–Ъ–ї–Є–љ–Є—З–µ—Б–Ї–∞—П —Д–∞—А–Љ–∞–Ї–Њ–ї–Њ–≥–Є—П –Љ–Є–Њ—А–µ–ї–∞–Ї—Б–∞–љ—В–Њ–≤
- –Р–љ–љ–Њ—В–∞—Ж–Є—П
- –°—В–∞—В—М—П
- –°—Б—Л–ї–Ї–Є
- English

–Т–≤–µ–і–µ–љ–Є–µ
–Ь–Є–Њ—А–µ–ї–∞–Ї—Б–∞–љ—В—Л —И–Є—А–Њ–Ї–Њ –Є—Б–њ–Њ–ї—М–Ј—Г—О—В—Б—П –≤¬†–Ї–Њ–Љ–њ–ї–µ–Ї—Б–љ–Њ–Љ –ї–µ—З–µ–љ–Є–Є —Б–њ–∞—Б—В–Є—З–љ–Њ—Б—В–Є –њ—А–Є —А–∞—Б—Б–µ—П–љ–љ–Њ–Љ —Б–Ї–ї–µ—А–Њ–Ј–µ –Є–ї–Є –њ–Њ—Б–ї–µ –Є–љ—Б—Г–ї—М—В–∞, –∞¬†—В–∞–Ї–ґ–µ –±–Њ–ї–µ–≤—Л—Е —Б–Є–љ–і—А–Њ–Љ–Њ–≤.
–£—З–µ–љ—Л–µ –Є–Ј¬†–°–®–Р —Г—Б—В–∞–љ–Њ–≤–Є–ї–Є, —З—В–Њ –ї–Є—И—М —Г¬†16% –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†—А–∞—Б—Б–µ—П–љ–љ—Л–Љ —Б–Ї–ї–µ—А–Њ–Ј–Њ–Љ —Б–њ–∞—Б—В–Є—З–љ–Њ—Б—В—М –Њ—В—Б—Г—В—Б—В–≤–Њ–≤–∞–ї–∞, –≤¬†31% —Б–ї—Г—З–∞–µ–≤ –Њ–љ–∞ –±—Л–ї–∞ –≤—Л—А–∞–ґ–µ–љ–∞ –≤¬†–Љ–Є–љ–Є–Љ–∞–ї—М–љ–Њ–є —Б—В–µ–њ–µ–љ–Є, –≤¬†19%¬†вАУ –≤¬†–ї–µ–≥–Ї–Њ–є, –≤¬†17%¬†вАУ —Б—А–µ–і–љ–µ–є, –≤¬†13%¬†вАУ —В—П–ґ–µ–ї–Њ–є, –≤¬†4% —Б–ї—Г—З–∞–µ–≤¬†вАУ –љ–Њ—Б–Є–ї–∞ —В–Њ—В–∞–ї—М–љ—Л–є —Е–∞—А–∞–Ї—В–µ—А, –і–µ–ї–∞—П –љ–µ–≤–Њ–Ј–Љ–Њ–ґ–љ–Њ–є –њ–Њ–≤—Б–µ–і–љ–µ–≤–љ—Г—О –∞–Ї—В–Є–≤–љ–Њ—Б—В—М [1].
–Х—Й–µ –Њ–і–љ—Г¬†–≥—А—Г–њ–њ—Г –њ–∞—Ж–Є–µ–љ—В–Њ–≤, –Ї–Њ—В–Њ—А—Л–Љ —В—А–µ–±—Г–µ—В—Б—П –љ–∞–Ј–љ–∞—З–µ–љ–Є–µ –Љ–Є–Њ—А–µ–ї–∞–Ї—Б–∞–љ—В–Њ–≤, —Б–Њ—Б—В–∞–≤–ї—П—О—В –њ–µ—А–µ–љ–µ—Б—И–Є–µ –Є–љ—Б—Г–ї—М—В [2]. –Ґ–∞–Ї, –≤¬†–њ–µ—А–≤—Л–µ —В—А–Є –Љ–µ—Б—П—Ж–∞ –њ–Њ—Б–ї–µ –Є–љ—Б—Г–ї—М—В–∞, —З–∞—Й–µ¬†–≥–µ–Љ–Њ—А—А–∞–≥–Є—З–µ—Б–Ї–Њ–≥–Њ, —Б–њ–∞—Б—В–Є—З–љ–Њ—Б—В—М –љ–∞–±–ї—О–і–∞–ї–∞—Б—М —Г¬†17вАУ25% –±–Њ–ї—М–љ—Л—Е [3].
–Ь–Є–Њ—А–µ–ї–∞–Ї—Б–∞–љ—В—Л —В–∞–Ї–ґ–µ –њ—А–Є–Љ–µ–љ—П—О—В—Б—П –њ—А–Є –±–Њ–ї–Є –≤¬†–љ–Є–ґ–љ–µ–є —З–∞—Б—В–Є —Б–њ–Є–љ—Л, –њ—А–µ–Є–Љ—Г—Й–µ—Б—В–≤–µ–љ–љ–Њ –Њ—Б—В—А–Њ–є –Є¬†–∞—Б—Б–Њ—Ж–Є–Є—А–Њ–≤–∞–љ–љ–Њ–є —Б¬†–Љ—Л—И–µ—З–љ—Л–Љ —Б–њ–∞–Ј–Љ–Њ–Љ [4]. –°–Њ–≥–ї–∞—Б–љ–Њ –і–∞–љ–љ—Л–Љ —А–∞–Ј–љ—Л—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞—В–µ–ї–µ–є, —А–∞—Б–њ—А–Њ—Б—В—А–∞–љ–µ–љ–љ–Њ—Б—В—М –±–Њ–ї–Є –≤¬†–љ–Є–ґ–љ–µ–є —З–∞—Б—В–Є —Б–њ–Є–љ—Л –≤–∞—А—М–Є—А—Г–µ—В—Б—П –Њ—В¬†1,4 –і–Њ 20,0% [5]. –Т¬†–†–Њ—Б—Б–Є–є—Б–Ї–Њ–є –§–µ–і–µ—А–∞—Ж–Є–Є, –њ–Њ¬†–і–∞–љ–љ—Л–Љ –Т—Б–µ–Љ–Є—А–љ–Њ–є –Њ—А–≥–∞–љ–Є–Ј–∞—Ж–Є–Є –Ј–і—А–∞–≤–Њ–Њ—Е—А–∞–љ–µ–љ–Є—П, –±–Њ–ї—М –≤¬†–љ–Є–ґ–љ–µ–є —З–∞—Б—В–Є —Б–њ–Є–љ—Л —Б—А–µ–і–Є –ї–Є—Ж —Б—В–∞—А—И–µ 50 –ї–µ—В –≤—Б—В—А–µ—З–∞–µ—В—Б—П –≤¬†56% —Б–ї—Г—З–∞–µ–≤ [6]. –Т¬†—В–Њ –ґ–µ –≤—А–µ–Љ—П —Г—Б—В–∞–љ–Њ–≤–ї–µ–љ–Њ, —З—В–Њ –Њ—В¬†50 –і–Њ 80% –≤–Ј—А–Њ—Б–ї–Њ–≥–Њ –љ–∞—Б–µ–ї–µ–љ–Є—П —Б—В–∞–ї–Ї–Є–≤–∞–µ—В—Б—П —Б¬†—В–∞–Ї–Њ–є –±–Њ–ї—М—О —Е–Њ—В—П –±—Л —А–∞–Ј –≤¬†–ґ–Є–Ј–љ–Є [7, 8].
–Ф–ї—П —Б—А–∞–≤–љ–µ–љ–Є—П —Н—Д—Д–µ–Ї—В–Є–≤–љ–Њ—Б—В–Є –Љ–Є–Њ—А–µ–ї–∞–Ї—Б–∞–љ—В–Њ–≤ –њ—А–Є –±–Њ–ї–Є –≤¬†–љ–Є–ґ–љ–µ–є —З–∞—Б—В–Є —Б–њ–Є–љ—Л R. Chou –Є¬†—Б–Њ–∞–≤—В. –њ—А–Њ–≤–µ–ї–Є —Б–Є—Б—В–µ–Љ–∞—В–Є—З–µ—Б–Ї–Є–є –Њ–±–Ј–Њ—А [9]. –£—З–µ–љ—Л–µ –Њ—В–Љ–µ—В–Є–ї–Є, —З—В–Њ –і–ї—П –Њ—Ж–µ–љ–Ї–Є —В–∞–Ї–Њ–≤–Њ–є –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ –Ї–∞—З–µ—Б—В–≤–µ–љ–љ—Л—Е –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є [9].
–Я–Њ—Б–Ї–Њ–ї—М–Ї—Г –љ–∞–Є–±–Њ–ї–µ–µ —Н—Д—Д–µ–Ї—В–Є–≤–љ—Л–є –Љ–Є–Њ—А–µ–ї–∞–Ї—Б–∞–љ—В –њ—А–Є –ї–µ—З–µ–љ–Є–Є –±–Њ–ї–Є –≤¬†–љ–Є–ґ–љ–µ–є —З–∞—Б—В–Є —Б–њ–Є–љ—Л –љ–µ¬†–Њ–њ—А–µ–і–µ–ї–µ–љ, –њ—А–Є –≤—Л–±–Њ—А–µ –њ—А–µ–њ–∞—А–∞—В–∞ —Б–ї–µ–і—Г–µ—В —Г–і–µ–ї—П—В—М –≤–љ–Є–Љ–∞–љ–Є–µ –µ–≥–Њ –њ–µ—А–µ–љ–Њ—Б–Є–Љ–Њ—Б—В–Є –Є¬†–њ—А–Њ—Д–Є–ї—О –±–µ–Ј–Њ–њ–∞—Б–љ–Њ—Б—В–Є.
–Ю—Б–Њ–±–µ–љ–љ–Њ –≤–∞–ґ–љ–∞ —Е–Њ—А–Њ—И–∞—П –њ–µ—А–µ–љ–Њ—Б–Є–Љ–Њ—Б—В—М –њ—А–Є –ї–µ—З–µ–љ–Є–Є –≤¬†–∞–Љ–±—Г–ї–∞—В–Њ—А–љ—Л—Е —Г—Б–ї–Њ–≤–Є—П—Е, —В–∞–Ї –Ї–∞–Ї —А–∞–Ј–≤–Є—В–Є–µ –љ–µ–ґ–µ–ї–∞—В–µ–ї—М–љ—Л—Е –ї–µ–Ї–∞—А—Б—В–≤–µ–љ–љ—Л—Е —А–µ–∞–Ї—Ж–Є–є –Љ–Њ–ґ–µ—В —Б–Њ–њ—А–Њ–≤–Њ–ґ–і–∞—В—М—Б—П —Б–љ–Є–ґ–µ–љ–Є–µ–Љ –Ї–Њ–Љ–њ–ї–∞–µ–љ—В–љ–Њ—Б—В–Є –Є¬†–і–∞–ґ–µ –њ—А–µ–Ї—А–∞—Й–µ–љ–Є–µ–Љ –њ—А–Є–µ–Љ–∞ –њ—А–µ–њ–∞—А–∞—В–∞ [10].
–Т¬†—Б—В–∞—В—М–µ —А–∞—Б—Б–Љ–Њ—В—А–µ–љ–∞ —Д–∞—А–Љ–∞–Ї–Њ–ї–Њ–≥–Є—П –љ–∞–Є–±–Њ–ї–µ–µ –Є–Ј—Г—З–µ–љ–љ—Л—Е –Є¬†—И–Є—А–Њ–Ї–Њ –њ—А–Є–Љ–µ–љ—П–µ–Љ—Л—Е –≤¬†–Ї–ї–Є–љ–Є—З–µ—Б–Ї–Њ–є –њ—А–∞–Ї—В–Є–Ї–µ –Љ–Є–Њ—А–µ–ї–∞–Ї—Б–∞–љ—В–Њ–≤: —В–Є–Ј–∞–љ–Є–і–Є–љ–∞, –±–∞–Ї–ї–Њ—Д–µ–љ–∞ –Є¬†—В–Њ–ї–њ–µ—А–Є–Ј–Њ–љ–∞.
