–Я–∞—В–Њ—Д–Є–Ј–Є–Њ–ї–Њ–≥–Є—П –Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є—П –∞–љ—В–Є–Љ–Є—В–Њ—Е–Њ–љ–і—А–Є–∞–ї—М–љ—Л—Е –∞–љ—В–Є—В–µ–ї –њ—А–Є –њ–µ—А–≤–Є—З–љ–Њ–Љ –±–Є–ї–Є–∞—А–љ–Њ–Љ —Е–Њ–ї–∞–љ–≥–Є—В–µ. –Ш–Љ–µ–µ—В—Б—П –ї–Є –і–Є—Б—Д—Г–љ–Ї—Ж–Є—П –Є–Љ–Љ—Г–љ–љ–Њ–є —Б–Є—Б—В–µ–Љ—Л –њ—А–Є –Я–С–•?
- –Р–љ–љ–Њ—В–∞—Ж–Є—П
- –°—В–∞—В—М—П
- –°—Б—Л–ї–Ї–Є
- English
–Ю–±—А–∞–Ј–Њ–≤–∞–љ–Є–µ –Р–Ь–Р –≤ –і–Њ–Ї–ї–Є–љ–Є—З–µ—Б–Ї–Њ–є —Б—В–∞–і–Є–Є –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П –њ–Њ—Б–ї—Г–ґ–Є–ї–Њ –Њ—Б–љ–Њ–≤–∞–љ–Є–µ–Љ –і–ї—П —Г—В–≤–µ—А–ґ–і–µ–љ–Є—П, —З—В–Њ –Њ–љ–Є –Ј–∞–њ—Г—Б–Ї–∞—О—В –њ—А–Њ—Ж–µ—Б—Б –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є—П –Є –≥–Є–±–µ–ї–Є —Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В–Њ–≤, —З—В–Њ –њ–Њ–Ј–≤–Њ–ї—П–µ—В —Б—З–Є—В–∞—В—М –Я–С–• –Є—Б—В–Є–љ–љ—Л–Љ –∞—Г—В–Њ–Є–Љ–Љ—Г–љ–љ—Л–Љ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–µ–Љ. –Ю–і–љ–∞–Ї–Њ –љ–µ —Б—Г—Й–µ—Б—В–≤—Г–µ—В –љ–Є–Ї–∞–Ї–Є—Е –і–Њ–Ї–∞–Ј–∞—В–µ–ї—М—Б—В–≤ –њ–Њ–≤—А–µ–ґ–і–∞—О—Й–µ–≥–Њ –і–µ–є—Б—В–≤–Є—П –Р–Ь–Р –љ–∞ —Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В—Л, –≤—Л—Б—В–Є–ї–∞—О—Й–Є–µ —В–Њ–ї—М–Ї–Њ –Љ–µ–ї–Ї–Є–µ (–≤–љ—Г—В—А–Є–і–Њ–ї—М–Ї–Њ–≤—Л–µ, –Љ–µ–ґ–і–Њ–ї—М–Ї–Њ–≤—Л–µ –Є —Б–µ–њ—В–∞–ї—М–љ—Л–µ) –ґ–µ–ї—З–љ—Л–µ –њ—А–Њ—В–Њ–Ї–Є. –Я–Њ—П–≤–Є–≤—И–Є–µ—Б—П –љ–Њ–≤—Л–µ –љ–∞—Г—З–љ—Л–µ –і–∞–љ–љ—Л–µ –Њ –љ–∞—А—Г—И–µ–љ–Є–Є –≤—Л—А–∞–±–Њ—В–Ї–Є –±–Є–Ї–∞—А–±–Њ–љ–∞—В–∞ —Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В–∞–Љ–Є —Г–ґ–µ –≤ –∞—Б–Є–Љ–њ—В–Њ–Љ–∞—В–Є—З–µ—Б–Ї–Њ–є —Б—В–∞–і–Є–Є –Я–С–• –њ–Њ–Ј–≤–Њ–ї–Є–ї–Є –∞–≤—В–Њ—А–∞–Љ —Н—В–Њ–≥–Њ –Њ–±–Ј–Њ—А–∞ –≤–њ–µ—А–≤—Л–µ –≤—Л—Б–Ї–∞–Ј–∞—В—М —Б–ї–µ–і—Г—О—Й–Є–µ –њ—А–µ–і–њ–Њ–ї–Њ–ґ–µ–љ–Є—П:
- –њ–Њ–≤—А–µ–ґ–і–∞—О—Й–Є–Љ —Д–∞–Ї—В–Њ—А–Њ–Љ –і–ї—П –Љ–µ–ї–Ї–Є—Е —Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В–Њ–≤ —П–≤–ї—П—О—В—Б—П –ґ–µ–ї—З–љ—Л–µ –Ї–Є—Б–ї–Њ—В—Л, –∞ –љ–µ –Р–Ь–Р;
- –ґ–µ–ї—З–љ—Л–µ –Ї–Є—Б–ї–Њ—В—Л –Ј–∞–њ—Г—Б–Ї–∞—О—В –њ—А–Њ—Ж–µ—Б—Б —Б—В–∞—А–µ–љ–Є—П –Є –∞–њ–Њ–њ—В–Њ–Ј–∞ –≤ –Љ–µ–ї–Ї–Є—Е —Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В–∞—Е, —З—В–Њ –њ—А–Є–≤–Њ–і–Є—В –Ї —А–∞–Ј–≤–Є—В–Є—О –і—Г–Ї—В—Г–ї–Њ–њ–µ–љ–Є–Є;
- –ґ–µ–ї—З–љ—Л–µ –Ї–Є—Б–ї–Њ—В—Л —Б–Њ–Ј–і–∞—О—В —Г—Б–ї–Њ–≤–Є—П –і–ї—П –і–Њ—Б—В—Г–њ–∞ –Ї –≤–љ—Г—В—А–µ–љ–љ–µ–є –Љ–µ–Љ–±—А–∞–љ–µ –Љ–Є—В–Њ—Е–Њ–љ–і—А–Є–є –Љ–µ–ї–Ї–Є—Е —Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В–Њ–≤ –Є –Ї E2 –Я–Ф–У вАУ –Ї–Њ–Љ–њ–ї–µ–Ї—Б—Г;
- –љ–∞–ї–Є—З–Є–µ –ї–Є–њ–Њ–µ–≤–Њ–є –Ї–Є—Б–ї–Њ—В—Л –≤ –Х2-—Б—Г–±—К–µ–і–Є–љ–Є—Ж–µ –Я–Ф–У-–Ї–Њ–Љ–њ–ї–µ–Ї—Б–∞ –Є –µ–µ –≤–Ј–∞–Є–Љ–Њ–і–µ–є—Б—В–≤–Є–µ —Б –ґ–µ–ї—З–љ—Л–Љ–Є –Ї–Є—Б–ї–Њ—В–∞–Љ–Є –њ—А–Є–≤–Њ–і–Є—В –Ї –Є–Љ–Љ—Г–љ–Њ–Љ–Њ–і–Є—Д–Є–Ї–∞—Ж–Є–Є –Є –њ—А–Є–Њ–±—А–µ—В–µ–љ–Є—О –љ–µ–Њ–∞–љ—В–Є–≥–µ–љ–љ—Л—Е —Б–≤–Њ–є—Б—В–≤ E2 –Я–Ф–У –≤ –Љ–µ–ї–Ї–Є—Е –С–≠–Ъ;
- –њ—А–Є–Њ–±—А–µ—В–µ–љ–Є–µ –љ–µ–Њ–∞–љ—В–Є–≥–µ–љ–љ—Л—Е —Б–≤–Њ–є—Б—В–≤ E2 –Я–Ф–У вАУ –Ї–Њ–Љ–њ–ї–µ–Ї—Б–Њ–Љ –≤ –Љ–µ–ї–Ї–Є—Е, –њ–Њ–і–≤–µ—А–≥–∞—О—Й–Є—Е—Б—П –∞–њ–Њ–њ—В–Њ–Ј—Г —Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В–∞—Е –њ—А–Є–≤–Њ–і–Є—В –Ї —В–Њ–Љ—Г, —З—В–Њ –љ–Њ—А–Љ–∞–ї—М–љ–∞—П (–Ј–і–Њ—А–Њ–≤–∞—П) –Є–Љ–Љ—Г–љ–љ–∞—П —Б–Є—Б—В–µ–Љ–∞ —А–∞—Б–њ–Њ–Ј–љ–∞–µ—В –µ–≥–Њ –Ї–∞–Ї —З—Г–ґ–µ—А–Њ–і–љ—Л–є –∞–љ—В–Є–≥–µ–љ, –Ј–∞–њ—Г—Б–Ї–∞—П –≤—Л—А–∞–±–Њ—В–Ї—Г –Р–Ь–Р. –Я–Њ—Н—В–Њ–Љ—Г –љ–∞–ї–Є—З–Є–µ –Р–Ь–Р –њ—А–Є –Я–С–• —П–≤–ї—П–µ—В—Б—П –њ—А–Є–Ј–љ–∞–Ї–Њ–Љ –Њ—А–≥–∞–љ–Њ—Б–њ–µ—Ж–Є—Д–Є—З–µ—Б–Ї–Њ–≥–Њ, –∞ –љ–µ –Є—Б—В–Є–љ–љ–Њ–≥–Њ –∞—Г—В–Њ–Є–Љ–Љ—Г–љ–љ–Њ–≥–Њ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П.
–Ю–±—А–∞–Ј–Њ–≤–∞–љ–Є–µ –Р–Ь–Р –≤ –і–Њ–Ї–ї–Є–љ–Є—З–µ—Б–Ї–Њ–є —Б—В–∞–і–Є–Є –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П –њ–Њ—Б–ї—Г–ґ–Є–ї–Њ –Њ—Б–љ–Њ–≤–∞–љ–Є–µ–Љ –і–ї—П —Г—В–≤–µ—А–ґ–і–µ–љ–Є—П, —З—В–Њ –Њ–љ–Є –Ј–∞–њ—Г—Б–Ї–∞—О—В –њ—А–Њ—Ж–µ—Б—Б –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є—П –Є –≥–Є–±–µ–ї–Є —Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В–Њ–≤, —З—В–Њ –њ–Њ–Ј–≤–Њ–ї—П–µ—В —Б—З–Є—В–∞—В—М –Я–С–• –Є—Б—В–Є–љ–љ—Л–Љ –∞—Г—В–Њ–Є–Љ–Љ—Г–љ–љ—Л–Љ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–µ–Љ. –Ю–і–љ–∞–Ї–Њ –љ–µ —Б—Г—Й–µ—Б—В–≤—Г–µ—В –љ–Є–Ї–∞–Ї–Є—Е –і–Њ–Ї–∞–Ј–∞—В–µ–ї—М—Б—В–≤ –њ–Њ–≤—А–µ–ґ–і–∞—О—Й–µ–≥–Њ –і–µ–є—Б—В–≤–Є—П –Р–Ь–Р –љ–∞ —Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В—Л, –≤—Л—Б—В–Є–ї–∞—О—Й–Є–µ —В–Њ–ї—М–Ї–Њ –Љ–µ–ї–Ї–Є–µ (–≤–љ—Г—В—А–Є–і–Њ–ї—М–Ї–Њ–≤—Л–µ, –Љ–µ–ґ–і–Њ–ї—М–Ї–Њ–≤—Л–µ –Є —Б–µ–њ—В–∞–ї—М–љ—Л–µ) –ґ–µ–ї—З–љ—Л–µ –њ—А–Њ—В–Њ–Ї–Є. –Я–Њ—П–≤–Є–≤—И–Є–µ—Б—П –љ–Њ–≤—Л–µ –љ–∞—Г—З–љ—Л–µ –і–∞–љ–љ—Л–µ –Њ –љ–∞—А—Г—И–µ–љ–Є–Є –≤—Л—А–∞–±–Њ—В–Ї–Є –±–Є–Ї–∞—А–±–Њ–љ–∞—В–∞ —Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В–∞–Љ–Є —Г–ґ–µ –≤ –∞—Б–Є–Љ–њ—В–Њ–Љ–∞—В–Є—З–µ—Б–Ї–Њ–є —Б—В–∞–і–Є–Є –Я–С–• –њ–Њ–Ј–≤–Њ–ї–Є–ї–Є –∞–≤—В–Њ—А–∞–Љ —Н—В–Њ–≥–Њ –Њ–±–Ј–Њ—А–∞ –≤–њ–µ—А–≤—Л–µ –≤—Л—Б–Ї–∞–Ј–∞—В—М —Б–ї–µ–і—Г—О—Й–Є–µ –њ—А–µ–і–њ–Њ–ї–Њ–ґ–µ–љ–Є—П:
- –њ–Њ–≤—А–µ–ґ–і–∞—О—Й–Є–Љ —Д–∞–Ї—В–Њ—А–Њ–Љ –і–ї—П –Љ–µ–ї–Ї–Є—Е —Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В–Њ–≤ —П–≤–ї—П—О—В—Б—П –ґ–µ–ї—З–љ—Л–µ –Ї–Є—Б–ї–Њ—В—Л, –∞ –љ–µ –Р–Ь–Р;
- –ґ–µ–ї—З–љ—Л–µ –Ї–Є—Б–ї–Њ—В—Л –Ј–∞–њ—Г—Б–Ї–∞—О—В –њ—А–Њ—Ж–µ—Б—Б —Б—В–∞—А–µ–љ–Є—П –Є –∞–њ–Њ–њ—В–Њ–Ј–∞ –≤ –Љ–µ–ї–Ї–Є—Е —Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В–∞—Е, —З—В–Њ –њ—А–Є–≤–Њ–і–Є—В –Ї —А–∞–Ј–≤–Є—В–Є—О –і—Г–Ї—В—Г–ї–Њ–њ–µ–љ–Є–Є;
- –ґ–µ–ї—З–љ—Л–µ –Ї–Є—Б–ї–Њ—В—Л —Б–Њ–Ј–і–∞—О—В —Г—Б–ї–Њ–≤–Є—П –і–ї—П –і–Њ—Б—В—Г–њ–∞ –Ї –≤–љ—Г—В—А–µ–љ–љ–µ–є –Љ–µ–Љ–±—А–∞–љ–µ –Љ–Є—В–Њ—Е–Њ–љ–і—А–Є–є –Љ–µ–ї–Ї–Є—Е —Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В–Њ–≤ –Є –Ї E2 –Я–Ф–У вАУ –Ї–Њ–Љ–њ–ї–µ–Ї—Б—Г;
- –љ–∞–ї–Є—З–Є–µ –ї–Є–њ–Њ–µ–≤–Њ–є –Ї–Є—Б–ї–Њ—В—Л –≤ –Х2-—Б—Г–±—К–µ–і–Є–љ–Є—Ж–µ –Я–Ф–У-–Ї–Њ–Љ–њ–ї–µ–Ї—Б–∞ –Є –µ–µ –≤–Ј–∞–Є–Љ–Њ–і–µ–є—Б—В–≤–Є–µ —Б –ґ–µ–ї—З–љ—Л–Љ–Є –Ї–Є—Б–ї–Њ—В–∞–Љ–Є –њ—А–Є–≤–Њ–і–Є—В –Ї –Є–Љ–Љ—Г–љ–Њ–Љ–Њ–і–Є—Д–Є–Ї–∞—Ж–Є–Є –Є –њ—А–Є–Њ–±—А–µ—В–µ–љ–Є—О –љ–µ–Њ–∞–љ—В–Є–≥–µ–љ–љ—Л—Е —Б–≤–Њ–є—Б—В–≤ E2 –Я–Ф–У –≤ –Љ–µ–ї–Ї–Є—Е –С–≠–Ъ;
- –њ—А–Є–Њ–±—А–µ—В–µ–љ–Є–µ –љ–µ–Њ–∞–љ—В–Є–≥–µ–љ–љ—Л—Е —Б–≤–Њ–є—Б—В–≤ E2 –Я–Ф–У вАУ –Ї–Њ–Љ–њ–ї–µ–Ї—Б–Њ–Љ –≤ –Љ–µ–ї–Ї–Є—Е, –њ–Њ–і–≤–µ—А–≥–∞—О—Й–Є—Е—Б—П –∞–њ–Њ–њ—В–Њ–Ј—Г —Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В–∞—Е –њ—А–Є–≤–Њ–і–Є—В –Ї —В–Њ–Љ—Г, —З—В–Њ –љ–Њ—А–Љ–∞–ї—М–љ–∞—П (–Ј–і–Њ—А–Њ–≤–∞—П) –Є–Љ–Љ—Г–љ–љ–∞—П —Б–Є—Б—В–µ–Љ–∞ —А–∞—Б–њ–Њ–Ј–љ–∞–µ—В –µ–≥–Њ –Ї–∞–Ї —З—Г–ґ–µ—А–Њ–і–љ—Л–є –∞–љ—В–Є–≥–µ–љ, –Ј–∞–њ—Г—Б–Ї–∞—П –≤—Л—А–∞–±–Њ—В–Ї—Г –Р–Ь–Р. –Я–Њ—Н—В–Њ–Љ—Г –љ–∞–ї–Є—З–Є–µ –Р–Ь–Р –њ—А–Є –Я–С–• —П–≤–ї—П–µ—В—Б—П –њ—А–Є–Ј–љ–∞–Ї–Њ–Љ –Њ—А–≥–∞–љ–Њ—Б–њ–µ—Ж–Є—Д–Є—З–µ—Б–Ї–Њ–≥–Њ, –∞ –љ–µ –Є—Б—В–Є–љ–љ–Њ–≥–Њ –∞—Г—В–Њ–Є–Љ–Љ—Г–љ–љ–Њ–≥–Њ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П.
–Т–≤–µ–і–µ–љ–Є–µ
–Я–µ—А–≤–Є—З–љ—Л–є –±–Є–ї–Є–∞—А–љ—Л–є —Е–Њ–ї–∞–љ–≥–Є—В (–Я–С–•)¬†вАУ —Н—В–Њ —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–µ —Е–Њ–ї–µ—Б—В–∞—В–Є—З–µ—Б–Ї–Њ–µ –њ—А–Њ–≥—А–µ—Б—Б–Є—А—Г—О—Й–µ–µ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–µ –њ–µ—З–µ–љ–Є, –њ—А–Њ—В–µ–Ї–∞—О—Й–µ–µ —Б¬†–і–µ—Б—В—А—Г–Ї—Ж–Є–µ–є, –∞–њ–Њ–њ—В–Њ–Ј–Њ–Љ –Є¬†–љ–µ–Ї—А–Њ–Ј–Њ–Љ —Н–њ–Є—В–µ–ї–Є—П –њ—А–µ–Є–Љ—Г—Й–µ—Б—В–≤–µ–љ–љ–Њ –Љ–µ–ї–Ї–Є—Е –≤–љ—Г—В—А–Є–і–Њ–ї—М–Ї–Њ–≤—Л—Е –Є¬†—Б–µ–њ—В–∞–ї—М–љ—Л—Е –ґ–µ–ї—З–љ—Л—Е –њ—А–Њ—В–Њ–Ї–Њ–≤, —Б¬†—А–∞–Ј–≤–Є—В–Є–µ–Љ –і—Г–Ї—В—Г–ї–Њ–њ–µ–љ–Є–Є –Є¬†—Е–Њ–ї–µ—Б—В–∞–Ј–∞, –≤¬†—В–µ—А–Љ–Є–љ–∞–ї—М–љ–Њ–є —Б—В–∞–і–Є–Є –Ї–Њ—В–Њ—А–Њ–≥–Њ —А–∞–Ј–≤–Є–≤–∞–µ—В—Б—П —Ж–Є—А—А–Њ–Ј –њ–µ—З–µ–љ–Є [1, 2].
–Ъ—А–∞–µ—Г–≥–Њ–ї—М–љ—Л–Љ –Ї–∞–Љ–љ–µ–Љ –≤¬†–і–Є–∞–≥–љ–Њ—Б—В–Є–Ї–µ –Я–С–• —П–≤–ї—П–µ—В—Б—П –Њ–±–љ–∞—А—Г–ґ–µ–љ–Є–µ –Р–Ь–Р –Є/–Є–ї–Є —Б–њ–µ—Ж–Є—Д–Є—З–µ—Б–Ї–Є—Е –і–ї—П –Я–С–• –∞–љ—В–Є–љ—Г–Ї–ї–µ–∞—А–љ—Л—Е –∞–љ—В–Є—В–µ–ї (–Р–Э–Р) –њ—А–Є –Њ–±—Б–ї–µ–і–Њ–≤–∞–љ–Є–Є –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†—Е–Њ–ї–µ—Б—В–∞—В–Є—З–µ—Б–Ї–Є–Љ–Є –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П–Љ–Є –њ–µ—З–µ–љ–Є [3, 4]. –°–њ–µ—Ж–Є—Д–Є—З–љ–Њ—Б—В—М –љ–∞–ї–Є—З–Є—П –≤¬†–њ–ї–∞–Ј–Љ–µ –Ї—А–Њ–≤–Є –∞–љ—В–Є–Љ–Є—В–Њ—Е–Њ–љ–і—А–Є–∞–ї—М–љ—Л—Е –∞—Г—В–Њ–∞–љ—В–Є—В–µ–ї (–Р–Ь–Р) –і–Њ—Б—В–Є–≥–∞–µ—В –±–Њ–ї–µ–µ 95% [5], –Є¬†—Н—В–Њ —Б–ї—Г–ґ–Є—В –Њ–і–љ–Є–Љ –Є–Ј –Њ–њ—А–µ–і–µ–ї—П—О—Й–Є—Е –њ—А–Є–Ј–љ–∞–Ї–Њ–≤ —А–∞–Ј–≤–Є–≤–∞—О—Й–µ–≥–Њ—Б—П –≤¬†–Љ–µ–ї–Ї–Є—Е —Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В–∞—Е –њ–∞—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–≥–Њ –њ—А–Њ—Ж–µ—Б—Б–∞ [5]. –Ь–µ—В–∞–∞–љ–∞–ї–Є–Ј 2014 –≥., –≤–Ї–ї—О—З–∞–≤—И–Є–є 24 –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П, –њ–Њ–Ї–∞–Ј–∞–ї, —З—В–Њ —Б—Г–Љ–Љ–∞—А–љ–∞—П —З—Г–≤—Б—В–≤–Є—В–µ–ї—М–љ–Њ—Б—В—М –Є¬†—Б–њ–µ—Ж–Є—Д–Є—З–љ–Њ—Б—В—М –Р–Ь–Р –≤¬†–і–Є–∞–≥–љ–Њ—Б—В–Є–Ї–µ –Я–С–• —Б–Њ—Б—В–∞–≤–ї—П–µ—В 84,5 –Є¬†97,8% —Б–Њ–Њ—В–≤–µ—В—Б—В–≤–µ–љ–љ–Њ [6]. –Р–Ь–Р –Њ–±–љ–∞—А—Г–ґ–Є–≤–∞—О—В—Б—П —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Г–ґ–µ –≤¬†–±–µ—Б—Б–Є–Љ–њ—В–Њ–Љ–љ–Њ–є —Б—В–∞–і–Є–Є –Я–С–•, —З—В–Њ —Г–Ї–∞–Ј—Л–≤–∞–µ—В –љ–∞ –Є—Е —Г—З–∞—Б—В–Є–µ –≤¬†–Љ–µ—Е–∞–љ–Є–Ј–Љ–µ —А–∞–Ј–≤–Є—В–Є—П –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П. –Э–∞–ї–Є—З–Є–µ –Р–Ь–Р –Є¬†–Р–Э–Р –њ–Њ—Б–ї—Г–ґ–Є–ї–Њ –њ–Њ–≤–Њ–і–Њ–Љ —Б—З–Є—В–∞—В—М –Я–С–• –њ—А–Њ—В–Њ—В–Є–њ–Њ–Љ –∞—Г—В–Њ–Є–Љ–Љ—Г–љ–љ–Њ–≥–Њ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П –Є¬†–Њ–њ–Є—Б—Л–≤–∞—В—М –µ–≥–Њ –Ї–∞–Ї ¬Ђ–њ–∞—А–∞–і–Є–≥–Љ–∞—В–Є—З–µ—Б–Ї—Г—О –Љ–Њ–і–µ–ї—М –∞—Г—В–Њ–Є–Љ–Љ—Г–љ–љ–Њ–≥–Њ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П¬ї [3, 7вАУ9]. –≠—В–Њ —Б–≤—П–Ј–∞–љ–Њ —Б¬†—В–µ–Љ, —З—В–Њ –Њ—Б–љ–Њ–≤—Г —Б–Њ–≤—А–µ–Љ–µ–љ–љ–Њ–є –і–Є–∞–≥–љ–Њ—Б—В–Є–Ї–Є –±–Њ–ї—М—И–Є–љ—Б—В–≤–∞ –∞—Г—В–Њ–Є–Љ–Љ—Г–љ–љ—Л—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є —Б–Њ—Б—В–∞–≤–ї—П–µ—В –Њ–±–љ–∞—А—Г–ґ–µ–љ–Є–µ –∞–љ—В–Є—В–µ–ї, –љ–∞–њ—А–∞–≤–ї–µ–љ–љ—Л—Е –њ—А–Њ—В–Є–≤ —Б–Њ–±—Б—В–≤–µ–љ–љ—Л—Е —В–Ї–∞–љ–µ–≤—Л—Е –∞–љ—В–Є–≥–µ–љ–Њ–≤ –Њ—А–≥–∞–љ–Є–Ј–Љ–∞ [10]. –Р—Г—В–Њ–∞–љ—В–Є—В–µ–ї–∞ —З–∞—Б—В–Њ –љ–∞–Ј—Л–≤–∞—О—В ¬Ђ—Б–≤–Є–і–µ—В–µ–ї—П–Љ–Є –∞—Г—В–Њ–Є–Љ–Љ—Г–љ–љ–Њ–≥–Њ –њ—А–Њ—Ж–µ—Б—Б–∞¬ї. –°—З–Є—В–∞–µ—В—Б—П, —З—В–Њ –љ–∞–ї–Є—З–Є–µ –≤—Л—Б–Њ–Ї–Є—Е —В–Є—В—А–Њ–≤ –Р–Ь–Р —Г¬†–±–Њ–ї—М—И–Є–љ—Б—В–≤–∞ (95%) –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–Я–С–• –Є¬†–Р–Э–Р –њ—А–Є–Љ–µ—А–љ–Њ —Г¬†50% –±–Њ–ї—М–љ—Л—Е —Г–Ї–∞–Ј—Л–≤–∞–µ—В –љ–∞ –њ–Њ—В–µ—А—О —В–Њ–ї–µ—А–∞–љ—В–љ–Њ—Б—В–Є –Т-–Ї–ї–µ—В–Њ–Ї –Ї¬†—Б–Њ–±—Б—В–≤–µ–љ–љ—Л–Љ –∞–љ—В–Є–≥–µ–љ–∞–Љ [11]. –Я–Њ—Н—В–Њ–Љ—Г –њ—А–Є–Љ–µ—А–љ–Њ —Б¬†—Б–µ—А–µ–і–Є–љ—Л 1960-—Е –≥–≥. –Я–С–• –Њ–њ–Є—Б—Л–≤–∞–µ—В—Б—П –Ї–∞–Ї –∞—Г—В–Њ–Є–Љ–Љ—Г–љ–љ–Њ–µ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–µ –њ–µ—З–µ–љ–Є [12]. –С–Њ–ї–µ–µ —В–Њ–≥–Њ, –Я–С–• –Њ–њ–Є—Б—Л–≤–∞—О—В –Ї–∞–Ї —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–µ —Е–Њ–ї–µ—Б—В–∞—В–Є—З–µ—Б–Ї–Њ–µ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–µ –њ–µ—З–µ–љ–Є, —Е–∞—А–∞–Ї—В–µ—А–Є–Ј—Г—О—Й–µ–µ—Б—П –Є–Љ–Љ—Г–љ–Њ–Њ–њ–Њ—Б—А–µ–і–Њ–≤–∞–љ–љ—Л–Љ (—Б –њ–Њ–Љ–Њ—Й—М—О –Р–Ь–Р) —А–∞–Ј—А—Г—И–µ–љ–Є–µ–Љ –С–≠–Ъ –Љ–µ–ї–Ї–Є—Е –Є¬†—Б—А–µ–і–љ–Є—Е –≤–љ—Г—В—А–Є–њ–µ—З–µ–љ–Њ—З–љ—Л—Е –ґ–µ–ї—З–љ—Л—Е –њ—А–Њ—В–Њ–Ї–Њ–≤ [1, 11]. –Я–С–• –Њ—В–љ–Њ—Б—П—В –Ї¬†–∞—Г—В–Њ–Є–Љ–Љ—Г–љ–љ–Њ–Љ—Г –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—О –µ—Й–µ –Є¬†–њ–Њ—В–Њ–Љ—Г, —З—В–Њ –Њ–љ–Њ –Є–Љ–µ–µ—В –Њ–±—Й–Є–µ —З–µ—А—В—Л —Б¬†–і—А—Г–≥–Є–Љ–Є –∞—Г—В–Њ–Є–Љ–Љ—Г–љ–љ—Л–Љ–Є –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П–Љ–Є. –≠—В–Њ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–µ –њ—А–µ–Є–Љ—Г—Й–µ—Б—В–≤–µ–љ–љ–Њ –њ–Њ—А–∞–ґ–∞–µ—В –ґ–µ–љ—Й–Є–љ, –∞¬†–∞—Г—В–Њ—А–µ–∞–Ї—В–Є–≤–љ—Л–µ –Ґ-–Ї–ї–µ—В–Ї–Є –Є–≥—А–∞—О—В –Њ–њ—А–µ–і–µ–ї–µ–љ–љ—Г—О —А–Њ–ї—М –≤¬†–њ–∞—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–Љ –њ—А–Њ—Ж–µ—Б—Б–µ [14вАУ18].
