Механизм развития лабораторных признаков при первичном билиарном холангите и их диагностическое значение
- Аннотация
- Статья
- Ссылки
- English
Цель данного обзора – обобщить имеющиеся литературные и собственные данные, касающиеся механизмов развития биохимических критериев ПБХ и их диагностического значения. Благодаря достижениям биохимии, молекулярной биологии и генетики стало возможным представить эти данные с учетом патофизиологических механизмов их развития.
Цель данного обзора – обобщить имеющиеся литературные и собственные данные, касающиеся механизмов развития биохимических критериев ПБХ и их диагностического значения. Благодаря достижениям биохимии, молекулярной биологии и генетики стало возможным представить эти данные с учетом патофизиологических механизмов их развития.

Введение
Первичный билиарный холангит (ПБХ), ранее известный как первичный билиарный цирроз, представляет собой хроническое холестатическое гранулематозное и деструктивное повреждение мелких интралобулярных и септальных желчных протоков с потенциальной тенденцией к прогрессированию в цирроз [1]. Запускающим фактором, инициирующим заболевание, считается нарушение выработки бикарбоната билиарными эпителиальными клетками (БЭК, холангиоцитами) мелких желчных протоков. Это приводит к поступлению и накоплению желчных кислот в мелких холангиоцитах с последующим повреждением мембранных структур, провоцирующих ускоренное старение и апоптоз БЭК мелких желчных протоков [2]. Накопившиеся в апоптотических холангиоцитах желчные кислоты разрушают клеточные мембранные структуры, в том числе внешнюю и внутреннюю мембраны митохондрий. Обладая детергентными свойствами, желчные кислоты взаимодействуют с липоевой кислотой Е2 субъединицы пируватдегидрогеназного комплекса (ПДГ), что приводит к изменению иммунологических свойств Е2 ПДГ. Образующийся в результате неоантиген инициирует выработку антимитохондриальных антител (АМА) [2]. Развитие апоптоза в мелких БЭК приводит в конечном итоге к развитию дуктулопении. Развивается постепенно прогрессирующий внутрипеченочный холестаз, сопровождающийся повреждением гепатоцитов [2].
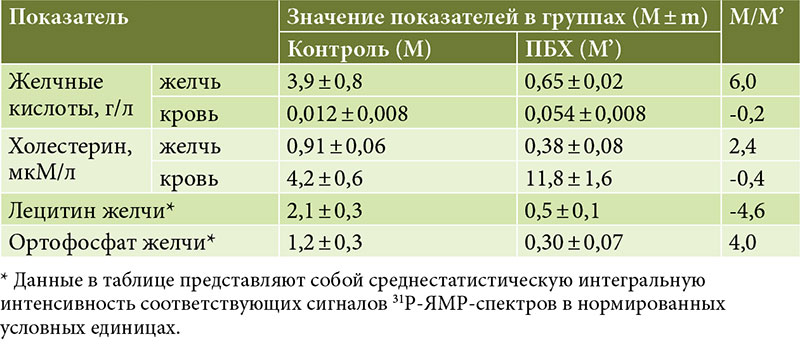
Холестаз при ПБХ связан с повреждением субклеточных структур БЭК мелких внутрипеченочных желчных протоков, что вызывает нарушение процессов желчевыделения и энтерогепатической циркуляции желчных кислот. Развивающийся внутрипеченочный холестаз приводит к недостаточному поступлению желчных кислот в двенадцатиперстную кишку и повышенному накоплению желчных кислот в гепатоцитах и плазме крови (рис. 1). Именно эти изменения в процессах желчеобразования и желчевыделения у пациентов с ПБХ следует рассматривать в качестве первопричины появления лабораторных признаков этого заболевания. Для ПБХ характерны изменения многих биохимических показателей [3]. В сыворотке крови пациентов выявляется увеличение активности щелочной фосфатазы (ЩФ), гамма-глутамилтрансферазы (ГГТ, γ-ГТ), лейцинаминопептидазы (ЛАП), 5′-нуклеотидазы (5′-НК), повышенное содержание желчных кислот, холестерина, фосфолипидов (ФЛ), билирубина, меди, γ-глобулинов, а также снижение уровня общего белка преимущественно за счет фракции альбуминов [3, 4].
Повышение активности ЩФ, γ-ГТ, 5′-НК, ЛАП при ПБХ
Первичному билиарному холангиту предшествует длительный бессимптомный период. В это время отсутствуют какие-либо физикальные признаки заболевания. Но при биохимическом исследовании сыворотки крови у этих больных отмечается повышение активности ферментов ЩФ, 5′-НК и γ-ГТ [4, 5]. Механизм развития гиперферментемии у пациентов с ПБХ тесно связан с повышением в плазме крови уровня ФЛ вследствие увеличения их синтеза в печени и регургитации из гепатоцитов и/или желчных капилляров в общий кровоток [6, 7]. Основу биосинтеза ФЛ (прежде всего лецитинов) составляют ортофосфат (Pi), глицерин, пальмитиновая и олеиновая жирные кислоты [8]. Уже на начальных стадиях развития заболевания в плазме крови пациентов с ПБХ выявляется повышенное содержание пальмитиновой и олеиновой жирных кислот, что сопровождается увеличением синтеза ФЛ и повышением их уровня в плазме крови [9, 10]. Образование Pi для синтеза ФЛ зависит от активности ферментов ЩФ и 5′-НК, участвующих в гидролизе фосфомоноэфиров с образованием Pi. ЩФ активирует гидролиз глицерофосфата, глюкозо-1-фосфата и глюкозо-6-фосфата с образованием соответствующих углеводов и Pi. Под влиянием 5′-НК осуществляется гидролиз рибонуклеотидов АМФ, ГМФ, ЦМФ и УМФ до рибонуклеозидов (аденозин, гуанозин, цимитидин, уридин) и Pi.
ЩФ присутствует во многих тканях организма. Изоферменты ЩФ обнаружены в эпителии желчных протоков (внутри- и внепеченочных), остеобластах, слизистой оболочке кишечника, плаценте и лактирующей молочной железе. Однако более 80% ЩФ, определяемой в сыворотке крови, выделяется из печени и костей [4]. Активность ЩФ у пациентов с ПБХ уже в асимптоматической стадии заболевания повышается в два и более раза выше верхней границы нормы [3]. Следует подчеркнуть, что при ПБХ отмечено увеличение активности преимущественно печеночной фракции изофермента ЩФ. Последняя вырабатывается как в гепатоцитах, так и в холангиоцитах, выстилающих желчные канальцы и протоки [11].
Повышение уровня ЩФ в сыворотке крови больных ПБХ происходит в ответ на повреждение желчными кислотами мембранных структур холангиоцитов уже в асимптоматической стадии заболевания [2, 3]. Механизм повышения уровня ЩФ и 5′-НК при ПБХ является предметом дискуссий. Существует теория, согласно которой ЩФ обеспечивает образование Pi там, где в нем возникает потребность [12]. Исходя из этой теории, увеличение активности ЩФ и 5′-НК при ПБХ свидетельствует о повышенной потребности и синтезе Pi в холангиоцитах и гепатоцитах. При этом его количество в печеночной порции желчи пациентов с ПБХ снижено (таблица) [13]. Секреция Pi в желчь из БЭК, выстилающих желчные протоки, и гепатоцитов осуществляется пассивно по градиенту концентрации [14, 15]. Следовательно, концентрация Pi в гепатоцитах и холангиоцитах также снижена. Все это свидетельствует об интенсивном использовании фосфорной группировки в метаболических процессах, происходящих в печеночных клетках и холангиоцитах желчных протоков при ПБХ. Одним из возможных путей использования Pi является биосинтез фосфолипидов БЭК, необходимых для репарации мембранных структур и нейтрализации желчных кислот, накапливающихся в холангиоцитах [2, 16]. Уровень 5′-НК относительно специфичен для заболеваний печени и сильно коррелирует с сывороточной ЩФ печеночного происхождения [4]. Умеренное повышение активности печеночной фракции ЩФ и 5′-НК уже в бессимптомной стадии ПБХ указывает на изменения в обмене фосфора [5].
Исследования убедительно показывают, что повышение активности ЩФ и 5′-НК при ПБХ связано с увеличением синтеза этих ферментов [4]. D. Hatoff и соавт. показали, что на пятый день после перевязки желчного протока у крыс начинает определяться активность ЩФ в эндоплазматическом ретикулуме и мембранах аппарата Гольджи [17]. Применение ингибитора трансляции и транскрипции белкового синтеза у крыс с лигированным желчным протоком не приводит к возрастанию уровня ЩФ в сыворотке крови [18]. Эти исследования указывают на то, что при хроническом холестазе имеет место процесс индукции синтеза ЩФ. Наряду с этим отмечено увеличение трансляции мРНК, ответственной за синтез ЩФ, с последующим увеличением секреции фермента в сыворотку крови [4].
Для повышенного биосинтеза ЩФ и 5′-НК при ПБХ требуется более активная доставка аминокислот в клетку. Роль γ-ГТ заключается в трансмембранном транспорте и доставке аминокислот в клетку, что позволяет активно использовать их в биосинтезе белков, включая ферменты [19]. Аналогично ЩФ, активность γ-ГТ повышается в пять и более раз выше верхней границы нормы при ПБХ [3]. При этом увеличение активности γ-ГТ предшествует повышению активности ЩФ и 5′-НК у пациентов с ПБХ, что может косвенно указывать на участие γ-ГТ в синтезе ферментов ЩФ и 5′-НК.
γ-ГТ представляет собой микросомальный фермент, который широко распространен в тканях человека, участвующих в секреторных и абсорбционных процессах, в том числе в БЭК желчных протоков [20]. Повышение сывороточного уровня γ-ГТ при ПБХ уже в асимптоматической стадии заболевания больше отражает повреждение холангиоцитов, а не гепатоцитов [4]. Повышение уровня γ-ГТ в этот период в сочетании с увеличением активности ЩФ и 5′-НК при ПБХ в значительной степени указывает на нарушение процессов желчевыделения, а не на повреждение печеночных клеток.
γ-ГТ является чувствительным, но не специфичным индикатором повреждения желчных канальцев и протоков. Повышение активности этого фермента показано уже в бессимптомной стадии заболевания, и это указывает на то, что ПБХ начинается с повреждения желчных канальцев и протоков [21]. Имеющиеся данные свидетельствуют о том, что по неизвестным причинам происходит повреждение холангиоцитов, выстилающих желчные протоки [22]. В норме холангиоциты защищены от токсического, детергентного воздействия желчных кислот «бикарбонатным зонтиком» [23]. Секреция бикарбонатных ионов защищает холангиоциты от неконтролируемого трансмембранного тока гликохенодезоксихолевой кислоты, которая может индуцировать апоптоз билиарных эпителиальных клеток [24]. При ПБХ «бикарбонатный зонтик» становится «негерметичным» и пропускает токсичные для холангиоцитов желчные кислоты, запуская процесс фиброгенеза. Последний приводит к развитию дуктулопении уже на ранних стадиях ПБХ. Значительное повышение активности γ-ГТ является признаком фиброза и может быть использовано в качестве косвенного маркера фиброза желчных протоков при ПБХ [25–27]. γ-ГТ является одним из первых биохимических признаков ПБХ, наряду с повышением активности ЩФ и появлением АМА, которые используются для диагностики этого заболевания [28].
Однако следует помнить, что γ-ГТ присутствует не только в печени, но и в почках, поджелудочной железе и кишечнике [19]. Индукция данного фермента в этих органах, как правило, обусловлена хроническим употреблением алкоголя [11]. В связи с тем что γ-ГT содержится в микросомах гепатоцитов, алкоголь и лекарственные препараты из группы индукторов микросомального окисления могут стимулировать ее активность. К индукторам микросомальных ферментов печени относятся снотворные средства (барбитураты, хлоралгидрат), транквилизаторы (диазепам, хлордиазепоксид, мепробамат), нейролептики (хлорпромазин, трифлуоперазин), противосудорожные (фенитоин), противовоспалительные (фенилбутазон), некоторые антибиотики (рифампицин), диуретики (спиронолактон) и другие лекарственные средства. При исключении влияния алкоголя или лекарственных препаратов выявление повышенного уровня γ-ГТ является весьма чувствительным для ранней диагностики ПБХ. Сывороточный уровень γ-ГТ может быть использован для повышения прогностической ценности измерения ЩФ при ПБХ [20].
ЛАП в самых высоких концентрациях содержится в печени, почках и тонкой кишке. Активность ЛАП повышается при повреждении эпителия желчных протоков при всех формах внутри- и внепеченочного холестаза. ЛАП имеет примерно такое же клиническое значение, как и ЩФ. Однако активность ЛАП при заболеваниях костной ткани практически не меняется и находится в пределах нормы. Поэтому при повышении активности ЩФ определение ЛАП используется для дифференциальной диагностики заболеваний гепатобилиарной системы и костной ткани. С момента внедрения в клиническую практику определения активности γ-ГТ интерес к исследованиям ЛАП заметно снизился. В клинической практике наиболее распространено определение активности ЩФ и γ-ГТ у пациентов с ПБХ. Европейская ассоциация по изучению печени (EASL) и Американская ассоциация по изучению заболеваний печени (AASLD) рекомендуют использовать определение активности ЩФ и γ-ГТ в сочетании с АМА в качестве ранних диагностических критериев ПБХ [29, 30].
Желчные кислоты, холестерин и лецитин при ПБХ
При ПБХ наблюдается снижение уровня желчных кислот, холестерина и лецитина в печеночной желчи и одновременное их повышение в гепатоцитах и крови (таблица) [13]. Это свидетельствует о том, что при ПБХ нарушена энтерогепатическая циркуляция желчных кислот.
Уменьшение секреции бикарбоната холангиоцитами, приводящее к повышенному поступлению и накоплению желчных кислот в БЭК с последующим повреждением мембранных структур, апоптозом и некрозом холангиоцитов, постепенно приводят к дуктулопении при ПБХ [2]. Увеличение количества запустевающих желчных протоков приводит к нарушению экскреции желчи и недостаточному поступлению желчных кислот в двенадцатиперстную кишку. Снижение уровня желчных кислот в кишечнике по механизму обратной связи вызывает компенсаторное увеличение биосинтеза желчных кислот и холестерина в гепатоцитах. Холестерин является основным субстратом для биосинтеза желчных кислот. Однако дуктулопения при этом не снижается, а, наоборот, нарастает, что способствует развитию внутрипеченочного холестаза. Концентрация желчных кислот в гепатоцитах постепенно повышается, а их поступление в просвет кишечника остается недостаточным, что порождает замкнутый порочный круг, приводящий к накоплению желчных кислот в клетках печени (рис. 1). Вследствие повышения уровня желчных кислот в гепатоцитах снижается их реабсорбция из портальной венозной крови, что приводит к поступлению и прогрессирующему накоплению желчных кислот в общем кровотоке. Холестаз в желчных капиллярах также приводит к рефлюксу желчи в системный кровоток через межгепатоцитарное пространство. Уже на ранних стадиях заболевания концентрация желчных кислот в сыворотке крови повышается у пациентов с ПБХ (таблица). Все фракции конъюгированных желчных кислот в заметных количествах присутствуют в крови у пациентов с ПБХ. Это сопровождается отложением желчных кислот в эпидермисе. Неконъюгированные желчные кислоты редко обнаруживаются в сыворотке крови больных ПБХ. Накопление желчных кислот в гепатоцитах вызывает их повреждение и индуцирует апоптоз. Эксперименты in vitro показали, что механизм развития последнего связан с образованием активных форм кислорода, генерируемых митохондриями [28, 31–33]. Но немаловажную роль в повреждении гепатоцитов играют детергентные свойства самих желчных кислот, что приводит к разрушению мембранных структур клетки [2].
Желчные кислоты, особенно неконъюгированные, являются сильными детергентами, способными оказывать раздражающее действие на нервные окончания, что способствует появлению кожного зуда – первого клинического признака заболевания [5]. Холевая (тригидроксихолановая) желчная кислота обладает меньшими детергентными свойствами, чем дезоксихолевая и хенодезоксихолевая (дигидроксихолановые) [15]. Конъюгация, сульфатирование и глюкуронирование желчных кислот направлены на уменьшение их детергентного и раздражающего действия на соматические клетки и нервные окончания, а также способствуют приобретению гидрофильных свойств и выведению из общего кровотока через кожу, почки и кишечник. Конъюгация желчных кислот с таурином более активно стимулирует образование мицелл с холестерином и фосфатидилхолином (лецитином), чем конъюгация с глицином [34]. Проникновение первичных желчных кислот, конъюгированных с глицином, в эпителиальные клетки, выстилающие желчные протоки, зависит от наличия «карбонатного зонтика» [28]. Недостаточный синтез бикарбоната холангиоцитами и его поступление в просвет желчного протока могут привести к повышенному проникновению конъюгированных с глицином желчных кислот в БЭК. Это способствует повреждению мембран, усилению окислительного стресса, ускоренному старению холангиоцитов и их апоптозу [2, 28]. В крови и желчи пациентов с ПБХ отмечается более высокое соотношение тригидрокси-/дигидроксихолановых желчных кислот и их конъюгатов с таурином. По мнению H. Greim и соавт. [35, 36], это связано с тем, что тригидроксихолановая (холевая) кислота более гидрофильна и обладает меньшими детергентными свойствами, чем более гидрофобные дезоксихолевая и хенодезоксихолевая желчные кислоты. Кроме того, увеличивается количество конъюгированных с глюкуроновой кислотой и сульфатированных желчных кислот. Повышение соотношения тригидрокси-/дигидроксихолановых желчных кислот, снижение коэффициента глицин/тауриновых конъюгатов желчных кислот и появление сульфатированных, глюкуронированных желчных кислот в общем кровотоке при ПБХ можно рассматривать как компенсаторную, детоксикационную реакцию организма в ответ на холестаз и поступление холановых кислот в плазму крови [37].
