≥—А–µ—Б—Б–Є—А—Г—О—Й–Є–є —Д–µ–љ–Њ—В–Є–њ —Д–Є–±—А–Њ–Ј–Є—А–Њ–≤–∞–љ–Є—П –ї–µ–≥–Ї–Є—Е —В–∞–Ї–ґ–µ –≤—Б—В—А–µ—З–∞–µ—В—Б—П —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–і—А—Г–≥–Є–Љ –Ш–Ч–Ы, –≤¬†—З–∞—Б—В–љ–Њ—Б—В–Є —А–∞–Ј–≤–Є–≤–∞—О—Й–Є–Љ—Б—П –љ–∞ —Д–Њ–љ–µ —А–µ–≤–Љ–∞—В–Њ–Є–і–љ–Њ–≥–Њ –∞—А—В—А–Є—В–∞ (–†–Р) [2, 9], —Б–Є—Б—В–µ–Љ–љ–Њ–є —Б–Ї–ї–µ—А–Њ–і–µ—А–Љ–Є–Є (–°–°–Ф) [3, 10], –і–µ—А–Љ–∞—В–Њ–Љ–Є–Њ–Ј–Є—В–∞/–њ–Њ–ї–Є–Љ–Є–Њ–Ј–Є—В–∞ (–Ф–Ь/–Я–Ь) [11], —Б–∞—А–Ї–Њ–Є–і–Њ–Ј–∞ [12], —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–≥–Њ –≥–Є–њ–µ—А—З—Г–≤—Б—В–≤–Є—В–µ–ї—М–љ–Њ–≥–Њ –њ–љ–µ–≤–Љ–Њ–љ–Є—В–∞ (–У–Я) [13] –Є¬†–Є–љ—Л—Е –≤–∞—А–Є–∞–љ—В–Њ–≤ –Ш–Ч–Ы [6].
–Я—А–Њ–≥—А–µ—Б—Б–Є—А—Г—О—Й–Є–є —Д–µ–љ–Њ—В–Є–њ –Ш–Ч–Ы –њ—А–Є —А–∞–Ј–ї–Є—З–љ—Л—Е —Б–Њ—Б—В–Њ—П–љ–Є—П—Е –Є–Љ–µ–µ—В –Љ–љ–Њ–≥–Њ –Њ–±—Й–Є—Е –њ–∞—В–Њ–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Є—Е –Љ–µ—Е–∞–љ–Є–Ј–Љ–Њ–≤, –Ї–Њ—В–Њ—А—Л–µ —Г–њ—А–∞–≤–ї—П—О—В –њ—А–Њ—Ж–µ—Б—Б–Њ–Љ —Д–Є–±—А–Њ–Ј–Є—А–Њ–≤–∞–љ–Є—П –Є¬†–њ—А–Є–≤–Њ–і—П—В –Ї¬†–љ–µ–Њ–±—А–∞—В–Є–Љ–Њ–є –њ–Њ—В–µ—А–µ —Ж–µ–ї–Њ—Б—В–љ–Њ—Б—В–Є —Н–њ–Є—В–µ–ї–Є–∞–ї—М–љ–Њ–≥–Њ/—Н–љ–і–Њ—В–µ–ї–Є–∞–ї—М–љ–Њ–≥–Њ –±–∞—А—М–µ—А–∞, —А–∞–Ј—А—Г—И–µ–љ–Є—О –∞—А—Е–Є—В–µ–Ї—В—Г—А—Л –ї–µ–≥–Ї–Њ–≥–Њ –Є¬†–њ–Њ—В–µ—А–µ –Є–Љ —Б–≤–Њ–µ–є —Д—Г–љ–Ї—Ж–Є–Є [6, 14, 15].
–Ъ–ї–Є–љ–Є—З–µ—Б–Ї–Њ–µ –Ј–љ–∞—З–µ–љ–Є–µ –њ—А–Є —А–µ–≤–Љ–∞—В–Є—З–µ—Б–Ї–Є—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П—Е
–Ш–љ—В–µ—А—Б—В–Є—Ж–Є–∞–ї—М–љ–Њ–µ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–µ –ї–µ–≥–Ї–Є—Е –≤—Б—В—А–µ—З–∞–µ—В—Б—П –њ—А–Є —А–∞–Ј–љ—Л—Е —А–µ–≤–Љ–∞—В–Є—З–µ—Б–Ї–Є—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П—Е, –њ—А–µ–Є–Љ—Г—Й–µ—Б—В–≤–µ–љ–љ–Њ –њ—А–Є –Ї–ї–∞—Б—Б–Є—З–µ—Б–Ї–Є—Е –∞—Г—В–Њ–Є–Љ–Љ—Г–љ–љ—Л—Е, –і–ї—П –Ї–Њ—В–Њ—А—Л—Е —Е–∞—А–∞–Ї—В–µ—А–љ–∞ –≤—Л—А–∞–±–Њ—В–Ї–∞ –∞—Г—В–Њ–∞–љ—В–Є—В–µ–ї. –Э–∞–Є–±–Њ–ї–µ–µ —В–Є–њ–Є—З–љ–Њ –Ш–Ч–Ы –і–ї—П –°–°–Ф, –†–Р, –Ф–Ь/–Я–Ь, —Б–Љ–µ—И–∞–љ–љ–Њ–≥–Њ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–Њ–є —В–Ї–∞–љ–Є (–°–Ч–°–Ґ), –±–Њ–ї–µ–Ј–љ–Є –®–µ–≥—А–µ–љ–∞ (–С–®), –Љ–µ–љ–µ–µ —В–Є–њ–Є—З–љ–Њ¬†вАУ –і–ї—П —Б–Є—Б—В–µ–Љ–љ–Њ–є –Ї—А–∞—Б–љ–Њ–є –≤–Њ–ї—З–∞–љ–Ї–Є (–°–Ъ–Т), —Б–Є—Б—В–µ–Љ–љ—Л—Е –≤–∞—Б–Ї—Г–ї–Є—В–Њ–≤ (–°–Т) (—А–Є—Б. 2) [5, 16, 17]. –Ю–і–љ–∞–Ї–Њ –Ш–Ч–Ы –љ–µ –µ–і–Є–љ—Б—В–≤–µ–љ–љ—Л–є –≤–∞—А–Є–∞–љ—В –њ–Њ—А–∞–ґ–µ–љ–Є—П –ї–µ–≥–Ї–Є—Е –њ—А–Є —Г–Ї–∞–Ј–∞–љ–љ—Л—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П—Е. –Ь–Њ–≥—Г—В —А–∞–Ј–≤–Є–≤–∞—В—М—Б—П –Є–љ—Д–Є–ї—М—В—А–∞—В–Є–≤–љ—Л–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П, —Б—Г—Е–Њ–є –Є¬†–≤—Л–њ–Њ—В–љ–Њ–є –њ–ї–µ–≤—А–Є—В, –ї–µ–≥–Њ—З–љ—Л–є –≤–∞—Б–Ї—Г–ї–Є—В, –≥–µ–Љ–Њ—А—А–∞–≥–Є—З–µ—Б–Ї–Є–є –∞–ї—М–≤–µ–Њ–ї–Є—В, –±—А–Њ–љ—Е–Є–Њ–ї–Є—В –Є¬†–і—А. –Ф–ї—П –Ш–Ч–Ы –љ–∞–Є–±–Њ–ї–µ–µ —Е–∞—А–∞–Ї—В–µ—А–љ–Њ —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–µ –њ—А–Њ–≥—А–µ—Б—Б–Є—А—Г—О—Й–µ–µ —В–µ—З–µ–љ–Є–µ —Б¬†—А–∞–Ј–≤–Є—В–Є–µ–Љ –љ–µ–Њ–±—А–∞—В–Є–Љ—Л—Е –Є–Ј–Љ–µ–љ–µ–љ–Є–є –≤¬†–ї–µ–≥–Ї–Є—Е.
–Я–∞—Ж–Є–µ–љ—В—Л —Б¬†—А–µ–≤–Љ–∞—В–Є—З–µ—Б–Ї–Є–Љ–Є –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П–Љ–Є –Є¬†–Ш–Ч–Ы —Б–Њ—Б—В–∞–≤–ї—П—О—В —Б—Г—Й–µ—Б—В–≤–µ–љ–љ—Г—О —З–∞—Б—В—М –±–Њ–ї—М–љ—Л—Е –Ш–Ч–Ы. –°–Њ–≥–ї–∞—Б–љ–Њ –і–∞–љ–љ—Л–Љ, –њ—А–µ–і—Б—В–∞–≤–ї–µ–љ–љ—Л–Љ P. Rivera-Ortega –Є¬†—Б–Њ–∞–≤—В. [18], –љ–∞ –Ш–Ч–Ы –њ—А–Є —А–µ–≤–Љ–∞—В–Є—З–µ—Б–Ї–Є—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П—Е –њ—А–Є—Е–Њ–і–Є—В—Б—П –Њ—В 2,9 –і–Њ 34,8%. –Ф–Њ–ї—П –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–Я–§-–Ш–Ч–Ы –љ–µ–Є–Ј–≤–µ—Б—В–љ–∞. –Ю–і–љ–∞–Ї–Њ, –њ–Њ –Њ—Ж–µ–љ–Ї–∞–Љ —Н–Ї—Б–њ–µ—А—В–Њ–≤ –Є¬†–і–∞–љ–љ—Л–Љ —Б–Є—Б—В–µ–Љ–∞—В–Є—З–µ—Б–Ї–Є—Е –Њ–±–Ј–Њ—А–Њ–≤, –Њ–љ–∞ –Љ–Њ–ґ–µ—В –Ї–Њ–ї–µ–±–∞—В—М—Б—П –Њ—В 13 –і–Њ 40% –Њ—В –Њ–±—Й–µ–≥–Њ —З–Є—Б–ї–∞ –±–Њ–ї—М–љ—Л—Е –Ш–Ч–Ы, –∞—Б—Б–Њ—Ж–Є–Є—А–Њ–≤–∞–љ–љ—Л–Љ —Б¬†—А–µ–≤–Љ–∞—В–Є—З–µ—Б–Ї–Є–Љ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–µ–Љ [5].
–°–Є—Б—В–µ–Љ–љ–∞—П —Б–Ї–ї–µ—А–Њ–і–µ—А–Љ–Є—П
–°–Є—Б—В–µ–Љ–љ–∞—П —Б–Ї–ї–µ—А–Њ–і–µ—А–Љ–Є—П¬†вАУ –∞—Г—В–Њ–Є–Љ–Љ—Г–љ–љ–Њ–µ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–µ —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–Њ–є —В–Ї–∞–љ–Є, –Ї–Њ—В–Њ—А–Њ–µ —Е–∞—А–∞–Ї—В–µ—А–Є–Ј—Г–µ—В—Б—П –њ—А–Њ–≥—А–µ—Б—Б–Є—А—Г—О—Й–Є–Љ –Є–Ј–±—Л—В–Њ—З–љ—Л–Љ —Д–Є–±—А–Њ–Ј–Њ–Љ –Ї–Њ–ґ–Є –Є¬†–≤–љ—Г—В—А–µ–љ–љ–Є—Е –Њ—А–≥–∞–љ–Њ–≤, –≤–∞–Ј–Њ—Б–њ–∞—Б—В–Є—З–µ—Б–Ї–Є–Љ–Є —А–µ–∞–Ї—Ж–Є—П–Љ–Є (—Д–µ–љ–Њ–Љ–µ–љ –†–µ–є–љ–Њ) —Б¬†—Д–Є–±—А–Њ–Є–љ—В–Є–Љ–∞–ї—М–љ–Њ–є –њ—А–Њ–ї–Є—Д–µ—А–∞—Ж–Є–µ–є –Љ–Є–Ї—А–Њ—Б–Њ—Б—Г–і–Њ–≤ [19]. –Ю–і–љ–Є–Љ –Є–Ј –љ–∞–Є–±–Њ–ї–µ–µ —Е–∞—А–∞–Ї—В–µ—А–љ—Л—Е –≤–Є—Б—Ж–µ—А–∞–ї—М–љ—Л—Е –њ—А–Њ—П–≤–ї–µ–љ–Є–є –°–°–Ф –љ–∞—А—П–і—Г —Б¬†–ї–µ–≥–Њ—З–љ–Њ–є –∞—А—В–µ—А–Є–∞–ї—М–љ–Њ–є –≥–Є–њ–µ—А—В–µ–љ–Ј–Є–µ–є (–Ы–Р–У) —Б—З–Є—В–∞–µ—В—Б—П –Ш–Ч–Ы.
–Т –Ї–Њ–љ—Ж–µ XX –≤. –њ—П—В–Є–ї–µ—В–љ—П—П –≤—Л–ґ–Є–≤–∞–µ–Љ–Њ—Б—В—М –њ—А–Є –°–°–Ф —Б–Њ—Б—В–∞–≤–ї—П–ї–∞ 77,9%, –і–µ—Б—П—В–Є–ї–µ—В–љ—П—П¬†вАУ 55,1%. –Ю–і–љ–∞–Ї–Њ –Њ—В–Љ–µ—З–µ–љ–∞ –Њ—В—З–µ—В–ї–Є–≤–∞—П —В–µ–љ–і–µ–љ—Ж–Є—П –Ї¬†–≤–Њ–Ј—А–∞—Б—В–∞–љ–Є—О —З–Є—Б–ї–∞ –ї–µ—В–∞–ї—М–љ—Л—Е –Є—Б—Е–Њ–і–Њ–≤ –≤¬†—Б–≤—П–Ј–Є —Б¬†–њ–Њ—А–∞–ґ–µ–љ–Є–µ–Љ –ї–µ–≥–Ї–Є—Е (–Ш–Ч–Ы –Є¬†–Ы–Р–У). –Т¬†–љ–∞—Б—В–Њ—П—Й–µ–µ –≤—А–µ–Љ—П –њ–Њ—А–∞–ґ–µ–љ–Є–µ –ї–µ–≥–Ї–Є—Е –њ—А–Є–Ј–љ–∞–љ–Њ –≤–µ–і—Г—Й–µ–є –њ—А–Є—З–Є–љ–Њ–є —Б–Љ–µ—А—В–Є –њ—А–Є –°–°–Ф [3].
–Э–µ—Б–Љ–Њ—В—А—П –љ–∞ –Ј–љ–∞—З–Є—В–µ–ї—М–љ—Л–µ —А–∞–Ј–ї–Є—З–Є—П –≤¬†—А–∞–Ј–љ—Л—Е –Ї–Њ–≥–Њ—А—В–∞—Е, —З–∞—Б—В–Њ—В–∞ –≤—Л—П–≤–ї–µ–љ–Є—П –Ш–Ч–Ы –њ—А–Є –°–°–Ф –і–Њ—Б—В–∞—В–Њ—З–љ–Њ –≤–µ–ї–Є–Ї–∞. –Я–Њ –і–∞–љ–љ—Л–Љ —А–µ–µ—Б—В—А–∞ CSRG, –Ш–Ч–Ы —Б¬†–њ–Њ–Љ–Њ—Й—М—О –Ї–Њ–Љ–њ—М—О—В–µ—А–љ–Њ–є —В–Њ–Љ–Њ–≥—А–∞—Д–Є–Є –≤—Л—Б–Њ–Ї–Њ–≥–Њ —А–∞–Ј—А–µ—И–µ–љ–Є—П (–Ъ–Ґ–Т–†) –±—Л–ї–Њ –і–Є–∞–≥–љ–Њ—Б—В–Є—А–Њ–≤–∞–љ–Њ —Г¬†64% –±–Њ–ї—М–љ—Л—Е –°–°–Ф [5]. –°–Њ–≥–ї–∞—Б–љ–Њ –Њ–±–Ј–Њ—А—Г –і–∞–љ–љ—Л—Е 21 —Ж–µ–љ—В—А–∞ –≤¬†–Ш—Б–њ–∞–љ–Є–Є, –Є–Ј 1374 –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–°–°–Ф —Г¬†43% –Њ–±–љ–∞—А—Г–ґ–µ–љ—Л –њ—А–Є–Ј–љ–∞–Ї–Є –ї–µ–≥–Њ—З–љ–Њ–≥–Њ —Д–Є–±—А–Њ–Ј–∞ –њ—А–Є —А–µ–љ—В–≥–µ–љ–Њ–≥—А–∞—Д–Є–Є –≥—А—Г–і–љ–Њ–є –Ї–ї–µ—В–Ї–Є –Є–ї–Є –Ъ–Ґ–Т–† [20]. –Я–Њ –і—А—Г–≥–Є–Љ –і–∞–љ–љ—Л–Љ, —З–∞—Б—В–Њ—В–∞ –≤—Л—П–≤–ї–µ–љ–Є—П –Ш–Ч–Ы –њ—А–Є –°–°–Ф —Б–Њ—Б—В–∞–≤–ї—П–µ—В –і–Њ 70вАУ80% [21, 22].
–Ъ —Д–∞–Ї—В–Њ—А–∞–Љ —А–Є—Б–Ї–∞ —А–∞–Ј–≤–Є—В–Є—П –Є–ї–Є –њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є—П –Ш–Ч–Ы —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–°–°–Ф –Њ—В–љ–Њ—Б—П—В—Б—П –і–Є—Д—Д—Г–Ј–љ–Њ–µ –њ–Њ—А–∞–ґ–µ–љ–Є–µ –Ї–Њ–ґ–Є (–і–Є—Д—Д—Г–Ј–љ–∞—П —Б–Ї–ї–µ—А–Њ–і–µ—А–Љ–Є—П), –њ—А–Є–љ–∞–і–ї–µ–ґ–љ–Њ—Б—В—М –Ї¬†–∞—Д—А–Њ–∞–Љ–µ—А–Є–Ї–∞–љ—Ж–∞–Љ, –њ–Њ–ґ–Є–ї–Њ–є –≤–Њ–Ј—А–∞—Б—В –≤¬†–і–µ–±—О—В–µ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П, –±–Њ–ї–µ–µ –Ї–Њ—А–Њ—В–Ї–∞—П –њ—А–Њ–і–Њ–ї–ґ–Є—В–µ–ї—М–љ–Њ—Б—В—М –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П, –љ–∞–ї–Є—З–Є–µ –∞–љ—В–Є—В–µ–ї –Ї¬†—В–Њ–њ–Њ–Є–Ј–Њ–Љ–µ—А–∞–Ј–µ 1 (–∞–љ—В–Є-Scl-70) –Є–ї–Є –Њ—В—Б—Г—В—Б—В–≤–Є–µ –∞–љ—В–Є—Ж–µ–љ—В—А–Њ–Љ–µ—А–љ—Л—Е –∞–љ—В–Є—В–µ–ї [22, 23].