–§–∞—А–Љ–∞–Ї–Њ–і–Є–љ–∞–Љ–Є–Ї–∞ –Є¬†—Д–∞—А–Љ–∞–Ї–Њ–Ї–Є–љ–µ—В–Є–Ї–∞
–Ґ–Є–Ј–∞–љ–Є–і–Є–љ
–Ґ–Є–Ј–∞–љ–Є–і–Є–љ –Њ—В–љ–Њ—Б–Є—В—Б—П –Ї¬†–Љ–Є–Њ—А–µ–ї–∞–Ї—Б–∞–љ—В–∞–Љ —Ж–µ–љ—В—А–∞–ї—М–љ–Њ–≥–Њ –і–µ–є—Б—В–≤–Є—П. –Я—А–µ–њ–∞—А–∞—В —П–≤–ї—П–µ—В—Б—П –∞–≥–Њ–љ–Є—Б—В–Њ–Љ –∞–ї—М—Д–∞-2-–∞–і—А–µ–љ–µ—А–≥–Є—З–µ—Б–Ї–Є—Е —А–µ—Ж–µ–њ—В–Њ—А–Њ–≤. –Ш—Е¬†—Б—В–Є–Љ—Г–ї—П—Ж–Є—П –≤¬†–Њ–±–ї–∞—Б—В–Є –њ—А–µ—Б–Є–љ–∞–њ—В–Є—З–µ—Б–Ї–Њ–є –Љ–µ–Љ–±—А–∞–љ—Л –њ—А–Є–≤–Њ–і–Є—В –Ї¬†—Г–Љ–µ–љ—М—И–µ–љ–Є—О —Б–µ–Ї—А–µ—Ж–Є–Є –∞–Љ–Є–љ–Њ–Ї–Є—Б–ї–Њ—В (–≥–ї—Г—В–∞–Љ–∞—В–∞ –Є¬†–∞—Б–њ–∞—А—В–∞—В–∞), –∞–Ї—В–Є–≤–Є—А—Г—О—Й–Є—Е –Љ–Њ—В–Њ–љ–µ–є—А–Њ–љ—Л —Б–њ–Є–љ–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞ [11, 12]. –Ъ–∞–Ї —Б–ї–µ–і—Б—В–≤–Є–µ, —Г–≥–љ–µ—В–∞—О—В—Б—П –њ–Њ–ї–Є—Б–Є–љ–∞–њ—В–Є—З–µ—Б–Ї–Є–µ —А–µ—Д–ї–µ–Ї—Б—Л —Б–њ–Є–љ–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞, –Њ—В–≤–µ—В—Б—В–≤–µ–љ–љ—Л–µ –Ј–∞¬†–≥–Є–њ–µ—А—В–Њ–љ—Г—Б –Љ—Л—И—Ж. –Э–∞—А—П–і—Г —Б¬†—Н—В–Є–Љ –Є–љ–≥–Є–±–Є—А–Њ–≤–∞–љ–Є–µ –Љ–µ–ґ–љ–µ–є—А–Њ–љ–∞–ї—М–љ–Њ–є –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є, —В–∞–Ї–ґ–µ –≤—Л–Ј–≤–∞–љ–љ–Њ–µ —Б—В–Є–Љ—Г–ї—П—Ж–Є–µ–є –∞–ї—М—Д–∞-2-–∞–і—А–µ–љ–µ—А–≥–Є—З–µ—Б–Ї–Є—Е —А–µ—Ж–µ–њ—В–Њ—А–Њ–≤, –Њ–±—Г—Б–ї–Њ–≤–ї–Є–≤–∞–µ—В –і–Њ–њ–Њ–ї–љ–Є—В–µ–ї—М–љ—Г—О –∞–љ—В–Є–љ–Њ—Ж–Є—Ж–µ–њ—В–Є–≤–љ—Г—О –Є¬†–њ—А–Њ—В–Є–≤–Њ—Б—Г–і–Њ—А–Њ–ґ–љ—Г—О –∞–Ї—В–Є–≤–љ–Њ—Б—В—М¬†[12].
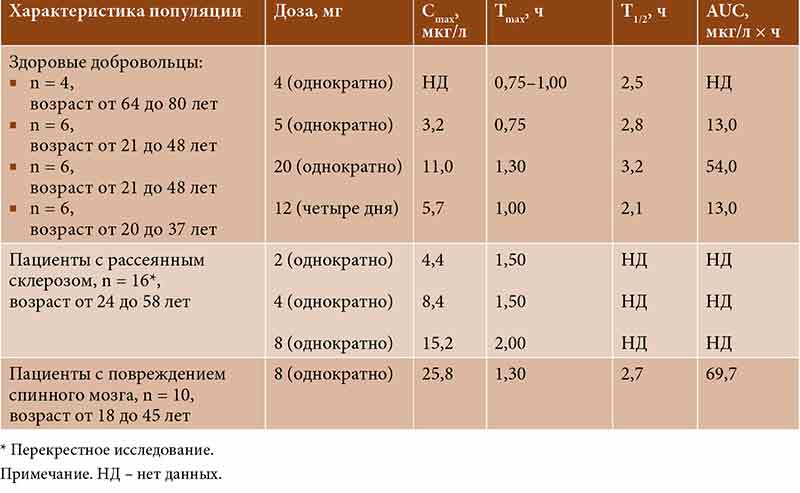
–§–∞—А–Љ–∞–Ї–Њ–Ї–Є–љ–µ—В–Є—З–µ—Б–Ї–Є–є –њ—А–Њ—Д–Є–ї—М —В–Є–Ј–∞–љ–Є–і–Є–љ–∞ –Є–Ј—Г—З–∞–ї—Б—П –≤¬†—А—П–і–µ –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є –Ї–∞–Ї —Б¬†—Г—З–∞—Б—В–Є–µ–Љ –Ј–і–Њ—А–Њ–≤—Л—Е –і–Њ–±—А–Њ–≤–Њ–ї—М—Ж–µ–≤, —В–∞–Ї –Є¬†—Б —Г—З–∞—Б—В–Є–µ–Љ –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–њ–Њ–≤—А–µ–ґ–і–µ–љ–Є–µ–Љ —Б–њ–Є–љ–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞ –Є–ї–Є —А–∞—Б—Б–µ—П–љ–љ—Л–Љ —Б–Ї–ї–µ—А–Њ–Ј–Њ–Љ. –£—Б—В–∞–љ–Њ–≤–ї–µ–љ–Њ, —З—В–Њ –њ—А–Є –њ–µ—А–Њ—А–∞–ї—М–љ–Њ–Љ –њ—А–Є–µ–Љ–µ —В–Є–Ј–∞–љ–Є–і–Є–љ –±—Л—Б—В—А–Њ –∞–±—Б–Њ—А–±–Є—А—Г–µ—В—Б—П (53вАУ66% –њ—А–Є–љ—П—В–Њ–є –і–Њ–Ј—Л). –Ь–∞–Ї—Б–Є–Љ–∞–ї—М–љ–∞—П –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є—П –≤¬†–њ–ї–∞–Ј–Љ–µ –Ї—А–Њ–≤–Є (Cmax) –њ—А–Є –Њ–і–љ–Њ–Ї—А–∞—В–љ–Њ–Љ –њ–µ—А–Њ—А–∞–ї—М–љ–Њ–Љ –њ—А–Є–µ–Љ–µ –≤¬†–і–Њ–Ј–∞—Е 5 –Є¬†8 –Љ–≥ –≤–∞—А—М–Є—А—Г–µ—В—Б—П –Њ—В¬†3,2 –і–Њ 25,8 –Љ–Ї–≥/–ї —Б–Њ–Њ—В–≤–µ—В—Б—В–≤–µ–љ–љ–Њ [13]. –Т—А–µ–Љ—П –і–Њ—Б—В–Є–ґ–µ–љ–Є—П –Љ–∞–Ї—Б–Є–Љ–∞–ї—М–љ–Њ–є –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є–Є (Tmax) –≤¬†–њ–ї–∞–Ј–Љ–µ –Ї—А–Њ–≤–Є —Б–Њ—Б—В–∞–≤–ї—П–µ—В 0,75вАУ2,00 —З–∞—Б–∞ (—В–∞–±–ї.¬†1) [13].
–Т¬†–Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–Є —Б¬†—Г—З–∞—Б—В–Є–µ–Љ –Ј–і–Њ—А–Њ–≤—Л—Е –і–Њ–±—А–Њ–≤–Њ–ї—М—Ж–µ–≤ –њ—А–Њ–і–µ–Љ–Њ–љ—Б—В—А–Є—А–Њ–≤–∞–љ–Њ, —З—В–Њ –њ—А–Є–µ–Љ –њ–Є—Й–Є –љ–µ¬†–Њ–Ї–∞–Ј—Л–≤–∞–ї –Ј–љ–∞—З–Є–Љ–Њ–≥–Њ –≤–ї–Є—П–љ–Є—П –љ–∞¬†—Д–∞—А–Љ–∞–Ї–Њ–Ї–Є–љ–µ—В–Є–Ї—Г —В–Є–Ј–∞–љ–Є–і–Є–љ–∞ [13].
–Р–±—Б–Њ–ї—О—В–љ–∞—П –±–Є–Њ–і–Њ—Б—В—Г–њ–љ–Њ—Б—В—М —В–Є–Ј–∞–љ–Є–і–Є–љ–∞ –њ—А–Є –њ–µ—А–Њ—А–∞–ї—М–љ–Њ–Љ –њ—А–Є–µ–Љ–µ —Б–Њ—Б—В–∞–≤–ї—П–µ—В¬† –Њ—В¬†20 –і–Њ 34%, —З—В–Њ –Њ–±—Г—Б–ї–Њ–≤–ї–µ–љ–Њ –≤—Л—А–∞–ґ–µ–љ–љ—Л–Љ —Н—Д—Д–µ–Ї—В–Њ–Љ –њ–µ—А–≤–Њ–≥–Њ –њ—А–Њ—Е–Њ–ґ–і–µ–љ–Є—П —З–µ—А–µ–Ј –њ–µ—З–µ–љ—М¬†[12].
–Ь–µ—В–∞–±–Њ–ї–Є–Ј–Є—А—Г–µ—В—Б—П —В–Є–Ј–∞–љ–Є–і–Є–љ –≤¬†–њ–µ—З–µ–љ–Є –њ—Г—В–µ–Љ –Њ–Ї–Є—Б–ї–µ–љ–Є—П –њ—А–Є —Г—З–∞—Б—В–Є–Є –Є–Ј–Њ—Д–µ—А–Љ–µ–љ—В–Њ–≤ —Б–Є—Б—В–µ–Љ—Л —Ж–Є—В–Њ—Е—А–Њ–Љ–∞ –†450 (CYP1A2) —Б¬†–Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є–µ–Љ –љ–µ–∞–Ї—В–Є–≤–љ—Л—Е –Љ–µ—В–∞–±–Њ–ї–Є—В–Њ–≤, 19вАУ23% –Є–Ј¬†–Ї–Њ—В–Њ—А—Л—Е –≤—Л–≤–Њ–і—П—В—Б—П —Б¬†—Д–µ–Ї–∞–ї–Є—П–Љ–Є, 53вАУ66%¬†вАУ —Б¬†–Љ–Њ—З–Њ–є. –Т¬†–љ–µ–Є–Ј–Љ–µ–љ–µ–љ–љ–Њ–Љ –≤–Є–і–µ —Б¬†–Љ–Њ—З–Њ–є –≤—Л–≤–Њ–і–Є—В—Б—П –Љ–µ–љ–µ–µ 3% –і–Њ–Ј—Л —В–Є–Ј–∞–љ–Є–і–Є–љ–∞ [14].
–Т¬†–±–Њ–ї—М—И–Є–љ—Б—В–≤–µ –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є –њ–Њ–Ї–∞–Ј–∞–љ–Њ, —З—В–Њ –њ–µ—А–Є–Њ–і –њ–Њ–ї—Г–≤—Л–≤–µ–і–µ–љ–Є—П (T1/2) —В–Є–Ј–∞–љ–Є–і–Є–љ–∞ —Б–Њ—Б—В–∞–≤–ї—П–µ—В 2,1вАУ4,2 —З–∞—Б–∞ [13]. –Я—А–Є –љ–∞—А—Г—И–µ–љ–Є–Є —Д—Г–љ–Ї—Ж–Є–Є –њ–Њ—З–µ–Ї (–Ї–ї–Є—А–µ–љ—Б –Ї—А–µ–∞—В–Є–љ–Є–љ–∞ –Љ–µ–љ–µ–µ 1,5 –ї/—З (25 –Љ–ї/–Љ–Є–љ)) –њ–µ—А–Є–Њ–і –њ–Њ–ї—Г–≤—Л–≤–µ–і–µ–љ–Є—П –њ—А–Є –Њ–і–љ–Њ–Ї—А–∞—В–љ–Њ–Љ –њ—А–Є–µ–Љ–µ –≤¬†–і–Њ–Ј–µ 4 –Љ–≥ —Г–≤–µ–ї–Є—З–Є–≤–∞–µ—В—Б—П –і–Њ 13,6 —З–∞—Б–∞ [13].
–С–∞–Ї–ї–Њ—Д–µ–љ
–С–∞–Ї–ї–Њ—Д–µ–љ —В–∞–Ї–ґ–µ –Њ—В–љ–Њ—Б–Є—В—Б—П –Ї¬†–Љ–Є–Њ—А–µ–ї–∞–Ї—Б–∞–љ—В–∞–Љ —Ж–µ–љ—В—А–∞–ї—М–љ–Њ–≥–Њ –і–µ–є—Б—В–≤–Є—П. –Я—А–µ–њ–∞—А–∞—В —П–≤–ї—П–µ—В—Б—П –ї–Є–њ–Њ—Д–Є–ї—М–љ—Л–Љ –њ—А–Њ–Є–Ј–≤–Њ–і–љ—Л–Љ –≥–∞–Љ–Љ–∞-–∞–Љ–Є–љ–Њ–Љ–∞—Б–ї—П–љ–Њ–є –Ї–Є—Б–ї–Њ—В—Л (–У–Р–Ь–Ъ) –Є¬†–Є–Љ–µ–µ—В –≤—Л—Б–Њ–Ї—Г—О –∞—Д—Д–Є–љ–љ–Њ—Б—В—М —Б¬†–µ–µ –Т-—А–µ—Ж–µ–њ—В–Њ—А–∞–Љ–Є. –Ю–љ –∞–Ї—В–Є–≤–Є—А—Г–µ—В —А–µ—Ж–µ–њ—В–Њ—А—Л –Т¬†–Ї –У–Р–Ь–Ъ –љ–∞¬†–Љ–Њ–љ–Њ- –Є¬†–њ–Њ–ї–Є—Б–Є–љ–∞–њ—В–Є—З–µ—Б–Ї–Є—Е –љ–µ–є—А–Њ–љ–∞—Е —Б–њ–Є–љ–љ–Њ–≥–Њ –Є¬†–≥–Њ–ї–Њ–≤–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞. –Т–Њ–Ј–і–µ–є—Б—В–≤—Г—П –љ–∞¬†–Т-—А–µ—Ж–µ–њ—В–Њ—А—Л, –±–∞–Ї–ї–Њ—Д–µ–љ –≤—Л–Ј—Л–≤–∞–µ—В —Г–Љ–µ–љ—М—И–µ–љ–Є–µ —Б–њ–∞—Б—В–Є—З–љ–Њ—Б—В–Є.