В период выраженного внутрипеченочного холестаза в крови и моче пациентов с ПБХ появляются атипичные, нефизиологические желчные кислоты [38]. Они обладают более мощным детергентным действием на клеточные мембраны и более сильным раздражающим действием на нервные рецепторы, чем первичные и вторичные желчные кислоты. Атипичные, нефизиологические желчные кислоты могут выделяться из общего кровотока через кожу, участвуя в механизме развития кожного зуда [5]. Интенсивность последнего может зависеть от количества атипичных желчных кислот в коже пациентов с ПБХ.
Появление желчных кислот в крови у пациентов с ПБХ приводит к увеличению экспрессии фактора роста фибробластов 19 (FGF19) через систему обратной связи. На мембранах гепатоцитов активируется рецептор фактора роста фибробластов 4 (FGFR4), что приводит к подавлению синтеза желчных кислот из холестерина. Это способствует развитию гиперхолестеринемии [39–42].
В печеночной желчи больных с ПБХ и группы сравнения соотношение липидных компонентов – желчные кислоты, лецитин и холестерин – составляет соответственно 6:4,6:2,4 (таблица), что свидетельствует о высокой литогенности желчи [13]. Желчнокаменная болезнь осложняет ПБХ в 35–40% случаев. Секреция лецитина печеночными клетками зависит от секреции желчных кислот. Снижение концентрации лецитина в печеночной желчи у пациентов с ПБХ связано с изменением относительного количества желчных кислот [43, 44]. Регуляторные эффекты желчных кислот на печень, желчевыводящие пути и секрецию фосфатидилхолина в желчь во многом зависят от гидрофобно-гидрофильного баланса рециркулирующего пула желчных кислот [45].
В то же время в мембранах гепатоцитов, полученных из печеночных биоптатов пациентов с ПБХ, наблюдалось полуторакратное увеличение содержания ФЛ (p = 0,044) [46]. Это сопровождается почти двукратным снижением соотношения холестерин/ФЛ. Одновременно в этих же мембранах снижается содержание лизофосфатидилхолина, сфингомиелина, фосфатидилсерина (ФС), фосфатидилинозитола и фосфатидилэтаноламина (ФЭ) [47, 48]. Это может отражаться на жидкокристаллической структуре мембраны, что изменяет функцию белков-транспортеров, в том числе и на апикальной мембране гепатоцитов. Процесс желчеобразования в желчном каналикуле может меняться.
Недостаточное поступление первичных желчных кислот в кишечник при ПБХ приводит к изменению состава микробиома, вызывая дисбиоз кишечника [48–52]. По данным J.K. DiBaise и F.F. Paustian [53], дисбиоз у пациентов с ПБХ определяет, наряду с недостаточным поступлением желчных кислот в кишечник, развитие стеатореи. Поэтому эти пациенты должны быть обязательно обследованы на предмет избыточного роста бактерий [49, 53].
Недостаточное поступление желчных кислот в кишечнике при ПБХ нарушает солюбилизацию и мицеллообразование пищевого холестерина, что снижает его всасывание в кишечнике. Через систему обратной связи стимулируется внутрипеченочный синтез холестерина и снижается поглощение печенью липопротеинов низкой плотности (ЛПНП) через их рецепторы [54] Повышенный печеночный синтез холестерина приводит к повышению его уровня в плазме крови.
Вследствие развивающегося и прогрессирующего холестаза происходит одновременное накопление желчных кислот в гепатоцитах и повышение их уровня в плазме крови. Для желчных кислот, попавших в системный кровоток у пациентов с ПБХ, требуется нейтрализации их детергентного действия на мембранные структуры клеток крови и эндотелиальных клеток сосудистой стенки. Это сопровождается нарушением липидного обмена у пациентов с ПБХ. Возникающая при этом дислипидемия связана прежде всего с изменениями синтеза и транспорта холестерина и ФЛ.
Механизм развития дислипидемии при ПБХ
У пациентов с ПБХ уже на ранних стадиях заболевания при биохимическом исследовании крови отмечается дислипидемия, проявляющаяся повышенным содержанием общего холестерина (ОХ), ФЛ, жирных кислот, холестерина липопротеинов низкой плотности (ХС ЛПНП) и холестерина липопротеинов высокой плотности (ХС ЛПВП) [55], а также появлением липопротеина X (ЛП-Х) [39]. Уровень триглицеридов при ПБХ практически не изменяется или повышен незначительно [56]. Считается, что небольшое повышение нейтральных липидов может быть связано со снижением уровня активности липопротеинлипазы (ЛПЛ) при ПБХ [56]. ЛПЛ расщепляет триглицериды самых крупных по размеру и богатых липидами липопротеинов плазмы крови – хиломикронов и липопротеинов очень низкой плотности (ЛПОНП).
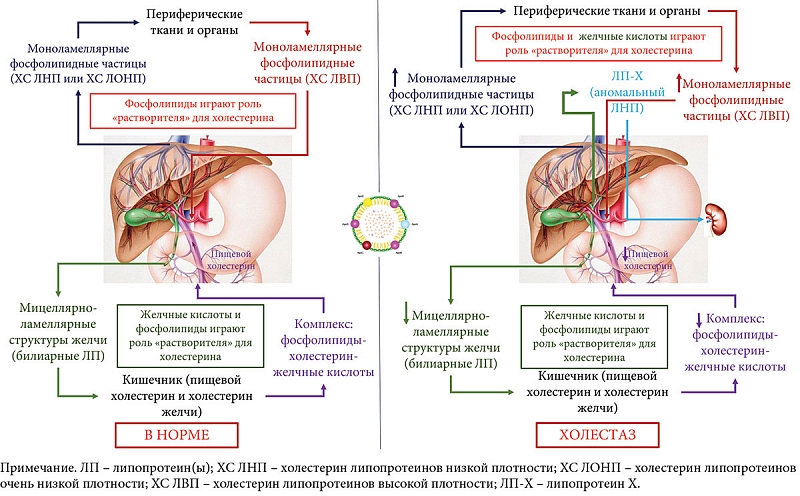
Поддержание постоянства уровня холестерина в плазме крови здорового человека зависит от его поступления, транспортировки и выведения из общего кровотока. Содержание холестерина в плазме крови пополняется вследствие всасывания в кишечнике пищевого и желчного холестерина, синтеза эндогенного холестерина, который синтезируется главным образом в печени, и секреции в кровоток частиц ЛПОНП и ЛПНП, содержащих холестерин [57]. В норме снижение уровня холестерина в плазме крови достигается благодаря поглощению ХС ЛПНП и ХС ЛПВП печенью, выведению холестерина с желчью и его катаболизму в печени с образованием первичных (холевая, хенодезоксихолевая) желчных кислот (рис. 2) [57, 58].
В условиях холестаза у пациентов с ПБХ происходит нарушение энтерогепатической циркуляции желчных кислот, что приводит, с одной стороны, к недостаточному поступлению желчных кислот в двенадцатиперстную кишку и, с другой стороны, к повышенному накоплению желчных кислот в гепатоцитах и плазме крови. Именно эти изменения в процессах желчеобразования и желчевыделения следует рассматривать в качестве первопричины нарушения липидного обмена при ПБХ. Количественные изменения одного из «растворителей» холестерина, желчных кислот в кишечнике (уменьшение) и плазме крови (появление и накопление) меняют условия для транспорта и выведения холестерина из организма (рис. 2) [58]. Возникающая при этом дислипидемия связана прежде всего с изменением синтеза и транспорта холестерина и ФЛ в результате развития внутрипеченочного холестаза и нарушения энтерогепатической циркуляции желчных кислот. Снижение секреции желчных кислот ухудшает солюбилизацию пищевого холестерина и образование мицелл, что приводит к уменьшению его всасывания из кишечника. Через систему обратной связи стимулируется внутрипеченочный синтез холестерина и снижается захват печенью ХС ЛПНП через рецепторы ЛПНП [39]. Наблюдается мальабсорбция жиров из-за недостаточного поступления желчных кислот в кишечник и развивается стеаторея [59]. Наряду с этим наблюдаются ингибирование липогенеза и активация окисления свободных жирных кислот, что связано с высокой экспрессией фактора роста фибробластов 19 [60]. Все это снижает накопление триглицеридов в печени и плазме крови [60].
