Интерстициальные заболевания легких и прогрессирующий фиброз: на каком этапе поставить знак равенства
- Аннотация
- Статья
- Ссылки
- English
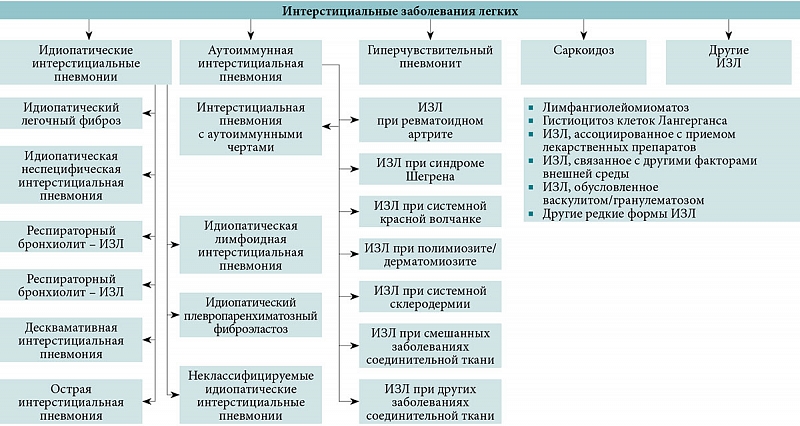
Интерстициальные заболевания легких (ИЗЛ) – гетерогенная группа заболеваний, характеризующихся поражением легочного интерстиция и дистальных отделов дыхательных путей. ИЗЛ охватывают множество разнообразных паренхиматозных болезней легких, включая заболевания неустановленной этиологии, известные как идиопатические интерстициальные пневмонии, а также заболевания, связанные с другими состояниями или воздействием окружающей среды (рисунок) [1].
В настоящее время среди всего многообразия ИЗЛ особое место занимает идиопатический легочный фиброз (ИЛФ), который встречается в основном у пациентов в возрасте старше 60 лет и характеризуется прогрессирующим фиброзированием, снижением функции легких и высокой смертностью [2]. Средняя продолжительность жизни пациента с ИЛФ составляет 3–5 лет. Другие варианты ИЗЛ также могут иметь фенотип прогрессирующего фиброзирования. К ним относятся ИЗЛ, ассоциированные с заболеваниями соединительной ткани, хронический гиперчувствительный пневмонит, неклассифицируемые ИЗЛ, идиопатическая неспецифическая интерстициальная пневмония и в редких случаях саркоидоз и ИЗЛ, связанные с профессиональной деятельностью [3, 4]. Для описания ИЗЛ, которое независимо от нозологической формы в определенный момент начинает проявлять признаки неуклонного прогрессирования фиброзирования, был предложен термин «прогрессирующий фиброз легких»». Это очень важно в аспекте тактики ведения пациента, поскольку данный фенотип в отсутствие антифибротической терапии может быть очень близок к ИЛФ по прогнозу, темпам снижения легочной функции, риску летального исхода и обострений [5–8].
Принято считать, что прогрессирование фиброзирующих ИЗЛ выражается в нарастании фиброза легких, выявляемого при проведении компьютерной томографии (КТ) органов грудной клетки, снижении форсированной жизненной емкости легких (ФЖЕЛ) и газообмена (диффузионной способности легких по монооксиду углерода (DLCO)), усилении респираторных симптомов, снижении толерантности к физической нагрузке, а также ухудшении качества жизни. В большинстве клинических и обсервационных исследований с участием пациентов с ИЗЛ определяли прогрессирование заболевания с точки зрения снижения ФЖЕЛ, измеряемой как изменение исходного уровня в миллилитрах или как процент от должного значения (обычно ≥ 10% от исходного) [10, 11]. Так, на основании результатов исследования INBUILD были сформулированы критерии прогрессирования фиброзирующих ИЗЛ, оцениваемых за 24 месяца:
- относительное снижение ФЖЕЛ на ≥ 10%долж;
- относительное снижение ФЖЕЛ на ≥ 5 < 10%долж и ухудшение респираторных симптомов;
- относительное снижение ФЖЕЛ на ≥ 5 < 10%долж и увеличение распространенности фиброза по данным КТ высокого разрешения;
- ухудшение респираторных симптомов и увеличение распространенности фиброза по данным КТ высокого разрешения [7, 11].
