–ü–Ķ—Ä—Ā–Ņ–Ķ–ļ—ā–ł–≤—č –Ņ—Ä–ł–ľ–Ķ–Ĺ–Ķ–Ĺ–ł—Ź —Ä–ĺ—Ā—Ā–ł–Ļ—Ā–ļ–ł—Ö –Ī–ł–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ł—Ö –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–ĺ–≤ –≤ —ā–Ķ—Ä–į–Ņ–ł–ł –∑–Ľ–ĺ–ļ–į—á–Ķ—Ā—ā–≤–Ķ–Ĺ–Ĺ—č—Ö –Ĺ–ĺ–≤–ĺ–ĺ–Ī—Ä–į–∑–ĺ–≤–į–Ĺ–ł–Ļ. –ü–Ķ—ā–Ķ—Ä–Ī—É—Ä–≥—Ā–ļ–ł–Ļ –ĺ–Ĺ–ļ–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ł–Ļ —Ą–ĺ—Ä—É–ľ ¬ę–Ď–Ķ–Ľ—č–Ķ –Ĺ–ĺ—á–ł ‚Äď 2015¬Ľ. –°–į—ā–Ķ–Ľ–Ľ–ł—ā–Ĺ—č–Ļ —Ā–ł–ľ–Ņ–ĺ–∑–ł—É–ľ –ļ–ĺ–ľ–Ņ–į–Ĺ–ł–ł BIOCAD
- –ź–Ĺ–Ĺ–ĺ—ā–į—Ü–ł—Ź
- –°—ā–į—ā—Ć—Ź
- –°—Ā—č–Ľ–ļ–ł






–†–į–∑—Ä–į–Ī–ĺ—ā–ļ–į –ĺ—ā–Ķ—á–Ķ—Ā—ā–≤–Ķ–Ĺ–Ĺ—č—Ö –≤—č—Ā–ĺ–ļ–ĺ—ā–Ķ—Ö–Ĺ–ĺ–Ľ–ĺ–≥–ł—á–Ĺ—č—Ö –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–ĺ–≤ –ļ–į–ļ —Ā–Ņ–ĺ—Ā–ĺ–Ī –Ņ–ĺ–≤—č—ą–Ķ–Ĺ–ł—Ź –ī–ĺ—Ā—ā—É–Ņ–Ĺ–ĺ—Ā—ā–ł –ī–ĺ—Ä–ĺ–≥–ĺ—Ā—ā–ĺ—Ź—Č–ł—Ö –Ľ–Ķ–ļ–į—Ä—Ā—ā–≤–Ķ–Ĺ–Ĺ—č—Ö —Ā—Ä–Ķ–ī—Ā—ā–≤
–ė–Ĺ–Ĺ–ĺ–≤–į—Ü–ł–ĺ–Ĺ–Ĺ–į—Ź –ĺ—ā–Ķ—á–Ķ—Ā—ā–≤–Ķ–Ĺ–Ĺ–į—Ź –Ī–ł–ĺ—Ą–į—Ä–ľ–į—Ü–Ķ–≤—ā–ł—á–Ķ—Ā–ļ–į—Ź –ļ–ĺ–ľ–Ņ–į–Ĺ–ł—Ź –Ņ–ĺ–Ľ–Ĺ–ĺ–≥–ĺ —Ü–ł–ļ–Ľ–į BIOCAD (Biopharmaceutical Company)¬†‚Äď –ĺ–ī–Ĺ–į –ł–∑¬†–Ĺ–Ķ–ľ–Ĺ–ĺ–≥–ł—Ö –ļ–ĺ–ľ–Ņ–į–Ĺ–ł–Ļ –≤¬†–†–ĺ—Ā—Ā–ł–ł, –ļ–ĺ—ā–ĺ—Ä–į—Ź –ĺ–Ī–Ľ–į–ī–į–Ķ—ā –ł–Ĺ—Ą—Ä–į—Ā—ā—Ä—É–ļ—ā—É—Ä–ĺ–Ļ, –Ĺ–Ķ–ĺ–Ī—Ö–ĺ–ī–ł–ľ–ĺ–Ļ –ī–Ľ—Ź –Ņ–ĺ–Ľ–Ĺ–ĺ–≥–ĺ —Ü–ł–ļ–Ľ–į —Ä–į–Ī–ĺ—ā –Ņ–嬆—Ā–ĺ–∑–ī–į–Ĺ–ł—é –ł¬†–Ņ—Ä–ĺ–ł–∑–≤–ĺ–ī—Ā—ā–≤—É –Ņ–Ķ—Ä–≤—č—Ö –≤¬†—Ā—ā—Ä–į–Ĺ–Ķ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–ĺ–≤ –Ĺ–į¬†–ĺ—Ā–Ĺ–ĺ–≤–Ķ –ľ–ĺ–Ĺ–ĺ–ļ–Ľ–ĺ–Ĺ–į–Ľ—Ć–Ĺ—č—Ö –į–Ĺ—ā–ł—ā–Ķ–Ľ –ī–Ľ—Ź –Ľ–Ķ—á–Ķ–Ĺ–ł—Ź –ĺ–Ĺ–ļ–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ł—Ö –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł–Ļ. –í–ł—Ü–Ķ-–Ņ—Ä–Ķ–∑–ł–ī–Ķ–Ĺ—ā –Ņ–嬆–Ī–ł–ĺ–ľ–Ķ–ī–ł—Ü–ł–Ĺ—Ā–ļ–ł–ľ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź–ľ –ł¬†—Ä–į–∑–≤–ł—ā–ł—é –ó–ź–ě¬†¬ę–Ď–ł–ĺ–ļ–į–ī¬Ľ –†–ĺ–ľ–į–Ĺ –ź–Ľ–Ķ–ļ—Ā–Ķ–Ķ–≤–ł—á –ė–í–ź–Ě–ě–í –≤¬†—Ā–≤–ĺ–Ķ–ľ –≤—č—Ā—ā—É–Ņ–Ľ–Ķ–Ĺ–ł–ł –ļ—Ä–į—ā–ļ–ĺ –ĺ—Ö–į—Ä–į–ļ—ā–Ķ—Ä–ł–∑–ĺ–≤–į–Ľ –ĺ—Ā–Ĺ–ĺ–≤–Ĺ—č–Ķ –ĺ—ā–Ľ–ł—á–ł—Ź –Ņ–ĺ–ī—Ö–ĺ–ī–ĺ–≤ –ļ¬†–ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź–ľ –≤–ĺ—Ā–Ņ—Ä–ĺ–ł–∑–≤–Ķ–ī–Ķ–Ĺ–Ĺ—č—Ö –ł¬†–ĺ—Ä–ł–≥–ł–Ĺ–į–Ľ—Ć–Ĺ—č—Ö –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–ĺ–≤, –į¬†—ā–į–ļ–∂–Ķ –Ī–ł–ĺ–į–Ĺ–į–Ľ–ĺ–≥–ĺ–≤.