При накоплении желчных кислот в плазме крови при ПБХ требуется нейтрализация их детергентного действия на мембранные структуры форменных элементов крови и эндотелиоцитов сосудистой стенки. Исходя из физико-химических свойств желчных кислот, сделать это можно посредством формирования в плазме крови мицеллярно-ламеллярных структур с ФЛ и холестерином, подобно желчным липопротеинам, которые образуются в желчи (рис. 2) [44]. В результате в плазме крови пациентов с холестазом при ПБХ уже на ранних стадиях заболевания повышается содержание ФЛ, ОХ, ХС ЛПОНП, ХС ЛПНП, ХС ЛПВП и определяется аномальный ЛП-Х [61]. На поздней, терминальной стадии ПБХ у пациентов с наличием ЛП-Х можно отметить уменьшение концентрации ХС ЛПВП вследствие значительного снижения белково-синтетической функции гепатоцитов.
ЛП-X считают аномальным липопротеином низкой плотности, который присутствует у пациентов с внутри- или внепеченочным холестазом [62]. ЛП-Х содержит желчные кислоты, альбумин, высокую долю неэстерифицированного холестерина и ФЛ [63]. К сожалению, информации о составе желчных кислот в ЛП-Х крайне мало. Считается, что литохолевая кислота является основным представителем желчных кислот в ЛП-Х [10]. В отличие от обычных липопротеинов, которые имеют один слой ФЛ, окружающий гидрофобное ядро из эфиров холестерина и триглицеридов, ЛП-Х имеет везикулярную структуру [63]. Вероятнее всего, такое строение ЛП-Х направлено на выведение избыточного количества холестерина и желчных кислот из общего кровотока пациентов с ПБХ минуя печень, неспособную в полной мере осуществлять желчевыделительную функцию [62]. Показано, что концентрация ЛП-Х в плазме определяется степенью холестаза и дефицитом лецитинхолестеринацилтрансферазы (ЛХАТ, или фосфатидилхолинстерол-O-трансфераза) [62]. Выявление дефицита ЛХАТ у пациентов с ПБХ требует проведения дифференциальной диагностики с первичным дефицитом ЛХАТ. При первичном дефиците ЛХАТ, врожденном дефекте, наличие ЛП-Х сопровождается низкой концентрацией ХС ЛПВП, анемией, помутнением роговицы и нарушениями функции почек.
Содержание сывороточного холестерина при ПБХ увеличивается из-за присутствия ЛП-Х, который имеет плотность, аналогичную плотности ЛПНП. Это делает ХС ЛП-Х неотличимым от ХС ЛПНП при его количественном определении. Из-за этого сходства в плотности ЛП-Х часто ответственен за ложное повышение уровня ХС ЛПНП при ПБХ. Поэтому при повышении ХС ЛПНП у пациентов с ПБХ требуется осторожная интерпретация и при необходимости проведение исследования на определение ЛП-Х [63]. Было показано in vitro и in vivo, что ЛП-Х не ингибирует синтез холестерина de novo в печени [64–66].
Патогенез появления ЛП-Х при холестазе полностью не выяснен, так же как и не определено место его образования. Высказывается предположение о регургитации желчи в результате холестаза в плазменный компартмент. В результате билиарные ЛП, попадая в плазму крови, не содержащую желчные кислоты и содержащую большую, чем в желчи, концентрацию альбумина, перестраиваются с образованием частиц ЛП-Х с везикулярной структурой [63]. Соотношение альбумин/желчные кислоты в плазме крови важно для формирования и поддержания структурной организации ЛП-Х. При этом концентрация ФЛ и неэстерифицированного холестерина, определяемая в липопротеиновых комплексах печеночной порции желчи, аналогична содержанию ФЛ и свободного холестерина в ЛП-Х [67]. Однако при этом во вновь образовавшихся ЛП-Х увеличивается количество альбумина, и происходит «разбавление» в них концентрации желчных кислот со снижением их количества (до < 0,01%) по сравнению с их количеством (~1–3%) в желчных липопротеинах [10]. Вероятно, происходит перераспределение желчных кислот между желчными и сывороточными липопротеинами. В работе S. Heimerl и соавт. сообщается об идентичном до < 0,01% содержании желчных кислот в ЛПНП у пациентов с холестазом и наличием ЛП-Х [10], в то время как у этих же пациентов в ЛПВП желчные кислоты не обнаруживаются. Подтверждением гипотезы об образовании ЛП-Х в результате регургитации желчи в кровь служат данные, полученные E. Manzato и соавт., которые показали, что липопротеин желчи может быть преобразован в «ЛП-Х-подобный» материал in vitro путем добавления альбумина или сыворотки к нативной желчи [67]. «ЛП-Х-подобный» материал, образованный in vitro, имеет физико-химические и химические характеристики, аналогичные или идентичные ЛП-Х, выделенному из сыворотки. И наоборот, инкубация in vitro ЛП-Х с желчными кислотами может преобразовывать их в частицы, подобные билиарным липопротеинам [67]. В связи с этим считается, что ЛП-Х представляет собой комбинацию желчного липопротеина и альбумина.
По-видимому, желчные кислоты при ПБХ могут попадать в общий кровоток не только с билиарными липопротеинами. Но их поступление и наличие в крови обязательно будут инициировать образование комплексов с ФЛ и холестерином вследствие мощных детергентных свойств желчных кислот. Образование таких комплексов требует дополнительного синтеза как ФЛ, так и свободного (неэстерифицированного) холестерина. Для синтеза ФЛ необходимы жирные кислоты [9, 10]. Синтез пальмитиновой и олеиновой жирных кислот увеличивается при ПБХ. Эти жирные кислоты являются основными компонентами ФЛ (лецитинов, фосфатидилхолинов) желчи [9, 10]. Также отмечено значительное увеличение уровня ФЛ в плазме крови этих пациентов по сравнению с контролем [68].
ФЛ и неэстерифицированный холестерин в достаточном количестве содержатся в ЛПНП, которые синтезируются в печени и являются основными структурами, транспортирующими холестерин из печени к периферическим тканям и органам. Это позволяет желчным кислотам, попавшим в общий кровоток в результате холестаза, инициировать солюбилизацию ФЛ и холестерина из ЛПНП с образованием мицеллярно-ламеллярных комплексов с вовлечением в этот процесс альбумина плазмы [8]. Жирные кислоты, попавшие в общий кровоток, могут сами встраиваться в ЛПНП. Оба этих механизма могут быть ответственными за повышенный синтез ЛПНП в печени и увеличение их содержания в плазме крови у пациентов с ПБХ. Липидный состав ЛПНП в образцах плазмы крови от пациентов с синдромом холестаза очень похож на липидный состав ЛП-Х с заметным увеличением мононенасыщенных молекул фосфатидилхолина (ФХ 32:1, ФХ 34:1), фосфатидилэтаноламина (ФЭ 32:1, ФЭ 34:1), свободного холестерина, при одновременном снижении уровня эcтерифицированного холестерина. Повышенный синтез жирных кислот и ФЛ в печени играет важную роль в образовании ЛП-Х при холестазе [9, 10].
Выявляемое повышение ОХ у пациентов с ПБХ, прежде всего, пытаются рассматривать с точки зрения его влияния на развитие атеросклероза и патологических изменений со стороны сердечно-сосудистых заболеваний у этих больных [56, 69, 70]. Однако повышение уровня холестерина у пациентов с ПБХ так же, как увеличение содержания ФЛ, направлено на нейтрализацию детергентного действия желчных кислот, попавших в общий кровоток по мере нарастания холестаза. В этом плане гиперхолестеринемия при ПБХ является «аномальной» и представляет собой компенсаторный ответ организма на появление желчных кислот в общем кровотоке [39, 55, 62, 69, 70]. Поэтому у пациентов с ПБХ, несмотря на увеличение ОХ в плазме крови, выявляется низкая степень стеатоза печени, повышенный уровень ЛПВП, появление в плазме крови аномального ЛП-Х и низкий риск развития атеросклероза и сердечно-сосудистых событий [39, 55]. У пациентов с ПБХ и высоким уровнем ЛП-Х не происходит возрастания частоты сердечно-сосудистых событий [62]. Исследования
Y. Zhang и соавт. показывают, что у пациентов с ПБХ степень стеатоза печени не только самая низкая среди пациентов с хроническими заболеваниями печени, но и ниже, чем у здоровых людей [39].
Наличие ЛП-Х при заболеваниях печени имеет важное клиническое значение, поскольку его обнаружение считается наиболее чувствительным и специфичным биохимическим маркером холестаза [71]. Положительный тест на ЛП-Х показывает более чем 95% соответствие с гистологическими методами, используемыми для подтверждения синдрома холестаза [72]. Однако в связи со сложностью методики определения ЛП-Х и высокой информативностью биохимических маркеров – ЩФ и γ-ГТ – тест на определение ЛП-Х для диагностики состояния холестаза используется редко.
