Механизм повреждения мелких холангиоцитов при первичном билиарном холангите
- Аннотация
- Статья
- Ссылки
- English



Введение
Первичный билиарный холангит (ПБХ) – это хроническое холестатическое прогрессирующее заболевание печени, протекающее с деструкцией, некрозом и/или апоптозом эпителия мелких внутридольковых и септальных желчных протоков, с формированием аутоиммунного компонента, в терминальной стадии которого развивается цирроз печени [1–3]. Первичному билиарному холангиту предшествует длительный бессимптомный период [1, 2]. В это время отсутствуют какие-либо физикальные признаки заболевания. Клинически может проявляться слабостью и недомоганием. Обнаружение в этот период антимитохондриальных аутоантител (АМА) в сыворотке крови в титре 1:40 и выше служит патогномоничным маркером развития ПБХ. Тот факт, что АМА обнаруживаются за много месяцев до появления клинических признаков ПБХ, указывает на их первичную патогенетическую роль, а не на вторичное явление, возникающее как следствие холестаза [4, 5]. При этом большинство исследователей подчеркивают отсутствие корреляции титра АМА с активностью и продолжительностью заболевания [4–7]. Раскрытие причин и механизмов повреждения мелких холангиоцитов и механизмов образования АМА может способствовать пониманию патогенеза развития клинических, морфологических, биохимических и иммунологических признаков ПБХ.
Антимитохондриальные антитела не являются строго специфичными для ПБХ [4]. Их относят к иммуноглобулинам класса М (IgM), которые реагируют с множеством антигенов в митохондриях, обозначенных как M1–M9 [8]. Высокочувствительными и наиболее часто
(> 95%) встречаемыми аутоантителами при ПБХ являются анти-М2 [8]. Доказано, что у пациентов с классическим течением ПБХ антигенные компоненты АМА относятся к дигидролипоилтрансацетилазе (Е2-субъединица) пируватдегидрогеназного (ПДГ) комплекса (Е2 ПДГ), который локализуется на внутренней мембране митохондрий [8]. Согласно экспериментальным данным по иммунизации лабораторных животных, E2 ПДГ в качестве рекомбинантного полипептида приводит к образованию АМА, но не к повреждению холангиоцитов [9]. Это свидетельствует о том, что АМА не являются фактором, запускающим деструкцию билиарных эпителиальных клеток (БЭК, холангиоцитов).
До сих пор неясно, каким образом антиген Е2 ПДГ, находясь на внутренней мембране митохондрий, может быть мишенью иммунных эффекторных механизмов. Крайне важно понять, почему в этот процесс вовлечен антиген Е2 ПДГ клеток желчного эпителия, причем расположенного в желчных протоках мелкого и среднего размера [4]. Четкого ответа на эти вопросы до настоящего времени не существует. Ранее обсуждалась теория антигенной мимикрии.
АМА и теория антигенной мимикрии
Пируватдегидрогеназный комплекс у прокариотов имеет структурное сходство с подобным комплексом эукариотов [10]. Было показано, что антитела, полученные из сыворотки пациентов с ПБХ, вступают в реакцию с дрожжевыми и бактериальными белками [11, 12]. В связи с этим было высказано предположение, что АМА при ПБХ возникают вследствие перекрестной реактивности к экзогенным бактериальным антигенам (антигенная мимикрия) [13, 14] и что заболевание, возможно, имеет бактериальное происхождение [15]. Но никому не удалось найти четких доказательств наличия какого-либо инфекционного агента [4]. Кроме того, при классическом бактериальном антигенном воздействии на организм в первую очередь вырабатываются и повышаются в крови IgM. Спустя три-четыре недели на их место приходят иммуноглобулины G (IgG). IgM при этом могут сохраняться в организме больного до трех месяцев с последующим их снижением. Но при ПБХ уровень АМА, относящихся к IgM, не исчезает и даже не уменьшается на протяжении многолетнего развития заболевания, что в полной мере не объясняет нарушения иммунной системы и не очень укладывается в бактериальную природу антигенов, запускающих выработку АМА. Хотя при продолжительном воздействии тимуснезависимых антигенов синтез IgM может приобрести стабильный характер [16]. Но при этом требуется постоянное наличие тимуснезависимого антигена в организме пациента и снижение иммунотолерантности к нему. Вероятность того, что бактериальный антиген постоянно присутствует в организме пациентов с ПБХ и запускает выработку АМА, невелика [10].
Логичнее предположить, что это антиген собственных тканей организма человека, а именно эпителия желчевыводящих путей. Но тогда для выработки АМА у пациентов с ПБХ необходимо, чтобы Е2 ПДГ стала иммуноизмененным антигеном, покинула митохондрию, холангиоцит и встретилась с иммунокомпетентными клетками, которые запустят выработку аутоантител. До настоящего времени остаются неизвестными триггеры и механизмы, запускающие эти процессы в холангиоцитах.