–Ъ–∞–Ї –Є–Ј–≤–µ—Б—В–љ–Њ, –У–Р–Ь–Ъ –≤–ї–Є—П–µ—В –љ–∞¬†–і–≤–∞ —В–Є–њ–∞ —А–µ—Ж–µ–њ—В–Њ—А–Њ–≤¬†вАУ –Р¬†–Є –Т¬†[15]. –†–µ—Ж–µ–њ—В–Њ—А—Л –Р¬†—П–≤–ї—П—О—В—Б—П –Є–Њ–љ–Њ—В—А–Њ–њ–љ—Л–Љ–Є. –Я—А–Є —Б–≤—П–Ј—Л–≤–∞–љ–Є–Є —Б¬†–љ–Є–Љ–Є –У–Р–Ь–Ъ –≤¬†–Љ–µ–Љ–±—А–∞–љ–µ –љ–µ—А–≤–љ–Њ–є –Ї–ї–µ—В–Ї–Є –Њ—В–Ї—А—Л–≤–∞–µ—В—Б—П –Є–Њ–љ–љ—Л–є –Ї–∞–љ–∞–ї, –Є–Њ–љ—Л —Е–ї–Њ—А–∞ –њ—А–Є–љ–Є–Ї–∞—О—В –≤¬†–Ї–ї–µ—В–Ї—Г, —Б–љ–Є–ґ–∞—П –µ–µ —А–µ–∞–Ї—В–Є–≤–љ–Њ—Б—В—М. –Ф–∞–љ–љ—Л–є —В–Є–њ —А–µ—Ж–µ–њ—В–Њ—А–Њ–≤ —П–≤–ї—П–µ—В—Б—П –Љ–Є—И–µ–љ—М—О –±–µ–љ–Ј–Њ–і–Є–∞–Ј–µ–њ–Є–љ–Њ–≤, –±–∞—А–±–Є—В—Г—А–∞—В–Њ–≤, –ї–µ—В—Г—З–Є—Е –∞–љ–µ—Б—В–µ—В–Є–Ї–Њ–≤ –Є¬†–∞–ї–Ї–Њ–≥–Њ–ї—П [15]. –†–µ—Ж–µ–њ—В–Њ—А—Л –Т¬†–Њ—В–љ–Њ—Б—П—В—Б—П –Ї¬†–Љ–µ—В–∞–±–Њ—В—А–Њ–њ–љ—Л–Љ. –Ю–њ–Њ—Б—А–µ–і–Њ–≤–∞–љ–љ–Њ, —З–µ—А–µ–Ј —Б–Є—Б—В–µ–Љ—Г G-–±–µ–ї–Ї–Њ–≤, –Њ–љ–Є —Б–љ–Є–ґ–∞—О—В —Г—А–Њ–≤–µ–љ—М –≤–Њ–Ј–±—Г–ґ–і–µ–љ–Є—П –≤¬†–Ї–ї–µ—В–Ї–µ. –£–Љ–µ–љ—М—И–µ–љ–Є–µ —З—Г–≤—Б—В–≤–Є—В–µ–ї—М–љ–Њ—Б—В–Є –Ї–ї–µ—В–Ї–Є –Ї¬†–≤–Њ–Ј–±—Г–ґ–і–∞—О—Й–µ–Љ—Г –≤–Њ–Ј–і–µ–є—Б—В–≤–Є—О –Њ–±–µ—Б–њ–µ—З–Є–≤–∞–µ—В—Б—П –±–ї–∞–≥–Њ–і–∞—А—П –≤–ї–Є—П–љ–Є—О –љ–∞¬†–Ї–∞–ї—М—Ж–Є–µ–≤—Л–µ –Є¬†–Ї–∞–ї–Є–µ–≤—Л–µ –Ї–∞–љ–∞–ї—Л. –Т-—А–µ—Ж–µ–њ—В–Њ—А—Л –Љ–Њ–≥—Г—В —А–∞—Б–њ–Њ–ї–∞–≥–∞—В—М—Б—П –Ї–∞–Ї –њ—А–µ—Б–Є–љ–∞–њ—В–Є—З–µ—Б–Ї–Є, —В–∞–Ї –Є¬†–њ–Њ—Б—В—Б–Є–љ–∞–њ—В–Є—З–µ—Б–Ї–Є [16].
–°—З–Є—В–∞–µ—В—Б—П, —З—В–Њ —В–Њ—А–Љ–Њ–Ј—П—Й–µ–µ –≤–ї–Є—П–љ–Є–µ –љ–∞¬†–≥–Њ–ї–Њ–≤–љ–Њ–є –Љ–Њ–Ј–≥ –њ—А–Є –∞–Ї—В–Є–≤–∞—Ж–Є–Є –Т-—А–µ—Ж–µ–њ—В–Њ—А–Њ–≤ —А–µ–∞–ї–Є–Ј—Г–µ—В—Б—П –Ј–∞ —Б—З–µ—В —Г–Љ–µ–љ—М—И–µ–љ–Є—П –≤—Л—Б–≤–Њ–±–Њ–ґ–і–µ–љ–Є—П –≤–Њ–Ј–±—Г–ґ–і–∞—О—Й–Є—Е –Ї–ї–µ—В–Ї—Г –∞–Љ–Є–љ–Њ–Ї–Є—Б–ї–Њ—В (–љ–∞–њ—А–Є–Љ–µ—А,¬†–≥–ї—Г—В–∞–Љ–∞—В–∞), –∞¬†—В–∞–Ї–ґ–µ –Ј–∞ —Б—З–µ—В¬†–≥–Є–њ–µ—А–њ–Њ–ї—П—А–Є–Ј–∞—Ж–Є–Є –њ–Њ—Б—В—Б–Є–љ–∞–њ—В–Є—З–µ—Б–Ї–Є—Е –љ–µ–є—А–Њ–љ–Њ–≤ [16].
–°–ї–µ–і—Г–µ—В –Њ—В–Љ–µ—В–Є—В—М, —З—В–Њ –і–ї—П –±–∞–Ї–ї–Њ—Д–µ–љ–∞ —Е–∞—А–∞–Ї—В–µ—А–µ–љ —Б–Є–љ–і—А–Њ–Љ –Њ—В–Љ–µ–љ—Л. –≠—В–Њ –Љ–Њ–ґ–µ—В –њ—А–Њ—П–≤–ї—П—В—М—Б—П –≤¬†–≤–Є–і–µ¬†–≥–∞–ї–ї—О—Ж–Є–љ–∞—Ж–Є–Є, –і–µ–Ј–Њ—А–Є–µ–љ—В–∞—Ж–Є–Є, —Б–њ—Г—В–∞–љ–љ–Њ—Б—В–Є —Б–Њ–Ј–љ–∞–љ–Є—П, —В–∞—Е–Є–Ї–∞—А–і–Є–Є, —В—А–µ–Љ–Њ—А–∞, —Б—Г–і–Њ—А–Њ–≥ –Є¬†—А–Є–≥–Є–і–љ–Њ—Б—В–Є –Љ—Л—И—Ж, –∞¬†—В–∞–Ї–ґ–µ –≤¬†–≤–Є–і–µ¬†–≥–Є–њ–µ—А—В–µ–љ–Ј–Є–Є –Є¬†–≥–Є–њ–µ—А—В–µ—А–Љ–Є–Є. –†–∞–Ј–≤–Є—В–Є–µ –і–∞–љ–љ—Л—Е —Б–Њ—Б—В–Њ—П–љ–Є–є –Њ–±—Г—Б–ї–Њ–≤–ї–µ–љ–Њ –±—Л—Б—В—А—Л–Љ –њ—А–µ–Ї—А–∞—Й–µ–љ–Є–µ–Љ —В–Њ—А–Љ–Њ–Ј—П—Й–µ–≥–Њ –≤–Њ–Ј–і–µ–є—Б—В–≤–Є—П –њ—А–µ–њ–∞—А–∞—В–∞ –љ–∞¬†—А–µ—Ж–µ–њ—В–Њ—А—Л –У–Р–Ь–Ъ. –§–∞–Ї—В–Њ—А–∞–Љ–Є —А–Є—Б–Ї–∞ –≤–Њ–Ј–љ–Є–Ї–љ–Њ–≤–µ–љ–Є—П —Б–Є–љ–і—А–Њ–Љ–∞ –Њ—В–Љ–µ–љ—Л —Б—З–Є—В–∞—О—В—Б—П –њ—А–Є–µ–Љ –≤—Л—Б–Њ–Ї–Є—Е –і–Њ–Ј, –і–ї–Є—В–µ–ї—М–љ–∞—П —В–µ—А–∞–њ–Є—П –Є¬†–±—Л—Б—В—А–∞—П –µ–µ –Њ—В–Љ–µ–љ–∞ [17].
–Я—А–Є –њ–µ—А–Њ—А–∞–ї—М–љ–Њ–Љ –њ—А–Є–Љ–µ–љ–µ–љ–Є–Є –±–∞–Ї–ї–Њ—Д–µ–љ –±—Л—Б—В—А–Њ –≤—Б–∞—Б—Л–≤–∞–µ—В—Б—П –Є–Ј¬†–ґ–µ–ї—Г–і–Њ—З–љ–Њ-–Ї–Є—И–µ—З–љ–Њ–≥–Њ —В—А–∞–Ї—В–∞, –±–Є–Њ–і–Њ—Б—В—Г–њ–љ–Њ—Б—В—М —Б–Њ—Б—В–∞–≤–ї—П–µ—В 70вАУ85% [18]. –Ю–і–љ–∞–Ї–Њ –њ—А–Є —Г–≤–µ–ї–Є—З–µ–љ–Є–Є –і–Њ–Ј—Л –∞–±—Б–Њ—А–±—Ж–Є—П –Љ–Њ–ґ–µ—В —Б–љ–Є–ґ–∞—В—М—Б—П. –Я—А–Є –Њ–і–љ–Њ–Ї—А–∞—В–љ–Њ–Љ –њ–µ—А–Њ—А–∞–ї—М–љ–Њ–Љ –њ—А–Є–µ–Љ–µ –≤¬†–і–Њ–Ј–µ 50 –Љ–≥ Cmax —Г¬†–Ј–і–Њ—А–Њ–≤—Л—Е –і–Њ–±—А–Њ–≤–Њ–ї—М—Ж–µ–≤ —Б–Њ—Б—В–∞–≤–ї—П–ї–∞ 737,6 –љ–≥/–Љ–ї, Tmax¬†вАУ 1,9 ¬± 0,3 —З–∞—Б–∞ [19].
–Э–µ–Њ–±—Е–Њ–і–Є–Љ–Њ –Њ—В–Љ–µ—В–Є—В—М, —З—В–Њ —А–µ–Ј—Г–ї—М—В–∞—В—Л –њ—А–Њ—Б–њ–µ–Ї—В–Є–≤–љ–Њ–≥–Њ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П, –≤¬†–Ї–Њ—В–Њ—А–Њ–Љ –њ—А–Є–љ–Є–Љ–∞–ї–Є —Г—З–∞—Б—В–Є–µ 15¬†–і–Њ–±—А–Њ–≤–Њ–ї—М—Ж–µ–≤ –Љ—Г–ґ—Б–Ї–Њ–≥–Њ –њ–Њ–ї–∞ –≤¬†–≤–Њ–Ј—А–∞—Б—В–µ –Њ—В¬†19 –і–Њ 30 –ї–µ—В [20], –њ–Њ–Ј–≤–Њ–ї–Є–ї–Є –њ—А–µ–і–њ–Њ–ї–Њ–ґ–Є—В—М –Њ—В—Б—Г—В—Б—В–≤–Є–µ –Ј–љ–∞—З–Є–Љ—Л—Е —Д–∞—А–Љ–∞–Ї–Њ–Ї–Є–љ–µ—В–Є—З–µ—Б–Ї–Є—Е –≤–Ј–∞–Є–Љ–Њ–і–µ–є—Б—В–≤–Є–є –Љ–µ–ґ–і—Г –±–∞–Ї–ї–Њ—Д–µ–љ–Њ–Љ –Є¬†—В–Є–Ј–∞–љ–Є–і–Є–љ–Њ–Љ. –Ш—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ —Б–Њ—Б—В–Њ—П–ї–Њ –Є–Ј¬†—В—А–µ—Е –њ–µ—А–Є–Њ–і–Њ–≤: –њ–µ—А–≤—Л–є¬†вАУ –њ—А–Є–µ–Љ —В–Є–Ј–∞–љ–Є–і–Є–љ–∞ –њ–Њ¬†4 –Љ–≥ —В—А–Є —А–∞–Ј–∞ –≤¬†—Б—Г—В–Ї–Є –≤¬†—В–µ—З–µ–љ–Є–µ —Б–µ–Љ–Є –і–љ–µ–є, –≤—В–Њ—А–Њ–є¬†вАУ –њ—А–Є–µ–Љ –±–∞–Ї–ї–Њ—Д–µ–љ–∞ –њ–Њ¬†10¬†–Љ–≥ —В—А–Є —А–∞–Ј–∞ –≤¬†—Б—Г—В–Ї–Є –≤¬†—В–µ—З–µ–љ–Є–µ —Б–µ–Љ–Є –і–љ–µ–є, —В—А–µ—В–Є–є¬†вАУ —Б–Њ–≤–Љ–µ—Б—В–љ—Л–є –њ—А–Є–µ–Љ —В–Є–Ј–∞–љ–Є–і–Є–љ–∞ –Є¬†–±–∞–Ї–ї–Њ—Д–µ–љ–∞ –≤¬†–≤—Л—И–µ–њ–µ—А–µ—З–Є—Б–ї–µ–љ–љ—Л—Е –і–Њ–Ј–∞—Е —В–∞–Ї–ґ–µ –≤¬†—В–µ—З–µ–љ–Є–µ —Б–µ–Љ–Є –і–љ–µ–є. –Ь–µ–ґ–і—Г –і–∞–љ–љ—Л–Љ–Є –њ–µ—А–Є–Њ–і–∞–Љ–Є –њ—А–Њ–≤–Њ–і–Є–ї–Є—Б—М –њ–µ—А–Є–Њ–і—Л –Њ—В–Љ—Л–≤–Ї–Є –і–ї–Є—В–µ–ї—М–љ–Њ—Б—В—М—О —З–µ—В—Л—А–µ –і–љ—П. Cmax –±–∞–Ї–ї–Њ—Д–µ–љ–∞ –њ—А–Є –µ–≥–Њ –Њ—В–і–µ–ї—М–љ–Њ–Љ –њ—А–Є–µ–Љ–µ –і–Њ—Б—В–Є–≥–∞–ї–∞ 211 –љ–≥/–Љ–ї, –њ—А–Є —Б–Њ–≤–Љ–µ—Б—В–љ–Њ–Љ —Б¬†—В–Є–Ј–∞–љ–Є–і–Є–љ–Њ–Љ¬†вАУ 208¬†–љ–≥/–Љ–ї [20].