–Р–љ—В–Є–≥–µ–љ–Њ–Љ –і–ї—П –њ—А–Њ–і—Г–Ї—Ж–Є–Є –Р–Ь–Р –њ—А–Є –Я–С–• —П–≤–ї—П–µ—В—Б—П –і–Є–≥–Є–і—А–Њ–ї–Є–њ–Њ–Є–ї—В—А–∞–љ—Б–∞—Ж–µ—В–Є–ї–∞–Ј–∞ (E2) –њ–Є—А—Г–≤–∞—В–і–µ–≥–Є–і—А–Њ–≥–µ–љ–∞–Ј–љ–Њ–≥–Њ (–Я–Ф–У) –Ї–Њ–Љ–њ–ї–µ–Ї—Б–∞ (E2 –Я–Ф–У¬†вАУ –Ї–Њ–Љ–њ–ї–µ–Ї—Б), –Ї–Њ—В–Њ—А—Л–є –ї–Њ–Ї–∞–ї–Є–Ј—Г–µ—В—Б—П –љ–∞ –≤–љ—Г—В—А–µ–љ–љ–µ–є –Љ–µ–Љ–±—А–∞–љ–µ –Љ–Є—В–Њ—Е–Њ–љ–і—А–Є–є. –Э–µ—Б–Љ–Њ—В—А—П –љ–∞ —В–Њ —З—В–Њ —З–µ—В–Ї–Њ –Њ–њ—А–µ–і–µ–ї–µ–љ –∞–љ—В–Є–≥–µ–љ, –Ї¬†–Ї–Њ—В–Њ—А–Њ–Љ—Г —А–∞–Ј–≤–Є–≤–∞—О—В—Б—П –Р–Ь–Р [7, 8, 11, 19], –і–Њ –љ–∞—Б—В–Њ—П—Й–µ–≥–Њ –≤—А–µ–Љ–µ–љ–Є –Њ—Б—В–∞–µ—В—Б—П –Њ—В–Ї—А—Л—В—Л–Љ –≤–Њ–њ—А–Њ—Б, –Ї–∞—Б–∞—О—Й–Є–є—Б—П —Д–∞–Ї—В–Њ—А–Њ–≤, –Ј–∞–њ—Г—Б–Ї–∞—О—Й–Є—Е –Љ–µ—Е–∞–љ–Є–Ј–Љ –Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є—П –Р–Ь–Р, –Є¬†–Є—Е –Ј–љ–∞—З–µ–љ–Є–µ –≤¬†—А–∞–Ј–≤–Є—В–Є–Є –њ–∞—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–≥–Њ –њ—А–Њ—Ж–µ—Б—Б–∞ –њ—А–Є –Я–С–•. –Э–µ—П—Б–љ–Њ, –Ї–∞–Ї–Є–Љ –Њ–±—А–∞–Ј–Њ–Љ –∞–љ—В–Є–≥–µ–љ E2 –Я–Ф–У, —А–∞—Б–њ–Њ–ї–Њ–ґ–µ–љ–љ—Л–є –љ–∞ –≤–љ—Г—В—А–µ–љ–љ–µ–є –Љ–Є—В–Њ—Е–Њ–љ–і—А–Є–∞–ї—М–љ–Њ–є –Љ–µ–Љ–±—А–∞–љ–µ, —Б—В–∞–љ–Њ–≤–Є—В—Б—П –Љ–Є—И–µ–љ—М—О –і–ї—П –Є–Љ–Љ—Г–љ–Њ–Ї–Њ–Љ–њ–µ—В–µ–љ—В–љ—Л—Е –Ї–ї–µ—В–Њ–Ї. –§–µ—А–Љ–µ–љ—В –Я–Ф–У —Б–Њ—Б—В–Њ–Є—В –Є–Ј —В—А–µ—Е —Б—Г–±—К–µ–і–Є–љ–Є—Ж: E1 (–њ–Є—А—Г–≤–∞—В–і–µ–≥–Є–і—А–Њ–≥–µ–љ–∞–Ј–∞), E2 –Є¬†E3 (–і–Є–≥–Є–і—А–Њ–ї–Є–њ–Њ–Є–ї–і–µ–≥–Є–і—А–Њ–≥–µ–љ–∞–Ј–∞), –љ–Њ –љ–µ–Є–Ј–≤–µ—Б—В–љ–Њ, –њ–Њ—З–µ–Љ—Г –њ—А–Є –Я–С–• –≤—Л—А–∞–±–∞—В—Л–≤–∞—О—В—Б—П –∞—Г—В–Њ–∞–љ—В–Є—В–µ–ї–∞ —В–Њ–ї—М–Ї–Њ –њ—А–Њ—В–Є–≤ –Њ–і–љ–Њ–є —Б—Г–±—К–µ–і–Є–љ–Є—Ж—Л –Х2. –Я—А–Є –Є–Љ–Љ—Г–љ–Є–Ј–∞—Ж–Є–Є —Н–Ї—Б–њ–µ—А–Є–Љ–µ–љ—В–∞–ї—М–љ—Л—Е –ґ–Є–≤–Њ—В–љ—Л—Е –∞–љ—В–Є–≥–µ–љ–Њ–Љ –Х2 –≤¬†–Ї–∞—З–µ—Б—В–≤–µ –Ї–Њ–Љ–±–Є–љ–Є—А–Њ–≤–∞–љ–љ–Њ–≥–Њ –њ–Њ–ї–Є–њ–µ–њ—В–Є–і–∞ –њ—А–Њ–Є—Б—Е–Њ–і–Є—В –Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є–µ –Р–Ь–Р. –Ю–і–љ–∞–Ї–Њ –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є—П —Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В–Њ–≤ –љ–µ –љ–∞–±–ї—О–і–∞–µ—В—Б—П, –њ–Њ—Н—В–Њ–Љ—Г –≤–Њ–Ј–љ–Є–Ї–∞–µ—В –≤–Њ–њ—А–Њ—Б, —П–≤–ї—П—О—В—Б—П –ї–Є –Р–Ь–Р —Д–∞–Ї—В–Њ—А–Њ–Љ, –Ј–∞–њ—Г—Б–Ї–∞—О—Й–Є–Љ —А–∞–Ј—А—Г—И–µ–љ–Є–µ –С–≠–Ъ. –Ґ–∞–Ї–ґ–µ –љ–µ—В –Њ—В–≤–µ—В–∞ –љ–∞ –≤–Њ–њ—А–Њ—Б, –њ–Њ—З–µ–Љ—Г –≤¬†—Н—В–Њ—В –њ—А–Њ—Ж–µ—Б—Б –≤–Њ–≤–ї–µ—З–µ–љ E2-–∞–љ—В–Є–≥–µ–љ –Я–Ф–У —В–Њ–ї—М–Ї–Њ –Љ–µ–ї–Ї–Є—Е –С–≠–Ъ. –Ю—В–≤–µ—В—Л –љ–∞ —Н—В–Є –≤–Њ–њ—А–Њ—Б—Л –Є–Љ–µ—О—В –њ—А–Є–љ—Ж–Є–њ–Є–∞–ї—М–љ–Њ–µ –Ј–љ–∞—З–µ–љ–Є–µ –і–ї—П –њ–Њ–љ–Є–Љ–∞–љ–Є—П –≤–Њ–≤–ї–µ—З–µ–љ–љ–Њ—Б—В–Є –Є–Љ–Љ—Г–љ–љ–Њ–є —Б–Є—Б—В–µ–Љ—Л –≤¬†–њ–∞—В–Њ–≥–µ–љ–µ–Ј –Я–С–•.
–•–∞—А–∞–Ї—В–µ—А–Є—Б—В–Є–Ї–∞ –∞—Г—В–Њ–Є–Љ–Љ—Г–љ–љ—Л—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є
–Р—Г—В–Њ–Є–Љ–Љ—Г–љ–љ—Л–µ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П –њ—А–µ–і—Б—В–∞–≤–ї—П—О—В —Б–Њ–±–Њ–є –Њ–±—И–Є—А–љ—Л–є –Ї–ї–∞—Б—Б —А–∞–Ј–љ–Њ—А–Њ–і–љ—Л—Е –њ–Њ –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є–Љ –њ—А–Њ—П–≤–ї–µ–љ–Є—П–Љ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є, —А–∞–Ј–≤–Є–≤–∞—О—Й–Є—Е—Б—П –≤—Б–ї–µ–і—Б—В–≤–Є–µ –љ–∞—А—Г—И–µ–љ–Є–є –≤¬†–Є–Љ–Љ—Г–љ–љ–Њ–є —Б–Є—Б—В–µ–Љ–µ, —Б–Њ–њ—А–Њ–≤–Њ–ґ–і–∞—О—Й–Є—Е—Б—П –њ–Њ—В–µ—А–µ–є —В–Њ–ї–µ—А–∞–љ—В–љ–Њ—Б—В–Є –Ї¬†–Њ–њ—А–µ–і–µ–ї–µ–љ–љ—Л–Љ —В–Ї–∞–љ—П–Љ –Є¬†–±–µ–ї–Ї–∞–Љ —Б¬†–њ–Њ—Б–ї–µ–і—Г—О—Й–µ–є –њ–∞—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–є –≤—Л—А–∞–±–Њ—В–Ї–Њ–є –∞—Г—В–Њ–Є–Љ–Љ—Г–љ–љ—Л—Е –∞–љ—В–Є—В–µ–ї. –°—З–Є—В–∞–µ—В—Б—П, —З—В–Њ –≤—Б–µ –∞—Г—В–Њ–Є–Љ–Љ—Г–љ–љ—Л–µ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П –Є–Љ–µ—О—В —Б—Е–Њ–і–љ—Л–µ –Љ–µ—Е–∞–љ–Є–Ј–Љ—Л —А–∞–Ј–≤–Є—В–Є—П [10]. –Т–Њ–Ј–љ–Є–Ї–∞—О—Й–Є–µ –њ—А–Є —Н—В–Њ–Љ –Ї–Њ–Љ–њ–ї–µ–Ї—Б—Л –∞–љ—В–Є–≥–µ–љ-–∞–љ—В–Є—В–µ–ї–Њ —Б–њ–Њ—Б–Њ–±–љ—Л –≤—Л–Ј—Л–≤–∞—В—М —А–∞–Ј–≤–Є—В–Є–µ –≤–Њ—Б–њ–∞–ї–µ–љ–Є—П –Є¬†–Њ–њ–Њ—Б—А–µ–і–Њ–≤–∞–љ–љ–Њ–≥–Њ —Н—В–Є–Љ–Є –Ї–Њ–Љ–њ–ї–µ–Ї—Б–∞–Љ–Є –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є—П —В–Ї–∞–љ–µ–є –Љ–Є—И–µ–љ–µ–є. –Я–Њ–Ї–∞–Ј–∞–љ–Њ, —З—В–Њ –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є–µ —В–Ї–∞–љ–µ–є –Љ–Є—И–µ–љ–µ–є –Љ–∞–Ї—А–Њ–Њ—А–≥–∞–љ–Є–Ј–Љ–∞ —П–≤–ї—П–µ—В—Б—П –Њ–±—Й–µ–є –Њ—Б–љ–Њ–≤–Њ–є –і–ї—П –Ј–∞–њ—Г—Б–Ї–∞ –љ–Њ–≤—Л—Е –Є–Љ–Љ—Г–љ–љ—Л—Е —А–µ–∞–Ї—Ж–Є–є [10]. –Я—А–Є —Н—В–Њ–Љ –≤¬†–Ї–∞—З–µ—Б—В–≤–µ –∞–і—К—О–≤–∞–љ—В–љ—Л—Е —Б—Г–±—Б—В–∞–љ—Ж–Є–є (—Г—Б–Є–ї–Є–≤–∞—О—Й–Є—Е –Є–Љ–Љ—Г–љ–љ—Л–є –Њ—В–≤–µ—В) –≤—Л—Б—В—Г–њ–∞—О—В –њ—А–Њ–і—Г–Ї—В—Л —А–∞–Ј—А—Г—И–µ–љ–Є—П —Б–Њ–±—Б—В–≤–µ–љ–љ—Л—Е –Ї–ї–µ—В–Њ–Ї –Є¬†—В–Ї–∞–љ–µ–є. –Т—Б–µ —Н—В–Њ –њ—А–Є–≤–Њ–і–Є—В –Ї¬†—А–∞–Ј–≤–Є—В–Є—О —Б–Є—Б—В–µ–Љ–љ—Л—Е (–њ–Њ–ї–Є–Њ—А–≥–∞–љ–љ—Л—Е) –љ–∞—А—Г—И–µ–љ–Є–є —Д—Г–љ–Ї—Ж–Є–є –≤–љ—Г—В—А–µ–љ–љ–Є—Е –Њ—А–≥–∞–љ–Њ–≤. –Я–Њ—Б—В–Њ—П–љ—Б—В–≤–Њ –і–Є—Б—Д—Г–љ–Ї—Ж–Є–Є –Є–Љ–Љ—Г–љ–љ–Њ–є —Б–Є—Б—В–µ–Љ—Л –Є¬†–і–µ—Б—В—А—Г–Ї—Ж–Є—П —В–Ї–∞–љ–µ–є —Б¬†–њ–Њ—Б–ї–µ–і—Г—О—Й–Є–Љ —А–∞—Б–њ–Њ–Ј–љ–∞–≤–∞–љ–Є–µ–Љ —В–Ї–∞–љ–µ–≤—Л—Е –∞–љ—В–Є–≥–µ–љ–Њ–≤ –∞–љ—В–Є–≥–µ–љ–њ—А–µ–Ј–µ–љ—В–Є—А—Г—О—Й–Є–Љ–Є –Ї–ї–µ—В–Ї–∞–Љ–Є –њ–Њ–і–і–µ—А–ґ–Є–≤–∞—О—В —Е—А–Њ–љ–Є–Ј–∞—Ж–Є—О —Н—В–Њ–≥–Њ –њ—А–Њ—Ж–µ—Б—Б–∞. –Я—А–Є —Н—В–Њ–Љ —В–µ—З–µ–љ–Є–µ –∞—Г—В–Њ–Є–Љ–Љ—Г–љ–љ—Л—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є –Љ–Њ–ґ–µ—В —Б–Њ–њ—А–Њ–≤–Њ–ґ–і–∞—В—М—Б—П —Б–∞–Љ–Њ–њ—А–Њ–Є–Ј–≤–Њ–ї—М–љ–Њ–є —А–µ–Љ–Є—Б—Б–Є–µ–є –њ–∞—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–≥–Њ —Б–Њ—Б—В–Њ—П–љ–Є—П, –Ї–Њ—В–Њ—А–∞—П —З–µ—А–µ–і—Г–µ—В—Б—П —Б¬†–Њ–±–Њ—Б—В—А–µ–љ–Є–µ–Љ. –Ґ—П–ґ–µ—Б—В—М —В–µ—З–µ–љ–Є—П –Є¬†–љ–∞—А—Г—И–µ–љ–Є–µ —Д—Г–љ–Ї—Ж–Є–Є –≤–љ—Г—В—А–µ–љ–љ–Є—Е –Њ—А–≥–∞–љ–Њ–≤ –њ—А–Є –∞—Г—В–Њ–Є–Љ–Љ—Г–љ–љ–Њ–Љ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–Є, –Ї–∞–Ї –њ—А–∞–≤–Є–ї–Њ, –Ї–Њ—А—А–µ–ї–Є—А—Г—О—В —Б¬†—В–Є—В—А–Њ–Љ (—Г—А–Њ–≤–љ–µ–Љ) –∞—Г—В–Њ–∞–љ—В–Є—В–µ–ї.
–Ю—Б–Њ–±–µ–љ–љ–Њ—Б—В–Є –Я–С–• –Ї–∞–Ї –∞—Г—В–Њ–Є–Љ–Љ—Г–љ–љ–Њ–≥–Њ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П
–Э–µ—Б–Љ–Њ—В—А—П –љ–∞ –њ–Њ—П–≤–ї–µ–љ–Є–µ –Р–Ь–Р —Г–ґ–µ –≤¬†–∞—Б–Є–Љ–њ—В–Њ–Љ–∞—В–Є—З–µ—Б–Ї–Њ–є —Б—В–∞–і–Є–Є –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П, –∞—Г—В–Њ–Є–Љ–Љ—Г–љ–љ–∞—П —В–µ–Њ—А–Є—П –Я–С–• –љ–µ –≤–њ–Њ–ї–љ–µ —Г–±–µ–і–Є—В–µ–ї—М–љ–∞, –њ–Њ—Б–Ї–Њ–ї—М–Ї—Г –Љ–љ–Њ–≥–Є–µ –њ—А–Є–Ј–љ–∞–Ї–Є, —Е–∞—А–∞–Ї—В–µ—А–љ—Л–µ –і–ї—П –∞—Г—В–Њ–Є–Љ–Љ—Г–љ–љ–Њ–є –њ–∞—В–Њ–ї–Њ–≥–Є–Є, —Г¬†—Н—В–Є—Е –њ–∞—Ж–Є–µ–љ—В–Њ–≤ –Њ—В—Б—Г—В—Б—В–≤—Г—О—В –Є–ї–Є –љ–µ –Є–Љ–µ—О—В —З–µ—В–Ї–Њ–≥–Њ –њ–Њ–і—В–≤–µ—А–ґ–і–µ–љ–Є—П (—В–∞–±–ї. 1).
–Я—А–Є–Ј–љ–∞–Ї–Є –Я–С–•, –Ї–Њ—В–Њ—А—Л–µ –љ–µ —Г–Ї–ї–∞–і—Л–≤–∞—О—В—Б—П –≤¬†–Ї—А–Є—В–µ—А–Є–Є –∞—Г—В–Њ¬≠–Є–Љ–Љ—Г–љ–љ–Њ–є —В–µ–Њ—А–Є–Є –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П:
- –њ—А–∞–Ї—В–Є—З–µ—Б–Ї–Є –≤—Б–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞—В–µ–ї–Є –Њ—В–Љ–µ—З–∞—О—В, —З—В–Њ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–µ –љ–µ –љ–Њ—Б–Є—В —Б–Є—Б—В–µ–Љ–љ—Л–є —Е–∞—А–∞–Ї—В–µ—А, –∞¬†–Њ–≥—А–∞–љ–Є—З–µ–љ–Њ –ї–Њ–Ї–∞–ї—М–љ—Л–Љ –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є–µ–Љ –Љ–µ–ї–Ї–Є—Е –≤–љ—Г—В—А–Є–њ–µ—З–µ–љ–Њ—З–љ—Л—Е –ґ–µ–ї—З–љ—Л—Е –њ—А–Њ—В–Њ–Ї–Њ–≤ [1, 7];
- –Я–С–• –љ–µ –≤–њ–Є—Б—Л–≤–∞–µ—В—Б—П –≤¬†—Б—Е–µ–Љ—Г –≤–Њ—Б–њ–∞–ї–µ–љ–Є—П, –Њ–њ–Њ—Б—А–µ–і–Њ–≤–∞–љ–љ–Њ–≥–Њ –Є–Љ–Љ—Г–љ–љ—Л–Љ–Є –Ї–Њ–Љ–њ–ї–µ–Ї—Б–∞–Љ–Є [1];
- –Љ–Є—В–Њ—Е–Њ–љ–і—А–Є–∞–ї—М–љ—Л–є –∞—Г—В–Њ–∞–љ—В–Є–≥–µ–љ –Х2 –Я–Ф–У, –Ї¬†–Ї–Њ—В–Њ—А–Њ–Љ—Г –Њ–±—А–∞–Ј—Г—О—В—Б—П –Р–Ь–Р, —А–∞—Б–њ–Њ–ї–Њ–ґ–µ–љ –љ–∞ –≤–љ—Г—В—А–µ–љ–љ–µ–є –Љ–µ–Љ–±—А–∞–љ–µ –Љ–Є—В–Њ—Е–Њ–љ–і—А–Є–є, –љ–µ —П–≤–ї—П–µ—В—Б—П —В–Ї–∞–љ–µ—Б–њ–µ—Ж–Є—Д–Є—З–љ—Л–Љ –Є¬†–Њ–±–ї–∞–і–∞–µ—В –≤—Л—Б–Њ–Ї–Њ–є —В–Њ–ї–µ—А–∞–љ—В–љ–Њ—Б—В—М—О –Ї¬†–Є–Љ–Љ—Г–љ–љ–Њ–є —Б–Є—Б—В–µ–Љ–µ [7];
- –Х2 –Я–Ф–У –≤¬†–Є–Ј–Њ–±–Є–ї–Є–Є —Б–Њ–і–µ—А–ґ–Є—В—Б—П –≤¬†–Љ–Є—В–Њ—Е–Њ–љ–і—А–Є—П—Е –≤—Б–µ—Е —П–і—А–Њ—Б–Њ–і–µ—А–ґ–∞—Й–Є—Е –Ї–ї–µ—В–Њ–Ї, –Њ–і–љ–∞–Ї–Њ —В–Њ–ї—М–Ї–Њ –Љ–µ–ї–Ї–Є–µ –С–≠–Ъ —П–≤–ї—П—О—В—Б—П –Љ–Є—И–µ–љ—М—О –∞—Г—В–Њ–Є–Љ–Љ—Г–љ–љ–Њ–є –∞—В–∞–Ї–Є –њ—А–Є –Я–С–• [1];
- –љ–Є —В–Є—В—А, –љ–Є —Е–∞—А–∞–Ї—В–µ—А –Р–Ь–Р –љ–µ –Ї–Њ—А—А–µ–ї–Є—А—Г—О—В —Б¬†–Ї–ї–Є–љ–Є—З–µ—Б–Ї–Њ–є –Ї–∞—А—В–Є–љ–Њ–є, —В—П–ґ–µ—Б—В—М—О –Є¬†–њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є–µ–Љ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П [8];
- —В–Є—В—А—Л –Р–Ь–Р –Є–ї–Є –Є—Е –Є–Ј–Љ–µ–љ–µ–љ–Є—П –≤¬†–і–Є–љ–∞–Љ–Є–Ї–µ –љ–µ —П–≤–ї—П—О—В—Б—П –њ—А–Њ–≥–љ–Њ—Б—В–Є—З–µ—Б–Ї–Є–Љ–Є –Љ–∞—А–Ї–µ—А–∞–Љ–Є –њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є—П –Я–С–• [29вАУ31];
- –Я–С–•, –±—Г–і—Г—З–Є —Е—А–Њ–љ–Є—З–µ—Б–Ї–Є–Љ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–µ–Љ –њ–µ—З–µ–љ–Є, –≤¬†—Ж–µ–ї–Њ–Љ —Е–∞—А–∞–Ї—В–µ—А–Є–Ј—Г–µ—В—Б—П –Љ–µ–і–ї–µ–љ–љ—Л–Љ –њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є–µ–Љ –±–µ–Ј –≤—Б–њ—Л—И–µ–Ї –Є¬†—А–µ–Љ–Є—Б—Б–Є–є –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є, —Е–∞—А–∞–Ї—В–µ—А–љ—Л—Е –і–ї—П –∞—Г—В–Њ–Є–Љ–Љ—Г–љ–љ—Л—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є;
- –Я–С–•, –≤¬†–Њ—В–ї–Є—З–Є–µ –Њ—В –і—А—Г–≥–Є—Е –∞—Г—В–Њ–Є–Љ–Љ—Г–љ–љ—Л—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є, –љ–µ –Є–Љ–µ–µ—В –∞–љ–∞–ї–Њ–≥–Њ–≤ —Г¬†–і–µ—В–µ–є [8];
- –љ–µ—Б–њ–Њ—Б–Њ–±–љ–Њ—Б—В—М –Є–Љ–Љ—Г–љ–Њ—Б—Г–њ—А–µ—Б—Б–Є–≤–љ–Њ–є —В–µ—А–∞–њ–Є–Є –Є¬†–≥–µ–љ–љ–Њ-–Є–љ–ґ–µ–љ–µ—А–љ—Л—Е (—В–∞—А–≥–µ—В–љ—Л—Е) –±–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є—Е –њ—А–µ–њ–∞—А–∞—В–Њ–≤ –Њ—Б—В–∞–љ–Њ–≤–Є—В—М –њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є–µ –Я–С–• [23вАУ25].