В последнее десятилетие появились научные данные о значении бикарбоната (HCO3-) в качестве «защитного зонтика» для холангиоцитов от токсического действия желчных кислот. Показано, что при ПБХ уменьшается выработка HCO3-, что приводит к повышенному поступлению желчных кислот в холангиоцит (теория «дырявого бикарбонатного зонтика»). Эти данные позволили нам высказать предположение, что происходящее при этом постепенное накопление желчных кислот в БЭК, уже в асимптоматической стадии ПБХ, может служить пусковым механизмом для повреждения мелких БЭК, развития дуктулопении, образования АМА и одного из ранних клинических признаков – слабости.
Факторы агрессии и защиты холангиоцитов
Желчь является агрессивной средой для холангиоцитов, выстилающих внутри- и внепеченочные желчные протоки. Наличие в желчи желчных кислот, обладающих мощными детергентными свойствами, способно вызывать повреждение клеточных мембран холангиоцитов. Гидрофобные желчные кислоты проявляют цитотоксичность ко многим типам клеток [17]. Однако эпителиальные клетки желчных протоков человека в физиологических условиях подвергаются воздействию очень высоких (миллимолярных) концентраций гидрофобных желчных кислот без признаков цитотоксичности [18]. Эта устойчивость подразумевает наличие механизмов, защищающих холангиоциты от токсического воздействия желчных кислот.
Конъюгирование желчных кислот и образование смешанных мицелл с холестерином и фосфолипидами рассматриваются в качестве защитных механизмов уже на уровне гепатоцитов, желчных капилляров и канальцев Геринга [18]. К известным факторам защиты, которые поступают в желчь в процессе ее прохождения по желчным протокам, относят выработку и секрецию муцина и бикарбоната [19]. В физиологических условиях основной функцией холангиоцитов является билиарная секреция HCO3- [20]. HCO3- вырабатывается холангиоцитами на всем протяжении билиарного дерева. Выработка муциновых гликопротеидов осуществляется перибилиарными железами (ПБЖ, железы желчных протоков) [21]. ПБЖ располагаются в стенке крупных внутри- и внепеченочных желчных протоков и непосредственно связаны с их просветом. Экспериментальные данные указывают на то, что гликокаликс, покрывающий апикальную поверхность мембран холангиоцитов, с гликозилированными муцинами и другими гликансодержащими мембранными гликопротеинами, стабилизирует «билиарный бикарбонатный зонтик», помогая таким образом защитить холангиоциты человека от токсичности желчных кислот [22]. Вырабатываемый ПБЖ муцин защищает холангиоциты только крупных желчных протоков [19]. Исходя из этого, холангиоциты крупных внутри- и внепеченочных желчных протоков имеют двойную защиту: муцин, вырабатываемый ПБЖ, и бикарбонат. Внутридольковые, междольковые и септальные желчные протоки перибилиарных желез не содержат, что сопровождается отсутствием в них муцина [21]. В результате на уровне внутридольковых, междольковых и септальных протоков фактором защиты БЭК служит только HCO3-.
В физиологических условиях складывается равновесие между факторами агрессии (желчные кислоты) и защиты (секреция бикарбоната и муцина).
Механизмы защиты холангиоцитов
Холангиоциты представляют собой поляризованные эпителиальные клетки, которые выстилают внутри- и внепеченочные желчные протоки, а также отвечают за регулирование объема желчи, модификацию желчи и поддержание рН (щелочности) желчи [23, 24]. Холангиоциты играют важную роль в изменении состава первичной желчи вследствие секреции воды, хлора (Cl-) и HCO3- [25], а также поглощения солей желчных кислот, аминокислот и глюкозы. Выделяют мелкие и крупные холангиоциты в зависимости от их размеров и расположения в мелких и крупных желчных протоках [26]. Они по-разному участвуют в процессах секреции и абсорбции [27]. Секреция HCO3- с желчью человека составляет 25–40% от общего объема выделяемой желчи и поддерживает физиологический рН в просвете внутрипеченочных желчных протоков [17, 28, 29].
В процессе желчеобразования в желчный капилляр поступают преимущественно конъюгированные желчные кислоты и минимальное количество неконъюгированных. В физиологических условиях как конъюгированные, так и неконъюгированные желчные кислоты секретируются в желчь гепатоцитами в анионной (депротонированной, ионизированной, имеющей отрицательный заряд) форме [30]. Бикарбонат, секретируемый холангиоцитами в просвет желчного протока, вследствие своих буферных свойств создает слабощелочной рН печеночной желчи. Это поддерживает желчные кислоты в депротонированном состоянии. Ионизированная форма желчных кислот не позволяет им проникать в БЭК в связи с наличием на апикальной поверхности цитоплазматической мембраны холангиоцитов отрицательно заряженных молекул HCO3- [30]. Таким образом, секреция бикарбонат-ионов защищает холангиоциты от неконтролируемого трансмембранного поступления желчных кислот, что получило название «билиарный бикарбонатный зонтик» [22]. Последний обеспечивает сохранность холангиоцитов и нормальное желчеотделение по билиарному дереву.