–С–∞–Ї–ї–Њ—Д–µ–љ –Њ–±–ї–∞–і–∞–µ—В –Њ—В–љ–Њ—Б–Є—В–µ–ї—М–љ–Њ –Ї–Њ—А–Њ—В–Ї–Є–Љ T1/2¬†вАУ –Њ—В¬†–і–≤—Г—Е –і–Њ —И–µ—Б—В–Є —З–∞—Б–Њ–≤.
–Ю–Ї–Њ–ї–Њ 15% –±–∞–Ї–ї–Њ—Д–µ–љ–∞ –Љ–µ—В–∞–±–Њ–ї–Є–Ј–Є—А—Г–µ—В—Б—П –≤¬†–њ–µ—З–µ–љ–Є –њ—Г—В–µ–Љ –і–µ–Ј–∞–Љ–Є–љ–Є—А–Њ–≤–∞–љ–Є—П. –Т¬†–Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П—Е —Б¬†–Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–љ–Є–µ–Љ –Љ–µ—З–µ–љ–љ–Њ–≥–Њ —А–∞–і–Є–Њ–∞–Ї—В–Є–≤–љ—Л–Љ –Є–Ј–Њ—В–Њ–њ–Њ–Љ –њ—А–µ–њ–∞—А–∞—В–∞ —Г—Б—В–∞–љ–Њ–≤–ї–µ–љ–Њ, —З—В–Њ –њ–Њ—А—П–і–Ї–∞ 85% –≤—Л–≤–Њ–і–Є—В—Б—П –≤¬†–љ–µ–Є–Ј–Љ–µ–љ–µ–љ–љ–Њ–Љ –≤–Є–і–µ –њ—А–µ–Є–Љ—Г—Й–µ—Б—В–≤–µ–љ–љ–Њ —Б¬†–Љ–Њ—З–Њ–є, –∞¬†—В–∞–Ї–ґ–µ —Б¬†—Д–µ–Ї–∞–ї–Є—П–Љ–Є¬†[18].
–Я–Њ–ї–љ–Њ–µ –≤—Л–≤–µ–і–µ–љ–Є–µ –њ—А–µ–њ–∞—А–∞—В–∞ –Є–Ј¬†–Њ—А–≥–∞–љ–Є–Ј–Љ–∞ –Њ—В–Љ–µ—З–∞–µ—В—Б—П —З–µ—А–µ–Ј 72 —З–∞—Б–∞ –њ–Њ—Б–ї–µ –њ—А–Є–µ–Љ–∞.
–Ґ–Њ–ї–њ–µ—А–Є–Ј–Њ–љ
–Ґ–Њ–ї–њ–µ—А–Є–Ј–Њ–љ¬†вАУ –Љ–Є–Њ—А–µ–ї–∞–Ї—Б–∞–љ—В —Ж–µ–љ—В—А–∞–ї—М–љ–Њ–≥–Њ –і–µ–є—Б—В–≤–Є—П, —Б—Е–Њ–ґ–Є–є –њ–Њ¬†—Е–Є–Љ–Є—З–µ—Б–Ї–Њ–є —Б—В—А—Г–Ї—В—Г—А–µ —Б¬†–Љ–µ—Б—В–љ—Л–Љ–Є –∞–љ–µ—Б—В–µ—В–Є–Ї–∞–Љ–Є (–ї–Є–і–Њ–Ї–∞–Є–љ–Њ–Љ). –Я—А–µ–њ–∞—А–∞—В –њ—А–µ–Є–Љ—Г—Й–µ—Б—В–≤–µ–љ–љ–Њ –њ—А–Є–Љ–µ–љ—П–µ—В—Б—П –≤¬†—Б—В—А–∞–љ–∞—Е –Х–≤—А–Њ–њ—Л. –Т¬†–°–®–Р –Њ–љ –љ–µ¬†–Ј–∞—А–µ–≥–Є—Б—В—А–Є—А–Њ–≤–∞–љ.
–Ь–µ—Е–∞–љ–Є–Ј–Љ –і–µ–є—Б—В–≤–Є—П —В–Њ–ї–њ–µ—А–Є–Ј–Њ–љ–∞ –і–Њ –Ї–Њ–љ—Ж–∞ –љ–µ¬†–Є–Ј—Г—З–µ–љ. –Ю–і–љ–∞–Ї–Њ —Г—Б—В–∞–љ–Њ–≤–ї–µ–љ–Њ, —З—В–Њ –Њ–љ –±–ї–Њ–Ї–Є—А—Г–µ—В –њ–Њ—В–µ–љ—Ж–Є–∞–ї-–Ј–∞–≤–Є—Б–Є–Љ—Л–µ –љ–∞—В—А–Є–µ–≤—Л–µ –Є¬†–Ї–∞–ї–Є–µ–≤—Л–µ –Ї–∞–љ–∞–ї—Л –љ–∞¬†—Г—А–Њ–≤–љ–µ —Б–њ–Є–љ–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞, –≤—Л–Ј—Л–≤–∞—П —В–Њ—А–Љ–Њ–ґ–µ–љ–Є–µ –њ—А–µ—Б–Є–љ–∞–њ—В–Є—З–µ—Б–Ї–Њ–≥–Њ –≤—Л—Б–≤–Њ–±–Њ–ґ–і–µ–љ–Є—П –љ–µ–є—А–Њ–Љ–µ–і–Є–∞—В–Њ—А–Њ–≤ [21]. –Ъ—А–Њ–Љ–µ —В–Њ–≥–Њ, –њ—А–µ–њ–∞—А–∞—В –њ–Њ–і–∞–≤–ї—П–µ—В –Љ–Њ–љ–Њ- –Є¬†–њ–Њ–ї–Є—Б–Є–љ–∞–њ—В–Є—З–µ—Б–Ї–Є–µ —А–µ—Д–ї–µ–Ї—Б—Л –љ–∞¬†—Г—А–Њ–≤–љ–µ —Б–њ–Є–љ–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞, –Њ–Ї–∞–Ј—Л–≤–∞–µ—В –ї–Є–і–Њ–Ї–∞–Є–љ–Њ–њ–Њ–і–Њ–±–љ–Њ–µ –Њ–±–µ–Ј–±–Њ–ї–Є–≤–∞–љ–Є–µ –Є¬†—Б—В–∞–±–Є–ї–Є–Ј–Є—А—Г–µ—В –Љ–µ–Љ–±—А–∞–љ—Л –љ–µ—А–≤–љ—Л—Е –Ї–ї–µ—В–Њ–Ї [21]. –Э–∞—А—П–і—Г —Б¬†—Н—В–Є–Љ —В–Њ–ї–њ–µ—А–Є–Ј–Њ–љ —Г—Б–Є–ї–Є–≤–∞–µ—В –њ–µ—А–Є—Д–µ—А–Є—З–µ—Б–Ї–Є–є –Ї—А–Њ–≤–Њ—В–Њ–Ї, –Њ–±–ї–µ–≥—З–∞–µ—В –њ—А–Њ–Є–Ј–≤–Њ–ї—М–љ—Л–µ –Љ—Л—И–µ—З–љ—Л–µ –і–≤–Є–ґ–µ–љ–Є—П [21].
–Ґ–Њ–ї–њ–µ—А–Є–Ј–Њ–љ –±—Л—Б—В—А–Њ –Є¬†–њ—А–∞–Ї—В–Є—З–µ—Б–Ї–Є –њ–Њ–ї–љ–Њ—Б—В—М—О –∞–±—Б–Њ—А–±–Є—А—Г–µ—В—Б—П –Є–Ј¬†–ґ–µ–ї—Г–і–Њ—З–љ–Њ-–Ї–Є—И–µ—З–љ–Њ–≥–Њ —В—А–∞–Ї—В–∞. –Ч–∞ —Б—З–µ—В —Н—Д—Д–µ–Ї—В–∞ –њ–µ—А–≤–Њ–≥–Њ –њ—А–Њ—Е–Њ–ґ–і–µ–љ–Є—П —З–µ—А–µ–Ј –њ–µ—З–µ–љ—М –µ–≥–Њ –∞–±—Б–Њ–ї—О—В–љ–∞—П –±–Є–Њ–і–Њ—Б—В—Г–њ–љ–Њ—Б—В—М —Б–Њ—Б—В–∞–≤–ї—П–µ—В 17вАУ20% [21, 22]. –Т—А–µ–Љ—П –і–Њ—Б—В–Є–ґ–µ–љ–Є—П –Љ–∞–Ї—Б–Є–Љ–∞–ї—М–љ–Њ–є –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є–Є¬†вАУ –Њ—В¬†0,5 –і–Њ 1,0 —З–∞—Б–∞ [21]. –Я–µ—А–Є–Њ–і –њ–Њ–ї—Г–≤—Л–≤–µ–і–µ–љ–Є—П –≤¬†—Б—А–µ–і–љ–µ–Љ —Б–Њ—Б—В–∞–≤–ї—П–µ—В 1,7 —З–∞—Б–∞ [22].
–Ю—В–ї–Є—З–Є—В–µ–ї—М–љ–Њ–є —З–µ—А—В–Њ–є —В–Њ–ї–њ–µ—А–Є–Ј–Њ–љ–∞ —П–≤–ї—П–µ—В—Б—П –Ј–љ–∞—З–Є—В–µ–ї—М–љ–∞—П –≤–∞—А–Є–∞–±–µ–ї—М–љ–Њ—Б—В—М —Д–∞—А–Љ–∞–Ї–Њ–Ї–Є–љ–µ—В–Є—З–µ—Б–Ї–Є—Е –њ–∞—А–∞–Љ–µ—В—А–Њ–≤ –≤¬†–Ј–∞–≤–Є—Б–Є–Љ–Њ—Б—В–Є –Њ—В¬†–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Є—Е –Њ—Б–Њ–±–µ–љ–љ–Њ—Б—В–µ–є. –Ґ–∞–Ї, –≤¬†–Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–Є M.¬†Pawlowska –Є¬†—Б–Њ–∞–≤—В. (2015¬†–≥.) [22] –њ—А–Є–љ—П–ї–Є —Г—З–∞—Б—В–Є–µ 28¬†–Ј–і–Њ—А–Њ–≤—Л—Е –Љ—Г–ґ—З–Є–љ –µ–≤—А–Њ–њ–µ–Њ–Є–і–љ–Њ–є —А–∞—Б—Л –≤¬†–≤–Њ–Ј—А–∞—Б—В–µ 27,3 ¬± 7,7¬†–≥–Њ–і–∞ [22]. –Ф–Њ–±—А–Њ–≤–Њ–ї—М—Ж—Л –Њ–і–љ–Њ–Ї—А–∞—В–љ–Њ –њ–µ—А–Њ—А–∞–ї—М–љ–Њ –њ–Њ–ї—Г—З–Є–ї–Є —В–Њ–ї–њ–µ—А–Є–Ј–Њ–љ –≤¬†–і–Њ–Ј–µ 150¬†–Љ–≥. –°¬†–њ–Њ–Љ–Њ—Й—М—О –њ–Њ–ї–Є–Љ–µ—А–∞–Ј–љ–Њ–є —Ж–µ–њ–љ–Њ–є —А–µ–∞–Ї—Ж–Є–Є –≤¬†—А–µ–ґ–Є–Љ–µ —А–µ–∞–ї—М–љ–Њ–≥–Њ –≤—А–µ–Љ–µ–љ–Є —Г¬†–љ–Є—Е –±—Л–ї–Њ –≤—Л—П–≤–ї–µ–љ–Њ –љ–Њ—Б–Є—В–µ–ї—М—Б—В–≤–Њ –∞–ї–ї–µ–ї–µ–є¬†–≥–µ–љ–Њ–≤ CYP2D6 –Є¬†CYP2C19 —Ж–Є—В–Њ—Е—А–Њ–Љ–∞ –†450, –Ї–Њ—В–Њ—А—Л–µ –Њ—В–≤–µ—В—Б—В–≤–µ–љ–љ—Л –Ј–∞ –Љ–µ—В–∞–±–Њ–ї–Є–Ј–Љ —В–Њ–ї–њ–µ—А–Є–Ј–Њ–љ–∞.