–ö–į–ļ –ł–∑–≤–Ķ—Ā—ā–Ĺ–ĺ, –Ī–ł–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ł–Ķ –Ľ–Ķ–ļ–į—Ä—Ā—ā–≤–Ķ–Ĺ–Ĺ—č–Ķ —Ā—Ä–Ķ–ī—Ā—ā–≤–į –Ī—č–≤–į—é—ā –ĺ—Ä–ł–≥–ł–Ĺ–į–Ľ—Ć–Ĺ—č–ľ–ł –ł¬†–≤–ĺ—Ā–Ņ—Ä–ĺ–ł–∑–≤–Ķ–ī–Ķ–Ĺ–Ĺ—č–ľ–ł. –í–ĺ—Ā–Ņ—Ä–ĺ–ł–∑–≤–Ķ–ī–Ķ–Ĺ–Ĺ—č–Ķ –Ī–ł–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ł–Ķ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā—č —Ā¬†–ī–ĺ–ļ–į–∑–į–Ĺ–Ĺ–ĺ–Ļ —ć–ļ–≤–ł–≤–į–Ľ–Ķ–Ĺ—ā–Ĺ–ĺ—Ā—ā—Ć—é –ĺ—Ä–ł–≥–ł–Ĺ–į–Ľ—Ć–Ĺ–ĺ–ľ—É –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā—É –Ĺ–į–∑—č–≤–į—é—ā—Ā—Ź –Ī–ł–ĺ–į–Ĺ–į–Ľ–ĺ–≥–į–ľ–ł, –Ņ–ĺ—Ā–ļ–ĺ–Ľ—Ć–ļ—É —ā–ĺ—á–Ĺ—É—é –ļ–ĺ–Ņ–ł—é –Ī–ł–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ĺ–≥–ĺ –Ľ–Ķ–ļ–į—Ä—Ā—ā–≤–Ķ–Ĺ–Ĺ–ĺ–≥–ĺ —Ā—Ä–Ķ–ī—Ā—ā–≤–į —Ā–ĺ–∑–ī–į—ā—Ć –Ĺ–Ķ–≤–ĺ–∑–ľ–ĺ–∂–Ĺ–ĺ. –í¬†–ĺ—ā–Ĺ–ĺ—ą–Ķ–Ĺ–ł–ł –Ī–ł–ĺ–į–Ĺ–į–Ľ–ĺ–≥–ĺ–≤ –Ņ—Ä–ł–ľ–Ķ–Ĺ—Ź—é—ā—Ā—Ź –ĺ—Ā–ĺ–Ī—č–Ķ –Ņ–ĺ–ī—Ö–ĺ–ī—č –ļ¬†–ī–ĺ–ļ–į–∑–į—ā–Ķ–Ľ—Ć—Ā—ā–≤—É –ł—Ö —ć–ļ–≤–ł–≤–į–Ľ–Ķ–Ĺ—ā–Ĺ–ĺ—Ā—ā–ł –ĺ—Ä–ł–≥–ł–Ĺ–į–Ľ—Ć–Ĺ—č–ľ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į–ľ.
