–Я–∞—В–Њ–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Њ–µ –Њ–±–Њ—Б–љ–Њ–≤–∞–љ–Є–µ –њ—А–Є–Љ–µ–љ–µ–љ–Є—П –∞–ї—М—Д–∞–Ї–∞–ї—М—Ж–Є–і–Њ–ї–∞ –і–ї—П –ї–µ—З–µ–љ–Є—П –Њ—Б—В–µ–Њ–њ–Њ—А–Њ–Ј–∞ –њ—А–Є –љ–µ–Ї–Њ—В–Њ—А—Л—Е —Н–љ–і–Њ–Ї—А–Є–љ–љ—Л—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П—Е
- –Р–љ–љ–Њ—В–∞—Ж–Є—П
- –°—В–∞—В—М—П
- –°—Б—Л–ї–Ї–Є
- English
–Т–≤–µ–і–µ–љ–Є–µ
–Ю—Б—В–µ–Њ–њ–Њ—А–Њ–Ј, –≤–Њ–Ј–љ–Є–Ї–∞—О—Й–Є–є –љ–∞¬†—Д–Њ–љ–µ —Н–љ–і–Њ–Ї—А–Є–љ–љ—Л—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є, –Њ—В–љ–Њ—Б–Є—В—Б—П –Ї¬†–≤—В–Њ—А–Є—З–љ—Л–Љ –Њ—Б—В–µ–Њ–њ–Њ—А–Њ–Ј–∞–Љ –Є¬†—А–∞—Б—Б–Љ–∞—В—А–Є–≤–∞–µ—В—Б—П –Ї–∞–Ї –Њ—Б–ї–Њ–ґ–љ–µ–љ–Є–µ –Њ—Б–љ–Њ–≤–љ–Њ–є –њ–∞—В–Њ–ї–Њ–≥–Є–Є. –Ш–Љ–µ–љ–љ–Њ –њ–Њ—Н—В–Њ–Љ—Г –≤–Њ–њ—А–Њ—Б–∞–Љ–Є –і–Є–∞–≥–љ–Њ—Б—В–Є–Ї–Є –Є¬†–ї–µ—З–µ–љ–Є—П —В–∞–Ї–Є—Е –њ–∞—Ж–Є–µ–љ—В–Њ–≤ –љ–µ—А–µ–і–Ї–Њ –Ј–∞–љ–Є–Љ–∞—О—В—Б—П —Н–љ–і–Њ–Ї—А–Є–љ–Њ–ї–Њ–≥–Є. –Ю—Б—В–µ–Њ–њ–Њ—А–Њ–Ј –Є¬†–Њ—Б—В–µ–Њ–њ–∞—В–Є–Є, –≤–Њ–Ј–љ–Є–Ї–∞—О—Й–Є–µ –љ–∞¬†—Д–Њ–љ–µ —А–∞–Ј–ї–Є—З–љ—Л—Е —Н–љ–і–Њ–Ї—А–Є–љ–љ—Л—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є, —Е–∞—А–∞–Ї—В–µ—А–Є–Ј—Г—О—В—Б—П –њ–∞—В–Њ–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Є–Љ–Є –Є¬†–Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є–Љ–Є –Њ—Б–Њ–±–µ–љ–љ–Њ—Б—В—П–Љ–Є, –Ј–љ–∞–љ–Є–µ –Ї–Њ—В–Њ—А—Л—Е –Є–≥—А–∞–µ—В –≤–∞–ґ–љ—Г—О —А–Њ–ї—М –њ—А–Є –Њ–њ—А–µ–і–µ–ї–µ–љ–Є–Є —В–∞–Ї—В–Є–Ї–Є –ї–µ—З–µ–љ–Є—П.
–У–ї—О–Ї–Њ–Ї–Њ—А—В–Є–Ї–Њ–Є–і–љ—Л–є –Њ—Б—В–µ–Њ–њ–Њ—А–Њ–Ј
–Я–Њ¬†—А–∞—Б–њ—А–Њ—Б—В—А–∞–љ–µ–љ–љ–Њ—Б—В–Є –Є¬†–Љ–µ–і–Є–Ї–Њ-—Б–Њ—Ж–Є–∞–ї—М–љ–Њ–є –Ј–љ–∞—З–Є–Љ–Њ—Б—В–Є –њ–µ—А–≤–Њ–µ –Љ–µ—Б—В–Њ —Б—А–µ–і–Є —А–∞–Ј–љ—Л—Е –≤–Є–і–Њ–≤ –≤—В–Њ—А–Є—З–љ–Њ–≥–Њ –Њ—Б—В–µ–Њ–њ–Њ—А–Њ–Ј–∞ –Ј–∞–љ–Є–Љ–∞–µ—В¬†–≥–ї—О–Ї–Њ–Ї–Њ—А—В–Є–Ї–Њ–Є–і–љ—Л–є –Њ—Б—В–µ–Њ–њ–Њ—А–Њ–Ј, –Ї–Њ—В–Њ—А—Л–є —А–∞–Ј–≤–Є–≤–∞–µ—В—Б—П –≤—Б–ї–µ–і—Б—В–≤–Є–µ –і–ї–Є—В–µ–ї—М–љ–Њ–≥–Њ –њ—А–Є–µ–Љ–∞¬†–≥–ї—О–Ї–Њ–Ї–Њ—А—В–Є–Ї–Њ–Є–і–Њ–≤ (–У–Ъ) –ї–Є–±–Њ —Н–љ–і–Њ–≥–µ–љ–љ–Њ–є¬†–≥–Є–њ–µ—А—Б–µ–Ї—А–µ—Ж–Є–Є –У–Ъ –љ–∞–і–њ–Њ—З–µ—З–љ–Є–Ї–∞–Љ–Є.
–≠–љ–і–Њ–≥–µ–љ–љ—Л–є¬†–≥–Є–њ–µ—А–Ї–Њ—А—В–Є—Ж–Є–Ј–Љ –≤—Б—В—А–µ—З–∞–µ—В—Б—П –Њ—З–µ–љ—М —А–µ–і–Ї–Њ. –Ю—Б—В–µ–Њ–њ–Њ—А–Њ–Ј –Њ—В–Љ–µ—З–∞—О—В —Г¬†60вАУ90% –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–±–Њ–ї–µ–Ј–љ—М—О –Ш—Ж–µ–љ–Ї–Њ¬†вАУ –Ъ—Г—И–Є–љ–≥–∞ –Є–ї–Є —Б–Є–љ–і—А–Њ–Љ–Њ–Љ –Ъ—Г—И–Є–љ–≥–∞, –∞¬†–њ–µ—А–µ–ї–Њ–Љ—Л –њ–Њ–Ј–≤–Њ–љ–Ї–Њ–≤¬†вАУ —Г¬†76% [1].
–Т¬†–љ–∞—Б—В–Њ—П—Й–µ–µ –≤—А–µ–Љ—П –Њ—Б–љ–Њ–≤–љ–Њ–є –њ—А–Є—З–Є–љ–Њ–є —А–∞–Ј–≤–Є—В–Є—П –≤—В–Њ—А–Є—З–љ–Њ–≥–Њ –Њ—Б—В–µ–Њ–њ–Њ—А–Њ–Ј–∞ –Ї–∞–Ї —Г¬†–ґ–µ–љ—Й–Є–љ, —В–∞–Ї –Є¬†—Г –Љ—Г–ґ—З–Є–љ —Б—З–Є—В–∞–µ—В—Б—П –њ—А–Є–µ–Љ —Б–Є—Б—В–µ–Љ–љ—Л—Е –У–Ъ. –Ґ–∞–Ї, —П—В—А–Њ–≥–µ–љ–љ—Л–є¬†–≥–ї—О–Ї–Њ–Ї–Њ—А—В–Є–Ї–Њ–Є–і–љ—Л–є –Њ—Б—В–µ–Њ–њ–Њ—А–Њ–Ј —А–∞–Ј–≤–Є–≤–∞–µ—В—Б—П –≤¬†—Б—А–µ–і–љ–µ–Љ —Г¬†30вАУ50% –њ–∞—Ж–Є–µ–љ—В–Њ–≤ [1]. –У–Ъ —И–Є—А–Њ–Ї–Њ –њ—А–Є–Љ–µ–љ—П—О—В—Б—П –і–ї—П –ї–µ—З–µ–љ–Є—П —А–∞–Ј–ї–Є—З–љ—Л—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є, –≤¬†–њ–µ—А–≤—Г—О –Њ—З–µ—А–µ–і—М –Њ–±—Б—В—А—Г–Ї—В–Є–≤–љ—Л—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є –ї–µ–≥–Ї–Є—Е –Є¬†–±–Њ–ї–µ–Ј–љ–µ–є –Ї–Њ—Б—В–љ–Њ-–Љ—Л—И–µ—З–љ–Њ–є —Б–Є—Б—В–µ–Љ—Л. –Т¬†–Љ–Є—А–µ –≤¬†—Б—А–µ–і–љ–µ–Љ 2,7вАУ4,6% –ґ–µ–љ—Й–Є–љ 55¬†–ї–µ—В –Є¬†—Б—В–∞—А—И–µ –њ—А–Є–љ–Є–Љ–∞—О—В –њ–µ—А–Њ—А–∞–ї—М–љ—Л–µ –У–Ъ –њ–Њ—Б—В–Њ—П–љ–љ–Њ [2], —Б—А–µ–і–Є –ґ–Є—В–µ–ї—М–љ–Є—Ж –Т–µ–ї–Є–Ї–Њ–±—А–Є—В–∞–љ–Є–Є –і–∞–љ–љ–Њ–≥–Њ –≤–Њ–Ј—А–∞—Б—В–∞¬†вАУ 1,4вАУ1,7% [3].
–Я—А–Є–µ–Љ –У–Ъ –њ—А–Є–≤–Њ–і–Є—В –Ї¬†–±—Л—Б—В—А–Њ–Љ—Г —Б–љ–Є–ґ–µ–љ–Є—О –Љ–Є–љ–µ—А–∞–ї—М–љ–Њ–є –њ–ї–Њ—В–љ–Њ—Б—В–Є –Ї–Њ—Б—В–Є (–Ь–Я–Ъ), –љ–∞–Є–±–Њ–ї–µ–µ –≤—Л—А–∞–ґ–µ–љ–љ–Њ–Љ—Г –≤¬†–њ–µ—А–≤—Л–є¬†–≥–Њ–і –ї–µ—З–µ–љ–Є—П. –£–ґ–µ –≤¬†–њ–µ—А–≤—Л–µ —В—А–Є¬†вАУ —И–µ—Б—В—М –Љ–µ—Б—П—Ж–µ–≤ —В–µ—А–∞–њ–Є–Є –њ–Њ–≤—Л—И–∞–µ—В—Б—П —А–Є—Б–Ї¬†–њ–µ—А–µ–ї–Њ–Љ–Њ–≤ [4]. –Ъ—А–Њ–Љ–µ —В–Њ–≥–Њ, –У–Ъ –љ–µ–≥–∞—В–Є–≤–љ–Њ –≤–ї–Є—П—О—В –љ–∞¬†–Љ—Л—И–µ—З–љ—Г—О —Б–Є–ї—Г, —З—В–Њ —Г–≤–µ–ї–Є—З–Є–≤–∞–µ—В —А–Є—Б–Ї¬†–њ–∞–і–µ–љ–Є–є [5вАУ7]. –Ь–µ—В–∞–∞–љ–∞–ї–Є–Ј –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є (–њ–Њ—А—П–і–Ї–∞ 42 000 –њ–∞—Ж–Є–µ–љ—В–Њ–≤) –њ–Њ–Ї–∞–Ј–∞–ї, —З—В–Њ –љ–∞¬†—Д–Њ–љ–µ –њ—А–µ–і—И–µ—Б—В–≤—Г—О—Й–µ–≥–Њ –Є–ї–Є —В–µ–Ї—Г—Й–µ–≥–Њ –њ—А–Є–µ–Љ–∞ –У–Ъ —А–Є—Б–Ї¬†–њ–µ—А–µ–ї–Њ–Љ–Њ–≤ —Г¬†–Љ—Г–ґ—З–Є–љ –Є¬†–ґ–µ–љ—Й–Є–љ –≤¬†–≤–Њ–Ј—А–∞—Б—В–µ 50 –ї–µ—В –Є¬†—Б—В–∞—А—И–µ –њ–Њ–≤—Л—И–∞–µ—В—Б—П –≤¬†—А–∞–≤–љ–Њ–є —Б—В–µ–њ–µ–љ–Є: –Њ—В–љ–Њ—Б–Є—В–µ–ї—М–љ—Л–є —А–Є—Б–Ї¬†(–Ю–†) –і–ї—П –ї—О–±–Њ–≥–Њ –њ–µ—А–µ–ї–Њ–Љ–∞ –≤–∞—А—М–Є—А—Г–µ—В –Њ—В¬†1,98 (–≤¬†50 –ї–µ—В) –і–Њ 1,66¬†(–≤¬†85¬†–ї–µ—В), –і–ї—П –ї—О–±–Њ–≥–Њ –Њ—Б—В–µ–Њ–њ–Њ—А–Њ—В–Є—З–µ—Б–Ї–Њ–≥–Њ –њ–µ—А–µ–ї–Њ–Љ–∞ –Є¬†–њ–µ—А–µ–ї–Њ–Љ–∞ –њ—А–Њ–Ї—Б–Є–Љ–∞–ї—М–љ–Њ–≥–Њ –Њ—В–і–µ–ї–∞ –±–µ–і—А–∞¬†вАУ 2,63вАУ1,71¬†–Є¬†2,48вАУ4,42¬†—Б–Њ–Њ—В–≤–µ—В—Б—В–≤–µ–љ–љ–Њ [8]. –Я–Њ–ї—Г—З–µ–љ—Л –і–∞–љ–љ—Л–µ, —З—В–Њ –љ–∞¬†—Д–Њ–љ–µ –њ—А–Є–µ–Љ–∞ –њ–µ—А–Њ¬≠—А–∞–ї—М–љ—Л—Е –У–Ъ –≤¬†–њ–µ—А–Є–Њ–і –њ–Њ—Б—В–Љ–µ–љ–Њ–њ–∞—Г–Ј—Л –њ–µ—А–µ–ї–Њ–Љ—Л –њ—А–Њ–Є—Б—Е–Њ–і—П—В –њ—А–Є –±–Њ–ї–µ–µ –≤—Л—Б–Њ–Ї–Є—Е –Ј–љ–∞—З–µ–љ–Є—П—Е –Ь–Я–Ъ, —З–µ–Љ –њ—А–Є –њ–Њ—Б—В–Љ–µ–љ–Њ–њ–∞—Г–Ј–∞–ї—М–љ–Њ–Љ –Њ—Б—В–µ–Њ–њ–Њ—А–Њ–Ј–µ [8].