–Э–µ—Б–Љ–Њ—В—А—П –љ–∞ —В–Њ —З—В–Њ –і–ї—П –°–°–Ф, –Ї–∞–Ї –Є¬†–і–ї—П –±–Њ–ї—М—И–Є–љ—Б—В–≤–∞ –і—А—Г–≥–Є—Е —А–µ–≤–Љ–∞—В–Є—З–µ—Б–Ї–Є—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є, –Ј–∞ –Є—Б–Ї–ї—О—З–µ–љ–Є–µ–Љ –†–Р, —Е–∞—А–∞–Ї—В–µ—А–µ–љ –њ–∞—В—В–µ—А–љ –љ–µ—Б–њ–µ—Ж–Є—Д–Є—З–µ—Б–Ї–Њ–є –Є–љ—В–µ—А—Б—В–Є—Ж–Є–∞–ї—М–љ–Њ–є –њ–љ–µ–≤–Љ–Њ–љ–Є–Є [24, 25], –њ—А–Њ–≥—А–µ—Б—Б–Є—А—Г—О—Й–µ–µ —В–µ—З–µ–љ–Є–µ –Ш–Ч–Ы —П–≤–ї—П–µ—В—Б—П —В–Є–њ–Є—З–љ—Л–Љ. –°–Ї–Њ—А–µ–µ –≤—Б–µ–≥–Њ, —Н—В–Њ —Б–≤—П–Ј–∞–љ–Њ —Б¬†–Њ–њ—А–µ–і–µ–ї–µ–љ–љ—Л–Љ —Б—Г–±—В–Є–њ–Њ–Љ –°–°–Ф, –∞—Б—Б–Њ—Ж–Є–Є—А–Њ–≤–∞–љ–љ—Л–Љ —Б¬†–љ–∞–ї–Є—З–Є–µ–Љ –∞–љ—В–Є-Scl-70 [3, 23, 25]. –Ш–Ч–Ы –Њ–±—Л—З–љ–Њ —А–∞–Ј–≤–Є–≤–∞–µ—В—Б—П –≤¬†—В–µ—З–µ–љ–Є–µ –њ–µ—А–≤—Л—Е —В—А–µ—Е –ї–µ—В –Њ—В –і–µ–±—О—В–∞ –°–°–Ф, —Г¬†40вАУ75% –њ–∞—Ж–Є–µ–љ—В–Њ–≤ –Њ—В–Љ–µ—З–∞–µ—В—Б—П —Б–љ–Є–ґ–µ–љ–Є–µ —Д—Г–љ–Ї—Ж–Є–Є –ї–µ–≥–Ї–Є—Е [26]. –Т¬†—А–∞–Љ–Ї–∞—Е –њ—А–Њ–µ–Ї—В–∞ EUSTAR (European League Against Rheumatism (EULAR) Scleroderma Trials and Research Group) —Г—Б—В–∞–љ–Њ–≤–ї–µ–љ–Њ, —З—В–Њ –њ—А–Є–Љ–µ—А–љ–Њ —Г¬†—В—А–µ—В–Є –Є–Ј 695 –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–°–°–Ф –њ—А–Њ–≥–љ–Њ–Ј–Є—А—Г–µ–Љ—Л–є —Г—А–Њ–≤–µ–љ—М –і–Є—Д—Д—Г–Ј–Є–Њ–љ–љ–Њ–є —Б–њ–Њ—Б–Њ–±–љ–Њ—Б—В–Є –ї–µ–≥–Ї–Є—Е –њ–Њ –Љ–Њ–љ–Њ–Њ–Ї—Б–Є–і—Г —Г–≥–ї–µ—А–Њ–і–∞ (DLCO) —Б–љ–Є–ґ–∞–µ—В—Б—П –±–Њ–ї–µ–µ —З–µ–Љ –љ–∞ 50% –≤¬†—В–µ—З–µ–љ–Є–µ —В—А–µ—Е –ї–µ—В –њ–Њ—Б–ї–µ –њ–Њ—П–≤–ї–µ–љ–Є—П –њ–µ—А–≤—Л—Е —Б–Є–Љ–њ—В–Њ–Љ–Њ–≤ (—Д–µ–љ–Њ–Љ–µ–љ–∞ –†–µ–є–љ–Њ) [27]. –Т¬†—Ж–µ–ї–Њ–Љ –Я–§-–Ш–Ч–Ы –≤—Б—В—А–µ—З–∞–µ—В—Б—П —Г¬†25вАУ30% –±–Њ–ї—М–љ—Л—Е –°–°–Ф –≤¬†–Ј–∞–≤–Є—Б–Є–Љ–Њ—Б—В–Є –Њ—В –Ї—А–Є—В–µ—А–Є–µ–≤ –і–Є–∞–≥–љ–Њ—Б—В–Є–Ї–Є [22].
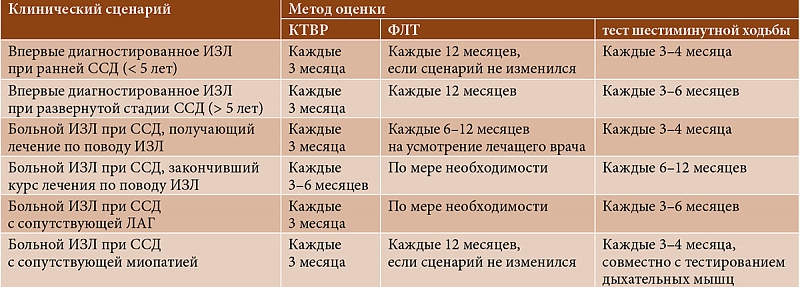
–Ф–ї—П –Љ–Њ–љ–Є—В–Њ—А–Є–љ–≥–∞ —Б–Њ—Б—В–Њ—П–љ–Є—П —В–∞–Ї–Є—Е –±–Њ–ї—М–љ—Л—Е –њ—А–µ–і–ї–Њ–ґ–µ–љ –∞–ї–≥–Њ—А–Є—В–Љ –Ї–ї–Є–љ–Є–Ї–Њ-–Є–љ—Б—В—А—Г–Љ–µ–љ—В–∞–ї—М–љ—Л—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є –≤¬†–Ј–∞–≤–Є—Б–Є–Љ–Њ—Б—В–Є –Њ—В –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Њ–≥–Њ —Б—Ж–µ–љ–∞—А–Є—П (—В–∞–±–ї. 1) [26]. –Ф–ї—П –љ–µ–≥–Њ —Е–∞—А–∞–Ї—В–µ—А–љ–∞ –Ї–Њ–Љ–њ–ї–µ–Ї—Б–љ–Њ—Б—В—М –Њ–±—Б–ї–µ–і–Њ–≤–∞–љ–Є—П –Є¬†–≤—Л—Б–Њ–Ї–∞—П —З–∞—Б—В–Њ—В–∞ –њ—А–Њ–≤–µ–і–µ–љ–Є—П –Ъ–Ґ–Т–†.
–Э–µ–Њ–±—Е–Њ–і–Є–Љ–Њ –Њ—В–Љ–µ—В–Є—В—М, —З—В–Њ –і–ї—П –≤—Л—П–≤–ї–µ–љ–Є—П –Я–§-–Ш–Ч–Ы –њ—А–Є –°–°–Ф –±—Л–ї–Є –њ—А–µ–і–њ—А–Є–љ—П—В—Л –њ–Њ–њ—Л—В–Ї–Є –Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞—В—М –Ъ–Ґ–Т–† –Є¬†—Д—Г–љ–Ї—Ж–Є–Њ–љ–∞–ї—М–љ—Л–µ –ї–µ–≥–Њ—З–љ—Л–µ —В–µ—Б—В—Л (–§–Ы–Ґ). –Т¬†–Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П—Е SLS I (NCT00004563) –Є¬†II (NCT00883129) –±–Њ–ї–µ–µ –≤—Л—Б–Њ–Ї–Є–є –Є—Б—Е–Њ–і–љ—Л–є –Ї–Њ–ґ–љ—Л–є —Б—З–µ—В, –њ–Њ–ґ–Є–ї–Њ–є –≤–Њ–Ј—А–∞—Б—В, —Б–љ–Є–ґ–µ–љ–Є–µ —Д–Њ—А—Б–Є—А–Њ–≤–∞–љ–љ–Њ–є –ґ–Є–Ј–љ–µ–љ–љ–Њ–є –µ–Љ–Ї–Њ—Б—В–Є –ї–µ–≥–Ї–Є—Е (–§–Ц–Х–Ы) –Є¬†DLCO –≤¬†—В–µ—З–µ–љ–Є–µ –і–≤—Г—Е –ї–µ—В –±—Л–ї–Є –љ–µ–Ј–∞–≤–Є—Б–Є–Љ–Њ —Б–≤—П–Ј–∞–љ—Л —Б¬†–њ–Њ–≤—Л—И–µ–љ–љ—Л–Љ —А–Є—Б–Ї–Њ–Љ —Б–Љ–µ—А—В–Є [28]. –°–Є—Б—В–µ–Љ–∞, —З–∞—Б—В–Њ –љ–∞–Ј—Л–≤–∞–µ–Љ–∞—П –Ї—А–Є—В–µ—А–Є—П–Љ–Є Goh, –≤—Л–і–µ–ї—П–µ—В –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†—А–∞—Б–њ—А–Њ—Б—В—А–∞–љ–µ–љ–љ—Л–Љ (–±–Њ–ї–µ–µ 20% –њ–Њ—А–∞–ґ–µ–љ–Є—П –ї–µ–≥–Ї–Є—Е –њ–Њ –і–∞–љ–љ—Л–Љ –Ъ–Ґ–Т–† –Є–ї–Є 10вАУ20% –њ–Њ –Ъ–Ґ–Т–† –Є¬†–§–Ц–Х–Ы –Љ–µ–љ–µ–µ 70% –Њ—В –њ—А–Њ–≥–љ–Њ–Ј–Є—А—Г–µ–Љ–Њ–є) –Є–ї–Є –Њ–≥—А–∞–љ–Є—З–µ–љ–љ—Л–Љ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–µ–Љ (–Љ–µ–љ–µ–µ 20% –њ–Њ—А–∞–ґ–µ–љ–Є—П –ї–µ–≥–Ї–Є—Е –њ–Њ –і–∞–љ–љ—Л–Љ –Ъ–Ґ–Т–† –Є–ї–Є 10вАУ20% –њ–Њ –Ъ–Ґ–Т–† –Є¬†–§–Ц–Х–Ы 70% –Є¬†–±–Њ–ї–µ–µ –Њ—В –њ—А–Њ–≥–љ–Њ–Ј–Є—А—Г–µ–Љ–Њ–є). –Э–∞–ї–Є—З–Є–µ —Г–Ї–∞–Ј–∞–љ–љ—Л—Е –Ї—А–Є—В–µ—А–Є–µ–≤ –±—Л–ї–Њ –њ—А–µ–і–Є–Ї—В–Њ—А–Њ–Љ —Б–Љ–µ—А—В–љ–Њ—Б—В–Є (–Њ—В–љ–Њ—И–µ–љ–Є–µ —А–Є—Б–Ї–Њ–≤ (–Ю–†) 3,46 –њ—А–Є 95%-–љ–Њ–Љ –і–Њ–≤–µ—А–Є—В–µ–ї—М–љ–Њ–Љ –Є–љ—В–µ—А–≤–∞–ї–µ (–Ф–Ш) 2,19вАУ5,46) [29]. –Т¬†–і—А—Г–≥–Њ–Љ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–Є –љ–∞–Є–±–Њ–ї–µ–µ —В–Њ—З–љ—Л–Љ –њ—А–µ–і–Є–Ї—В–Њ—А–Њ–Љ —Б–Љ–µ—А—В–љ–Њ—Б—В–Є –±—Л–ї–Њ –Њ—В–љ–Њ—Б–Є—В–µ–ї—М–љ–Њ–µ —Б–љ–Є–ґ–µ–љ–Є–µ –§–Ц–Х–Ы –љ–∞ 10% –Є¬†–±–Њ–ї–µ–µ –Є–ї–Є –Њ—В–љ–Њ—Б–Є—В–µ–ї—М–љ–Њ–µ —Б–љ–Є–ґ–µ–љ–Є–µ –§–Ц–Х–Ы –љ–∞ 5вАУ9% —Б¬†–Њ—В–љ–Њ—Б–Є—В–µ–ї—М–љ—Л–Љ —Б–љ–Є–ґ–µ–љ–Є–µ–Љ DLCO –±–Њ–ї–µ–µ 15% [30]. –Ю–і–љ–∞–Ї–Њ —И–Є—А–Њ–Ї–Є—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є –Ї—А–Є—В–µ—А–Є–µ–≤ –Я–§-–Ш–Ч–Ы –њ—А–Є –°–°–Ф –љ–µ –њ—А–Њ–≤–Њ–і–Є–ї–Њ—Б—М [22].
–Ш–љ—В–µ—А—Б—В–Є—Ж–Є–∞–ї—М–љ–Њ–µ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–µ –ї–µ–≥–Ї–Є—Е –њ—А–Є –°–°–Ф –∞—Б—Б–Њ—Ж–Є–Є—А—Г–µ—В—Б—П —Б¬†—А–∞–љ–љ–µ–є —Б–Љ–µ—А—В—М—О –±–Њ–ї—М–љ—Л—Е. –Т¬†–Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–Є –њ—А–Є—З–Є–љ —Б–Љ–µ—А—В–Є 1508 –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–°–°–Ф –Є–Ј —Ж–µ–љ—В—А–∞ –≤¬†–°–®–Р —Б–Љ–µ—А—В–љ–Њ—Б—В—М, —Б–≤—П–Ј–∞–љ–љ–∞—П —Б¬†—Д–Є–±—А–Њ–Ј–Њ–Љ –ї–µ–≥–Ї–Є—Е, —Г–≤–µ–ї–Є—З–Є–ї–∞—Б—М —Б¬†6% –≤¬†1972вАУ1976 –≥–≥. –і–Њ 33% –≤¬†1997вАУ2001 –≥–≥. [31]. –Я—А–Є –∞–љ–∞–ї–Є–Ј–µ –±–∞–Ј—Л –і–∞–љ–љ—Л—Е EUSTAR –≤¬†–Њ—В–љ–Њ—И–µ–љ–Є–Є 5850 –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Г—Б—В–∞–љ–Њ–≤–ї–µ–љ–Њ, —З—В–Њ —Б¬†2004 –њ–Њ 2008 –≥. –Є–Ј-–Ј–∞ –ї–µ–≥–Њ—З–љ–Њ–≥–Њ —Д–Є–±—А–Њ–Ј–∞ —Г–Љ–µ—А–ї–Њ 35% [32]. –Т–Њ –§—А–∞–љ—Ж–Є–Є —Б¬†2000 –њ–Њ 2011 –≥. –Є–Ј 2719 —Г–Љ–µ—А—И–Є—Е –±–Њ–ї—М–љ—Л—Е –°–°–Ф –њ–Њ—З—В–Є —Г¬†–њ–Њ–ї–Њ–≤–Є–љ—Л —Б–Љ–µ—А—В—М –±—Л–ї–∞ –≤—Л–Ј–≤–∞–љ–∞ —Б–µ—А–і–µ—З–љ—Л–Љ–Є –Є–ї–Є —А–µ—Б–њ–Є—А–∞—В–Њ—А–љ—Л–Љ–Є –њ—А–Є—З–Є–љ–∞–Љ–Є. –Я—А–Є —Н—В–Њ–Љ —З–∞—Б—В–Њ—В–∞ —Б–Љ–µ—А—В–µ–ї—М–љ—Л—Е —Б–ї—Г—З–∞–µ–≤, —Б–≤—П–Ј–∞–љ–љ—Л—Е —Б¬†–Ш–Ч–Ы, –Ј–∞ —Н—В–Њ—В –њ–µ—А–Є–Њ–і —Г–≤–µ–ї–Є—З–Є–ї–∞—Б—М [33].