–Т —Б–≤—П–Ј–Є —Б¬†—Н—В–Є–Љ –≤¬†–љ–∞—Б—В–Њ—П—Й–µ–µ –≤—А–µ–Љ—П –≤—Л—Б–Ї–∞–Ј—Л–≤–∞—О—В—Б—П —Б–Њ–Љ–љ–µ–љ–Є—П –≤¬†—В–Њ–Љ, —П–≤–ї—П–µ—В—Б—П –ї–Є –Я–С–• –Є—Б—В–Є–љ–љ—Л–Љ –∞—Г—В–Њ–Є–Љ–Љ—Г–љ–љ—Л–Љ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–µ–Љ [1]. –Ф–ї—П –њ–Њ–ї—Г—З–µ–љ–Є—П –Њ—В–≤–µ—В–∞ –љ–∞ —Н—В–Њ—В –≤–Њ–њ—А–Њ—Б –≤–∞–ґ–љ–Њ –Є–Љ–µ—В—М —З–µ—В–Ї–Њ–µ –њ—А–µ–і—Б—В–∞–≤–ї–µ–љ–Є–µ –Њ¬†–Љ–µ—Е–∞–љ–Є–Ј–Љ–µ –Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є—П –Р–Ь–Р. –Ш–Љ–µ–љ–љ–Њ –њ–Њ—Н—В–Њ–Љ—Г —Г—Б–Є–ї–Є—П –Љ–љ–Њ–≥–Є—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞—В–µ–ї–µ–є –љ–∞–њ—А–∞–≤–ї–µ–љ—Л –љ–∞ –≤—Л—П–≤–ї–µ–љ–Є–µ —Д–∞–Ї—В–Њ—А–Њ–≤, –Ј–∞–њ—Г—Б–Ї–∞—О—Й–Є—Е —Н—В–Њ—В –њ—А–Њ—Ж–µ—Б—Б, –Є¬†–љ–∞ –≤—Л—П—Б–љ–µ–љ–Є–µ –њ–∞—В–Њ—Д–Є–Ј–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є—Е –Љ–µ—Е–∞–љ–Є–Ј–Љ–Њ–≤, –ї–µ–ґ–∞—Й–Є—Е –≤¬†–Њ—Б–љ–Њ–≤–µ –Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є—П –∞–љ—В–Є–Љ–Є—В–Њ—Е–Њ–љ–і—А–Є–∞–ї—М–љ—Л—Е –∞—Г—В–Њ–∞–љ—В–Є—В–µ–ї [1, 11]. –Я–Њ–њ—Л—В–Ї–Є –і–Њ–Ї–∞–Ј–∞—В—М –Є–ї–Є –Њ–њ—А–Њ–≤–µ—А–≥–љ—Г—В—М –∞—Г—В–Њ–Є–Љ–Љ—Г–љ–љ—Г—О —В–µ–Њ—А–Є—О –Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є—П –Р–Ь–Р –ї–µ–ґ–∞—В –≤¬†–Њ—Б–љ–Њ–≤–µ —Н—В–Є—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є.
–Ф–ї—П —В–Њ–≥–Њ —З—В–Њ–±—Л –Њ—В–≤–µ—В–Є—В—М –љ–∞ —Н—В–Є –≤–Њ–њ—А–Њ—Б—Л, –≤–∞–ґ–љ–Њ –њ–Њ–љ—П—В—М, —З—В–Њ –ї–µ–ґ–Є—В –≤¬†–Њ—Б–љ–Њ–≤–µ –Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є—П –Р–Ь–Р:
- –і–Є—Б—Д—Г–љ–Ї—Ж–Є—П –Є–Љ–Љ—Г–љ–љ–Њ–є —Б–Є—Б—В–µ–Љ—Л, –њ—А–Є–≤–Њ–і—П—Й–∞—П –Ї¬†–њ–Њ—В–µ—А–µ –µ–µ —В–Њ–ї–µ—А–∞–љ—В–љ–Њ—Б—В–Є –Ї¬†–љ–µ–Є–Ј–Љ–µ–љ–µ–љ–љ–Њ–Љ—Г –Х2 –Я–Ф–У –±–µ–ї–Ї—Г-–∞–љ—В–Є–≥–µ–љ—Г?
–Є–ї–Є
- –љ–∞—А—Г—И–µ–љ–Є—П –≤¬†—Б—В—А—Г–Ї—В—Г—А–µ –Є/–Є–ї–Є –Ї–Њ–љ—Д–Њ—А–Љ–∞—Ж–Є–Є –∞–љ—В–Є–≥–µ–љ–∞ –Х2 –Я–Ф–У, –Ї–Њ—В–Њ—А—Л–µ –њ—А–Є–≤–Њ–і—П—В –Ї¬†–Є–Ј–Љ–µ–љ–µ–љ–Є—О –µ–≥–Њ –Є–Љ–Љ—Г–љ–љ—Л—Е —Б–≤–Њ–є—Б—В–≤, —З—В–Њ –≤–Њ—Б–њ—А–Є–љ–Є–Љ–∞–µ—В—Б—П –љ–µ–Є–Ј–Љ–µ–љ–µ–љ–љ–Њ–є (–Ј–і–Њ—А–Њ–≤–Њ–є) –Є–Љ–Љ—Г–љ–љ–Њ–є —Б–Є—Б—В–µ–Љ–Њ–є –Ї–∞–Ї —З—Г–ґ–µ—А–Њ–і–љ—Л–є –∞–љ—В–Є–≥–µ–љ?
–Ш–Љ–µ–µ—В—Б—П –ї–Є –і–Є—Б—Д—Г–љ–Ї—Ж–Є—П –Є–Љ–Љ—Г–љ–љ–Њ–є —Б–Є—Б—В–µ–Љ—Л –њ—А–Є –Я–С–•?
–Х—Б–ї–Є –Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є–µ –Р–Ь–Р —П–≤–ї—П–µ—В—Б—П —А–µ–Ј—Г–ї—М—В–∞—В–Њ–Љ –і–Є—Б—Д—Г–љ–Ї—Ж–Є–Є –Є–Љ–Љ—Г–љ–љ–Њ–є —Б–Є—Б—В–µ–Љ—Л, –њ—А–Є–≤–Њ–і—П—Й–µ–є –Ї¬†–њ–Њ—В–µ—А–µ —В–Њ–ї–µ—А–∞–љ—В–љ–Њ—Б—В–Є –Ї¬†–љ–µ–Є–Ј–Љ–µ–љ–µ–љ–љ–Њ–Љ—Г (–љ–µ–Љ–Њ–і–Є—Д–Є—Ж–Є—А–Њ–≤–∞–љ–љ–Њ–Љ—Г) –∞–љ—В–Є–≥–µ–љ—Г, —В–Њ —Н—В–Њ, —Б–Ї–Њ—А–µ–µ –≤—Б–µ–≥–Њ, —Б–ї–µ–і—Г–µ—В —А–∞—Б—Б–Љ–∞—В—А–Є–≤–∞—В—М –Ї–∞–Ї –њ—А–Є–Ј–љ–∞–Ї –∞—Г—В–Њ–Є–Љ–Љ—Г–љ–љ–Њ–≥–Њ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П. –Э–Њ –µ—Б–ї–Є –Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є–µ –Р–Ь–Р —П–≤–ї—П–µ—В—Б—П —Б–ї–µ–і—Б—В–≤–Є–µ–Љ –њ—А–Є–Њ–±—А–µ—В–µ–љ–Є—П –љ–Њ–≤—Л—Е –Є–Љ–Љ—Г–љ–љ—Л—Е —Б–≤–Њ–є—Б—В–≤ –∞–љ—В–Є–≥–µ–љ–Њ–Љ –Х2 –Я–Ф–У, –Ї–Њ—В–Њ—А—Л–µ –µ—Б—В–µ—Б—В–≤–µ–љ–љ—Л–Љ –Њ–±—А–∞–Ј–Њ–Љ —А–∞—Б–њ–Њ–Ј–љ–∞—О—В—Б—П ¬Ђ–Ј–і–Њ—А–Њ–≤–Њ–є¬ї –Є–Љ–Љ—Г–љ–љ–Њ–є —Б–Є—Б—В–µ–Љ–Њ–є –Ї–∞–Ї —З—Г–ґ–µ—А–Њ–і–љ—Л–є –∞–љ—В–Є–≥–µ–љ (–љ–µ–Њ–∞–љ—В–Є–≥–µ–љ), —В–Њ, –≤–µ—А–Њ—П—В–љ–µ–µ –≤—Б–µ–≥–Њ, –Я–С–• —Б–ї–Њ–ґ–љ–Њ —А–∞—Б—Б–Љ–∞—В—А–Є–≤–∞—В—М –Ї–∞–Ї –Є—Б—В–Є–љ–љ–Њ–µ –∞—Г—В–Њ–Є–Љ–Љ—Г–љ–љ–Њ–µ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–µ.
–Ъ–∞–Ї —Г–ґ–µ –Њ—В–Љ–µ—З–∞–ї–Њ—Б—М —А–∞–љ–µ–µ, —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–Ї–ї–∞—Б—Б–Є—З–µ—Б–Ї–Є–Љ —В–µ—З–µ–љ–Є–µ–Љ –Я–С–• –Њ–±—А–∞–Ј—Г—О—В—Б—П –Р–Ь–Р –Ї¬†–∞–љ—В–Є–≥–µ–љ–љ–Њ–Љ—Г –Ї–Њ–Љ–њ–Њ–љ–µ–љ—В—Г, –Њ—В–љ–Њ—Б—П—Й–µ–Љ—Г—Б—П –Ї¬†–Х2 –Я–Ф–У, –Ї–Њ—В–Њ—А—Л–є –ї–Њ–Ї–∞–ї–Є–Ј—Г–µ—В—Б—П –љ–∞ –≤–љ—Г—В—А–µ–љ–љ–µ–є –Љ–µ–Љ–±—А–∞–љ–µ –Љ–Є—В–Њ—Е–Њ–љ–і—А–Є–є [7, 8, 11, 19]. –С–Њ–ї—М—И–Є–љ—Б—В–≤–Њ –њ—А–Њ–≤–µ–і–µ–љ–љ—Л—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є –∞–њ—А–Є–Њ—А–Є –њ–Њ—Б—В—Г–ї–Є—А—Г—О—В, —З—В–Њ –њ—А–Є –Я–С–• –Є–Љ–µ–µ—В –Љ–µ—Б—В–Њ –і–Є—Б—Д—Г–љ–Ї—Ж–Є—П –Є–Љ–Љ—Г–љ–љ–Њ–є —Б–Є—Б—В–µ–Љ—Л, –Ї–Њ—В–Њ—А–∞—П –њ—А–Є–≤–Њ–і–Є—В –Ї¬†–Є–Ј–Љ–µ–љ–µ–љ–Є—О –µ–µ —В–Њ–ї–µ—А–∞–љ—В–љ–Њ—Б—В–Є –Ї¬†–Х2 –Я–Ф–У [1, 11, 32]. –Ш—Б—Б–ї–µ–і–Њ–≤–∞—В–µ–ї–Є –љ–∞–њ—А–∞–≤–ї—П—О—В —Б–≤–Њ–Є —Г—Б–Є–ї–Є—П –љ–∞ —В–Њ, —З—В–Њ–±—Л –њ–Њ–љ—П—В—М –Љ–µ—Е–∞–љ–Є–Ј–Љ —А–∞–Ј–≤–Є—В–Є—П –Є¬†–љ–∞—А—Г—И–µ–љ–Є—П —Н—В–Њ–є —В–Њ–ї–µ—А–∞–љ—В–љ–Њ—Б—В–Є [1, 11, 32]. –°—З–Є—В–∞–µ—В—Б—П, —З—В–Њ –њ–Њ—В–µ—А—П –Є–Љ–Љ—Г–љ–љ–Њ–є —В–Њ–ї–µ—А–∞–љ—В–љ–Њ—Б—В–Є –Ї¬†–Х2 –Я–Ф–У –њ—А–Є –Я–С–• —П–≤–ї—П–µ—В—Б—П –љ–∞—З–∞–ї—М–љ—Л–Љ —Б–Њ–±—Л—В–Є–µ–Љ –≤¬†–Љ–µ—Е–∞–љ–Є–Ј–Љ–µ –Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є—П –Р–Ь–Р –Є¬†–Є–≥—А–∞–µ—В –≤–∞–ґ–љ—Г—О —А–Њ–ї—М –≤¬†–Ї–∞—З–µ—Б—В–≤–µ –Ј–∞–њ—Г—Б–Ї–∞—О—Й–µ–≥–Њ —Д–∞–Ї—В–Њ—А–∞ –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є—П —Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В–Њ–≤ [1, 11, 32]. –Ь–љ–µ–љ–Є–µ –Њ–± –Є–Љ–Љ—Г–љ–Њ–Њ–њ–Њ—Б—А–µ–і–Њ–≤–∞–љ–љ–Њ–Љ —А–∞–Ј—А—Г—И–µ–љ–Є–Є –Љ–µ–ї–Ї–Є—Е –Є¬†—Б—А–µ–і–љ–Є—Е –≤–љ—Г—В—А–Є–њ–µ—З–µ–љ–Њ—З–љ—Л—Е –ґ–µ–ї—З–љ—Л—Е –њ—А–Њ—В–Њ–Ї–Њ–≤ –Њ–±—Б—Г–ґ–і–∞–µ—В—Б—П –Љ–љ–Њ–≥–Є–Љ–Є –Є—Б—Б–ї–µ–і–Њ–≤–∞—В–µ–ї—П–Љ–Є [1, 13, 33]. –Я—А–Є —Н—В–Њ–Љ —З–µ—В–Ї–Є—Е –і–Њ–Ї–∞–Ј–∞—В–µ–ї—М—Б—В–≤ –њ–Њ–≤—А–µ–ґ–і–∞—О—Й–µ–≥–Њ –і–µ–є—Б—В–≤–Є—П –Є–Љ–Љ—Г–љ–љ–Њ–≥–Њ –Ї–Њ–Љ–њ–ї–µ–Ї—Б–∞ –Х2 –Я–Ф–У-–∞–љ—В–Є–≥–µ–љ-–Р–Ь–Р –љ–∞ —Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В—Л –љ–µ—В, –Є¬†–Љ–µ—Е–∞–љ–Є–Ј–Љ —Н—В–Њ–≥–Њ –њ–Њ–≤—А–µ–ґ–і–∞—О—Й–µ–≥–Њ –і–µ–є—Б—В–≤–Є—П –Њ—Б—В–∞–µ—В—Б—П –љ–µ–Є–Ј–≤–µ—Б—В–љ—Л–Љ.
–Э–Њ –Є–Ј–Љ–µ–љ–µ–љ–Є–µ —В–Њ–ї–µ—А–∞–љ—В–љ–Њ—Б—В–Є –Є–Љ–Љ—Г–љ–љ–Њ–є —Б–Є—Б—В–µ–Љ—Л –Ї¬†–≤—Л—Б–Њ–Ї–Њ–Ї–Њ–љ—Б–µ—А–≤–∞—В–Є–≤–љ–Њ–Љ—Г –Є¬†¬Ђ–≤–µ–Ј–і–µ—Б—Г—Й–µ–Љ—Г¬ї –±–µ–ї–Ї—Г –Х2 –Я–Ф–У –і–Њ–ї–ґ–љ–Њ –њ—А–Є–≤–Њ–і–Є—В—М –Ї¬†—А–∞–Ј–≤–Є—В–Є—О –љ–∞—А—Г—И–µ–љ–Є–є, –Ї–Њ—В–Њ—А—Л–µ –±—Г–і—Г—В –љ–Њ—Б–Є—В—М —Б–Є—Б—В–µ–Љ–љ—Л–є —Е–∞—А–∞–Ї—В–µ—А. –Р¬†–њ—А–Є –Я–С–• –Є–Ј–Љ–µ–љ–µ–љ–Є—П –Њ–≥—А–∞–љ–Є—З–µ–љ—Л –ї–Њ–Ї–∞–ї—М–љ—Л–Љ –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є–µ–Љ –Љ–µ–ї–Ї–Є—Е –Є¬†—Б—А–µ–і–љ–Є—Е –≤–љ—Г—В—А–Є–њ–µ—З–µ–љ–Њ—З–љ—Л—Е –ґ–µ–ї—З–љ—Л—Е –њ—А–Њ—В–Њ–Ї–Њ–≤. –Ш¬†—Н—В–Њ –±–Њ–ї—М—И–µ —Г–Ї–∞–Ј—Л–≤–∞–µ—В –љ–∞ —В–Њ, —З—В–Њ –њ—А–Є –Я–С–•, –≤–µ—А–Њ—П—В–љ–µ–µ –≤—Б–µ–≥–Њ, –њ—А–Њ–Є—Б—Е–Њ–і—П—В –Є–Ј–Љ–µ–љ–µ–љ–Є—П —Б¬†—Б–∞–Љ–Є–Љ –∞–љ—В–Є–≥–µ–љ–Њ–Љ –Х2 –Я–Ф–У, –Ї–Њ—В–Њ—А—Л–µ –Љ–µ–љ—П—О—В –µ–≥–Њ –Є–Љ–Љ—Г–љ–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–µ —Б–≤–Њ–є—Б—В–≤–∞ –і–Њ —В–∞–Ї–Њ–є —Б—В–µ–њ–µ–љ–Є, —З—В–Њ –љ–µ–Є–Ј–Љ–µ–љ–µ–љ–љ–∞—П –Є–Љ–Љ—Г–љ–љ–∞—П —Б–Є—Б—В–µ–Љ–∞ –љ–∞—З–Є–љ–∞–µ—В –≤–Њ—Б–њ—А–Є–љ–Є–Љ–∞—В—М –µ–≥–Њ –Ї–∞–Ї —З—Г–ґ–µ—А–Њ–і–љ—Л–є –љ–µ–Њ–∞–љ—В–Є–≥–µ–љ. –Ш¬†–њ—А–Њ–Є—Б—Е–Њ–і–Є—В —Н—В–Њ —Б¬†–∞–љ—В–Є–≥–µ–љ–Њ–Љ –Х2 –Я–Ф–У, —А–∞—Б–њ–Њ–ї–Њ–ґ–µ–љ–љ—Л–Љ –≤¬†–Љ–Є—В–Њ—Е–Њ–љ–і—А–Є—П—Е –С–≠–Ъ, –≤—Л—Б—В–Є–ї–∞—О—Й–Є—Е —В–Њ–ї—М–Ї–Њ –Љ–µ–ї–Ї–Є–µ –Є¬†—Б—А–µ–і–љ–Є–µ –ґ–µ–ї—З–љ—Л–µ –њ—А–Њ—В–Њ–Ї–Є. –Ш¬†–Є–Љ–µ–љ–љ–Њ —Н—В–Є –Є–Ј–Љ–µ–љ–µ–љ–Є—П —Б¬†–∞–љ—В–Є–≥–µ–љ–Њ–Љ –Х2 –Я–Ф–У, —А–∞—Б–њ–Њ–ї–Њ–ґ–µ–љ–љ—Л–Љ –љ–∞ –≤–љ—Г—В—А–µ–љ–љ–µ–є –Љ–µ–Љ–±—А–∞–љ–µ –Љ–Є—В–Њ—Е–Њ–љ–і—А–Є–є –Љ–µ–ї–Ї–Є—Е —Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В–Њ–≤, –і–Њ–ї–ґ–љ—Л –њ–Њ—Б–ї—Г–ґ–Є—В—М –Ј–∞–њ—Г—Б–Ї–∞—О—Й–Є–Љ –Љ–Њ–Љ–µ–љ—В–Њ–Љ –і–ї—П –Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є—П –Р–Ь–Р. –Я–Њ—Б–ї–µ–і–љ–Є–µ –±—Г–і—Г—В —А–µ–∞–≥–Є—А–Њ–≤–∞—В—М —Б¬†–љ–µ–Њ–∞–љ—В–Є–≥–µ–љ–Њ–Љ –Х2 –Я–Ф–У —В–Њ–ї—М–Ї–Њ –≤¬†—Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В–∞—Е –Љ–µ–ї–Ї–Є—Е –Є¬†—Б—А–µ–і–љ–Є—Е –ґ–µ–ї—З–љ—Л—Е –њ—А–Њ—В–Њ–Ї–Њ–≤, –љ–Њ –љ–µ –±—Г–і—Г—В —А–µ–∞–≥–Є—А–Њ–≤–∞—В—М —Б¬†–љ–Њ—А–Љ–∞–ї—М–љ—Л–Љ–Є, –љ–µ–Є–Ј–Љ–µ–љ–µ–љ–љ—Л–Љ–Є –Х2 –Я–Ф–У –≤¬†–Љ–Є—В–Њ—Е–Њ–љ–і—А–Є—П—Е –і—А—Г–≥–Є—Е –Ї–ї–µ—В–Њ–Ї.
–Р–љ–∞–ї–Є–Ј –Є–Љ–µ—О—Й–Є—Е—Б—П –љ–∞—Г—З–љ—Л—Е –і–∞–љ–љ—Л—Е –њ–Њ–Ї–∞–Ј—Л–≤–∞–µ—В, —З—В–Њ, –≤–µ—А–Њ—П—В–љ–µ–µ –≤—Б–µ–≥–Њ, –ї–Њ–Ї–∞–ї—М–љ—Л–є —Е–∞—А–∞–Ї—В–µ—А –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є–є, –њ—А–Њ–Є—Б—Е–Њ–і—П—Й–Є–є –≤¬†—Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В–∞—Е, –≤—Л—Б—В–Є–ї–∞—О—Й–Є—Е –Љ–µ–ї–Ї–Є–µ –Є¬†—Б—А–µ–і–љ–Є–µ –ґ–µ–ї—З–љ—Л–µ –њ—А–Њ—В–Њ–Ї–Є –њ—А–Є –Я–С–•, –Њ–±—Г—Б–ї–Њ–≤–ї–µ–љ –Є–Ј–Љ–µ–љ–µ–љ–Є–µ–Љ –Є–Љ–Љ—Г–љ–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є—Е —Б–≤–Њ–є—Б—В–≤ –∞–љ—В–Є–≥–µ–љ–∞ –Х2 –Я–Ф–У –Љ–Є—В–Њ—Е–Њ–љ–і—А–Є–є –Љ–µ–ї–Ї–Є—Е –С–≠–Ъ. –Х—Б–ї–Є —Н—В–Њ —В–∞–Ї, —В–Њ –Ї—А–∞–є–љ–µ –≤–∞–ґ–љ–Њ –њ—А–µ–і—Б—В–∞–≤–ї—П—В—М –Љ–µ—Е–∞–љ–Є–Ј–Љ –Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є—П —В–∞–Ї–Њ–≥–Њ –Є–Љ–Љ—Г–љ–Њ–Є–Ј–Љ–µ–љ–µ–љ–љ–Њ–≥–Њ –∞–љ—В–Є–≥–µ–љ–∞ –Є¬†–≤–∞–ґ–љ–Њ –Ј–љ–∞—В—М, –Ї–∞–Ї–Њ–є —Д–∞–Ї—В–Њ—А –Љ–Њ–ґ–µ—В –Ј–∞–њ—Г—Б—В–Є—В—М –њ—А–Њ—Ж–µ—Б—Б –њ—А–µ–≤—А–∞—Й–µ–љ–Є—П –Х2 –Я–Ф–У –≤¬†–љ–µ–Њ–∞–љ—В–Є–≥–µ–љ. –°¬†—Г—З–µ—В–Њ–Љ —В–Њ–≥–Њ, —З—В–Њ –Х2 –Я–Ф–У¬†вАУ —Н—В–Њ –ї–Є—И—М —З–∞—Б—В—М –Я–Ф–У-–Ї–Њ–Љ–њ–ї–µ–Ї—Б–∞, —В–Њ –љ–µ–Њ–±—Е–Њ–і–Є–Љ–Њ –њ–Њ–љ—П—В—М –Є¬†–Њ–±—К—П—Б–љ–Є—В—М, –њ–Њ—З–µ–Љ—Г –Є–Љ–µ–љ–љ–Њ —Н—В–∞ —З–∞—Б—В—М (—Б—Г–±—К–µ–і–Є–љ–Є—Ж–∞) –Я–Ф–У-–Ї–Њ–Љ–њ–ї–µ–Ї—Б–∞ –њ—А–Є–Њ–±—А–µ—В–∞–µ—В —Б–≤–Њ–є—Б—В–≤–∞ –љ–µ–Њ–∞–љ—В–Є–≥–µ–љ–∞.
–Ъ—А–Њ–Љ–µ —В–Њ–≥–Њ, –і–ї—П –Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є—П –Р–Ь–Р –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ –њ—А–µ–≤—А–∞—Й–µ–љ–Є—П –Х2 –Я–Ф–У –≤¬†–Є–Љ–Љ—Г–љ–Њ–Є–Ј–Љ–µ–љ–µ–љ–љ—Л–є –љ–µ–Њ–∞–љ—В–Є–≥–µ–љ. –Т–∞–ґ–љ–Њ —В–∞–Ї–ґ–µ –њ–Њ–љ–Є–Љ–∞—В—М, –Ї–∞–Ї —Н—В–Њ—В –љ–µ–Њ–∞–љ—В–Є–≥–µ–љ –њ–Њ–Ї–Є–і–∞–µ—В –≤–љ—Г—В—А–µ–љ–љ—О—О –Љ–µ–Љ–±—А–∞–љ—Г –Љ–Є—В–Њ—Е–Њ–љ–і—А–Є–є, —Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В–Њ–≤ –Є¬†–Ї–∞–Ї –њ—А–Њ–Є—Б—Е–Њ–і–Є—В –µ–≥–Њ –≤—Б—В—А–µ—З–∞ —Б¬†–Є–Љ–Љ—Г–љ–Њ–Ї–Њ–Љ–њ–µ—В–µ–љ—В–љ—Л–Љ–Є –Ї–ї–µ—В–Ї–∞–Љ–Є, –Њ—В–≤–µ—З–∞—О—Й–Є–Љ–Є –Ј–∞ –≤—Л—А–∞–±–Њ—В–Ї—Г –∞—Г—В–Њ–∞–љ—В–Є—В–µ–ї. –Ґ–∞–Ї–ґ–µ –Ї—А–∞–є–љ–µ –≤–∞–ґ–љ–Њ –њ–Њ–љ—П—В—М, –њ–Њ—З–µ–Љ—Г –≤¬†—Н—В–Њ—В –њ—А–Њ—Ж–µ—Б—Б –≤–Њ–≤–ї–µ—З–µ–љ –∞–љ—В–Є–≥–µ–љ –Х2 –Я–Ф–У –Ї–ї–µ—В–Њ–Ї –±–Є–ї–Є–∞—А–љ–Њ–≥–Њ —Н–њ–Є—В–µ–ї–Є—П, —А–∞—Б–њ–Њ–ї–Њ–ґ–µ–љ–љ–Њ–≥–Њ –≤¬†–ґ–µ–ї—З–љ—Л—Е –њ—А–Њ—В–Њ–Ї–∞—Е –Љ–µ–ї–Ї–Њ–≥–Њ –Є¬†—Б—А–µ–і–љ–µ–≥–Њ —А–∞–Ј–Љ–µ—А–Њ–≤ [8]. –Ґ—А–Є–≥–≥–µ—А—Л –Є¬†–Љ–µ—Е–∞–љ–Є–Ј–Љ—Л, –Ї–Њ—В–Њ—А—Л–µ –Љ–Њ–≥—Г—В –Ј–∞–њ—Г—Б—В–Є—В—М —В–∞–Ї–Є–µ –њ—А–Њ—Ж–µ—Б—Б—Л –≤¬†—Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В–∞—Е –Љ–µ–ї–Ї–Є—Е –Є¬†—Б—А–µ–і–љ–Є—Е –ґ–µ–ї—З–љ—Л—Е –њ—А–Њ—В–Њ–Ї–Њ–≤, –љ–µ–Є–Ј–≤–µ—Б—В–љ—Л.