–°max –њ—А–µ–њ–∞—А–∞—В–∞ –≤–∞—А—М–Є—А–Њ–≤–∞–ї–∞—Б—М –Њ—В¬†4,47 –і–Њ 336,43 –љ–≥/–Љ–ї, –≤¬†—Б—А–µ–і–љ–µ–Љ –Њ–љ–∞ —Б–Њ—Б—В–∞–≤–ї—П–ї–∞ 90,88 ¬± 86,04 –љ–≥/–Љ–ї (—В–∞–±–ї.¬†2) [22].
–Т¬†–Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–Є —В–∞–Ї–ґ–µ –±—Л–ї–Њ —Г—Б—В–∞–љ–Њ–≤–ї–µ–љ–Њ, —З—В–Њ —З–∞—Б—В–Њ—В–∞¬†–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Є—Е –њ–Њ–ї–Є–Љ–Њ—А—Д–Є–Ј–Љ–Њ–≤¬†–≥–µ–љ–Њ–≤, –Њ–±–µ—Б–њ–µ—З–Є–≤–∞—О—Й–Є—Е –Љ–µ–і–ї–µ–љ–љ—Л–є/–Ј–∞–Љ–µ–і–ї–µ–љ–љ—Л–є –Љ–µ—В–∞–±–Њ–ї–Є–Ј–Љ —В–Њ–ї–њ–µ—А–Є–Ј–Њ–љ–∞, —Г¬†–Ј–і–Њ—А–Њ–≤—Л—Е –і–Њ–±—А–Њ–≤–Њ–ї—М—Ж–µ–≤ –і–ї—П –Є–Ј–Њ—Д–µ—А–Љ–µ–љ—В–Њ–≤ CYP2D6 –Є¬†CYP2C19 —Б–Њ—Б—В–∞–≤–ї—П–ї–∞ 46,43¬†–Є¬†0% —Б–Њ–Њ—В–≤–µ—В—Б—В–≤–µ–љ–љ–Њ [22]. –†–µ–Ј—Г–ї—М—В–∞—В—Л –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П Z. Desta –Є¬†—Б–Њ–∞–≤—В. —Б–≤–Є–і–µ—В–µ–ї—М—Б—В–≤—Г—О—В, —З—В–Њ –Ї–Њ–ї–Є—З–µ—Б—В–≤–Њ –ї–Є—Ж –µ–≤—А–Њ–њ–µ–Њ–Є–і–љ–Њ–є —А–∞—Б—Л —Б¬†–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Є –Њ–±—Г—Б–ї–Њ–≤–ї–µ–љ–љ—Л–Љ –Љ–µ–і–ї–µ–љ–љ—Л–Љ –Љ–µ—В–∞–±–Њ–ї–Є–Ј–Љ–Њ–Љ —В–Њ–ї–њ–µ—А–Є–Ј–Њ–љ–∞ –љ–µ¬†–њ—А–µ–≤—Л—И–∞–µ—В 6% [23]. –Ф–ї—П –њ–Њ–ї—Г—З–µ–љ–Є—П —В–Њ—З–љ–Њ–є –Є–љ—Д–Њ—А–Љ–∞—Ж–Є–Є —В—А–µ–±—Г–µ—В—Б—П –±–Њ–ї–µ–µ —И–Є—А–Њ–Ї–Њ–µ –Є–Ј—Г—З–µ–љ–Є–µ –њ—А–Њ–±–ї–µ–Љ—Л –≤¬†—А–∞–Ј–љ—Л—Е –њ–Њ–њ—Г–ї—П—Ж–Є—П—Е –Є¬†—Б—В—А–∞–љ–∞—Е.
–Р–љ–∞–ї–Њ–≥–Є—З–љ—Л–µ –і–∞–љ–љ—Л–µ –Њ¬†–≤–∞—А–Є–∞–±–µ–ї—М–љ–Њ—Б—В–Є —Д–∞—А–Љ–∞–Ї–Њ–Ї–Є–љ–µ—В–Є—З–µ—Б–Ї–Є—Е –њ–∞—А–∞–Љ–µ—В—А–Њ–≤ —В–Њ–ї–њ–µ—А–Є–Ј–Њ–љ–∞ –њ—А–µ–і—Б—В–∞–≤–Є–ї–Є J.W. Bae –Є¬†—Б–Њ–∞–≤—В. (2007¬†–≥.) [24]. –Ш—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ –њ—А–Њ–≤–Њ–і–Є–ї–Њ—Б—М —Б¬†—Г—З–∞—Б—В–Є–µ–Љ 15 –Ј–і–Њ—А–Њ–≤—Л—Е –і–Њ–±—А–Њ–≤–Њ–ї—М—Ж–µ–≤ –Љ–Њ–љ–≥–Њ–ї–Њ–Є–і–љ–Њ–є —А–∞—Б—Л (–Ї–Њ—А–µ–є—Ж—Л). –°—А–µ–і–љ–Є–є –≤–Њ–Ј—А–∞—Б—В –Љ—Г–ґ—З–Є–љ¬†вАУ 23,6 ¬± 1,3¬†–≥–Њ–і–∞. –Ґ–Њ–ї–њ–µ—А–Є–Ј–Њ–љ –љ–∞–Ј–љ–∞—З–∞–ї—Б—П –≤–љ—Г—В—А—М –≤¬†–і–Њ–Ј–µ 450 –Љ–≥ –Њ–і–љ–Њ–Ї—А–∞—В–љ–Њ. Cmax –≤–∞—А—М–Є—А–Њ–≤–∞–ї–∞—Б—М –Њ—В¬†64,2 –і–Њ 784,9 –љ–≥/–Љ–ї. –Я–ї–Њ—Й–∞–і—М –њ–Њ–і —Д–∞—А–Љ–∞–Ї–Њ–Ї–Є–љ–µ—В–Є—З–µ—Б–Ї–Њ–є –Ї—А–Є–≤–Њ–є (AUC), –љ–∞—З–Є–љ–∞—П —Б¬†–љ—Г–ї–µ–≤–Њ–≥–Њ –Ј–љ–∞—З–µ–љ–Є—П –і–Њ –±–µ—Б–Ї–Њ–љ–µ—З–љ–Њ—Б—В–Є (AUC0-вИЮ),¬†вАУ –Њ—В¬†125,9 –і–Њ¬†1241,3 –љ–≥ √Ч —З/–Љ–ї [24].
–Ґ–∞–Ї–Є–Љ –Њ–±—А–∞–Ј–Њ–Љ, –≤¬†–Ј–∞–≤–Є—Б–Є–Љ–Њ—Б—В–Є –Њ—В¬†–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Є—Е –Њ—Б–Њ–±–µ–љ–љ–Њ—Б—В–µ–є —З–µ–ї–Њ–≤–µ–Ї–∞ –Љ–∞–Ї—Б–Є–Љ–∞–ї—М–љ–∞—П –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є—П —В–Њ–ї–њ–µ—А–Є–Ј–Њ–љ–∞ –≤¬†–њ–ї–∞–Ј–Љ–µ –Ї—А–Њ–≤–Є –Љ–Њ–ґ–µ—В –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ –Ї–Њ–ї–µ–±–∞—В—М—Б—П. –Ъ–∞–Ї —Б–ї–µ–і—Б—В–≤–Є–µ, –ї–Є–±–Њ –љ–µ—Н—Д—Д–µ–Ї—В–Є–≤–љ–Њ—Б—В—М —В–µ—А–∞–њ–Є–Є (–њ—А–Є –љ–Є–Ј–Ї–Є—Е –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є—П—Е, –Њ–±—Г—Б–ї–Њ–≤–ї–µ–љ–љ—Л—Е –≤—Л—Б–Њ–Ї–Є–Љ —Г—А–Њ–≤–љ–µ–Љ –Љ–µ—В–∞–±–Њ–ї–Є–Ј–Љ–∞ —В–Њ–ї–њ–µ—А–Є–Ј–Њ–љ–∞), –ї–Є–±–Њ –≤—Л—Б–Њ–Ї–Є–є —А–Є—Б–Ї¬†—А–∞–Ј–≤–Є—В–Є—П –љ–µ–ґ–µ–ї–∞—В–µ–ї—М–љ—Л—Е –Є¬†—В–Њ–Ї—Б–Є—З–µ—Б–Ї–Є—Е —А–µ–∞–Ї—Ж–Є–є (–њ—А–Є –љ–Є–Ј–Ї–Њ–Љ —Г—А–Њ–≤–љ–µ –Љ–µ—В–∞–±–Њ–ї–Є–Ј–Љ–∞, –њ—А–Є–≤–Њ–і—П—Й–µ–Љ –Ї¬†–≤—Л—Б–Њ–Ї–Є–Љ –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є—П–Љ –њ—А–µ–њ–∞—А–∞—В–∞ –≤¬†–Ї—А–Њ–≤–Є).
–Т—Л–≤–Њ–і–Є—В—Б—П —В–Њ–ї–њ–µ—А–Є–Ј–Њ–љ –њ—А–µ–Є–Љ—Г—Й–µ—Б—В–≤–µ–љ–љ–Њ –њ–Њ—З–Ї–∞–Љ–Є (85%). –Я—А–Є —Н—В–Њ–Љ 98% –њ—А–µ–њ–∞—А–∞—В–∞ —Н–ї–Є–Љ–Є–љ–Є—А—Г–µ—В—Б—П –Є–Ј¬†–Њ—А–≥–∞–љ–Є–Ј–Љ–∞ –≤¬†—В–µ—З–µ–љ–Є–µ 24 —З–∞—Б–Њ–≤ –њ–Њ—Б–ї–µ –њ—А–Є–µ–Љ–∞ [21].
–Э–µ–ґ–µ–ї–∞—В–µ–ї—М–љ—Л–µ¬† —А–µ–∞–Ї—Ж–Є–Є
–Ґ–Є–Ј–∞–љ–Є–і–Є–љ
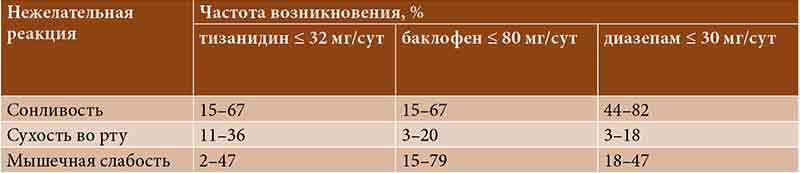
–Ґ–Є–Ј–∞–љ–Є–і–Є–љ, –Ї–∞–Ї –њ—А–∞–≤–Є–ї–Њ, —Е–Њ—А–Њ—И–Њ –њ–µ—А–µ–љ–Њ—Б–Є—В—Б—П. –°–Њ–≥–ї–∞—Б–љ–Њ —А–µ–Ј—Г–ї—М—В–∞—В–∞–Љ –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є, —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б–Њ¬†—Б–њ–∞—Б—В–Є—З–љ–Њ—Б—В—М—О, –њ—А–Є–љ–Є–Љ–∞–≤—И–Є—Е —В–Є–Ј–∞–љ–Є–і–Є–љ вЙ§ 36 –Љ–≥/—Б—Г—В, –љ–∞–Є–±–Њ–ї–µ–µ —З–∞—Б—В—Л–Љ–Є –љ–µ–ґ–µ–ї–∞—В–µ–ї—М–љ—Л–Љ–Є —А–µ–∞–Ї—Ж–Є—П–Љ–Є –±—Л–ї–Є —Б—Г—Е–Њ—Б—В—М –≤–Њ —А—В—Г (23вАУ57%), —Б–Њ–љ–ї–Є–≤–Њ—Б—В—М (24вАУ48%), –Љ—Л—И–µ—З–љ–∞—П —Б–ї–∞–±–Њ—Б—В—М (18вАУ48%) –Є¬†–≥–Њ–ї–Њ–≤–Њ–Ї—А—Г–ґ–µ–љ–Є–µ (10вАУ19%). –Т¬†5вАУ7% —Б–ї—Г—З–∞–µ–≤ –Њ—В–Љ–µ—З–∞–ї–Њ—Б—М –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є –Ј–љ–∞—З–Є–Љ–Њ–µ –њ–Њ–≤—Л—И–µ–љ–Є–µ —Г—А–Њ–≤–љ—П –њ–µ—З–µ–љ–Њ—З–љ—Л—Е —Д–µ—А–Љ–µ–љ—В–Њ–≤, —А–∞–Ј—А–µ—И–∞–≤—И–µ–µ—Б—П –њ–Њ—Б–ї–µ –Њ—В–Љ–µ–љ—Л –њ—А–µ–њ–∞—А–∞—В–∞ [25вАУ27].