–†–µ–≤–Љ–∞—В–Њ–Є–і–љ—Л–є –∞—А—В—А–Є—В
–†–µ–≤–Љ–∞—В–Њ–Є–і–љ—Л–є –∞—А—В—А–Є—В –њ—А–Є–Ј–љ–∞–љ –љ–∞–Є–±–Њ–ї–µ–µ —А–∞—Б–њ—А–Њ—Б—В—А–∞–љ–µ–љ–љ—Л–Љ —А–µ–≤–Љ–∞—В–Є—З–µ—Б–Ї–Є–Љ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–µ–Љ¬†вАУ –њ–Њ—А—П–і–Ї–∞ 1000 —Б–ї—Г—З–∞–µ–≤ –љ–∞ 100 —В—Л—Б. –љ–∞—Б–µ–ї–µ–љ–Є—П, –Є–ї–Є 1% –Њ–±—Й–µ–є —З–Є—Б–ї–µ–љ–љ–Њ—Б—В–Є, —З—В–Њ –љ–∞ –њ–Њ—А—П–і–Њ–Ї –њ—А–µ–≤–Њ—Б—Е–Њ–і–Є—В –≤—Б—В—А–µ—З–∞–µ–Љ–Њ—Б—В—М –°–°–Ф [17, 25]. –Ш–Ч–Ы¬†вАУ –Њ–і–љ–Њ –Є–Ј —В–Є–њ–Є—З–љ—Л—Е —Б–Є—Б—В–µ–Љ–љ—Л—Е –њ—А–Њ—П–≤–ї–µ–љ–Є–є –†–Р [34]. –Я–Њ –љ–µ–Ї–Њ—В–Њ—А—Л–Љ –і–∞–љ–љ—Л–Љ, –њ—А–Є–±–ї–Є–Ј–Є—В–µ–ї—М–љ–Њ —Г¬†10% –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–†–Р –љ–∞–±–ї—О–і–∞—О—В—Б—П –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є–µ —Б–Є–Љ–њ—В–Њ–Љ—Л –Ш–Ч–Ы, —Г¬†30%¬†вАУ —Б—Г–±–Ї–ї–Є–љ–Є—З–µ—Б–Ї–Њ–µ —В–µ—З–µ–љ–Є–µ [35, 36]. –Т¬†—А–∞–Ј–љ—Л—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П—Е –Ј–∞–±–Њ–ї–µ–≤–∞–µ–Љ–Њ—Б—В—М –Ш–Ч–Ы –≤–∞—А—М–Є—А–Њ–≤–∞–ї–∞—Б—М –≤¬†–Ј–∞–≤–Є—Б–Є–Љ–Њ—Б—В–Є –Њ—В –Є—Б—Б–ї–µ–і—Г–µ–Љ–Њ–є –њ–Њ–њ—Г–ї—П—Ж–Є–Є –Є¬†–Ї—А–Є—В–µ—А–Є–µ–≤ –µ–≥–Њ –Њ–њ—А–µ–і–µ–ї–µ–љ–Є—П [5]. –Т¬†—Ж–µ–ї–Њ–Љ –њ—А–Є –њ—А–Є–Љ–µ–љ–µ–љ–Є–Є –Ъ–Ґ–Т–† –≤—Л—П–≤–ї—П–µ–Љ–Њ—Б—В—М –Ш–Ч–Ы –њ—А–Є –†–Р –і–Њ—Б—В–∞—В–Њ—З–љ–Њ –≤—Л—Б–Њ–Ї–∞—П. –Ґ–∞–Ї, –≤¬†–Њ–і–љ–Њ–Љ –±—А–Є—В–∞–љ—Б–Ї–Њ–Љ —Ж–µ–љ—В—А–µ –њ—А–Є –∞–љ–∞–ї–Є–Ј–µ –і–∞–љ–љ—Л—Е 150 –∞–Љ–±—Г–ї–∞—В–Њ—А–љ—Л—Е –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–†–Р —Г—Б—В–∞–љ–Њ–≤–ї–µ–љ–Њ, —З—В–Њ —Г¬†28 (19%) –Ш–Ч–Ы –±—Л–ї–Њ –≤—Л—П–≤–ї–µ–љ–Њ –њ–Њ—Б–ї–µ –њ—А–Њ–≤–µ–і–µ–љ–Є—П –Ъ–Ґ–Т–† [36]. –Р–љ–∞–ї–Њ–≥–Є—З–љ—Л–µ —А–µ–Ј—Г–ї—М—В–∞—В—Л –њ–Њ–ї—Г—З–µ–љ—Л –Є—В–∞–ї—М—П–љ—Б–Ї–Є–Љ–Є –Є—Б—Б–ї–µ–і–Њ–≤–∞—В–µ–ї—П–Љ–Є [37]. –Т¬†–Ї–Є—В–∞–є—Б–Ї–Њ–Љ —А–µ—В—А–Њ—Б–њ–µ–Ї—В–Є–≤–љ–Њ–Љ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–Є —Б¬†—Г—З–∞—Б—В–Є–µ–Љ 550 –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–†–Р, –Ї–Њ—В–Њ—А—Л–Љ –±—Л–ї–∞ –≤—Л–њ–Њ–ї–љ–µ–љ–∞ –Ъ–Ґ–Т–†, –њ–Њ–Ї–∞–Ј–∞–љ–Њ, —З—В–Њ –і–∞–љ–љ—Л–є –Љ–µ—В–Њ–і –њ–Њ–Ј–≤–Њ–ї–Є–ї –≤—Л—П–≤–Є—В—М –Ш–Ч–Ы —Г¬†43,1%. –Я—А–Є —Н—В–Њ–Љ —Г¬†13,5% –±–Њ–ї—М–љ—Л—Е –Њ–љ–Њ –±—Л–ї–Њ –≤—Л—П–≤–ї–µ–љ–Њ –і–Њ –љ–∞—З–∞–ї–∞ –†–Р, —Г¬†69,6%¬†вАУ –≤¬†—В–µ—З–µ–љ–Є–µ –і–µ—Б—П—В–Є –ї–µ—В –Њ—В –љ–∞—З–∞–ї–∞ –†–Р, —Г¬†16,9%¬†вАУ –±–Њ–ї–µ–µ —З–µ–Љ —З–µ—А–µ–Ј –і–µ—Б—П—В—М –ї–µ—В –Њ—В –і–µ–±—О—В–∞ –†–Р [38].
–Я–Њ —А–∞—Б—З–µ—В–љ—Л–Љ –і–∞–љ–љ—Л–Љ, —А–∞—Б–њ—А–Њ—Б—В—А–∞–љ–µ–љ–љ–Њ—Б—В—М –Ш–Ч–Ы, –∞—Б—Б–Њ—Ж–Є–Є—А–Њ–≤–∞–љ–љ–Њ–≥–Њ —Б¬†–†–Р, –њ—А–Є –∞–Ї—В–Є–≤–љ–Њ–Љ –Њ–±—Б–ї–µ–і–Њ–≤–∞–љ–Є–Є –Љ–Њ–ґ–µ—В —Б–Њ—Б—В–∞–≤–ї—П—В—М –Њ—В 100 –і–Њ 400 —Б–ї—Г—З–∞–µ–≤ –љ–∞ 100 —В—Л—Б. –љ–∞—Б–µ–ї–µ–љ–Є—П, —З—В–Њ –Љ–љ–Њ–≥–Њ–Ї—А–∞—В–љ–Њ –њ—А–µ–≤—Л—И–∞–µ—В —В–∞–Ї–Њ–≤—Г—О –њ—А–Є –і—А—Г–≥–Є—Е –≤–∞—А–Є–∞–љ—В–∞—Е –Ш–Ч–Ы.
–Ф–ї–Є—В–µ–ї—М–љ–Њ–µ –≤—А–µ–Љ—П –Њ–±—Б—Г–ґ–і–∞–µ—В—Б—П —А–Њ–ї—М –Љ–µ—В–Њ—В—А–µ–Ї—Б–∞—В–∞, —И–Є—А–Њ–Ї–Њ –њ—А–Є–Љ–µ–љ—П–µ–Љ–Њ–≥–Њ –њ—А–Є –†–Р, –≤¬†—А–∞–Ј–≤–Є—В–Є–Є –Ш–Ч–Ы. –Э–µ–Њ–±—Е–Њ–і–Є–Љ–Њ –њ–Њ–і—З–µ—А–Ї–љ—Г—В—М, —З—В–Њ –њ–љ–µ–≤–Љ–Њ–љ–Є—В, –∞—Б—Б–Њ—Ж–Є–Є—А–Њ–≤–∞–љ–љ—Л–є —Б¬†—В–µ—А–∞–њ–Є–µ–є –Љ–µ—В–Њ—В—А–µ–Ї—Б–∞—В–Њ–Љ, –Њ—В–ї–Є—З–∞–µ—В—Б—П –Њ—В –Ш–Ч–Ы —Е–∞—А–∞–Ї—В–µ—А–Њ–Љ —В–µ—З–µ–љ–Є—П [41]. –Я–Њ—Б–Ї–Њ–ї—М–Ї—Г –Љ–µ—В–Њ—В—А–µ–Ї—Б–∞—В¬†вАУ –Њ–і–Є–љ –Є–Ј –Ї–ї—О—З–µ–≤—Л—Е –њ—А–µ–њ–∞—А–∞—В–Њ–≤ –і–ї—П –і–Њ—Б—В–Є–ґ–µ–љ–Є—П —Ж–µ–ї–µ–є —В–µ—А–∞–њ–Є–Є –†–Р –Є¬†—А–∞—Б–њ—А–Њ—Б—В—А–∞–љ–µ–љ–љ–Њ—Б—В—М –њ–Њ—В–µ–љ—Ж–Є–∞–ї—М–љ–Њ —Б–≤—П–Ј–∞–љ–љ—Л—Е —Б¬†–љ–Є–Љ –Њ—Б–ї–Њ–ґ–љ–µ–љ–Є–є –љ–µ–≤–µ–ї–Є–Ї–∞, —А–µ—И–µ–љ–Є–µ –Њ¬†–µ–≥–Њ –љ–∞–Ј–љ–∞—З–µ–љ–Є–Є –і–Њ–ї–ґ–љ–Њ –±—Л—В—М –≤–Ј–≤–µ—И–µ–љ–љ—Л–Љ [42]. –Я–Њ—Б–ї–µ–і–љ–Є–µ –і–∞–љ–љ—Л–µ –Љ–љ–Њ–≥–Њ—Ж–µ–љ—В—А–Њ–≤—Л—Е –љ–∞–±–ї—О–і–∞—В–µ–ї—М–љ—Л—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є ERAS –Є¬†ERAN —Б–≤–Є–і–µ—В–µ–ї—М—Б—В–≤—Г—О—В –Њ–± –Њ—В—Б—Г—В—Б—В–≤–Є–Є –њ—А—П–Љ–Њ–є —Б–≤—П–Ј–Є –Љ–µ–ґ–і—Г –љ–∞–Ј–љ–∞—З–µ–љ–Є–µ–Љ –Љ–µ—В–Њ—В—А–µ–Ї—Б–∞—В–∞ –Є¬†—А–∞–Ј–≤–Є—В–Є–µ–Љ –Ш–Ч–Ы –њ—А–Є –†–Р [43]. –≠—В–Њ –њ–Њ–і—В–≤–µ—А–ґ–і–∞—О—В –Є¬†—А–µ–Ј—Г–ї—М—В–∞—В—Л –љ–∞–±–ї—О–і–µ–љ–Є—П –Ј–∞ –±–Њ–ї—М—И–Њ–є –Ї–Њ–≥–Њ—А—В–Њ–є BRASS (n = 1419) [44]. –Ъ—А–Њ–Љ–µ —В–Њ–≥–Њ, –њ–Њ–ї—Г—З–µ–љ—Л –і–∞–љ–љ—Л–µ –Њ¬†–≤–Њ–Ј–Љ–Њ–ґ–љ–Њ—Б—В–Є –Љ–µ—В–Њ—В—А–µ–Ї—Б–∞—В–∞ –њ—А–µ–і–Њ—В–≤—А–∞—Й–∞—В—М —А–∞–Ј–≤–Є—В–Є–µ –Ш–Ч–Ы [43] –Є¬†—Г–≤–µ–ї–Є—З–Є–≤–∞—В—М –≤—Л–ґ–Є–≤–∞–µ–Љ–Њ—Б—В—М –±–Њ–ї—М–љ—Л—Е –†–Р —Б¬†–Ш–Ч–Ы [45].
–Ю—Б–љ–Њ–≤–љ—Л–Љ–Є —Д–∞–Ї—В–Њ—А–∞–Љ–Є —А–Є—Б–Ї–∞ —А–∞–Ј–≤–Є—В–Є—П –Ш–Ч–Ы –њ—А–Є –†–Р —Б—З–Є—В–∞—О—В—Б—П –Ї—Г—А–µ–љ–Є–µ, –Љ—Г–ґ—Б–Ї–Њ–є –њ–Њ–ї, –њ–Њ–ґ–Є–ї–Њ–є –≤–Њ–Ј—А–∞—Б—В, –≤—Л—Б–Њ–Ї–Є–µ —В–Є—В—А—Л —А–µ–≤–Љ–∞—В–Њ–Є–і–љ–Њ–≥–Њ —Д–∞–Ї—В–Њ—А–∞ –Є¬†–∞–љ—В–Є—В–µ–ї –Ї¬†—Ж–Є–Ї–ї–Є—З–µ—Б–Ї–Њ–Љ—Г —Ж–Є—В—А—Г–ї–ї–Є–љ–Є—А–Њ–≤–∞–љ–љ–Њ–Љ—Г –њ–µ–њ—В–Є–і—Г, –≤—Л—Б–Њ–Ї–∞—П –∞–Ї—В–Є–≤–љ–Њ—Б—В—М –†–Р [2, 37, 43, 44].
–Ъ–∞–Ї –±—Л–ї–Њ –Њ—В–Љ–µ—З–µ–љ–Њ —А–∞–љ–µ–µ, –њ—А–Є –†–Р –≤¬†—Б—Г—Й–µ—Б—В–≤–µ–љ–љ–Њ–Љ –Ї–Њ–ї–Є—З–µ—Б—В–≤–µ —Б–ї—Г—З–∞–µ–≤ –Ш–Ч–Ы –і–ї–Є—В–µ–ї—М–љ–Њ –њ—А–Њ—В–µ–Ї–∞–µ—В —Б—Г–±–Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є [2]. –Ф–ї—П –†–Р –≤¬†–Њ—В–ї–Є—З–Є–µ –Њ—В –і—А—Г–≥–Є—Е —А–µ–≤–Љ–∞—В–Є—З–µ—Б–Ї–Є—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є —Е–∞—А–∞–Ї—В–µ—А–µ–љ –њ–∞—В—В–µ—А–љ –Њ–±—Л—З–љ–Њ–є –Є–љ—В–µ—А—Б—В–Є—Ж–Є–∞–ї—М–љ–Њ–є –њ–љ–µ–≤–Љ–Њ–љ–Є–Є (–Ю–Ш–Я), —З—В–Њ —Б–∞–Љ–Њ –њ–Њ —Б–µ–±–µ –Њ–њ—А–µ–і–µ–ї—П–µ—В –±–Њ–ї—М—И—Г—О —Б–Ї–ї–Њ–љ–љ–Њ—Б—В—М –Ї¬†–њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є—О –ї–µ–≥–Њ—З–љ–Њ–≥–Њ —Д–Є–±—А–Њ–Ј–∞ –Є¬†–љ–µ–±–ї–∞–≥–Њ–њ—А–Є—П—В–љ–Њ–Љ—Г –њ—А–Њ–≥–љ–Њ–Ј—Г [24, 25, 46]. –Я—А–Њ–≥—А–µ—Б—Б–Є—А—Г—О—Й–µ–µ —Г—Е—Г–і—И–µ–љ–Є–µ –§–Ы–Ґ –љ–∞–±–ї—О–і–∞–ї–Њ—Б—М –≤¬†—В–µ—З–µ–љ–Є–µ –њ—П—В–Є –ї–µ—В —Г¬†8вАУ11% –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–Ш–Ч–Ы [5]. –Т¬†—Г–ґ–µ —Г–њ–Њ–Љ–Є–љ–∞–≤—И–µ–Љ—Б—П —А–∞–љ–µ–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–Є ERAS –љ–∞–ї–Є—З–Є–µ –Ш–Ч–Ы —Г¬†–±–Њ–ї—М–љ—Л—Е —А–∞–љ–љ–Є–Љ –†–Р –∞—Б—Б–Њ—Ж–Є–Є—А–Њ–≤–∞–ї–Њ—Б—М —Б¬†–љ–µ–±–ї–∞–≥–Њ–њ—А–Є—П—В–љ—Л–Љ –ґ–Є–Ј–љ–µ–љ–љ—Л–Љ –њ—А–Њ–≥–љ–Њ–Ј–Њ–Љ. –Ґ–∞–Ї, –Љ–µ–і–Є–∞–љ–∞ –≤—Л–ґ–Є–≤–∞–µ–Љ–Њ—Б—В–Є —Б–Њ—Б—В–∞–≤–Є–ї–∞ —В—А–Є –≥–Њ–і–∞ [47]. –Т¬†–∞–Љ–µ—А–Є–Ї–∞–љ—Б–Ї–Њ–Љ –љ–∞–±–ї—О–і–∞—В–µ–ї—М–љ–Њ–Љ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–Є –њ–∞—Ж–Є–µ–љ—В—Л —Б¬†–Ш–Ч–Ы –њ—А–Є –†–Р —Б¬†–њ–∞—В—В–µ—А–љ–Њ–Љ –Ю–Ш–Я –њ–Њ –і–∞–љ–љ—Л–Љ –Ъ–Ґ–Т–† –Є–Љ–µ–ї–Є —Е—Г–і—И—Г—О –≤—Л–ґ–Є–≤–∞–µ–Љ–Њ—Б—В—М –њ–Њ —Б—А–∞–≤–љ–µ–љ–Є—О —Б¬†–і—А—Г–≥–Є–Љ–Є –±–Њ–ї—М–љ—Л–Љ–Є –†–Р¬†вАУ 3,2 –њ—А–Њ—В–Є–≤ 6,6 –≥–Њ–і–∞ [48]. –Т¬†–Њ—В–љ–Њ—Б–Є—В–µ–ї—М–љ–Њ –љ–µ–±–Њ–ї—М—И–Њ–є —Д–Є–љ—Б–Ї–Њ–є –Ї–Њ–≥–Њ—А—В–µ (n = 59), –љ–∞—Е–Њ–і–Є–≤—И–µ–є—Б—П –њ–Њ–і –і–ї–Є—В–µ–ї—М–љ—Л–Љ –љ–∞–±–ї—О–і–µ–љ–Є–µ–Љ, —Г¬†–±–Њ–ї—М–љ—Л—Е –Ш–Ч–Ы –њ—А–Є –†–Р –Њ—В–Љ–µ—З–µ–љ–Њ —А–∞–Ј–ї–Є—З–Є–µ –≤¬†–Њ—В–љ–Њ—И–µ–љ–Є–Є –Ї–Њ–ї–Є—З–µ—Б—В–≤–∞ –≥–Њ—Б–њ–Є—В–∞–ї–Є–Ј–∞—Ж–Є–є –њ–Њ —А–µ—Б–њ–Є—А–∞—В–Њ—А–љ—Л–Љ –њ—А–Є—З–Є–љ–∞–Љ (1,9 ¬± 2,6 —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–Ю–Ш–Я –њ—А–Њ—В–Є–≤ 0,5 ¬± 0,9 —Г¬†–±–Њ–ї—М–љ—Л—Е –±–µ–Ј –Ю–Ш–Я, p = 0,004), –Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–љ–Є—П –Ї–Є—Б–ї–Њ—А–Њ–і–љ–Њ–є —В–µ—А–∞–њ–Є–Є (22,9 –њ—А–Њ—В–Є–≤ 0%, p = 0,016), –Ї–Њ–ї–Є—З–µ—Б—В–≤–∞ —Б–Љ–µ—А—В–µ–є (65,7 –њ—А–Њ—В–Є–≤ 41,7%, p = 0,046) –Є¬†—Б–љ–Є–ґ–µ–љ–Є—П DLCO (56 ¬± 20,6 –њ—А–Њ—В–Є–≤ 69 ¬± 20,2%, p = 0,021). –Ш–Ч–Ы –±—Л–ї–Њ –љ–∞–Є–±–Њ–ї–µ–µ —З–∞—Б—В–Њ–є –њ—А–Є—З–Є–љ–Њ–є —Б–Љ–µ—А—В–Є –±–Њ–ї—М–љ—Л—Е –†–Р¬†вАУ 39,4% [49].