–Т –њ–Њ—Б–ї–µ–і–љ–Є–µ –і–µ—Б—П—В–Є–ї–µ—В–Є—П –њ–Њ—П–≤–Є–ї–Є—Б—М –љ–Њ–≤—Л–µ –љ–∞—Г—З–љ—Л–µ –і–∞–љ–љ—Л–µ, –њ–Њ–Ј–≤–Њ–ї—П—О—Й–Є–µ –њ—А–Њ—П—Б–љ–Є—В—М –њ–∞—В–Њ–≥–µ–љ–µ–Ј –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є—П —Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В–Њ–≤ [34, 35]. –≠—В–Є –і–∞–љ–љ—Л–µ —Б–≤–Є–і–µ—В–µ–ї—М—Б—В–≤—Г—О—В –Њ¬†—В–Њ–Љ, —З—В–Њ –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є–µ –Љ–µ–ї–Ї–Є—Е —Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В–Њ–≤ —Б–≤—П–Ј–∞–љ–Њ –љ–µ —Б¬†–Є–Љ–Љ—Г–љ–Њ–Њ–њ–Њ—Б—А–µ–і–Њ–≤–∞–љ–љ—Л–Љ–Є —А–µ–∞–Ї—Ж–Є—П–Љ–Є, –∞¬†—Б –і–µ—Д–µ–Ї—В–Њ–Љ –±–Є–ї–Є–∞—А–љ–Њ–≥–Њ –±–Є–Ї–∞—А–±–Њ–љ–∞—В–љ–Њ–≥–Њ ¬Ђ–Ј–Њ–љ—В–Є–Ї–∞¬ї. –Ш–Љ–µ–љ–љ–Њ –Њ–љ –Ј–∞—Й–Є—Й–∞–µ—В –С–≠–Ъ –Њ—В –і–µ—В–µ—А–≥–µ–љ—В–љ–Њ–≥–Њ –і–µ–є—Б—В–≤–Є—П —В–Њ–Ї—Б–Є—З–љ—Л—Е –ґ–µ–ї—З–љ—Л—Е –Ї–Є—Б–ї–Њ—В –≤¬†—Д–Є–Ј–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є—Е —Г—Б–ї–Њ–≤–Є—П—Е [34вАУ37]. –Я–Њ–Ї–∞–Ј–∞–љ–Њ, —З—В–Њ –њ—А–Є –Я–С–• —Г–Љ–µ–љ—М—И–∞–µ—В—Б—П –≤—Л—А–∞–±–Њ—В–Ї–∞ –±–Є–Ї–∞—А–±–Њ–љ–∞—В–∞ (HCO3-), —З—В–Њ –њ—А–Є–≤–Њ–і–Є—В –Ї¬†–њ–Њ—Б—В—Г–њ–ї–µ–љ–Є—О –Є¬†–љ–∞–Ї–Њ–њ–ї–µ–љ–Є—О –ґ–µ–ї—З–љ—Л—Е –Ї–Є—Б–ї–Њ—В –≤¬†—Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В–∞—Е (—В–µ–Њ—А–Є—П ¬Ђ–і—Л—А—П–≤–Њ–≥–Њ¬ї –±–Є–Ї–∞—А–±–Њ–љ–∞—В–љ–Њ–≥–Њ ¬Ђ–Ј–Њ–љ—В–Є–Ї–∞¬ї) [34вАУ37]. –≠—В–Є –і–∞–љ–љ—Л–µ –њ–Њ–Ј–≤–Њ–ї–Є–ї–Є –љ–∞–Љ –≤—Л—Б–Ї–∞–Ј–∞—В—М –њ—А–µ–і–њ–Њ–ї–Њ–ґ–µ–љ–Є–µ, —З—В–Њ –њ—А–Њ–Є—Б—Е–Њ–і—П—Й–µ–µ –њ—А–Є —Н—В–Њ–Љ –њ–Њ—Б—В–µ–њ–µ–љ–љ–Њ–µ –љ–∞–Ї–Њ–њ–ї–µ–љ–Є–µ –ґ–µ–ї—З–љ—Л—Е –Ї–Є—Б–ї–Њ—В –≤¬†–С–≠–Ъ, —Г–ґ–µ –≤¬†–∞—Б–Є–Љ–њ—В–Њ–Љ–∞—В–Є—З–µ—Б–Ї–Њ–є —Б—В–∞–і–Є–Є –Я–С–•, –Љ–Њ–ґ–µ—В —Б–ї—Г–ґ–Є—В—М –њ—Г—Б–Ї–Њ–≤—Л–Љ –Љ–µ—Е–∞–љ–Є–Ј–Љ–Њ–Љ –і–ї—П –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є—П –Љ–µ–ї–Ї–Є—Е —Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В–Њ–≤, —А–∞–Ј–≤–Є—В–Є—П –і—Г–Ї—В—Г–ї–Њ–њ–µ–љ–Є–Є –Є¬†–Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є—П –Р–Ь–Р [2]. –Ф–ї—П —Н—В–Њ–≥–Њ –≤–∞–ґ–љ–Њ –њ—А–µ–і—Б—В–∞–≤–ї—П—В—М –Љ–µ—Е–∞–љ–Є–Ј–Љ—Л –Ј–∞—Й–Є—В—Л –Љ–µ–ї–Ї–Є—Е –Є¬†–Ї—А—Г–њ–љ—Л—Е –С–≠–Ъ –Њ—В —В–Њ–Ї—Б–Є—З–µ—Б–Ї–Њ–≥–Њ (–і–µ—В–µ—А–≥–µ–љ—В–љ–Њ–≥–Њ) –і–µ–є—Б—В–≤–Є—П –ґ–µ–ї—З–љ—Л—Е –Ї–Є—Б–ї–Њ—В.
–Ь–µ—Е–∞–љ–Є–Ј–Љ—Л –Ј–∞—Й–Є—В—Л —Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В–Њ–≤ –Њ—В —В–Њ–Ї—Б–Є—З–µ—Б–Ї–Њ–≥–Њ –і–µ–є—Б—В–≤–Є—П –ґ–µ–ї—З–љ—Л—Е –Ї–Є—Б–ї–Њ—В
–Т—Л–і–µ–ї—П—О—В –Љ–µ–ї–Ї–Є–µ –Є¬†–Ї—А—Г–њ–љ—Л–µ —Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В—Л –≤¬†–Ј–∞–≤–Є—Б–Є–Љ–Њ—Б—В–Є –Њ—В –Є—Е —А–∞–Ј–Љ–µ—А–Њ–≤ –Є¬†—А–∞—Б–њ–Њ–ї–Њ–ґ–µ–љ–Є—П –≤¬†–Љ–µ–ї–Ї–Є—Е –Є¬†–Ї—А—Г–њ–љ—Л—Е –ґ–µ–ї—З–љ—Л—Е –њ—А–Њ—В–Њ–Ї–∞—Е [38]. –Ю–љ–Є –њ–Њ-—А–∞–Ј–љ–Њ–Љ—Г —Г—З–∞—Б—В–≤—Г—О—В –≤¬†–њ—А–Њ—Ж–µ—Б—Б–∞—Е —Б–µ–Ї—А–µ—Ж–Є–Є –Є¬†–∞–±—Б–Њ—А–±—Ж–Є–Є [39]. –Э–Њ –ґ–µ–ї—З–љ—Л–µ –Ї–Є—Б–ї–Њ—В—Л –Њ–Ї–∞–Ј—Л–≤–∞—О—В —В–Њ–Ї—Б–Є—З–µ—Б–Ї–Њ–µ –і–µ–є—Б—В–≤–Є–µ –Ї–∞–Ї –љ–∞ –Љ–µ–ї–Ї–Є–µ, —В–∞–Ї –Є¬†–љ–∞ –Ї—А—Г–њ–љ—Л–µ –С–≠–Ъ. –Я—А–Є —Н—В–Њ–Љ –Њ–±–∞ —В–Є–њ–∞ –С–≠–Ъ –Є–Љ–µ—О—В —А–∞–Ј–ї–Є—З–љ—Л–µ –Ј–∞—Й–Є—В–љ—Л–µ –Љ–µ—Е–∞–љ–Є–Ј–Љ—Л –і–ї—П –њ—А–Њ—В–Є–≤–Њ–і–µ–є—Б—В–≤–Є—П –њ–Њ–≤—А–µ–ґ–і–∞—О—Й–µ–Љ—Г (—В–Њ–Ї—Б–Є—З–µ—Б–Ї–Њ–Љ—Г) –і–µ–є—Б—В–≤–Є—О –ґ–µ–ї—З–љ—Л—Е –Ї–Є—Б–ї–Њ—В. –Ъ¬†–Є–Ј–≤–µ—Б—В–љ—Л–Љ —Д–∞–Ї—В–Њ—А–∞–Љ –Ј–∞—Й–Є—В—Л —Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В–Њ–≤ –Њ—В–љ–Њ—Б—П—В –≤—Л—А–∞–±–Њ—В–Ї—Г –Є¬†—Б–µ–Ї—А–µ—Ж–Є—О –Љ—Г—Ж–Є–љ–∞ –Є¬†–±–Є–Ї–∞—А–±–Њ–љ–∞—В–∞ –С–≠–Ъ [40, 41]. –Т¬†—Д–Є–Ј–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є—Е —Г—Б–ї–Њ–≤–Є—П—Е –Њ—Б–љ–Њ–≤–љ–Њ–є —Д—Г–љ–Ї—Ж–Є–µ–є —Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В–Њ–≤ —П–≤–ї—П–µ—В—Б—П –±–Є–ї–Є–∞—А–љ–∞—П —Б–µ–Ї—А–µ—Ж–Є—П HCO3- [41]. –Э–∞—Г—З–љ—Л–µ –і–∞–љ–љ—Л–µ —Б–≤–Є–і–µ—В–µ–ї—М—Б—В–≤—Г—О—В –Њ¬†—В–Њ–Љ, —З—В–Њ HCO3- –Є–≥—А–∞–µ—В –Ї—А–Є—В–Є—З–µ—Б–Ї–Є –≤–∞–ґ–љ—Г—О —А–Њ–ї—М –≤¬†–Ј–∞—Й–Є—В–µ –С–≠–Ъ (–Ј–∞—Й–Є—В–љ—Л–є –±–Є–ї–Є–∞—А–љ—Л–є –±–Є–Ї–∞—А–±–Њ–љ–∞—В–љ—Л–є ¬Ђ–Ј–Њ–љ—В–Є–Ї¬ї) –Њ—В —В–Њ–Ї—Б–Є—З–µ—Б–Ї–Њ–≥–Њ –≤–Њ–Ј–і–µ–є—Б—В–≤–Є—П –ґ–µ–ї—З–љ—Л—Е –Ї–Є—Б–ї–Њ—В [36, 37]. –Э–∞ –і–Њ–ї—О HCO3- –њ—А–Є—Е–Њ–і–Є—В—Б—П –Њ—В 25 –і–Њ 40% –Њ–±—Й–µ–≥–Њ –Њ–±—К–µ–Љ–∞ –≤—Л–і–µ–ї—П–µ–Љ–Њ–є –ґ–µ–ї—З–Є [36, 42, 43]. –£¬†—З–µ–ї–Њ–≤–µ–Ї–∞ —Б–µ–Ї—А–µ—Ж–Є—П HCO3- —Б¬†–ґ–µ–ї—З—М—О –њ–Њ–і–і–µ—А–ґ–Є–≤–∞–µ—В —Д–Є–Ј–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є –љ–µ–є—В—А–∞–ї—М–љ—Л–є –Є–ї–Є —Б–ї–∞–±–Њ—Й–µ–ї–Њ—З–љ–Њ–є —Г—А–Њ–≤–µ–љ—М pH –≤¬†–њ—А–Њ—Б–≤–µ—В–µ –ґ–µ–ї—З–љ—Л—Е –њ—А–Њ—В–Њ–Ї–Њ–≤ [36, 40, 42]. –•–Њ—А–Њ—И–Њ –Є–Ј–≤–µ—Б—В–љ–Њ, —З—В–Њ HCO3- –≤—Л—А–∞–±–∞—В—Л–≤–∞–µ—В—Б—П –Ї–∞–Ї –Љ–µ–ї–Ї–Є–Љ–Є, —В–∞–Ї –Є¬†–Ї—А—Г–њ–љ—Л–Љ–Є —Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В–∞–Љ–Є, —В–Њ –µ—Б—В—М –љ–∞ –≤—Б–µ–Љ –њ—А–Њ—В—П–ґ–µ–љ–Є–Є –±–Є–ї–Є–∞—А–љ–Њ–≥–Њ –і–µ—А–µ–≤–∞. –Ю–і–љ–∞–Ї–Њ –Љ–µ—Е–∞–љ–Є–Ј–Љ—Л –≤—Л—А–∞–±–Њ—В–Ї–Є –Є¬†—Б–µ–Ї—А–µ—Ж–Є–Є HCO3- –Љ–µ–ї–Ї–Є–Љ–Є –Є¬†–Ї—А—Г–њ–љ—Л–Љ–Є –С–≠–Ъ –Њ—Б—Г—Й–µ—Б—В–≤–ї—П—О—В—Б—П –њ–Њ-—А–∞–Ј–љ–Њ–Љ—Г, —З—В–Њ –њ–Њ–і—А–Њ–±–љ–Њ —А–∞—Б—Б–Љ–Њ—В—А–µ–љ–Њ –≤¬†—Б—В–∞—В—М–µ [39] –Є¬†–Њ–±–Ј–Њ—А–µ [2].
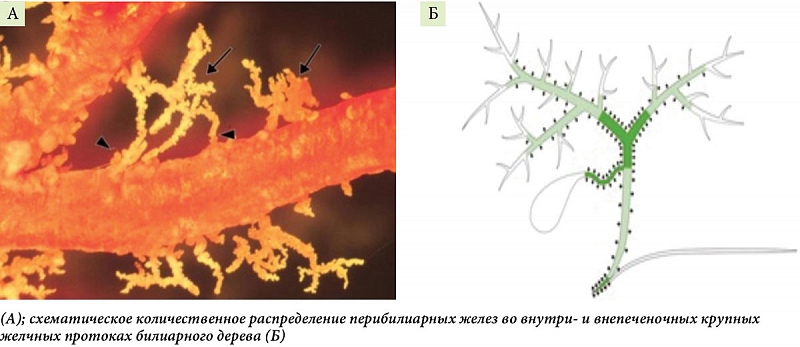
–Т—Л—А–∞–±–Њ—В–Ї–∞ –Љ—Г—Ж–Є–љ–Њ–≤—Л—Е –≥–ї–Є–Ї–Њ–њ—А–Њ—В–µ–Є–і–Њ–≤ –Њ—Б—Г—Й–µ—Б—В–≤–ї—П–µ—В—Б—П –њ–µ—А–Є–±–Є–ї–Є–∞—А–љ—Л–Љ–Є –ґ–µ–ї–µ–Ј–∞–Љ–Є (–Я–С–Ц) [44]. –Я–Њ—Б–ї–µ–і–љ–Є–µ —А–∞—Б–њ–Њ–ї–∞–≥–∞—О—В—Б—П –≤¬†—Б—В–µ–љ–Ї–∞—Е –Ї—А—Г–њ–љ—Л—Е –≤–љ—Г—В—А–Є- –Є¬†–≤–љ–µ–њ–µ—З–µ–љ–Њ—З–љ—Л—Е –ґ–µ–ї—З–љ—Л—Е –њ—А–Њ—В–Њ–Ї–Њ–≤ –Є¬†–љ–µ–њ–Њ—Б—А–µ–і—Б—В–≤–µ–љ–љ–Њ —Б–≤—П–Ј–∞–љ—Л —Б¬†–Є—Е –њ—А–Њ—Б–≤–µ—В–Њ–Љ (—А–Є—Б. 1 –Р). –≠–Ї—Б–њ–µ—А–Є–Љ–µ–љ—В–∞–ї—М–љ—Л–µ –і–∞–љ–љ—Л–µ —Г–Ї–∞–Ј—Л–≤–∞—О—В –љ–∞ —В–Њ, —З—В–Њ –≥–ї–Є–Ї–Њ–Ї–∞–ї–Є–Ї—Б, –њ–Њ–Ї—А—Л–≤–∞—О—Й–Є–є –∞–њ–Є–Ї–∞–ї—М–љ—Г—О –њ–Њ–≤–µ—А—Е–љ–Њ—Б—В—М –Љ–µ–Љ–±—А–∞–љ –Ї—А—Г–њ–љ—Л—Е —Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В–Њ–≤, —Б¬†–≥–ї–Є–Ї–Њ–Ј–Є–ї–Є—А–Њ–≤–∞–љ–љ—Л–Љ–Є –Љ—Г—Ж–Є–љ–∞–Љ–Є –Є¬†–і—А—Г–≥–Є–Љ–Є –≥–ї–Є–Ї–∞–љ—Б–Њ–і–µ—А–ґ–∞—Й–Є–Љ–Є –Љ–µ–Љ–±—А–∞–љ–љ—Л–Љ–Є –≥–ї–Є–Ї–Њ–њ—А–Њ—В–µ–Є–љ–∞–Љ–Є —Б—В–∞–±–Є–ї–Є–Ј–Є—А—Г–µ—В –±–Є–ї–Є–∞—А–љ—Л–є –±–Є–Ї–∞—А–±–Њ–љ–∞—В–љ—Л–є ¬Ђ–Ј–Њ–љ—В–Є–Ї¬ї, –њ–Њ–Љ–Њ–≥–∞—П —В–∞–Ї–Є–Љ –Њ–±—А–∞–Ј–Њ–Љ –Ј–∞—Й–Є—В–Є—В—М –Ї—А—Г–њ–љ—Л–µ —Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В—Л —З–µ–ї–Њ–≤–µ–Ї–∞ –Њ—В —В–Њ–Ї—Б–Є—З–µ—Б–Ї–Њ–≥–Њ (–і–µ—В–µ—А–≥–µ–љ—В–љ–Њ–≥–Њ) –і–µ–є—Б—В–≤–Є—П –ґ–µ–ї—З–љ—Л—Е –Ї–Є—Б–ї–Њ—В [37]. –°–ї–µ–і–Њ–≤–∞—В–µ–ї—М–љ–Њ, –≤—Л—А–∞–±–∞—В—Л–≤–∞–µ–Љ—Л–є –Я–С–Ц –Љ—Г—Ж–Є–љ –Ј–∞—Й–Є—Й–∞–µ—В —Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В—Л —В–Њ–ї—М–Ї–Њ –Ї—А—Г–њ–љ—Л—Е –ґ–µ–ї—З–љ—Л—Е –њ—А–Њ—В–Њ–Ї–Њ–≤ [40]. –Ш—Б—Е–Њ–і—П –Є–Ј —Н—В–Њ–≥–Њ —Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В—Л –Ї—А—Г–њ–љ—Л—Е –≤–љ—Г—В—А–Є- –Є¬†–≤–љ–µ–њ–µ—З–µ–љ–Њ—З–љ—Л—Е –ґ–µ–ї—З–љ—Л—Е –њ—А–Њ—В–Њ–Ї–Њ–≤ –Є–Љ–µ—О—В –і–≤–Њ–є–љ—Г—О –Ј–∞—Й–Є—В—Г: –Љ—Г—Ж–Є–љ, –≤—Л—А–∞–±–∞—В—Л–≤–∞–µ–Љ—Л–є –Я–С–Ц, –Є¬†–±–Є–Ї–∞—А–±–Њ–љ–∞—В.
–Ь–µ–ї–Ї–Є–µ –ґ–µ–ї—З–љ—Л–µ –њ—А–Њ—В–Њ–Ї–Є –њ–µ—А–Є–±–Є–ї–Є–∞—А–љ—Л—Е –ґ–µ–ї–µ–Ј –љ–µ —Б–Њ–і–µ—А–ґ–∞—В (—А–Є—Б. 1 –С), —З—В–Њ —Б–Њ–њ—А–Њ–≤–Њ–ґ–і–∞–µ—В—Б—П –Њ—В—Б—Г—В—Б—В–≤–Є–µ–Љ –≤¬†–љ–Є—Е –Љ—Г—Ж–Є–љ–∞ [40, 44]. –Т¬†—А–µ–Ј—Г–ї—М—В–∞—В–µ –љ–∞ —Г—А–Њ–≤–љ–µ –≤–љ—Г—В—А–Є–і–Њ–ї—М–Ї–Њ–≤—Л—Е, –Љ–µ–ґ–і–Њ–ї—М–Ї–Њ–≤—Л—Е –Є¬†—Б–µ–њ—В–∞–ї—М–љ—Л—Е –ґ–µ–ї—З–љ—Л—Е –њ—А–Њ—В–Њ–Ї–Њ–≤ —Д–∞–Ї—В–Њ—А–Њ–Љ –Ј–∞—Й–Є—В—Л –С–≠–Ъ —Б–ї—Г–ґ–Є—В —В–Њ–ї—М–Ї–Њ –±–Є–Ї–∞—А–±–Њ–љ–∞—В. –Ь–µ—Е–∞–љ–Є–Ј–Љ—Л –≤—Л—А–∞–±–Њ—В–Ї–Є –Є¬†—Б–µ–Ї—А–µ—Ж–Є–Є –±–Є–Ї–∞—А–±–Њ–љ–∞—В–∞ –Љ–µ–ї–Ї–Є–Љ–Є –Є¬†–Ї—А—Г–њ–љ—Л–Љ–Є —Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В–∞–Љ–Є –Њ—В–ї–Є—З–∞—О—В—Б—П, —З—В–Њ —А–∞—Б—Б–Љ–∞—В—А–Є–≤–∞–µ—В—Б—П –≤¬†–Њ–±–Ј–Њ—А–µ [2]. –Ш–Љ–µ–љ–љ–Њ —А–∞–Ј–ї–Є—З–Є—П –≤¬†–Љ–µ—Е–∞–љ–Є–Ј–Љ–∞—Е –Ј–∞—Й–Є—В—Л –Љ–µ–ї–Ї–Є—Е –Є¬†–Ї—А—Г–њ–љ—Л—Е —Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В–Њ–≤ –Њ—В —В–Њ–Ї—Б–Є—З–µ—Б–Ї–Њ–≥–Њ –і–µ–є—Б—В–≤–Є—П –ґ–µ–ї—З–љ—Л—Е –Ї–Є—Б–ї–Њ—В –Є–≥—А–∞—О—В –≤–∞–ґ–љ—Г—О —А–Њ–ї—М –≤¬†–Љ–µ—Е–∞–љ–Є–Ј–Љ–µ —А–∞–Ј–≤–Є—В–Є—П –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є–є –Љ–µ–ї–Ї–Є—Е (–љ–Њ –љ–µ –Ї—А—Г–њ–љ—Л—Е) –С–≠–Ъ –њ—А–Є –Я–С–•.