–Я—А–Є —Н—В–Њ–Љ —Г—Б—В–∞–љ–Њ–≤–ї–µ–љ –і–Њ–Ј–Њ–Ј–∞–≤–Є—Б–Є–Љ—Л–є —Н—Д—Д–µ–Ї—В –У–Ъ –љ–∞¬†—А–Є—Б–Ї –њ–µ—А–µ–ї–Њ–Љ–Њ–≤: —Г¬†–±–Њ–ї—М–љ—Л—Е, –њ—А–Є–љ–Є–Љ–∞–≤—И–Є—Е –У–Ъ –≤¬†–і–Њ–Ј–µ вЙ• 7,5 –Љ–≥/—Б—Г—В –≤¬†–њ—А–µ–і–љ–Є–Ј–Њ–ї–Њ–љ–Њ–≤–Њ–Љ —Н–Ї–≤–Є–≤–∞–ї–µ–љ—В–µ, –Ю–† –Ї–Њ–Љ–њ—А–µ—Б—Б–Є–Њ–љ–љ—Л—Е –њ–µ—А–µ–ї–Њ–Љ–Њ–≤ –њ–Њ–Ј–≤–Њ–љ–Ї–Њ–≤ —Б–Њ—Б—В–∞–≤–Є–ї 5,18 –њ—А–Є 95%-–љ–Њ–Љ –і–Њ–≤–µ—А–Є—В–µ–ї—М–љ–Њ–Љ –Є–љ—В–µ—А–≤–∞–ї–µ (–Ф–Ш) 4,25вАУ6,31, –і–ї—П –љ–µ–њ–Њ–Ј–≤–Њ–љ–Њ—З–љ—Л—Е –њ–µ—А–µ–ї–Њ–Љ–Њ–≤¬†вАУ 2,27¬†(95% –Ф–Ш 2,16вАУ3,10) [6]. –Т¬†–њ–µ—А–Є–Њ–і –њ–Њ—Б—В–Љ–µ–љ–Њ–њ–∞—Г–Ј—Л –љ–∞¬†—Д–Њ–љ–µ —В–µ—А–∞–њ–Є–Є –У–Ъ –њ—А–Є —Б–љ–Є–ґ–µ–љ–Є–Є –Ь–Я–Ъ –љ–∞¬†–Њ–і–љ–Њ —Б—В–∞–љ–і–∞—А—В–љ–Њ–µ –Њ—В–Ї–ї–Њ–љ–µ–љ–Є–µ —А–Є—Б–Ї¬†–і–µ—Д–Њ—А–Љ–∞—Ж–Є–Є –њ–Њ–Ј–≤–Њ–љ–Ї–Њ–≤ –≤–Њ–Ј—А–∞—Б—В–∞–µ—В –љ–∞¬†85%, –∞¬†–≤ —Б–ї—Г—З–∞–µ —Г–≤–µ–ї–Є—З–µ–љ–Є—П —Б—Г—В–Њ—З–љ–Њ–є –і–Њ–Ј—Л –љ–∞¬†–Ї–∞–ґ–і—Л–µ 10¬†–Љ–≥¬†вАУ –љ–∞¬†62% [4].
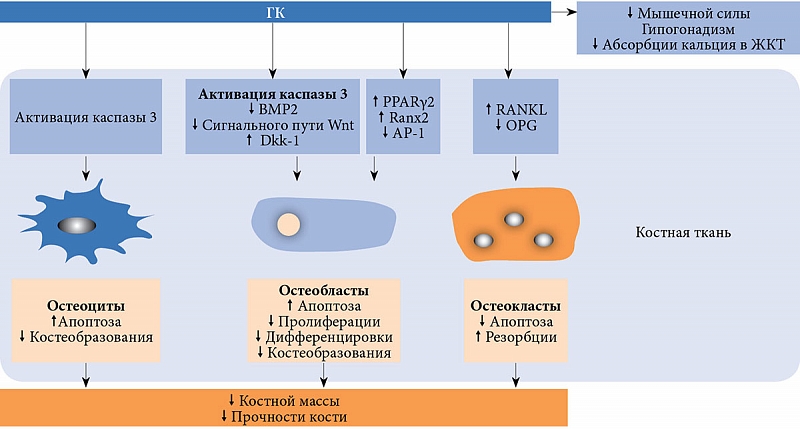
–Ю—Б–љ–Њ–≤–љ–Њ–µ –≤–ї–Є—П–љ–Є–µ –У–Ъ –љ–∞¬†–Ї–Њ—Б—В–љ–Њ–µ —А–µ–Љ–Њ–і–µ–ї–Є—А–Њ–≤–∞–љ–Є–µ¬†вАУ –њ–Њ–і–∞–≤–ї–µ–љ–Є–µ –Ї–Њ—Б—В–µ–Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є—П –≤¬†—А–µ–Ј—Г–ї—М—В–∞—В–µ —Б–љ–Є–ґ–µ–љ–Є—П –њ–Њ–Ї–∞–Ј–∞—В–µ–ї–µ–є –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є –Њ—Б—В–µ–Њ–±–ї–∞—Б—В–Њ–≤, –∞¬†—В–∞–Ї–ґ–µ –њ–Њ–≤—Л—И–µ–љ–Є–µ –∞–њ–Њ–њ—В–Њ–Ј–∞ –Њ—Б—В–µ–Њ–±–ї–∞—Б—В–Њ–≤ –Є¬†–Њ—Б—В–µ–Њ—Ж–Є—В–Њ–≤, —З—В–Њ —Б—З–Є—В–∞–µ—В—Б—П –Њ—Б–љ–Њ–≤–љ–Њ–є –њ—А–Є—З–Є–љ–Њ–є —А–∞–Ј–≤–Є—В–Є—П –Њ—Б—В–µ–Њ–њ–Њ—А–Њ–Ј–∞ [5вАУ7]. –Ъ—А–Њ–Љ–µ —В–Њ–≥–Њ, –У–Ъ —Б–њ–Њ—Б–Њ–±—Б—В–≤—Г—О—В –∞–Ї—В–Є–≤–∞—Ж–Є–Є –Ї–∞—Б–њ–∞–Ј—Л 3¬†вАУ –≤–∞–ґ–љ–Њ–≥–Њ —В—А–Є–≥–≥–µ—А–∞ –∞–њ–Њ–њ—В–Њ–Ј–∞ –Њ—Б—В–µ–Њ–±–ї–∞—Б—В–Њ–≤ –Є¬†–Њ—Б—В–µ–Њ—Ж–Є—В–Њ–≤ [9]. –°–љ–Є–ґ–µ–љ–Є–µ —Д—Г–љ–Ї—Ж–Є–Є –Њ—Б—В–µ–Њ–±–ї–∞—Б—В–Њ–≤ –Љ–Њ–ґ–µ—В –±—Л—В—М —Б–≤—П–Ј–∞–љ–Њ –Є¬†—Б –∞–Ї—В–Є–≤–∞—Ж–Є–µ–є¬†–≥–ї–Є–Ї–Њ–≥–µ–љ-—Б–Є–љ—В–∞–Ј—Л-–Ї–Є–љ–∞–Ј—Л-3ќ≤¬†вАУ –њ—А–Њ—В–µ–Є–љ–∞, –≤–Њ–≤–ї–µ—З–µ–љ–љ–Њ–≥–Њ –≤¬†—Б–Є–≥–љ–∞–ї—М–љ—Л–є –њ—Г—В—М Wnt –Є¬†–Є–≥—А–∞—О—Й–µ–≥–Њ –Ї–ї—О—З–µ–≤—Г—О —А–Њ–ї—М –≤¬†–Њ—Б—В–µ–Њ–±–ї–∞—Б—В–Њ–≥–µ–љ–µ–Ј–µ [10]. –У–Ъ –∞–Ї—В–Є–≤–Є—А—Г—О—В –≤—Л—А–∞–±–Њ—В–Ї—Г —Б–Є–≥–љ–∞–ї—М–љ—Л—Е –Љ–Њ–ї–µ–Ї—Г–ї Dickkopf-1 (Dkk-1) –Є¬†—Б–Ї–ї–µ—А–Њ—Б—В–Є–љ–∞¬†вАУ –Є–љ–≥–Є–±–Є—В–Њ—А–Њ–≤ —Б–Є–≥–љ–∞–ї—М–љ–Њ–≥–Њ –њ—Г—В–Є Wnt. –Ъ–∞–Ї —Б–ї–µ–і—Б—В–≤–Є–µ, –њ–Њ–≤—Л—И–∞–µ—В—Б—П –∞–Ї—В–Є–≤–љ–Њ—Б—В—М ќ≤-–Ї–∞—В–µ–њ—Б–Є–љ–∞ –Є¬†–Ј–∞–Љ–µ–і–ї—П–µ—В—Б—П –њ—А–Њ–і—Г–Ї—Ж–Є—П –Ї–Њ–ї–ї–∞–≥–µ–љ–∞ 1 –Ј–∞ —Б—З–µ—В –±–ї–Њ–Ї–Є—А–Њ–≤–∞–љ–Є—П —В—А–∞–љ—Б–Ї—А–Є–њ—Ж–Є–Є —Б–Њ–Њ—В–≤–µ—В—Б—В–≤—Г—О—Й–Є—Е¬†–≥–µ–љ–Њ–≤ [5, 7, 10]. –У–Ъ –Љ–Њ–≥—Г—В —В–∞–Ї–ґ–µ —Б–Љ–µ—Й–∞—В—М –љ–∞–њ—А–∞–≤–ї–µ–љ–Є–µ –і–Є—Д—Д–µ—А–µ–љ—Ж–Є—А–Њ–≤–Ї–Є —Б—В—А–Њ–Љ–∞–ї—М–љ—Л—Е –Ї–ї–µ—В–Њ–Ї –Ї–Њ—Б—В–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞ (–њ—А–µ–і—И–µ—Б—В–≤–µ–љ–љ–Є—Ж –Њ—Б—В–µ–Њ–±–ї–∞—Б—В–Њ–≤) –≤¬†—Б—В–Њ—А–Њ–љ—Г –Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є—П –∞–і–Є–њ–Њ—Ж–Є—В–Њ–≤ –њ—Г—В–µ–Љ —Г–≥–љ–µ—В–µ–љ–Є—П –Р–†-1 (–Рctivator Protein¬†1¬†вАУ –∞–Ї—В–Є–≤–Є—А—Г—О—Й–Є–є –њ—А–Њ—В–µ–Є–љ 1), –њ–Њ–≤—Л—И–µ–љ–Є—П –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є PPARќ≥2 (Peroxisome Proliferator-Activated Receptors¬†вАУ —А–µ—Ж–µ–њ—В–Њ—А—Л, –∞–Ї—В–Є–≤–Є—А—Г–µ–Љ—Л–µ –њ–µ—А–Њ–Ї—Б–Є—Б–Њ–Љ–љ—Л–Љ–Є –њ—А–Њ–ї–Є—Д–µ—А–∞—В–Њ—А–∞–Љ–Є) –Є¬†—Г–≥–љ–µ—В–µ–љ–Є—П Ranx2 (—А–Є—Б.¬†1) [7, 10].
–Ь–µ—Е–∞–љ–Є–Ј–Љ, —Г—Б–Є–ї–Є–≤–∞—О—Й–Є–є –Ї–Њ—Б—В–љ—Г—О —А–µ–Ј–Њ—А–±—Ж–Є—О –≤¬†—А–µ–Ј—Г–ї—М—В–∞—В–µ –≤–Њ–Ј–і–µ–є—Б—В–≤–Є—П –У–Ъ, –і–Њ –Ї–Њ–љ—Ж–∞ –љ–µ¬†–Є–Ј—Г—З–µ–љ. –Ю–і–љ–∞–Ї–Њ –Є–Ј–≤–µ—Б—В–љ–Њ, —З—В–Њ –У–Ъ —Б—В–Є–Љ—Г–ї–Є—А—Г—О—В —Б–Њ–Ј—А–µ–≤–∞–љ–Є–µ –Є¬†–∞–Ї—В–Є–≤–љ–Њ—Б—В—М –Њ—Б—В–µ–Њ–Ї–ї–∞—Б—В–Њ–≤ —З–µ—А–µ–Ј –њ–Њ–≤—Л—И–µ–љ–Є–µ —Г—А–Њ–≤–љ—П RANKL (Receptor Activator of¬†Nuclear Factor Kappa-B Ligand¬†вАУ –ї–Є–≥–∞–љ–і —А–µ—Ж–µ–њ—В–Њ—А–∞ –∞–Ї—В–Є–≤–∞—В–Њ—А–∞ —П–і–µ—А–љ–Њ–≥–Њ —Д–∞–Ї—В–Њ—А–∞ –Ї–∞–њ–њ–∞-–Т) –Ї¬†–Ю–†G (osteoprotegerin¬†вАУ –Њ—Б—В–µ–Њ–њ—А–Њ—В–µ–≥–µ—А–Є–љ) [5, 7]. –Ю—В–Љ–µ—З–∞–µ—В—Б—П —В–∞–Ї–ґ–µ —Б–љ–Є–ґ–µ–љ–Є–µ –њ—А–Њ–і—Г–Ї—Ж–Є–Є –њ–Њ–ї–Њ–≤—Л—Е¬†–≥–Њ—А–Љ–Њ–љ–Њ–≤, —Б–Њ–њ—А–Њ–≤–Њ–ґ–і–∞—О—Й–µ–µ—Б—П –∞–Ї—В–Є–≤–∞—Ж–Є–µ–є –њ—А–Њ—Ж–µ—Б—Б–Њ–≤ –Ї–Њ—Б—В–љ–Њ–≥–Њ —А–µ–Љ–Њ–і–µ–ї–Є—А–Њ–≤–∞–љ–Є—П [5, 6]. –У–Ъ —Б–љ–Є–ґ–∞—О—В –∞–±—Б–Њ—А–±—Ж–Є—О –Ї–∞–ї—М—Ж–Є—П –≤¬†–Ї–Є—И–µ—З–љ–Є–Ї–µ –Є¬†–µ–≥–Њ —В—Г–±—Г–ї—П—А–љ—Г—О —А–µ–∞–±—Б–Њ—А–±—Ж–Є—О, —З—В–Њ –њ—А–Є–≤–Њ–і–Є—В –Ї¬†–њ–Њ–≤—Л—И–µ–љ–Є—О —Б–µ–Ї—А–µ—Ж–Є–Є –њ–∞—А–∞—В–Є—А–µ–Њ–Є–і–љ–Њ–≥–Њ¬†–≥–Њ—А–Љ–Њ–љ–∞ (–Я–Ґ–У) (—А–Є—Б.¬†1). –Ш–Ј–±—Л—В–Њ—З–љ–Њ–µ —Б–Њ–і–µ—А–ґ–∞–љ–Є–µ –У–Ъ –њ—А–Є–≤–Њ–і–Є—В –Ї –њ–Њ–і–∞–≤–ї–µ–љ–Є—О —Н–Ї—Б–њ—А–µ—Б—Б–Є–Є VDR (Vitamin D Receptor¬†вАУ —А–µ—Ж–µ–њ—В–Њ—А –≤–Є—В–∞–Љ–Є–љ–∞ D) –Є¬†–Њ–њ–Њ—Б—А–µ–і–Њ–≤–∞–љ–љ–Њ –љ–µ–≥–∞—В–Є–≤–љ–Њ –≤–ї–Є—П–µ—В –љ–∞¬†–Љ—Л—И–µ—З–љ—Г—О —Б–Є–ї—Г (—Б–ї–µ–і—Б—В–≤–Є–µ –≤–Њ–Ј–і–µ–є—Б—В–≤–Є—П –љ–∞¬†–Ї–ї–µ—В–Ї–Є —Б–Ї–µ–ї–µ—В–љ—Л—Е –Љ—Л—И—Ж –њ–Њ–≤—Л—И–µ–љ–љ–Њ–≥–Њ —Г—А–Њ–≤–љ—П –Я–Ґ–У, —Б–љ–Є–ґ–µ–љ–љ–Њ–≥–Њ —Г—А–Њ–≤–љ—П IGF-1 (Insulinlike Growth Factor 1¬†вАУ –Є–љ—Б—Г–ї–Є–љ–Њ–њ–Њ–і–Њ–±–љ—Л–є —Д–∞–Ї—В–Њ—А —А–Њ—Б—В–∞ 1) –Є¬†–і–µ—Д–Є—Ж–Є—В–∞ VDR) [11].