–Ф—А—Г–≥–Є–µ —Б–Є—Б—В–µ–Љ–љ—Л–µ —А–µ–≤–Љ–∞—В–Є—З–µ—Б–Ї–Є–µ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П
–Я—А–Є –і—А—Г–≥–Є—Е —А–µ–≤–Љ–∞—В–Є—З–µ—Б–Ї–Є—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П—Е, —В–∞–Ї –ґ–µ –Ї–∞–Ї –њ—А–Є –°–°–Ф –Є¬†–†–Р, –і–Њ—Б—В–∞—В–Њ—З–љ–Њ –≤–µ–ї–Є–Ї–∞ –і–Њ–ї—П —Б–ї—Г—З–∞–µ–≤ —А–∞–Ј–≤–Є—В–Є—П –Я–§-–Ш–Ч–Ы. –Т—Л—Б–Њ–Ї–∞—П —З–∞—Б—В–Њ—В–∞ –Ш–Ч–Ы –љ–∞–±–ї—О–і–∞–µ—В—Б—П –њ—А–Є –С–®. –Ґ–∞–Ї, –≤¬†–Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–Є P. Guisado-Vasco –Є¬†—Б–Њ–∞–≤—В. –Є–Ј 102 –њ–Њ—Б–ї–µ–і–Њ–≤–∞—В–µ–ї—М–љ—Л—Е –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–С–® —Г¬†35,3% –і–Є–∞–≥–љ–Њ—Б—В–Є—А–Њ–≤–∞–љ–Њ –Ш–Ч–Ы. –Я—А–Є —Н—В–Њ–Љ —Г¬†–Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ–є —З–∞—Б—В–Є –Њ—В–Љ–µ—З–∞–ї–Њ—Б—М –њ—А–Њ–≥—А–µ—Б—Б–Є—А—Г—О—Й–µ–µ —В–µ—З–µ–љ–Є–µ [50]. –Т¬†–Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–Є, –њ—А–Њ–≤–µ–і–µ–љ–љ–Њ–Љ J.G. Parambil –Є¬†—Б–Њ–∞–≤—В., –њ—А–Є –∞–љ–∞–ї–Є–Ј–µ –і–∞–љ–љ—Л—Е 18 –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–С–® –Є¬†–Ш–Ч–Ы, –љ–∞–±–ї—О–і–∞–≤—И–Є—Е—Б—П –≤¬†—Б—А–µ–і–љ–µ–Љ –≤¬†—В–µ—З–µ–љ–Є–µ 38 –Љ–µ—Б—П—Ж–µ–≤, —Г¬†–њ—П—В–Є (28%) –±—Л–ї–Њ –њ—А–Њ–≥–љ–Њ–Ј–Є—А—Г–µ–Љ–Њ–µ —Б–љ–Є–ґ–µ–љ–Є–µ –§–Ц–Х–Ы вЙ• 10% –Є–ї–Є –њ—А–Њ–≥–љ–Њ–Ј–Є—А—Г–µ–Љ–Њ–µ —Б–љ–Є–ґ–µ–љ–Є–µ DLCO вЙ• 15%, –љ–µ—Б–Љ–Њ—В—А—П –љ–∞ –Є–Љ–Љ—Г–љ–Њ—Б—Г–њ—А–µ—Б—Б–Є—О [51]. –°–Њ–≥–ї–∞—Б–љ–Њ –і–∞–љ–љ—Л–Љ Y. Enomoto –Є¬†—Б–Њ–∞–≤—В., —Г¬†—В–∞–Ї–Є—Е –њ–∞—Ж–Є–µ–љ—В–Њ–≤ –њ—А–Њ–≥–љ–Њ–Ј –≤¬†—Ж–µ–ї–Њ–Љ –Љ–Њ–ґ–µ—В –±—Л—В—М –Њ—В–љ–Њ—Б–Є—В–µ–ї—М–љ–Њ –±–ї–∞–≥–Њ–њ—А–Є—П—В–љ—Л–Љ [52]. –Я—П—В–Є–ї–µ—В–љ—П—П –≤—Л–ґ–Є–≤–∞–µ–Љ–Њ—Б—В—М —Б–Њ—Б—В–∞–≤–Є–ї–∞ 87,3% –Њ—В –Њ–±—Й–µ–є –њ–Њ–њ—Г–ї—П—Ж–Є–Є –њ–∞—Ж–Є–µ–љ—В–Њ–≤ –љ–µ–Ј–∞–≤–Є—Б–Є–Љ–Њ –Њ—В –њ–∞—В—В–µ—А–љ–∞ –њ–Њ—А–∞–ґ–µ–љ–Є—П –ї–µ–≥–Ї–Є—Е –њ–Њ –і–∞–љ–љ—Л–Љ –Ъ–Ґ–Т–†. –Т¬†–Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–Є I. Marie –Є¬†—Б–Њ–∞–≤—В. —Г—Б—В–∞–љ–Њ–≤–ї–µ–љ–Њ, —З—В–Њ –Є–Ј 107 –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–Ш–Ч–Ы, –∞—Б—Б–Њ—Ж–Є–Є—А–Њ–≤–∞–љ–љ—Л–Љ —Б¬†–Я–Ь/–Ф–Ь, –њ–Њ–ї—Г—З–∞–≤—И–Є—Е –Є–Љ–Љ—Г–љ–Њ—Б—Г–њ—А–µ—Б—Б–∞–љ—В—Л, —Г¬†16% –±—Л–ї–Њ –њ—А–Њ–≥–љ–Њ–Ј–Є—А—Г–µ–Љ–Њ–µ —Б–љ–Є–ґ–µ–љ–Є–µ –§–Ц–Х–Ы вЙ• 10% –Є/–Є–ї–Є —Б–љ–Є–ґ–µ–љ–Є–µ DLCO вЙ• 15% –≤¬†—В–µ—З–µ–љ–Є–µ –њ–µ—А–Є–Њ–і–∞ –љ–∞–±–ї—О–і–µ–љ–Є—П –≤¬†—Б—А–µ–і–љ–µ–Љ 34 –Љ–µ—Б—П—Ж–∞ [11]. H. Yamakawa –Є¬†—Б–Њ–∞–≤—В. –њ—А–Є –∞–љ–∞–ї–Є–Ј–µ –і–∞–љ–љ—Л—Е 75 –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–∞–љ—В–Є—Б–Є–љ—В–µ—В–∞–Ј–љ—Л–Љ —Б–Є–љ–і—А–Њ–Љ–Њ–Љ –Є¬†–Ш–Ч–Ы, –њ–Њ–ї—Г—З–∞–≤—И–Є—Е –њ—А–Њ—В–Є–≤–Њ–≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ—Г—О —В–µ—А–∞–њ–Є—О, –≤—Л—П–≤–Є–ї–Є, —З—В–Њ —Г¬†—И–µ—Б—В–Є (8%) –±–Њ–ї—М–љ—Л—Е –љ–∞–±–ї—О–і–∞–ї–Њ—Б—М —Б–љ–Є–ґ–µ–љ–Є–µ –§–Ц–Х–Ы > 10% –Є/–Є–ї–Є DLCO > 15% –Њ—В –њ—А–Њ–≥–љ–Њ–Ј–Є—А—Г–µ–Љ–Њ–≥–Њ —З–µ—А–µ–Ј –≥–Њ–і –њ–Њ—Б–ї–µ –і–Є–∞–≥–љ–Њ—Б—В–Є–Ї–Є –Ш–Ч–Ы [53]. –Ш–Ј 36 –њ–∞—Ж–Є–µ–љ—В–Њ–≤, —Г¬†–Ї–Њ—В–Њ—А—Л—Е –Њ–ґ–Є–і–∞–ї–Њ—Б—М —Г–≤–µ–ї–Є—З–µ–љ–Є–µ –§–Ц–Х–Ы > 10% –Є/–Є–ї–Є DLCO > 15% —З–µ—А–µ–Ј –≥–Њ–і, —Г¬†12 (33%) –љ–∞–±–ї—О–і–∞–ї–Њ—Б—М —Г—Е—Г–і—И–µ–љ–Є–µ –≤¬†—В–µ—З–µ–љ–Є–µ —Б–ї–µ–і—Г—О—Й–Є—Е –і–≤—Г—Е –ї–µ—В [53].
–Ґ—А–∞–і–Є—Ж–Є–Њ–љ–љ—Л–µ –Љ–µ—В–Њ–і—Л –Љ–µ–і–Є–Ї–∞–Љ–µ–љ—В–Њ–Ј–љ–Њ–є —В–µ—А–∞–њ–Є–Є
–Т–Њ–Ј–Љ–Њ–ґ–љ–Њ—Б—В–Є —В—А–∞–і–Є—Ж–Є–Њ–љ–љ—Л—Е –Є–Љ–Љ—Г–љ–Њ—Б—Г–њ—А–µ—Б—Б–Є–≤–љ—Л—Е –Є¬†–њ—А–Њ—В–Є–≤–Њ–≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ—Л—Е –њ—А–µ–њ–∞—А–∞—В–Њ–≤ –њ—А–Є –Ш–Ч–Ы, –∞—Б—Б–Њ—Ж–Є–Є—А–Њ–≤–∞–љ–љ–Њ–Љ —Б¬†—А–µ–≤–Љ–∞—В–Є—З–µ—Б–Ї–Є–Љ–Є –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П–Љ–Є, –≤–µ—Б—М–Љ–∞ –Њ–≥—А–∞–љ–Є—З–µ–љ—Л (—В–∞–±–ї. 2) [2, 5, 17, 54вАУ59].
–Ъ–ї–Є–љ–Є—З–µ—Б–Ї–∞—П –њ—А–∞–Ї—В–Є–Ї–∞ –Њ—Б–љ–Њ–≤—Л–≤–∞–µ—В—Б—П –љ–∞ —А–µ–Ј—Г–ї—М—В–∞—В–∞—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є –≤¬†–Њ—В–љ–Њ—И–µ–љ–Є–Є –°–°–Ф. –Я–Њ–ї–Њ–ґ–Є—В–µ–ї—М–љ—Л–є —Н—Д—Д–µ–Ї—В —Ж–Є–Ї–ї–Њ—Д–Њ—Б—Д–∞–Љ–Є–і–∞ –Є¬†–Љ–Є–Ї–Њ—Д–µ–љ–Њ–ї–∞—В–∞ –Љ–Њ—Д–µ—В–Є–ї–∞ —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–°–°–Ф –і–Њ–Ї–∞–Ј–∞–љ –≤¬†—А–∞–љ–і–Њ–Љ–Є–Ј–Є—А–Њ–≤–∞–љ–љ—Л—Е –Ї–Њ–љ—В¬≠—А–Њ–ї–Є—А—Г–µ–Љ—Л—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П—Е (–†–Ъ–Ш) [57вАУ59]. –Ф—А—Г–≥–Є–µ –њ—А–µ–њ–∞—А–∞—В—Л, –Ї–Њ—В–Њ—А—Л–µ –Њ—В–љ–Њ—Б–Є—В–µ–ї—М–љ–Њ —Г—Б–њ–µ—И–љ–Њ –њ—А–Є–Љ–µ–љ—П—О—В—Б—П –і–ї—П –ї–µ—З–µ–љ–Є—П –Ш–Ч–Ы –њ—А–Є —А–µ–≤–Љ–∞—В–Є—З–µ—Б–Ї–Є—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П—Е, —В–∞–Ї–Є–µ –Ї–∞–Ї –∞–Ј–∞—В–Є–Њ–њ—А–Є–љ, –Љ–µ—В–Њ—В—А–µ–Ї—Б–∞—В, —А–Є—В—Г–Ї—Б–Є–Љ–∞–±, –Њ—Ж–µ–љ–Є–≤–∞–ї–Є—Б—М —В–Њ–ї—М–Ї–Њ –≤¬†–љ–∞–±–ї—О–і–∞—В–µ–ї—М–љ—Л—Е –Є¬†—А–µ—В—А–Њ—Б–њ–µ–Ї—В–Є–≤–љ—Л—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П—Е —Б¬†–љ–Є–Ј–Ї–Є–Љ —Г—А–Њ–≤–љ–µ–Љ –і–Њ–Ї–∞–Ј–∞—В–µ–ї—М–љ–Њ—Б—В–Є [5, 60, 61]. –≠—В–Њ –Ї–∞—Б–∞–µ—В—Б—П –Ї–∞–Ї –°–°–Ф, —В–∞–Ї –Є¬†–†–Р, –С–®, –Ф–Ь/–Я–Ь, –°–Т. –Ф—А—Г–≥–Є–µ –Љ–µ—В–Њ–і—Л –Љ–µ–і–Є–Ї–∞–Љ–µ–љ—В–Њ–Ј–љ–Њ–є —В–µ—А–∞–њ–Є–Є –Њ–Ї–∞–Ј–∞–ї–Є—Б—М –љ–µ—Н—Д—Д–µ–Ї—В–Є–≤–љ—Л–Љ–Є [56]. –Ш–Љ–µ–љ–љ–Њ –њ–Њ—Н—В–Њ–Љ—Г –≤¬†—А–µ–Ї–Њ–Љ–µ–љ–і–∞—Ж–Є–Є EULAR –њ–Њ –ї–µ—З–µ–љ–Є—О –°–°–Ф –і–ї—П —Б—Г–±—В–Є–њ–∞ —Б¬†–Ш–Ч–Ы —Д–∞–Ї—В–Є—З–µ—Б–Ї–Є –≤–Њ—И–µ–ї —В–Њ–ї—М–Ї–Њ —Ж–Є–Ї–ї–Њ—Д–Њ—Б—Д–∞–Љ–Є–і. –†–Њ—Б—Б–Є–є—Б–Ї–Є–µ –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є–µ —А–µ–Ї–Њ–Љ–µ–љ–і–∞—Ж–Є–Є —Б–Њ–і–µ—А–ґ–∞—В —В–∞–Ї–ґ–µ —Г–Ї–∞–Ј–∞–љ–Є–µ –љ–∞ –≤–Њ–Ј–Љ–Њ–ґ–љ–Њ—Б—В—М –њ—А–Є–Љ–µ–љ–µ–љ–Є—П –Љ–Є–Ї–Њ—Д–µ–љ–Њ–ї–∞—В–∞ –Љ–Њ—Д–µ—В–Є–ї–∞, –∞–Ј–∞—В–Є–Њ–њ—А–Є–љ–∞ –Є¬†—А–Є—В—Г–Ї—Б–Є–Љ–∞–±–∞.
–Ю–і–љ–∞–Ї–Њ —Г–Ї–∞–Ј–∞–љ–љ—Л–µ –њ—А–µ–њ–∞—А–∞—В—Л –Є–Љ–µ—О—В –і–Њ—Б—В–∞—В–Њ—З–љ–Њ –Њ–≥—А–∞–љ–Є—З–µ–љ–љ—Л–є —В–µ—А–∞–њ–µ–≤—В–Є—З–µ—Б–Ї–Є–є –њ–Њ—В–µ–љ—Ж–Є–∞–ї –≤¬†–Њ—В–љ–Њ—И–µ–љ–Є–Є —В–Њ—А–Љ–Њ–ґ–µ–љ–Є—П –ї–µ–≥–Њ—З–љ–Њ–≥–Њ —Д–Є–±—А–Њ–Ј–∞, –±—Г–і—Г—З–Є –±–Њ–ї–µ–µ –Є–Љ–Љ—Г–љ–Њ—Б—Г–њ—А–µ—Б—Б–Є–≤–љ—Л–Љ–Є, —З–µ–Љ –∞–љ—В–Є–њ—А–Њ–ї–Є—Д–µ—А–∞—В–Є–≤–љ—Л–Љ–Є. –Ъ—А–Њ–Љ–µ —В–Њ–≥–Њ, —З–∞—Б—В–Њ –≤–Њ–Ј–љ–Є–Ї–∞—О—В –њ—А–Њ–±–ї–µ–Љ—Л —Б¬†–Є—Е –њ–µ—А–µ–љ–Њ—Б–Є–Љ–Њ—Б—В—М—О (—Ж–Є–Ї–ї–Њ—Д–Њ—Б—Д–∞–Љ–Є–і) –Є–ї–Є –і–Њ—Б—В—Г–њ–љ–Њ—Б—В—М—О (–∞–Ј–∞—В–Є–Њ–њ—А–Є–љ).
–Ъ–Њ–љ–Ї—А–µ—В–љ—Л—Е —А–µ–Ї–Њ–Љ–µ–љ–і–∞—Ж–Є–є –≤—А–∞—З–µ–±–љ—Л—Е —Б–Њ–Њ–±—Й–µ—Б—В–≤ –њ–Њ –ї–µ—З–µ–љ–Є—О –Ш–Ч–Ы –њ—А–Є –†–Р –њ–Њ–Ї–∞ –љ–µ —Б—Г—Й–µ—Б—В–≤—Г–µ—В.