Основные регуляторы выработки и секреции бикарбоната холангиоцитами
Колебания рН в желчных протоках зависят от скорости выработки бикарбоната холангиоцитами. В крупных и малых холангиоцитах сигнальные пути, регулирующие секрецию HCO3-, различаются [31]. В малых холангиоцитах активация секреции бикарбоната происходит вследствие билиарного АТФ, секретируемого из вышележащих гепатоцитов канальцев Геринга (рис. 1). Холангиоциты экспрессируют апикальные мембранные белки семейства пуринергических рецепторов (P2YR), которые стимулируются аденозинтрифосфатами (АТФ) [32]. Люминальный АТФ мелких желчных протоков связывается с P2YR, стимулируя внутриклеточное высвобождение ионов Ca2+ через инозитол-1,4,5-трисфосфатный рецептор третьего типа (InsP3R3, ITPR3) [33]. В холангиоцитах InsP3R3 является основной изоформой рецептора, которая локализуется в апикальной области [31, 34] и участвует в InsP3R3-опосредованной передаче сигналов в клетке и секреции Ca2+ [35]. InsP3R3 являются единственными рецепторами, способствующими открытию внутриклеточных кальциевых каналов и высвобождению ионов кальция [34]. Кальций является одним из мессенджеров в холангиоците, который модулирует и регулирует такие разнообразные функции клеток, как активация ионных каналов, секреция, клеточная пролиферация, апоптоз и др. [34, 36]. Высвобождение Ca2+ из субапикальных запасов в эндоплазматическом ретикулуме запускает и локально активирует трансмембранные 16А хлоридные (Cl-) каналы (TMEM16A) на апикальной мембране холангиоцитов (рис. 1) [36–38]. Возникающий градиент концентрации Cl- на апикальной мембране активирует хлор/карбонатный (Cl-/HCO3-) анионообменник 2 (AE2, также Slc4A2), что приводит к секреции HCO3- в просвет желчного протока.
В крупных холангиоцитах, кроме Ca2+-зависимого пути секреции бикарбоната, существует дополнительный механизм, функционирующий с участием гормонов секретина и соматостатина [39] (рис. 1). Секретин вырабатывается S-клетками слизистой оболочки двенадцатиперстной кишки и стимулирует выработку HCO3- не только слизистой оболочкой самой кишки, но и холангиоцитами, а также эпителиальными клетками протоков поджелудочной железы [39]. Секретин регулирует секрецию крупными холангиоцитами HCO3- и Cl- в желчь посредством взаимодействия с рецепторами секретина (SRs), расположенными на базолатеральной мембране БЭК [39–42] (рис. 1).
В результате такого взаимодействия через G-белок стимулируется образование циклического АМФ (цАМФ). Последний через аденилатциклазу (АС) активирует регулятор трансмембранной проводимости при муковисцидозе (CFTR, Cystic Fibrosis Transmembrane conductance Regulator), вызывая секрецию ионов Cl- в желчный проток [28]. Возникающий градиент концентрации Cl- на апикальной мембране холангиоцита активирует хлор/карбонатный анионообменник 2, что приводит к секреции HCO3- в просвет желчного протока взамен на внутриклеточное поступление ионов Cl- в холангиоцит [20, 24, 26, 43]. Параллельно происходит поступление АТФ из холангиоцита в просвет желчного протока вследствие экзоцитоза, что стимулирует секрецию HCO3- по Са2+-зависимому механизму [44].
Рецепторы к секретину и регулятор трансмембранной проводимости при муковисцидозе (CFTR) не обнаруживаются в малых холангиоцитах. Поэтому секретин не способен стимулировать в малых холангиоцитах секрецию и поступление в желчь HCO3- и Cl- [24]. При этом сигнальный Ca2+-зависимый механизм секреции бикарбоната присутствует как в малых, так и в крупных типах холангиоцитов (рис. 1) [20, 24].
Соматостатин, связываясь с рецептором соматостатина (SSTR), противодействует стимулирующему действию секретина, ингибирует секрецию жидкости, тормозит выработку и поступление HCO3- из холангиоцитов в просвет желчного протока [45].
Механизмы протонирования-депротонирования желчных кислот и проникновения в холангиоциты
Неконтролируемое, независимое от переносчиков, пассивное диффундирование внутрь БЭК неконъюгированных первичных желчных кислот определяется их полярностью и степенью протонирования [18, 46, 47].
Протонирование желчных кислот является экспоненциальной функцией от рН. При закислении рН печеночной желчи желчные кислоты могут подвергаться протонированию. Степень протонирования желчных кислот зависит как от их константы диссоциации (pKa), так и от рН желчи. Значения pKa для неконъюгированных первичных желчных кислот составляют 5–6 [48–50]. Конъюгация первичных желчных кислот с аминокислотами снижает pKa до значений 4–5 для конъюгатов с глицином и до 1–2 – для конъюгатов с таурином, что улучшает их растворимость в воде и снижает липофильность [48–50]. Низкие значения pKa тауриновых конъюгатов первичных желчных кислот свидетельствуют о том, что они являются более сильными кислотами, чем глициновые конъюгаты. Поэтому конъюгированные с таурином желчные кислоты будут находиться в диссоциированной (депротонированной) форме даже при кислых значениях рН желчи, в то время как глициновые конъюгаты, с более высокими значениями pKa, являются слабыми кислотами и при малейшем закислении желчи будут быстро переходить в протонированное состояние [50].