Изменение активности АСТ, АЛТ при ПБХ
На начальной стадии ПБХ основной мишенью для запуска патологического процесса являются холангиоциты. Однако по мере развития дуктулопении и внутрипеченочного холестаза в патологический процесс вовлекаются и гепатоциты, что приводит к постепенному их повреждению [73]. Поэтому в период формирования ПБХ активность аланиновой (АЛТ) и аспарагиновой (АСТ) аминотрансфераз сыворотки крови, в отличие от γ-ГТ, ЩФ и 5'-НК, длительное время не превышает верхней границы нормы или увеличивается незначительно. АЛТ присутствует только в цитоплазме гепатоцитов, тогда как АСТ присутствует как в цитоплазме гепатоцитов, так и в митохондриях [74]. Обе аминотрансферазы относятся к классу ферментов трансаминаз и играют важную роль в метаболизме аминокислот. Они участвуют в ферментативном переаминировании по следующей суммарной реакции: аминокислота1 + кетокислота2 аминотрансфераза аминокислота2 + кетокислота1.
У здорового человека эти ферменты обычно высвобождаются из клеток с постоянной скоростью в результате запрограммированной гибели (апоптоз) клеток печени с последующим выбросом ферментов в плазму и их клиренсом из организма [74]. Имеется равновесие между нормальным жизненным циклом гепатоцитов и выведением ферментов из плазмы [74]. Повышенный выброс АЛТ и АСТ из клеток печени в кровоток связан обычно либо с повреждением цитоплазматической мембраны, либо с повышенной гибелью гепатоцитов. Сохранение в пределах нормы уровня АСТ и АЛТ на ранних стадиях ПБХ свидетельствует о целостности цитоплазматической мембраны гепатоцитов и нормальной скорости апоптоза печеночных клеток [6, 74]. Отмечено, что уровень активности АЛТ при ПБХ выше у мужчин, чем у женщин [75, 76]. Предполагается, что это связано с биологическими и физиологическими особенностями, с уровнем мужских и женских половых гормонов [75, 77, 78].
В клинической практике соотношение АСТ:АЛТ (соотношение, коэффициент Де Ритиса) часто используют для дифференциальной диагностики. В сочетании с холестатической картиной соотношение АСТ:АЛТ < 1,5 указывает на внепеченочную обструкцию [79]. При холестатических заболеваниях печени коэффициент Де Ритиса имеет значение на поздних (цирроз) стадиях развития заболевания или для мониторинга эффективности лечения пациентов с ПБХ препаратами урсодезоксихолевой кислоты. Повышенное отношение АСТ:АЛТ коррелирует со стадией заболевания и превосходит другие негистологические показатели для оценки наличия цирроза при ПБХ, но при этом имеет низкую чувствительность и специфичность – 65–79% [80]. Развитие фиброза на ранних стадиях заболевания ограничено областью мелких и средних желчных протоков. При этом клетки печени довольно долго остаются интактными. Поэтому на ранних стадиях заболевания, до развития выраженных клинических признаков дуктулопении и холестаза, АЛТ и АСТ не могут быть использованы в качестве косвенных маркеров ранних стадий фиброза печени. На стадии развившегося выраженного холестаза желчные кислоты, накапливаясь в гепатоцитах, запускают процессы повреждения их цитоплазматических мембран, апоптоза и фиброза. В этот период АЛТ и АСТ могут быть использованы для оценки всех этих повреждений. Для прогнозирования развития фиброза предлагается использовать индекс АСТ к тромбоцитам (APRI – AST to platelet ratio index) [81]. Было показано, что параметр APRI имеет определенное значение для диагностики фиброза у пациентов с ПБХ [82].
Несмотря на то что цитоплазматические мембраны гепатоцитов при ПБХ довольно длительное время сохраняют свою целостность, данные электронно-микроскопических исследований показывают нарушение межклеточных контактов [83, 84]. Важной структурой, обеспечивающей механическое сцепление клеток и поддерживающих барьерные свойства тканей, являются плотные контактные комплексы (tight junction). Зона плотного контакта между гепатоцитами ограничивает просвет желчного капилляра, не позволяя желчи проникать в межгепатоцитарное пространство и в область синусоида. Нарушение целостности плотного соединения между печеночными клетками приводит к попаданию компонентов желчи в общий кровоток.
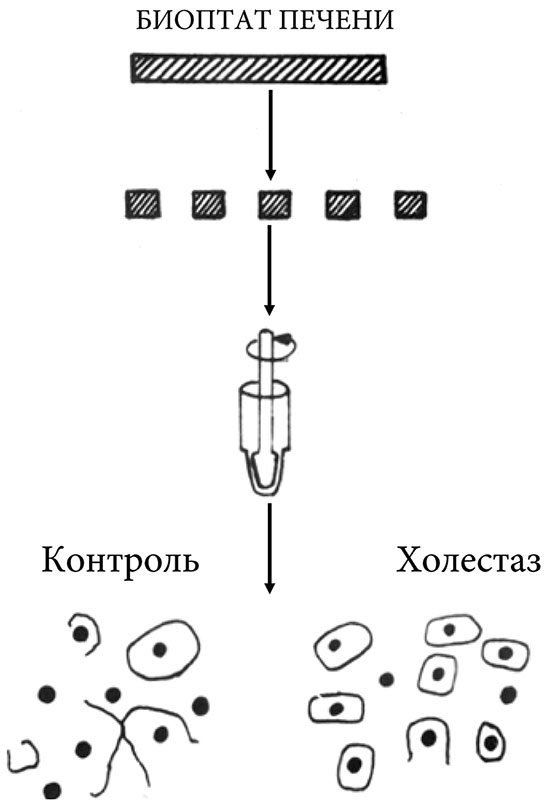
На модели экспериментально индуцированного холестаза у животных обнаружены выраженные изменения в области плотного соединения, приводящие к уменьшению площади контактирующих поверхностей гепатоцитов в два раза [85]. Снижение взаимодействия между клетками печени при ПБХ может определять более высокие значения соотношения АСТ:АЛТ. При ПБХ выявляется снижение межгепатоцитарных взаимодействий в месте плотного соединения, что связано с повышением давления в желчных капиллярах вследствие холестаза [86]. У пациентов с ПБХ отмечается нарушение межгепатоцитарного взаимодействия, выявленное при электронно-микроскопическом исследовании и адгезиометрическом методе [86]. У пациентов с ПБХ и явной клинической картиной холестаза отмечено нарушение межгепатоцитарного взаимодействия [86]. Ослабление межклеточных контактов приводит к тому, что при механическом диспергировании гепатобиоптата ткани печени гепатоциты отходят друг от друга без разрыва цитоплазматической мембраны (рис. 3). В результате происходит выделение большого числа одиночных клеток и значительно меньше определяется под микроскопом одиночных ядер, образовавшихся в результате механического разрыва мембран вследствие сцепления двух гепатоцитов плотными контактными комплексами (рис. 3).
Использование адгезиометрического метода позволяет количественно определить коэффициент разобщенности (Кр) гепатоцитов при холестазе. Кр представляет собой отношение числа одиночных клеток к сумме одиночных клеток и клеточных ядер, выделенных после диспергирования ткани печени: Кр = Nкл/(Nкл+ Nя).
Чем выше значение Кр, тем сильнее ослаблены межгепатоцитарные контакты. Исследование гепатобиоптатов показало статистически достоверное увеличение (в 4–6 раз) значения Кр у пациентов с ПБХ по сравнению с другими группами больных с хроническими заболеваниями печени без холестаза [86]. Увеличение значения Кр при ПБХ связано с ослаблением межклеточных контактов вследствие повышения давления в желчных капиллярах, связанного с холестазом. Из-за сдавления клеток отмечалось уменьшение в 1,5–2,0 раза размеров гепатоцитов у пациентов с ПБХ.
Гипербилирубинемия
В связи с вовлечением в патологический процесс гепатоцитов после повреждения холангиоцитов и развития дуктулопении гипербилирубинемия не является характерным признаком начальных стадий ПБХ [87]. В асимптоматической стадии заболевания общий билирубин обычно находится в пределах нормы [3, 79]. Содержание общего билирубина в крови пациентов с ПБХ постепенно возрастает на стадии клинически выраженного холестаза и редко достигает высоких цифр [51]. Повышение уровня билирубина в сыворотке крови у пациентов с ПБХ происходит в основном за счет конъюгированной фракции [88]. Эти данные свидетельствуют о том, что глюкуронирование билирубина в гепатоцитах остается сохранным, а процесс его экскреции нарушается [5].
Причиной развития конъюгированной гипербилирубинемии при ПБХ является затруднение оттока желчи по желчным протокам, связанного с дуктулопенией, или изменение активности белков-транспортеров билирубина на апикальной мембране гепатоцитов [88]. Поскольку при ПБХ нарушается целостность межгепатоцитарного взаимодействия и развивается дуктулопения, конъюгированная гипербилирубинемия ассоциируется с затруднением оттока желчи [88]. Последнее связано с дуктулопенией мелких и средних желчных протоков [87]. Возникающий застой желчи приводит к повышению давления в желчных капиллярах. Происходят ослабление межклеточных взаимодействий и рефлюкс компонентов желчи, в том числе конъюгированного билирубина, через плотные контактные комплексы в синусоидальное пространство, а затем в общий кровоток.