–Т¬†—Б—А–∞–≤–љ–Є—В–µ–ї—М–љ—Л—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П—Е —Б—А–µ–і–Є –љ–∞–Є–±–Њ–ї–µ–µ —З–∞—Б—В—Л—Е –њ–Њ–±–Њ—З–љ—Л—Е —Н—Д—Д–µ–Ї—В–Њ–≤ –љ–∞¬†—Д–Њ–љ–µ –њ—А–Є–µ–Љ–∞ —В–Є–Ј–∞–љ–Є–і–Є–љ–∞ вЙ§ 32 –Љ–≥/—Б—Г—В —В–∞–Ї–ґ–µ —Г–Ї–∞–Ј—Л–≤–∞–ї–Є—Б—М —Б–Њ–љ–ї–Є–≤–Њ—Б—В—М, —Б—Г—Е–Њ—Б—В—М –≤–Њ —А—В—Г –Є¬†–Љ—Л—И–µ—З–љ–∞—П —Б–ї–∞–±–Њ—Б—В—М [13]. –Я—А–Є —Н—В–Њ–Љ —З–∞—Б—В–Њ—В–∞ —А–∞–Ј–≤–Є—В–Є—П —Б–Њ–љ–ї–Є–≤–Њ—Б—В–Є —Г¬†–њ–Њ–ї—Г—З–∞–≤—И–Є—Е —В–Є–Ј–∞–љ–Є–і–Є–љ –Є¬†–њ—А–Є–љ–Є–Љ–∞–≤—И–Є—Е –±–∞–Ї–ї–Њ—Д–µ–љ –±—Л–ї–∞ —Б–Њ–њ–Њ—Б—В–∞–≤–Є–Љ–Њ–є¬†вАУ –Њ—В¬†15 –і–Њ¬†67% (—В–∞–±–ї.¬†3) [13].
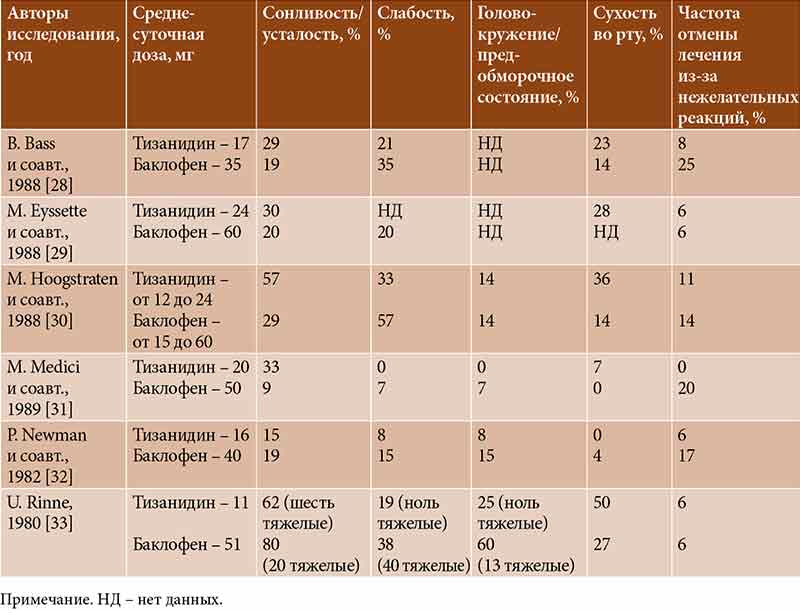
–°–ї–µ–і—Г–µ—В –Њ—В–Љ–µ—В–Є—В—М, —З—В–Њ –љ–∞–Є–±–Њ–ї–µ–µ –≤—Л—Б–Њ–Ї–Њ–є —З–∞—Б—В–Њ—В–∞ –Њ—В–Љ–µ–љ—Л –њ—А–µ–њ–∞—А–∞—В–∞ –≤¬†—Б–≤—П–Ј–Є —Б¬†–Љ—Л—И–µ—З–љ–Њ–є —Б–ї–∞–±–Њ—Б—В—М—О –±—Л–ї–∞ —Г¬†–±–∞–Ї–ї–Њ—Д–µ–љ–∞ (0вАУ25%), –і–∞–ї–µ–µ –њ–Њ¬†—Г–Љ–µ–љ—М—И–µ–љ–Є—О –њ–Њ–Ї–∞–Ј–∞—В–µ–ї—П —Б–ї–µ–і–Њ–≤–∞–ї–Є –і–Є–∞–Ј–µ–њ–∞–Љ (4вАУ6%) –Є¬†—В–Є–Ј–∞–љ–Є–і–Є–љ (0вАУ6%) (—В–∞–±–ї.¬†4) [28вАУ33].
–°–µ—А—М–µ–Ј–љ—Л–µ –љ–µ–ґ–µ–ї–∞—В–µ–ї—М–љ—Л–µ —А–µ–∞–Ї—Ж–Є–Є –љ–∞¬†—Д–Њ–љ–µ —В–µ—А–∞–њ–Є–Є —В–Є–Ј–∞–љ–Є–і–Є–љ–Њ–Љ –Ј–∞—Д–Є–Ї—Б–Є—А–Њ–≤–∞–љ—Л —Г¬†0,18% –њ–∞—Ж–Є–µ–љ—В–Њ–≤ [34]. –Ґ–∞–Ї–Њ–≤—Л–µ –±—Л–ї–Є –њ—А–µ–і—Б—В–∞–≤–ї–µ–љ—Л¬†–≥–∞–ї–ї—О—Ж–Є–љ–∞—Ж–Є—П–Љ–Є –Є¬†—В—П–ґ–µ–ї—Л–Љ–Є –љ–∞—А—Г—И–µ–љ–Є—П–Љ–Є —Д—Г–љ–Ї—Ж–Є–Є –њ–µ—З–µ–љ–Є. –Т¬†–±–Њ–ї—М—И–Є–љ—Б—В–≤–µ —Б–ї—Г—З–∞–µ–≤ –њ–Њ—А–∞–ґ–µ–љ–Є–µ –њ–µ—З–µ–љ–Є –љ–∞¬†—Д–Њ–љ–µ —В–µ—А–∞–њ–Є–Є —В–Є–Ј–∞–љ–Є–і–Є–љ–Њ–Љ –њ—А–Њ—П–≤–ї—П–µ—В—Б—П –≤¬†–≤–Є–і–µ –њ–Њ–≤—Л—И–µ–љ–Є—П —Г—А–Њ–≤–љ—П –њ–µ—З–µ–љ–Њ—З–љ—Л—Е —Д–µ—А–Љ–µ–љ—В–Њ–≤ (–∞–ї–∞–љ–Є–љ–∞–Љ–Є–љ–Њ—В—А–∞–љ—Б—Д–µ—А–∞–Ј—Л –Є¬†–∞—Б–њ–∞—А—В–∞—В–∞–Љ–Є–љ–Њ—В—А–∞–љ—Б—Д–µ—А–∞–Ј—Л)¬†вАУ 5% –±–Њ–ї—М–љ—Л—Е.
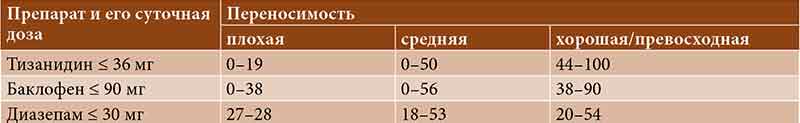
–Я–Њ–Љ–Є–Љ–Њ –њ—А–Њ—Д–Є–ї—П –±–µ–Ј–Њ–њ–∞—Б–љ–Њ—Б—В–Є –≤–∞–ґ–љ–Њ–є —Е–∞—А–∞–Ї—В–µ—А–Є—Б—В–Є–Ї–Њ–є –ї—О–±–Њ–≥–Њ –њ—А–µ–њ–∞—А–∞—В–∞ —Б—З–Є—В–∞–µ—В—Б—П –њ–µ—А–µ–љ–Њ—Б–Є–Љ–Њ—Б—В—М. –Ю–њ—А–Њ—Б –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б–Њ¬†—Б–њ–∞—Б—В–Є—З–љ–Њ—Б—В—М—О, –њ–Њ–ї—Г—З–∞–≤—И–Є—Е —В–Є–Ј–∞–љ–Є–і–Є–љ, –њ–Њ–Ї–∞–Ј–∞–ї, —З—В–Њ –Њ–±—Й–∞—П –њ–µ—А–µ–љ–Њ—Б–Є–Љ–Њ—Б—В—М –њ—А–µ–њ–∞—А–∞—В–∞ –±—Л–ї–∞ —Е–Њ—А–Њ—И–µ–є –Є–ї–Є –њ—А–µ–≤–Њ—Б—Е–Њ–і–љ–Њ–є –≤¬†44вАУ100% —Б–ї—Г—З–∞–µ–≤ (—В–∞–±–ї.¬†5) [13].
–Т¬†—Б—А–∞–≤–љ–Є—В–µ–ї—М–љ—Л—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П—Е –њ–µ—А–µ–љ–Њ—Б–Є–Љ–Њ—Б—В—М —В–Є–Ј–∞–љ–Є–і–Є–љ–∞ –±—Л–ї–∞ –ї—Г—З—И–µ, —З–µ–Љ –±–∞–Ї–ї–Њ—Д–µ–љ–∞ –Є¬†–і–Є–∞–Ј–µ–њ–∞–Љ–∞, —З—В–Њ –Љ–Њ–ґ–µ—В –≤–ї–Є—П—В—М –љ–∞¬†–њ—А–Є–≤–µ—А–ґ–µ–љ–љ–Њ—Б—В—М —В–µ—А–∞–њ–Є–Є, –Њ—Б–Њ–±–µ–љ–љ–Њ –≤¬†–∞–Љ–±—Г–ї–∞—В–Њ—А–љ—Л—Е —Г—Б–ї–Њ–≤–Є—П—Е [13].
–С–∞–Ї–ї–Њ—Д–µ–љ
–І–∞—Б—В–Њ—В–∞ –љ–µ–ґ–µ–ї–∞—В–µ–ї—М–љ—Л—Е —А–µ–∞–Ї—Ж–Є–є –њ—А–Є –њ–µ—А–Њ—А–∞–ї—М–љ–Њ–Љ –њ—А–Є–µ–Љ–µ –±–∞–Ї–ї–Њ—Д–µ–љ–∞ –Ї–Њ–ї–µ–±–ї–µ—В—Б—П –Њ—В¬†10 –і–Њ 75% [35]. –Ъ¬†–љ–∞–Є–±–Њ–ї–µ–µ —А–∞—Б–њ—А–Њ—Б—В—А–∞–љ–µ–љ–љ—Л–Љ –Њ—В–љ–Њ—Б—П—В—Б—П —Б–Њ–љ–ї–Є–≤–Њ—Б—В—М, —З—А–µ–Ј–Љ–µ—А–љ–∞—П –Њ–±—Й–∞—П —Б–ї–∞–±–Њ—Б—В—М, –≤—Л—А–∞–ґ–µ–љ–љ–∞—П –Љ—Л—И–µ—З–љ–∞—П —Б–ї–∞–±–Њ—Б—В—М –Є¬†–≥–Њ–ї–Њ–≤–Њ–Ї—А—Г–ґ–µ–љ–Є–µ.