Ионизированные (депротонированные, имеющие отрицательный заряд) желчные кислоты не способны преодолевать «бикарбонатный зонтик» на внешнем лепестке апикальной цитоплазматической мембраны холангиоцитов [18, 47]. В норме незначительное количество неконъюгированных протонированных первичных желчных кислот попадает в холангиоцит. Нейтральный внутриклеточный рН способствует транспортировке неконъюгированных протонированных первичных желчных кислот далее в перибилиарное сосудистое сплетение с последующим возвращением в гепатоциты и повторным выделением в желчные капилляры [51]. Такой желчнопеченочный шунт направлен на предотвращение накопления в холангиоцитах токсичных, обладающих сильными детергентными свойствами желчных кислот [18, 47].
Конъюгированные желчные кислоты могут транспортироваться через апикальную и базолатеральную мембраны холангиоцитов с помощью специфических транспортеров [26, 52–56].
Конъюгаты желчных кислот с глицином в печеночной желчи человека составляют 3/4 всех конъюгированных желчных кислот и имеют значение pKa, близкое к 4 [57]. При физиологическом рН 7,4 глициновые конъюгаты первичных желчных кислот, являясь относительно слабыми кислотами, будут частично протонированы (станут неполярными), что способствует их проникновению в микромолярных количествах в холангиоциты. Небольшое смещение локального рН в кислую среду в желчевыводящих протоках приведет к увеличению количества протонированных глициновых конъюгатов первичных желчных кислот. Существенное увеличение соотношения протонированных: депротонированных конъюгированных с глицином желчных кислот приведет к повышенному поступлению их в холангиоциты.
Конъюгаты желчных кислот с таурином в печеночной желчи составляют 1/4 часть всех конъюгированных желчных кислот. Они являются более сильными кислотами и имеют pKa 1–2 [57, 58], поэтому изменения рН желчевыводящих путей будут слабо влиять на их протонирование. Большая часть тауриновых конъюгатов желчных кислот будет находиться в анионной форме и не сможет поступать в холангиоциты. В связи с этим тауриновые конъюгаты первичных желчных кислот обладают меньшей токсичностью для холангиоцитов.
Активное функционирование транспортеров желчных кислот в базолатеральной мембране холангиоцита, как правило, приводит к быстрому выведению гидрофобных желчных кислот из внутриклеточного пространства и обратной доставке их в гепатоциты [59], поэтому накопление токсичных, обладающих детергентными свойствами желчных кислот в холангиоците в норме не происходит.
Теория «дырявого бикарбонатного зонтика» при ПБХ
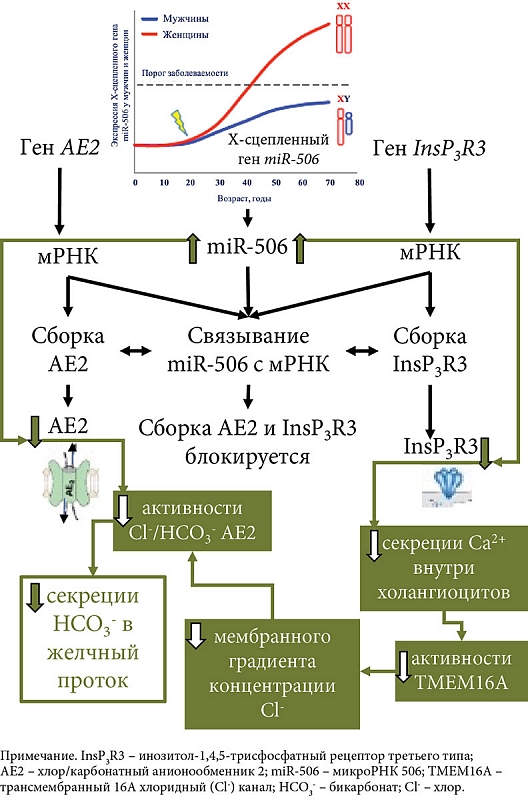
Исследования последнего десятилетия показывают снижение защитной роли бикарбоната при ПБХ. Активно обсуждается теория «дырявого бикарбонатного зонтика» [1, 17, 22]. Эта теория основана на ряде клинических и экспериментальных работ, показывающих недостаточное поступление HCO3- в желчные протоки при ПБХ, что приводит к смещению рН внутрипротоковой (печеночной) желчи в слабокислую область, а рН внутри холангиоцита – в слабощелочную область. Причины недостаточной выработки HCO3- холангиоцитами до настоящего времени остаются неизвестными. Обсуждается вовлеченность в этот процесс рецепторов InsP3R3 (ITPR3) и анионообменника AE2. Показано, что в биоптатах печени и мононуклеарных клетках крови пациентов с ПБХ снижена экспрессия генов InsP3R3 и AE2, что свидетельствует об их дисфункции и вовлечении в патогенез этого заболевания [60, 61]. Снижение активности InsP3R3 и AE2, а также нарушение их секреторной функции связывают с микро-РНК 506 (miR-506) [62]. Микро-РНК представляют собой небольшие некодирующие РНК длиной 22–23 нуклеотида, которые ингибируют экспрессию генов путем полного или частичного спаривания с начальными последовательностями, расположенными в 3'-нетранслируемых областях (3'-UTR) мРНК [62].