–Э–Є–љ—В–µ–і–∞–љ–Є–±¬†вАУ –љ–Њ–≤—Л–є –њ—А–µ–њ–∞—А–∞—В –і–ї—П –ї–µ—З–µ–љ–Є—П –Є–љ—В–µ—А—Б—В–Є—Ж–Є–∞–ї—М–љ–Њ–≥–Њ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П –ї–µ–≥–Ї–Є—Е
–Ф–ї—П —В–Њ–≥–Њ —З—В–Њ–±—Л –Ј–∞–Љ–µ–і–ї–Є—В—М –њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є–µ —Д–Є–±—А–Њ–Ј–Є—А—Г—О—Й–µ–≥–Њ –Ш–Ч–Ы —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†—Б–Є—Б—В–µ–Љ–љ—Л–Љ–Є –∞—Г—В–Њ–Є–Љ–Љ—Г–љ–љ—Л–Љ–Є –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П–Љ–Є, —Ж–µ–ї–µ—Б–Њ–Њ–±—А–∞–Ј–љ–Њ –і–Њ–±–∞–≤–Є—В—М –Ї¬†–Є–Љ–Љ—Г–љ–Њ—Б—Г–њ—А–µ—Б—Б–∞–љ—В–∞–Љ —Б–Њ–±—Б—В–≤–µ–љ–љ–Њ –∞–љ—В–Є–њ—А–Њ–ї–Є—Д–µ—А–∞—В–Є–≤–љ—Л–µ/–∞–љ—В–Є—Д–Є–±—А–Њ–Ј–љ—Л–µ –њ—А–µ–њ–∞—А–∞—В—Л. –Ъ–∞–Ї —Г–ґ–µ —Г–њ–Њ–Љ–Є–љ–∞–ї–Њ—Б—М, —Г¬†–Ш–Ч–Ы, –∞—Б—Б–Њ—Ж–Є–Є—А–Њ–≤–∞–љ–љ–Њ–≥–Њ —Б¬†–∞—Г—В–Њ–Є–Љ–Љ—Г–љ–љ—Л–Љ–Є –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П–Љ–Є, –Є¬†–Ш–Ч–Ы, –≤—Л–Ј–≤–∞–љ–љ–Њ–≥–Њ –і—А—Г–≥–Є–Љ–Є –њ—А–Є—З–Є–љ–∞–Љ–Є, –≤–Ї–ї—О—З–∞—П –Ш–Ы–§, –µ—Б—В—М –Њ–±—Й–Є–µ –Љ–µ—Е–∞–љ–Є–Ј–Љ—Л, —Г–њ—А–∞–≤–ї—П—О—Й–Є–µ –њ—А–Њ–≥—А–µ—Б—Б–Є—А—Г—О—Й–Є–Љ —Д–Є–±—А–Њ–Ј–Њ–Љ [5]. –°–Њ–≥–ї–∞—Б–љ–Њ —Б—Г—Й–µ—Б—В–≤—Г—О—Й–Є–Љ –њ—А–µ–і—Б—В–∞–≤–ї–µ–љ–Є—П–Љ, –Я–§-–Ш–Ч–Ы —А–∞–Ј–≤–Є–≤–∞–µ—В—Б—П –љ–∞ —Д–Њ–љ–µ –њ–Њ–≤—В–Њ—А–љ—Л—Е –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є–є –∞–ї—М–≤–µ–Њ–ї—П—А–љ–Њ–≥–Њ —Н–њ–Є—В–µ–ї–Є—П –Є–ї–Є –Љ–Є–Ї—А–Њ—Б–Њ—Б—Г–і–Є—Б—В–Њ–≥–Њ —Н–љ–і–Њ—В–µ–ї–Є—П, —З—В–Њ –њ—А–Є–≤–Њ–і–Є—В –Ї¬†—А–∞–Ј—А—Г—И–µ–љ–Є—О –Ї–ї–µ—В–Њ–Ї –Є¬†–љ–µ—А–µ–≥—Г–ї–Є—А—Г–µ–Љ–Њ–Љ—Г –Є—Е –≤–Њ—Б—Б—В–∞–љ–Њ–≤–ї–µ–љ–Є—О. –§–Є–±—А–Њ–±–ї–∞—Б—В—Л, —А–∞—Б–њ–Њ–ї–Њ–ґ–µ–љ–љ—Л–µ –≤¬†–Љ–µ—Б—В–∞—Е –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є—П, –∞–Ї—В–Є–≤–Є—А—Г—О—В—Б—П, –њ—А–µ–≤—А–∞—Й–∞—О—В—Б—П –≤¬†–Љ–Є–Њ—Д–Є–±—А–Њ–±–ї–∞—Б—В—Л, –Ї–Њ—В–Њ—А—Л–µ —Б–µ–Ї—А–µ—В–Є—А—Г—О—В —З—А–µ–Ј–Љ–µ—А–љ–Њ–µ –Ї–Њ–ї–Є—З–µ—Б—В–≤–Њ –≤–љ–µ–Ї–ї–µ—В–Њ—З–љ–Њ–≥–Њ –Љ–∞—В—А–Є–Ї—Б–∞. –Ъ–∞–Ї —Б–ї–µ–і—Б—В–≤–Є–µ, —Г–≤–µ–ї–Є—З–Є–≤–∞–µ—В—Б—П –ґ–µ—Б—В–Ї–Њ—Б—В—М —В–Ї–∞–љ–Є, —З—В–Њ –≤¬†—Б–≤–Њ—О –Њ—З–µ—А–µ–і—М –і–Њ–њ–Њ–ї–љ–Є—В–µ–ї—М–љ–Њ –∞–Ї—В–Є–≤–Є—А—Г–µ—В –Є¬†—Б—В–Є–Љ—Г–ї–Є—А—Г–µ—В —Д–Є–±—А–Њ–±–ї–∞—Б—В—Л [62, 63]. –Ь–∞–Ї—А–Њ—Д–∞–≥–Є –Є¬†–ї–Є–Љ—Д–Њ—Ж–Є—В—Л, –њ—А–Є–≤–ї–µ—З–µ–љ–љ—Л–µ –Ї¬†–Љ–µ—Б—В—Г –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є—П, –≤—Л—Б–≤–Њ–±–Њ–ґ–і–∞—О—В –Ї–∞–Ї –њ—А–Њ–≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ—Л–µ –Љ–µ–і–Є–∞—В–Њ—А—Л, —В–∞–Ї –Є¬†–Љ–µ–і–Є–∞—В–Њ—А—Л, —Б—В–Є–Љ—Г–ї–Є—А—Г—О—Й–Є–µ –њ—А–Њ–ї–Є—Д–µ—А–∞—Ж–Є—О (—Д–∞–Ї—В–Њ—А—Л —А–Њ—Б—В–∞) [62]. –≠—В–Њ—В –њ—А–Њ—Ж–µ—Б—Б —Г—Б–Є–ї–Є–≤–∞–µ—В—Б—П –Ј–∞ —Б—З–µ—В –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є—П —Б–Њ—Б—Г–і–Њ–≤ –Є¬†–њ—А–Є–≤–Њ–і–Є—В –Ї¬†–∞–Ї—В–Є–≤–∞—Ж–Є–Є –Є¬†–і–µ–≥—А–∞–љ—Г–ї—П—Ж–Є–Є —В—А–Њ–Љ–±–Њ—Ж–Є—В–Њ–≤ [64]. –Я–Њ–і –≤–Њ–Ј–і–µ–є—Б—В–≤–Є–µ–Љ —Г–Ї–∞–Ј–∞–љ–љ—Л—Е —Д–∞–Ї—В–Њ—А–Њ–≤ —А–∞–Ј–≤–Є–≤–∞–µ—В—Б—П —Б–∞–Љ–Њ–њ–Њ–і–і–µ—А–ґ–Є–≤–∞—О—Й–Є–є—Б—П –њ—А–Њ—Ж–µ—Б—Б –њ—А–Њ–≥—А–µ—Б—Б–Є—А—Г—О—Й–µ–≥–Њ —Д–Є–±—А–Њ–Ј–∞. –Я—А–Є –љ–∞–Ї–Њ–њ–ї–µ–љ–Є–Є –≤–љ–µ–Ї–ї–µ—В–Њ—З–љ–Њ–≥–Њ –Љ–∞—В—А–Є–Ї—Б–∞ —Г–≤–µ–ї–Є—З–Є–≤–∞–µ—В—Б—П —А–∞—Б—Б—В–Њ—П–љ–Є–µ –і–Є—Д—Д—Г–Ј–Є–Є –Љ–µ–ґ–і—Г –Ї—А–Њ–≤–µ–љ–Њ—Б–љ—Л–Љ–Є —Б–Њ—Б—Г–і–∞–Љ–Є –Є¬†–Ї–ї–µ—В–Ї–∞–Љ–Є. –Т¬†—А–µ–Ј—Г–ї—М—В–∞—В–µ —Б–љ–Є–ґ–∞–µ—В—Б—П —Б–љ–∞–±–ґ–µ–љ–Є–µ —В–Ї–∞–љ–µ–є –Ї–Є—Б–ї–Њ—А–Њ–і–Њ–Љ. –Ґ–Ї–∞–љ–µ–≤–∞—П –≥–Є–њ–Њ–Ї—Б–Є—П –Љ–Њ–ґ–µ—В —Б—В–Є–Љ—Г–ї–Є—А–Њ–≤–∞—В—М –і–∞–ї—М–љ–µ–є—И—Г—О –њ—А–Њ–і—Г–Ї—Ж–Є—О –±–µ–ї–Ї–Њ–≤ –Љ–∞—В—А–Є–Ї—Б–∞ [65]. –Ф–Њ–њ–Њ–ї–љ–Є—В–µ–ї—М–љ–Њ–µ –Ј–љ–∞—З–µ–љ–Є–µ –Љ–Њ–ґ–µ—В –Є–Љ–µ—В—М –њ–µ—А—Б–Є—Б—В–µ–љ—Ж–Є—П –∞–љ—В–Є–≥–µ–љ–Њ–≤, —Б—В–Є–Љ—Г–ї–Є—А—Г—О—Й–Є—Е –Є–Љ–Љ—Г–љ–љ—Л–є –ї–Њ–Ї–∞–ї—М–љ—Л–є –Њ—В–≤–µ—В. –Я—А–Є –†–Р —В–∞–Ї–Њ–≤—Л–Љ–Є —Б—З–Є—В–∞—О—В—Б—П —Ж–Є—В—А—Г–ї–ї–Є–љ–Є—А–Њ–≤–∞–љ–љ—Л–µ –њ–µ–њ—В–Є–і—Л, —Б–њ–Њ—Б–Њ–±—Б—В–≤—Г—О—Й–Є–µ —А–∞–Ј–≤–Є—В–Є—О –Є–Љ–Љ—Г–љ–љ–Њ–≥–Њ –≤–Њ—Б–њ–∞–ї–µ–љ–Є—П –≤¬†—Б—Г—Б—В–∞–≤–∞—Е –Є¬†–њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є—О –њ—А–Њ–ї–Є—Д–µ—А–∞—В–Є–≤–љ—Л—Е –њ—А–Њ—Ж–µ—Б—Б–Њ–≤ –≤¬†–ї–µ–≥–Ї–Є—Е [62, 66]. –Ъ—А–Њ–Љ–µ —В–Њ–≥–Њ, –≤¬†–њ–Њ—Б–ї–µ–і–љ–Є—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞—В–µ–ї—М—Б–Ї–Є—Е —А–∞–±–Њ—В–∞—Е —Г¬†–±–Њ–ї—М–љ—Л—Е –Ш–Ы–§ –≤—Л—П–≤–ї–µ–љ–Њ –њ–Њ–≤—Л—И–µ–љ–Є–µ —Г—А–Њ–≤–љ—П –∞–љ—В–Є—Ж–Є—В—А—Г–ї–ї–Є–љ–Њ–≤—Л—Е –∞–љ—В–Є—В–µ–ї [67].
–Ґ–∞–Ї–Є–Љ –Њ–±—А–∞–Ј–Њ–Љ, –Њ–±—Й–љ–Њ—Б—В—М –њ–∞—В–Њ–≥–µ–љ–µ–Ј–∞ –Є–і–Є–Њ–њ–∞—В–Є—З–µ—Б–Ї–Є—Е –Є¬†—Б–≤—П–Ј–∞–љ–љ—Л—Е —Б¬†—А–µ–≤–Љ–∞—В–Є—З–µ—Б–Ї–Є–Љ–Є –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П–Љ–Є –Є–љ—В–µ—А—Б—В–Є—Ж–Є–∞–ї—М–љ—Л—Е —Д–Є–±—А–Њ—В–Є—З–µ—Б–Ї–Є—Е –њ—А–Њ—Ж–µ—Б—Б–Њ–≤ –≤¬†–ї–µ–≥–Ї–Є—Е –Њ–±–Њ—Б–љ–Њ–≤—Л–≤–∞–µ—В —Ж–µ–ї–µ—Б–Њ–Њ–±—А–∞–Ј–љ–Њ—Б—В—М –њ—А–Є–Љ–µ–љ–µ–љ–Є—П –∞–љ—В–Є—Д–Є–±—А–Њ–Ј–љ–Њ–є/–∞–љ—В–Є–њ—А–Њ–ї–Є—Д–µ—А–∞—В–Є–≤–љ–Њ–є —В–µ—А–∞–њ–Є–Є —Г¬†–і–∞–љ–љ–Њ–є –Ї–∞—В–µ–≥–Њ—А–Є–Є –±–Њ–ї—М–љ—Л—Е.
–Т –љ–∞—Б—В–Њ—П—Й–µ–µ –≤—А–µ–Љ—П –љ–∞–Є–±–Њ–ї–µ–µ —П—А–Ї–Є–Љ –њ—А–µ–і—Б—В–∞–≤–Є—В–µ–ї–µ–Љ –њ—А–µ–њ–∞—А–∞—В–Њ–≤ —Г–Ї–∞–Ј–∞–љ–љ–Њ–є –≥—А—Г–њ–њ—Л —П–≤–ї—П–µ—В—Б—П –љ–Є–љ—В–µ–і–∞–љ–Є–± (–Т–∞—А–≥–∞—В–µ—Д, –љ–Њ–Љ–µ—А —А–µ–≥–Є—Б—В—А–∞—Ж–Є–Њ–љ–љ–Њ–≥–Њ —Г–і–Њ—Б—В–Њ–≤–µ—А–µ–љ–Є—П –Ы–Я-002830).
–Э–Є–љ—В–µ–і–∞–љ–Є–±¬†вАУ –Є–љ–≥–Є–±–Є—В–Њ—А —В–Є—А–Њ–Ј–Є–љ–Ї–Є–љ–∞–Ј—Л, –Њ—В–љ–Њ—Б—П—Й–Є–є—Б—П –Ї¬†–Ї–ї–∞—Б—Б—Г –Љ–∞–ї—Л—Е –Љ–Њ–ї–µ–Ї—Г–ї. –Ю–љ –±–ї–Њ–Ї–Є—А—Г–µ—В –Ї–Є–љ–∞–Ј–љ—Г—О –∞–Ї—В–Є–≤–љ–Њ—Б—В—М —А–µ—Ж–µ–њ—В–Њ—А–Њ–≤ —Д–∞–Ї—В–Њ—А–∞ —А–Њ—Б—В–∞ —Н–љ–і–Њ—В–µ–ї–Є—П —Б–Њ—Б—Г–і–Њ–≤ 1вАУ3 (VEGFR 1вАУ3), —А–µ—Ж–µ–њ—В–Њ—А–Њ–≤ —В—А–Њ–Љ–±–Њ—Ж–Є—В–∞—А–љ–Њ–≥–Њ —Д–∞–Ї—В–Њ—А–∞ —А–Њ—Б—В–∞ ќ±, ќ≤ (PDGFR ќ±, ќ≤) –Є¬†—А–µ—Ж–µ–њ—В–Њ—А–Њ–≤ —Д–∞–Ї—В–Њ—А–∞ —А–Њ—Б—В–∞ —Д–Є–±—А–Њ–±–ї–∞—Б—В–Њ–≤ 1вАУ3 (FGFR 1вАУ3). –Ъ—А–Њ–Љ–µ —В–Њ–≥–Њ, –Є–љ–≥–Є–±–Є—А—Г—О—В—Б—П Fms-–њ–Њ–і–Њ–±–љ–∞—П –њ—А–Њ—В–µ–Є–љ—В–Є—А–Њ–Ј–Є–љ–Ї–Є–љ–∞–Ј–∞ (Flt-3), –ї–Є–Љ—Д–Њ—Ж–Є—В-—Б–њ–µ—Ж–Є—Д–Є—З–µ—Б–Ї–∞—П –њ—А–Њ—В–µ–Є–љ—В–Є—А–Њ–Ј–Є–љ–Ї–Є–љ–∞–Ј–∞ (Lck), –њ—А–Њ—В–Њ–Њ–љ–Ї–Њ–≥–µ–љ–љ–∞—П –њ—А–Њ—В–µ–Є–љ—В–Є—А–Њ–Ј–Є–љ–Ї–Є–љ–∞–Ј–∞ (Src) –Є¬†–Ї–Є–љ–∞–Ј—Л —А–µ—Ж–µ–њ—В–Њ—А–∞ –Ї–Њ–ї–Њ–љ–Є–µ—Б—В–Є–Љ—Г–ї–Є—А—Г—О—Й–µ–≥–Њ —Д–∞–Ї—В–Њ—А–∞ 1 (CSF1R). –Э–Є–љ—В–µ–і–∞–љ–Є–± –Ї–Њ–љ–Ї—Г—А–µ–љ—В–љ–Њ –≤–Ј–∞–Є–Љ–Њ–і–µ–є—Б—В–≤—Г–µ—В —Б¬†–∞–і–µ–љ–Њ–Ј–Є–љ—В—А–Є—Д–Њ—Б—Д–∞—В-—Б–≤—П–Ј—Л–≤–∞—О—Й–Є–Љ —Г—З–∞—Б—В–Ї–Њ–Љ —Н—В–Є—Е –Ї–Є–љ–∞–Ј –Є¬†–±–ї–Њ–Ї–Є—А—Г–µ—В –Ї–∞—Б–Ї–∞–і—Л –≤–љ—Г—В—А–Є–Ї–ї–µ—В–Њ—З–љ–Њ–є –њ–µ—А–µ–і–∞—З–Є —Б–Є–≥–љ–∞–ї–Њ–≤, –Ї–Њ—В–Њ—А—Л–µ —Г—З–∞—Б—В–≤—Г—О—В –≤¬†—А–µ–Љ–Њ–і–µ–ї–Є—А–Њ–≤–∞–љ–Є–Є —Д–Є–±—А–Њ–Ј–љ–Њ–є —В–Ї–∞–љ–Є –њ—А–Є –Є–љ—В–µ—А—Б—В–Є—Ж–Є–∞–ї—М–љ–Њ–Љ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–Є –ї–µ–≥–Ї–Є—Е [68].