–Ф–Є–∞–±–µ—В–Є—З–µ—Б–Ї–∞—П –Њ—Б—В–µ–Њ–њ–∞—В–Є—П
–°–∞—Е–∞—А–љ—Л–є –і–Є–∞–±–µ—В (–°–Ф)¬†вАУ –Њ–і–љ–∞ –Є–Ј¬†–≤–∞–ґ–љ–µ–є—И–Є—Е –њ—А–Њ–±–ї–µ–Љ —Б–Њ–≤—А–µ–Љ–µ–љ–љ–Њ–≥–Њ –Ј–і—А–∞–≤–Њ–Њ—Е—А–∞–љ–µ–љ–Є—П. –Ч–∞ –њ–Њ—Б–ї–µ–і–љ–Є–µ 20 –ї–µ—В —З–Є—Б–ї–µ–љ–љ–Њ—Б—В—М –±–Њ–ї—М–љ—Л—Е –°–Ф¬†–≤¬†–Љ–Є—А–µ –≤—Л—А–Њ—Б–ї–∞ –±–Њ–ї–µ–µ —З–µ–Љ –≤¬†–і–≤–∞ —А–∞–Ј–∞ –Є¬†–і–Њ—Б—В–Є–≥–ї–∞ 366 –Љ–ї–љ (7% –љ–∞—Б–µ–ї–µ–љ–Є—П). –Я—А–Є —Н—В–Њ–Љ –Њ–Ї–Њ–ї–Њ 50% —Б–ї—Г—З–∞–µ–≤ –њ—А–Є—Е–Њ–і–Є—В—Б—П –љ–∞¬†–≤–Њ–Ј—А–∞—Б—В–љ—Г—О –Ї–∞—В–µ–≥–Њ—А–Є—О –Њ—В¬†50 –і–Њ 69 –ї–µ—В [12]. –Ь–µ–і–Є–Ї–Њ-—Б–Њ—Ж–Є–∞–ї—М–љ–Њ–µ –Ј–љ–∞—З–µ–љ–Є–µ –°–Ф¬†–Њ–±—Г—Б–ї–Њ–≤–ї–µ–љ–Њ —А–∞–Ј–≤–Є—В–Є–µ–Љ —Б–Є—Б—В–µ–Љ–љ—Л—Е —Б–Њ—Б—Г–і–Є—Б—В—Л—Е –Њ—Б–ї–Њ–ґ–љ–µ–љ–Є–є, –Ї–Њ—В–Њ—А—Л–µ —П–≤–ї—П—О—В—Б—П –Њ—Б–љ–Њ–≤–љ–Њ–є –њ—А–Є—З–Є–љ–Њ–є –Є–љ–≤–∞–ї–Є–і–Є–Ј–∞—Ж–Є–Є –Є¬†—Б–Љ–µ—А—В–Є –њ–∞—Ж–Є–µ–љ—В–Њ–≤. –Ъ–∞–ґ–і—Л–µ 5 —Б–µ–Ї—Г–љ–і –≤¬†–Љ–Є—А–µ —Г–Љ–Є—А–∞–µ—В –Њ–і–Є–љ –±–Њ–ї—М–љ–Њ–є, –Ј–∞¬†–≥–Њ–і —Н—В–∞ —Ж–Є—Д—А–∞ —Б–Њ—Б—В–∞–≤–ї—П–µ—В 4,6 –Љ–ї–љ, –≤¬†–†–Њ—Б—Б–Є–Є¬†вАУ 66 —В—Л—Б. [12]. –°–µ–≥–Њ–і–љ—П –≤—Б–µ –±–Њ–ї—М—И–Є–є –Є–љ—В–µ—А–µ—Б –≤—Л–Ј—Л–≤–∞–µ—В –Љ–∞–ї–Њ–Є–Ј—Г—З–µ–љ–љ–Њ–µ –Њ—Б–ї–Њ–ґ–љ–µ–љ–Є–µ –°–Ф¬†вАУ –Њ—Б—В–µ–Њ–њ–Њ—А–Њ–Ј, –Є–ї–Є –і–Є–∞–±–µ—В–Є—З–µ—Б–Ї–∞—П –Њ—Б—В–µ–Њ–њ–∞—В–Є—П, –∞—Б—Б–Њ—Ж–Є–Є—А—Г—О—Й–Є–є—Б—П —Б¬†–њ–Њ–≤—Л—И–µ–љ–љ—Л–Љ —А–Є—Б–Ї–Њ–Љ –њ–µ—А–µ–ї–Њ–Љ–Њ–≤, –Є¬†–≤¬†–њ–µ—А–≤—Г—О –Њ—З–µ—А–µ–і—М –њ–µ—А–µ–ї–Њ–Љ–∞ –њ—А–Њ–Ї—Б–Є–Љ–∞–ї—М–љ–Њ–≥–Њ –Њ—В–і–µ–ї–∞ –±–µ–і—А–∞.
–Я—А–Є—З–Є–љ–∞ —А–∞–Ј–≤–Є—В–Є—П –і–Є–∞–±–µ—В–Є—З–µ—Б–Ї–Њ–є –Њ—Б—В–µ–Њ–њ–∞—В–Є–Є¬†вАУ –љ–∞—А—Г—И–µ–љ–Є–µ –±–∞–ї–∞–љ—Б–∞ –њ—А–Њ—Ж–µ—Б—Б–Њ–≤ –Ї–Њ—Б—В–љ–Њ–≥–Њ —А–µ–Љ–Њ–і–µ–ї–Є—А–Њ–≤–∞–љ–Є—П: —Б–Ї–Њ—А–Њ—Б—В—М –Ї–Њ—Б—В–љ–Њ–є —А–µ–Ј–Њ—А–±—Ж–Є–Є –њ–Њ–≤—Л—И–∞–µ—В—Б—П, –∞¬†–Ї–Њ—Б—В–µ–Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є—П —Б–љ–Є–ґ–∞–µ—В—Б—П. –£–≤–µ–ї–Є—З–µ–љ–Є–µ –Ї–Њ–ї–Є—З–µ—Б—В–≤–∞ –Њ—Б—В–µ–Њ–Ї–ї–∞—Б—В–Њ–≤ –Є¬†—Г—Б–Є–ї–µ–љ–Є–µ —А–µ–Ј–Њ—А–±—Ж–Є–Є –Ї–Њ—Б—В–љ–Њ–є —В–Ї–∞–љ–Є –њ—А–Њ–Є—Б—Е–Њ–і—П—В –≤¬†—А–µ–Ј—Г–ї—М—В–∞—В–µ –њ–Њ–≤—Л—И–µ–љ–Є—П —Н–Ї—Б–њ—А–µ—Б—Б–Є–Є RANK, —Б–Є–љ—В–µ–Ј–∞ RANKL¬†вАУ –Њ—Б–љ–Њ–≤–љ–Њ–≥–Њ –Љ–µ–і–Є–∞—В–Њ—А–∞ –і–Є—Д—Д–µ—А–µ–љ—Ж–Є—А–Њ–≤–Ї–Є, —Б–Њ–Ј—А–µ–≤–∞–љ–Є—П –Є¬†—Д—Г–љ–Ї—Ж–Є–Є –Њ—Б—В–µ–Њ–Ї–ї–∞—Б—В–Њ–≤, —Д–∞–Ї—В–Њ—А–∞ –љ–µ–Ї—А–Њ–Ј–∞ –Њ–њ—Г—Е–Њ–ї–Є ќ± –Є¬†–Ї–Њ–ї–Њ–љ–Є–µ—Б—В–Є–Љ—Г–ї–Є—А—Г—О—Й–µ–≥–Њ —Д–∞–Ї—В–Њ—А–∞ –Љ–∞–Ї—А–Њ—Д–∞–≥–Њ–≤ [13].
–°–љ–Є–ґ–µ–љ–Є–µ –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є –Ї–Њ—Б—В–µ–Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є—П —Б–≤—П–Ј–∞–љ–Њ –≤¬†–њ–µ—А–≤—Г—О –Њ—З–µ—А–µ–і—М —Б¬†—Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–є¬†–≥–Є–њ–µ—А–≥–ї–Є–Ї–µ–Љ–Є–µ–є. –Т—Л—Б–Њ–Ї–Є–є —Г—А–Њ–≤–µ–љ—М¬†–≥–ї—О–Ї–Њ–Ј—Л –≤¬†–Ї—А–Њ–≤–Є —В–Њ—А–Љ–Њ–Ј–Є—В —Б–Њ–Ј—А–µ–≤–∞–љ–Є–µ –Њ—Б—В–µ–Њ–±–ї–∞—Б—В–Њ–≤ –Є¬†–њ—А–Њ—Ж–µ—Б—Б –Љ–Є–љ–µ—А–∞–ї–Є–Ј–∞—Ж–Є–Є, —З—В–Њ –њ—А–Њ—П–≤–ї—П–µ—В—Б—П –≤¬†—Б–љ–Є–ґ–µ–љ–Є–Є –±–Є–Њ—Е–Є–Љ–Є—З–µ—Б–Ї–Є—Е –њ–Њ–Ї–∞–Ј–∞—В–µ–ї–µ–є –Ї–Њ—Б—В–µ–Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є—П¬†вАУ –Њ—Б—В–µ–Њ–Ї–∞–ї—М—Ж–Є–љ–∞ –Є¬†–Ї–Њ–ї–ї–∞–≥–µ–љ–∞ 1 [14]. –Я—А–Њ—З–љ–Њ—Б—В—М –Ї–Њ—Б—В–љ–Њ–є —В–Ї–∞–љ–Є —Б–љ–Є–ґ–∞–µ—В—Б—П —В–∞–Ї–ґ–µ –≤¬†—А–µ–Ј—Г–ї—М—В–∞—В–µ –і–µ–≥—А–∞–і–∞—Ж–Є–Є –Ї–Њ–ї–ї–∞–≥–µ–љ–∞ 1 AGE (–Рssay of¬†Advanced Glycation Endproducts¬†вАУ –Ї–Њ–љ–µ—З–љ—Л–µ –њ—А–Њ–і—Г–Ї—В—Л¬†–≥–ї–Є–Ї–Є—А–Њ–≤–∞–љ–Є—П) [15]. –Я–Њ–Љ–Є–Љ–Њ –њ—А—П–Љ–Њ–≥–Њ —Г–≥–љ–µ—В–∞—О—Й–µ–≥–Њ –≤–Њ–Ј–і–µ–є—Б—В–≤–Є—П –љ–∞¬†—Д—Г–љ–Ї—Ж–Є—О –Њ—Б—В–µ–Њ–±–ї–∞—Б—В–Њ–≤¬†–≥–Є–њ–µ—А–≥–ї–Є–Ї–µ–Љ–Є—П —Б—В–Є–Љ—Г–ї–Є—А—Г–µ—В –љ–∞–Ї–Њ–њ–ї–µ–љ–Є–µ –∞–і–Є–њ–Њ—Ж–Є—В–Њ–≤ –≤¬†–Ї–Њ—Б—В–љ–Њ–Љ –Љ–Њ–Ј–≥–µ –і–ї–Є–љ–љ—Л—Е —В—А—Г–±—З–∞—В—Л—Е –Ї–Њ—Б—В–µ–є, —З—В–Њ –њ—А–Є–≤–Њ–і–Є—В –Ї¬†—Г–Љ–µ–љ—М—И–µ–љ–Є—О –Ї–Њ–ї–Є—З–µ—Б—В–≤–∞ –Њ—Б—В–µ–Њ–±–ї–∞—Б—В–Њ–≤ –Є, –Ї–∞–Ї —Б–ї–µ–і—Б—В–≤–Є–µ, –Є—Б—В–Њ–љ—З–µ–љ–Є—О –Ї–Њ—А—В–Є–Ї–∞–ї—М–љ–Њ–≥–Њ —Б–ї–Њ—П [16]. –Т–∞–ґ–љ—Л–Љ –Љ–µ—Е–∞–љ–Є–Ј–Љ–Њ–Љ –Ј–∞–Љ–µ–і–ї–µ–љ–Є—П –Ї–Њ—Б—В–µ–Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є—П –њ—А–Є –°–Ф¬†—В–Є–њ–∞ 1 —П–≤–ї—П–µ—В—Б—П —Б–љ–Є–ґ–µ–љ–Є–µ —Н–љ–і–Њ–≥–µ–љ–љ–Њ–є —Б–µ–Ї—А–µ—Ж–Є–Є –Є–љ—Б—Г–ї–Є–љ–∞ –Є¬†IGF-1 [17].