Области 3'-UTR мРНК InsP3R-3 [62] и 3'UTR мРНК AE2 [63] содержат сайты связывания для miRNA-506. Последняя, связываясь с 3'UTR-областью мРНК InsP3R3 и 3'UTR мРНК AE2, предотвращает трансляцию этих белков. Вследствие этого miR-506 является регулятором экспрессии InsP3R3 и AE2 (рис. 2). Экспрессия miR-506, вероятно, подвержена эпигенетической регуляции и может варьировать у разных индивидуумов в результате полиморфизмов в сигнальном пути NF-κB (Nuclear Factor kappa-light-chain-enhancer of activated B cells, ядерный фактор каппа – усилитель легкой цепи активированных В-клеток) [30].
В холангиоцитах пациентов с ПБХ отмечается повышение количества и активности miR-506 [63]. Открытие повышенной активности Х-сцепленной miR-506 в холангиоцитах пациентов с ПБХ приводит к снижению экспрессии и активности InsP3R3 и АЕ2, а также потенциально объясняет преобладание этого заболевания у женщин [30] (рис. 2).
Снижение экспрессии и активности InsP3R3 в холангиоцитах при ПБХ [64] ухудшает внутриклеточную секрецию ионов Ca2+i и использование его в качестве мессенджера в передаче сигналов на трансмембранный Cl- канал TMEM16A [44]. О нарушении передачи сигналов Ca2+ в холангиоцитах при ПБХ свидетельствуют данные об отсутствии АТФ-стимуляции пуринергических рецепторов (P2YR) на апикальной мембране [36]. Снижение кальций-зависимой активности TMEM16A на апикальной мембране холангиоцитов приводит к уменьшению секреции ионов Cl- в просвет желчных протоков, что сопровождается снижением активности хлор/бикарбонатного анионообменника 2 и нарушением секреции HCO3- БЭК. В моделях холангиоцитов, экспрессирующих miRNA-506, показано опосредованное InsP3R3 уменьшение внутриклеточного высвобождения Ca2+ и снижение секреции жидкости и HCO3- в желчные протоки [36, 44]. Связывание miR-506 с 3'UTR мРНК AE2 также способствует снижению активности хлор/бикарбонатного анионообменника и уменьшению секреции HCO3- холангиоцитами (рис. 2). Холангиоциты человека, выделенные из биоптатов пациентов с ПБХ, показывают снижение активности АЕ2 [65]. Вследствие этого гомеостаз внутриклеточного рН (pHi) в холангиоцитах и рН в желчных протоках у пациентов с ПБХ может подвергаться изменению [30]. Изменения внутри- и внеклеточного рН при ПБХ, связанные с потерей InsP3R3 и снижением активности АЕ2, способствуют протонированию желчных кислот, поступлению их в холангиоциты и развитию повреждения последних [44].
Деструкция билиарного эпителия мелких внутрипеченочных желчных протоков уже на ранней асимптоматической стадии ПБХ, вероятнее всего, связана с дисбалансом между факторами агрессии (желчными кислотами) и факторами защиты («бикарбонатный зонтик») холангиоцитов. Так как внутридольковые, междольковые и септальные желчные протоки, которые повреждаются при ПБХ, не содержат перибилиарные железы, вырабатывающие муциновые гликопротеины [21], этот механизм защиты малых холангиоцитов (муциновый надэпителиальный слой), вероятнее всего, не играет патогенетической роли в развитии ПБХ.
Механизм повреждения холангиоцитов при «дырявом бикарбонатном зонтике»
Уменьшение поступления HCO3- в желчные протоки вследствие снижения активности InsP3R3 и АЕ2 будет смещать рН в просвете желчного протока в кислую среду [66]. Одновременно с этим из-за задержки и накопления HCO3- в цитозоле холангиоцитов будет происходить постепенное защелачивание внутриклеточного pHi у пациентов с ПБХ [30, 65, 67]. Полный дефицит AE2 приведет к внутриклеточному алкалозу холангиоцитов [61], но у пациентов с ПБХ наблюдается снижение (а не отсутствие) экспрессии генов InsP3R3 и AE2 [61].