–Я–Њ –Њ—Б–љ–Њ–≤–љ–Њ–Љ—Г –Љ–µ—Е–∞–љ–Є–Ј–Љ—Г –і–µ–є—Б—В–≤–Є—П –љ–Є–љ—В–µ–і–∞–љ–Є–± –њ—А–µ–і—Б—В–∞–≤–ї—П–µ—В —Б–Њ–±–Њ–є —В—А–Њ–є–љ–Њ–є –Є–љ–≥–Є–±–Є—В–Њ—А —В–Є—А–Њ–Ј–Є–љ–Ї–Є–љ–∞–Ј. –Ю–љ –Њ–±–ї–∞–і–∞–µ—В –∞–љ—В–Є—Д–Є–±—А–Њ–ї–Є—В–Є—З–µ—Б–Ї–Є–Љ–Є, –њ—А–Њ—В–Є–≤–Њ–≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ—Л–Љ–Є —Б–≤–Њ–є—Б—В–≤–∞–Љ–Є –Є¬†—Н—Д—Д–µ–Ї—В–∞–Љ–Є, –љ–∞–њ—А–∞–≤–ї–µ–љ–љ—Л–Љ–Є –љ–∞ —Б–Њ—Б—Г–і–Є—Б—В–Њ–µ —А–µ–Љ–Њ–і–µ–ї–Є—А–Њ–≤–∞–љ–Є–µ.
–Я–µ—А–≤–Њ–љ–∞—З–∞–ї—М–љ–Њ –љ–Є–љ—В–µ–і–∞–љ–Є–± –њ—А–Є–Љ–µ–љ—П–ї–Є –і–ї—П –ї–µ—З–µ–љ–Є—П –љ–µ–Љ–µ–ї–Ї–Њ–Ї–ї–µ—В–Њ—З–љ–Њ–≥–Њ —А–∞–Ї–∞ –ї–µ–≥–Ї–Њ–≥–Њ. –Я—А–∞–Ї—В–Є—З–µ—Б–Ї–Є –Њ–і–љ–Њ–≤—А–µ–Љ–µ–љ–љ–Њ —Б¬†—Н—В–Є–Љ –≤¬†2014 –≥. –±—Л–ї–Є –Ј–∞–≤–µ—А—И–µ–љ—Л 52-–љ–µ–і–µ–ї—М–љ—Л–µ –і–≤–Њ–є–љ—Л–µ —Б–ї–µ–њ—Л–µ –њ–ї–∞—Ж–µ–±–Њ–Ї–Њ–љ—В—А–Њ–ї–Є—А—Г–µ–Љ—Л–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П INPULSIS-1 –Є¬†-2, –≤¬†–Ї–Њ—В–Њ—А—Л—Е –љ–∞ –±–Њ–ї—М—И–Њ–є –≥—А—Г–њ–њ–µ –±–Њ–ї—М–љ—Л—Е –Ш–Ы–§ (n = 1066) –±—Л–ї–∞ –і–Њ–Ї–∞–Ј–∞–љ–∞ –µ–≥–Њ —Б–њ–Њ—Б–Њ–±–љ–Њ—Б—В—М –Ј–∞–Љ–µ–і–ї—П—В—М –њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є–µ –Є–і–Є–Њ–њ–∞—В–Є—З–µ—Б–Ї–Њ–≥–Њ –ї–µ–≥–Њ—З–љ–Њ–≥–Њ —Д–Є–±—А–Њ–Ј–∞ [69, 70]. –Я–Њ—Н—В–Њ–Љ—Г –њ—А–µ–њ–∞—А–∞—В –±—Л–ї –Ј–∞—А–µ–≥–Є—Б—В—А–Є—А–Њ–≤–∞–љ –њ–Њ –њ–Њ–Ї–∞–Ј–∞–љ–Є—О ¬Ђ–Є–і–Є–Њ–њ–∞—В–Є—З–µ—Б–Ї–Є–є –ї–µ–≥–Њ—З–љ—Л–є —Д–Є–±—А–Њ–Ј (–Є–і–Є–Њ–њ–∞—В–Є—З–µ—Б–Ї–Є–є —Д–Є–±—А–Њ–Ј–Є—А—Г—О—Й–Є–є –∞–ї—М–≤–µ–Њ–ї–Є—В)¬ї.
–Т –љ–∞—Б—В–Њ—П—Й–µ–µ –≤—А–µ–Љ—П —Г–ґ–µ –љ–∞–Ї–Њ–њ–ї–µ–љ —А–Њ—Б—Б–Є–є—Б–Ї–Є–є –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є–є –Њ–њ—Л—В –њ—А–Є–Љ–µ–љ–µ–љ–Є—П –љ–Є–љ—В–µ–і–∞–љ–Є–±–∞. –Ю–љ –њ–Њ–і—В–≤–µ—А–ґ–і–∞–µ—В —Н—Д—Д–µ–Ї—В–Є–≤–љ–Њ—Б—В—М –і–∞–љ–љ–Њ–≥–Њ –њ—А–µ–њ–∞—А–∞—В–∞ [70, 71].
–Я—А–Є–Љ–µ–љ–µ–љ–Є–µ –љ–Є–љ—В–µ–і–∞–љ–Є–±–∞ –њ—А–Є —Б–Є—Б—В–µ–Љ–љ–Њ–є —Б–Ї–ї–µ—А–Њ–і–µ—А–Љ–Є–Є
–°–ї–µ–і—Г—О—Й–Є–Љ —Н—В–∞–њ–Њ–Љ –≤–љ–µ–і—А–µ–љ–Є—П —Б—В–∞–ї–Њ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ –љ–Є–љ—В–µ–і–∞–љ–Є–±–∞ –њ—А–Є –°–°–Ф –Ї–∞–Ї –µ—Б—В–µ—Б—В–≤–µ–љ–љ–Њ–є –Љ–Њ–і–µ–ї–Є —Д–Є–±—А–Њ—В–Є—З–µ—Б–Ї–Є—Е –њ—А–Њ—Ж–µ—Б—Б–Њ–≤ –љ–∞ —Д–Њ–љ–µ –∞—Г—В–Њ–Є–Љ–Љ—Г–љ–љ–Њ–є –њ–∞—В–Њ–ї–Њ–≥–Є–Є. –Т¬†2019 –≥. –Њ–њ—Г–±–ї–Є–Ї–Њ–≤–∞–љ—Л —А–µ–Ј—Г–ї—М—В–∞—В—Л –і–≤–Њ–є–љ–Њ–≥–Њ —Б–ї–µ–њ–Њ–≥–Њ –њ–ї–∞—Ж–µ–±–Њ–Ї–Њ–љ—В—А–Њ–ї–Є—А—Г–µ–Љ–Њ–≥–Њ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П SENCSIS [72]. –Т¬†–љ–µ–≥–Њ –±—Л–ї–Є –≤–Ї–ї—О—З–µ–љ—Л 576 –±–Њ–ї—М–љ—Л—Е –°–°–Ф, —А–∞–љ–і–Њ–Љ–Є–Ј–Є—А–Њ–≤–∞–љ–љ—Л—Е –љ–∞ –≥—А—Г–њ–њ—Г –љ–Є–љ—В–µ–і–∞–љ–Є–±–∞ –≤¬†–і–Њ–Ј–µ 150 –Љ–≥ –і–≤–∞ —А–∞–Ј–∞ –≤¬†–і–µ–љ—М –њ–µ—А–Њ—А–∞–ї—М–љ–Њ –Є¬†–≥—А—Г–њ–њ—Г –њ–ї–∞—Ж–µ–±–Њ. –Ъ—А–Є—В–µ—А–Є—П–Љ–Є –≤–Ї–ї—О—З–µ–љ–Є—П –≤—Л–±—А–∞–љ—Л –≤–Њ–Ј—А–∞—Б—В –њ–∞—Ж–Є–µ–љ—В–∞ вЙ• 18 –ї–µ—В, –њ–Њ–і—В–≤–µ—А–ґ–і–µ–љ–љ—Л–є –і–Є–∞–≥–љ–Њ–Ј –°–°–Ф –њ–Њ –Ї—А–Є—В–µ—А–Є—П–Љ ACR/EULAR 2013 –≥. [73], –њ—А–Є –љ–∞—З–∞–ї–µ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П –њ–µ—А–≤—Л–є —Б–Є–Љ–њ—В–Њ–Љ, –Њ—В–ї–Є—З–љ—Л–є –Њ—В —Д–µ–љ–Њ–Љ–µ–љ–∞ –†–µ–є–љ–Њ, –љ–µ —А–∞–љ–µ–µ —З–µ–Љ –Ј–∞ —Б–µ–Љ—М –ї–µ—В –і–Њ —Б–Ї—А–Є–љ–Є–љ–≥–∞, –Ш–Ч–Ы –њ–Њ –і–∞–љ–љ—Л–Љ –Ъ–Ґ–Т–† –≥—А—Г–і–љ–Њ–є –Ї–ї–µ—В–Ї–Є –љ–µ —А–∞–љ–µ–µ —З–µ–Љ –Ј–∞ 12 –Љ–µ—Б—П—Ж–µ–≤ –і–Њ —Б–Ї—А–Є–љ–Є–љ–≥–∞ –њ—А–Є —Б—В–µ–њ–µ–љ–Є –Њ—Е–≤–∞—В–∞ –ї–µ–≥–Ї–Њ–≥–Њ —Д–Є–±—А–Њ–Ј–Њ–Љ вЙ• 10%, –§–Ц–Х–Ы вЙ• 40%, DLCO –Њ—В 30 –і–Њ 89%. –Т¬†–Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ –љ–µ –≤–Ї–ї—О—З–∞–ї–Є –±–Њ–ї—М–љ—Л—Е —Б¬†—Г—А–Њ–≤–љ—П–Љ–Є –∞–ї–∞–љ–Є–љ¬≠–∞–Љ–Є–љ–Њ—В—А–∞–љ—Б—Д–µ—А–∞–Ј—Л/–∞—Б–њ–∞—А—В–∞—В–∞–Љ–Є–љ–Њ—В—А–∞–љ—Б—Д–µ—А–∞–Ј—Л/–±–Є–ї–Є—А—Г–±–Є–љ–∞, –±–Њ–ї–µ–µ —З–µ–Љ –≤¬†–њ–Њ–ї—В–Њ—А–∞ —А–∞–Ј–∞ –њ—А–µ–≤—Л—И–∞—О—Й–Є–Љ–Є –љ–Њ—А–Љ—Г, —А–Є—Б–Ї–Њ–Љ –Ї—А–Њ–≤–Њ—В–µ—З–µ–љ–Є—П, –Є–љ—Д–∞—А–Ї—В–Њ–Љ –Љ–Є–Њ–Ї–∞—А–і–∞ –Є–ї–Є –љ–µ—Б—В–∞–±–Є–ї—М–љ–Њ–є —Б—В–µ–љ–Њ–Ї–∞—А–і–Є–µ–є –Љ–µ–љ–µ–µ —З–µ–Љ –Ј–∞ —И–µ—Б—В—М –Љ–µ—Б—П—Ж–µ–≤ –і–Њ —Б–Ї—А–Є–љ–Є–љ–≥–∞, —В—А–Њ–Љ–±–Њ—В–Є—З–µ—Б–Ї–Є–Љ —Б–Њ–±—Л—В–Є–µ–Љ –≤¬†–∞–љ–∞–Љ–љ–µ–Ј–µ –љ–µ —А–∞–љ–µ–µ —З–µ–Љ –Ј–∞ 12 –Љ–µ—Б—П—Ж–µ–≤ –і–Њ —Б–Ї—А–Є–љ–Є–љ–≥–∞, –±–Њ–ї–µ–µ —З–µ–Љ —В—А–µ–Љ—П –Њ—З–∞–≥–∞–Љ–Є –Є–Ј—К—П–Ј–≤–ї–µ–љ–Є—П –њ–∞–ї—М—Ж–µ–≤ –Є–ї–Є –Є—Е –љ–µ–Ї—А–Њ–Ј–Њ–Љ –≤¬†–∞–љ–∞–Љ–љ–µ–Ј–µ –≤¬†—В—П–ґ–µ–ї–Њ–є —Д–Њ—А–Љ–µ, –њ–Њ—В—А–µ–±–Њ–≤–∞–≤—И–Є–Љ –≥–Њ—Б–њ–Є—В–∞–ї–Є–Ј–∞—Ж–Є–Є, –≤—Л—А–∞–ґ–µ–љ–љ–Њ–є –ї–µ–≥–Њ—З–љ–Њ–є –≥–Є–њ–µ—А—В–µ–љ–Ј–Є–µ–є, —Б–Ї–ї–µ—А–Њ–і–µ—А–Љ–Є—З–µ—Б–Ї–Є–Љ –њ–Њ—З–µ—З–љ—Л–Љ –Ї—А–Є–Ј–Њ–Љ –≤¬†–∞–љ–∞–Љ–љ–µ–Ј–µ.
–Ф–Њ–њ—Г—Б–Ї–∞–≤—И–∞—П—Б—П –≤¬†–Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–Є —В–µ—А–∞–њ–Є—П –Њ—Б–љ–Њ–≤–љ–Њ–≥–Њ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П –≤–Ї–ї—О—З–∞–ї–∞ –њ—А–µ–і–љ–Є–Ј–Њ–ї–Њ–љ –≤¬†–і–Њ–Ј–µ 10 –Љ–≥/—Б—Г—В –Є¬†–Љ–µ–љ–µ–µ –Є–ї–Є –µ–≥–Њ —Н–Ї–≤–Є–≤–∞–ї–µ–љ—В. –Ъ—Г—А—Б —Б—В–∞–±–Є–ї—М–љ–Њ–є —В–µ—А–∞–њ–Є–Є –Љ–Є–Ї–Њ—Д–µ–љ–Њ–ї–∞—В–∞ –Љ–Њ—Д–µ—В–Є–ї–Њ–Љ –Є–ї–Є –Љ–µ—В–Њ—В—А–µ–Ї—Б–∞—В–Њ–Љ –Ј–∞ —И–µ—Б—В—М –Љ–µ—Б—П—Ж–µ–≤ –Є¬†–±–Њ–ї–µ–µ –і–Њ —А–∞–љ–і–Њ–Љ–Є–Ј–∞—Ж–Є–Є.
–Я–µ—А–≤–Є—З–љ–Њ–є –Ї–Њ–љ–µ—З–љ–Њ–є —В–Њ—З–Ї–Њ–є –≤—Л–±—А–∞–љ–Њ —Б–љ–Є–ґ–µ–љ–Є–µ –§–Ц–Х–Ы –Ј–∞ –≥–Њ–і —Б¬†–Њ—Ж–µ–љ–Ї–Њ–є –≤¬†—В–µ—З–µ–љ–Є–µ 52 –љ–µ–і–µ–ї—М. –Ъ–ї—О—З–µ–≤—Л–µ –≤—В–Њ—А–Є—З–љ—Л–µ –Ї–Њ–љ–µ—З–љ—Л–µ —В–Њ—З–Ї–Є –≤–Ї–ї—О—З–∞–ї–Є –∞–±—Б–Њ–ї—О—В–љ–Њ–µ –Є–Ј–Љ–µ–љ–µ–љ–Є–µ –Љ–Њ–і–Є—Д–Є—Ж–Є—А–Њ–≤–∞–љ–љ–Њ–≥–Њ –Ї–Њ–ґ–љ–Њ–≥–Њ —Б—З–µ—В–∞ –†–Њ–і–љ–∞–љ–∞ [17] –Є¬†–∞–±—Б–Њ–ї—О—В–љ–Њ–µ –Є–Ј–Љ–µ–љ–µ–љ–Є–µ –Ї–Њ–Љ–њ–ї–µ–Ї—Б–љ–Њ–є –Њ—Ж–µ–љ–Ї–Є –њ–Њ –Њ–њ—А–Њ—Б–љ–Є–Ї—Г SGRQ [74] –љ–∞ 52-–є –љ–µ–і–µ–ї–µ –њ–Њ —Б—А–∞–≤–љ–µ–љ–Є—О —Б¬†–Є—Б—Е–Њ–і–љ—Л–Љ —Г—А–Њ–≤–љ–µ–Љ.
–Т –Њ–±—Й–µ–є —Б–ї–Њ–ґ–љ–Њ—Б—В–Є 576 –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–°–°–Ф –≤¬†177 —Ж–µ–љ—В—А–∞—Е –Є–Ј 32 —Б—В—А–∞–љ –њ–Њ–ї—Г—З–Є–ї–Є –њ–Њ –Ї—А–∞–є–љ–µ–є –Љ–µ—А–µ –Њ–і–љ—Г –і–Њ–Ј—Г –љ–Є–љ—В–µ–і–∞–љ–Є–±–∞ (–љ–∞–Ј–љ–∞—З–∞–ї—Б—П –≤–љ—Г—В—А—М –њ–Њ 150 –Љ–≥ –і–≤–∞ —А–∞–Ј–∞ –≤¬†–і–µ–љ—М) –Є–ї–Є –њ–ї–∞—Ж–µ–±–Њ.
–£ 51,9% –Є–Љ–µ–ї–∞ –Љ–µ—Б—В–Њ –і–Є—Д—Д—Г–Ј–љ–∞—П —Б–Ї–ї–µ—А–Њ–і–µ—А–Љ–Є—П, 48,4% –Є—Б—Е–Њ–і–љ–Њ –њ–Њ–ї—Г—З–∞–ї–Є –Љ–Є–Ї–Њ—Д–µ–љ–Њ–ї–∞—В–∞ –Љ–Њ—Д–µ—В–Є–ї.