–Э–µ–Њ–±—Е–Њ–і–Є–Љ–Њ –Њ—В–Љ–µ—В–Є—В—М, —З—В–Њ –њ—А–Є –°–Ф¬†—В–Є–њ–∞ 2 —Б–љ–Є–ґ–µ–љ–Є–µ –њ—А–Њ—З–љ–Њ—Б—В–Є –Ї–Њ—Б—В–љ–Њ–є —В–Ї–∞–љ–Є –Є¬†–њ–Њ–≤—Л—И–µ–љ–Є–µ —А–Є—Б–Ї–∞ –њ–µ—А–µ–ї–Њ–Љ–Њ–≤ –љ–µ¬†–≤—Б–µ–≥–і–∞ –∞—Б—Б–Њ—Ж–Є–Є—А—Г—О—В—Б—П —Б¬†—Г–Љ–µ–љ—М—И–µ–љ–Є–µ–Љ –Ь–Я–Ъ –Є¬†–Њ—Б—В–µ–Њ–њ–µ–љ–Є—З–µ—Б–Ї–Є–Љ —Б–Є–љ–і—А–Њ–Љ–Њ–Љ [18, 19]. –Т¬†—В–Њ –ґ–µ –≤—А–µ–Љ—П –њ—А–Є –°–Ф¬†—В–Є–њ–∞ 1, –Њ—Б–Њ–±–µ–љ–љ–Њ –≤¬†—Б–ї—Г—З–∞–µ —А–∞–љ–љ–µ–≥–Њ –і–µ–±—О—В–∞ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П, –і–µ—Д–Є—Ж–Є—В –Ь–Я–Ъ –Є¬†–Њ—Б—В–µ–Њ–њ–Њ—А–Њ–Ј –≤—Б—В—А–µ—З–∞—О—В—Б—П —З–∞—Й–µ [20вАУ22]. –Я—А–Є—З–Є–љ–∞ вАУ –љ–∞—А—Г—И–µ–љ–Є–µ —Д–Њ—А–Љ–Є—А–Њ–≤–∞–љ–Є—П –∞–і–µ–Ї–≤–∞—В–љ–Њ–≥–Њ –њ–Є–Ї–∞ –Ї–Њ—Б—В–љ–Њ–є –Љ–∞—Б—Б—Л –≤¬†–њ–Њ–і—А–Њ—Б—В–Ї–Њ–≤–Њ–Љ –њ–µ—А–Є–Њ–і–µ [23]. –£¬†–ґ–µ–љ—Й–Є–љ —Б¬†–°–Ф¬†—В–Є–њ–∞ 1 —А–Є—Б–Ї¬†–њ–µ—А–µ–ї–Њ–Љ–∞ –њ—А–Њ–Ї—Б–Є–Љ–∞–ї—М–љ–Њ–≥–Њ –Њ—В–і–µ–ї–∞ –±–µ–і—А–∞ —Г–≤–µ–ї–Є—З–Є–≤–∞–µ—В—Б—П –≤¬†12¬†—А–∞–Ј –њ–Њ¬†—Б—А–∞–≤–љ–µ–љ–Є—О —Б¬†–ґ–µ–љ—Й–Є–љ–∞–Љ–Є –±–µ–Ј –і–Є–∞–±–µ—В–∞ [24]. –Я—А–Є –°–Ф¬†—В–Є–њ–∞ 2 —В–∞–Ї–Њ–≤–Њ–є –њ–Њ–≤—Л—И–∞–µ—В—Б—П –≤¬†–і–≤–∞ —А–∞–Ј–∞ [24, 25], –∞¬†—В–∞–Ї–ґ–µ –љ–µ—Б–Ї–Њ–ї—М–Ї–Њ —Г–≤–µ–ї–Є—З–Є–≤–∞–µ—В—Б—П —А–Є—Б–Ї¬†–њ–µ—А–µ–ї–Њ–Љ–Њ–≤ –њ–Њ–Ј–≤–Њ–љ–Ї–Њ–≤, –і–Є—Б—В–∞–ї—М–љ–Њ–≥–Њ –Њ—В–і–µ–ї–∞ –њ—А–µ–і–њ–ї–µ—З—М—П, –њ–ї–µ—З–µ–≤–Њ–є –Ї–Њ—Б—В–Є –Є¬†–Ї–Њ—Б—В–µ–є —Б—В–Њ–њ¬†[26].
–Я–Њ–≤—Л—И–µ–љ–Є–µ —А–Є—Б–Ї–∞ –њ–µ—А–µ–ї–Њ–Љ–Њ–≤ —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–°–Ф¬†–Њ–±—К—П—Б–љ—П–µ—В—Б—П –љ–µ¬†—В–Њ–ї—М–Ї–Њ —Б–љ–Є–ґ–µ–љ–Є–µ–Љ –Ї–Њ—Б—В–љ–Њ–є –Љ–∞—Б—Б—Л –Є¬†–њ—А–Њ—З–љ–Њ—Б—В–Є –Ї–Њ—Б—В–µ–є, –љ–Њ¬†–Є –≤–Њ–Ј—А–∞—Б—В–∞–љ–Є–µ–Љ —А–Є—Б–Ї–∞ –њ–∞–і–µ–љ–Є–є –≤—Б–ї–µ–і—Б—В–≤–Є–µ¬†–≥–Є–њ–Њ–≥–ї–Є–Ї–µ–Љ–Є–є –Є¬†–Њ—Б–ї–Њ–ґ–љ–µ–љ–Є–є –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П: –і–Є–∞–±–µ—В–Є—З–µ—Б–Ї–Њ–є —А–µ—В–Є–љ–Њ–њ–∞—В–Є–Є, –Ї–∞—В–∞—А–∞–Ї—В—Л –Є¬†—В.–і., –Є–Ј-–Ј–∞ —З–µ–≥–Њ –Љ–Њ–ґ–µ—В —Б–љ–Є–ґ–∞—В—М—Б—П –њ–µ—А–Є—Д–µ—А–Є—З–µ—Б–Ї–Њ–µ —Б—Г–Љ–µ—А–µ—З–љ–Њ–µ –Ј—А–µ–љ–Є–µ [27]. –Я–µ—А–Є—Д–µ—А–Є—З–µ—Б–Ї–∞—П –љ–µ–є—А–Њ–њ–∞—В–Є—П —В–∞–Ї–ґ–µ —П–≤–ї—П–µ—В—Б—П –Ј–љ–∞—З–Є–Љ—Л–Љ —Д–∞–Ї—В–Њ—А–Њ–Љ —А–Є—Б–Ї–∞ –њ–∞–і–µ–љ–Є–є [28]. –Э–∞¬†–µ–µ —Д–Њ–љ–µ –Њ—В–Љ–µ—З–∞–µ—В—Б—П –њ–Њ–≤—Л—И–µ–љ–Є–µ —З–∞—Б—В–Њ—В—Л –њ–µ—А–Є—Д–µ—А–Є—З–µ—Б–Ї–Є—Е –љ–Є–Ј–Ї–Њ—Н–љ–µ—А–≥–µ—В–Є—З–µ—Б–Ї–Є—Е –њ–µ—А–µ–ї–Њ–Љ–Њ–≤ —Г¬†–±–Њ–ї—М–љ—Л—Е –°–Ф¬†[29]. –°–ї–µ–і—Г–µ—В –Њ—В–Љ–µ—В–Є—В—М, —З—В–Њ –њ–µ—А–Є—Д–µ—А–Є—З–µ—Б–Ї–∞—П –љ–µ–є—А–Њ–њ–∞—В–Є—П –њ—А–Є –°–Ф¬†—П–≤–ї—П–µ—В—Б—П —Б–∞–Љ–Њ—Б—В–Њ—П—В–µ–ї—М–љ—Л–Љ —Д–∞–Ї—В–Њ—А–Њ–Љ —Г–Љ–µ–љ—М—И–µ–љ–Є—П –Ь–Я–Ъ –њ–µ—А–Є—Д–µ—А–Є—З–µ—Б–Ї–Њ–≥–Њ —Б–Ї–µ–ї–µ—В–∞¬†[30].
–Я–Њ¬†—Б—А–∞–≤–љ–µ–љ–Є—О —Б–Њ¬†–Ј–і–Њ—А–Њ–≤–Њ–є –њ–Њ–њ—Г–ї—П—Ж–Є–µ–є —Г¬†–±–Њ–ї—М–љ—Л—Е –°–Ф¬†—З–∞—Б—В–Њ –љ–∞–±–ї—О–і–∞–µ—В—Б—П –і–µ—Д–Є—Ж–Є—В –≤–Є—В–∞–Љ–Є–љ–∞ D [31]. –Ъ—А–Њ–Љ–µ —В–Њ–≥–Њ, –Љ–µ—В–∞–±–Њ–ї–Є–Ј–Љ –≤–Є—В–∞–Љ–Є–љ–∞ D –і–Њ –∞–Ї—В–Є–≤–љ–Њ–≥–Њ D-–≥–Њ—А–Љ–Њ–љ–∞ –љ–∞—А—Г—И–∞–µ—В—Б—П –≤—Б–ї–µ–і—Б—В–≤–Є–µ —А–∞–Ј–≤–Є—В–Є—П –і–Є–∞–±–µ—В–Є—З–µ—Б–Ї–Њ–є –љ–µ—Д—А–Њ–њ–∞—В–Є–Є. –Ф–µ—Д–Є—Ж–Є—В –≤–Є—В–∞–Љ–Є–љ–∞ D –∞—Б—Б–Њ—Ж–Є–Є—А—Г–µ—В—Б—П —Б¬†–Љ—Л—И–µ—З–љ–Њ–є —Б–ї–∞–±–Њ—Б—В—М—О, –Ј–∞–Љ–µ–і–ї–µ–љ–Є–µ–Љ —Б–Ї–Њ—А–Њ—Б—В–Є –Є¬†–Ї–∞—З–µ—Б—В–≤–∞ —Е–Њ–і—М–±—Л, —Г–Љ–µ–љ—М—И–µ–љ–Є–µ–Љ –Љ–∞—Б—Б—Л –Є¬†–∞—В—А–Њ—Д–Є–µ–є –Љ—Л—И–µ—З–љ–Њ–є —В–Ї–∞–љ–Є [32]. –Я–Њ—Н—В–Њ–Љ—Г –≤–Њ—Б–њ–Њ–ї–љ–µ–љ–Є–µ –і–µ—Д–Є—Ж–Є—В–∞ –≤–Є—В–∞–Љ–Є–љ–∞¬†D –Є¬†–і–∞–ї—М–љ–µ–є—И–µ–µ –њ—А–Є–Љ–µ–љ–µ–љ–Є–µ –µ–≥–Њ –∞–Ї—В–Є–≤–љ—Л—Е –Љ–µ—В–∞–±–Њ–ї–Є—В–Њ–≤ —П–≤–ї—П—О—В—Б—П –≤–∞–ґ–љ—Л–Љ –Ј–≤–µ–љ–Њ–Љ –≤¬†–њ—А–Њ—Д–Є–ї–∞–Ї—В–Є–Ї–µ –њ–∞–і–µ–љ–Є–є –Є¬†–њ–µ—А–µ–ї–Њ–Љ–Њ–≤ —Г¬†—В–∞–Ї–Є—Е –±–Њ–ї—М–љ—Л—Е.
–Я–Њ—З–µ—З–љ–∞—П –Њ—Б—В–µ–Њ–і–Є—Б—В—А–Њ—Д–Є—П
–Я–Њ—З–Ї–Є, —В–∞–Ї –ґ–µ –Ї–∞–Ї –Є¬†–Ї–Є—И–µ—З–љ–Є–Ї, –њ–∞—А–∞—Й–Є—В–Њ–≤–Є–і–љ—Л–µ –ґ–µ–ї–µ–Ј—Л –Є¬†—Б–Ї–µ–ї–µ—В, –∞–Ї—В–Є–≤–љ–Њ —Г—З–∞—Б—В–≤—Г—О—В –≤¬†—А–µ–≥—Г–ї—П—Ж–Є–Є –Љ–Є–љ–µ—А–∞–ї—М–љ–Њ–≥–Њ¬†–≥–Њ–Љ–µ–Њ—Б—В–∞–Ј–∞. –Ф–ї—П —А–µ–≥—Г–ї—П—Ж–Є–Є –Ї–∞–ї—М—Ж–Є–є-—Д–Њ—Б—Д–Њ—А–љ–Њ–≥–Њ –Њ–±–Љ–µ–љ–∞ –≤–∞–ґ–љ—Л –Ї–∞–Ї —Н–Ї—Б–Ї—А–µ—В–Њ—А–љ–∞—П, —В–∞–Ї –Є¬†–Љ–µ—В–∞–±–Њ–ї–Є—З–µ—Б–Ї–Є–µ —Д—Г–љ–Ї—Ж–Є–Є –њ–Њ—З–µ–Ї. –Ю—Б–љ–Њ–≤–љ–∞—П –њ—А–Є—З–Є–љ–∞ —А–∞–Ј–≤–Є—В–Є—П –њ–Њ—З–µ—З–љ–Њ–є –Њ—Б—В–µ–Њ–і–Є—Б—В—А–Њ—Д–Є–Є¬†вАУ —В—П–ґ–µ–ї–∞—П —Е—А–Њ–љ–Є—З–µ—Б–Ї–∞—П –њ–Њ—З–µ—З–љ–∞—П –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В—М, —В—Г–±—Г–ї–Њ–њ–∞—В–Є–Є, –Ї–∞–љ–∞–ї—М—Ж–µ–≤—Л–є –∞—Ж–Є–і–Њ–Ј –Є –і—А—Г–≥–Є–µ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П, —Б–Њ–њ—А–Њ–≤–Њ–ґ–і–∞—О—Й–Є–µ—Б—П –љ–∞—А—Г—И–µ–љ–Є–µ–Љ —Д—Г–љ–Ї—Ж–Є–Є –њ–Њ—З–µ—З–љ—Л—Е –Ї–∞–љ–∞–ї—М—Ж–µ–≤. –Ю—Б–Њ–±–Њ–µ –Љ–µ—Б—В–Њ —Б—А–µ–і–Є –њ—А–Є—З–Є–љ –њ–Њ—З–µ—З–љ–Њ–є –Њ—Б—В–µ–Њ–і–Є—Б—В—А–Њ—Д–Є–Є –Ј–∞–љ–Є–Љ–∞–µ—В –і–Є–∞–±–µ—В–Є—З–µ—Б–Ї–∞—П –љ–µ—Д—А–Њ–њ–∞—В–Є—П. –Х–ґ–µ–≥–Њ–і–љ–Њ –≤¬†–Љ–Є—А–µ 500¬†—В—Л—Б. –±–Њ–ї—М–љ—Л—Е –°–Ф¬†–љ–∞—З–Є–љ–∞—О—В –њ–Њ–ї—Г—З–∞—В—М –Ј–∞–Љ–µ—Б—В–Є—В–µ–ї—М–љ—Г—О –њ–Њ—З–µ—З–љ—Г—О —В–µ—А–∞–њ–Є—О –≤—Б–ї–µ–і—Б—В–≤–Є–µ —А–∞–Ј–≤–Є—В–Є—П –њ–Њ—З–µ—З–љ–Њ–є –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В–Є –і–Њ —В–µ—А–Љ–Є–љ–∞–ї—М–љ–Њ–є —Б—В–∞–і–Є–Є [12].
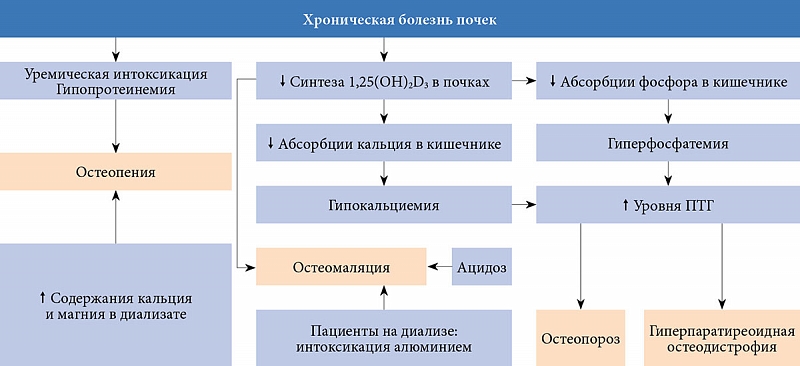
–Э–∞ —В–µ—А–Љ–Є–љ–∞–ї—М–љ–Њ–є —Б—В–∞–і–Є–Є —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–є –±–Њ–ї–µ–Ј–љ–Є –њ–Њ—З–µ–Ї (–•–С–Я) –љ–∞–±–ї—О–і–∞–µ—В—Б—П –Ї–Њ–Љ–њ–ї–µ–Ї—Б –Љ–µ—В–∞–±–Њ–ї–Є—З–µ—Б–Ї–Є—Е –љ–∞—А—Г—И–µ–љ–Є–є, –њ—А–Є–≤–Њ–і—П—Й–Є—Е –Ї¬†–њ–Њ–≤—Л—И–µ–љ–Є—О —Г—А–Њ–≤–љ—П –Я–Ґ–У, –Є–Ј–Љ–µ–љ–µ–љ–Є—О –Ї–∞—З–µ—Б—В–≤–∞ –Є¬†–њ—А–Њ—З–љ–Њ—Б—В–Є –Ї–Њ—Б—В–љ–Њ–є —В–Ї–∞–љ–Є. –Ъ¬†–љ–Є–Љ –Њ—В–љ–Њ—Б—П—В—Б—П¬†–≥–Є–њ–Њ–Ї–∞–ї—М—Ж–Є–µ–Љ–Є—П,¬†–≥–Є–њ–µ—А—Д–Њ—Б—Д–∞—В–µ–Љ–Є—П, –і–µ—Д–Є—Ж–Є—В –∞–Ї—В–Є–≤–љ–Њ–≥–Њ –Љ–µ—В–∞–±–Њ–ї–Є—В–∞ –≤–Є—В–∞–Љ–Є–љ–∞ D¬†вАУ 1,25(OH)2D3, –љ–∞—А—Г—И–µ–љ–Є–µ –Њ–±—А–∞—В–љ–Њ–є —А–µ–≥—Г–ї—П—Ж–Є–Є —Д—Г–љ–Ї—Ж–Є–Є VDR, —З—Г–≤—Б—В–≤–Є—В–µ–ї—М–љ–Њ—Б—В–Є –Є¬†—Н–Ї—Б–њ—А–µ—Б—Б–Є–Є CaR (–Ї–∞–ї—М—Ж–Є–є-—З—Г–≤—Б—В–≤–Є—В–µ–ї—М–љ—Л—Е —А–µ—Ж–µ–њ—В–Њ—А–Њ–≤) [13]. –У–ї–∞–≤–љ—Л–є –њ–∞—В–Њ–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Є–є —Д–∞–Ї—В–Њ—А –њ–Њ—З–µ—З–љ–Њ–є –Њ—Б—В–µ–Њ–і–Є—Б—В—А–Њ—Д–Є–Є¬†вАУ–љ–∞—А—Г—И–µ–љ–Є–µ –њ–Њ—З–µ—З–љ–Њ–≥–Њ —Б–Є–љ—В–µ–Ј–∞ –Є¬†–Є–Ј–Љ–µ–љ–µ–љ–Є–µ –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є–Є –≤¬†—Б—Л–≤–Њ—А–Њ—В–Ї–µ –Ї—А–Њ–≤–Є –∞–Ї—В–Є–≤–љ—Л—Е –Љ–µ—В–∞–±–Њ–ї–Є—В–Њ–≤ –≤–Є—В–∞–Љ–Є–љ–∞ D¬†вАУ 1,25(OH)2D3 –Є¬†24,25(OH)2D3 –Є–Ј-–Ј–∞ –≤—Л—А–∞–ґ–µ–љ–љ–Њ–≥–Њ –і–µ—Д–Є—Ж–Є—В–∞ —Д–µ—А–Љ–µ–љ—В–Њ–≤ –њ–Њ—З–µ–Ї¬†вАУ 1ќ±-–≥–Є–і—А–Њ–Ї—Б–Є–ї–∞–Ј—Л –Є¬†24-–≥–Є–і—А–Њ–Ї—Б–Є–ї–∞–Ј—Л.
–•—А–Њ–љ–Є—З–µ—Б–Ї–Є–є –і–µ—Д–Є—Ж–Є—В 1,25(OH)2D3 –њ—А–Є–≤–Њ–і–Є—В –Ї¬†–њ–Њ–≤—Л—И–µ–љ–Є—О —Б–µ–Ї—А–µ—Ж–Є–Є –Я–Ґ–У –Є¬†—Д–Њ—А–Љ–Є—А–Њ–≤–∞–љ–Є—О —Б–Є–Љ–њ—В–Њ–Љ–Њ–Ї–Њ–Љ–њ–ї–µ–Ї—Б–∞ –≤—В–Њ—А–Є—З–љ–Њ–≥–Њ¬†–≥–Є–њ–µ—А–њ–∞—А–∞—В–Є—А–µ–Њ–Ј–∞. –Я—А–Є —Г—А–µ–Љ–Є–Є –≤¬†–њ–∞—А–∞—Й–Є—В–Њ–≤–Є–і–љ—Л—Е –ґ–µ–ї–µ–Ј–∞—Е –Љ–Њ–ґ–µ—В —В–∞–Ї–ґ–µ –њ—А–Њ–Є—Б—Е–Њ–і–Є—В—М –Ї–ї–Њ–љ–∞–ї—М–љ–∞—П —Н–Ї—Б–њ–∞–љ—Б–Є—П –Ї–ї–µ—В–Њ–Ї¬†вАУ –љ–Њ—Б–Є—В–µ–ї–µ–є¬†–≥–µ–љ–љ—Л—Е –Љ—Г—В–∞—Ж–Є–є, —Б–њ–Њ—Б–Њ–±—Б—В–≤—Г—О—Й–Є—Е —А–Њ—Б—В—Г —Н—В–Є—Е –ґ–µ–ї–µ–Ј –Є¬†—Д–Њ—А–Љ–Є—А–Њ–≤–∞–љ–Є—О –≤¬†–љ–Є—Е —Г–Ј–µ–ї–Ї–Њ–≤–Њ–є¬†–≥–Є–њ–µ—А–њ–ї–∞–Ј–Є–Є [34]. –Ъ—А–Њ–Љ–µ —В–Њ–≥–Њ, –њ—А–Є –≤—В–Њ—А–Є—З–љ–Њ–Љ¬†–≥–Є–њ–µ—А–њ–∞—А–∞—В–Є—А–µ–Њ–Ј–µ –Њ—В–Љ–µ—З–∞—О—В—Б—П —Б–љ–Є–ґ–µ–љ–Є–µ —Г—А–Њ–≤–љ—П –Ї–∞–ї—М—Ж–Є–є-—Б–≤—П–Ј—Л–≤–∞—О—Й–µ–≥–Њ –њ—А–Њ—В–µ–Є–љ–∞ –≤¬†—Б—Л–≤–Њ—А–Њ—В–Ї–µ –Ї—А–Њ–≤–Є –Є¬†—Н–Ї—Б–њ—А–µ—Б—Б–Є—П CaR, —З—В–Њ –Њ—Б–ї–∞–±–ї—П–µ—В –Є–љ–≥–Є–±–Є—А—Г—О—Й–µ–µ –≤–Њ–Ј–і–µ–є—Б—В–≤–Є–µ –Ї–∞–ї—М—Ж–Є—П –љ–∞¬†–њ—А–Њ–ї–Є—Д–µ—А–∞—Ж–Є—О –Ї–ї–µ—В–Њ–Ї –њ–∞—А–∞—Й–Є—В–Њ–≤–Є–і–љ—Л—Е –ґ–µ–ї–µ–Ј [34]. –Т–∞–ґ–љ—Л–Љ –њ–∞—В–Њ–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Є–Љ –Ј–≤–µ–љ–Њ–Љ –Є–Ј–Љ–µ–љ–µ–љ–Є–є, –њ—А–Њ–Є—Б—Е–Њ–і—П—Й–Є—Е –≤¬†–Ї–Њ—Б—В–љ–Њ–є —В–Ї–∞–љ–Є —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†—В–µ—А–Љ–Є–љ–∞–ї—М–љ–Њ–є —Б—В–∞–і–Є–µ–є –•–С–Я, —П–≤–ї—П–µ—В—Б—П —Д–Њ—А–Љ–Є—А–Њ–≤–∞–љ–Є–µ —Д–Є–±—А–Њ–Ј–љ–Њ-–Ї–Є—Б—В–Њ–Ј–љ–Њ–≥–Њ –Њ—Б—В–µ–Є—В–∞ –≤ —А–µ–Ј—Г–ї—М—В–∞—В–µ –Є–Ј–±—Л—В–Ї–∞ –Я–Ґ–У, —А–∞–Ј–≤–Є—В–Є–µ –Њ—Б—В–µ–Њ–Љ–∞–ї—П—Ж–Є–Є –љ–∞¬†—Д–Њ–љ–µ –≤—Л—А–∞–ґ–µ–љ–љ–Њ–≥–Њ –і–µ—Д–Є—Ж–Є—В–∞ 1,25(OH)2D3 –Є¬†–Є–љ—В–Њ–Ї—Б–Є–Ї–∞—Ж–Є—П –∞–ї—О–Љ–Є–љ–Є–µ–Љ –њ—А–Є –і–ї–Є—В–µ–ї—М–љ–Њ–Љ –љ–∞—Е–Њ–ґ–і–µ–љ–Є–Є –љ–∞¬†–≥–µ–Љ–Њ–і–Є–∞–ї–Є–Ј–µ (—А–Є—Б.¬†2).
–Т—В–Њ—А–Є—З–љ—Л–є¬†–≥–Є–њ–µ—А–њ–∞—А–∞—В–Є—А–µ–Њ–Ј
–Т—В–Њ—А–Є—З–љ—Л–є¬†–≥–Є–њ–µ—А–њ–∞—А–∞—В–Є—А–µ–Њ–Ј —Е–∞—А–∞–Ї—В–µ—А–Є–Ј—Г–µ—В—Б—П¬†–≥–Є–њ–µ—А—Д—Г–љ–Ї—Ж–Є–µ–є –њ–∞—А–∞—Й–Є—В–Њ–≤–Є–і–љ—Л—Е –ґ–µ–ї–µ–Ј –≤¬†–Њ—В–≤–µ—В –љ–∞¬†—Е—А–Њ–љ–Є—З–µ—Б–Ї—Г—О¬†–≥–Є–њ–Њ–Ї–∞–ї—М—Ж–Є–µ–Љ–Є—О. –Э–∞–Є–±–Њ–ї–µ–µ —З–∞—Б—В–∞—П –њ—А–Є—З–Є–љ–∞ –µ–≥–Њ —А–∞–Ј–≤–Є—В–Є—П¬†вАУ —В–µ—А–Љ–Є–љ–∞–ї—М–љ–∞—П —Б—В–∞–і–Є—П –•–С–Я. –Т—В–Њ—А–Є—З–љ—Л–є¬†–≥–Є–њ–µ—А–њ–∞—А–∞—В–Є—А–µ–Њ–Ј —В–∞–Ї–ґ–µ –љ–∞–±–ї—О–і–∞–µ—В—Б—П –њ—А–Є D-–і–µ—Д–Є—Ж–Є—В–љ—Л—Е –Є¬†D-—А–µ–Ј–Є—Б—В–µ–љ—В–љ—Л—Е —Б–Њ—Б—В–Њ—П–љ–Є—П—Е [33]:
-
–Њ—Б—В–µ–Њ–Љ–∞–ї—П—Ж–Є–Є;
-
–і–µ—Д–Є—Ж–Є—В–µ –≤–Є—В–∞–Љ–Є–љ–∞ D, –≤–Њ–Ј–љ–Є–Ї–∞—О—Й–µ–Љ –Є–Ј-–Ј–∞ –µ–≥–Њ –љ–µ—Е–≤–∞—В–Ї–Є –≤¬†–њ–Є—Й–µ –Є–ї–Є –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ–є –Є–љ—Б–Њ–ї—П—Ж–Є–Є, —Б–Є–љ–і—А–Њ–Љ–∞ –Љ–∞–ї—М–∞–±—Б–Њ—А–±—Ж–Є–Є –Є¬†–і—А.;
-
–њ—А–Є–µ–Љ–µ –∞–љ—В–Є—А–µ–Ј–Њ—А–±—В–Є–≤–љ—Л—Е –њ—А–µ¬≠–њ–∞—А–∞—В–Њ–≤¬†вАУ –±–Є—Б—Д–Њ—Б—Д–Њ–љ–∞—В–Њ–≤, –і–µ–љ–Њ—Б—Г¬≠–Љ–∞–±–∞;
-
–љ–∞—А—Г—И–µ–љ–Є–Є –Љ–µ—В–∞–±–Њ–ї–Є–Ј–Љ–∞ –≤–Є—В–∞–Љ–Є–љ–∞ D –љ–∞¬†—Д–Њ–љ–µ –ї–µ—З–µ–љ–Є—П –∞–љ—В–Є–Ї–Њ–љ–≤—Г–ї—М—Б–∞–љ—В–∞–Љ–Є, —Ж–Є—В–Њ—Б—В–∞—В–Є–Ї–∞–Љ–Є;
-
–њ–µ—А–≤–Є—З–љ–Њ–Љ –і–µ—Д–Є—Ж–Є—В–µ 1ќ±-–≥–Є–і—А–Њ¬≠–Ї—Б–Є–ї–∞–Ј—Л;
-
–њ–µ—А–≤–Є—З–љ–Њ–Љ –±–Є–ї–Є–∞—А–љ–Њ–Љ —Ж–Є—А—А–Њ–Ј–µ –Є¬†–і—А—Г–≥–Њ–є —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–є –њ–∞—В–Њ–ї–Њ–≥–Є–Є –њ–µ—З–µ–љ–Є;
-
–Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П—Е, —Б–Њ–њ—А–Њ–≤–Њ–ґ–і–∞—О—Й–Є—Е¬≠—Б—П –љ–∞—А—Г—И–µ–љ–Є–µ–Љ –≤—Б–∞—Б—Л–≤–∞–љ–Є—П –Ї–∞–ї—М¬≠—Ж–Є—П –≤¬†–ґ–µ–ї—Г–і–Њ—З–љ–Њ-–Ї–Є—И–µ—З–љ–Њ–Љ —В—А–∞–Ї—В–µ (–Ц–Ъ–Ґ),¬†вАУ —А–µ–Ј–µ–Ї—Ж–Є—П –ґ–µ–ї—Г–і–Ї–∞ –Є–ї–Є –Ї–Є—И–µ—З–љ–Є–Ї–∞, —Б–љ–Є–ґ–µ–љ–Є–µ –ґ–µ–ї—Г–і–Њ—З–љ–Њ–є —Б–µ–Ї—А–µ—Ж–Є–Є –Є¬†–і—А.;
-
–њ—А–Є–µ–Љ–µ –∞–ї—О–Љ–Є–љ–Є–є—Б–Њ–і–µ—А–ґ–∞—Й–Є—Е –∞–љ¬≠—В–∞—Ж–Є–і–Њ–≤;¬†
-
–≥–Є–њ–µ—А—Д–Њ—Б—Д–∞—В–µ–Љ–Є–Є;
-
–њ–µ—А–≤–Є—З–љ–Њ–є¬†–≥–Є–њ–µ—А–Ї–∞–ї—М—Ж–Є—Г—А–Є–Є;
-
–Љ–∞—Б—Б–Є–≤–љ—Л—Е¬†–≥–µ–Љ–Њ—В—А–∞–љ—Б—Д—Г–Ј–Є—П—Е –Ј–∞ —Б—З–µ—В –і–Њ–±–∞–≤–ї–µ–љ–Є—П —Ж–Є—В—А–∞—В–∞.