Смещение рН в слабокислую область в просвете желчных протоков увеличит количество протонированных неконъюгированных и конъюгированных с глицином первичных желчных кислот. Это приведет к повышенному поступлению их в малые (внутридольковые, междольковые, септальные) холангиоциты желчных протоков. Попадая в слабощелочное pHi внутри холангиоцитов, протонированные желчные кислоты будут подвергаться депротонированию. Защелачивание pHi и ионизация конъюгированных с глицином и неконъюгированных первичных желчных кислот внутри холангиоцитов снижают процесс их диффундирования из внутриклеточного в перибилиарное пространство. В результате происходит задержка и постепенное накопление конъюгированных с глицином и неконъюгированных первичных желчных кислот в малых холангиоцитах. Теория «дырявого бикарбонатного зонтика» позволяет объяснить внутриклеточное неконтролируемое повышенное проникновение и накопление желчных кислот в малых БЭК.
Наличие на апикальной поверхности крупных холангиоцитов муцинсодержащего гликокаликсного слоя защищает их от проникновения и повреждающего действия протонированных конъюгированных и неконъюгированных желчных кислот.
Внутриклеточное накопление гидрофобных желчных кислот является предпосылкой для их цитотоксических эффектов [68]. Являясь сильными детергентами, они способны солюбилизировать фосфолипиды и холестерин из мембранных структур холангиоцитов, что приводит к повреждению и деструкции цитоплазматической мембраны и мембран клеточных органелл (рис. 3). При этом попадание и накопление в малых холангиоцитах неконъюгированных желчных кислот, обладающих более сильными детергентными свойствами, является более токсичным для клетки, чем накопление конъюгированных желчных кислот.
Хроническое повреждающее действие желчных кислот на мембранные структуры запускает ускоренное старение, некроз и/или апоптоз БЭК [69]. Происходит разрушение желчными кислотами мембран клеточных органелл и ядерной мембраны в холангиоцитах с выходом апоптогенных факторов. Нарушается барьерная функция желчного эпителия, что приводит к сопутствующему повреждению, воспалению и окислительному стрессу. Цитокины, хемокины и провоспалительные медиаторы, высвобождаемые холангиоцитом, вероятно, стимулируют апоптотические и пролиферативные реакции, активируют фиброгенез [70]. Желчные кислоты также опосредуют свои токсические, апоптотические эффекты через специфические сигнальные пути на внутриклеточном уровне. Активируется внутренний апоптотический путь, включающий митохондриальную транслокацию BAX (BCL2-associated X protein), высвобождение цитохрома С из митохондрий, активацию каспазы 3, расщепление PARP (Poly (ADP-ribose) polymerase) и фрагментацию ДНК [71]. Имеются данные, что miRNA-506 активирует путь апоптоза при стимуляции токсичными желчными кислотами [66]. Провоспалительные цитокины дополнительно увеличивают экспрессию miR-506 [30]. Развивается порочный круг, который поддерживает старение, апоптоз и пролиферацию холангиоцитов, а в конечном счете – дуктулопению [30]. Все это отражает прямое воздействие желчных кислот на холангиоциты, а не на неспецифические эффекты, возникающие в результате перипортального воспаления [31].
С патофизиологической точки зрения общим для всех холангиопатий является сосуществование гибели и пролиферации холангиоцитов, а также различных степеней портального воспаления и фиброза [70]. Гибель клеток индуцирует активацию воспалительных и профиброгенных путей, которые запускают развитие и прогрессирование фиброза, что постепенно приводит к развитию дуктулопении мелких желчных протоков [72]. Концептуальные механизмы развития этих процессов описаны в обзорах [69, 72].
Нарушение апоптоза считается триггером ПБХ и уже в асимптоматической стадии приводит к развитию дуктулопении мелких желчных протоков, одного из ранних морфологических признаков заболевания [73].
Апоптоз зависит от пермеабилизации митохондрий, связанной с избыточным внутриклеточным накоплением желчных кислот [74–76]. Кроме того, желчные кислоты и неполный апоптоз БЭК, перенаправленный в некроз, могут приводить к патогенному воздействию на внутриклеточные компоненты с последующей генерацией AMA [77].
Механизм пермеабилизации митохондрий и образования АМА
Солюбилизация желчными кислотами фосфолипидов и холестерина с внешней мембраны митохондрий приводит к нарушению их проницаемости (пермеабилизации) [78]. Происходит увеличение проницаемости внешней мембраны митохондрий для ионов и растворов [71, 78], возникает утечка содержимого межмембранного пространства в цитозоль и потеря мембранного потенциала, происходят набухание митохондрий, разрыв их внешней мембраны и выход апоптогенных факторов [78]. Открывается внутренняя митохондриальная мембрана, основная мишень для образования AMA. Дальнейшая деструкция митохондрий и солюбилизация желчными кислотами фосфолипидов и холестерина с их внутренней мембраны могут приводить к высвобождению и деградации пируватдегидрогеназного комплекса (ПДГ). Последний включает в себя три фермента: пируватдегидрогеназу (Е1 ПДГ), дигидролипоилтрансацетилазу (Е2 ПДГ) и дигидролипоилдегидрогеназу (Е3 ПДГ) [73]. Каждый из этих ферментов, кроме белковой части, имеет кофакторы: Е1 ПДГ в качестве кофактора содержит тиаминпирофосфат, Е2 ПДГ – липоевую кислоту и коэнзим А, Е3 ПДГ – ФАД и НАД. Е1 ПДГ и Е3 ПДГ являются белковыми комплексами, которые не содержат липидных компонентов, поэтому отсутствует вероятность влияния на них накопившихся в холангиоците желчных кислот, воздействующих на липидные компоненты. Сыворотки пациентов с ПБХ не проявляют серологически определяемой реактивности против Е1- и E3-компонентов ПДГ [79].