Повышенное содержание конъюгированного билирубина в желчном капилляре приводит к снижению активности белков-транспортеров билирубина на апикальной мембране гепатоцитов. В результате конъюгированный билирубин накапливается в клетках печени, что было отмечено в биоптатах печени у пациентов с ПБХ [89].
Гипербилирубинемия на поздних стадиях ПБХ может быть дополнительно обусловлена усиленным гемолизом эритроцитов. Поступление в кровь значительного количества желчных кислот вызывает гемолиз эритроцитов вследствие детергентного действия на цитоплазматические мембраны эритроцитов. В результате усиления гемолиза в крови пациентов с ПБХ на поздних стадиях заболевания повышается и неконъюгированная фракция билирубина. Детергентному действию желчных кислот подвергаются и другие клетки крови: лейкоциты, тромбоциты.
Конъюгированный билирубин может также определяться в моче. При этом уробилиноген экскретируется с мочой пропорционально количеству желчи, поступающей в двенадцатиперстную кишку. У пациентов с ПБХ отмечаются выраженные индивидуальные колебания показателей билирубина. Однако в целом его содержание соответствует стадии заболевания и степени активности патологического процесса [55]. Показано, что билирубин является важным биомаркером для оценки тяжести заболевания [55]. В связи с этим эксперты Американской ассоциации по изучению заболеваний печени (AASLD) считают, что уровень билирубина в сыворотке крови больных ПБХ является наиболее важным фактором для прогнозирования развития заболевания [90]. Чтобы выделить пациентов с высоким риском развития осложнений, у пациентов с ПБХ используется несколько неинвазивных прогностических шкал, таких как классификация по шкале Чайлда – Пью, модель терминальной стадии заболевания печени (MELD), оценка риска по Мейо (Mayo risk score) и модель Newcastle [91–94]. Все эти шкалы включают уровень билирубина в сыворотке крови [95].
Билирубин является потенциальным ранним маркером печеночной дисфункции [55, 96]. В сочетании с низким содержанием альбумина и низким содержанием тромбоцитов повышенный уровень билирубина в сыворотке крови может быть предвестником прогрессирования заболевания, развития цирроза и портальной гипертензии [55]. Разработан и предложен новый простой метод оценки функции печени, который рассчитывается с использованием только значений сывороточного альбумина и билирубина, – альбумин-билирубиновый показатель (ALBI) [95, 97–99]. Преимущество ALBI заключается в том, что его можно просто рассчитать с помощью объективных анализов крови без субъективных факторов или инвазивных процедур. Альбумин-билирубиновый показатель и шкала Мейо обладают самой высокой прогностической ценностью при прогнозировании исходов заболевания, определении стратегии лечения и сроков проведения трансплантации печени у пациентов с ПБХ [95, 97].
Прогрессирующее повышение уровня билирубина должно вызывать беспокойство, так как является плохим прогностическим признаком и используется как один из важных признаков при определении сроков проведения трансплантации печени у пациента с ПБХ [3, 97]. Считается, что любое повышение уровня билирубина у пациентов с ПБХ является важным неблагоприятным прогностическим фактором с необходимостью оптимизации ведения этих пациентов [97, 100, 101].
Изменение метаболизма меди при ПБХ
При осмотре больных с ПБХ обращают на себя внимание потеря эластичности кожи, ее сухость, а также гиперпигментация. Последняя связана с нарушением обмена меди. Поступающая с пищей медь метаболизируется в печени с образованием комплекса с церулоплазмином [102]. В результате образования комплекса «медь – церулоплазмин» снижаются токсические свойства меди, повышается ее растворимость в водной среде (кровь, желчь), облегчается ее транспорт и выведение из организма. Большая часть меди (около 80%) выводится из организма с желчью. Содержание меди в клетках печени у больных ПБХ значительно возрастает и может достигать 25 мг/100 г сухой ткани печени (в норме до 6 мг/100 г) [103, 104]. Это связано с рефлюксом компонентов желчи в общий кровоток в связи с развитием холестаза, что сопровождается повышением уровня меди в плазме крови. Наряду с этим в связи с внутрипеченочным холестазом происходит снижение скорости экскреции меди через апикальную мембрану гепатоцита в желчный капилляр и ее накопление в печеночных клетках [105]. Накопление меди в гепатоцитах снижает ее всасывание из венозной крови воротной вены, что также приводит к повышению уровня меди в плазме крови. При ПБХ повышенное поступление меди в плазму крови приводит к активации медьсодержащего фермента тирозиназы, что сопровождается повышенным синтезом меланина. Наряду с этим организм пытается вывести избыток меди не только через почки, но и через кожу. Это приводит к отложению меди в эпидермисе, что придает гиперпигментированной коже бронзовый оттенок. Считается, что накопление меди в гепатоцитах у пациентов с ПБХ не оказывает клинически значимого токсического действия на клетки печени, так как выработка церулоплазмина и его связывание с медью не нарушены [105]. Поэтому типичных гистологических признаков токсического действия меди на клетки печени у больных ПБХ не наблюдается. Так как при ПБХ медь попадает в плазму крови в связанном с церулоплазмином виде, то и кольцо Кайзера – Флейшера у этих пациентов отсутствует. Уровень церулоплазмина в плазме крови сохраняется в пределах нормы [102]. Все это свидетельствует о сохранении процессов синтеза церулоплазмина и его конъюгации с медью в гепатоцитах, а также о нарушении выведения комплекса «медь – церулоплазмин» с желчью в результате холестаза. Но происходящее при ПБХ повышение концентрации меди в клетках печени коррелирует со стадией холестаза [105, 106].
Снижение белково-синтетической функции печени. Асцит
Содержание альбуминов и глобулинов в крови больных ПБХ на ранних стадиях заболевания длительное время остается в пределах физиологической нормы [5, 107]. Вместе с тем в сыворотке крови пациентов уже в асимптоматической стадии обнаруживают АМА в диагностическом титре 1:40 и выше. По мере прогрессирования заболевания наблюдается повышение уровня γ-глобулинов, прежде всего класса IgM [5, 108]. Развитие печеночно-клеточной недостаточности в далеко зашедшей стадии ПБХ приводит к снижению различных функций печени, в первую очередь белково-синтетической. Биохимическими признаками этих нарушений в терминальной стадии заболевания являются гипоальбуминемия, уменьшение содержания в сыворотке крови уровня протромбина, фибриногена, V и VII факторов свертывания крови, снижение концентрации холестерина, гипербилирубинемия. Альбумин – основной синтезируемый в печеночных клетках белок плазмы крови. Главная роль альбумина – участие в поддержании коллоидно-осмотического (онкотического) давления плазмы и объема циркулирующей крови, а также транспорт и депонирование различных веществ. Появление гипоальбуминемии при ПБХ указывает на нарушение белково-синтетической функции гепатоцитов [52]. Исследование содержания щелочной фракции альбумина относится к тестам высокой чувствительности для определения печеночно-клеточной недостаточности [109]. В норме щелочная фракция составляет около 3% от всего альбумина. Период полувыведения щелочной фракции более длительный, чем у других фракций альбумина, в связи с чем при нарушении его синтеза в печени процентное содержание щелочной фракции возрастает (при терминальном циррозе печени – до 50%), в то время как при потерях альбумина с мочой и калом без повреждения печени удельный вес щелочной фракции не изменяется [109].
Развитию гипоальбуминемии при ПБХ способствует гибель значительного (50% и более) количества гепатоцитов, а также нарастание процессов катаболизма вследствие увеличения скорости метаболизма в состоянии покоя и общего термогенеза [9, 52]. Создается метаболическая ситуация перераспределения ресурсов, которая усиливается по мере нарастания холестаза и развития печеночно-клеточной недостаточности. Наряду с уменьшением синтеза белков сыворотки крови в печени пациентов с ПБХ снижается скорость превращения аммиака в мочевину. Все это приводит к резкому снижению уровня циркулирующих в плазме альбуминов и увеличению экскреции азота с мочой [72, 110].