–Ґ–µ–Њ—А–Є—П ¬Ђ–і—Л—А—П–≤–Њ–≥–Њ¬ї –±–Є–ї–Є–∞—А–љ–Њ–≥–Њ –±–Є–Ї–∞—А–±–Њ–љ–∞—В–љ–Њ–≥–Њ ¬Ђ–Ј–Њ–љ—В–Є–Ї–∞¬ї –њ—А–Є –Я–С–•
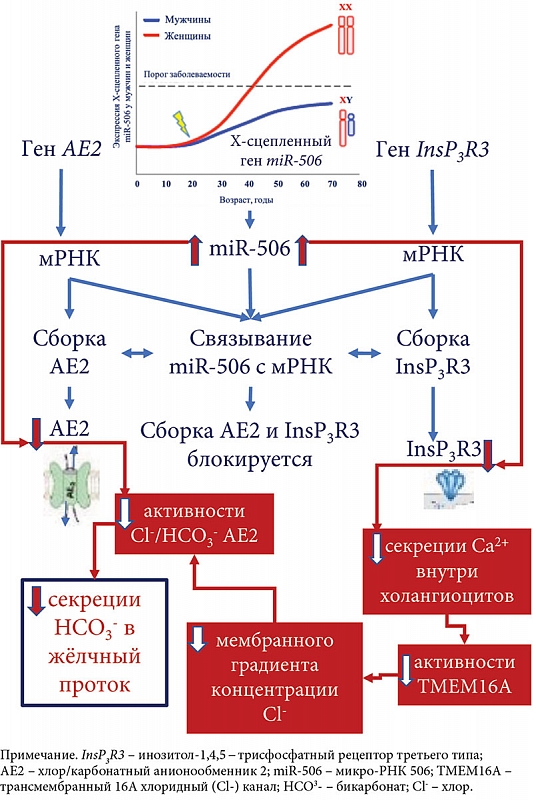
–Ґ–µ–Њ—А–Є—П ¬Ђ–і—Л—А—П–≤–Њ–≥–Њ¬ї –±–Є–ї–Є–∞—А–љ–Њ–≥–Њ –±–Є–Ї–∞—А–±–Њ–љ–∞—В–љ–Њ–≥–Њ ¬Ђ–Ј–Њ–љ—В–Є–Ї–∞¬ї –Њ—Б–љ–Њ–≤–∞–љ–∞ –љ–∞ —А—П–і–µ –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є—Е –Є¬†—Н–Ї—Б–њ–µ—А–Є–Љ–µ–љ—В–∞–ї—М–љ—Л—Е —А–∞–±–Њ—В, –њ–Њ–Ї–∞–Ј—Л–≤–∞—О—Й–Є—Е –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ–µ –њ–Њ—Б—В—Г–њ–ї–µ–љ–Є–µ HCO3- –≤¬†–ґ–µ–ї—З–љ—Л–µ –њ—А–Њ—В–Њ–Ї–Є –њ—А–Є –Я–С–•, —З—В–Њ –њ—А–Є–≤–Њ–і–Є—В –Ї¬†—Б–Љ–µ—Й–µ–љ–Є—О —А–Э –≤–љ—Г—В—А–Є–њ—А–Њ—В–Њ–Ї–Њ–≤–Њ–є (–њ–µ—З–µ–љ–Њ—З–љ–Њ–є) –ґ–µ–ї—З–Є –≤¬†—Б–ї–∞–±–Њ–Ї–Є—Б–ї—Г—О –Њ–±–ї–∞—Б—В—М –Є¬†—Г–≤–µ–ї–Є—З–µ–љ–Є–µ —А–Э –≤–љ—Г—В—А–Є —Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В–∞ –≤¬†—Б–ї–∞–±–Њ—Й–µ–ї–Њ—З–љ—Г—О –Њ–±–ї–∞—Б—В—М [45, 46]. –Ю–±—Б—Г–ґ–і–∞–µ—В—Б—П –≤–Њ–≤–ї–µ—З–µ–љ–љ–Њ—Б—В—М –≤¬†—Н—В–Њ—В –њ—А–Њ—Ж–µ—Б—Б InsP3R3 –Є¬†AE2 [45вАУ47]. –Я–Њ–Ї–∞–Ј–∞–љ–Њ, —З—В–Њ –≤¬†–±–Є–Њ–њ—В–∞—В–∞—Е –њ–µ—З–µ–љ–Є –Є¬†–Љ–Њ–љ–Њ–љ—Г–Ї–ї–µ–∞—А–љ—Л—Е –Ї–ї–µ—В–Ї–∞—Е –Ї—А–Њ–≤–Є –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–Я–С–• —Б–љ–Є–ґ–µ–љ–∞ —Н–Ї—Б–њ—А–µ—Б—Б–Є—П –≥–µ–љ–Њ–≤ InsP3R3 –Є¬†AE2, —З—В–Њ —Б–≤–Є–і–µ—В–µ–ї—М—Б—В–≤—Г–µ—В –Њ–± –Є—Е –і–Є—Б—Д—Г–љ–Ї—Ж–Є–Є –Є¬†–≤–Њ–≤–ї–µ—З–µ–љ–Є–Є –≤¬†–њ–∞—В–Њ–≥–µ–љ–µ–Ј –Я–С–• [45, 46]. –°–љ–Є–ґ–µ–љ–Є–µ –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є InsP3R3 –Є¬†AE2, –∞¬†—В–∞–Ї–ґ–µ –љ–∞—А—Г—И–µ–љ–Є–µ –Є—Е —Б–µ–Ї—А–µ—В–Њ—А–љ–Њ–є —Д—Г–љ–Ї—Ж–Є–Є —Б–≤—П–Ј—Л–≤–∞—О—В —Б¬†–Љ–Є–Ї—А–Њ-–†–Э–Ъ 506 (miR-506) [47].
–Т —Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В–∞—Е –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–Я–С–• –Њ—В–Љ–µ—З–∞–µ—В—Б—П –њ–Њ–≤—Л—И–µ–љ–Є–µ –Ї–Њ–ї–Є—З–µ—Б—В–≤–∞ –Є¬†–∞–Ї—В–Є–≤–љ–Њ—Б—В–Є miR-506 [48]. –Я–Њ–Ї–∞–Ј–∞–љ–Њ, —З—В–Њ –њ–Њ–≤—Л—И–µ–љ–Є–µ –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є –•-—Б—Ж–µ–њ–ї–µ–љ–љ–Њ–є miR-506 –≤¬†—Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В–∞—Е –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–Я–С–• –њ—А–Є–≤–Њ–і–Є—В –Ї¬†—Б–љ–Є–ґ–µ–љ–Є—О —Н–Ї—Б–њ—А–µ—Б—Б–Є–Є –Є¬†–∞–Ї—В–Є–≤–љ–Њ—Б—В–Є InsP3R3 –Є¬†–Р–Х2, –∞¬†—В–∞–Ї–ґ–µ –њ–Њ—В–µ–љ—Ж–Є–∞–ї—М–љ–Њ –Њ–±—К—П—Б–љ—П–µ—В –њ—А–µ–Њ–±–ї–∞–і–∞–љ–Є–µ —Н—В–Њ–≥–Њ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П —Г¬†–ґ–µ–љ—Й–Є–љ [2, 48, 49] (—А–Є—Б. 2). –•–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В—Л —З–µ–ї–Њ–≤–µ–Ї–∞, –≤—Л–і–µ–ї–µ–љ–љ—Л–µ –Є–Ј –±–Є–Њ–њ—В–∞—В–Њ–≤ –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–Я–С–•, –њ–Њ–Ї–∞–Ј—Л–≤–∞—О—В —Б–љ–Є–ґ–µ–љ–Є–µ –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є InsP3R3 –Є¬†–Р–Х2 [50]. –Т—Б–ї–µ–і—Б—В–≤–Є–µ —Н—В–Њ–≥–Њ –≥–Њ–Љ–µ–Њ—Б—В–∞–Ј –≤–љ—Г—В—А–Є–Ї–ї–µ—В–Њ—З–љ–Њ–≥–Њ —А–Э (pHi) –≤¬†—Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В–∞—Е –Є¬†—А–Э –≤¬†–ґ–µ–ї—З–љ—Л—Е –њ—А–Њ—В–Њ–Ї–∞—Е —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–Я–С–• –Љ–Њ–ґ–µ—В –њ–Њ–і–≤–µ—А–≥–∞—В—М—Б—П –Є–Ј–Љ–µ–љ–µ–љ–Є—О [49]. –Ш–Ј–Љ–µ–љ–µ–љ–Є—П –≤–љ—Г—В—А–Є- –Є¬†–≤–љ–µ–Ї–ї–µ—В–Њ—З–љ–Њ–≥–Њ —А–Э –њ—А–Є –Я–С–•, —Б–≤—П–Ј–∞–љ–љ—Л–µ —Б¬†–њ–Њ—В–µ—А–µ–є InsP3R3 –Є¬†—Б–љ–Є–ґ–µ–љ–Є–µ–Љ –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є –Р–Х2, —Б–њ–Њ—Б–Њ–±—Б—В–≤—Г—О—В –њ—А–Њ—В–Њ–љ–Є—А–Њ–≤–∞–љ–Є—О –ґ–µ–ї—З–љ—Л—Е –Ї–Є—Б–ї–Њ—В. –≠—В–Њ –њ–Њ–Ј–≤–Њ–ї—П–µ—В –ґ–µ–ї—З–љ—Л–Љ –Ї–Є—Б–ї–Њ—В–∞–Љ –њ—А–µ–Њ–і–Њ–ї–µ–≤–∞—В—М –±–Є–ї–Є–∞—А–љ—Л–є –±–Є–Ї–∞—А–±–Њ–љ–∞—В–љ—Л–є ¬Ђ–Ј–Њ–љ—В–Є–Ї¬ї –Є¬†–њ—А–Њ–љ–Є–Ї–∞—В—М –≤¬†—Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В—Л, —З—В–Њ, –≤¬†–Ї–Њ–љ–µ—З–љ–Њ–Љ –Є—В–Њ–≥–µ, –њ—А–Є–≤–Њ–і–Є—В –Ї¬†–Є—Е –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є—О [1, 2, 51, 52]. –Ь–µ—Е–∞–љ–Є–Ј–Љ—Л –њ—А–Њ—В–Њ–љ–Є—А–Њ–≤–∞–љ–Є—П-–і–µ–њ—А–Њ—В–Њ–љ–Є—А–Њ–≤–∞–љ–Є—П –ґ–µ–ї—З–љ—Л—Е –Ї–Є—Б–ї–Њ—В –Є¬†–њ—А–Њ–љ–Є–Ї–љ–Њ–≤–µ–љ–Є—П –Є—Е –≤¬†—Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В—Л –њ–Њ–і—А–Њ–±–љ–Њ –Њ–њ–Є—Б–∞–љ—Л –≤¬†–Њ–±–Ј–Њ—А–∞—Е [2, 53]. –Э–µ–Њ–±—Е–Њ–і–Є–Љ–Њ –њ–Њ–і—З–µ—А–Ї–љ—Г—В—М, —З—В–Њ –Њ–њ–Є—Б–∞–љ–љ—Л–є –Љ–µ—Е–∞–љ–Є–Ј–Љ –њ—А–Њ–љ–Є–Ї–љ–Њ–≤–µ–љ–Є—П –ґ–µ–ї—З–љ—Л—Е –Ї–Є—Б–ї–Њ—В –≤¬†—Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В—Л —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–Я–С–• –њ—А–Њ–Є—Б—Е–Њ–і–Є—В –Є–Љ–µ–љ–љ–Њ –љ–∞ —Г—А–Њ–≤–љ–µ –Љ–µ–ї–Ї–Є—Е –С–≠–Ъ. –≠—В–Њ—В —Д–µ–љ–Њ–Љ–µ–љ —Б–≤—П–Ј–∞–љ —Б¬†–љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ–є –њ—А–Њ–і—Г–Ї—Ж–Є–µ–є HCO3- –і–∞–љ–љ—Л–Љ–Є –Ї–ї–µ—В–Ї–∞–Љ–Є –Є¬†–Њ—В—Б—Г—В—Б—В–≤–Є–µ–Љ –Љ—Г—Ж–Є–љ–Њ–≤–Њ–≥–Њ –Ї–Њ–Љ–њ–Њ–љ–µ–љ—В–∞, –Ї–Њ—В–Њ—А—Л–є —Б–ї—Г–ґ–Є—В –≤¬†–Ї–∞—З–µ—Б—В–≤–µ –Ј–∞—Й–Є—В–љ–Њ–≥–Њ –±–∞—А—М–µ—А–∞ —В–Њ–ї—М–Ї–Њ –і–ї—П –Ї—А—Г–њ–љ—Л—Е –С–≠–Ъ [2, 53].
–Ь–µ—Е–∞–љ–Є–Ј–Љ –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є—П –Љ–µ–ї–Ї–Є—Е —Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В–Њ–≤ –њ—А–Є ¬Ђ–і—Л—А—П–≤–Њ–Љ¬ї –±–Є–ї–Є–∞—А–љ–Њ–Љ –±–Є–Ї–∞—А–±–Њ–љ–∞—В–љ–Њ–Љ ¬Ђ–Ј–Њ–љ—В–Є–Ї–µ¬ї
–Ґ–µ–Њ—А–Є—П –і–µ—Б—В–∞–±–Є–ї–Є–Ј–∞—Ж–Є–Є –±–Є–ї–Є–∞—А–љ–Њ–≥–Њ –±–Є–Ї–∞—А–±–Њ–љ–∞—В–љ–Њ–≥–Њ ¬Ђ–Ј–Њ–љ—В–Є–Ї–∞¬ї –ї–µ–ґ–Є—В –≤¬†–Њ—Б–љ–Њ–≤–µ –љ–µ–Ї–Њ–љ—В—А–Њ–ї–Є—А—Г–µ–Љ–Њ–≥–Њ –њ–Њ—Б—В—Г–њ–ї–µ–љ–Є—П –ґ–µ–ї—З–љ—Л—Е –Ї–Є—Б–ї–Њ—В –≤–љ—Г—В—А—М –Љ–µ–ї–Ї–Є—Е —Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В–Њ–≤ [1, 51, 52]. –≠—В–Њ, –≤¬†—Б–≤–Њ—О –Њ—З–µ—А–µ–і—М, –Ј–∞–њ—Г—Б–Ї–∞–µ—В –Љ–µ—Е–∞–љ–Є–Ј–Љ –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є—П –±–Є–ї–Є–∞—А–љ—Л—Е —Н–њ–Є—В–µ–ї–Є–∞–ї—М–љ—Л—Е –Ї–ї–µ—В–Њ–Ї. –Я–Њ–њ–∞–і–∞—П –≤¬†–С–≠–Ъ —Б–Њ —Б–ї–∞–±–Њ—Й–µ–ї–Њ—З–љ—Л–Љ —А–Э, –ґ–µ–ї—З–љ—Л–µ –Ї–Є—Б–ї–Њ—В—Л –њ–Њ–і–≤–µ—А–≥–∞—О—В—Б—П –і–µ–њ—А–Њ—В–Њ–љ–Є—А–Њ–≤–∞–љ–Є—О, —З—В–Њ —Б–њ–Њ—Б–Њ–±—Б—В–≤—Г–µ—В –Є—Е –љ–∞–Ї–Њ–њ–ї–µ–љ–Є—О –≤¬†–Љ–µ–ї–Ї–Є—Е —Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В–∞—Е [2, 53]. –Ц–µ–ї—З–љ—Л–µ –Ї–Є—Б–ї–Њ—В—Л, –Њ–±–ї–∞–і–∞—О—Й–Є–µ —Б–Є–ї—М–љ—Л–Љ–Є –і–µ—В–µ—А–≥–µ–љ—В–љ—Л–Љ–Є —Б–≤–Њ–є—Б—В–≤–∞–Љ–Є, —Б–њ–Њ—Б–Њ–±–љ—Л —А–∞—Б—В–≤–Њ—А—П—В—М —Д–Њ—Б—Д–Њ–ї–Є–њ–Є–і—Л –Є¬†—Е–Њ–ї–µ—Б—В–µ—А–Є–љ –Є–Ј –Љ–µ–Љ–±—А–∞–љ–љ—Л—Е —Б—В—А—Г–Ї—В—Г—А –Љ–µ–ї–Ї–Є—Е –С–≠–Ъ. –≠—В–Њ—В –њ—А–Њ—Ж–µ—Б—Б –њ—А–Є–≤–Њ–і–Є—В –Ї¬†–њ–Њ–≤—А–µ–ґ–і–µ–љ–Є—О –Є¬†—А–∞–Ј—А—Г—И–µ–љ–Є—О —Ж–Є—В–Њ–њ–ї–∞–Ј–Љ–∞—В–Є—З–µ—Б–Ї–Њ–є –Љ–µ–Љ–±—А–∞–љ—Л –Є¬†–Љ–µ–Љ–±—А–∞–љ –Ї–ї–µ—В–Њ—З–љ—Л—Е –Њ—А–≥–∞–љ–µ–ї–ї. –Э–∞—А—Г—И–µ–љ–Є–µ —Ж–µ–ї–Њ—Б—В–љ–Њ—Б—В–Є —П–і–µ—А–љ–Њ–є –Є¬†–Љ–Є—В–Њ—Е–Њ–љ–і—А–Є–∞–ї—М–љ–Њ–є –Љ–µ–Љ–±—А–∞–љ –Љ–µ–ї–Ї–Є—Е —Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В–Њ–≤ –њ—А–Є –Я–С–• –њ—А–Є–≤–Њ–і–Є—В –Ї¬†–≤—Л—Б–≤–Њ–±–Њ–ґ–і–µ–љ–Є—О —Д–∞–Ї—В–Њ—А–Њ–≤, –Є–љ–і—Г—Ж–Є—А—Г—О—Й–Є—Е –∞–њ–Њ–њ—В–Њ–Ј. –С—Л–ї–Њ –њ–Њ–Ї–∞–Ј–∞–љ–Њ, —З—В–Њ —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–µ –њ–Њ–≤—А–µ–ґ–і–∞—О—Й–µ–µ –і–µ–є—Б—В–≤–Є–µ –ґ–µ–ї—З–љ—Л—Е –Ї–Є—Б–ї–Њ—В –љ–∞ –Љ–µ–Љ–±—А–∞–љ–љ—Л–µ —Б—В—А—Г–Ї—В—Г—А—Л –њ—А–Є–≤–Њ–і–Є—В –Ї¬†—Г—Б–Ї–Њ—А–µ–љ–љ–Њ–Љ—Г —Б—В–∞—А–µ–љ–Є—О, –∞–њ–Њ–њ—В–Њ–Ј—Г –Є/–Є–ї–Є –љ–µ–Ї—А–Њ–Ј—Г –Љ–µ–ї–Ї–Є—Е –С–≠–Ъ, –≤—Л—Б—В–Є–ї–∞—О—Й–Є—Е –Є–љ—В—А–∞–ї–Њ–±—Г–ї—П—А–љ—Л–µ, –Є–љ—В–µ—А–ї–Њ–±—Г–ї—П—А–љ—Л–µ –Є¬†—Б–µ–њ—В–∞–ї—М–љ—Л–µ –ґ–µ–ї—З–љ—Л–µ –њ—А–Њ—В–Њ–Ї–Є [1, 54]. –Ш–Ј–±—Л—В–Њ—З–љ—Л–є –∞–њ–Њ–њ—В–Њ–Ј –њ—А–Њ–≤–Њ—Ж–Є—А—Г–µ—В –љ–µ–Ї—А–Њ—В–Є—З–µ—Б–Ї–Є–є –њ—Г—В—М –≥–Є–±–µ–ї–Є –Ї–ї–µ—В–Њ–Ї (—В–∞–Ї –љ–∞–Ј—Л–≤–∞–µ–Љ—Л–є –≤—В–Њ—А–Є—З–љ—Л–є –љ–µ–Ї—А–Њ–Ј) –Є¬†—Б–њ–Њ—Б–Њ–±—Б—В–≤—Г–µ—В —Д–Њ—А–Љ–Є—А–Њ–≤–∞–љ–Є—О –≤–Њ—Б–њ–∞–ї–µ–љ–Є—П [10]. –Ш–љ–і—Г–Ї—Ж–Є—П —Г—Б–Ї–Њ—А–µ–љ–љ–Њ–≥–Њ –Ї–ї–µ—В–Њ—З–љ–Њ–≥–Њ –∞–њ–Њ–њ—В–Њ–Ј–∞ –Є¬†–≤—В–Њ—А–Є—З–љ–Њ–≥–Њ –љ–µ–Ї—А–Њ–Ј–∞ –њ—А–Є–≤–Њ–і–Є—В –Ї¬†–∞–Ї—В–Є–≤–∞—Ж–Є–Є –≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ—Л—Е –Є¬†–њ—А–Њ—Д–Є–±—А–Њ–≥–µ–љ–љ—Л—Е –њ—Г—В–µ–є, –Ї–Њ—В–Њ—А—Л–µ, –≤¬†—Б–≤–Њ—О –Њ—З–µ—А–µ–і—М, –Ј–∞–њ—Г—Б–Ї–∞—О—В —А–∞–Ј–≤–Є—В–Є–µ –Є¬†–њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є–µ —Д–Є–±—А–Њ–Ј–∞ [1, 54, 56]. –Э–∞—А—Г—И–∞–µ—В—Б—П –±–∞—А—М–µ—А–љ–∞—П —Д—Г–љ–Ї—Ж–Є—П —Н–њ–Є—В–µ–ї–Є—П, –≤—Л—Б—В–Є–ї–∞—О—Й–µ–≥–Њ –Љ–µ–ї–Ї–Є–µ –Є–љ—В—А–∞–ї–Њ–±—Г–ї—П—А–љ—Л–µ, –Є–љ—В–µ—А–ї–Њ–±—Г–ї—П—А–љ—Л–µ –Є¬†—Б–µ–њ—В–∞–ї—М–љ—Л–µ –ґ–µ–ї—З–љ—Л–µ –њ—А–Њ—В–Њ–Ї–Є. –¶–Є—В–Њ–Ї–Є–љ—Л, —Е–µ–Љ–Њ–Ї–Є–љ—Л –Є¬†–њ—А–Њ–≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ—Л–µ –Љ–µ–і–Є–∞—В–Њ—А—Л, –≤—Л–і–µ–ї—П–µ–Љ—Л–µ —Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В–∞–Љ–Є, –≤¬†—Б–≤–Њ—О –Њ—З–µ—А–µ–і—М, —Б—В–Є–Љ—Г–ї–Є—А—Г—О—В –∞–њ–Њ–њ—В–Њ—В–Є—З–µ—Б–Ї–Є–µ –Є¬†–њ—А–Њ–ї–Є—Д–µ—А–∞—В–Є–≤–љ—Л–µ —А–µ–∞–Ї—Ж–Є–Є –Є¬†–∞–Ї—В–Є–≤–Є—А—Г—О—В —Д–Є–±—А–Њ–≥–µ–љ–µ–Ј [57]. –Т¬†–Њ–±–Ј–Њ—А–∞—Е –ї–Є—В–µ—А–∞—В—Г—А—Л [54, 55] –њ—А–µ–і—Б—В–∞–≤–ї–µ–љ—Л –Њ–њ–Є—Б–∞–љ–Є—П –Ї–Њ–љ—Ж–µ–њ—В—Г–∞–ї—М–љ—Л—Е –Љ–µ—Е–∞–љ–Є–Ј–Љ–Њ–≤, –ї–µ–ґ–∞—Й–Є—Е –≤¬†–Њ—Б–љ–Њ–≤–µ —Н—В–Є—Е –њ—А–Њ—Ж–µ—Б—Б–Њ–≤. –Т¬†–Ї–Њ–љ–µ—З–љ–Њ–Љ –Є—В–Њ–≥–µ —А–∞–Ј–≤–Є–≤–∞–µ—В—Б—П –і—Г–Ї—В—Г–ї–Њ–њ–µ–љ–Є—П, –Ї–Њ—В–Њ—А–∞—П —Б–Њ–њ—А–Њ–≤–Њ–ґ–і–∞–µ—В—Б—П —Д–Њ—А–Љ–Є—А–Њ–≤–∞–љ–Є–µ–Љ –њ–Њ—Б—В–µ–њ–µ–љ–љ–Њ –њ—А–Њ–≥—А–µ—Б—Б–Є—А—Г—О—Й–µ–≥–Њ –≤–љ—Г—В—А–Є–њ–µ—З–µ–љ–Њ—З–љ–Њ–≥–Њ —Е–Њ–ї–µ—Б—В–∞–Ј–∞ [49, 55].
–У–Є—Б—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–Я–С–• –љ–∞–±–ї—О–і–∞–µ—В—Б—П –њ—А–Њ–≥—А–µ—Б—Б–Є—А—Г—О—Й–µ–µ –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є–µ –±–Є–ї–Є–∞—А–љ–Њ–≥–Њ —Н–њ–Є—В–µ–ї–Є—П —Б¬†–Є–љ—Д–Є–ї—М—В—А–∞—Ж–Є–µ–є –Љ–Њ–љ–Њ–љ—Г–Ї–ї–µ–∞—А–љ—Л–Љ–Є –Ї–ї–µ—В–Ї–∞–Љ–Є –≤–Њ–Ї—А—Г–≥ –С–≠–Ъ –ґ–µ–ї—З–љ—Л—Е –њ—А–Њ—В–Њ–Ї–Њ–≤ –Љ–∞–ї–Њ–≥–Њ –Є–ї–Є —Б—А–µ–і–љ–µ–≥–Њ —А–∞–Ј–Љ–µ—А–Њ–≤. –Ъ–∞–Ї –њ—А–∞–≤–Є–ї–Њ, –≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ—Л–µ –Ї–ї–µ—В–Ї–Є –њ—А–Є–ї–µ–≥–∞—О—В –Ї¬†—Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В–∞–Љ –њ–Њ–≤—А–µ–ґ–і–µ–љ–љ—Л—Е –ґ–µ–ї—З–љ—Л—Е –њ—А–Њ—В–Њ–Ї–Њ–≤ [58]. –Ш–љ–Є—Ж–Є–∞—Ж–Є—О –∞–њ–Њ–њ—В–Њ–Ј–∞ –ґ–µ–ї—З–љ—Л–Љ–Є –Ї–Є—Б–ї–Њ—В–∞–Љ–Є –Љ–Њ–ґ–љ–Њ —Б—З–Є—В–∞—В—М —В—А–Є–≥–≥–µ—А–Њ–Љ, –Ј–∞–њ—Г—Б–Ї–∞—О—Й–Є–Љ —А–∞–Ј–≤–Є—В–Є–µ –і—Г–Ї—В—Г–ї–Њ–њ–µ–љ–Є–Є –Љ–µ–ї–Ї–Є—Е –ґ–µ–ї—З–љ—Л—Е –њ—А–Њ—В–Њ–Ї–Њ–≤ –њ—А–Є –Я–С–• —Г–ґ–µ –≤¬†–∞—Б–Є–Љ–њ—В–Њ–Љ–∞—В–Є—З–µ—Б–Ї–Њ–є —Б—В–∞–і–Є–Є –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П [59, 60]. –Я—А–µ–і—Б—В–∞–≤–ї–µ–љ–љ—Л–є –Љ–µ—Е–∞–љ–Є–Ј–Љ –Њ—В—А–∞–ґ–∞–µ—В –њ—А—П–Љ–Њ–µ –њ–Њ–≤—А–µ–ґ–і–∞—О—Й–µ–µ –і–µ–є—Б—В–≤–Є–µ –ґ–µ–ї—З–љ—Л—Е –Ї–Є—Б–ї–Њ—В –љ–∞ –Љ–µ–ї–Ї–Є–µ —Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В—Л —Б¬†—А–∞–Ј–≤–Є—В–Є–µ–Љ —Б—В–∞—А–µ–љ–Є—П, –∞–њ–Њ–њ—В–Њ–Ј–∞, –≤—В–Њ—А–Є—З–љ–Њ–≥–Њ –љ–µ–Ї—А–Њ–Ј–∞ –Є¬†–≥–Є–±–µ–ї–Є –Љ–µ–ї–Ї–Є—Е –С–≠–Ъ.
–Э–∞–ї–Є—З–Є–µ –љ–∞ –∞–њ–Є–Ї–∞–ї—М–љ–Њ–є –њ–Њ–≤–µ—А—Е–љ–Њ—Б—В–Є –Ї—А—Г–њ–љ—Л—Е —Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В–Њ–≤ –Љ—Г—Ж–Є–љ—Б–Њ–і–µ—А–ґ–∞—Й–µ–≥–Њ –≥–ї–Є–Ї–Њ–Ї–∞–ї–Є–Ї—Б–љ–Њ–≥–Њ —Б–ї–Њ—П –Ј–∞—Й–Є—Й–∞–µ—В –Є—Е –Њ—В –њ—А–Њ–љ–Є–Ї–љ–Њ–≤–µ–љ–Є—П –Є¬†–њ–Њ–≤—А–µ–ґ–і–∞—О—Й–µ–≥–Њ –і–µ–є—Б—В–≤–Є—П –њ—А–Њ—В–Њ–љ–Є—А–Њ–≤–∞–љ–љ—Л—Е –Ї–Њ–љ—К—О–≥–Є—А–Њ–≤–∞–љ–љ—Л—Е –Є¬†–љ–µ–Ї–Њ–љ—К—О–≥–Є—А–Њ–≤–∞–љ–љ—Л—Е –ґ–µ–ї—З–љ—Л—Е –Ї–Є—Б–ї–Њ—В.