–í¬†–Ĺ–į—Ā—ā–ĺ—Ź—Č–Ķ–Ķ –≤—Ä–Ķ–ľ—Ź –í—Ā–Ķ–ľ–ł—Ä–Ĺ–į—Ź –ĺ—Ä–≥–į–Ĺ–ł–∑–į—Ü–ł—Ź –∑–ī—Ä–į–≤–ĺ–ĺ—Ö—Ä–į–Ĺ–Ķ–Ĺ–ł—Ź (–í–ě–ó), –ē–≤—Ä–ĺ–Ņ–Ķ–Ļ—Ā–ļ–ĺ–Ķ –į–≥–Ķ–Ĺ—ā—Ā—ā–≤–ĺ –Ņ–嬆–Ľ–Ķ–ļ–į—Ä—Ā—ā–≤–Ķ–Ĺ–Ĺ—č–ľ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į–ľ (European Medicines Agency¬†‚Äď EMA) –ł¬†–£–Ņ—Ä–į–≤–Ľ–Ķ–Ĺ–ł–Ķ –Ņ–嬆—Ā–į–Ĺ–ł—ā–į—Ä–Ĺ–ĺ–ľ—É –Ĺ–į–ī–∑–ĺ—Ä—É –∑–į –ļ–į—á–Ķ—Ā—ā–≤–ĺ–ľ –Ņ–ł—Č–Ķ–≤—č—Ö –Ņ—Ä–ĺ–ī—É–ļ—ā–ĺ–≤ –ł¬†–ľ–Ķ–ī–ł–ļ–į–ľ–Ķ–Ĺ—ā–ĺ–≤ –°–®–ź (Food and Drug Administration¬†‚Äď FDA) —Ä–į–∑—Ä–į–Ī–ĺ—ā–į–Ľ–ł —Ā–≤–ĺ–ī —Ä–Ķ–ļ–ĺ–ľ–Ķ–Ĺ–ī–į—Ü–ł–Ļ –≤¬†–ĺ—ā–Ĺ–ĺ—ą–Ķ–Ĺ–ł–ł –ĺ–Ī—ä–Ķ–ľ–į –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–Ļ –Ī–ł–ĺ–į–Ĺ–į–Ľ–ĺ–≥–ĺ–≤ –ī–Ľ—Ź –Ņ–ĺ–ī—ā–≤–Ķ—Ä–∂–ī–Ķ–Ĺ–ł—Ź –ł—Ö —Ā–ĺ–ĺ—ā–≤–Ķ—ā—Ā—ā–≤–ł—Ź –ĺ—Ä–ł–≥–ł–Ĺ–į–Ľ—Ć–Ĺ—č–ľ –Ľ–Ķ–ļ–į—Ä—Ā—ā–≤–Ķ–Ĺ–Ĺ—č–ľ —Ā—Ä–Ķ–ī—Ā—ā–≤–į–ľ. –†–Ķ–ļ–ĺ–ľ–Ķ–Ĺ–ī–į—Ü–ł–ł –Ņ—Ä–Ķ–ī—É—Ā–ľ–į—ā—Ä–ł–≤–į—é—ā –Ņ—Ä–Ķ–∂–ī–Ķ –≤—Ā–Ķ–≥–ĺ –Ī–ĺ–Ľ—Ć—ą–ĺ–Ļ –ĺ–Ī—ä–Ķ–ľ —Ā—Ä–į–≤–Ĺ–ł—ā–Ķ–Ľ—Ć–Ĺ—č—Ö –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–Ļ —Ą–ł–∑–ł–ļ–ĺ-—Ö–ł–ľ–ł—á–Ķ—Ā–ļ–ł—Ö –ł¬†–Ī–ł–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ł—Ö —Ā–≤–ĺ–Ļ—Ā—ā–≤.
–ě—Ā–Ĺ–ĺ–≤–Ĺ–ĺ–Ļ –į–ļ—Ü–Ķ–Ĺ—ā –ī–Ķ–Ľ–į–Ķ—ā—Ā—Ź –Ĺ–į¬†—Ā—Ä–į–≤–Ĺ–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ–ľ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–ł —Ö–į—Ä–į–ļ—ā–Ķ—Ä–ł—Ā—ā–ł–ļ –ļ–į—á–Ķ—Ā—ā–≤–į, —Ā–Ņ–ĺ—Ā–ĺ–Ī–Ĺ—č—Ö –ĺ–ļ–į–∑—č–≤–į—ā—Ć –≤–Ľ–ł—Ź–Ĺ–ł–Ķ –Ĺ–į¬†–Ī–ł–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ł–Ķ —Ā–≤–ĺ–Ļ—Ā—ā–≤–į –ľ–ĺ–Ľ–Ķ–ļ—É–Ľ¬†‚Äď —Ā—ā—Ä—É–ļ—ā—É—Ä—É –Ī–Ķ–Ľ–ļ–į, –Ņ–ĺ—Ā—ā—ā—Ä–į–Ĺ—Ā–Ľ—Ź—Ü–ł–ĺ–Ĺ–Ĺ—č–Ķ –ľ–ĺ–ī–ł—Ą–ł–ļ–į—Ü–ł–ł, —á–ł—Ā—ā–ĺ—ā—É –ł¬†–Ņ—Ä–ĺ—Ą–ł–Ľ—Ć –Ņ—Ä–ł–ľ–Ķ—Ā–Ķ–Ļ, —Ą—É–Ĺ–ļ—Ü–ł–ĺ–Ĺ–į–Ľ—Ć–Ĺ—č–Ķ —Ö–į—Ä–į–ļ—ā–Ķ—Ä–ł—Ā—ā–ł–ļ–ł. –ė–ī–Ķ–Ĺ—ā–ł—á–Ĺ–ĺ—Ā—ā—Ć —Ā—ā—Ä—É–ļ—ā—ɗė謆‚Äď —ć—ā–ĺ –Ĺ–Ķ¬†—ā–ĺ–Ľ—Ć–ļ–ĺ –Ņ–ĺ¬≠—Ā–Ľ–Ķ–ī–ĺ–≤–į—ā–Ķ–Ľ—Ć–Ĺ–ĺ—Ā—ā—Ć –į–ľ–ł–Ĺ–ĺ–ļ–ł—Ā–Ľ–ĺ—ā. –≠—ā–ĺ –Ķ—Č–Ķ –ł¬†—Ā–Ľ–ĺ–∂–Ĺ–į—Ź –Ņ—Ä–ĺ—Ā—ā—Ä–į–Ĺ—Ā—ā–≤–Ķ–Ĺ–Ĺ–į—Ź –ĺ—Ä–ł–Ķ–Ĺ—ā–į—Ü–ł—Ź –Ī–Ķ–Ľ–ļ–ĺ–≤—č—Ö —Ü–Ķ–Ņ–Ķ–Ļ –ł¬†–ľ–ĺ–Ľ–Ķ–ļ—É–Ľ, —Ā–ĺ—Ā—ā–į–≤–Ľ—Ź—é—Č–į—Ź –≤—ā–ĺ—Ä–ł—á–Ĺ—É—é –ł¬†—ā—Ä–Ķ—ā–ł—á–Ĺ—É—é —Ā—ā—Ä—É–ļ—ā—É—Ä—č –Ī–Ķ–Ľ–ļ–į. –≠—ā–ĺ —Ö–į—Ä–į–ļ—ā–Ķ—Ä–ł—Ā—ā–ł–ļ–ł, —Ā–≤—Ź–∑–į–Ĺ–Ĺ—č–Ķ —Ā¬†–∑–į—Ä—Ź–ī–ĺ–ľ –Ī–Ķ–Ľ–ļ–ĺ–≤–ĺ–Ļ –ľ–ĺ–Ľ–Ķ–ļ—É–Ľ—č, –ĺ—Ä–ł–Ķ–Ĺ—ā–į—Ü–ł–Ķ–Ļ –≤¬†–Ņ—Ä–ĺ—Ā—ā—Ä–į–Ĺ—Ā—ā–≤–Ķ –ł¬†—ā.–ī. –Ě–Ķ¬†—Ā–Ľ—É—á–į–Ļ–Ĺ–ĺ –ī–Ľ—Ź –ī–ĺ–ļ–į–∑–į—ā–Ķ–Ľ—Ć—Ā—ā–≤–į –ł–ī–Ķ–Ĺ—ā–ł—á–Ĺ–ĺ—Ā—ā–ł —Ā—ā—Ä—É–ļ—ā—É—Ä—č –Ņ—Ä–ł–ľ–Ķ–Ĺ—Ź–Ķ—ā—Ā—Ź –ļ–ĺ–ľ–Ņ–Ľ–Ķ–ļ—Ā –ł–∑¬†–ī–Ķ—Ā—Ź—ā–ļ–ĺ–≤ –ľ–Ķ—ā–ĺ–ī–ĺ–≤, –ī–ĺ–Ņ–ĺ–Ľ–Ĺ—Ź—é—Č–ł—Ö –ī—Ä—É–≥ –ī—Ä—É–≥–į.