–Т—Б–µ —Н—В–Є –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П –Є¬†—Б–Њ—Б—В–Њ—П–љ–Є—П —Б–Њ–њ—А–Њ–≤–Њ–ґ–і–∞—О—В—Б—П –і–ї–Є—В–µ–ї—М–љ—Л–Љ —Е—А–Њ–љ–Є—З–µ—Б–Ї–Є–Љ –і–µ—Д–Є—Ж–Є—В–Њ–Љ –∞–Ї—В–Є–≤–љ–Њ–≥–Њ –Љ–µ—В–∞–±–Њ–ї–Є—В–∞ 1,25(OH)2D3, —Б–љ–Є–ґ–µ–љ–Є–µ–Љ –∞–±—Б–Њ—А–±—Ж–Є–Є –Ї–∞–ї—М—Ж–Є—П –≤¬†–Ї–Є—И–µ—З–љ–Є–Ї–µ –Є¬†—Г—А–Њ–≤–љ—П –Ї–∞–ї—М—Ж–Є—П –≤¬†–Ї—А–Њ–≤–Є. –Э–∞¬†—Г–Љ–µ–љ—М—И–µ–љ–Є–µ —Г—А–Њ–≤–љ—П –Є–Њ–љ–Є–Ј–Є—А–Њ–≤–∞–љ–љ–Њ–≥–Њ –Ї–∞–ї—М—Ж–Є—П —А–µ–∞–≥–Є—А—Г—О—В —А–µ—Ж–µ–њ—В–Њ—А—Л CaR –њ–∞—А–∞—Й–Є—В–Њ–≤–Є–і–љ—Л—Е –ґ–µ–ї–µ–Ј, —З—В–Њ –≤¬†–Ї–Њ–Љ–њ–ї–µ–Ї—Б–µ –њ—А–Є–≤–Њ–і–Є—В –Ї¬†—Б—В–Њ–є–Ї–Њ–Љ—Г –њ–Њ–≤—Л—И–µ–љ–Є—О —Б–µ–Ї—А–µ—Ж–Є–Є –Я–Ґ–У, –≤–ї–Є—П—О—Й–µ–≥–Њ –љ–∞¬†—В—А–Є –Њ—Б–љ–Њ–≤–љ—Л–µ –Љ–Є—И–µ–љ–Є¬†вАУ –њ–Њ—З–Ї–Є, –Ї–Є—И–µ—З–љ–Є–Ї –Є¬†–Ї–Њ—Б—В–љ—Г—О —В–Ї–∞–љ—М. –Т¬†–њ–Њ—З–Ї–∞—Е –Я–Ґ–У –∞–Ї—В–Є–≤–Є—А—Г–µ—В 1ќ±-–≥–Є–і—А–Њ–Ї—Б–Є–ї–∞–Ј—Г –Є¬†–≤—Л—А–∞–±–Њ—В–Ї—Г 1,25(OH)2D3. –Т¬†—А–µ–Ј—Г–ї—М—В–∞—В–µ –њ–Њ–≤—Л—И–∞–µ—В—Б—П –∞–±—Б–Њ—А–±—Ж–Є—П –Ї–∞–ї—М—Ж–Є—П –Є¬†—Д–Њ—Б—Д–∞—В–∞ –≤¬†–Ї–Є—И–µ—З–љ–Є–Ї–µ. –Ъ—А–Њ–Љ–µ —В–Њ–≥–Њ, –≤¬†–њ–Њ—З–Ї–∞—Е –Я–Ґ–У —Г–≤–µ–ї–Є—З–Є–≤–∞–µ—В —А–µ–∞–±—Б–Њ—А–±—Ж–Є—О –Ї–∞–ї—М—Ж–Є—П –Є¬†–њ–Њ–≤—Л—И–∞–µ—В —Н–Ї—Б–Ї—А–µ—Ж–Є—О —Д–Њ—Б—Д–∞—В–∞ —Б¬†–Љ–Њ—З–Њ–є. –Т¬†–Ї–Њ—Б—В–љ–Њ–є —В–Ї–∞–љ–Є –Њ–љ —Б—В–Є–Љ—Г–ї–Є—А—Г–µ—В —Д—Г–љ–Ї—Ж–Є—О –Њ—Б—В–µ–Њ–Ї–ї–∞—Б—В–Њ–≤ –Є¬†–∞–Ї—В–Є–≤–љ–Њ—Б—В—М –Ї–Њ—Б—В–љ–Њ–є —А–µ–Ј–Њ—А–±—Ж–Є–Є, –±–ї–∞–≥–Њ–і–∞—А—П —З–µ–Љ—Г –Ї–∞–ї—М—Ж–Є–є –Є¬†—Д–Њ—Б—Д–∞—В –Ї–Њ—Б—В–љ–Њ–≥–Њ¬†–≥–Є–і—А–Њ–Ї—Б–Є–∞–њ–∞—В–Є—В–∞ –њ–Њ–њ–∞–і–∞—О—В –≤¬†–Ї—А–Њ–≤—М. –Т—Б–µ —Н—Д—Д–µ–Ї—В—Л –Я–Ґ–У –љ–∞–њ—А–∞–≤–ї–µ–љ—Л –љ–∞¬†–њ–Њ–≤—Л—И–µ–љ–Є–µ –њ–Њ—Б—В—Г–њ–ї–µ–љ–Є—П –Ї–∞–ї—М—Ж–Є—П –≤–Њ –≤–љ–µ–Ї–ї–µ—В–Њ—З–љ—Г—О –ґ–Є–і–Ї–Њ—Б—В—М –Є¬†–њ–Њ–і–і–µ—А–ґ–∞–љ–Є–µ –µ–≥–Њ –љ–Њ—А–Љ–∞–ї—М–љ–Њ–є –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є–Є –≤¬†–Ї—А–Њ–≤–Є [33, 34].
–Ф–ї–Є—В–µ–ї—М–љ–∞—П¬†–≥–Є–њ–µ—А—Б—В–Є–Љ—Г–ї—П—Ж–Є—П —Д—Г–љ–Ї—Ж–Є–Є –њ–∞—А–∞—Й–Є—В–Њ–≤–Є–і–љ—Л—Е –ґ–µ–ї–µ–Ј –Є¬†–њ–Њ–≤—Л—И–µ–љ–љ—Л–є —Г—А–Њ–≤–µ–љ—М –Я–Ґ–У —Б–њ–Њ—Б–Њ–±—Б—В–≤—Г—О—В —Д–Њ—А–Љ–Є—А–Њ–≤–∞–љ–Є—О —Б–Є–Љ–њ—В–Њ–Љ–Њ–Ї–Њ–Љ–њ–ї–µ–Ї—Б–∞ –≤—В–Њ—А–Є—З–љ–Њ–≥–Њ¬†–≥–Є–њ–µ—А–њ–∞—А–∞—В–Є—А–µ–Њ–Ј–∞. –Ю–љ —Е–∞—А–∞–Ї—В–µ—А–Є–Ј—Г–µ—В—Б—П –±—Л—Б—В—А–Њ–є –њ–Њ—В–µ—А–µ–є –Ї–Њ—Б—В–љ–Њ–є –Љ–∞—Б—Б—Л, —А–∞–Ј–≤–Є—В–Є–µ–Љ –Њ—Б—В–µ–Њ–њ–Њ—А–Њ–Ј–∞ –Є¬†–њ–µ—А–µ–ї–Њ–Љ–Њ–≤, –Њ—Б—В–µ–Њ–Љ–∞–ї—П—Ж–Є–µ–є, –њ—А–Њ—П–≤–ї—П—О—Й–µ–є—Б—П –Љ—Л—И–µ—З–љ–Њ–є —Б–ї–∞–±–Њ—Б—В—М—О, –±–Њ–ї—П–Љ–Є –≤¬†–Ї–Њ—Б—В—П—Е, —Б–ї–∞–±–Њ—Б—В—М—О –њ—А–Њ–Ї—Б–Є–Љ–∞–ї—М–љ—Л—Е –Љ—Л—И–µ—З–љ—Л—Е¬†–≥—А—Г–њ–њ, —В—А—Г–і–љ–Њ—Б—В—П–Љ–Є –њ—А–Є –њ–Њ–і—К–µ–Љ–µ –њ–Њ¬†–ї–µ—Б—В–љ–Є—Ж–µ –Є–ї–Є –≤—Б—В–∞–≤–∞–љ–Є–Є —Б–Њ¬†—Б—В—Г–ї–∞, –≤¬†—В—П–ґ–µ–ї—Л—Е —Б–ї—Г—З–∞—П—Е¬†вАУ —Д–Њ—А–Љ–Є—А–Њ–≤–∞–љ–Є–µ–Љ –≤–љ–µ–Ї–Њ—Б—В–љ—Л—Е –Ї–∞–ї—М—Ж–Є—Д–Є–Ї–∞—В–Њ–≤ [34].
–Ы–µ—З–µ–љ–Є–µ –Њ—Б—В–µ–Њ–њ–Њ—А–Њ–Ј–∞ –њ—А–Є¬†—Н–љ–і–Њ–Ї—А–Є–љ–љ—Л—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П—Е: —А–Њ–ї—М –∞–ї—М—Д–∞–Ї–∞–ї—М—Ж–Є–і–Њ–ї–∞
–Т¬†–љ–∞—Б—В–Њ—П—Й–µ–µ –≤—А–µ–Љ—П –і–ї—П –ї–µ—З–µ–љ–Є—П –Њ—Б—В–µ–Њ–њ–Њ—А–Њ–Ј–∞ –Є—Б–њ–Њ–ї—М–Ј—Г–µ—В—Б—П —И–Є—А–Њ–Ї–Є–є –∞—А—Б–µ–љ–∞–ї –њ—А–µ–њ–∞—А–∞—В–Њ–≤, –Њ—Б–љ–Њ–≤–љ–Њ–є —Ж–µ–ї—М—О –љ–∞–Ј–љ–∞—З–µ–љ–Є—П –Ї–Њ—В–Њ—А—Л—Е —П–≤–ї—П–µ—В—Б—П —Б–љ–Є–ґ–µ–љ–Є–µ —А–Є—Б–Ї–∞ –њ–µ—А–µ–ї–Њ–Љ–Њ–≤. –Т¬†–Ї–∞—З–µ—Б—В–≤–µ —Б–∞–Љ–Њ—Б—В–Њ—П—В–µ–ї—М–љ–Њ–є —В–µ—А–∞–њ–Є–Є —Б–Є—Б—В–µ–Љ–љ–Њ–≥–Њ –Њ—Б—В–µ–Њ–њ–Њ—А–Њ–Ј–∞ –њ—А–Є–Љ–µ–љ—П—О—В—Б—П –±–Є—Б—Д–Њ—Б—Д–Њ–љ–∞—В—Л, –і–µ–љ–Њ—Б—Г–Љ–∞–±, —В–µ—А–Є–њ–∞—А–∞—В–Є–і –Є¬†—Б—В—А–Њ–љ—Ж–Є—П —А–∞–љ–µ–ї–∞—В (–≤—В–Њ—А–∞—П –ї–Є–љ–Є—П –ї–µ—З–µ–љ–Є—П). –Т—Б–µ –∞–љ—В–Є–Њ—Б—В–µ–Њ–њ–Њ—А–Њ—В–Є—З–µ—Б–Ї–Є–µ –њ—А–µ–њ–∞—А–∞—В—Л –љ–∞–Ј–љ–∞—З–∞—О—В—Б—П –љ–∞¬†—Д–Њ–љ–µ –±–∞–Ј–Њ–≤–Њ–є —В–µ—А–∞–њ–Є–Є —Б–Њ–ї—П–Љ–Є –Ї–∞–ї—М—Ж–Є—П (–≤¬†—Б–ї—Г—З–∞–µ –µ–≥–Њ –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ–≥–Њ –њ–Є—Й–µ–≤–Њ–≥–Њ –њ–Њ—В—А–µ–±–ї–µ–љ–Є—П), –≤–Є—В–∞–Љ–Є–љ–Њ–Љ D –Є–ї–Є –µ–≥–Њ –∞–Ї—В–Є–≤–љ—Л–Љ –Љ–µ—В–∞–±–Њ–ї–Є—В–Њ–Љ¬†вАУ –∞–ї—М—Д–∞–Ї–∞–ї—М—Ж–Є–і–Њ–ї–Њ–Љ.
–Р–ї—М—Д–∞–Ї–∞–ї—М—Ж–Є–і–Њ–ї 1ќ±(–Ю–Э)D3 (–Р–ї—М—Д–∞ –Ф3-–Ґ–µ–≤–∞¬Ѓ)¬†вАУ —Б–Є–љ—В–µ—В–Є—З–µ—Б–Ї–Є–є –∞–љ–∞–ї–Њ–≥ –µ—Б—В–µ—Б—В–≤–µ–љ–љ—Л—Е –∞–Ї—В–Є–≤–љ—Л—Е –Љ–µ—В–∞–±–Њ–ї–Є—В–Њ–≤ –≤–Є—В–∞–Љ–Є–љ–∞ D. –Т¬†–Њ—В–ї–Є—З–Є–µ –Њ—В¬†–љ–∞—В–Є–≤–љ–Њ–≥–Њ –≤–Є—В–∞–Љ–Є–љ–∞ D –Њ–љ –њ—А–µ–≤—А–∞—Й–∞–µ—В—Б—П –≤¬†D-–≥–Њ—А–Љ–Њ–љ 1,25(–Ю–Э)2D3 –Ј–∞ –Њ–і–љ—Г –Љ–µ—В–∞–±–Њ–ї–Є—З–µ—Б–Ї—Г—О —Б—В–∞–і–Є—О (–Љ–Є–љ—Г—П –њ–Њ—З–Ї–Є) –њ—А–Є –њ–Њ–Љ–Њ—Й–Є –њ–µ—З–µ–љ–Њ—З–љ–Њ–≥–Њ —Д–µ—А–Љ–µ–љ—В–∞ 25-–≥–Є–і—А–Њ¬≠–Ї—Б–Є–ї–∞–Ј—Л. –°–Ї–Њ—А–Њ—Б—В—М –Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є—П 1,25(–Ю–Э)2D3 –Є–Ј¬†–∞–ї—М—Д–∞–Ї–∞–ї—М—Ж–Є–і–Њ–ї–∞ –љ–∞–Љ–љ–Њ–≥–Њ –≤—Л—И–µ, —З–µ–Љ –Є–Ј¬†–љ–∞—В–Є–≤–љ–Њ–≥–Њ –≤–Є—В–∞–Љ–Є–љ–∞ D, –њ–Њ—Н—В–Њ–Љ—Г –±–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–µ —Н—Д—Д–µ–Ї—В—Л –њ–µ—А–≤–Њ–≥–Њ –њ—А–Њ—П–≤–ї—П—О—В—Б—П –±—Л—Б—В—А–µ–µ –Є¬†–Є–љ—В–µ–љ—Б–Є–≤–љ–µ–µ.