Прогрессирующий фиброз (ПФ) легких может формироваться на различных этапах естественного течения многих ИЗЛ. Так, по данным исследования PROGRESS, 27% неИЛФ-ИЗЛ соответствует критериям прогрессирования. В недавно опубликованных обновленных рекомендациях ATS/ERS/JRS/ALAT по ИЛФ и ПФ-ИЗЛ экспертами были предложены следующие критерии прогрессирующего легочного фиброза [9].
У пациентов с ИЗЛ независимо от известной или неизвестной этиологии, отличной от ИЛФ, у которых имеются рентгенологические признаки легочного фиброза, ПФ легких диагностируется при наличии по крайней мере двух из трех критериев, возникших в течение последнего года:
- ухудшение респираторных симптомов;
- физиологические признаки прогрессирования заболевания (любое из следующего): а) абсолютное снижение ФЖЕЛ ≥ 5% от прогнозируемого в течение года наблюдения; б) абсолютное снижение DlCO (с поправкой на Hb) ≥ 10% от прогнозируемого в течение года наблюдения);
- рентгенологические признаки прогрессирования заболевания (один или несколько из следующих признаков): а) увеличенная степень или тяжесть тракционных бронхоэктазов; б) появление новых участков повышенной плотности легочной ткани по типу «матового стекла» с тракционными бронхоэктазами; в) появление тонких ретикулярных изменений; г) увеличение степени или повышение грубости ретикулярных изменений; д) появление или увеличение «сотовых» структур; ж) прогрессирующее уменьшение объема доли легкого.
Среди всех ИЗЛ с высоким риском прогрессирующего фиброза, возможностью формирования фибротического фенотипа на втором месте по частоте встречаемости после ИЛФ находится неспецифическая интерстициальная пневмония (НсИП).
НсИП – это интерстициальная пневмония, генез которой может существенно различаться. КТ-паттерн НсИП может наблюдаться как в рамках первичного процесса – идиопатическая НсИП, так и в рамках других патологических состояний, по отношению к которым ИЗЛ является вторичным, что требует исключения других возможных причин, главным образом системных заболеваний соединительной ткани (СЗСТ) [13].
Распространенность идиопатической НсИП, по данным нескольких ретроспективных когортных исследований, оценивается от одного до девяти случаев на 100 тыс. человек [14], а заболеваемость – примерно три случая на 1 млн человек в год [15].
Согласно общенациональному исследованию, проведенному Корейской академией туберкулеза и респираторных заболеваний в 2008 г., НсИП – второе по распространенности ИЗЛ после идиопатического легочного фиброза – 11,9% из 2186 пациентов с идиопатическими ИЗЛ [16].
Между тем в разных странах показатели распространенности НсИП отличаются. Так, в когорте университетской больницы в Дании был проанализирован 431 случай интерстициального заболевания легких с 2003 по 2009 г. Из всех ИЗЛ на НсИП приходилось 7%, что ставило НсИП на четвертое место по распространенности после ИЛФ, ИЗЛ, ассоциированных с СЗСТ, и гиперчувствительного пневмонита.
НСИП чаще встречается у женщин, некурящих и возникает в более раннем возрасте, чем ИЛФ [17, 18]. Клинические проявления включают подострую или хроническую одышку и кашель, длящиеся в среднем шесть месяцев. При аускультации у пациентов с НсИП выслушиваются двусторонние трескучие хрипы в конце вдоха в базальных отделах грудной клетки, но большинство физикальных признаков неспецифичны.
Функциональные тесты демонстрируют рестриктивный тип нарушений вентиляционной способности легких [19].
НсИП является наиболее распространенной морфологической картиной поражения легких при различных СЗСТ. Поэтому для исключения системных иммуновоспалительных заболеваний критически важно обращать внимание на экстрапульмональные проявления: феномен Рейно, артралгию или артрит, кожную сыпь, сухость во рту и сухость глаз, а также результаты лабораторных маркеров. Если не удалось подтвердить конкретное СЗСТ или некоторые клинические признаки сходны с таковыми при ЗСТ, может быть поставлен диагноз идиопатической НСИП [20].