–Ш–Ј–±—Л—В–Њ—З–љ–Њ–µ –љ–∞–Ї–Њ–њ–ї–µ–љ–Є–µ –ґ–µ–ї—З–љ—Л—Е –Ї–Є—Б–ї–Њ—В –≤–љ—Г—В—А–Є –Љ–∞–ї—Л—Е –С–≠–Ъ —Б–Њ–њ—А–Њ–≤–Њ–ґ–і–∞–µ—В—Б—П —А–∞–Ј–≤–Є—В–Є–µ–Љ –њ–Њ–≤—Л—И–µ–љ–љ–Њ–є –њ—А–Њ–љ–Є—Ж–∞–µ–Љ–Њ—Б—В–Є (–њ–µ—А–Љ–µ–∞–±–Є–ї–Є–Ј–∞—Ж–Є–Є) –≤–љ–µ—И–љ–µ–є –Љ–µ–Љ–±—А–∞–љ—Л –Љ–Є—В–Њ—Е–Њ–љ–і—А–Є–є [61вАУ64]. –Я–µ—А–Љ–µ–∞–±–Є–ї–Є–Ј–∞—Ж–Є—П –Љ–Є—В–Њ—Е–Њ–љ–і—А–Є–є –Љ–Њ–ґ–µ—В —Б–Њ–Ј–і–∞–≤–∞—В—М –±–ї–∞–≥–Њ–њ—А–Є—П—В–љ—Л–µ —Г—Б–ї–Њ–≤–Є—П –і–ї—П –Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є—П –Р–Ь–Р [60, 65].
–Ь–µ—Е–∞–љ–Є–Ј–Љ –њ–µ—А–Љ–µ–∞–±–Є–ї–Є–Ј–∞—Ж–Є–Є –Љ–Є—В–Њ—Е–Њ–љ–і—А–Є–є –Є¬†–Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є–µ –Р–Ь–Р
–Я–Њ—Б—В—Г–њ–∞—О—Й–Є–µ –Є¬†–љ–∞–Ї–∞–њ–ї–Є–≤–∞—О—Й–Є–µ—Б—П –≤¬†–Љ–µ–ї–Ї–Є—Е —Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В–∞—Е –ґ–µ–ї—З–љ—Л–µ –Ї–Є—Б–ї–Њ—В—Л —Б–Њ–ї—О–±–Є–ї–Є–Ј–Є—А—Г—О—В —Д–Њ—Б—Д–Њ–ї–Є–њ–Є–і—Л –Є¬†—Е–Њ–ї–µ—Б—В–µ—А–Є–љ —Б¬†–≤–љ–µ—И–љ–µ–є –Љ–µ–Љ–±—А–∞–љ—Л –Љ–Є—В–Њ—Е–Њ–љ–і—А–Є–є, —З—В–Њ —Б–Њ–њ—А–Њ–≤–Њ–ґ–і–∞–µ—В—Б—П –љ–∞—А—Г—И–µ–љ–Є–µ–Љ –Є—Е –њ—А–Њ–љ–Є—Ж–∞–µ–Љ–Њ—Б—В–Є¬†вАУ –њ–µ—А–Љ–µ–∞–±–Є–ї–Є–Ј–∞—Ж–Є–µ–є [60]. –Я–Њ–≤—Л—И–µ–љ–љ–∞—П –њ—А–Њ–љ–Є—Ж–∞–µ–Љ–Њ—Б—В—М –≤–љ–µ—И–љ–µ–є –Љ–µ–Љ–±—А–∞–љ—Л –Љ–Є—В–Њ—Е–Њ–љ–і—А–Є–є –і–ї—П –Є–Њ–љ–Њ–≤ –Є¬†—А–∞—Б—В–≤–Њ—А–Њ–≤ –њ—А–Є–≤–Њ–і–Є—В –Ї¬†–њ–Њ—В–µ—А–µ –Љ–µ–Љ–±—А–∞–љ–љ–Њ–≥–Њ –њ–Њ—В–µ–љ—Ж–Є–∞–ї–∞ –Є¬†—Г—В–µ—З–Ї–µ –≤¬†—Ж–Є—В–Њ–Ј–Њ–ї—М —Б–Њ–і–µ—А–ґ–Є–Љ–Њ–≥–Њ –Љ–µ–ґ–Љ–µ–Љ–±—А–∞–љ–љ–Њ–≥–Њ –њ—А–Њ—Б—В—А–∞–љ—Б—В–≤–∞ [64, 66]. –Я—А–Њ–Є—Б—Е–Њ–і–Є—В –љ–∞–±—Г—Е–∞–љ–Є–µ –Љ–Є—В–Њ—Е–Њ–љ–і—А–Є–є —Б¬†–Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є–µ–Љ –њ—Б–µ–≤–і–Њ–њ–Њ–і–Є–є, –љ–∞—А—Г—И–µ–љ–Є–µ —Ж–µ–ї–Њ—Б—В–љ–Њ—Б—В–Є –Є—Е –≤–љ–µ—И–љ–µ–є –Љ–µ–Љ–±—А–∞–љ—Л –Є¬†–≤—Л—Е–Њ–і –∞–њ–Њ–њ—В–Њ–≥–µ–љ–љ—Л—Е —Д–∞–Ї—В–Њ—А–Њ–≤ [64], –≤–Ї–ї—О—З–∞—О—Й–Є—Е—Б—П –≤¬†–њ—А–Њ—Ж–µ—Б—Б—Л –∞–њ–Њ–њ—В–Њ–Ј–∞ –Љ–µ–ї–Ї–Є—Е –С–≠–Ъ. –Ю—В–Ї—А—Л–≤–∞–µ—В—Б—П –≤–љ—Г—В—А–µ–љ–љ—П—П –Љ–Є—В–Њ—Е–Њ–љ–і—А–Є–∞–ї—М–љ–∞—П –Љ–µ–Љ–±—А–∞–љ–∞¬†вАУ –Њ—Б–љ–Њ–≤–љ–∞—П –Љ–Є—И–µ–љ—М –і–ї—П –Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є—П AMA. –°–Њ–ї—О–±–Є–ї–Є–Ј–∞—Ж–Є—П –ґ–µ–ї—З–љ—Л–Љ–Є –Ї–Є—Б–ї–Њ—В–∞–Љ–Є —Д–Њ—Б—Д–Њ–ї–Є–њ–Є–і–Њ–≤ –Є¬†—Е–Њ–ї–µ—Б—В–µ—А–Є–љ–∞ —Б¬†–≤–љ—Г—В—А–µ–љ–љ–µ–є –Љ–µ–Љ–±—А–∞–љ—Л —Б–Њ–њ—А–Њ–≤–Њ–ґ–і–∞–µ—В—Б—П –і–∞–ї—М–љ–µ–є—И–µ–є –і–µ—Б—В—А—Г–Ї—Ж–Є–µ–є –Љ–Є—В–Њ—Е–Њ–љ–і—А–Є–є —Б¬†–≤—Л—Б–≤–Њ–±–Њ–ґ–і–µ–љ–Є–µ–Љ –Є¬†–і–µ–≥—А–∞–і–∞—Ж–Є–µ–є –њ–Є—А—Г–≤–∞—В–і–µ–≥–Є–і—А–Њ–≥–µ–љ–∞–Ј–љ–Њ–≥–Њ –Ї–Њ–Љ–њ–ї–µ–Ї—Б–∞. –Я–Ф–У –Ї–Њ–Љ–њ–ї–µ–Ї—Б –≤–Ї–ї—О—З–∞–µ—В –≤¬†—Б–µ–±—П —В—А–Є —Д–µ—А–Љ–µ–љ—В–∞ [–њ–Є—А—Г–≤–∞—В–і–µ–≥–Є–і—А–Њ–≥–µ–љ–∞–Ј–∞ (–Х1 –Я–Ф–У), –і–Є–≥–Є–і—А–Њ–ї–Є–њ–Њ–Є–ї—В—А–∞–љ—Б–∞—Ж–µ—В–Є–ї–∞–Ј–∞ (–Х2 –Я–Ф–У), –і–Є–≥–Є–і—А–Њ–ї–Є–њ–Њ–Є–ї–і–µ–≥–Є–і—А–Њ–≥–µ–љ–∞–Ј–∞ (–Х3 –Я–Ф–У)] –Є¬†–і–≤–∞ –≤—Б–њ–Њ–Љ–Њ–≥–∞—В–µ–ї—М–љ—Л—Е –±–µ–ї–Ї–∞ [59]. –Ф–ї—П –µ–≥–Њ —Д—Г–љ–Ї—Ж–Є–Њ–љ–Є—А–Њ–≤–∞–љ–Є—П –љ–µ–Њ–±—Е–Њ–і–Є–Љ—Л –њ—П—В—М –Ї–Њ—Д–∞–Ї—В–Њ—А–Њ–≤: –Ї–Њ—Н–љ–Ј–Є–Љ –Р, NAD+, —В–Є–∞–Љ–Є–љ–њ–Є—А–Њ—Д–Њ—Б—Д–∞—В (–≤–Є—В–∞–Љ–Є–љ –Т1), FAD –Є¬†–ї–Є–њ–Њ–µ–≤–∞—П –Ї–Є—Б–ї–Њ—В–∞. –Т–∞–ґ–љ–Њ –Њ—В–Љ–µ—В–Є—В—М, —З—В–Њ –Х1 –Я–Ф–У –Є¬†–Х3 –Я–Ф–У —П–≤–ї—П—О—В—Б—П –±–µ–ї–Ї–Њ–≤—Л–Љ–Є –Ї–Њ–Љ–њ–ї–µ–Ї—Б–∞–Љ–Є, –Ї–Њ—В–Њ—А—Л–µ –љ–µ —Б–Њ–і–µ—А–ґ–∞—В –ї–Є–њ–Є–і–љ—Л—Е –Ї–Њ–Љ–њ–Њ–љ–µ–љ—В–Њ–≤. –Я–Њ–Ї–∞–Ј–∞–љ–Њ, —З—В–Њ —Б—Л–≤–Њ—А–Њ—В–Ї–Є –њ–∞—Ж–Є–µ–љ—В–Њ–≤ –Я–С–• –љ–µ –њ—А–Њ—П–≤–ї—П—О—В —Б–µ—А–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є –Њ–њ—А–µ–і–µ–ї—П–µ–Љ–Њ–є —А–µ–∞–Ї—В–Є–≤–љ–Њ—Б—В–Є –њ—А–Њ—В–Є–≤ –Х1 –Є¬†E3 –Ї–Њ–Љ–њ–Њ–љ–µ–љ—В–Њ–≤ –Я–Ф–У [67].
–Р –≤–Њ—В –Х2 –Я–Ф–У —П–≤–ї—П–µ—В—Б—П –ї–Є–њ–Њ–њ—А–Њ—В–µ–Є–і–Њ–Љ –Є¬†–Є–Љ–µ–µ—В –і–≤–∞ —Б–∞–є—В–∞ —Б–≤—П–Ј—Л–≤–∞–љ–Є—П –ї–Є–њ–Њ–µ–≤–Њ–є –Ї–Є—Б–ї–Њ—В—Л [19]. –Х2 –Я–Ф–У —Б–Њ–і–µ—А–ґ–∞—В –љ–µ–Ј–∞–Љ–µ–љ–Є–Љ—Л–є –Њ—Б—В–∞—В–Њ–Ї –ї–Є–Ј–Є–љ–∞ –≤¬†–ї–Є–њ–Њ–∞–Љ–Є–і–љ–Њ–Љ –і–Њ–Љ–µ–љ–µ, –Ї¬†–Ї–Њ—В–Њ—А–Њ–Љ—Г –Ї–Њ–≤–∞–ї–µ–љ—В–љ–Њ –њ—А–Є—Б–Њ–µ–і–Є–љ–µ–љ–∞ –ї–Є–њ–Њ–µ–≤–∞—П –Ї–Є—Б–ї–Њ—В–∞ [19]. –Ы–Є–њ–Њ–µ–≤–Њ-–ї–Є–Ј–Є–љ–Њ–≤–∞—П —Б–≤—П–Ј—М –≤¬†–њ–Њ–ї–Њ–ґ–µ–љ–Є–Є 173 –≤—Л—Б–Њ–Ї–Њ –Ї–Њ–љ—Б–µ—А–≤–∞—В–Є–≤–љ–∞ —Г¬†—А–∞–Ј–љ—Л—Е –≤–Є–і–Њ–≤ –Є¬†–љ–µ–Њ–±—Е–Њ–і–Є–Љ–∞ –і–ї—П —А–∞—Б–њ–Њ–Ј–љ–∞–≤–∞–љ–Є—П –∞–љ—В–Є–≥–µ–љ–∞ [68]. –Т—Б–µ –Є–Љ–Љ—Г–љ–Њ–і–Њ–Љ–Є–љ–∞–љ—В–љ—Л–µ —Н–њ–Є—В–Њ–њ—Л ¬≠–Х2 –Я–Ф–У –≤–Ї–ї—О—З–∞—О—В –ї–Є–њ–Њ–µ–≤—Г—О –Ї–Є—Б–ї–Њ—В—Г, –Ї–Њ—В–Њ—А–∞—П –љ–µ–Њ–±—Е–Њ–і–Є–Љ–∞ –і–ї—П –Ґ-–Ї–ї–µ—В–Њ—З–љ–Њ–≥–Њ —А–∞—Б–њ–Њ–Ј–љ–∞–≤–∞–љ–Є—П –∞–љ—В–Є–≥–µ–љ–∞ [69]. –†—П–і –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є, –≤—Л–њ–Њ–ї–љ–µ–љ–љ—Л—Е –≤¬†–Ї–Њ–љ—Ж–µ –њ—А–Њ—И–ї–Њ–≥–Њ —Б—В–Њ–ї–µ—В–Є—П, –њ–Њ–Ї–∞–Ј–∞–ї, —З—В–Њ –Њ—Б–љ–Њ–≤–љ–∞—П –Є–Љ–Љ—Г–љ–Њ–≥–µ–љ–љ–∞—П –Њ–±–ї–∞—Б—В—М –љ–∞ E2 –Я–Ф–У, —А–∞—Б–њ–Њ–Ј–љ–∞–≤–∞–µ–Љ–∞—П –њ—А–Є –≤—Л–њ–Њ–ї–љ–µ–љ–Є–Є –Є–Љ–Љ—Г–љ–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є—Е —А–µ–∞–Ї—Ж–Є–є —Б¬†—Б—Л–≤–Њ—А–Њ—В–Ї–∞–Љ–Є –Њ—В –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–Я–С–•, –ї–Њ–Ї–∞–ї–Є–Ј–Њ–≤–∞–љ–∞ –≤¬†–ї–Є–њ–Є–і—Б–Њ–і–µ—А–ґ–∞—Й–µ–Љ –і–Њ–Љ–µ–љ–µ [70вАУ73]. AMA –љ–∞—Ж–µ–ї–µ–љ—Л –љ–∞ –Є–Љ–Љ—Г–љ–Њ–і–Њ–Љ–Є–љ–∞–љ—В–љ—Л–µ —Н–њ–Є—В–Њ–њ—Л –Х2 –Я–Ф–У, —Б–Њ–і–µ—А–ґ–∞—Й–Є–µ –ї–Є–њ–Њ–µ–≤—Г—О –Ї–Є—Б–ї–Њ—В—Г.
–Я–∞—В–Њ—Д–Є–Ј–Є–Њ–ї–Њ–≥–Є—П –Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є—П –Р–Ь–Р
–Я—А–µ–і—Б—В–∞–≤–ї–µ–љ–љ—Л–µ –≤—Л—И–µ –і–∞–љ–љ—Л–µ –і–∞—О—В –Њ—Б–љ–Њ–≤–∞–љ–Є—П –і–ї—П –≤—Л–і–≤–Є–ґ–µ–љ–Є—П –њ—А–µ–і–њ–Њ–ї–Њ–ґ–µ–љ–Є—П (–≥–Є–њ–Њ—В–µ–Ј—Л) –Њ¬†—В–Њ–Љ, —З—В–Њ –Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є–µ –Р–Ь–Р –њ—А–Њ—В–Є–≤ –Х2 –Я–Ф–У –њ—А–Є –Я–С–•, —Б–Ї–Њ—А–µ–µ –≤—Б–µ–≥–Њ, —П–≤–ї—П–µ—В—Б—П —Б–ї–µ–і—Б—В–≤–Є–µ–Љ –≤–Њ–Ј–і–µ–є—Б—В–≤–Є—П –ґ–µ–ї—З–љ—Л—Е –Ї–Є—Б–ї–Њ—В –љ–∞ –ї–Є–њ–Є–і–љ—Л–є –Ї–Њ–Љ–њ–Њ–љ–µ–љ—В –Я–Ф–У –Є¬†–µ–≥–Њ –Є–Љ–Љ—Г–љ–Њ–Љ–Њ–і–Є—Д–Є–Ї–∞—Ж–Є–Є. –Ц–µ–ї—З–љ—Л–µ –Ї–Є—Б–ї–Њ—В—Л —Б¬†–≤—Л—А–∞–ґ–µ–љ–љ—Л–Љ–Є –і–µ—В–µ—А–≥–µ–љ—В–љ—Л–Љ–Є —Б–≤–Њ–є—Б—В–≤–∞–Љ–Є —Б–њ–Њ—Б–Њ–±–љ—Л –≤–Ј–∞–Є–Љ–Њ–і–µ–є—Б—В–≤–Њ–≤–∞—В—М —Б¬†–ї–Є–њ–Њ–µ–≤–Њ–є –Ї–Є—Б–ї–Њ—В–Њ–є –Х2 –Я–Ф–У, —З—В–Њ –Љ–Њ–ґ–µ—В –њ—А–Є–≤–Њ–і–Є—В—М –Ї¬†–Ї–Њ–љ—Д–Њ—А–Љ–∞—Ж–Є–Њ–љ–љ—Л–Љ –Є/–Є–ї–Є —Б—В—А—Г–Ї—В—Г—А–љ—Л–Љ –Є–Ј–Љ–µ–љ–µ–љ–Є—П–Љ –Є–Љ–Љ—Г–љ–Њ–і–Њ–Љ–Є–љ–∞–љ—В–љ–Њ–≥–Њ –ї–Є–њ–Є–і–љ–Њ–≥–Њ —Н–њ–Є—В–Њ–њ–∞ –Є¬†–Љ–µ–љ—П—В—М –Є–Љ–Љ—Г–љ–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–µ —Б–≤–Њ–є—Б—В–≤–∞ –Х2 –Я–Ф–У –∞–љ—В–Є–≥–µ–љ–∞. –Ъ–Њ–љ—Д–Њ—А–Љ–∞—Ж–Є–Њ–љ–љ—Л–µ –Є, –≤–Њ–Ј–Љ–Њ–ґ–љ–Њ, –Њ—В—З–∞—Б—В–Є —Б—В—А—Г–Ї—В—Г—А–љ—Л–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П –ї–Є–њ–Є–і–љ–Њ–≥–Њ –і–Њ–Љ–µ–љ–∞ –Х2 –Я–Ф–У –Є–Ј-–Ј–∞ —Е–Є–Љ–Є—З–µ—Б–Ї–Њ–є –Љ–Њ–і–Є—Д–Є–Ї–∞—Ж–Є–Є –ґ–µ–ї—З–љ—Л–Љ–Є –Ї–Є—Б–ї–Њ—В–∞–Љ–Є –Љ–Њ–≥—Г—В —Б–њ–Њ—Б–Њ–±—Б—В–≤–Њ–≤–∞—В—М –њ—А–Є–Њ–±—А–µ—В–µ–љ–Є—О –љ–Њ–≤—Л—Е –љ–µ–Њ–∞–љ—В–Є–≥–µ–љ–љ—Л—Е —Б–≤–Њ–є—Б—В–≤, —А–∞—Б—Б–Љ–∞—В—А–Є–≤–∞–µ–Љ—Л—Е –Є–Љ–Љ—Г–љ–љ–Њ–є —Б–Є—Б—В–µ–Љ–Њ–є –≤¬†–Ї–∞—З–µ—Б—В–≤–µ —З—Г–ґ–µ—А–Њ–і–љ–Њ–≥–Њ –±–µ–ї–Ї–∞.
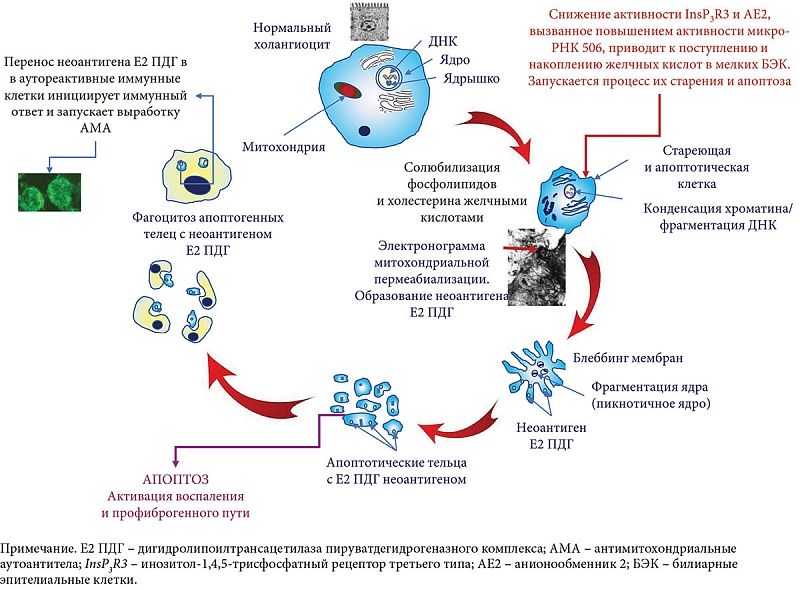
–Р–њ–Њ–њ—В–Њ–Ј –Ї–ї–µ—В–Њ–Ї –±–Є–ї–Є–∞—А–љ–Њ–≥–Њ —Н–њ–Є—В–µ–ї–Є—П –±—Л–ї –њ—А–µ–і–ї–Њ–ґ–µ–љ –≤¬†–Ї–∞—З–µ—Б—В–≤–µ –њ–Њ—В–µ–љ—Ж–Є–∞–ї—М–љ–Њ–≥–Њ –Є—Б—В–Њ—З–љ–Є–Ї–∞ –Х2 –Я–Ф–У –љ–µ–Њ–∞–љ—В–Є–≥–µ–љ–Њ–≤, –Њ—В–≤–µ—В—Б—В–≤–µ–љ–љ—Л—Е –Ј–∞ –∞–Ї—В–Є–≤–∞—Ж–Є—О –∞—Г—В–Њ—А–µ–∞–Ї—В–Є–≤–љ—Л—Е –ї–Є–Љ—Д–Њ—Ж–Є—В–Њ–≤ [74]. –£–ґ–µ –≤¬†–∞—Б–Є–Љ–њ—В–Њ–Љ–∞—В–Є—З–µ—Б–Ї–Њ–є —Б—В–∞–і–Є–Є –Я–С–• –∞—Г—В–Њ—А–µ–∞–Ї—В–Є–≤–љ—Л–µ –Т-–ї–Є–Љ—Д–Њ—Ж–Є—В—Л —Б–њ–Њ—Б–Њ–±–љ—Л —А–∞—Б–њ–Њ–Ј–љ–∞–≤–∞—В—М –Є–Љ–Љ—Г–љ–Њ–Љ–Њ–і–Є—Д–Є—Ж–Є—А–Њ–≤–∞–љ–љ—Л–є –љ–µ–Њ–∞–љ—В–Є–≥–µ–љ –Х2 –Я–Ф–У, –њ–Њ–ї—Г—З–µ–љ–љ—Л–є –Є–Ј –∞–њ–Њ–њ—В–Њ—В–Є—З–µ—Б–Ї–Є—Е –Љ–µ–ї–Ї–Є—Е –С–≠–Ъ –≤¬†–Ї–∞—З–µ—Б—В–≤–µ —З—Г–ґ–µ—А–Њ–і–љ–Њ–≥–Њ –±–µ–ї–Ї–∞ [60]. –Я—А–µ–Ј–µ–љ—В–∞—Ж–Є—П –ї–Є–Љ—Д–Њ—Ж–Є—В–∞–Љ –Є–Љ–Љ—Г–љ–Њ–Љ–Њ–і–Є—Д–Є—Ж–Є—А–Њ–≤–∞–љ–љ–Њ–≥–Њ –Х2 –Я–Ф–У –љ–µ–Њ–∞–љ—В–Є–≥–µ–љ–∞ –Љ–Њ–ґ–µ—В –њ—А–Є–≤–µ—Б—В–Є –Ї¬†—Б—В–Є–Љ—Г–ї–Є—А–Њ–≤–∞–љ–Є—О —Б—Г–±–њ–Њ–њ—Г–ї—П—Ж–Є–Є –Ґ-–Ї–ї–µ—В–Њ–Ї –Є¬†—Б–њ–µ—Ж–Є—Д–Є—З–µ—Б–Ї–Њ–є –њ—А–Њ–і—Г–Ї—Ж–Є–Є –Р–Ь–Р [20, 75вАУ77]. –Э–∞ —А–∞–љ–љ–Є—Е —Б—В–∞–і–Є—П—Е –Я–С–• –∞—Г—В–Њ—А–µ–∞–Ї—В–Є–≤–љ—Л–µ –Т-–Ї–ї–µ—В–Ї–Є –Љ–Њ–≥—Г—В —Д—Г–љ–Ї—Ж–Є–Њ–љ–Є—А–Њ–≤–∞—В—М –Ї–∞–Ї –Ї—А–Є—В–Є—З–µ—Б–Ї–Є–µ –∞–љ—В–Є–≥–µ–љ-–њ—А–µ–Ј–µ–љ—В–Є—А—Г—О—Й–Є–µ –Ї–ї–µ—В–Ї–Є, –њ–Њ–≥–ї–Њ—Й–∞—П –Є¬†–њ—А–µ–і—Б—В–∞–≤–ї—П—П –љ–µ–Њ–∞–љ—В–Є–≥–µ–љ—Л –Х2 –Я–Ф–У –Ґ-–Ї–ї–µ—В–Ї–∞–Љ [11, 60, 78]. –Я–Њ–≤—А–µ–ґ–і–µ–љ–љ—Л–µ –ґ–µ–ї—З–љ—Л–Љ–Є –Ї–Є—Б–ї–Њ—В–∞–Љ–Є –С–≠–Ъ (—Б—В–∞—А–µ—О—Й–Є–µ –Є¬†–њ–Њ–і–≤–µ—А–≥–∞—О—Й–Є–µ—Б—П —Г—Б–Ї–Њ—А–µ–љ–љ–Њ–Љ—Г –∞–њ–Њ–њ—В–Њ–Ј—Г) –Љ–Њ–≥—Г—В —В—А–∞–љ—Б–ї–Њ—Ж–Є—А–Њ–≤–∞—В—М –Є–Љ–Љ—Г–љ–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є –Љ–Њ–і–Є—Д–Є—Ж–Є—А–Њ–≤–∞–љ–љ—Л–є –љ–µ–Њ–∞–љ—В–Є–≥–µ–љ –Х2 –Я–Ф–У —Б¬†–∞–њ–Њ–њ—В–Њ—В–Є—З–µ—Б–Ї–Є–Љ–Є —В–µ–ї—М—Ж–∞–Љ–Є –Є¬†–њ–µ—А–µ–і–∞–≤–∞—В—М –µ–≥–Њ –∞—Г—В–Њ—А–µ–∞–Ї—В–Є–≤–љ—Л–Љ –Є–Љ–Љ—Г–љ–љ—Л–Љ –Ї–ї–µ—В–Ї–∞–Љ [11]. A. Lleo –Є¬†—Б–Њ–∞–≤—В. –≤–њ–µ—А–≤—Л–µ —Б–Њ–Њ–±—Й–Є–ї–Є –Њ¬†–њ—А–Є—Б—Г—В—Б—В–≤–Є–Є –Х2 –Я–Ф–У –≤¬†–∞–њ–Њ–њ—В–Њ—В–Є—З–µ—Б–Ї–Є—Е —В–µ–ї—М—Ж–∞—Е –Ї–ї–µ—В–Њ–Ї –≤–љ—Г—В—А–Є–њ–µ—З–µ–љ–Њ—З–љ—Л—Е –ґ–µ–ї—З–љ—Л—Е –њ—А–Њ—В–Њ–Ї–Њ–≤ —З–µ–ї–Њ–≤–µ–Ї–∞, –њ–Њ–і–≤–µ—А–≥—И–Є—Е—Б—П –∞–њ–Њ–њ—В–Њ–Ј—Г [79]. –С—Л–ї–Њ –њ–Њ–Ї–∞–Ј–∞–љ–Њ, —З—В–Њ –∞—Г—В–Њ–∞–љ—В–Є–≥–µ–љ—Л –Х2 –Я–Ф–У, –Њ–±–љ–∞—А—Г–ґ–µ–љ–љ—Л–µ –≤¬†–∞–њ–Њ–њ—В–Њ—В–Є—З–µ—Б–Ї–Є—Е —В–µ–ї—М—Ж–∞—Е (–∞–њ–Њ—В–Њ–њ—Л), –≤–њ–Њ—Б–ї–µ–і—Б—В–≤–Є–Є –±—Л–ї–Є –њ–Њ–≥–ї–Њ—Й–µ–љ—Л –Љ–∞–Ї—А–Њ—Д–∞–≥–∞–Љ–Є [79]. –Р–њ–Њ–њ—В–Њ—В–Є—З–µ—Б–Ї–Є–µ —В–µ–ї—М—Ж–∞ —Б–Њ–≤–Љ–µ—Б—В–љ–Њ —Б¬†–Љ–∞–Ї—А–Њ—Д–∞–≥–∞–Љ–Є –Љ–Њ–≥—Г—В –Є–љ–і—Г—Ж–Є—А–Њ–≤–∞—В—М –љ–µ —В–Њ–ї—М–Ї–Њ —Б–Є–љ—В–µ–Ј –Љ–µ—Б—В–љ–Њ–і–µ–є—Б—В–≤—Г—О—Й–Є—Е –њ—А–Њ–≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ—Л—Е —Ж–Є—В–Њ–Ї–Є–љ–Њ–≤, –љ–Њ –Є¬†–њ–µ—А–µ–і–∞–≤–∞—В—М –љ–µ–Њ–∞–љ—В–Є–≥–µ–љ –Х2 –Я–Ф–У –∞—Г—В–Њ—А–µ–∞–Ї—В–Є–≤–љ—Л–Љ –Є–Љ–Љ—Г–љ–љ—Л–Љ –Ї–ї–µ—В–Ї–∞–Љ, –Є–љ–Є—Ж–Є–Є—А—Г—П —В–∞–Ї–Є–Љ –Њ–±—А–∞–Ј–Њ–Љ –Є–Љ–Љ—Г–љ–љ—Л–є –Њ—В–≤–µ—В –Є¬†–Ј–∞–њ—Г—Б–Ї–∞—П –≤—Л—А–∞–±–Њ—В–Ї—Г –Р–Ь–Р –њ—А–Є –Я–С–• [80] (—А–Є—Б. 3). –Я—А–Є —Н—В–Њ–Љ –ґ–µ–ї—З–љ—Л–µ –Ї–Є—Б–ї–Њ—В—Л –љ–µ –Љ–Њ–≥—Г—В –≤–ї–Є—П—В—М –љ–∞ —З–Є—Б—В–Њ –±–µ–ї–Ї–Њ–≤—Л–µ –Х1 –Є¬†–Х3 —Б—Г–±—К–µ–і–Є–љ–Є—Ж—Л –Я–Ф–У, —В–∞–Ї –Ї–∞–Ї –Њ–љ–Є –љ–µ –Є–Љ–µ—О—В –≤¬†—Б–≤–Њ–µ–Љ —Б–Њ—Б—В–∞–≤–µ –ї–Є–њ–Є–і–љ—Л—Е –Ї–Њ–Љ–њ–Њ–љ–µ–љ—В–Њ–≤.