–ú–ĺ–Ľ–Ķ–ļ—É–Ľ—č —ā–Ķ—Ä–į–Ņ–Ķ–≤—ā–ł—á–Ķ—Ā–ļ–ł—Ö –Ī–Ķ–Ľ–ļ–ĺ–≤, —Ā–ł–Ĺ—ā–Ķ–∑–ł—Ä—É–Ķ–ľ—č–Ķ –ļ–Ľ–Ķ—ā–ļ–į–ľ–ł –ľ–Ľ–Ķ–ļ–ĺ–Ņ–ł—ā–į—é—Č–ł—Ö, –ł–ľ–Ķ—é—ā —É–Ĺ–ł–ļ–į–Ľ—Ć–Ĺ—č–Ļ —Ā–ĺ—Ā—ā–į–≤ —Ā–į—Ö–į—Ä–ĺ–≤¬†‚Äď —ā–į–ļ –Ĺ–į–∑—č–≤–į–Ķ–ľ—č–Ļ –Ņ—Ä–ĺ—Ą–ł–Ľ—Ƭ†–≥–Ľ–ł–ļ–ĺ–∑–ł–Ľ–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź, —Ö–į—Ä–į–ļ—ā–Ķ—Ä–ł—Ā—ā–ł–ļ–į –ļ–ĺ—ā–ĺ—Ä–ĺ–≥–ĺ –≤–ĺ –ľ–Ĺ–ĺ–≥–ĺ–ľ –∑–į–≤–ł—Ā–ł—ā –ĺ—ā¬†—Ā–Ņ–ĺ—Ā–ĺ–Ī–į –Ņ—Ä–ĺ–ł–∑–≤–ĺ–ī—Ā—ā–≤–į –Ī–Ķ–Ľ–ļ–į, —É—Ā–Ľ–ĺ–≤–ł–Ļ –ļ—É–Ľ—Ć—ā–ł–≤–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź –ļ–Ľ–Ķ—ā–ĺ–ļ-–Ņ—Ä–ĺ–ī—É—Ü–Ķ–Ĺ—ā–ĺ–≤. –ď–Ľ–ł–ļ–ĺ–∑–ł–Ľ–ł—Ä–ĺ–≤–į–Ĺ–ł–Ķ –≤–Ľ–ł—Ź–Ķ—ā –Ĺ–į¬†—ā–į–ļ–ł–Ķ –≤–į–∂–Ĺ—č–Ķ –Ī–ł–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ł–Ķ —Ā–≤–ĺ–Ļ—Ā—ā–≤–į –ľ–ĺ–Ĺ–ĺ–ļ–Ľ–ĺ–Ĺ–į–Ľ—Ć–Ĺ–ĺ–≥–ĺ –į–Ĺ—ā–ł—ā–Ķ–Ľ–į, –ļ–į–ļ —Ü–ł—ā–ĺ—ā–ĺ–ļ—Ā–ł—á–Ĺ–ĺ—Ā—ā—Ć, —Ą–į—Ä–ľ–į–ļ–ĺ–ļ–ł–Ĺ–Ķ—ā–ł–ļ–į, –ł–ľ–ľ—É–Ĺ–ĺ–≥–Ķ–Ĺ–Ĺ–ĺ—Ā—ā—Ć. –ė–ľ–Ķ–Ĺ–Ĺ–ĺ –Ņ–ĺ—ć—ā–ĺ–ľ—É –ī–Ľ—Ź –Ņ–ĺ–ī—ā–≤–Ķ—Ä–∂–ī–Ķ–Ĺ–ł—Ź –ł–ī–Ķ–Ĺ—ā–ł—á–Ĺ–ĺ—Ā—ā–ł –Ņ—Ä–ĺ—Ą–ł–Ľ—Ź¬†–≥–Ľ–ł–ļ–ĺ–∑–ł–Ľ–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź –Ņ—Ä–ł–ľ–Ķ–Ĺ—Ź–Ķ—ā—Ā—Ź —Ü–Ķ–Ľ—č–Ļ –ļ–ĺ–ľ–Ņ–Ľ–Ķ–ļ—Ā –ľ–Ķ—ā–ĺ–ī–ĺ–≤. –Ę—Č–į—ā–Ķ–Ľ—Ć–Ĺ—č–ľ –ĺ–Ī—Ä–į–∑–ĺ–ľ –ļ–ĺ–Ĺ—ā—Ä–ĺ–Ľ–ł—Ä—É—é—ā—Ā—Ź –ł¬†–Ņ—Ä–ł–ľ–Ķ—Ā–ł, –ļ–į–ļ —Ā–≤—Ź–∑–į–Ĺ–Ĺ—č–Ķ —Ā¬†–ī–Ķ–≥—Ä–į–ī–į—Ü–ł–Ķ–Ļ –Ī–Ķ–Ľ–ļ–ĺ–≤–ĺ–Ļ –ľ–ĺ–Ľ–Ķ–ļ—É–Ľ—č, —ā–į–ļ –ł¬†–≤–ĺ–∑–Ĺ–ł–ļ–į—é—Č–ł–Ķ –≤¬†–Ņ—Ä–ĺ—Ü–Ķ—Ā—Ā–Ķ –ĺ—á–ł—Ā—ā–ļ–ł.