–Ъ¬†–Њ—Б–љ–Њ–≤–љ—Л–Љ –±–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–Љ –Є¬†–Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є–Љ —Н—Д—Д–µ–Ї—В–∞–Љ –∞–ї—М—Д–∞–Ї–∞–ї—М—Ж–Є–і–Њ–ї–∞ –Њ—В–љ–Њ—Б—П—В—Б—П [35]:
1) —Г—Б–Є–ї–µ–љ–Є–µ –∞–±—Б–Њ—А–±—Ж–Є–Є –Ї–∞–ї—М—Ж–Є—П –≤¬†–Ц–Ъ–Ґ –Є¬†–њ–Њ–≤—Л—И–µ–љ–Є–µ –Ї–∞–ї—М—Ж–Є–µ–Љ–Є–Є;
2) –њ–Њ–і–∞–≤–ї–µ–љ–Є–µ¬†–≥–Є–њ–µ—А–њ—А–Њ–і—Г–Ї—Ж–Є–Є –Я–Ґ–У –њ–∞—А–∞—Й–Є—В–Њ–≤–Є–і–љ—Л–Љ–Є –ґ–µ–ї–µ–Ј–∞–Љ–Є –Є¬†—Б–љ–Є–ґ–µ–љ–Є–µ –±–ї–∞–≥–Њ–і–∞—А—П —Н—В–Њ–Љ—Г –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є –Ї–Њ—Б—В–љ–Њ–є —А–µ–Ј–Њ—А–±—Ж–Є–Є;
3) —Б—В–Є–Љ—Г–ї—П—Ж–Є—П –Ї–Њ—Б—В–љ–Њ–≥–Њ —А–µ–Љ–Њ–і–µ–ї–Є—А–Њ–≤–∞–љ–Є—П –Є¬†—Б–Є–љ—В–µ–Ј–∞ –Ї–Њ—Б—В–љ–Њ–≥–Њ –Љ–∞—В—А–Є–Ї—Б–∞ –њ—Г—В–µ–Љ –њ—А—П–Љ–Њ–≥–Њ –≤–Њ–Ј–і–µ–є—Б—В–≤–Є—П –љ–∞¬†VDR –Њ—Б—В–µ–Њ–±–ї–∞—Б—В–Њ–≤, –њ–Њ–≤—Л—И–µ–љ–Є—П –Є—Е –і–Є—Д—Д–µ—А–µ–љ—Ж–Є—А–Њ–≤–Ї–Є –Є¬†—Д—Г–љ–Ї—Ж–Є–Њ–љ–∞–ї—М–љ–Њ–є –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є (–≤–∞–ґ–љ—Л–є —Н—Д—Д–µ–Ї—В –њ—А–Є –њ–Њ–і–∞–≤–ї–µ–љ–Є–Є –Ї–Њ—Б—В–µ–Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є—П —Г¬†–±–Њ–ї—М–љ—Л—Е –°–Ф¬†–Є¬†–њ—А–Є–љ–Є–Љ–∞—О—Й–Є—Е –У–Ъ);
4) —Г–ї—Г—З—И–µ–љ–Є–µ –Ї–∞—З–µ—Б—В–≤–∞ –Ї–Њ—Б—В–Є –≤—Б–ї–µ–і—Б—В–≤–Є–µ –њ–Њ–і–∞–≤–ї–µ–љ–Є—П –њ–µ—А—Д–Њ—А–∞—Ж–Є–Є —В—А–∞–±–µ–Ї—Г–ї—П—А–љ—Л—Е –њ–ї–∞—Б—В–Є–љ, —Г—Б–Є–ї–µ–љ–Є—П —А–µ–њ–∞—А–∞—Ж–Є–Є –Ї–Њ—Б—В–µ–є, –њ–Њ–≤—Л—И–µ–љ–Є—П —Б–Є–љ—В–µ–Ј–∞ –Ї–Њ—Б—В–љ–Њ–≥–Њ –Љ–∞—В—А–Є–Ї—Б–∞ –Є¬†–Љ–µ—Б—В–љ—Л—Е —А–Њ—Б—В–Њ–≤—Л—Е —Д–∞–Ї—В–Њ—А–Њ–≤;
5) —Б–љ–Є–ґ–µ–љ–Є–µ —А–Є—Б–Ї–∞ –њ–∞–і–µ–љ–Є–є –≤¬†—А–µ–Ј—Г–ї—М—В–∞—В–µ –њ–Њ–≤—Л—И–µ–љ–Є—П –Љ—Л—И–µ—З–љ–Њ–є —Б–Є–ї—Л.
–Э–µ–Њ–±—Е–Њ–і–Є–Љ–Њ –Њ—В–Љ–µ—В–Є—В—М, —З—В–Њ –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є –Ј–љ–∞—З–Є–Љ—Л–µ —Н—Д—Д–µ–Ї—В—Л –љ–∞—В–Є–≤–љ–Њ–≥–Њ –≤–Є—В–∞–Љ–Є–љ–∞ D –њ—А–Њ—П–≤–ї—П—О—В—Б—П —В–Њ–ї—М–Ї–Њ —Г¬†–ї–Є—Ж —Б¬†–µ–≥–Њ –Є—Б—Е–Њ–і–љ—Л–Љ –і–µ—Д–Є—Ж–Є—В–Њ–Љ –њ—А–Є –љ–Њ—А–Љ–∞–ї—М–љ–Њ–є —Д—Г–љ–Ї—Ж–Є–Є –њ–Њ—З–µ–Ї, –≤¬†—В–Њ –≤—А–µ–Љ—П –Ї–∞–Ї –∞–ї—М—Д–∞–Ї–∞–ї—М—Ж–Є–і–Њ–ї –∞–Ї—В–Є–≤–љ–Њ –њ—А–µ–≤—А–∞—Й–∞–µ—В—Б—П –≤¬†D-–≥–Њ—А–Љ–Њ–љ –≤–љ–µ –Ј–∞–≤–Є—Б–Є–Љ–Њ—Б—В–Є –Њ—В¬†—Г—А–Њ–≤–љ—П 25(–Ю–Э)D3 –Є¬†—Д—Г–љ–Ї—Ж–Є–Є –њ–Њ—З–µ–Ї [36]. –Ю–і–љ–∞–Ї–Њ –њ—А–Є –ї–∞–±–Њ—А–∞—В–Њ—А–љ–Њ –њ–Њ–і—В–≤–µ—А–ґ–і–µ–љ–љ–Њ–Љ —Б–љ–Є–ґ–µ–љ–Є–Є —Г—А–Њ–≤–љ—П 25(–Ю–Э)D3 –љ–∞–Ј–љ–∞—З–∞—В—М –∞–ї—М—Д–∞–Ї–∞–ї—М—Ж–Є–і–Њ–ї –і–ї—П –µ–≥–Њ –≤–Њ—Б–њ–Њ–ї–љ–µ–љ–Є—П –љ–µ—Ж–µ–ї–µ—Б–Њ–Њ–±—А–∞–Ј–љ–Њ, —В–∞–Ї –Ї–∞–Ї –Њ–љ –ї–Є—И—М –љ–µ–Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ –њ–Њ–≤—Л—И–∞–µ—В —Г—А–Њ–≤–µ–љ—М 25(–Ю–Э)D3 –≤¬†—Б—Л–≤–Њ—А–Њ—В–Ї–µ –Ї—А–Њ–≤–Є –Ј–∞ —Б—З–µ—В –њ–∞—Б—Б–Є–≤–љ–Њ–≥–Њ —Б–љ–Є–ґ–µ–љ–Є—П —А–∞—Б—Е–Њ–і–∞ –µ–≥–Њ –Ј–∞–њ–∞—Б–Њ–≤ –≤¬†–ґ–Є—А–Њ–≤–Њ–є —В–Ї–∞–љ–Є –і–ї—П —Б–Є–љ—В–µ–Ј–∞ D-–≥–Њ—А–Љ–Њ–љ–∞. –Э–µ—Б–Љ–Њ—В—А—П –љ–∞¬†—В–Њ —З—В–Њ 25(–Ю–Э)D3 —Б–∞–Љ –њ–Њ¬†—Б–µ–±–µ –љ–µ¬†–Њ–Ї–∞–Ј—Л–≤–∞–µ—В –≤—Л—А–∞–ґ–µ–љ–љ—Л—Е –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є—Е —Н—Д—Д–µ–Ї—В–Њ–≤, —Б–љ–∞—З–∞–ї–∞ —Б–ї–µ–і—Г–µ—В –љ–Њ—А–Љ–∞–ї–Є–Ј–Њ–≤–∞—В—М –µ–≥–Њ –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є—О –≤¬†–Ї—А–Њ–≤–Є (–≤—Л—И–µ 30 –љ–≥/–Љ–ї), –∞¬†–њ–Њ—В–Њ–Љ –ї–µ—З–Є—В—М –њ–∞—Ж–Є–µ–љ—В–∞ –∞–Ї—В–Є–≤–љ—Л–Љ –Љ–µ—В–∞–±–Њ–ї–Є—В–Њ–Љ –≤–Є—В–∞–Љ–Є–љ–∞ D¬†вАУ –∞–ї—М—Д–∞–Ї–∞–ї—М—Ж–Є–і–Њ–ї–Њ–Љ.
–Р–ї—М—Д–∞–Ї–∞–ї—М—Ж–Є–і–Њ–ї —Г—Б–њ–µ—И–љ–Њ –њ—А–Є–Љ–µ–љ—П–µ—В—Б—П –і–ї—П –ї–µ—З–µ–љ–Є—П –Є¬†–њ—А–Њ—Д–Є–ї–∞–Ї—В–Є–Ї–Є¬†–≥–ї—О–Ї–Њ–Ї–Њ—А—В–Є–Ї–Њ–Є–і–љ–Њ–≥–Њ –Њ—Б—В–µ–Њ–њ–Њ—А–Њ–Ј–∞. –Я–∞—В–Њ–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–∞—П –Њ—Б–љ–Њ–≤–∞¬†вАУ —Д–Њ—А–Љ–Є—А–Њ–≤–∞–љ–Є–µ –Њ—В—А–Є—Ж–∞—В–µ–ї—М–љ–Њ–≥–Њ –±–∞–ї–∞–љ—Б–∞ –Ї–∞–ї—М—Ж–Є—П –Є¬†–њ–Њ–≤—Л—И–µ–љ–Є–µ –њ—А–Њ–і—Г–Ї—Ж–Є–Є –Я–Ґ–У, —Г–Љ–µ–љ—М—И–µ–љ–Є–µ —Н–Ї—Б–њ—А–µ—Б—Б–Є–Є VDR –Є¬†—А–∞–Ј–≤–Є—В–Є–µ –Љ—Л—И–µ—З–љ–Њ–є —Б–ї–∞–±–Њ—Б—В–Є –љ–∞¬†—Д–Њ–љ–µ –њ—А–Є–µ–Љ–∞ –У–Ъ. –Т¬†–њ–µ—А–µ—З–Є—Б–ї–µ–љ–љ—Л—Е —Б–Є—В—Г–∞—Ж–Є—П—Е –∞–ї—М—Д–∞–Ї–∞–ї—М—Ж–Є–і–Њ–ї –Њ–Ї–∞–Ј—Л–≤–∞–µ—В –±–Њ–ї–µ–µ –≤—Л—А–∞–ґ–µ–љ–љ—Л–є —Н—Д—Д–µ–Ї—В –љ–∞¬†–Ь–Я–Ъ –Є¬†—А–Є—Б–Ї –њ–µ—А–µ–ї–Њ–Љ–∞ –њ–Њ–Ј–≤–Њ–љ–Ї–Њ–≤, —З–µ–Љ –љ–∞—В–Є–≤–љ—Л–є –≤–Є—В–∞–Љ–Є–љ D. –Т¬†–Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–Є J.D.¬†Ringe –Є¬†—Б–Њ–∞–≤—В. (2004) —Г—З–∞—Б—В–≤–Њ–≤–∞–ї–Њ 204¬†–±–Њ–ї—М–љ—Л—Е, –Ї–Њ—В–Њ—А—Л–µ –њ–Њ—Б—В–Њ—П–љ–љ–Њ –њ–Њ–ї—Г—З–∞–ї–Є –У–Ъ –Є¬†–Ї–Њ—В–Њ—А—Л–Љ –±—Л–ї –њ–Њ—Б—В–∞–≤–ї–µ–љ –і–Є–∞–≥–љ–Њ–Ј ¬Ђ–≥–ї—О–Ї–Њ–Ї–Њ—А—В–Є–Ї–Њ–Є–і–љ—Л–є –Њ—Б—В–µ–Њ–њ–Њ—А–Њ–Ј¬ї. –Т¬†—В–µ—З–µ–љ–Є–µ —В—А–µ—Е –ї–µ—В –њ–∞—Ж–Є–µ–љ—В—Л –њ–Њ–ї—Г—З–∞–ї–Є –∞–ї—М—Д–∞–Ї–∞–ї—М—Ж–Є–і–Њ–ї –≤¬†–і–Њ–Ј–µ 1¬†–Љ–Ї–≥/—Б—Г—В –Є–ї–Є –≤–Є—В–∞–Љ–Є–љ D3 –≤¬†–і–Њ–Ј–µ 1000 –Ь–Х/—Б—Г—В. –†–µ–Ј—Г–ї—М—В–∞—В—Л¬† –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П –њ—А–Њ–і–µ–Љ–Њ–љ—Б—В—А–Є—А–Њ–≤–∞–ї–Є –±–Њ–ї–µ–µ –≤—Л—А–∞–ґ–µ–љ–љ–Њ–µ –њ–Њ–≤—Л—И–µ–љ–Є–µ –Ь–Я–Ъ –≤¬†–≥—А—Г–њ–њ–µ –∞–ї—М—Д–∞–Ї–∞–ї—М—Ж–Є–і–Њ–ї–∞ –њ–Њ¬†—Б—А–∞–≤–љ–µ–љ–Є—О —Б¬†–≥—А—Г–њ–њ–Њ–є –≤–Є—В–∞–Љ–Є–љ–∞ D3: –≤¬†–њ–Њ–Ј–≤–Њ–љ–Њ—З–љ–Є–Ї–µ¬†вАУ –љ–∞¬†3,2% (—А < 0,0001), –≤¬†—И–µ–є–Ї–µ –±–µ–і—А–∞¬†вАУ –љ–∞¬†1,8% (—А < 0,01). –Ъ—А–Њ–Љ–µ —В–Њ–≥–Њ, —Г¬†–±–Њ–ї—М–љ—Л—Е, –њ–Њ–ї—Г—З–∞–≤—И–Є—Е –∞–ї—М—Д–∞–Ї–∞–ї—М—Ж–Є–і–Њ–ї, –Њ—В–Љ–µ—З–µ–љ–Њ —Б—В–∞—В–Є—Б—В–Є—З–µ—Б–Ї–Є –±–Њ–ї–µ–µ –Ј–љ–∞—З–Є–Љ–Њ–µ —Б–љ–Є–ґ–µ–љ–Є–µ —А–Є—Б–Ї–∞ –њ–µ—А–µ–ї–Њ–Љ–Њ–≤ –њ–Њ–Ј–≤–Њ–љ–Ї–Њ–≤¬†вАУ –љ–∞¬†39% (—А = 0,005) –Є¬†—А–Є—Б–Ї–∞ –≤—Б–µ—Е —В–Є–њ–Њ–≤ –њ–µ—А–µ–ї–Њ–Љ–Њ–≤¬†вАУ –љ–∞¬†48% (—А = 0,001) [37].