–Я–Њ—Б—В–Њ—П–љ–љ–∞—П –і–µ—Б—В–∞–±–Є–ї–Є–Ј–∞—Ж–Є—П –±–Є–ї–Є–∞—А–љ–Њ–≥–Њ –±–Є–Ї–∞—А–±–Њ–љ–∞—В–љ–Њ–≥–Њ ¬Ђ–Ј–Њ–љ—В–Є–Ї–∞¬ї –њ—А–Є –Я–С–• —Б–њ–Њ—Б–Њ–±—Б—В–≤—Г–µ—В –љ–µ–њ—А–µ—А—Л–≤–љ–Њ–Љ—Г –љ–∞–Ї–Њ–њ–ї–µ–љ–Є—О –Є¬†–і–µ—В–µ—А–≥–µ–љ—В–љ–Њ–Љ—Г –і–µ–є—Б—В–≤–Є—О –ґ–µ–ї—З–љ—Л—Е –Ї–Є—Б–ї–Њ—В –љ–∞ –Љ–µ–ї–Ї–Є–µ –С–≠–Ъ. –≠—В–Њ—В –њ—А–Њ—Ж–µ—Б—Б —Е–∞—А–∞–Ї—В–µ—А–Є–Ј—Г–µ—В—Б—П –Љ–µ–і–ї–µ–љ–љ–Њ –њ—А–Њ–≥—А–µ—Б—Б–Є—А—Г—О—Й–Є–Љ –љ–µ–њ—А–µ—А—Л–≤–љ—Л–Љ —А–∞–Ј—А—Г—И–µ–љ–Є–µ–Љ –Љ–µ–ї–Ї–Є—Е —Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В–Њ–≤, –Ї–Њ—В–Њ—А—Л–µ –≤—Л—Б—В–Є–ї–∞—О—В –Є–љ—В—А–∞–ї–Њ–±—Г–ї—П—А–љ—Л–µ, –Є–љ—В–µ—А–ї–Њ–±—Г–ї—П—А–љ—Л–µ –Є¬†—Б–µ–њ—В–∞–ї—М–љ—Л–µ –ґ–µ–ї—З–љ—Л–µ –њ—А–Њ—В–Њ–Ї–Є. –Т¬†—А–µ–Ј—Г–ї—М—В–∞—В–µ —А–∞–Ј—А—Г—И–µ–љ–Є—П –Љ–µ–ї–Ї–Є—Е –С–≠–Ъ –њ—А–Њ–Є—Б—Е–Њ–і–Є—В –њ–Њ—Б—В–Њ—П–љ–љ–∞—П –≤—Л—А–∞–±–Њ—В–Ї–∞ –Р–Ь–Р. –Я–Њ—Б–Ї–Њ–ї—М–Ї—Г –Р–Ь–Р –љ–∞—Ж–µ–ї–µ–љ—Л –љ–∞ –Є–Љ–Љ—Г–љ–Њ–Љ–Њ–і–Є—Д–Є—Ж–Є—А–Њ–≤–∞–љ–љ—Л–є –љ–µ–Њ–∞–љ—В–Є–≥–µ–љ E2 –Я–Ф–У, –Њ–љ–Є –≤–Ј–∞–Є–Љ–Њ–і–µ–є—Б—В–≤—Г—О—В –Є—Б–Ї–ї—О—З–Є—В–µ–ї—М–љ–Њ —Б¬†–С–≠–Ъ, –Ї–Њ—В–Њ—А—Л–µ –њ–Њ–≤—А–µ–ґ–і–µ–љ—Л –ґ–µ–ї—З–љ—Л–Љ–Є –Ї–Є—Б–ї–Њ—В–∞–Љ–Є (—В–Њ –µ—Б—В—М —Б—В–∞—А–µ—О—Й–Є–Љ–Є –Є/–Є–ї–Є –∞–њ–Њ–њ—В–Њ—В–Є—З–µ—Б–Ї–Є–Љ–Є) –Є¬†—Б–Њ–і–µ—А–ґ–∞—В –љ–µ–Њ–∞–љ—В–Є–≥–µ–љ E2 –Я–Ф–У. –≠—В–Њ —Б–њ–Њ—Б–Њ–±—Б—В–≤—Г–µ—В –њ–Њ–і–і–µ—А–ґ–∞–љ–Є—О –≤–Њ—Б–њ–∞–ї–µ–љ–Є—П –Є¬†—Д–Є–±—А–Њ–Ј–∞. –Т¬†—В–Њ –ґ–µ –≤—А–µ–Љ—П –Р–Ь–Р –љ–µ –Њ–Ї–∞–Ј—Л–≤–∞—О—В –Є–Љ–Љ—Г–љ–Њ–њ–Њ–≤—А–µ–ґ–і–∞—О—Й–µ–≥–Њ –і–µ–є—Б—В–≤–Є—П –љ–∞ –Ј–і–Њ—А–Њ–≤—Л–µ (–љ–Њ—А–Љ–∞–ї—М–љ—Л–µ) –Ї–ї–µ—В–Ї–Є, —Б–Њ–і–µ—А–ґ–∞—Й–Є–µ –љ–µ–Є–Ј–Љ–µ–љ–µ–љ–љ—Л–є –∞–љ—В–Є–≥–µ–љ E2 –Я–Ф–У [8, 60]. –Р–љ—В–Є–≥–µ–љ E2 –Я–Ф–У –і—А—Г–≥–Є—Е –Ј–і–Њ—А–Њ–≤—Л—Е (–љ–Њ—А–Љ–∞–ї—М–љ—Л—Е) –Ї–ї–µ—В–Њ–Ї –Њ—Б—В–∞–µ—В—Б—П –љ–µ–Є–Ј–Љ–µ–љ–µ–љ–љ—Л–Љ –Є¬†—А–∞—Б–њ–Њ–ї–∞–≥–∞–µ—В—Б—П –љ–∞ –≤–љ—Г—В—А–µ–љ–љ–µ–є –Љ–Є—В–Њ—Е–Њ–љ–і—А–Є–∞–ї—М–љ–Њ–є –Љ–µ–Љ–±—А–∞–љ–µ, —З—В–Њ —В–∞–Ї–ґ–µ –і–µ–ї–∞–µ—В –µ–≥–Њ –љ–µ–і–Њ—Б—В—Г–њ–љ—Л–Љ –і–ї—П –Р–Ь–Р.
–Я—А–µ–і—Б—В–∞–≤–ї–µ–љ–љ—Л–µ –і–∞–љ–љ—Л–µ, –±–µ–Ј—Г—Б–ї–Њ–≤–љ–Њ, —В—А–µ–±—Г—О—В –і–∞–ї—М–љ–µ–є—И–Є—Е –≤—Б–µ—Б—В–Њ—А–Њ–љ–љ–Є—Е –Ї–Њ–Љ–њ–ї–µ–Ї—Б–љ—Л—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є –і–ї—П –њ–Њ–і—В–≤–µ—А–ґ–і–µ–љ–Є—П –≤—Л—Б–Ї–∞–Ј–∞–љ–љ—Л—Е –њ—А–µ–і–њ–Њ–ї–Њ–ґ–µ–љ–Є–є. –Ю–і–љ–∞–Ї–Њ –Є¬†–Є–Љ–µ—О—Й–Є–µ—Б—П –љ–∞—Г—З–љ—Л–µ –і–∞–љ–љ—Л–µ, –љ–∞ –љ–∞—И –≤–Ј–≥–ї—П–і, —З–∞—Б—В–Є—З–љ–Њ –њ–Њ–і—В–≤–µ—А–ґ–і–∞—О—В –Є–Ј–ї–Њ–ґ–µ–љ–љ—Л–µ –≥–Є–њ–Њ—В–µ—В–Є—З–µ—Б–Ї–Є–µ —Г—В–≤–µ—А–ґ–і–µ–љ–Є—П –Њ¬†–Љ–µ—Е–∞–љ–Є–Ј–Љ–µ –њ—А–Є–Њ–±—А–µ—В–µ–љ–Є—П E2 –Я–Ф–У –љ–µ–Њ–∞–љ—В–Є–≥–µ–љ–љ—Л—Е —Б–≤–Њ–є—Б—В–≤ –њ–Њ–і –≤–ї–Є—П–љ–Є–µ–Љ –ґ–µ–ї—З–љ—Л—Е –Ї–Є—Б–ї–Њ—В. –Т¬†—А–µ–Ј—Г–ї—М—В–∞—В–µ –њ—А–Њ–≤–µ–і–µ–љ–Є—П –≤¬†–ї–∞–±–Њ—А–∞—В–Њ—А–Є–Є M.E. Gershwin [81вАУ87] —Н–Ї—Б–њ–µ—А–Є–Љ–µ–љ—В–∞–ї—М–љ–Њ–є –Є–Љ–Љ—Г–љ–Є–Ј–∞—Ж–Є–Є –ґ–Є–≤–Њ—В–љ—Л—Е –±—Л–ї–Є –њ–Њ–ї—Г—З–µ–љ—Л —Б–ї–µ–і—Г—О—Й–Є–µ –і–∞–љ–љ—Л–µ:
- –Р–љ—В–Є—В–µ–ї–∞ —Б¬†—А–µ–∞–Ї—В–Є–≤–љ–Њ—Б—В—М—О, –Є–і–µ–љ—В–Є—З–љ–Њ–є —А–µ–∞–Ї—В–Є–≤–љ–Њ—Б—В–Є –Њ–±—А–∞–Ј—Ж–Њ–≤ —Б—Л–≤–Њ—А–Њ—В–Ї–Є –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–Я–С–•, –љ–µ –Љ–Њ–≥—Г—В –±—Л—В—М –њ–Њ–ї—Г—З–µ–љ—Л –њ—Г—В–µ–Љ —Н–Ї—Б–њ–µ—А–Є–Љ–µ–љ—В–∞–ї—М–љ–Њ–є –Є–Љ–Љ—Г–љ–Є–Ј–∞—Ж–Є–Є [81]. –Т–µ—А–Њ—П—В–љ–Њ, —Н—В–Њ –љ–µ—Б–Њ–Њ—В–≤–µ—В—Б—В–≤–Є–µ –Љ–Њ–ґ–љ–Њ –Њ–±—К—П—Б–љ–Є—В—М –Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–љ–Є–µ–Љ –≤¬†–Є–Љ–Љ—Г–љ–Є–Ј–∞—Ж–Є–Є –ґ–Є–≤–Њ—В–љ—Л—Е –љ–∞—В–Є–≤–љ–Њ–≥–Њ E2 –Я–Ф–У, –∞¬†–љ–µ E2 –Я–Ф–У-–љ–µ–Њ–∞–љ—В–Є–≥–µ–љ–∞, –њ–Њ–ї—Г—З–µ–љ–љ–Њ–≥–Њ –њ–Њ—Б–ї–µ –≤–Ј–∞–Є–Љ–Њ–і–µ–є—Б—В–≤–Є—П —Б¬†–ґ–µ–ї—З–љ—Л–Љ–Є –Ї–Є—Б–ї–Њ—В–∞–Љ–Є, –Ї–∞–Ї —Н—В–Њ –љ–∞–±–ї—О–і–∞–µ—В—Б—П –њ—А–Є –Я–С–•.
- –≠–Ї—Б–њ–µ—А–Є–Љ–µ–љ—В–∞–ї—М–љ—Л–µ –і–∞–љ–љ—Л–µ –њ–Њ –Є–Љ–Љ—Г–љ–Є–Ј–∞—Ж–Є–Є –ї–∞–±–Њ—А–∞—В–Њ—А–љ—Л—Е –ґ–Є–≤–Њ—В–љ—Л—Е E2 –Я–Ф–У –≤¬†–≤–Є–і–µ —А–µ–Ї–Њ–Љ–±–Є–љ–∞–љ—В–љ–Њ–≥–Њ –њ–Њ–ї–Є–њ–µ–њ—В–Є–і–∞ —Б–≤–Є–і–µ—В–µ–ї—М—Б—В–≤—Г—О—В –Њ¬†—В–Њ–Љ, —З—В–Њ –Њ–љ –њ—А–Є–≤–Њ–і–Є—В –Ї¬†–Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є—О –Р–Ь–Р, –љ–Њ –љ–µ –Ї¬†–њ–Њ–≤—А–µ–ґ–і–µ–љ–Є—О —Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В–Њ–≤ [82]. –≠—В–Њ –Љ–Њ–ґ–µ—В —Б–≤–Є–і–µ—В–µ–ї—М—Б—В–≤–Њ–≤–∞—В—М –Њ¬†—В–Њ–Љ, —З—В–Њ –Р–Ь–Р –љ–µ —П–≤–ї—П—О—В—Б—П —Д–∞–Ї—В–Њ—А–Њ–Љ, –Ј–∞–њ—Г—Б–Ї–∞—О—Й–Є–Љ –і–µ—Б—В—А—Г–Ї—Ж–Є—О –С–≠–Ъ.
- –Я—А–Њ–Є–Ј–≤–Њ–і–љ—Л–µ –Њ–Ї—В–∞–љ–Њ–≤–Њ–є (–ґ–Є—А–љ–Њ–є) –Ї–Є—Б–ї–Њ—В—Л –±—Л–ї–Є —Б–њ–Њ—Б–Њ–±–љ—Л –≤—Л–Ј—Л–≤–∞—В—М –Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є–µ –Р–Ь–Р –Є¬†–Я–С–•-–њ–Њ–і–Њ–±–љ–Њ–µ –њ–Њ—А–∞–ґ–µ–љ–Є–µ –њ–µ—З–µ–љ–Є —Г¬†–Љ–Њ—А—Б–Ї–Є—Е —Б–≤–Є–љ–Њ–Ї –Є¬†–Љ—Л—И–µ–є [83вАУ87]. –≠—В–Є –і–∞–љ–љ—Л–µ –њ–Њ–і—З–µ—А–Ї–Є–≤–∞—О—В –Ї–ї—О—З–µ–≤—Г—О —А–Њ–ї—М –Њ—Б—В–∞—В–Ї–∞ –ґ–Є—А–љ–Њ–є –Ї–Є—Б–ї–Њ—В—Л –Є¬†–µ–µ –Љ–Њ–і–Є—Д–Є–Ї–∞—Ж–Є–Є –≤¬†–∞–љ—В–Є–≥–µ–љ–µ E2 –Я–Ф–У –і–ї—П –Є–љ–і—Г–Ї—Ж–Є–Є –≤—Л—А–∞–±–Њ—В–Ї–Є –Р–Ь–Р –Є¬†–њ–Њ–≤—А–µ–ґ–і–µ–љ–Є—П —Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В–Њ–≤.
- –Ш–Љ–Љ—Г–љ–Є–Ј–∞—Ж–Є—П –Ї—А–Њ–ї–Є–Ї–Њ–≤ –ї–Є–њ–Є–і–љ—Л–Љ –Ї—Б–µ–љ–Њ–±–Є–Њ—В–Є–Ї–Њ–Љ, –Ї–Њ–љ—К—О–≥–Є—А–Њ–≤–∞–љ–љ—Л–Љ —Б¬†–±—Л—З—М–Є–Љ —Б—Л–≤–Њ—А–Њ—В–Њ—З–љ—Л–Љ –∞–ї—М–±—Г–Љ–Є–љ–Њ–Љ, –≤—Л–Ј—Л–≤–∞–µ—В –Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є–µ AMA, –±–µ–Ј –њ–µ–њ—В–Є–і–љ–Њ–є –Њ—Б–љ–Њ–≤—Л –∞–љ—В–Є–≥–µ–љ–∞ E2 –Я–Ф–У [87]. –≠—В–Њ —В–∞–Ї–ґ–µ –Ї–Њ—Б–≤–µ–љ–љ—Л–Љ –Њ–±—А–∞–Ј–Њ–Љ —Б–≤–Є–і–µ—В–µ–ї—М—Б—В–≤—Г–µ—В –Њ¬†—В–Њ–Љ, —З—В–Њ –і–ї—П –≤—Л—А–∞–±–Њ—В–Ї–Є –Р–Ь–Р –≤–∞–ґ–љ—Г—О —А–Њ–ї—М –Є–≥—А–∞–µ—В –Љ–Њ–і–Є—Д–Є–Ї–∞—Ж–Є—П –ґ–Є—А–љ–Њ–Ї–Є—Б–ї–Њ—В–љ–Њ–є, –∞¬†–љ–µ –±–µ–ї–Ї–Њ–≤–Њ–є —З–∞—Б—В–Є –∞–љ—В–Є–≥–µ–љ–∞ –Х2 –Я–Ф–У. –Я–Њ—Б–ї–µ –њ—А–µ–Ї—А–∞—Й–µ–љ–Є—П –і–µ–є—Б—В–≤–Є—П —Б—В–Є–Љ—Г–ї–∞ —Н—В–Є –∞—Г—В–Њ–∞–љ—В–Є—В–µ–ї–∞ –Є—Б—З–µ–Ј–∞–ї–Є [87]. –Я—А–Є –Я–С–• –њ–Њ–≤—А–µ–ґ–і–∞—О—Й–µ–µ –і–µ–є—Б—В–≤–Є–µ –ґ–µ–ї—З–љ—Л—Е –Ї–Є—Б–ї–Њ—В –љ–∞ –Љ–Є—В–Њ—Е–Њ–љ–і—А–Є–Є –Љ–µ–ї–Ї–Є—Е —Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В–Њ–≤ –Њ–±–µ—Б–њ–µ—З–Є–≤–∞–µ—В –њ–Њ—Б—В–Њ—П–љ—Б—В–≤–Њ –≤—Л—Б–≤–Њ–±–Њ–ґ–і–µ–љ–Є—П –Є¬†–Є–Љ–Љ—Г–љ–Њ–Љ–Њ–і–Є—Д–Є–Ї–∞—Ж–Є–Є –Х2 –Я–Ф–У, —З—В–Њ, –≤¬†—Б–≤–Њ—О –Њ—З–µ—А–µ–і—М, –њ—А–Є–≤–Њ–і–Є—В –Ї¬†–љ–µ–њ—А–µ—А—Л–≤–љ–Њ–є –њ—А–Њ–і—Г–Ї—Ж–Є–Є –Р–Ь–Р.
–Т–µ—А–Њ—П—В–љ–Њ, –њ—А–Њ—Ж–µ—Б—Б—Л –Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є—П –Р–Ь–Р –Є¬†–і–µ—Б—В—А—Г–Ї—Ж–Є–Є –Љ–µ–ї–Ї–Є—Е –С–≠–Ъ —П–≤–ї—П—О—В—Б—П –љ–µ–Ј–∞–≤–Є—Б–Є–Љ—Л–Љ–Є, –Њ–±—К–µ–і–Є–љ—П–µ—В –Є—Е –Ј–∞–≤–Є—Б–Є–Љ–Њ—Б—В—М –Њ—В –њ–Њ–≤—А–µ–ґ–і–∞—О—Й–µ–≥–Њ –і–µ–є—Б—В–≤–Є—П –ґ–µ–ї—З–љ—Л—Е –Ї–Є—Б–ї–Њ—В, –Ї–Њ—В–Њ—А—Л–µ –Є–Ј-–Ј–∞ –љ–∞—А—Г—И–µ–љ–Є—П –≤—Л—А–∞–±–Њ—В–Ї–Є HCO3- –њ–Њ—Б—В—Г–њ–∞—О—В –≤¬†–Љ–µ–ї–Ї–Є–µ —Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В—Л –Є¬†–љ–∞–Ї–∞–њ–ї–Є–≤–∞—О—В—Б—П –≤¬†–љ–Є—Е. –≠—В–Њ –њ—А–Є–≤–Њ–і–Є—В –Ї–∞–Ї –Ї¬†–њ–Њ–≤—А–µ–ґ–і–µ–љ–Є—О —Б–∞–Љ–Є—Е —Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В–Њ–≤, —В–∞–Ї –Є¬†–Ї –Ї–Њ–љ—Д–Њ—А–Љ–∞—Ж–Є–Њ–љ–љ—Л–Љ –Є/–Є–ї–Є —Б—В—А—Г–Ї—В—Г—А–љ—Л–Љ –Є–Ј–Љ–µ–љ–µ–љ–Є—П–Љ –≤¬†–ї–Є–њ–Њ—Д–Є–ї—М–љ–Њ–Љ –і–Њ–Љ–µ–љ–µ E2 –Я–Ф–У, —З—В–Њ —Б–Њ–њ—А–Њ–≤–Њ–ґ–і–∞–µ—В—Б—П –≤—Л—А–∞–±–Њ—В–Ї–Њ–є –Р–Ь–Р [2, 53, 60].