Постепенно в период декомпенсации у пациентов с ПБХ развивается белково-энергетическая недостаточность (БЭН) питания. Клинические проявления нарушения трофологического статуса у пациентов с ПБХ в терминальной стадии заболевания приобретают промежуточную форму БЭН – маразм-квашиоркор [59]. Развитию БЭН способствует снижение всасывания белков в кишечнике. Портальная гипертензия, приводящая к циркуляторной гипоксии слизистой оболочки кишечника и увеличению ее проницаемости, также вызывает повышенную потерю белков. При этом необходимы сокращение приема соли, жидкости и, если отсутствуют признаки печеночной энцефалопатии, включение в рацион питания продуктов с повышенным содержанием белка [59]. Нутритивная поддержка в этот период должна включать белковые модули с преимущественным содержанием аминокислот с разветвленной цепью и минимальным количеством ароматических аминокислот, а также с разным количеством и соотношением заменимых и незаменимых аминокислот [59, 111]. Следует избегать длительного голодания. Для предотвращения катаболизма белка и поддержания баланса азота рекомендуют прием пищи, содержащей 50 г углеводов, перед сном. Заметное улучшение статуса питания пациентов с ПБХ на стадии развития цирроза и резистентного асцита отмечено после успешного лечения последнего, что подчеркивает важность применения нутритивной поддержки у этих пациентов.
Снижение белково-синтетической функции печеночных клеток и усиление процессов катаболизма белков приводят к развитию недостаточности висцерального пула белков с последующим развитием отеков и асцита. Определенная роль в развитии асцита принадлежит портальной гипертензии. При портальной гипертензии повышается фильтрационное давление в капиллярах, вследствие чего жидкость пропотевает в брюшную полость. Появление асцита у больных ПБХ на стадии развития цирроза сопровождается выраженной задержкой и накоплением натрия в межклеточном пространстве. Происходит постепенное увеличение объема живота, может появиться одышка. Причиной увеличения живота является не только скопление асцитической жидкости, но и раздутые газом петли кишечника в результате дисбиоза. Наиболее ранним симптомом асцита является притупление перкуторного звука в боковых отделах живота. Развитие асцита при ПБХ свидетельствует о печеночно-клеточной недостаточности с портальной гипертензией.
Изменения свертывающей системы крови при ПБХ
Прокоагуляционные, антикоагуляционные и фибринолитические лабораторные показатели на ранних стадиях ПБХ остаются в пределах нормальных значений или изменения могут быть минимальными и практически не выявляться [112–114]. В процессе развития заболевания, как правило, сохраняется равновесие между свертывающей и противосвертывающей системами, но часто со сниженным балансом [112]. Сохраняющаяся в это время белково-синтетическая функция гепатоцитов позволяет поддерживать в равновесии прокоагуляционную и антикоагулянтную систему свертывания крови [115]. В терминальной стадии печеночно-клеточной недостаточности баланс легко нарушается в ту либо другую сторону, что может приводить как к кровотечениям, так и к развитию тромбозов [116, 117]. У части пациентов с ПБХ снижение белково-синтетической функции гепатоцитов приводит к уменьшению синтеза факторов свертывания (факторов II, VII, IX, X, XIII) в меньшей степени, чем естественных антикоагулянтов (протеина S, C и/или антитромбина III) [114, 118]. Возникает дисбаланс между снижением уровня естественных антикоагулянтов и снижением содержания факторов свертывания крови. Более низкая концентрация витамин-К-зависимых гликопротеинов С и S может способствовать повышенному тромбообразованию.
Гиперкоагуляция определяется у части пациентов с ПБХ с помощью тромбоэластографии (ТЭГ) задолго до стадии развития цирроза [119–121]. То есть активация коагуляции является одним из частых признаков нарушения системы свертывания крови при холестатических заболеваниях печени в период его развития [119, 120, 122, 123]. ТЭГ и тромбоэластометрия (ROTEM) являются общепринятыми тестами коагуляции, которые позволяют оценить суммарные свойства свертывающей системы плазмы крови с учетом клеточных и плазменных ее компонентов [124, 125]. ТЭГ позволяет определять взаимодействие плазменных факторов свертывания, тромбоцитов и эндотелиоцитов сосудов и благодаря этому оценить состояние гипо- и гиперкоагуляции. На основе параметров тромбоэластограммы Z. Ben-Ari и соавт. показали, что у пациентов с ПБХ гиперкоагуляция наблюдается чаще, чем у здоровых добровольцев (p = 0,005) [119]. До настоящего времени остается неясным механизм развития гиперкоагуляции у части пациентов с ПБХ.
Гемостатическая система находится в тонком равновесии между протромботическими и антитромботическими процессами, направленными на предотвращение спонтанного тромбообразования [116, 117]. При инициации свертывания крови необходимо высокоспецифичное взаимодействие между факторами свертывания и компонентами клеточной мембраны тромбоцитов и эндотелиоцитов сосудов [126–131].
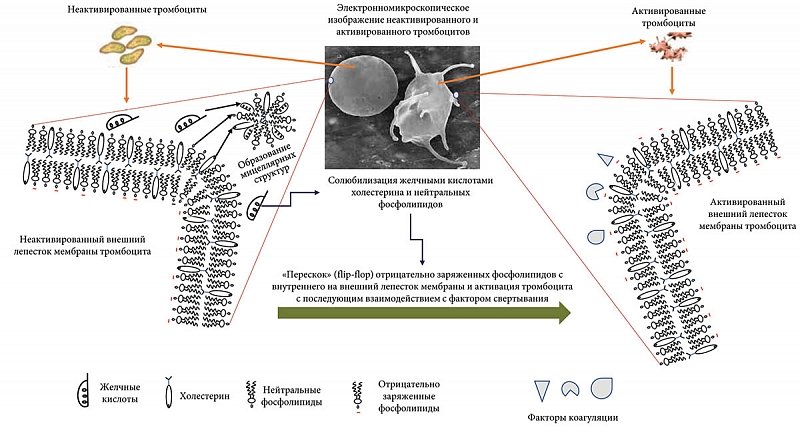
Гемостаз существенно зависит от ФЛ клеточных мембран, взаимодействующих с факторами свертывания крови [132–134]. В норме цитоплазматическая мембрана тромбоцитов и эндотелиальных клеток, выстилающих сосуды, имеет поперечную асимметрию: в наружном лепестке цитоплазматической мембраны содержатся преимущественно нейтральные ФЛ (фосфатидилхолин и сфингомиелин), а во внутреннем – отрицательно заряженные ФЭ и ФС [132–134]. То есть в норме внешняя поверхность плазматических мембран клеток не заряжена [135].
Наибольшей коагуляционной активностью обладают отрицательно заряженные ФЛ – ФС и ФЭ, которые в больших концентрациях присутствуют на внутреннем лепестке цитоплазматической мембраны [134], что затрудняет их контакт с факторами свертывания. Мембранные ферменты: флоппаза, скрамблаза и аминофосфолипидная транслоказа (флиппаза) – регулируют движение ФЛ и поддерживают эту динамическую асимметрию в устойчивом состоянии. При повреждении клеточной мембраны ФС и ФЭ становятся доступными для взаимодействия с факторами свертываемости, инициируя процесс коагуляции.
Инициаторами развития гиперкоагуляции уже на ранних стадиях ПБХ могут выступать желчные кислоты, попадающие в общий кровоток в результате внутрипеченочного холестаза. Попадая в общий кровоток, желчные кислоты способны влиять на фосфолипидный состав цитоплазматических мембран форменных элементов крови, в частности тромбоцитов и эндотелия сосудов, нарушая поперечную асимметрию мембран. Желчные кислоты, являясь сильными детергентами, могут захватывать нейтральные ФЛ с внешнего лепестка цитоплазматической мембраны форменных элементов крови, прежде всего тромбоцитов и эндотелиоцитов (рис. 4). При этом отрицательно заряженные ФЛ могут «перескакивать» по механизму flip-flop с внутреннего лепестка цитоплазматической мембраны на внешний.
При нарушении поперечной асимметрии мембран тромбоцитов и эндотелиальных клеток сосудов на их поверхности формируются отрицательно заряженные (тромбогенные) участки и экспонируется апопротеин III тканевого фактора (ТФ). Появление отрицательно заряженных ФЛ и ТФ на внешнем лепестке цитоплазматической мембраны приводит к активации тромбоцитов, а эндотелиоциты становятся доступными для плазменных факторов свертывания крови. ТФ является основным физиологическим инициатором коагуляции in vivo вследствие его взаимодействия с протеазой фактора свертывания VII/VIIa и экспрессируется большинством клеточных компонентов сосудистой стенки [136]. На поверхности цитоплазматической мембраны может формироваться первый ферментный комплекс прокоагулянтного пути свертывания крови, состоящий из фактора VII свертывания, ТФ и катионов кальция Са2+ (VII-ТФ-Са2+). Все это может способствовать адгезии активированных тромбоцитов на сосудистой стенке и запускать процесс тромбообразования (рис. 4).