Третий по частоте встречаемости среди ИЗЛ – гиперчувствительный пневмонит (ГП). ГП – это иммуноопосредованное заболевание, развивающееся при ингаляционном воздействии разнообразных органических и неорганических антигенов (индукторов) у восприимчивых людей.
По данным Y. Lacasse и соавт., доля ГП среди пациентов с ИЗЛ составляет около 29% [13, 21]. Распространенность ГП наиболее высока среди людей старшего возраста (65 лет и старше). При этом чаще диагноз пациенту с ГП устанавливается на пятом или шестом десятилетии жизни. Тем не менее заболеванию подвержены и другие возрастные группы. Распространенность ГП зависит от региональных различий в климате, профессиональных воздействий и влияния окружающей среды. Согласно имеющимся данным, заболеваемость ГП составляет от 0,3 до 0,9 на 100 тыс. человек, но может быть и выше. Показано, что пациенты с фиброзирующим фенотипом ГП имеют более низкую жизненную емкость легких, диффузионную способность и процент лимфоцитов в жидкости бронхоальвеолярного лаважа, чем пациенты с нефиброзирующим вариантом [8].
Фиброз развивается у многих пациентов с хроническим ГП. Из-за сходства ИЛФ и хронического фиброзирующего ГП их порой трудно дифференцировать [21–25]. При фиброзирующем фенотипе ГП нередко определяется картина обычной интерстициальной пневмонии (ОИП) на КТ органов грудной клетки (ОГК) [26]. Потенциальные рентгенологические симптомы, по которым можно отличить фибротический вариант ГП от ИЛФ, включают один или несколько из следующих признаков: участки повышения плотности легочной ткани по типу «матового стекла», бронхоцентрический характер выявленных изменений, мозаичная плотность (при нативном исследовании) и «воздушные ловушки» (при экспираторном исследовании), картина трех плотностей – так называемый синдром «головки сыра» и иногда наличие центрилобулярных очагов мягкотканной плотности (то есть КТ-признаков поражения малых бронхов) [9, 27].
Хронический фиброзирующий ГП развивается в течение периода от нескольких месяцев до нескольких лет и обычно является следствием продолжительного воздействия ингаляционного антигена. При этом в значительном числе случаев этиологический агент остается невыясненым. У пациентов с хроническим ГП качество жизни может быть даже значительно хуже, чем у пациентов с ИЛФ [31]. Выраженность фиброза на КТ ОГК у пациентов с хроническим ГП служит прогностическим фактором смертности [24]. ГП как с картиной ОИП, по данным КТ ОГК, так и с картиной НСИП может ассоциироваться с показателями выживаемости, аналогичными показателям при ИЛФ [28]. Медиана выживаемости после постановки диагноза хронического фиброзирующего ГП составляет около пяти лет [29].
В современную классификацию ИЗЛ, приводящих к фиброзу, также входит саркоидоз [5]. Саркоидоз – системное воспалительное заболевание, характеризующееся образованием неказеифицирующихся гранулем и активацией Т-клеток в месте гранулематозного воспаления с высвобождением различных хемокинов и цитокинов [30]. Примерно в 70% случаев заболевание протекает благоприятно. Однако в последние годы возрос интерес к фибротическому фенотипу саркоидоза, который сопровождается развитием фиброзно-кистозных изменений в легких, а также нарушением их вентиляционной функции. Существует мнение, что развитие фиброза при саркоидозе не всегда является признаком хронического заболевания. С одной стороны, фиброз может возникать и на ранней стадии заболевания, с другой – наличие фиброза не обязательно означает, что воспаление хронически активное. И хронический активный фиброзирующий процесс, и остаточное фиброзное повреждение приводит к долгосрочным нарушениям и попадает под категорию хронического саркоидоза [28]. Среди всех пациентов с саркоидозом поражение легких выявляется приблизительно у 90%. Признаки и симптомы саркоидоза легких неспецифичны и включают непродуктивный кашель, одышку при физической нагрузке, стеснение в груди, гипоксемию и снижение функции легких [32, 33].
Приблизительно у 20% пациентов с саркоидозом легких развивается фиброз (IV стадия саркоидоза), который ассоциируется со значительной заболеваемостью и смертностью [34]. Легочный фиброз поражает преимущественно верхние доли (задние сегменты) и локализуется в дыхательных путях без образования воздушных ловушек [45]. Степень и тип фиброза в популяции заметно варьируются. На КТ ОГК определяются ретикулярные изменения – от слабых и еле заметных до плотных линейных полос, кистозная трансформация, тракционные бронхоэктазы и деформация бронхов [33]. Можно также наблюдать обширную паренхиматозную деструкцию. У пациентов с фиброзирующим саркоидозом легких редко имеет место картина ОИП, хотя иногда в верхних отделах легких можно увидеть сотовую структуру [34].
Не до конца изучены темпы снижения функции легких у пациентов с фибротическим фенотипом саркоидоза [34]. В некоторых публикациях сообщается о 16%-ной смертности в течение десяти лет среди пациентов с фиброзирующим саркоидозом легких [36]. С учетом сопутствующих заболеваний и других потенциальных причин смерти это может свидетельствовать о том, что лишь небольшая часть пациентов имеет прогрессирующий фиброзирующий фенотип, несмотря на проводимую терапию.
По данным рандомизированных плацебо-контролируемых исследований INBUILD и INPULSIS, пациенты в исследовании INBUILD, у которых был диагностирован фиброзирующий фенотип ИЗЛ, отличный от ИЛФ, и которые соответствовали критериям прогрессирования ИЗЛ в течение предыдущих 24 месяцев, имели течение заболевания, сходное с таковым у пациентов с ИЛФ. Темпы снижения ФЖЕЛ и смертности в течение 52 недель были сопоставимы в исследованиях INBUILD и INPULSIS.
Другие исследования показали, что у пациентов с фиброзирующими ИЗЛ наличие ОИП-подобной картины на КТ ОГК ассоциируется с более быстрым прогрессированием заболевания [3, 6, 37–40]. Исходя из этого, скорость снижения ФЖЕЛ и смертность в течение 52 недель в исследовании INBUILD были выше у пациентов с паттерном ОИП на КТ ОГК, чем у пациентов с другими фиброзными паттернами. Тем не менее скорость снижения ФЖЕЛ у субъектов с признаками фиброза на КТ высокого разрешения была значительной (160 мл/год), причем почти у 50% этих пациентов наблюдалось относительное снижение ФЖЕЛ > 10% от прогнозируемого в течение 52 недель, аналогично пациентам с ИЛФ.
Анализ данных исследований INBUILD и INPULSIS позволяет предположить, что прогрессирующие фиброзирующие ИЗЛ, отличные от ИЛФ, имеют клиническое течение, сходное с ИЛФ, независимо от основного диагноза ИЗЛ или признаков фиброза на КТ ОГК [10]. Прогрессирующие фиброзирующие ИЗЛ характеризуются патофизиологически схожими процессами с ИЛФ, что указывает на возможность применения общих подходов к лечению. Пациентам с ИЛФ рекомендуется использование антифибротических препаратов (пирфенидон или нинтеданиб). В большинстве случаев фиброзирующих ИЗЛ, отличных от ИЛФ, показана противовоспалительная терапия системными глюкокортикостероидами, цитостатическими препаратами либо их комбинацией. Обычно она используется в качестве терапии первой линии при подозрении на заболевание, вызванное воспалением.
Хотя в исследовании INBUILD не предполагалось оценивать эффекты нинтеданиба у пациентов с конкретными диагнозами ИЗЛ, по данным K. Kevin и соавт., у пациентов со степенью фиброза > 10%, по данным КТ ОГК, и клиническими признаками прогрессирования, несмотря на лечение, скорость снижения ФЖЕЛ была одинаковой в подгруппах с разными диагнозами. Это говорит о необходимости в кратчайшие сроки определить фенотип ИЗЛ для назначения оптимального лечения и замедления прогрессирования заболевания.