Е2 ПДГ является липопротеидом и имеет два сайта связывания липоевой кислоты [8]. Е2 ПДГ содержит незаменимый остаток лизина в липоильном домене, к которому ковалентно присоединена липоевая кислота [8]. Липоево-лизиновая связь в положении 173 высоко консервативна у разных видов и необходима для распознавания антигена [80]. AMA нацелены на иммунодоминантные эпитопы, содержащие липоевую кислоту.
Ранее было показано значение химических ксенобиотиков, способных модифицировать липоевую кислоту в Е2 ПДГ, для появления серологической реактивности этого комплекса [81, 82]. Изменение конформационной структуры липоильного домена Е2 ПДГ вследствие химической модификации липоевой кислоты может способствовать потере иммунной толерантности [83, 84]. Вероятнее всего, такими химическими модификаторами при ПБХ выступают желчные кислоты, накапливающиеся в холангиоцитах при потере защитных свойств «бикарбонатного зонтика». Желчные кислоты, обладающие мощными детергентными свойствами, могут взаимодействовать с липоевой кислотой антигенраспознаваемого участка Е2 ПДГ. Результатом такого взаимодействия может быть иммуномодификация Е2 ПДГ-комплекса с приобретением аутоантигенных свойств и потерей иммунной толерантности [66]. Подтверждением этого предположения служат ряд исследований, выполненных в конце прошлого столетия. В этих работах было показано, что основная иммуногенная область на E2 ПДГ, распознаваемая сыворотками от пациентов с ПБХ, локализована в липоилсодержащем домене [85–87]. При этом считается, что содержание липоевой кислоты в E2 ПДГ играет роль мощного адъюванта [88]. Презентация лимфоцитам иммуномодифицированного Е2 ПДГ-комплекса может привести к стимулированию субпопуляции Т-клеток и специфическому продуцированию АМА [66, 88, 89].
«Дырявый бикарбонатный зонтик» запускает постоянный и бесконечный процесс накопления и детергентного воздействия желчных кислот на малые холангиоциты с образованием АМА. Так как нарушение поступления бикарбоната в просвет желчного протока является постоянным, то и выработка АМА будет непрерывной. В результате в плазме крови больных ПБХ будет постоянно поддерживаться повышенный уровень IgM (М2).
Появление АМА в сыворотке крови является еще одним ранним иммунологическим патогномоничным признаком ПБХ, который появляется уже в асимптоматической стадии заболевания.
Дисфункция ПДГ-комплекса и первые клинические признаки асимптоматической стадии ПБХ
Детекция АМА в асимптоматической стадии заболевания сопровождается появлением первых субъективных клинических признаков: слабость, недомогание, усталость, снижение работоспособности [90]. Усталость является наиболее распространенным симптомом ПБХ в асимптоматической и ранней стадии заболевания [91–93]. Сообщается, что около 40–80% пациентов сталкиваются с усталостью в качестве симптома ПБХ [94, 95], однако нет никакой корреляции между усталостью и тяжестью или продолжительностью заболевания [95–98]. Механизм развития усталости тесно связан с постепенно прогрессирующей энергетической недостаточностью [99]. Последняя скорее всего связана с вовлечением пируватдегидрогеназного комплекса в патологический процесс развития ПБХ. Комплекс пируватдегидрогеназы является очень важным метаболическим энзимом. ПДГ функционирует в каждой клетке и необходим для превращения пирувата в ацетил-КоА, который включается в цикл Кребса и крайне важен для получения организмом энергии в форме АТФ [73]. По мере пермеабилизации митохондрий в холангиоцитах и вовлечения ПДГ в выработку АМА происходит постепенное и постоянное снижение синтеза АТФ. Это приводит к развитию локальной энергетической недостаточности, что, в свою очередь, способствует усилению процессов старения и апоптоза мелких БЭК, инициированных желчными кислотами. Возникает порочный замкнутый круг, способствующий прогрессированию дуктулопении и образованию АМА. Аутоантитела при этом способны реагировать с полипептидами, относящимися к Е2 ПДГ, в митохондриях практически любых клеток. Показано, что антитела к ПБХ перекрестно реагируют с полипептидами в митохондриях говяжьего сердца, предположительно относящимися к Е2 ПДГ [100]. Происходит снижение выработки АТФ и развитие энергетической недостаточности во всем организме.