–Э–µ–Ї–Њ—В–Њ—А—Л–µ –њ–∞—Ж–Є–µ–љ—В—Л —Б¬†–Я–С–• —П–≤–ї—П—О—В—Б—П –Р–Ь–Р-–Њ—В—А–Є—Ж–∞—В–µ–ї—М–љ—Л–Љ–Є. –Ґ–µ–Љ –љ–µ –Љ–µ–љ–µ–µ –њ–∞—Ж–Є–µ–љ—В—Л, —Г¬†–Ї–Њ—В–Њ—А—Л—Е –Њ—В—Б—Г—В—Б—В–≤—Г—О—В –Р–Ь–Р, –і–µ–Љ–Њ–љ—Б—В—А–Є—А—Г—О—В —В–∞–Ї–Є–µ –ґ–µ –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є–µ —Е–∞—А–∞–Ї—В–µ—А–Є—Б—В–Є–Ї–Є –≤¬†–Њ—В–љ–Њ—И–µ–љ–Є–Є —В–µ—З–µ–љ–Є—П –Є¬†–њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є—П –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П, –Ї–∞–Ї –Є¬†—В–µ, –Ї—В–Њ —П–≤–ї—П—О—В—Б—П –Р–Ь–Р-–њ–Њ–Ј–Є—В–Є–≤–љ—Л–Љ–Є. –Я–Њ-–≤–Є–і–Є–Љ–Њ–Љ—Г, —Г¬†5% –Р–Ь–Р-–љ–µ–≥–∞—В–Є–≤–љ—Л—Е –њ–∞—Ж–Є–µ–љ—В–Њ–≤ –≤¬†—Б–Є–ї—Г –Ї–∞–Ї–Є—Е-—В–Њ –Є–љ–і–Є–≤–Є–і—Г–∞–ї—М–љ—Л—Е –Њ—Б–Њ–±–µ–љ–љ–Њ—Б—В–µ–є –љ–∞–Ї–Њ–њ–ї–µ–љ–Є–µ –ґ–µ–ї—З–љ—Л—Е –Ї–Є—Б–ї–Њ—В –≤¬†–Љ–µ–ї–Ї–Є—Е –С–≠–Ъ –љ–µ —Б–Њ–њ—А–Њ–≤–Њ–ґ–і–∞–µ—В—Б—П –Є–Љ–Љ—Г–љ–љ–Њ–є –Љ–Њ–і–Є—Д–Є–Ї–∞—Ж–Є–µ–є –∞–љ—В–Є–≥–µ–љ–∞ E2 –Я–Ф–У. –Э–Њ –њ—А–Є —Н—В–Њ–Љ –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є–µ –Љ–µ–ї–Ї–Є—Е –С–≠–Ъ, —Б–≤—П–Ј–∞–љ–љ–Њ–µ —Б¬†–љ–∞–Ї–Њ–њ–ї–µ–љ–Є–µ–Љ –≤¬†–љ–Є—Е –ґ–µ–ї—З–љ—Л—Е –Ї–Є—Б–ї–Њ—В, —А–∞–Ј–≤–Є–≤–∞–µ—В—Б—П, –≤–µ—А–Њ—П—В–љ–µ–µ –≤—Б–µ–≥–Њ, –њ–Њ —В–Њ–Љ—Г –ґ–µ –Љ–µ—Е–∞–љ–Є–Ј–Љ—Г, —З—В–Њ –Є¬†—Г –Р–Ь–Р-–њ–Њ–ї–Њ–ґ–Є—В–µ–ї—М–љ—Л—Е –њ–∞—Ж–Є–µ–љ—В–Њ–≤. –≠—В–Њ –Њ–±—К—П—Б–љ—П–µ—В —Б—Е–Њ–і–љ—Г—О –Ї–ї–Є–љ–Є—З–µ—Б–Ї—Г—О –Ї–∞—А—В–Є–љ—Г, —В–µ—З–µ–љ–Є–µ –Є¬†–њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є–µ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П —Г¬†–Р–Ь–Р-–љ–µ–≥–∞—В–Є–≤–љ—Л—Е –Є¬†–Р–Ь–Р-–њ–Њ–ї–Њ–ґ–Є—В–µ–ї—М–љ—Л—Е –њ–∞—Ж–Є–µ–љ—В–Њ–≤.
–Ч–∞–Љ–µ—В–љ—Л–Љ –Њ–≥—А–∞–љ–Є—З–µ–љ–Є–µ–Љ —Б—Г—Й–µ—Б—В–≤—Г—О—Й–Є—Е –љ–∞—Г—З–љ—Л—Е –ї–Є—В–µ—А–∞—В—Г—А–љ—Л—Е –і–∞–љ–љ—Л—Е —П–≤–ї—П–µ—В—Б—П –Њ—В—Б—Г—В—Б—В–≤–Є–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є, –љ–∞–њ—А–∞–≤–ї–µ–љ–љ—Л—Е –љ–∞ –≤—Л—П–≤–ї–µ–љ–Є–µ –љ–Њ–≤—Л—Е –љ–µ–Њ–∞–љ—В–Є–≥–µ–љ–љ—Л—Е —Б–≤–Њ–є—Б—В–≤ E2 –Я–Ф–У. –Ш—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П –±—Л–ї–Є –љ–∞–њ—А–∞–≤–ї–µ–љ—Л –љ–∞ –≤—Л—П–≤–ї–µ–љ–Є–µ –Љ–µ—Е–∞–љ–Є–Ј–Љ–Њ–≤, –ї–µ–ґ–∞—Й–Є—Е –≤¬†–Њ—Б–љ–Њ–≤–µ –њ–Њ—В–µ—А–Є —В–Њ–ї–µ—А–∞–љ—В–љ–Њ—Б—В–Є –Є–Љ–Љ—Г–љ–љ–Њ–є —Б–Є—Б—В–µ–Љ—Л –Ї¬†–љ–µ–Є–Ј–Љ–µ–љ–µ–љ–љ–Њ–Љ—Г (–љ–Њ—А–Љ–∞–ї—М–љ–Њ–Љ—Г) –∞–љ—В–Є–≥–µ–љ—Г E2 –Я–Ф–У [73, 75, 76].
–Т —Б–≤—П–Ј–Є —Б¬†–≤—Л—И–µ–Є–Ј–ї–Њ–ґ–µ–љ–љ—Л–Љ –љ–∞–ї–Є—З–Є–µ –Р–Ь–Р –њ—А–Є –Я–С–• —Б–Ї–Њ—А–µ–µ —Г–Ї–∞–Ј—Л–≤–∞–µ—В –љ–∞ –∞—Г—В–Њ–Є–Љ–Љ—Г–љ–Њ–њ–Њ–і–Њ–±–љ–Њ–µ —Б–Њ—Б—В–Њ—П–љ–Є–µ, —З–µ–Љ –љ–∞ —В–Њ, —З—В–Њ –Я–С–• —П–≤–ї—П–µ—В—Б—П –Є—Б—В–Є–љ–љ—Л–Љ –Ї–ї–∞—Б—Б–Є—З–µ—Б–Ї–Є–Љ –∞—Г—В–Њ–Є–Љ–Љ—Г–љ–љ—Л–Љ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–µ–Љ.
–Ч–∞–Ї–ї—О—З–µ–љ–Є–µ
–Ю—В–Ї—А—Л—В–Є–µ –Р–Ь–Р –Є¬†–Є—Е —Г—Б–њ–µ—И–љ–Њ–µ –њ—А–Є–Љ–µ–љ–µ–љ–Є–µ –≤¬†–і–Є–∞–≥–љ–Њ—Б—В–Є–Ї–µ –Я–С–• —Б–њ–Њ—Б–Њ–±—Б—В–≤–Њ–≤–∞–ї–Њ –≤—Л—П–≤–ї–µ–љ–Є—О –±–µ—Б—Б–Є–Љ–њ—В–Њ–Љ–љ—Л—Е –Є¬†—А–∞–љ–љ–Є—Е —Б—В–∞–і–Є–є –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П, —Г–ї—Г—З—И–Є–≤ —В–∞–Ї–Є–Љ –Њ–±—А–∞–Ј–Њ–Љ –њ—А–Њ–≥–љ–Њ–Ј –і–ї—П –њ–∞—Ж–Є–µ–љ—В–Њ–≤ [3]. –Т¬†–љ–∞—Б—В–Њ—П—Й–µ–µ –≤—А–µ–Љ—П –і–Є–∞–≥–љ–Њ—Б—В–Є—З–µ—Б–Ї–∞—П —Ж–µ–љ–љ–Њ—Б—В—М –Р–Ь–Р –њ—А–Є –Я–С–• –Њ–±—Й–µ–њ—А–Є–Ј–љ–∞–љ–∞. –Ґ–µ–Љ –љ–µ –Љ–µ–љ–µ–µ –љ–µ–Њ–±—Е–Њ–і–Є–Љ—Л –і–∞–ї—М–љ–µ–є—И–Є–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П, —З—В–Њ–±—Л –њ–Њ–љ—П—В—М –Љ–µ—Е–∞–љ–Є–Ј–Љ—Л –Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є—П –Р–Ь–Р –Є¬†–Њ–њ—А–µ–і–µ–ї–Є—В—М –Є—Е –Ј–љ–∞—З–µ–љ–Є–µ –≤¬†–њ–∞—В–Њ–≥–µ–љ–µ–Ј–µ –Я–С–•. –Ш–Љ–µ—О—Й–Є–µ—Б—П –љ–∞—Г—З–љ—Л–µ –і–∞–љ–љ—Л–µ –њ–Њ–Ј–≤–Њ–ї–Є–ї–Є —Б—Д–Њ—А–Љ—Г–ї–Є—А–Њ–≤–∞—В—М –≥–Є–њ–Њ—В–µ–Ј—Г –Њ¬†–њ–Њ—В–µ–љ—Ж–Є–∞–ї—М–љ–Њ–є —А–Њ–ї–Є –ґ–µ–ї—З–љ—Л—Е –Ї–Є—Б–ї–Њ—В –Ї–∞–Ї —Д–∞–Ї—В–Њ—А–Њ–≤, —Б–њ–Њ—Б–Њ–±—Б—В–≤—Г—О—Й–Є—Е –Ј–∞–њ—Г—Б–Ї—Г –Љ–µ—Е–∞–љ–Є–Ј–Љ–∞ –Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є—П –Р–Ь–Р –≤¬†–Љ–µ–ї–Ї–Є—Е –С–≠–Ъ –њ—А–Є –Я–С–• [53]. –Я—А–µ–і—Б—В–∞–≤–ї–µ–љ–љ–∞—П –≤¬†–Њ–±–Ј–Њ—А–µ –≥–Є–њ–Њ—В–µ–Ј–∞ –њ–Њ–Ј–≤–Њ–ї—П–µ—В –Њ—В–≤–µ—В–Є—В—М –љ–∞ —А—П–і –љ–µ—А–µ—И–µ–љ–љ—Л—Е –≤–Њ–њ—А–Њ—Б–Њ–≤, –Ї–∞—Б–∞—О—Й–Є—Е—Б—П —Д–∞–Ї—В–Њ—А–Њ–≤, –Є–љ–Є—Ж–Є–Є—А—Г—О—Й–Є—Е –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є–µ –Љ–∞–ї—Л—Е –С–≠–Ъ, –Љ–µ—Е–∞–љ–Є–Ј–Љ–∞ –Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є—П –Р–Ь–Р, –Є—Е –Ј–љ–∞—З–Є–Љ–Њ—Б—В–Є –≤¬†—А–∞–Ј–≤–Є—В–Є–Є –Я–С–• –Є¬†—Б—В–µ–њ–µ–љ–Є –≤–Њ–≤–ї–µ—З–µ–љ–љ–Њ—Б—В–Є –Є–Љ–Љ—Г–љ–љ–Њ–є —Б–Є—Б—В–µ–Љ—Л –≤¬†—Н—В–Њ—В –њ—А–Њ—Ж–µ—Б—Б [60]. –Я—А–µ–і—Б—В–∞–≤–ї–µ–љ–љ—Л–µ –≤¬†–Њ–±–Ј–Њ—А–µ –і–∞–љ–љ—Л–µ —А–∞—Б–Ї—А—Л–≤–∞—О—В —А—П–і –≤–Њ–њ—А–Њ—Б–Њ–≤, –Ї–Њ—В–Њ—А—Л–µ –і–Њ —Б–Є—Е –њ–Њ—А –Њ—Б—В–∞–≤–∞–ї–Є—Б—М –љ–µ—П—Б–љ—Л–Љ–Є.
–°—В–∞–љ–Њ–≤–Є—В—Б—П –њ–Њ–љ—П—В–љ—Л–Љ –Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є–µ –Р–Ь–Р –Є–Љ–µ–љ–љ–Њ –Ї¬†–∞–љ—В–Є–≥–µ–љ—Г E2 –Я–Ф–У, –љ–Њ –љ–µ –Ї¬†—Б—Г–±—К–µ–і–Є–љ–Є—Ж–∞–Љ E1 –Є¬†E3 –Ї–Њ–Љ–њ–ї–µ–Ї—Б–∞ –Я–Ф–У.
–Я—А–Њ–і–µ–Љ–Њ–љ—Б—В—А–Є—А–Њ–≤–∞–љ–∞ –њ–Њ—В–µ–љ—Ж–Є–∞–ї—М–љ–∞—П –≤–Њ–Ј–Љ–Њ–ґ–љ–Њ—Б—В—М —Е–Є–Љ–Є—З–µ—Б–Ї–Њ–є –Љ–Њ–і–Є—Д–Є–Ї–∞—Ж–Є–Є –ї–Є–њ–Є–і–љ–Њ–≥–Њ –і–Њ–Љ–µ–љ–∞ E2 –Я–Ф–У –∞–љ—В–Є–≥–µ–љ–∞ –ґ–µ–ї—З–љ—Л–Љ–Є –Ї–Є—Б–ї–Њ—В–∞–Љ–Є –≤¬†–Љ–µ–ї–Ї–Є—Е –С–≠–Ъ —Б¬†–њ–Њ—Б–ї–µ–і—Г—О—Й–µ–є —В—А–∞–љ—Б—Д–Њ—А–Љ–∞—Ж–Є–µ–є –≤¬†–љ–µ–Њ–∞–љ—В–Є–≥–µ–љ. –Т¬†–і–∞–љ–љ–Њ–Љ —Б–ї—Г—З–∞–µ –Є—Б–Ї–ї—О—З–∞–µ—В—Б—П –і–Є—Б—Д—Г–љ–Ї—Ж–Є—П –Є–Љ–Љ—Г–љ–љ–Њ–є —Б–Є—Б—В–µ–Љ—Л.
–Я—А–µ–і—Б—В–∞–≤–ї–µ–љ –њ–Њ—В–µ–љ—Ж–Є–∞–ї—М–љ—Л–є –Љ–µ—Е–∞–љ–Є–Ј–Љ —Г—З–∞—Б—В–Є—П –ґ–µ–ї—З–љ—Л—Е –Ї–Є—Б–ї–Њ—В –≤¬†—Д–Њ—А–Љ–Є—А–Њ–≤–∞–љ–Є–Є –і–Њ—Б—В—Г–њ–∞ –Є–Љ–Љ—Г–љ–Њ–Ї–Њ–Љ–њ–µ—В–µ–љ—В–љ—Л—Е –Ї–ї–µ—В–Њ–Ї –Ї¬†E2 –Я–Ф–У –∞–љ—В–Є–≥–µ–љ—Г, —А–∞—Б–њ–Њ–ї–Њ–ґ–µ–љ–љ–Њ–Љ—Г –љ–∞ –≤–љ—Г—В—А–µ–љ–љ–µ–є –Љ–µ–Љ–±—А–∞–љ–µ –Љ–Є—В–Њ—Е–Њ–љ–і—А–Є–є –Љ–µ–ї–Ї–Є—Е –С–≠–Ъ, —З—В–Њ –≤–њ–Њ—Б–ї–µ–і—Б—В–≤–Є–Є –њ—А–Є–≤–Њ–і–Є—В –Ї¬†–Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є—О AMA.
–°—В–Њ–є–Ї–Њ–µ –Є¬†–њ—А–Њ–і–Њ–ї–ґ–Є—В–µ–ї—М–љ–Њ–µ –њ–Њ–≤—А–µ–ґ–і–∞—О—Й–µ–µ –і–µ–є—Б—В–≤–Є–µ –ґ–µ–ї—З–љ—Л—Е –Ї–Є—Б–ї–Њ—В –љ–∞ –Љ–µ–ї–Ї–Є–µ —Е–Њ–ї–∞–љ–≥–Є–Њ—Ж–Є—В—Л —П–≤–ї—П–µ—В—Б—П –Њ–њ—А–µ–і–µ–ї—П—О—Й–Є–Љ —Д–∞–Ї—В–Њ—А–Њ–Љ –≤¬†–њ–µ—А—Б–Є—Б—В–µ–љ—Ж–Є–Є –Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є—П E2 –Я–Ф–У –љ–µ–Њ–∞–љ—В–Є–≥–µ–љ–∞, —З—В–Њ, –≤¬†—Б–≤–Њ—О –Њ—З–µ—А–µ–і—М, –њ–Њ–і–і–µ—А–ґ–Є–≤–∞–µ—В —Г—Б—В–Њ–є—З–Є–≤—Л–є —В–Є—В—А –Р–Ь–Р –≤¬†–Ї—А–Њ–≤–Є –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–Я–С–•.
–Ю–±—А–∞–Ј–Њ–≤–∞–љ–Є–µ –Р–Ь–Р –≤¬†–Њ—В–≤–µ—В –љ–∞ E2 –Я–Ф–У –љ–µ–Њ–∞–љ—В–Є–≥–µ–љ –Є—Б–Ї–ї—О—З–∞–µ—В –µ–≥–Њ —Ж–Є—В–Њ—В–Њ–Ї—Б–Є—З–µ—Б–Ї–Њ–µ –і–µ–є—Б—В–≤–Є–µ –љ–∞ –≤—Б–µ –і—А—Г–≥–Є–µ –Ї–ї–µ—В–Ї–Є, —Б–Њ–і–µ—А–ґ–∞—Й–Є–µ –Љ–Є—В–Њ—Е–Њ–љ–і—А–Є–Є –Є¬†–љ–µ–Є–Ј–Љ–µ–љ–µ–љ–љ—Л–є E2 –Я–Ф–У.
–Ю–±—А–∞–Ј–Њ–≤–∞–љ–Є–µ –Р–Ь–Р –≤¬†–Њ—В–≤–µ—В –љ–∞ E2 –Я–Ф–У –љ–µ–Њ–∞–љ—В–Є–≥–µ–љ —В–Њ–ї—М–Ї–Њ –≤¬†–Љ–µ–ї–Ї–Є—Е –С–≠–Ъ –Њ–њ—А–µ–і–µ–ї—П–µ—В –Є—Е –і–Є–∞–≥–љ–Њ—Б—В–Є—З–µ—Б–Ї—Г—О, –љ–Њ –љ–µ –њ—А–Њ–≥–љ–Њ—Б—В–Є—З–µ—Б–Ї—Г—О —Ж–µ–љ–љ–Њ—Б—В—М.
–£—З–∞—Б—В–Є–µ –ґ–µ–ї—З–љ—Л—Е –Ї–Є—Б–ї–Њ—В –≤¬†–Ї–∞—З–µ—Б—В–≤–µ –њ–µ—А–≤–Њ–њ—А–Є—З–Є–љ—Л –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є—П –Љ–µ–ї–Ї–Є—Е –С–≠–Ъ –Њ–њ—А–µ–і–µ–ї—П–µ—В —В–µ—З–µ–љ–Є–µ –Я–С–• –Ї–∞–Ї –љ–µ–≥–љ–Њ–є–љ–Њ–≥–Њ –і–µ—Б—В—А—Г–Ї—В–Є–≤–љ–Њ–≥–Њ —Е–Њ–ї–∞–љ–≥–Є—В–∞, –Ї–Њ—В–Њ—А—Л–є –љ–µ —Б–Њ–Њ—В–≤–µ—В—Б—В–≤—Г–µ—В —В–Є–њ–Є—З–љ–Њ–є –Ї–∞—А—В–Є–љ–µ –Є–Љ–Љ—Г–љ–Њ–Њ–њ–Њ—Б—А–µ–і–Њ–≤–∞–љ–љ–Њ–≥–Њ –≤–Њ—Б–њ–∞–ї–µ–љ–Є—П.
–Я—А–µ–і—Б—В–∞–≤–ї—П–µ—В—Б—П –Њ–±–Њ—Б–љ–Њ–≤–∞–љ–љ—Л–Љ –њ—А–µ–і–њ–Њ–ї–Њ–ґ–µ–љ–Є–µ, —З—В–Њ —Г—З–∞—Б—В–Є–µ –ґ–µ–ї—З–љ—Л—Е –Ї–Є—Б–ї–Њ—В –≤¬†–Ї–∞—З–µ—Б—В–≤–µ –њ–µ—А–≤–Њ–њ—А–Є—З–Є–љ—Л –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є—П –Љ–µ–ї–Ї–Є—Е –С–≠–Ъ –Љ–Њ–ґ–µ—В –Њ–±—К—П—Б–љ–Є—В—М –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ—Г—О —Н—Д—Д–µ–Ї—В–Є–≤–љ–Њ—Б—В—М –Є–Љ–Љ—Г–љ–Њ—Б—Г–њ—А–µ—Б—Б–Є–≤–љ—Л—Е –њ—А–µ–њ–∞—А–∞—В–Њ–≤.
–†–∞—Б–Ї—А—Л—В–Є–µ –љ–Њ–≤—Л—Е —Н—В–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є—Е —Д–∞–Ї—В–Њ—А–Њ–≤ –Є¬†–і–µ—В–∞–ї—М–љ—Л—Е –њ–∞—В–Њ—Д–Є–Ј–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є—Е –Љ–µ—Е–∞–љ–Є–Ј–Љ–Њ–≤ —А–∞–Ј–≤–Є—В–Є—П –Я–С–• –Љ–Њ–ґ–µ—В –њ–Њ—Б–ї—Г–ґ–Є—В—М –≤–∞–ґ–љ–Њ–є –Њ—Б–љ–Њ–≤–Њ–є –і–ї—П —А–∞–Ј—А–∞–±–Њ—В–Ї–Є –љ–Њ–≤—Л—Е –ї–µ–Ї–∞—А—Б—В–≤–µ–љ–љ—Л—Е –њ—А–µ–њ–∞—А–∞—В–Њ–≤ –Є¬†–Љ–µ—В–Њ–і–Њ–≤ –ї–µ—З–µ–љ–Є—П —Н—В–Є—Е –њ–∞—Ж–Є–µ–љ—В–Њ–≤ –≤¬†–±—Г–і—Г—Й–µ–Љ.
V.I. Reshetnyak, PhD, Prof., I.V. Maev, PhD, Prof., Academician of the RAS
Russian University of Medicine, Moscow
Contact person: Vasily I. Reshetnyak, vasiliy.reshetnyak@yandex.ru
Primary biliary cholangitis (PBC) is a chronic cholestatic progressive liver disease that falls within the category of cholangiopathies. The identification of antimitochondrial antibodies (AMAs) is a crucial aspect of the diagnosis of classic PBC. It has been demonstrated that AMAs are formed in response to the dihydrolipoyl transacetylase of the pyruvate dehydrogenase complex (E2 PDH), which is situated on the inner mitochondrial membrane. Loss of immune tolerance to E2 PDH in PBC is considered to be the factor that triggers the mechanism of AMA formation and the subsequent immune-mediated destruction of biliary epithelial cells (BECs, cholangiocytes) lining the small and medium-sized intrahepatic bile ducts. It is well established that the E2 PDH complex is present not only in BECs, but also in the mitochondria of all nucleus-containing cells. This raises the question of why only the E2 PDH of small cholangiocytes becomes a target for autoimmune attack.
The formation of AMAs in the asymptomatic stage of the disease has provided the basis for the claim that they trigger the process of cholangiocyte damage and death, thereby allowing PBC to be considered a true autoimmune disease. However, there is no evidence of damaging effects of AMAs on cholangiocytes lining small (intra-lobular, inter-lobular and septal) bile ducts. Emerging new scientific evidence of impaired bicarbonate production by cholangiocytes already in the asymptomatic stage of PBC allowed the authors of this review to suggest for the first time that:
- The damaging factor for small cholangiocytes is bile acids and not AMA;
- Bile acids trigger senescence and apoptosis in small cholangiocytes, which ultimately leads to the development of ductulopenia;
- Bile acids create conditions for access to the inner mitochondrial membrane of small cholangiocytes and to the E2 PDH complex;
- The presence of lipoic acid in the E2 subunit of the PDH complex and its interaction with bile acids, leads to immunomodification and acquisition of neoantigenic properties of E2 PDH in small BECs;
- The acquisition of neoantigenic properties of the E2 PDG complex in small cholangiocytes undergoing apoptosis results in the recognition of this complex by the normal ('healthy') immune system as a foreign antigen, thereby triggering the production of AMA. It can therefore be concluded that the presence of AMAs in PBC is indicative of an organ-specific rather than a true autoimmune disease.
–£–≤–∞–ґ–∞–µ–Љ—Л–є –њ–Њ—Б–µ—В–Є—В–µ–ї—М uMEDp!
–£–≤–µ–і–Њ–Љ–ї—П–µ–Љ –Т–∞—Б –Њ —В–Њ–Љ, —З—В–Њ –Ј–і–µ—Б—М —Б–Њ–і–µ—А–ґ–Є—В—Б—П –Є–љ—Д–Њ—А–Љ–∞—Ж–Є—П, –њ—А–µ–і–љ–∞–Ј–љ–∞—З–µ–љ–љ–∞—П –Є—Б–Ї–ї—О—З–Є—В–µ–ї—М–љ–Њ –і–ї—П —Б–њ–µ—Ж–Є–∞–ї–Є—Б—В–Њ–≤ –Ј–і—А–∞–≤–Њ–Њ—Е—А–∞–љ–µ–љ–Є—П.
–Х—Б–ї–Є –Т—Л –љ–µ —П–≤–ї—П–µ—В–µ—Б—М —Б–њ–µ—Ж–Є–∞–ї–Є—Б—В–Њ–Љ –Ј–і—А–∞–≤–Њ–Њ—Е—А–∞–љ–µ–љ–Є—П, –∞–і–Љ–Є–љ–Є—Б—В—А–∞—Ж–Є—П –љ–µ –љ–µ—Б–µ—В –Њ—В–≤–µ—В—Б—В–≤–µ–љ–љ–Њ—Б—В–Є –Ј–∞ –≤–Њ–Ј–Љ–Њ–ґ–љ—Л–µ –Њ—В—А–Є—Ж–∞—В–µ–ї—М–љ—Л–µ –њ–Њ—Б–ї–µ–і—Б—В–≤–Є—П, –≤–Њ–Ј–љ–Є–Ї—И–Є–µ –≤ —А–µ–Ј—Г–ї—М—В–∞—В–µ —Б–∞–Љ–Њ—Б—В–Њ—П—В–µ–ї—М–љ–Њ–≥–Њ –Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–љ–Є—П –Т–∞–Љ–Є –Є–љ—Д–Њ—А–Љ–∞—Ж–Є–Є —Б –њ–Њ—А—В–∞–ї–∞ –±–µ–Ј –њ—А–µ–і–≤–∞—А–Є—В–µ–ї—М–љ–Њ–є –Ї–Њ–љ—Б—Г–ї—М—В–∞—Ж–Є–Є —Б –≤—А–∞—З–Њ–Љ.
–Э–∞–ґ–Є–Љ–∞—П –љ–∞ –Ї–љ–Њ–њ–Ї—Г ¬Ђ–Т–Њ–є—В–Є¬ї, –Т—Л –њ–Њ–і—В–≤–µ—А–ґ–і–∞–µ—В–µ, —З—В–Њ —П–≤–ї—П–µ—В–µ—Б—М –≤—А–∞—З–Њ–Љ –Є–ї–Є —Б—В—Г–і–µ–љ—В–Њ–Љ –Љ–µ–і–Є—Ж–Є–љ—Б–Ї–Њ–≥–Њ –≤—Г–Ј–∞.