–Я–∞—Ж–Є–µ–љ—В–∞–Љ —Б¬†–љ–∞—А—Г—И–µ–љ–љ–Њ–є —Д—Г–љ–Ї—Ж–Є–µ–є –њ–Њ—З–µ–Ї –њ—А–µ–њ–∞—А–∞—В –і–Њ–ї–ґ–µ–љ –љ–∞–Ј–љ–∞—З–∞—В—М—Б—П —Б¬†–Њ—Б—В–Њ—А–Њ–ґ–љ–Њ—Б—В—М—О –≤¬†—Б–≤—П–Ј–Є —Б¬†–±–Њ–ї–µ–µ –≤—Л—Б–Њ–Ї–Є–Љ —А–Є—Б–Ї–Њ–Љ —А–∞–Ј–≤–Є—В–Є—П –љ–µ–ґ–µ–ї–∞—В–µ–ї—М–љ—Л—Е —А–µ–∞–Ї—Ж–Є–є. –Ф–∞–љ–љ—Л–µ –Њ–±¬†—Н—В–Њ–Љ –±—Л–ї–Є –њ–Њ–ї—Г—З–µ–љ—Л F.T. Muanda –Є¬†—Б–Њ–∞–≤—В. –њ—А–Є –њ—А–Њ–≤–µ–і–µ–љ–Є–Є –Ї—А—Г–њ–љ–Њ–≥–Њ —А–µ—В—А–Њ—Б–њ–µ–Ї—В–Є–≤–љ–Њ–≥–Њ –Ї–Њ–≥–Њ—А—В–љ–Њ–≥–Њ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П —Б¬†—Г—З–∞—Б—В–Є–µ–Љ 15 942 –њ–Њ–ґ–Є–ї—Л—Е –њ–∞—Ж–Є–µ–љ—В–Њ–≤ (66¬†–ї–µ—В –Є¬†—Б—В–∞—А—И–µ), –Є–Љ–µ–≤—И–Є—Е —Е—А–Њ–љ–Є—З–µ—Б–Ї—Г—О –±–Њ–ї–µ–Ј–љ—М –њ–Њ—З–µ–Ї (—А–∞—Б—З–µ—В–љ–∞—П —Б–Ї–Њ—А–Њ—Б—В—М –Ї–ї—Г–±–Њ—З–Ї–Њ–≤–Њ–є —Д–Є–ї—М—В—А–∞—Ж–Є–Є (—А–°–Ъ–§) < 60 –Љ–ї/–Љ–Є–љ/1,73¬†–Љ2), –љ–Њ¬†–љ–µ¬†–љ–∞—Е–Њ–і–Є–≤—И–Є—Е—Б—П –љ–∞¬†–≥–µ–Љ–Њ–і–Є–∞–ї–Є–Ј–µ [36]. –£—З–µ–љ—Л–µ –Њ—Ж–µ–љ–Є–≤–∞–ї–Є 30-–і–љ–µ–≤–љ—Л–є —А–Є—Б–Ї¬†—А–∞–Ј–≤–Є—В–Є—П —Н–љ—Ж–µ—Д–∞–ї–Њ–њ–∞—В–Є–Є –љ–∞¬†—Д–Њ–љ–µ –њ—А–Є–µ–Љ–∞ –±–∞–Ї–ї–Њ—Д–µ–љ–∞ –≤¬†–і–Њ–Ј–∞—Е вЙ•¬†20 –Є¬†< 20 –Љ–≥/—Б—Г—В. –°—А–µ–і–Є –≤–Ї–ї—О—З–µ–љ–љ—Л—Е –≤¬†–Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ 61% –±—Л–ї–Є –ґ–µ–љ—Й–Є–љ—Л. –°—А–µ–і–љ–Є–є –≤–Њ–Ј—А–∞—Б—В —Г—З–∞—Б—В–љ–Є–Ї–Њ–≤¬†вАУ 77 –ї–µ—В. –°—В–∞—А—В–Њ–≤–∞—П –і–Њ–Ј–∞ –±–∞–Ї–ї–Њ—Д–µ–љ–∞ 20 –Љ–≥/—Б—Г—В –Є¬†–±–Њ–ї–µ–µ –љ–∞–Ј–љ–∞—З–µ–љ–∞ 61% –њ–∞—Ж–Є–µ–љ—В–Њ–≤, –Љ–µ–љ–µ–µ 20 –Љ–≥/—Б—Г—В¬†вАУ 39%. –І–∞—Б—В–Њ—В–∞¬†–≥–Њ—Б–њ–Є—В–∞–ї–Є–Ј–∞—Ж–Є–є –њ–Њ¬†–њ—А–Є—З–Є–љ–µ —Н–љ—Ж–µ—Д–∞–ї–Њ–њ–∞—В–Є–Є —Б—А–µ–і–Є –њ—А–Є–љ–Є–Љ–∞–≤—И–Є—Е –±–∞–Ї–ї–Њ—Д–µ–љ –≤¬†–і–Њ–Ј–µ 20 –Љ–≥/—Б—Г—В –Є¬†–±–Њ–ї–µ–µ —Б–Њ—Б—В–∞–≤–Є–ї–∞ 1,11%, –Љ–µ–љ–µ–µ 20 –Љ–≥/–і–µ–љ—М¬†вАУ 0,42% [36]. –І–∞—Б—В–Њ—В–∞¬†–≥–Њ—Б–њ–Є—В–∞–ї–Є–Ј–∞—Ж–Є–є —Г–≤–µ–ї–Є—З–Є–≤–∞–ї–∞—Б—М –њ–Њ¬†–Љ–µ—А–µ —Г—Е—Г–і—И–µ–љ–Є—П —Д—Г–љ–Ї—Ж–Є–Є –њ–Њ—З–µ–Ї. –Я—А–Є —А–°–Ъ–§ 45вАУ59¬†–Љ–ї/–Љ–Є–љ/1,73 –Љ2 –Њ–љ–∞ —Б–Њ—Б—В–∞–≤–ї—П–ї–∞ 0,42%, —А–°–Ъ–§ 30вАУ44 –Љ–ї/–Љ–Є–љ/1,73 –Љ2¬†вАУ 1,23%, —А–°–Ъ–§ < 30 –Љ–ї/–Љ–Є–љ/1,73 –Љ2¬†вАУ 2,90%.
–°—А–∞–≤–љ–Є–≤–∞–ї–Є —В–∞–Ї–ґ–µ –Њ—В–љ–Њ—Б–Є—В–µ–ї—М–љ—Л–є —А–Є—Б–Ї¬†–≥–Њ—Б–њ–Є—В–∞–ї–Є–Ј–∞—Ж–Є–є –њ–Њ¬†–њ—А–Є—З–Є–љ–µ —Н–љ—Ж–µ—Д–∞–ї–Њ–њ–∞—В–Є–Є —Г¬†–њ—А–Є–љ–Є–Љ–∞–≤—И–Є—Е –Є¬†–љ–µ –њ—А–Є–љ–Є–Љ–∞–≤—И–Є—Е –±–∞–Ї–ї–Њ—Д–µ–љ (n = 284 263). –Т–Ј–≤–µ—И–µ–љ–љ—Л–є –Њ—В–љ–Њ—Б–Є—В–µ–ї—М–љ—Л–є —А–Є—Б–Ї¬†–≥–Њ—Б–њ–Є—В–∞–ї–Є–Ј–∞—Ж–Є–є –љ–∞¬†—Д–Њ–љ–µ –њ—А–Є–µ–Љ–∞ –њ—А–µ–њ–∞—А–∞—В–∞ –≤¬†–і–Њ–Ј–µ –Љ–µ–љ–µ–µ 20 –Љ–≥/—Б—Г—В —Б–Њ—Б—В–∞–≤–Є–ї 5,9 (–њ—А–Є 95%-–љ–Њ–Љ –і–Њ–≤–µ—А–Є—В–µ–ї—М–љ–Њ–Љ –Є–љ—В–µ—А–≤–∞–ї–µ (–Ф–Ш) 3,59вАУ9,70), –≤¬†–і–Њ–Ј–µ 20 –Љ–≥/—Б—Г—В –Є¬†–±–Њ–ї–µ–µ¬†вАУ 19,8 (95% –Ф–Ш 14,0вАУ28,0).
–Ґ–∞–Ї–Є–Љ –Њ–±—А–∞–Ј–Њ–Љ, –±—Л–ї–Њ —Г—Б—В–∞–љ–Њ–≤–ї–µ–љ–Њ, —З—В–Њ —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–љ–∞—А—Г—И–µ–љ–Є–µ–Љ —Д—Г–љ–Ї—Ж–Є–Є –њ–Њ—З–µ–Ї –±–∞–Ї–ї–Њ—Д–µ–љ –≤¬†—В–µ—З–µ–љ–Є–µ –њ–µ—А–≤—Л—Е 30 –і–љ–µ–є –њ—А–Є–µ–Љ–∞ —Б–њ–Њ—Б–Њ–±—Б—В–≤–Њ–≤–∞–ї —А–∞–Ј–≤–Є—В–Є—О —Н–љ—Ж–µ—Д–∞–ї–Њ–њ–∞—В–Є–Є, —В—А–µ–±—Г—О—Й–µ–є¬†–≥–Њ—Б–њ–Є—В–∞–ї–Є–Ј–∞—Ж–Є–Є, –њ–Њ—Н—В–Њ–Љ—Г –њ—А–Є –µ–≥–Њ –љ–∞–Ј–љ–∞—З–µ–љ–Є–Є —В–∞–Ї–Є–Љ –±–Њ–ї—М–љ—Л–Љ —Б–ї–µ–і—Г–µ—В –Њ—Ж–µ–љ–Є—В—М —Б–Њ–Њ—В–љ–Њ—И–µ–љ–Є–µ ¬Ђ–њ–Њ–ї—М–Ј–∞/—А–Є—Б–Ї¬ї [36].
–Ґ–Њ–ї–њ–µ—А–Є–Ј–Њ–љ
–Э–∞–Є–±–Њ–ї–µ–µ —З–∞—Б—В–Њ –љ–∞¬†—Д–Њ–љ–µ —В–µ—А–∞–њ–Є–Є —В–Њ–ї–њ–µ—А–Є–Ј–Њ–љ–Њ–Љ –љ–∞–±–ї—О–і–∞—О—В—Б—П —Б–ї–∞–±–Њ—Б—В—М –Є¬†–±–Њ–ї—М –≤¬†–Љ—Л—И—Ж–∞—Е,¬†–≥–Њ–ї–Њ–≤–Њ–Ї—А—Г–ґ–µ–љ–Є–µ, –Ї—А–∞–њ–Є–≤–љ–Є—Ж–∞, —В–Њ—И–љ–Њ—В–∞, —А–≤–Њ—В–∞, —Б—Г—Е–Њ—Б—В—М –≤–Њ —А—В—Г [21]. –Ф–∞–љ–љ—Л–µ —А–µ–∞–Ї—Ж–Є–Є –Њ–±—Л—З–љ–Њ –љ–Є–≤–µ–ї–Є—А—Г—О—В—Б—П –њ–Њ—Б–ї–µ –Њ—В–Љ–µ–љ—Л –њ—А–µ–њ–∞—А–∞—В–∞. –Ю–і–љ–∞–Ї–Њ –≤¬†–ї–Є—В–µ—А–∞—В—Г—А–µ –Њ–њ–Є—Б–∞–љ—Л –Є¬†—В—П–ґ–µ–ї—Л–µ –љ–µ–ґ–µ–ї–∞—В–µ–ї—М–љ—Л–µ —А–µ–∞–Ї—Ж–Є–Є, —В–∞–Ї–Є–µ –Ї–∞–Ї –∞–љ–∞—Д–Є–ї–∞–Ї—В–Є—З–µ—Б–Ї–Є–є —И–Њ–Ї [37]. –°–ї–µ–і—Г–µ—В –Њ—В–Љ–µ—В–Є—В—М, —З—В–Њ —В–Њ–ї–њ–µ—А–Є–Ј–Њ–љ –љ–µ¬†–Њ–±–ї–∞–і–∞–µ—В —Б–µ–і–∞—В–Є–≤–љ—Л–Љ —Н—Д—Д–µ–Ї—В–Њ–Љ [6].
–°–Њ–≥–ї–∞—Б–љ–Њ –і–∞–љ–љ—Л–Љ –њ–Њ—Б—В—А–µ–≥–Є—Б—В—А–∞—Ж–Є–Њ–љ–љ—Л—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є –Є¬†—Б–Є—Б—В–µ–Љ—Л —Б–њ–Њ–љ—В–∞–љ–љ—Л—Е —Б–Њ–Њ–±—Й–µ–љ–Є–є, –њ—А–Є–Љ–µ—А–љ–Њ –≤¬†50% —Б–ї—Г—З–∞–µ–≤ –≤–Њ–Ј–љ–Є–Ї–∞–ї–Є –∞–ї–ї–µ—А–≥–Є—З–µ—Б–Ї–Є–µ —А–µ–∞–Ї—Ж–Є–Є, –і–∞–ї–µ–µ —Б–ї–µ–і–Њ–≤–∞–ї–Є –љ–µ–ґ–µ–ї–∞—В–µ–ї—М–љ—Л–µ —А–µ–∞–Ї—Ж–Є–Є —Б–Њ¬†—Б—В–Њ—А–Њ–љ—Л –ґ–µ–ї—Г–і–Њ—З–љ–Њ-–Ї–Є—И–µ—З–љ–Њ–≥–Њ —В—А–∞–Ї—В–∞ [38]. –°—А–µ–і–Є –∞–ї–ї–µ—А–≥–Є—З–µ—Б–Ї–Є—Е —А–µ–∞–Ї—Ж–Є–є –љ–∞–Є–±–Њ–ї–µ–µ —З–∞—Б—В–Њ –Њ—В–Љ–µ—З–∞–ї–Є—Б—М –Ї—А–∞–њ–Є–≤–љ–Є—Ж–∞ (5,1%), –Ј—Г–і (4,8%), –Њ–і—Л—И–Ї–∞ (4,2%), –∞–љ–≥–Є–Њ–љ–µ–≤—А–Њ—В–Є—З–µ—Б–Ї–Є–є –Њ—В–µ–Ї (3%), —Н—А–Є—В–µ–Љ–∞ (2,6%), —Б—Л–њ—М (1,8%) –Є¬†–∞–љ–∞—Д–Є–ї–∞–Ї—В–Є—З–µ—Б–Ї–Є–є —И–Њ–Ї (1,2%). –Я–Њ—А—П–і–Ї–∞ 30% –∞–ї–ї–µ—А–≥–Є—З–µ—Б–Ї–Є—Е —А–µ–∞–Ї—Ж–Є–є —А–∞–Ј–≤–Є–≤–∞–ї–Є—Б—М –њ—А–Є –њ—А–Є–Љ–µ–љ–µ–љ–Є–Є —В–Њ–ї–њ–µ—А–Є–Ј–Њ–љ–∞ —Б¬†–і—А—Г–≥–Є–Љ–Є –њ—А–µ–њ–∞—А–∞—В–∞–Љ–Є, –љ–∞–Є–±–Њ–ї–µ–µ —З–∞—Б—В–Њ —Б¬†–љ–µ—Б—В–µ—А–Њ–Є–і–љ—Л–Љ–Є –њ—А–Њ—В–Є–≤–Њ–≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ—Л–Љ–Є –њ—А–µ–њ–∞—А–∞—В–∞–Љ–Є [38]. –Р–ї–ї–µ—А–≥–Є—З–µ—Б–Ї–Є–µ —А–µ–∞–Ї—Ж–Є–Є –љ–∞¬†—Д–Њ–љ–µ —В–µ—А–∞–њ–Є–Є —В–Њ–ї–њ–µ—А–Є–Ј–Њ–љ–Њ–Љ —Б—В–∞–ї–Є –Њ—Б–љ–Њ–≤–∞–љ–Є–µ–Љ –і–ї—П –Х–≤—А–Њ–њ–µ–є—Б–Ї–Њ–≥–Њ –Љ–µ–і–Є—Ж–Є–љ—Б–Ї–Њ–≥–Њ –∞–≥–µ–љ—В—Б—В–≤–∞ —А–µ–Ї–Њ–Љ–µ–љ–і–Њ–≤–∞—В—М –≤–Њ–Ј–і–µ—А–ґ–∞—В—М—Б—П –Њ—В¬†–љ–∞–Ј–љ–∞—З–µ–љ–Є—П –њ–µ—А–Њ—А–∞–ї—М–љ—Л—Е —Д–Њ—А–Љ —В–Њ–ї–њ–µ—А–Є–Ј–Њ–љ–∞, –Ј–∞ –Є—Б–Ї–ї—О—З–µ–љ–Є–µ–Љ –≤–Ј—А–Њ—Б–ї—Л—Е –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б–Њ¬†—Б–њ–∞—Б—В–Є—З–љ–Њ—Б—В—М—О –њ–Њ—Б–ї–µ –Є–љ—Б—Г–ї—М—В–∞ (2012¬†–≥.) [39]. –≠—В–Њ –Ј–∞—П–≤–ї–µ–љ–Є–µ –±—Л–ї–Њ —Б–і–µ–ї–∞–љ–Њ¬†–њ–Њ—Б–ї–µ –Њ—Ж–µ–љ–Ї–Є —Б–Њ–Њ—В–љ–Њ—И–µ–љ–Є—П ¬Ђ–њ–Њ–ї—М–Ј–∞/—А–Є—Б–Ї¬ї –њ—А–Є —А–∞–Ј–љ—Л—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П—Е.