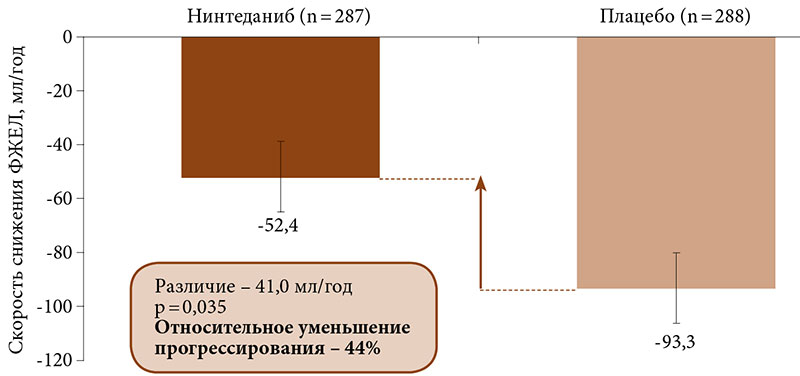
–Я—А–Є –њ–µ—А–≤–Є—З–љ–Њ–Љ –∞–љ–∞–ї–Є–Ј–µ –Ї–Њ–љ–µ—З–љ—Л—Е —В–Њ—З–µ–Ї —Б–Ї–Њ—А—А–µ–Ї—В–Є—А–Њ–≤–∞–љ–љ–∞—П —Б–Ї–Њ—А–Њ—Б—В—М –Є–Ј–Љ–µ–љ–µ–љ–Є—П –§–Ц–Х–Ы –≤¬†–≥—А—Г–њ–њ–µ –љ–Є–љ—В–µ–і–∞–љ–Є–±–∞ —Б–Њ—Б—В–∞–≤–Є–ї–∞ 52,4 –Љ–ї/–≥–Њ–і, –≤¬†–≥—А—Г–њ–њ–µ –њ–ї–∞—Ж–µ–±–Њ¬†вАУ 93,3 –Љ–ї/–≥–Њ–і. –†–∞–Ј–љ–Є—Ж–∞¬†вАУ 41,0 –Љ–ї/–≥–Њ–і (95% –Ф–Ш 2,9вАУ79,0, p = 0,04) (—А–Є—Б. 3). –Ъ–Њ–ґ–љ—Л–є —Б—З–µ—В –†–Њ–і–љ–∞–љ–∞ –Є¬†–Њ–±—Й–∞—П –Њ—Ж–µ–љ–Ї–∞ –њ–Њ SGRQ —З–µ—А–µ–Ј 52 –љ–µ–і–µ–ї–Є —Б—Г—Й–µ—Б—В–≤–µ–љ–љ–Њ –љ–µ —А–∞–Ј–ї–Є—З–∞–ї–Є—Б—М.
–Ґ–∞–Ї–Є–Љ –Њ–±—А–∞–Ј–Њ–Љ, –ї–µ—З–µ–љ–Є–µ –љ–Є–љ—В–µ–і–∞–љ–Є–±–Њ–Љ –і–Њ—Б—В–Њ–≤–µ—А–љ–Њ —В–Њ—А–Љ–Њ–Ј–Є–ї–Њ —А–∞–Ј–≤–Є—В–Є–µ –ї–µ–≥–Њ—З–љ–Њ–≥–Њ —Д–Є–±—А–Њ–Ј–∞ –Є, —Б–Њ–Њ—В–≤–µ—В—Б—В–≤–µ–љ–љ–Њ, —Б–љ–Є–ґ–µ–љ–Є–µ –ї–µ–≥–Њ—З–љ–Њ–є —Д—Г–љ–Ї—Ж–Є–Є —Г¬†–±–Њ–ї—М–љ—Л—Е –°–°–Ф.
–Я–Њ–ї—Г—З–µ–љ–љ—Л–µ —А–µ–Ј—Г–ї—М—В–∞—В—Л —Б—В–∞–ї–Є –Њ—Б–љ–Њ–≤–∞–љ–Є–µ–Љ –і–ї—П —А–µ–≥–Є—Б—В—А–∞—Ж–Є–Є –њ—А–µ–њ–∞—А–∞—В–∞ –њ–Њ –њ–Њ–Ї–∞–Ј–∞–љ–Є—О ¬Ђ–Є–љ—В–µ—А—Б—В–Є—Ж–Є–∞–ї—М–љ—Л–µ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П –ї–µ–≥–Ї–Є—Е –њ—А–Є —Б–Є—Б—В–µ–Љ–љ–Њ–є —Б–Ї–ї–µ—А–Њ–і–µ—А–Љ–Є–Є¬ї.
–Э–Є–љ—В–µ–і–∞–љ–Є–± –Є¬†–і—А—Г–≥–Є–µ –Є–Љ–Љ—Г–љ–Њ–≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ—Л–µ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П
–£—Б–њ–µ—Е –љ–Є–љ—В–µ–і–∞–љ–Є–±–∞ –њ—А–Є –°–°–Ф —Б–њ–Њ—Б–Њ–±—Б—В–≤–Њ–≤–∞–ї –Є–љ–Є—Ж–Є–∞—Ж–Є–Є –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є –≤¬†–Њ—В–љ–Њ—И–µ–љ–Є–Є –і—А—Г–≥–Є—Е –Є–Љ–Љ—Г–љ–Њ–≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ—Л—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є. –Я—А–µ–і–≤–∞—А–Є—В–µ–ї—М–љ–Њ –±—Л–ї–Є –њ–Њ–ї—Г—З–µ–љ—Л —Н–Ї—Б–њ–µ—А–Є–Љ–µ–љ—В–∞–ї—М–љ—Л–µ –і–∞–љ–љ—Л–µ, –њ–Њ–і–і–µ—А–ґ–Є–≤–∞—О—Й–Є–µ –≥–Є–њ–Њ—В–µ–Ј—Г –Њ–± —Н—Д—Д–µ–Ї—В–Є–≤–љ–Њ—Б—В–Є –љ–Є–љ—В–µ–і–∞–љ–Є–±–∞ –њ—А–Є —А–∞–Ј–ї–Є—З–љ—Л—Е –Ш–Ч–Ы. –Ґ–∞–Ї, E.F. Redente –Є¬†—Б–Њ–∞–≤—В. –Њ—Ж–µ–љ–Є–ї–Є –≤–ї–Є—П–љ–Є–µ –љ–Є–љ—В–µ–і–∞–љ–Є–±–∞ –љ–∞ —А–∞–Ј–≤–Є—В–Є–µ —Д–Є–±—А–Њ–Ј–∞ –ї–µ–≥–Ї–Є—Е –Є¬†–Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є —Б—Г—Б—В–∞–≤–Њ–≤ —Г¬†—Б–∞–Љ–Њ–Ї –Љ—Л—И–µ–є –ї–Є–љ–Є–Є SKG, —Г¬†–Ї–Њ—В–Њ—А—Л—Е –∞—А—В—А–Є—В –±—Л–ї –≤—Л–Ј–≤–∞–љ –≤–љ—Г—В—А–Є–±—А—О—И–Є–љ–љ–Њ–є –Є–љ—К–µ–Ї—Ж–Є–µ–є –Ј–Є–Љ–Њ–Ј–∞–љ–∞ –≤¬†–і–Њ–Ј–µ 5 –Љ–≥ [75]. –Я–Њ–Ї–∞–Ј–∞—В–µ–ї–Є –Є—Б—Е–Њ–і–Њ–≤ –≤¬†–Њ—В–љ–Њ—И–µ–љ–Є–Є –∞—А—В—А–Є—В–∞ –Є¬†—Д–Є–±—А–Њ–Ј–∞ –ї–µ–≥–Ї–Є—Е –Њ–њ—А–µ–і–µ–ї—П–ї–Є —З–µ—А–µ–Ј —И–µ—Б—В—М –љ–µ–і–µ–ї—М –ї–µ—З–µ–љ–Є—П. –Ч–љ–∞—З–Є—В–µ–ї—М–љ–Њ–µ —Б–љ–Є–ґ–µ–љ–Є–µ —Г—А–Њ–≤–љ—П –Ї–Њ–ї–ї–∞–≥–µ–љ–∞ –≤¬†–ї–µ–≥–Ї–Є—Е, —Г—Б—В–∞–љ–Њ–≤–ї–µ–љ–љ–Њ–µ —Б¬†–њ–Њ–Љ–Њ—Й—М—О –Є–Ј–Љ–µ—А–µ–љ–Є—П –≥–Є–і—А–Њ–Ї—Б–Є–њ—А–Њ–ї–Є–љ–∞ –Є¬†–Њ–Ї—А–∞—И–Є–≤–∞–љ–Є—П –љ–∞ –Ї–Њ–ї–ї–∞–≥–µ–љ, –љ–∞–±–ї—О–і–∞–ї–Њ—Б—М —Г¬†–њ–Њ–ї—Г—З–∞–≤—И–Є—Е –љ–Є–љ—В–µ–і–∞–љ–Є–±. –†–∞–љ–љ–µ–µ –≤–Љ–µ—И–∞—В–µ–ї—М—Б—В–≤–Њ —Б¬†–њ—А–Є–Љ–µ–љ–µ–љ–Є–µ–Љ –љ–Є–љ—В–µ–і–∞–љ–Є–±–∞ –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ —Б–љ–Є–Ј–Є–ї–Њ —А–Є—Б–Ї —А–∞–Ј–≤–Є—В–Є—П –∞—А—В—А–Є—В–∞.
–≠—Д—Д–µ–Ї—В–Є–≤–љ–Њ—Б—В—М –љ–Є–љ—В–µ–і–∞–љ–Є–±–∞ –њ—А–Є —А–∞–Ј–љ—Л—Е –њ—А–Њ–≥—А–µ—Б—Б–Є—А—Г—О—Й–Є—Е –Ш–Ч–Ы –±—Л–ї–∞ –њ—А–Њ–і–µ–Љ–Њ–љ—Б—В—А–Є—А–Њ–≤–∞–љ–∞ –≤¬†–Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–Є INBUILD [76]. –Т¬†—Н—В–Њ–Љ –і–≤–Њ–є–љ–Њ–Љ —Б–ї–µ–њ–Њ–Љ –њ–ї–∞—Ж–µ–±–Њ–Ї–Њ–љ—В—А–Њ–ї–Є—А—Г–µ–Љ–Њ–Љ –†–Ъ–Ш —Д–∞–Ј—Л III, –њ—А–Њ–≤–µ–і–µ–љ–љ–Њ–Љ –≤¬†15 —Б—В—А–∞–љ–∞—Е, —Г—З–∞—Б—В–≤–Њ–≤–∞–ї–Є –њ–∞—Ж–Є–µ–љ—В—Л —Б¬†—Д–Є–±—А–Њ–Ј–Є—А—Г—О—Й–Є–Љ–Є –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П–Љ–Є –ї–µ–≥–Ї–Є—Е, –Ї–Њ—В–Њ—А—Л–Љ –љ–∞–Ј–љ–∞—З–∞–ї–Є –љ–Є–љ—В–µ–і–∞–љ–Є–± –≤¬†–і–Њ–Ј–µ 150 –Љ–≥ –і–≤–∞ —А–∞–Ј–∞ –≤¬†–і–µ–љ—М –њ–µ—А–Њ—А–∞–ї—М–љ–Њ –Є–ї–Є –њ–ї–∞—Ж–µ–±–Њ. –Т¬†–Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ –≤–Ї–ї—О—З–∞–ї–Є –ї–Є—Ж —Б¬†–њ—А–Є–Ј–љ–∞–Ї–∞–Љ–Є —Д–Є–±—А–Њ–Ј–∞ –ї–µ–≥–Ї–Є—Е –±–Њ–ї–µ–µ 10% –њ–Њ –і–∞–љ–љ—Л–Љ –Ъ–Ґ–Т–†, –§–Ц–Х–Ы 45% –Є–ї–Є –±–Њ–ї–µ–µ –Њ—В –њ—А–Њ–≥–љ–Њ–Ј–Є—А—Г–µ–Љ–Њ–є –Є¬†DLCO –Њ—В 30 –і–Њ 80% –Њ—В –њ—А–Њ–≥–љ–Њ–Ј–Є—А—Г–µ–Љ–Њ–≥–Њ. –Т¬†–Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ –љ–µ –≤–Ї–ї—О—З–∞–ї–Є –њ–Њ–ї—Г—З–∞–≤—И–Є—Е –∞–Ј–∞—В–Є–Њ–њ—А–Є–љ, —Ж–Є–Ї–ї–Њ—Б–њ–Њ—А–Є–љ, –Љ–Є–Ї–Њ—Д–µ–љ–Њ–ї–∞—В–∞ –Љ–Њ—Д–µ—В–Є–ї, —В–∞–Ї—А–Њ–ї–Є–Љ—Г—Б, —А–Є—В—Г–Ї—Б–Є–Љ–∞–±, —Ж–Є–Ї–ї–Њ—Д–Њ—Б—Д–∞–Љ–Є–і –Є–ї–Є –њ–µ—А–Њ—А–∞–ї—М–љ—Л–µ –≥–ї—О–Ї–Њ–Ї–Њ—А—В–Є–Ї–Њ—Б—В–µ—А–Њ–Є–і—Л –≤¬†–і–Њ–Ј–µ –±–Њ–ї–µ–µ 20 –Љ–≥/—Б—Г—В.
–Я–µ—А–≤–Є—З–љ–Њ–є –Ї–Њ–љ–µ—З–љ–Њ–є —В–Њ—З–Ї–Њ–є —Б—В–∞–ї–∞ —Б–Ї–Њ—А–Њ—Б—В—М —Б–љ–Є–ґ–µ–љ–Є—П –§–Ц–Х–Ы –≤¬†–≥–Њ–і –њ–Њ –Њ—Ж–µ–љ–Ї–µ –Ј–∞ 52 –љ–µ–і–µ–ї–Є.
–°–њ–Є—А–Њ–Љ–µ—В—А–Є—П –≤—Л–њ–Њ–ї–љ—П–ї–∞—Б—М –Є—Б—Е–Њ–і–љ–Њ, –љ–∞ –≤—В–Њ—А–Њ–є, —З–µ—В–≤–µ—А—В–Њ–є, —И–µ—Б—В–Њ–є, 12, 24, 36 –Є¬†52-–є –љ–µ–і–µ–ї—П—Е.
–†–∞–љ–і–Њ–Љ–Є–Ј–∞—Ж–Є—О –њ—А–Њ—И–ї–Є –Є¬†–њ–Њ–ї—Г—З–Є–ї–Є —Е–Њ—В—П –±—Л –Њ–і–љ—Г –і–Њ–Ј—Г –љ–Є–љ—В–µ–і–∞–љ–Є–±–∞ –Є–ї–Є –њ–ї–∞—Ж–µ–±–Њ 663 –њ–∞—Ж–Є–µ–љ—В–∞ (332 –≤¬†–≥—А—Г–њ–њ–µ –љ–Є–љ—В–µ–і–∞–љ–Є–±–∞ –Є¬†331 –≤¬†–≥—А—Г–њ–њ–µ –њ–ї–∞—Ж–µ–±–Њ). –°—А–µ–і–љ–Є–є –≤–Њ–Ј—А–∞—Б—В —Б–Њ—Б—В–∞–≤–Є–ї 65,8 ¬± 9,8 –≥–Њ–і–∞, –§–Ц–Х–Ы¬†вАУ 69,0 ¬± 15,6%, DLCO¬†вАУ 46,1 ¬± 13,6% –Њ—В –њ—А–Њ–≥–љ–Њ–Ј–Є—А—Г–µ–Љ–Њ–≥–Њ.
–Ш–Ј 663 –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Г¬†412 (62,1%) –Є–Љ–µ–ї –Љ–µ—Б—В–Њ –њ–∞—В—В–µ—А–љ –Ю–Ш–Я. –°–∞–Љ—Л–Љ–Є —З–∞—Б—В—Л–Љ–Є –і–Є–∞–≥–љ–Њ–Ј–∞–Љ–Є –±—Л–ї–Є —Е—А–Њ–љ–Є—З–µ—Б–Ї–Є–є –У–Я (26,1%) –Є¬†–∞—Г—В–Њ–Є–Љ–Љ—Г–љ–љ—Л–µ –Ш–Ч–Ы (25,6%). –£¬†170 –±–Њ–ї—М–љ—Л—Е¬† (82 –≤ –≥—А—Г–њ–њ–µ –љ–Є–љ—В–µ–і–∞–љ–Є–±–∞ –Є 88 –≤ –≥—А—Г–њ–њ–µ –њ–ї–∞—Ж–µ–±–Њ) –Њ—В–Љ–µ—З–µ–љ—Л —Б–Є—Б—В–µ–Љ–љ—Л–µ —А–µ–≤–Љ–∞—В–Є—З–µ—Б–Ї–Є–µ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П, —В–∞–Ї–Є–µ –Ї–∞–Ї –°–°–Ф, –†–Р, –°–Ч–°–Ґ.