Кроме того, в плазме крови пациентов, находящихся в предтромботическом состоянии, зарегистрировано повышенное содержание микрочастиц (МЧ), которые на своей поверхности содержат активированные ТФ+ и ФС+ [137, 138]. МЧ являются внеклеточными везикулами и представляют собой субмикронные, неповрежденные мембранные везикулы, которые высвобождаются из клеток после их активации или во время апоптоза [139]. Микрочастицы – это везикулы клеточного происхождения размером 0,1–1 мкм, которые не имеют ядра, не обладают синтетической способностью, но могут содержать белки цитоскелета, а на внешнем лепестке мембраны – некоторое количество ФС [140]. МЧ присутствуют в кровотоке как у здоровых людей, так и при различных заболеваниях [141]. Нормальные базальные уровни МЧ отражают строго контролируемый баланс между выживанием, пролиферацией и гибелью клеток [141]. МЧ участвуют в различных жизненно важных биологических функциях, таких как гемостаз, коагуляция, воспаление и ангиогенез [142]. МЧ считаются циркулирующим в крови хранилищем биоактивных молекул, которые при правильной интерпретации могут свидетельствовать о состоянии организма и быть использованы в качестве биомаркеров различных патологических состояний [135–141]. МЧ могут образовываться в результате формирования пузырьков (блеббинга) на мембранах форменных элементов крови и эндотелиоцитов с последующим отрывом пузырька, образованием и высвобождением МЧ [137, 138]. МЧ могут высвобождаться из инактивированных тромбоцитов [143]. В этом смысле МЧ могут действовать как сигнальные молекулы для системы гемостаза благодаря экспрессии различных белков и липидов на их поверхности, таких как ТФ и ФС [144]. Известно, что ряд веществ, таких как эндотоксины или цитокины, могут выступать в качестве специфических стимуляторов индукции образования МЧ [145]. Можно предположить, что у пациентов с ПБХ индукторами образования МЧ, обогащенных ТФ и отрицательно заряженным ФС, служат желчные кислоты, попавшие в системный кровоток в результате холестаза. При этом потеря асимметрии ФЛ с экспозицией ФС на внешний лепесток клеточной мембраны представляется важным для формирования таких МЧ с наличием на их поверхности одновременно активированных ТФ+ и ФС+ [137, 146]. Блеббинг, являющийся интегральным показателем влияния индуцирующих и повреждающих факторов на мембраны клеток, а также потеря асимметрии ФЛ в цитоплазматических мембранах форменных элементов крови, в частности тромбоцитов, могут приводить к образованию и высвобождению МЧ [137]. Показано, что полученные из тромбоцитов МЧ обладают в 50–100 раз большей прокоагулянтной активностью, чем сами активированные тромбоциты [147]. Как экспозицией ФС, так и наличием ТФ в мембранах МЧ можно объяснить их прокоагулянтные свойства: ФС действует как каталитическая поверхность для сборки ферментативных комплексов коагуляции, а ТФ считается основным инициатором каскада коагуляции во внешнем пути [137, 148]. Клеточные МЧ и их предполагаемые функции в отношении коагуляции, ангиогенеза, воспаления и сосудистой функции изложены в обзоре H. El‐Gamal и соавт. [141]. Однако на сегодняшний день отсутствуют данные об участии МЧ в дисбалансе системы гемостаза при развитии гиперкоагуляции у пациентов с холестатическими заболеваниями печени. Изучение МЧ при холестатических заболеваниях печени может помочь понять механизм развития склонности к тромбообразованию у этих пациентов.
Заключение
Развивающееся нарушение процессов желчеотделения и энтерогепатической циркуляции желчных кислот у пациентов с ПБХ уже на ранних стадиях приводит к поступлению желчных кислот в общий кровоток, что по мере нарастания холестаза сопровождается изменением биохимических показателей: повышается количество желчных кислот, холестерина, ФЛ, жирных кислот, ХС ЛПНП и ХС ЛПВП, билирубина, меди, γ-глобулинов; появляется ЛП-Х; снижается уровень общего белка в основном вследствие альбуминовой фракции; повышается активность ферментов ЩФ, γ-ГТ, ЛАП и 5'-НК. Поступление желчных кислот в плазму крови больных ПБХ является одной из причин развития зуда уже в бессимптомной стадии заболевания и нарушений липидного обмена. В крови больных ПБХ повышается уровень ОХ, ФЛ, жирных кислот, ХС ЛПНП, ХС ЛПВП, появляется аномальный ЛП-Х. Последний представляет собой липопротеиновую частицу, в которой помимо ФЛ и холестерина присутствуют желчные кислоты, попавшие в общий кровоток в результате холестаза. ЛП-Х выполняет транспортную функцию в плазме крови подобно другим липопротеинам крови. Синтез ЛП-Х у больных ПБХ является важным механизмом инактивации желчных кислот в плазме крови. Содержание ЛП-Х хорошо коррелирует с уровнем желчных кислот в плазме крови, но не с уровнем ОХ. Образование ЛП-Х при ПБХ можно рассматривать как защитный механизм в ответ на развивающийся холестаз. Гиперхолестеролемия при ПБХ не сопровождается увеличением частоты сердечно-сосудистых событий и поэтому называется «аномальной».
Ортофосфат, необходимый для повышенного синтеза ФЛ, образуется в результате увеличения активности ферментов ЩФ и 5'-НК. Повышение активности этих ферментов происходит вследствие увеличения их синтеза в печени и желчных протоках, что обуславливает более активную доставку аминокислот в гепатоциты и эндотелиальные клетки желчных капилляров. В связи с этим уже в бессимптомной стадии ПБХ наблюдается повышение активности γ-ГТ. Основное клиническое значение измерения уровня ЩФ, 5'-НК и γ-ГТ в сыворотке крови заключается в ранней диагностике ПБХ. Активность ЛАП при наличии повышенной активности ЩФ определяется с целью проведения дифференциальной диагностики заболеваний гепатобилиарной системы и заболеваний костной ткани.
Развивающееся нарушение процессов желчеотделения уже на ранних стадиях заболевания вызывает повышение давления в желчных капиллярах, которое увеличивается по мере нарастания внутрипеченочного холестаза, что сопровождается нарушением межклеточных контактов при сохраняющейся целостности цитоплазматических мембран гепатоцитов. На поздних стадиях заболевания накопление желчных кислот в гепатоцитах сопровождается нарушением целостности и проницаемости их цитоплазматических мембран, что проявляется повышением активности АЛТ и АСТ, а также нарушением обмена меди и билирубина. Последнее проявляется гиперпигментацией кожи и конъюгированной гипербилирубинемией с желтухой. При ПБХ развитие стадии декомпенсированного цирроза может приводить к повышению уровня и неконъюгированного билирубина. Развитие цирроза печени на поздней стадии ПБХ сопровождается нарушением белково-синтетической функции клеток печени. Это приводит к развитию дефицита висцерального пула белков, а изменение онкотического давления – к развитию отеков и асцита. Белково-синтетическая дисфункция клеток печени способствует снижению синтеза белков свертывающей и антисвертывающей систем крови. Смещение баланса между этими системами может сопровождаться как гипо-, так и гиперкоагуляцией. Дисбаланс, связанный с более низкой концентрацией витамин-К-зависимых гликопротеинов С и S по сравнению со сниженным уровнем факторов свертывания крови, может способствовать увеличению тромбообразования. Выраженность гипербилирубинемии и гипоальбуминемии позволяет оценить гепатоцеллюлярную недостаточность, что используется в прогностических шкалах.
Авторы заявляют об отсутствии конфликта интересов.
I.V. Maev, PhD, Prof., Academician of the RAS, V.I. Reshetnyak, PhD, Prof.
Russian University of Medicine, Moscow
Contact person: Vasiliy I. Reshetnyak, vasiliy.reshetnyak@yandex.ru
Primary biliary cholangitis (PBC), previously known as primary biliary cirrhosis until 2015, is a chronic cholestatic progressive liver disease related to cholangiopathies. Damage to cholangiocytes triggers the development of intrahepatic cholestasis, progression of which leads to liver cirrhosis in the terminal stage of the disease. The developing disturbance of biliary excretion and enterohepatic circulation of bile acids in patients with PBC already at early stages leads to the appearance of laboratory signs of the disease. Understanding of pathophysiological mechanisms of development of these signs is important both for diagnostics and treatment of patients with PBC. Diagnosis of early stages of the disease contributes to its more effective treatment. New scientific data on the problem of PBC have been accumulated. The purpose of this review is to summarise the available literature and authors' own data concerning the mechanisms of development of biochemical criteria of PBC and their diagnostic value. Thanks to the advances
in biochemistry, molecular biology and genetics, it has become possible to present these data taking into account the pathophysiological mechanisms of their development.
Уважаемый посетитель uMEDp!
Уведомляем Вас о том, что здесь содержится информация, предназначенная исключительно для специалистов здравоохранения.
Если Вы не являетесь специалистом здравоохранения, администрация не несет ответственности за возможные отрицательные последствия, возникшие в результате самостоятельного использования Вами информации с портала без предварительной консультации с врачом.
Нажимая на кнопку «Войти», Вы подтверждаете, что являетесь врачом или студентом медицинского вуза.