Развитие энергетической недостаточности сопровождается повышением гликогенолиза и снижением гликогеногенеза. Представленные J.H. Green и соавт. данные указывают на то, что уже на начальных стадиях ПБХ в печени постепенно происходит снижение запасов гликогена, связанное с повышением гликогенолиза и снижением гликогеногенеза [101]. Авторы убедительно показали, что у пациентов с ПБХ значительно (вплоть до нуля) падает активность глюкокиназы, что свидетельствует о снижении образования гликогена в печени [101]. При этом гексокиназа (производит фосфорилирование гексоз), отвечающая за синтез гликогена преимущественно в мышцах, у пациентов с ПБХ в этот период достоверно увеличивается по сравнению со здоровыми лицами [101].
В результате развивающейся энергетической недостаточности в асимптоматической стадии заболевания появляются первые клинические признаки выраженной слабости, быстрой утомляемости, снижения работоспособности, функционального статуса и качества жизни пациентов с ПБХ [90, 102–104].
Заключение
Растущий объем знаний о молекулярных механизмах развития повреждений холангиоцитов у пациентов с ПБХ позволяет выдвинуть гипотезу, объясняющую патогенез возникновения первых морфологических (дуктулопения), иммунологических (АМА) и клинических (слабость, недомогание, быструю утомляемость) признаков заболевания в асимптоматической стадии [105] (рис. 4).
Имеющиеся данные свидетельствуют о том, что у восприимчивых индивидуумов первоначальный неизвестный триггер вызывает Х-сцепленное эпигенетическое изменение, что приводит к реактивации гена и увеличению экспрессии miR-506 [30]. Запуск повышенного синтеза и активации miR-506 приводит к ингибированию трансляции InsP3R3 и AE2 [106]. В результате уменьшается поступление бикарбоната в просвет желчного протока и происходит накопление HCO3- в цитозоле холангиоцитов [30]. Изменение вне- и внутриклеточного рН влияет на состояние протонирования (в просвете желчного протока) и депротонирования (внутри холангиоцита) желчных кислот, увеличиваются неконтролируемое поступление и накопление неконъюгированных и конъюгированных с глицином желчных кислот в БЭК.
Детергентные свойства желчных кислот запускают процесс разрушения клеточных мембран, старение и апоптоз холангиоцитов, пермеабилизацию митохондрий, деструкцию и иммуномодификацию Е2 ПДГ с последующим образованием АМА [105]. Старение, апоптоз и пролиферация холангиоцитов приводят к постепенному развитию дуктулопении. Вовлечение в патологический процесс ПДГ способствует недостаточному синтезу АТФ, развитию энергетической недостаточности, появлению неспецифического клинического признака – слабости. Развитие дуктулопении сопровождается развитием внутрипеченочного холестаза [107].
Холангиоциты являются основной мишенью на начальной стадии ПБХ. Но как только развивается холестаз, гепатоциты также вовлекаются в патологический процесс, что приводит к их повреждению [30, 108].
Авторы заявляют об отсутствии конфликта интересов.
V.I. Reshetnyak, PhD, Prof., I.V. Maev, PhD, Prof., Academician of the RAS
Russian University of Medicine, Moscow
Contact person: Vasilii I. Reshetnyak, vasiliy.reshetnyak@yandex.ru
Primary biliary cholangitis (PBC) is a chronic cholestatic progressive liver disease and one of the most important progressive cholangiopathies in adults. Damage to cholangiocytes triggers the development of intrahepatic cholestasis, which progresses to cirrhosis in the terminal stage of the disease. Accumulating data indicate that damage to biliary epithelial cells [(BECs), cholangiocytes] is most likely associated with the intracellular accumulation of bile acids, which have potent detergent properties and damaging effects on cell membranes.
The mechanisms underlying uncontrolled bile acid intake into BECs in PBC are associated with pH change in the bile duct lumen, which is controlled by the bicarbonate (HCO3-) buffer system "biliary HCO3- umbrella". The impaired production and entry of HCO3- from BECs into the bile duct lumen is due to epigenetic changes in expression of the X-linked microRNA 506. Based on the growing body of knowledge on the molecular mechanisms of cholangiocyte damage in patients with PBC, we propose a hypothesis explaining the pathogenesis of the first morphologic (ductulopenia), immunologic (antimitochondrial autoantibodies) and clinical (weakness, malaise, rapid fatigue) signs of the disease in the asymptomatic stage. This review focuses on the consideration
of these mechanisms.
Уважаемый посетитель uMEDp!
Уведомляем Вас о том, что здесь содержится информация, предназначенная исключительно для специалистов здравоохранения.
Если Вы не являетесь специалистом здравоохранения, администрация не несет ответственности за возможные отрицательные последствия, возникшие в результате самостоятельного использования Вами информации с портала без предварительной консультации с врачом.
Нажимая на кнопку «Войти», Вы подтверждаете, что являетесь врачом или студентом медицинского вуза.