–Т –Њ–±—Й–µ–є –њ–Њ–њ—Г–ї—П—Ж–Є–Є —Б–Ї–Њ—А—А–µ–Ї—В–Є—А–Њ–≤–∞–љ–љ–∞—П —Б–Ї–Њ—А–Њ—Б—В—М —Б–љ–Є–ґ–µ–љ–Є—П –§–Ц–Х–Ы —Б–Њ—Б—В–∞–≤–Є–ї–∞ 80,8 –Љ–ї/–≥–Њ–і –≤¬†–≥—А—Г–њ–њ–µ –љ–Є–љ—В–µ–і–∞–љ–Є–±–∞ –Є¬†187,8 –Љ–ї/–≥–Њ–і –≤¬†–≥—А—Г–њ–њ–µ –њ–ї–∞—Ж–µ–±–Њ. –†–∞–Ј–љ–Є—Ж–∞ –Љ–µ–ґ–і—Г –≥—А—Г–њ–њ–∞–Љ–Є¬†вАУ 107,0 –Љ–ї/–≥–Њ–і (95% –Ф–Ш 65,4вАУ148,5, p < 0,001). –£¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–њ–∞—В—В–µ—А–љ–Њ–Љ –њ–Њ—А–∞–ґ–µ–љ–Є—П –ї–µ–≥–Ї–Є—Е –њ–Њ —В–Є–њ—Г –Ю–Ш–Я —Б–Ї–Њ—А—А–µ–Ї—В–Є—А–Њ–≤–∞–љ–љ–∞—П —Б–Ї–Њ—А–Њ—Б—В—М —Б–љ–Є–ґ–µ–љ–Є—П –§–Ц–Х–Ы —Б–Њ—Б—В–∞–≤–Є–ї–∞ 82,9 –Љ–ї/–≥–Њ–і –і–ї—П –≥—А—Г–њ–њ—Л –љ–Є–љ—В–µ–і–∞–љ–Є–±–∞ –Є¬†211,1 –Љ–ї/–≥–Њ–і –і–ї—П –≥—А—Г–њ–њ—Л –њ–ї–∞—Ж–µ–±–Њ. –†–∞–Ј–љ–Є—Ж–∞¬†вАУ 128,2 –Љ–ї/–≥–Њ–і (95% –Ф–Ш 70,8вАУ185,6, p < 0,001).
–Ґ–∞–Ї–Є–Љ –Њ–±—А–∞–Ј–Њ–Љ, –≤¬†–±–Њ–ї—М—И–Њ–є –≥—А—Г–њ–њ–µ –±–Њ–ї—М–љ—Л—Е —Б¬†–њ—А–Њ–≥—А–µ—Б—Б–Є—А—Г—О—Й–Є–Љ –Ш–Ы–§ –µ–ґ–µ–≥–Њ–і–љ–∞—П —Б–Ї–Њ—А–Њ—Б—В—М —Б–љ–Є–ґ–µ–љ–Є—П –§–Ц–Х–Ы –±—Л–ї–∞ –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ –љ–Є–ґ–µ —Б—А–µ–і–Є –њ–Њ–ї—Г—З–∞–≤—И–Є—Е –љ–Є–љ—В–µ–і–∞–љ–Є–±, —З–µ–Љ —Б—А–µ–і–Є –њ—А–Є–Љ–µ–љ—П–≤—И–Є—Е –њ–ї–∞—Ж–µ–±–Њ.
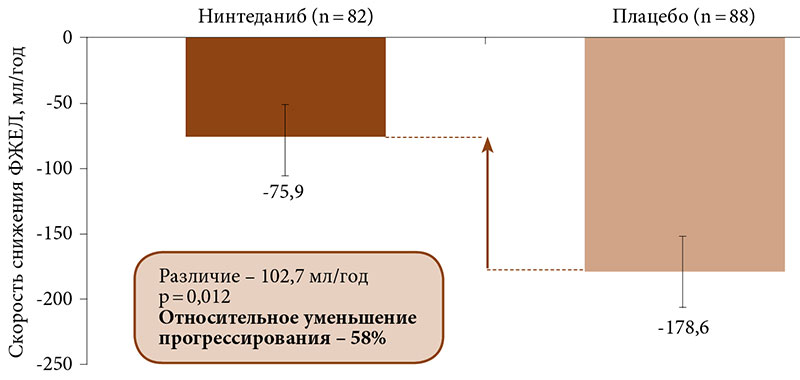
–Я—А–Є –њ—А–Њ–≤–µ–і–µ–љ–Є–Є —Б—Г–±–∞–љ–∞–ї–Є–Ј–∞ —А–µ–Ј—Г–ї—М—В–∞—В–Њ–≤ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П INBUILD –≤¬†–Ј–∞–≤–Є—Б–Є–Љ–Њ—Б—В–Є –Њ—В –Ї–Њ–љ–Ї—А–µ—В–љ–Њ–≥–Њ –і–Є–∞–≥–љ–Њ–Ј–∞ —Н—Д—Д–µ–Ї—В –љ–Є–љ—В–µ–і–∞–љ–Є–±–∞ –њ–Њ —Б—А–∞–≤–љ–µ–љ–Є—О —Б¬†–њ–ї–∞—Ж–µ–±–Њ –љ–∞ —Б–љ–Є–ґ–µ–љ–Є–µ —Б–Ї–Њ—А–Њ—Б—В–Є –§–Ц–Х–Ы –±—Л–ї –њ–Њ—Б—В–Њ—П–љ–љ—Л–Љ –≤–Њ –≤—Б–µ—Е –њ—П—В–Є –њ–Њ–і–≥—А—Г–њ–њ–∞—Е: –≥–Є–њ–µ—А—З—Г–≤—Б—В–≤–Є—В–µ–ї—М–љ—Л–є –њ–љ–µ–≤–Љ–Њ–љ–Є—В¬†вАУ 73,1 –Љ–ї/–≥–Њ–і (95% –Ф–Ш -8,6вАУ154,8), –∞—Г—В–Њ–Є–Љ–Љ—Г–љ–љ—Л–µ –Є–љ—В–µ—А—Б—В–Є—Ж–Є–∞–ї—М–љ—Л–µ –њ–љ–µ–≤–Љ–Њ–љ–Є–Є¬†вАУ 104,0 –Љ–ї/–≥–Њ–і (–Ф–Ш 21,1вАУ186,9), –Є–і–Є–Њ–њ–∞—В–Є—З–µ—Б–Ї–∞—П –љ–µ—Б–њ–µ—Ж–Є—Д–Є—З–µ—Б–Ї–∞—П –Є–љ—В–µ—А—Б—В–Є—Ж–Є–∞–ї—М–љ–∞—П –њ–љ–µ–≤–Љ–Њ–љ–Є—П¬†вАУ 141,6 –Љ–ї/–≥–Њ–і (–Ф–Ш 46,0вАУ237,2), –љ–µ–Ї–ї–∞—Б—Б–Є—Д–Є—Ж–Є—А—Г–µ–Љ–∞—П –Є–і–Є–Њ–њ–∞—В–Є—З–µ—Б–Ї–∞—П –Є–љ—В–µ—А—Б—В–Є—Ж–Є–∞–ї—М–љ–∞—П –њ–љ–µ–≤–Љ–Њ–љ–Є—П¬†вАУ 68,3 –Љ–ї/–≥–Њ–і (–Ф–Ш -31,4вАУ168,1), –і—А—Г–≥–Є–µ –Ш–Ч–Ы¬†вАУ 197,1 –Љ–ї/–≥–Њ–і (–Ф–Ш 77,6вАУ316,7) (p = 0,41 –Љ–µ–ґ–і—Г –≥—А—Г–њ–њ–∞–Љ–Є) [77]. –І—В–Њ –Ї–∞—Б–∞–µ—В—Б—П —А–µ–≤–Љ–∞—В–Є—З–µ—Б–Ї–Є—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є, —В–Њ —А–∞–Ј–љ–Є—Ж–∞ –њ–Њ –≤—Л—А–∞–ґ–µ–љ–љ–Њ—Б—В–Є —Б–љ–Є–ґ–µ–љ–Є—П –§–Ц–Х–Ы –Љ–µ–ґ–і—Г –≥—А—Г–њ–њ–Њ–є –∞–Ї—В–Є–≤–љ–Њ–≥–Њ –ї–µ—З–µ–љ–Є—П –Є¬†–≥—А—Г–њ–њ–Њ–є –њ–ї–∞—Ж–µ–±–Њ —Б–Њ—Б—В–∞–≤–Є–ї–∞ 102,7 –Љ–ї/–≥–Њ–і, —З—В–Њ –Њ–Ј–љ–∞—З–∞–µ—В –Њ—В–љ–Њ—Б–Є—В–µ–ї—М–љ–Њ–µ —Г–Љ–µ–љ—М—И–µ–љ–Є–µ –њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є—П –љ–∞ 58% (p = 0,012) (—А–Є—Б. 4).
–Т–∞–ґ–љ–Њ, —З—В–Њ –≤¬†–Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П—Е –љ–Є–љ—В–µ–і–∞–љ–Є–±–∞ —З–∞—Б—В–Њ—В–∞ —Б–µ—А—М–µ–Ј–љ—Л—Е –љ–µ–ґ–µ–ї–∞—В–µ–ї—М–љ—Л—Е —П–≤–ї–µ–љ–Є–є –±—Л–ї–∞ –њ—А–Є–µ–Љ–ї–µ–Љ–Њ–є –Є¬†–і–Њ—Б—В–Њ–≤–µ—А–љ–Њ –љ–µ –Њ—В–ї–Є—З–∞–ї–∞—Б—М –Њ—В –≥—А—Г–њ–њ—Л –њ–ї–∞—Ж–µ–±–Њ. –І–∞—Й–µ –Њ—В–Љ–µ—З–∞–ї–Є—Б—М —В—А–∞–љ–Ј–Є—В–Њ—А–љ—Л–µ –і–Є—Б–њ–µ–њ—В–Є—З–µ—Б–Ї–Є–µ —П–≤–ї–µ–љ–Є—П –Є¬†–і–Є–∞—А–µ—П [69, 72, 76].
–Т –Є—О–ї–µ 2020 –≥. –љ–Є–љ—В–µ–і–∞–љ–Є–± –Ј–∞—А–µ–≥–Є—Б—В—А–Є—А–Њ–≤–∞–љ –њ–Њ –њ–Њ–Ї–∞–Ј–∞–љ–Є—О ¬Ђ–і—А—Г–≥–Є–µ —Е—А–Њ–љ–Є—З–µ—Б–Ї–Є–µ —Д–Є–±—А–Њ–Ј–Є—А—Г—О—Й–Є–µ –Є–љ—В–µ—А—Б—В–Є—Ж–Є–∞–ї—М–љ—Л–µ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П –ї–µ–≥–Ї–Є—Е (–Ш–Ч–Ы) —Б¬†–њ—А–Њ–≥—А–µ—Б—Б–Є—А—Г—О—Й–Є–Љ —Д–µ–љ–Њ—В–Є–њ–Њ–Љ¬ї, —З—В–Њ –њ—А–Є –љ–∞–ї–Є—З–Є–Є –Њ—Б–љ–Њ–≤–∞–љ–Є–є –њ–Њ–Ј–≤–Њ–ї—П–µ—В –љ–∞–Ј–љ–∞—З–∞—В—М –µ–≥–Њ –њ–∞—Ж–Є–µ–љ—В–∞–Љ —Б¬†–†–Р –Є¬†–і—А—Г–≥–Є–Љ–Є —А–µ–≤–Љ–∞—В–Є—З–µ—Б–Ї–Є–Љ–Є –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П–Љ–Є, —Б–Њ–њ—А–Њ–≤–Њ–ґ–і–∞—О—Й–Є–Љ–Є—Б—П –Я–§-–Ш–Ч–Ы.
–Ч–∞–Ї–ї—О—З–µ–љ–Є–µ
–Э–∞ —Б–µ–≥–Њ–і–љ—П—И–љ–Є–є –і–µ–љ—М –љ–Є–љ—В–µ–і–∞–љ–Є–± —П–≤–ї—П–µ—В—Б—П –µ–і–Є–љ—Б—В–≤–µ–љ–љ—Л–Љ –њ—А–µ–њ–∞—А–∞—В–Њ–Љ —Б—А–µ–і–Є –њ—А–Є–Љ–µ–љ—П—О—Й–Є—Е—Б—П –і–ї—П –ї–µ—З–µ–љ–Є—П –Ш–Ч–Ы –њ—А–Є —А–µ–≤–Љ–∞—В–Є—З–µ—Б–Ї–Є—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П—Е. –Ю–љ —Е–∞—А–∞–Ї—В–µ—А–Є–Ј—Г–µ—В—Б—П –Ї–∞–Ї –∞–љ—В–Є–њ—А–Њ–ї–Є—Д–µ—А–∞—В–Є–≤–љ—Л–Љ, —В–∞–Ї –Є¬†–∞–љ—В–Є—Д–Є–±—А–Њ—В–Є—З–µ—Б–Ї–Є–Љ –і–µ–є—Б—В–≤–Є–µ–Љ. –С–ї–∞–≥–Њ–і–∞—А—П —В–∞–Ї–Њ–є —Г–љ–Є–Ї–∞–ї—М–љ–Њ–є –Њ—Б–Њ–±–µ–љ–љ–Њ—Б—В–Є –љ–Є–љ—В–µ–і–∞–љ–Є–± —Б–њ–Њ—Б–Њ–±–µ–љ —Г–ї—Г—З—И–Є—В—М —Б–Њ—Б—В–Њ—П–љ–Є–µ –±–Њ–ї—М–љ—Л—Е —Б¬†–Љ–љ–Њ–ґ–µ—Б—В–≤–µ–љ–љ–Њ–є –ї–µ–Ї–∞—А—Б—В–≤–µ–љ–љ–Њ–є —А–µ–Ј–Є—Б—В–µ–љ—В–љ–Њ—Б—В—М—О, –∞¬†—В–∞–Ї–ґ–µ –ї–Є—Ж, –Є–Љ–µ—О—Й–Є—Е –њ—А–Њ—В–Є–≤–Њ–њ–Њ–Ї–∞–Ј–∞–љ–Є—П –і–ї—П –љ–∞–Ј–љ–∞—З–µ–љ–Є—П —Ж–Є—В–Њ—В–Њ–Ї—Б–Є—З–µ—Б–Ї–Њ–є –Є–Љ–Љ—Г–љ–Њ—Б—Г–њ—А–µ—Б—Б–Є–≤–љ–Њ–є —В–µ—А–∞–њ–Є–Є.
–Э–Є–љ—В–µ–і–∞–љ–Є–± –њ–Њ–њ–Њ–ї–љ–Є–ї –∞—А—Б–µ–љ–∞–ї —Б—А–µ–і—Б—В–≤ –і–ї—П –±–Њ—А—М–±—Л —Б¬†—В—П–ґ–µ–ї–Њ–є —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–є —А–µ–≤–Љ–∞—В–Є—З–µ—Б–Ї–Њ–є –њ–∞—В–Њ–ї–Њ–≥–Є–µ–є.
D.E. Karateev, MD, PhD, Prof., E.L. Luchikhina, PhD, A.R. Tangiyeva
Moscow Regional Research and Clinical Institute
Contact person: Dmitry E. Karateev, dekar@inbox.ru
Interstitial lung disease (ILD) is a heterogeneous group of diseases that includes autoimmune rheumatic diseases such as systemic scleroderma, rheumatoid arthritis, and others. ILD is associated with a severe course and the risk of early death of patients.
For a long time, the rheumatologistsвАЩ arsenal in terms of treatment of ILD was very limited by the use of several drugs, such as cyclophosphamide, mycophenolate mofetil, etc., which do not allow to control all clinical situations. Currently, after publication of the results of clinical trials SENCSIS and INBUILD for the treatment of ILD in systemic scleroderma and other rheumatic diseases, the antiproliferative and antifibrotic drug nintedanib has been approved, which significantly expands the therapeutic options.
The review details the pathogenetic basis of the use of nintedanib and the results of its use with a focus on ILD in rheumatic diseases.
–£–≤–∞–ґ–∞–µ–Љ—Л–є –њ–Њ—Б–µ—В–Є—В–µ–ї—М uMEDp!
–£–≤–µ–і–Њ–Љ–ї—П–µ–Љ –Т–∞—Б –Њ —В–Њ–Љ, —З—В–Њ –Ј–і–µ—Б—М —Б–Њ–і–µ—А–ґ–Є—В—Б—П –Є–љ—Д–Њ—А–Љ–∞—Ж–Є—П, –њ—А–µ–і–љ–∞–Ј–љ–∞—З–µ–љ–љ–∞—П –Є—Б–Ї–ї—О—З–Є—В–µ–ї—М–љ–Њ –і–ї—П —Б–њ–µ—Ж–Є–∞–ї–Є—Б—В–Њ–≤ –Ј–і—А–∞–≤–Њ–Њ—Е—А–∞–љ–µ–љ–Є—П.
–Х—Б–ї–Є –Т—Л –љ–µ —П–≤–ї—П–µ—В–µ—Б—М —Б–њ–µ—Ж–Є–∞–ї–Є—Б—В–Њ–Љ –Ј–і—А–∞–≤–Њ–Њ—Е—А–∞–љ–µ–љ–Є—П, –∞–і–Љ–Є–љ–Є—Б—В—А–∞—Ж–Є—П –љ–µ –љ–µ—Б–µ—В –Њ—В–≤–µ—В—Б—В–≤–µ–љ–љ–Њ—Б—В–Є –Ј–∞ –≤–Њ–Ј–Љ–Њ–ґ–љ—Л–µ –Њ—В—А–Є—Ж–∞—В–µ–ї—М–љ—Л–µ –њ–Њ—Б–ї–µ–і—Б—В–≤–Є—П, –≤–Њ–Ј–љ–Є–Ї—И–Є–µ –≤ —А–µ–Ј—Г–ї—М—В–∞—В–µ —Б–∞–Љ–Њ—Б—В–Њ—П—В–µ–ї—М–љ–Њ–≥–Њ –Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–љ–Є—П –Т–∞–Љ–Є –Є–љ—Д–Њ—А–Љ–∞—Ж–Є–Є —Б –њ–Њ—А—В–∞–ї–∞ –±–µ–Ј –њ—А–µ–і–≤–∞—А–Є—В–µ–ї—М–љ–Њ–є –Ї–Њ–љ—Б—Г–ї—М—В–∞—Ж–Є–Є —Б –≤—А–∞—З–Њ–Љ.
–Э–∞–ґ–Є–Љ–∞—П –љ–∞ –Ї–љ–Њ–њ–Ї—Г ¬Ђ–Т–Њ–є—В–Є¬ї, –Т—Л –њ–Њ–і—В–≤–µ—А–ґ–і–∞–µ—В–µ, —З—В–Њ —П–≤–ї—П–µ—В–µ—Б—М –≤—А–∞—З–Њ–Љ –Є–ї–Є —Б—В—Г–і–µ–љ—В–Њ–Љ –Љ–µ–і–Є—Ж–Є–љ—Б–Ї–Њ–≥–Њ –≤—Г–Ј–∞.