–Т¬†–і—А—Г–≥–Њ–Љ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–Є –њ–Њ–Ї–∞–Ј–∞–љ–Њ —Б–љ–Є–ґ–µ–љ–Є–µ —А–Є—Б–Ї–∞ –њ–µ—А–µ–ї–Њ–Љ–Њ–≤ –њ–Њ–Ј–≤–Њ–љ–Ї–Њ–≤ –њ—А–Є¬†–≥–ї—О–Ї–Њ–Ї–Њ—А—В–Є–Ї–Њ–Є–і–љ–Њ–Љ –Њ—Б—В–µ–Њ–њ–Њ—А–Њ–Ј–µ –≤¬†1,8 —А–∞–Ј–∞ –њ—А–Є –ї–µ—З–µ–љ–Є–Є –∞–Ї—В–Є–≤–љ—Л–Љ–Є –Љ–µ—В–∞–±–Њ–ї–Є—В–∞–Љ–Є –≤–Є—В–∞–Љ–Є–љ–∞ D –њ–Њ¬†—Б—А–∞–≤–љ–µ–љ–Є—О —Б¬†–њ–ї–∞—Ж–µ–±–Њ, –љ–∞—В–Є–≤–љ—Л–Љ –≤–Є—В–∞–Љ–Є–љ–Њ–Љ D –Є/–Є–ї–Є –Ї–∞–ї—М—Ж–Є–µ–Љ (–Ю–† 0,56 (95% –Ф–Ш 0,34вАУ0,92)) [38].
–Х—Й–µ –Њ–і–Є–љ –≤–∞–ґ–љ—Л–є –∞—Б–њ–µ–Ї—В –ї–µ—З–µ–љ–Є—П –Њ—Б—В–µ–Њ–њ–Њ—А–Њ–Ј–∞ –њ—А–Є —А–∞–Ј–ї–Є—З–љ—Л—Е —Н–љ–і–Њ–Ї—А–Є–љ–љ—Л—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П—Е¬†вАУ —Б–љ–Є–ґ–µ–љ–Є–µ —А–Є—Б–Ї–∞ –њ–∞–і–µ–љ–Є–є, –Ї–Њ—В–Њ—А—Л–є, –Ї–∞–Ї —Г–Ї–∞–Ј—Л–≤–∞–ї–Њ—Б—М —А–∞–љ–µ–µ, –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ –њ–Њ–≤—Л—И–∞–µ—В—Б—П –љ–∞¬†—Д–Њ–љ–µ –°–Ф, –≤—Л—А–∞–ґ–µ–љ–љ–Њ–є –њ–Њ—З–µ—З–љ–Њ–є –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В–Є –Є¬†–њ—А–Є –њ—А–Є–Љ–µ–љ–µ–љ–Є–Є –±–Њ–ї—М—И–Є—Е –і–Њ–Ј –У–Ъ. –Т¬†—А—П–і–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є –њ—А–Њ–і–µ–Љ–Њ–љ—Б—В—А–Є—А–Њ–≤–∞–љ–Њ –њ–Њ–ї–Њ–ґ–Є—В–µ–ї—М–љ–Њ–µ –≤–ї–Є—П–љ–Є–µ –∞–ї—М—Д–∞–Ї–∞–ї—М—Ж–Є–і–Њ–ї–∞ –љ–∞¬†–Љ—Л—И–µ—З–љ—Г—О —Б–Є–ї—Г, —А–∞–≤–љ–Њ–≤–µ—Б–Є–µ –Є¬†–Ї–Њ–Њ—А–і–Є–љ–∞—Ж–Є—О —В–µ–ї–∞ –Є¬†—Д—Г–љ–Ї—Ж–Є–Њ–љ–∞–ї—М–љ—Л–µ –≤–Њ–Ј–Љ–Њ–ґ–љ–Њ—Б—В–Є –њ–∞—Ж–Є–µ–љ—В–Њ–≤ [39]. –Ґ–∞–Ї, –ї–µ—З–µ–љ–Є–µ –∞–ї—М—Д–∞–Ї–∞–ї—М—Ж–Є–і–Њ–ї–Њ–Љ –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ —Б–љ–Є–ґ–∞–ї–Њ —А–Є—Б–Ї¬†–њ–∞–і–µ–љ–Є–є —Г¬†–њ–Њ–ґ–Є–ї—Л—Е –њ–∞—Ж–Є–µ–љ—В–Њ–≤ (–Ю–† 0,45 (95% –Ф–Ш 0,21вАУ0,97), p = 0,042) [40].
–Ф–ї—П –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–≤—В–Њ—А–Є—З–љ—Л–Љ¬†–≥–Є–њ–µ—А–њ–∞—А–∞—В–Є—А–µ–Њ–Ј–Њ–Љ –Є–ї–Є –њ–Њ—З–µ—З–љ–Њ–є –Њ—Б—В–µ–Њ–і–Є—Б—В—А–Њ—Д–Є–µ–є –∞–ї—М—Д–∞–Ї–∞–ї—М—Ж–Є–і–Њ–ї —П–≤–ї—П–µ—В—Б—П –њ—А–µ–њ–∞—А–∞—В–Њ–Љ –≤—Л–±–Њ—А–∞ –≤—Б–ї–µ–і—Б—В–≤–Є–µ —Ж–µ–ї–Њ–≥–Њ —А—П–і–∞ –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є—Е —Н—Д—Д–µ–Ї—В–Њ–≤: —Б–њ–Њ—Б–Њ–±–љ–Њ—Б—В–Є –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ –њ–Њ–≤—Л—И–∞—В—М –Ї–Є—И–µ—З–љ—Г—О –∞–±—Б–Њ—А–±—Ж–Є—О –Ї–∞–ї—М—Ж–Є—П, –њ–Њ–і–∞–≤–ї—П—В—М¬†–≥–Є–њ–µ—А–њ—А–Њ–і—Г–Ї—Ж–Є—О –Я–Ґ–У –Є¬†—Б—В–Є–Љ—Г–ї–Є—А–Њ–≤–∞—В—М –∞–Ї—В–Є–≤–љ–Њ—Б—В—М –Ї–Њ—Б—В–љ–Њ–≥–Њ —А–µ–Љ–Њ–і–µ–ї–Є—А–Њ–≤–∞–љ–Є—П. –£¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–њ–Њ—З–µ—З–љ–Њ–є –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В—М—О, –≤¬†—В–Њ–Љ —З–Є—Б–ї–µ —В–µ—А–Љ–Є–љ–∞–ї—М–љ–Њ–є —Б—В–∞–і–Є–µ–є –•–С–Я –љ–∞¬†–≥–µ–Љ–Њ–і–Є–∞–ї–Є–Ј–µ, —В–µ—А–∞–њ–Є—П –∞–ї—М—Д–∞–Ї–∞–ї—М—Ж–Є–і–Њ–ї–Њ–Љ –≤¬†–і–Њ–Ј–∞—Е –Њ—В¬†0,5¬†–Љ–Ї–≥ –≤ —Б—Г—В–Ї–Є –і–Њ 4 –Љ–Ї–≥ –і–≤–∞-—В—А–Є —А–∞–Ј–∞ –≤¬†–љ–µ–і–µ–ї—О –њ–Њ–≤—Л—И–∞–µ—В —Г—А–Њ–≤–µ–љ—М —Б—Л–≤–Њ—А–Њ—В–Њ—З–љ–Њ–≥–Њ –Ї–∞–ї—М—Ж–Є—П, —Б–љ–Є–ґ–∞–µ—В —Г—А–Њ–≤–µ–љ—М –Я–Ґ–У –Є¬†—Б–њ–Њ—Б–Њ–±—Б—В–≤—Г–µ—В —Г–≤–µ–ї–Є—З–µ–љ–Є—О –Ь–Я–Ъ, —Г–Љ–µ–љ—М—И–∞—П —В–∞–Ї–Є–Љ –Њ–±—А–∞–Ј–Њ–Љ –њ—А–Њ—П–≤–ї–µ–љ–Є—П –Њ—Б—В–µ–Њ–њ–µ–љ–Є–Є –Є¬†–Њ—Б—В–µ–Њ–Љ–∞–ї—П—Ж–Є–Є [41вАУ43]. –°–∞–Љ—Л–є —Б—Г—Й–µ—Б—В–≤–µ–љ–љ—Л–є —Н—Д—Д–µ–Ї—В –∞–ї—М—Д–∞–Ї–∞–ї—М—Ж–Є–і–Њ–ї–∞ –≤¬†–њ—А–Њ—Д–Є–ї–∞–Ї—В–Є–Ї–µ –њ–∞–і–µ–љ–Є–є¬†вАУ —Б–љ–Є–ґ–µ–љ–Є–µ —А–Є—Б–Ї–∞ –њ–Њ—Б–ї–µ–і–љ–Є—Е –љ–∞¬†71% –њ—А–Є –њ—А–Є–µ–Љ–µ –њ—А–µ–њ–∞—А–∞—В–∞ 1¬†–Љ–Ї–≥/—Б—Г—В¬†вАУ –љ–∞–±–ї—О–і–∞–ї—Б—П —В–∞–Ї–ґ–µ —Г¬†–њ–Њ–ґ–Є–ї—Л—Е –±–Њ–ї—М–љ—Л—Е —Б–Њ¬†—Б–љ–Є–ґ–µ–љ–љ–Њ–є —Д–Є–ї—М—В—А–∞—Ж–Є–Њ–љ–љ–Њ–є —Б–њ–Њ—Б–Њ–±–љ–Њ—Б—В—М—О –њ–Њ—З–µ–Ї [44]. –Я—А–Є —Н—В–Њ–Љ –∞–ї—М—Д–∞–Ї–∞–ї—М—Ж–Є–і–Њ–ї –њ—А–Њ–і–µ–Љ–Њ–љ—Б—В—А–Є—А–Њ–≤–∞–ї —Е–Њ—А–Њ—И–Є–є –њ—А–Њ—Д–Є–ї—М –±–µ–Ј–Њ–њ–∞—Б–љ–Њ—Б—В–Є: —З–∞—Б—В–Њ—В–∞¬†–≥–Є–њ–µ—А–Ї–∞–ї—М—Ж–Є–µ–Љ–Є–Є¬†вАУ 1,1%, —А–Є—Б–Ї¬†—А–∞–Ј–≤–Є—В–Є—П –љ–µ—Д—А–Њ–ї–Є—В–Є–∞–Ј–∞ –Њ—В—Б—Г—В—Б—В–≤–Њ–≤–∞–ї [45].
–Ч–∞–Ї–ї—О—З–µ–љ–Є–µ
–Ю—Б—В–µ–Њ–њ–Њ—А–Њ–Ј –Є¬†–Њ—Б—В–µ–Њ–њ–∞—В–Є–Є, –≤–Њ–Ј–љ–Є–Ї–∞—О—Й–Є–µ –љ–∞¬†—Д–Њ–љ–µ —А—П–і–∞ —Н–љ–і–Њ–Ї—А–Є–љ–љ—Л—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є, —Е–∞—А–∞–Ї—В–µ—А–Є–Ј—Г—О—В—Б—П –њ–∞—В–Њ–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Є–Љ–Є –Є¬†–Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є–Љ–Є –Њ—Б–Њ–±–µ–љ–љ–Њ—Б—В—П–Љ–Є, –Ї–Њ—В–Њ—А—Л–µ –Њ—В–Ї—А—Л–≤–∞—О—В —И–Є—А–Њ–Ї–Є–µ –≤–Њ–Ј–Љ–Њ–ґ–љ–Њ—Б—В–Є –Є¬†–њ–µ—А—Б–њ–µ–Ї—В–Є–≤—Л –і–ї—П –љ–∞–Ј–љ–∞—З–µ–љ–Є—П —В–µ—А–∞–њ–Є–Є –∞–Ї—В–Є–≤–љ—Л–Љ –Љ–µ—В–∞–±–Њ–ї–Є—В–Њ–Љ –≤–Є—В–∞–Љ–Є–љ–∞¬†D¬†вАУ –∞–ї—М—Д–∞–Ї–∞–ї—М—Ж–Є–і–Њ–ї–Њ–Љ.
L.A. Marchenkova
Russian Scientific Center of Medical Rehabilitation and Balneology
Contact person: Larisa Aleksandrovna Marchenkova, MarchenkovaLA@rncmrik.com
Benefits of active metabolite of vitamin D (alfacalcidol) over native vitamin D as well as its biological and clinical effects mediated via activity of 1,25(–Ю–Э)2D3 and discussed, which determine a rationale for its use in treatment of osteoporosis under various endocrine diseases. It is demonstrated that use of alfacalcidol (Alpha D3-Teva¬Ѓ) is among important links in prophylaxis of falls and fractures in such patients.
–£–≤–∞–ґ–∞–µ–Љ—Л–є –њ–Њ—Б–µ—В–Є—В–µ–ї—М uMEDp!
–£–≤–µ–і–Њ–Љ–ї—П–µ–Љ –Т–∞—Б –Њ —В–Њ–Љ, —З—В–Њ –Ј–і–µ—Б—М —Б–Њ–і–µ—А–ґ–Є—В—Б—П –Є–љ—Д–Њ—А–Љ–∞—Ж–Є—П, –њ—А–µ–і–љ–∞–Ј–љ–∞—З–µ–љ–љ–∞—П –Є—Б–Ї–ї—О—З–Є—В–µ–ї—М–љ–Њ –і–ї—П —Б–њ–µ—Ж–Є–∞–ї–Є—Б—В–Њ–≤ –Ј–і—А–∞–≤–Њ–Њ—Е—А–∞–љ–µ–љ–Є—П.
–Х—Б–ї–Є –Т—Л –љ–µ —П–≤–ї—П–µ—В–µ—Б—М —Б–њ–µ—Ж–Є–∞–ї–Є—Б—В–Њ–Љ –Ј–і—А–∞–≤–Њ–Њ—Е—А–∞–љ–µ–љ–Є—П, –∞–і–Љ–Є–љ–Є—Б—В—А–∞—Ж–Є—П –љ–µ –љ–µ—Б–µ—В –Њ—В–≤–µ—В—Б—В–≤–µ–љ–љ–Њ—Б—В–Є –Ј–∞ –≤–Њ–Ј–Љ–Њ–ґ–љ—Л–µ –Њ—В—А–Є—Ж–∞—В–µ–ї—М–љ—Л–µ –њ–Њ—Б–ї–µ–і—Б—В–≤–Є—П, –≤–Њ–Ј–љ–Є–Ї—И–Є–µ –≤ —А–µ–Ј—Г–ї—М—В–∞—В–µ —Б–∞–Љ–Њ—Б—В–Њ—П—В–µ–ї—М–љ–Њ–≥–Њ –Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–љ–Є—П –Т–∞–Љ–Є –Є–љ—Д–Њ—А–Љ–∞—Ж–Є–Є —Б –њ–Њ—А—В–∞–ї–∞ –±–µ–Ј –њ—А–µ–і–≤–∞—А–Є—В–µ–ї—М–љ–Њ–є –Ї–Њ–љ—Б—Г–ї—М—В–∞—Ж–Є–Є —Б –≤—А–∞—З–Њ–Љ.
–Э–∞–ґ–Є–Љ–∞—П –љ–∞ –Ї–љ–Њ–њ–Ї—Г ¬Ђ–Т–Њ–є—В–Є¬ї, –Т—Л –њ–Њ–і—В–≤–µ—А–ґ–і–∞–µ—В–µ, —З—В–Њ —П–≤–ї—П–µ—В–µ—Б—М –≤—А–∞—З–Њ–Љ –Є–ї–Є —Б—В—Г–і–µ–љ—В–Њ–Љ –Љ–µ–і–Є—Ж–Є–љ—Б–Ї–Њ–≥–Њ –≤—Г–Ј–∞.