Снижение ФЖЕЛ > 10% от должного ассоциируется с летальным исходом у пациентов с ИЛФ [41, 42] и другими хроническими фиброзирующими ИЗЛ [43–45]. Лица, включенные в исследование INBUILD, соответствовали определенным в протоколе критериям прогрессирования ИЗЛ за два года до скрининга. Пациенты с ИЛФ, включенные в исследование INPULSIS, не должны были соответствовать критериям прогрессирования заболевания для включения в исследование, поскольку ИЛФ по определению является прогрессирующим заболеванием [46]. В исследованиях INBUILD и INPULSIS приблизительно у половины пациентов группы плацебо наблюдалось относительное снижение ФЖЕЛ более чем на 10% от должного, а у 2/3 – относительное снижение > 5% от исходного в течение 52 недель. В исследовании INBUILD относительное снижение ФЖЕЛ более чем на 10% от должного было связано с более чем трехкратным увеличением риска смерти в течение 52 недель как в общей популяции, так и у субъектов с фиброзным паттерном, подобным ОИП, на КТ ОГК, который был сопоставим с тем, что наблюдалось в испытаниях INPULSIS. Эти данные свидетельствуют о том, что, как и в случае с ИЛФ, снижение ФЖЕЛ обусловлено повышенным риском ранней смерти у пациентов с фиброзирующим фенотипом ИЗЛ, не относящимися к ИЛФ, которые прогрессировали, несмотря на лечение.
■■■
ИЗЛ – обширная группа патологических состояний, проявляющихся воспалением и фибротическим ремоделированием легочного интерстиция, причем выраженность и взаимосвязь этих процессов могут существенно различаться при разных нозологических формах ИЗЛ. Так, воспаление играет ключевую роль в развитии гиперчувствительного пневмонита, аутоиммунных ИЗЛ и саркоидоза. Однако в патогенезе идиопатического легочного фиброза значение воспаления минимально.
Среди всех ИЗЛ наиболее прогностически неблагоприятным считается ИЛФ: медиана выживаемости едва превышает три года. Тем не менее, согласно данным post-hoc-анализа INBUILD, ИЗЛ с развитием прогрессирующего легочного фиброза могут быть прогностически неотличимыми от ИЛФ.
Таким образом, развитие фиброза легочного интерстиция не является облигатной характеристикой ИЗЛ, однако его развитие – важнейшая прогностическая детерминанта и определяющий фактор в выборе тактики лечения. Для замедления темпов фиброзообразования в терапевтическом арсенале существуют две молекулы – нинтеданиб и пирфенидон. Если для пирфенидона зарегистрировано одно единственное показание – ИЛФ, то для нинтеданиба – весь спектр ИЗЛ с прогрессирующим фиброзом легких. Собственно критерии INBUILD являются наиболее используемыми в клинической практике и научных исследованиях для определения ПФ-ИЗЛ.
В недавно опубликованных рекомендациях по ИЛФ и ПФ-ИЗЛ [9] предложены новые критерии. Но они требуют клинической валидации и проверки временем. Представляется критически необходимым поиск новых биологических маркеров, позволяющих выявлять пациентов с ИЗЛ, имеющих наибольший риск прогрессирования, для раннего назначения болезнь-модифицирующего лечения, а не констатации факта прогрессирования на запущенных стадиях заболевания, когда терапевтические интервенции едва ли способны нарушить неблагоприятное естественное течение болезни.
Ye.N. Adamovskaya, Ye.I. Shchepikhin, Ye.I. Shmelev, PhD, Prof.
Central Research Institute of Tuberculosis
Contact person: Yevgeny I. Shchepikhin, shhepikhin11@yandex.ru
Interstitial lung diseases are a large group of pathological conditions manifested by inflammation and fibrotic remodeling of the pulmonary interstitium, and the severity and relationship of these processes can vary significantly in various nosological forms of ILD. The severity of fibrosis in the territory of the pulmonary interstitium and the rate of its progression are the most important prognostic determinant and the determining factor in the choice of therapeutic strategies. To describe ILD, which, regardless of the nosological form, at a certain point in time begins to show signs of a steady progression of fibrosis, the term ‘progressive pulmonary fibrosis’ was created. This article will present the current state of the problem of ILD with a progressive fibrotic phenotype.
Уважаемый посетитель uMEDp!
Уведомляем Вас о том, что здесь содержится информация, предназначенная исключительно для специалистов здравоохранения.
Если Вы не являетесь специалистом здравоохранения, администрация не несет ответственности за возможные отрицательные последствия, возникшие в результате самостоятельного использования Вами информации с портала без предварительной консультации с врачом.
Нажимая на кнопку «Войти», Вы подтверждаете, что являетесь врачом или студентом медицинского вуза.