–Ф–∞–љ–љ—Л–µ –њ–Њ—Б—В—А–µ–≥–Є—Б—В—А–∞—Ж–Є–Њ–љ–љ—Л—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є –Є¬†—Б–Є—Б—В–µ–Љ—Л —Б–њ–Њ–љ—В–∞–љ–љ—Л—Е —Б–Њ–Њ–±—Й–µ–љ–Є–є —В–∞–Ї–ґ–µ –њ–Њ–Ј–≤–Њ–ї–Є–ї–Є –≤—Л—П–≤–Є—В—М —Б–µ—А—М–µ–Ј–љ—Л–µ –њ–Њ–±–Њ—З–љ—Л–µ —Н—Д—Д–µ–Ї—В—Л –≤¬†–≤–Є–і–µ —А–µ–∞–Ї—Ж–Є–є¬†–≥–Є–њ–µ—А—З—Г–≤—Б—В–≤–Є—В–µ–ї—М–љ–Њ—Б—В–Є, –∞¬†—В–∞–Ї–ґ–µ –љ–µ–і–Њ—Б—В–∞—В–Њ–Ї –і–∞–љ–љ—Л—Е, –њ–Њ–і—В–≤–µ—А–ґ–і–∞—О—Й–Є—Е —Н—Д—Д–µ–Ї—В–Є–≤–љ–Њ—Б—В—М —В–Њ–ї–њ–µ—А–Є–Ј–Њ–љ–∞ –њ—А–Є –ї–µ—З–µ–љ–Є–Є —А—П–і–∞ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є. –Т¬†—Б–≤—П–Ј–Є —Б¬†—Н—В–Є–Љ —Н–Ї—Б–њ–µ—А—В—Л –Х–≤—А–Њ–њ–µ–є—Б–Ї–Њ–≥–Њ –Љ–µ–і–Є—Ж–Є–љ—Б–Ї–Њ–≥–Њ –∞–≥–µ–љ—В—Б—В–≤–∞ —А–µ–Ї–Њ–Љ–µ–љ–і–Њ–≤–∞–ї–Є –Њ–≥—А–∞–љ–Є—З–Є—В—М –њ—А–Є–Љ–µ–љ–µ–љ–Є–µ –њ–µ—А–Њ—А–∞–ї—М–љ–Њ–≥–Њ —В–Њ–ї–њ–µ—А–Є–Ј–Њ–љ–∞, –Ј–∞ –Є—Б–Ї–ї—О—З–µ–љ–Є–µ–Љ –ї–µ—З–µ–љ–Є—П —Б–њ–∞—Б—В–Є—З–љ–Њ—Б—В–Є –њ–Њ—Б–ї–µ –Є–љ—Б—Г–ї—М—В–∞, –њ—А–Є –Ї–Њ—В–Њ—А–Њ–є –њ–Њ–ї—М–Ј–∞ –Њ—В¬†–њ—А–Є–Љ–µ–љ–µ–љ–Є—П –њ—А–µ–њ–∞—А–∞—В–∞ –њ–µ—А–µ–≤–µ—И–Є–≤–∞–µ—В –њ–Њ—В–µ–љ—Ж–Є–∞–ї—М–љ—Л–є —А–Є—Б–Ї¬†[39].
–Ч–∞–Ї–ї—О—З–µ–љ–Є–µ
–Э–∞¬†—Б–µ–≥–Њ–і–љ—П—И–љ–Є–є –і–µ–љ—М –Љ–Є–Њ—А–µ–ї–∞–Ї—Б–∞–љ—В—Л —И–Є—А–Њ–Ї–Њ –њ—А–Є–Љ–µ–љ—П—О—В—Б—П –≤¬†–ї–µ—З–µ–љ–Є–Є —Б–њ–∞—Б—В–Є—З–љ–Њ—Б—В–Є —А–∞–Ј–ї–Є—З–љ–Њ–є –њ—А–Є—А–Њ–і—Л –Є¬†–±–Њ–ї–Є –≤¬†–љ–Є–ґ–љ–µ–є —З–∞—Б—В–Є —Б–њ–Є–љ—Л. –Ф–∞–љ–љ—Л–µ –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є –љ–µ¬†–њ–Њ–Ј–≤–Њ–ї—П—О—В –і–Њ—Б—В–Њ–≤–µ—А–љ–Њ –≤—Л–і–µ–ї–Є—В—М –љ–∞–Є–±–Њ–ї–µ–µ —Н—Д—Д–µ–Ї—В–Є–≤–љ—Л–є –Љ–Є–Њ—А–µ–ї–∞–Ї—Б–∞–љ—В, –≤¬†—Б–≤—П–Ј–Є —Б¬†—З–µ–Љ –Њ–і–љ–Є–Љ–Є –Є–Ј¬†–Ї–ї—О—З–µ–≤—Л—Е —Д–∞–Ї—В–Њ—А–Њ–≤ –њ—А–Є –≤—Л–±–Њ—А–µ –њ—А–µ–њ–∞—А–∞—В–∞ —Б—В–∞–љ–Њ–≤—П—В—Б—П –њ—А–Њ—Д–Є–ї—М –±–µ–Ј–Њ–њ–∞—Б–љ–Њ—Б—В–Є –Є¬†–њ–µ—А–µ–љ–Њ—Б–Є–Љ–Њ—Б—В—М. –°–ї–µ–і—Г–µ—В –Њ—В–Љ–µ—В–Є—В—М, —З—В–Њ –≤—Л–±–Є—А–∞—В—М –Љ–Є–Њ—А–µ–ї–∞–Ї—Б–∞–љ—В —Б–ї–µ–і—Г–µ—В –Є–љ–і–Є–≤–Є–і—Г–∞–ї—М–љ–Њ, —Г—З–Є—В—Л–≤–∞—П —Б–њ–µ–Ї—В—А –≤–Њ–Ј–Љ–Њ–ґ–љ—Л—Е –љ–µ–ґ–µ–ї–∞—В–µ–ї—М–љ—Л—Е —А–µ–∞–Ї—Ж–Є–є, –њ–µ—А–µ–љ–Њ—Б–Є–Љ–Њ—Б—В—М –Є¬†–љ–∞–ї–Є—З–Є–µ —Б–Њ–њ—Г—В—Б—В–≤—Г—О—Й–µ–є –њ–∞—В–Њ–ї–Њ–≥–Є–Є, –њ—А–µ–і—А–∞—Б–њ–Њ–ї–∞–≥–∞—О—Й–µ–є –Ї¬†—А–∞–Ј–≤–Є—В–Є—О –љ–µ–ґ–µ–ї–∞—В–µ–ї—М–љ—Л—Е –ї–µ–Ї–∞—А—Б—В–≤–µ–љ–љ—Л—Е —А–µ–∞–Ї—Ж–Є–є.
–Ю–і–љ–Є–Љ –Є–Ј¬†–љ–∞–Є–±–Њ–ї–µ–µ –Є–Ј—Г—З–µ–љ–љ—Л—Е –Є¬†–Њ–±–ї–∞–і–∞—О—Й–Є—Е –±–ї–∞–≥–Њ–њ—А–Є—П—В–љ—Л–Љ –њ—А–Њ—Д–Є–ї–µ–Љ –±–µ–Ј–Њ–њ–∞—Б–љ–Њ—Б—В–Є –Љ–Є–Њ—А–µ–ї–∞–Ї—Б–∞–љ—В–Њ–≤ —П–≤–ї—П–µ—В—Б—П —В–Є–Ј–∞–љ–Є–і–Є–љ (–°–Є—А–і–∞–ї—Г–і¬Ѓ), –Ї–Њ—В–Њ—А—Л–є –њ—А–Є –њ–Њ—Б—В–µ–њ–µ–љ–љ–Њ–є —В–Є—В—А–∞—Ж–Є–Є –і–Њ–Ј—Л, –Ї–∞–Ї –њ—А–∞–≤–Є–ї–Њ, —Е–Њ—А–Њ—И–Њ –њ–µ—А–µ–љ–Њ—Б–Є—В—Б—П –њ–∞—Ж–Є–µ–љ—В–∞–Љ–Є –Є¬†–љ–µ –≤—Л–Ј—Л–≤–∞–µ—В —Б–µ—А—М–µ–Ј–љ—Л—Е –љ–µ–ґ–µ–ї–∞—В–µ–ї—М–љ—Л—Е —А–µ–∞–Ї—Ж–Є–є.
Ye.V. Dovgan, PhD
Smolensk Regional Clinical Hospital
Contact person: Yevgeny V. Dovgan, dovganrus@mail.ru
Muscle relaxants are currently widely used in the treatment of spasticity and various pain syndromes. This article reviews the clinical pharmacology of three muscle relaxants, such as: tizanidine, baclofen and tolperisone. Special attention is paid to the safety and tolerability of these drugs.
–£–≤–∞–ґ–∞–µ–Љ—Л–є –њ–Њ—Б–µ—В–Є—В–µ–ї—М uMEDp!
–£–≤–µ–і–Њ–Љ–ї—П–µ–Љ –Т–∞—Б –Њ —В–Њ–Љ, —З—В–Њ –Ј–і–µ—Б—М —Б–Њ–і–µ—А–ґ–Є—В—Б—П –Є–љ—Д–Њ—А–Љ–∞—Ж–Є—П, –њ—А–µ–і–љ–∞–Ј–љ–∞—З–µ–љ–љ–∞—П –Є—Б–Ї–ї—О—З–Є—В–µ–ї—М–љ–Њ –і–ї—П —Б–њ–µ—Ж–Є–∞–ї–Є—Б—В–Њ–≤ –Ј–і—А–∞–≤–Њ–Њ—Е—А–∞–љ–µ–љ–Є—П.
–Х—Б–ї–Є –Т—Л –љ–µ —П–≤–ї—П–µ—В–µ—Б—М —Б–њ–µ—Ж–Є–∞–ї–Є—Б—В–Њ–Љ –Ј–і—А–∞–≤–Њ–Њ—Е—А–∞–љ–µ–љ–Є—П, –∞–і–Љ–Є–љ–Є—Б—В—А–∞—Ж–Є—П –љ–µ –љ–µ—Б–µ—В –Њ—В–≤–µ—В—Б—В–≤–µ–љ–љ–Њ—Б—В–Є –Ј–∞ –≤–Њ–Ј–Љ–Њ–ґ–љ—Л–µ –Њ—В—А–Є—Ж–∞—В–µ–ї—М–љ—Л–µ –њ–Њ—Б–ї–µ–і—Б—В–≤–Є—П, –≤–Њ–Ј–љ–Є–Ї—И–Є–µ –≤ —А–µ–Ј—Г–ї—М—В–∞—В–µ —Б–∞–Љ–Њ—Б—В–Њ—П—В–µ–ї—М–љ–Њ–≥–Њ –Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–љ–Є—П –Т–∞–Љ–Є –Є–љ—Д–Њ—А–Љ–∞—Ж–Є–Є —Б –њ–Њ—А—В–∞–ї–∞ –±–µ–Ј –њ—А–µ–і–≤–∞—А–Є—В–µ–ї—М–љ–Њ–є –Ї–Њ–љ—Б—Г–ї—М—В–∞—Ж–Є–Є —Б –≤—А–∞—З–Њ–Љ.
–Э–∞–ґ–Є–Љ–∞—П –љ–∞ –Ї–љ–Њ–њ–Ї—Г ¬Ђ–Т–Њ–є—В–Є¬ї, –Т—Л –њ–Њ–і—В–≤–µ—А–ґ–і–∞–µ—В–µ, —З—В–Њ —П–≤–ї—П–µ—В–µ—Б—М –≤—А–∞—З–Њ–Љ –Є–ї–Є —Б—В—Г–і–µ–љ—В–Њ–Љ –Љ–µ–і–Є—Ж–Є–љ—Б–Ї–Њ–≥–Њ –≤—Г–Ј–∞.