–Ē–Ľ—Ź –≤—č—Ź–≤–Ľ–Ķ–Ĺ–ł—Ź –ľ–į–Ľ–Ķ–Ļ—ą–ł—Ö —Ä–į–∑–Ľ–ł—á–ł–Ļ –≤¬†–Ī–ł–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ĺ–Ļ –į–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā–ł –ľ–Ķ–∂–ī—É –Ī–ł–ĺ–į–Ĺ–į–Ľ–ĺ–≥–ĺ–ľ –ł¬†–ĺ—Ä–ł–≥–ł–Ĺ–į–Ľ—Ć–Ĺ—č–ľ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–ĺ–ľ –Ĺ–Ķ–ĺ–Ī—Ö–ĺ–ī–ł–ľ–ĺ –ł—Ā–Ņ–ĺ–Ľ—Ć–∑–ĺ–≤–į—ā—Ć –ī–į–Ĺ–Ĺ—č–Ķ –ľ–Ĺ–ĺ–≥–ĺ—á–ł—Ā–Ľ–Ķ–Ĺ–Ĺ—č—Ö —Ā—Ä–į–≤–Ĺ–ł—ā–Ķ–Ľ—Ć–Ĺ—č—Ö –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–Ļ in¬†vitro.
–Ę–ĺ–Ľ—Ć–ļ–ĺ –Ņ–ĺ—Ā–Ľ–Ķ –ī–ĺ–ļ–į–∑–į–Ĺ–Ĺ–ĺ–≥–ĺ –Ņ–ĺ–Ľ–Ĺ–ĺ–≥–ĺ —Ā–ĺ–ĺ—ā–≤–Ķ—ā—Ā—ā–≤–ł—Ź –≤—Ā–Ķ—Ö —Ö–į—Ä–į–ļ—ā–Ķ—Ä–ł—Ā—ā–ł–ļ –Ī–ł–ĺ–į–Ĺ–į–Ľ–ĺ–≥–į –ĺ—Ä–ł–≥–ł–Ĺ–į–Ľ—Ć–Ĺ–ĺ–ľ—É –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā—É –Ņ—Ä–ĺ–≤–ĺ–ī—Ź—ā—Ā—Ź —Ā—Ä–į–≤–Ĺ–ł—ā–Ķ–Ľ—Ć–Ĺ—č–Ķ –ī–ĺ–ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ł–Ķ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź –Ĺ–į¬†—Ä–Ķ–Ľ–Ķ–≤–į–Ĺ—ā–Ĺ—č—Ö –∂–ł–≤–ĺ—ā–Ĺ—č—Ö, –Ņ–ĺ–ī—Ä–ĺ–Ī–Ĺ—č–Ķ —Ā—Ä–į–≤–Ĺ–ł—ā–Ķ–Ľ—Ć–Ĺ—č–Ķ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź —Ą–į—Ä–ľ–į–ļ–ĺ–ļ–ł–Ĺ–Ķ—ā–ł–ļ–ł –ł¬†—Ą–į—Ä–ľ–į–ļ–ĺ–ī–ł–Ĺ–į–ľ–ł–ļ–ł –Ī–ł–ĺ–į–Ĺ–į–Ľ–ĺ–≥–į. –ó–į—ā–Ķ–ľ –≤—č–Ņ–ĺ–Ľ–Ĺ—Ź—é—ā—Ā—Ź —Ā—Ä–į–≤–Ĺ–ł—ā–Ķ–Ľ—Ć–Ĺ—č–Ķ –ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ł–Ķ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā–ł, –Ī–Ķ–∑–ĺ–Ņ–į—Ā–Ĺ–ĺ—Ā—ā–ł –ł¬†–ł–ľ–ľ—É–Ĺ–ĺ–≥–Ķ–Ĺ–Ĺ–ĺ—Ā—ā–ł –Ī–ł–ĺ–į–Ĺ–į–Ľ–ĺ–≥–į –Ņ–嬆–ĺ–ī–Ĺ–ĺ–ľ—É –Ņ–ĺ–ļ–į–∑–į–Ĺ–ł—é, –Ĺ–į–ł–Ī–ĺ–Ľ–Ķ–Ķ —á—É–≤—Ā—ā–≤–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ–ľ—É –≤¬†–ĺ—ā–Ĺ–ĺ—ą–Ķ–Ĺ–ł–ł –≤—č—Ź–≤–Ľ–Ķ–Ĺ–ł—Ź –≤–ĺ–∑–ľ–ĺ–∂–Ĺ—č—Ö —Ä–į–∑–Ľ–ł—á–ł–Ļ. –ē—Ā–Ľ–ł –ī–ĺ–ļ–į–∑–į–Ĺ–į —ć–ļ–≤–ł–≤–į–Ľ–Ķ–Ĺ—ā–Ĺ–ĺ—Ā—ā—Ć –Ņ–嬆–ĺ–ī–Ĺ–ĺ–ľ—É –Ĺ–į–ł–Ī–ĺ–Ľ–Ķ–Ķ —á—É–≤—Ā—ā–≤–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ–ľ—É –Ņ–ĺ–ļ–į–∑–į–Ĺ–ł—é, —Ā—á–ł—ā–į–Ķ—ā—Ā—Ź, —á—ā–ĺ –Ī–ł–ĺ–į–Ĺ–į–Ľ–ĺ–≥ –ľ–ĺ–∂–Ķ—ā –Ņ—Ä–ł–ľ–Ķ–Ĺ—Ź—ā—Ć—Ā—Ź –ł¬†–Ņ–ĺ –ī—Ä—É–≥–ł–ľ –Ņ–ĺ–ļ–į–∑–į–Ĺ–ł—Ź–ľ, –ĺ–ī–ĺ–Ī—Ä–Ķ–Ĺ–Ĺ—č–ľ –ī–Ľ—Ź –ĺ—Ä–ł–≥–ł–Ĺ–į–Ľ—Ć–Ĺ–ĺ–≥–ĺ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į (—ā–ĺ –Ķ—Ā—ā—Ć –≤–ĺ–∑–ľ–ĺ–∂–Ĺ–į —ć–ļ—Ā—ā—Ä–į–Ņ–ĺ–Ľ—Ź—Ü–ł—Ź –Ņ–ĺ–ļ–į–∑–į–Ĺ–ł–Ļ –ļ¬†–Ņ—Ä–ł–ľ–Ķ–Ĺ–Ķ–Ĺ–ł—é).
–ě—Ā–Ĺ–ĺ–≤–Ĺ–ĺ–Ļ —Ü–Ķ–Ľ—Ć—é —Ā—Ä–į–≤–Ĺ–ł—ā–Ķ–Ľ—Ć–Ĺ—č—Ö –ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ł—Ö –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–Ļ —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź –ī–Ķ–ľ–ĺ–Ĺ—Ā—ā—Ä–į—Ü–ł—Ź –ł–ī–Ķ–Ĺ—ā–ł—á–Ĺ–ĺ—Ā—ā–ł —ā–Ķ—Ä–į–Ņ–Ķ–≤—ā–ł—á–Ķ—Ā–ļ–ł—Ö —ć—Ą—Ą–Ķ–ļ—ā–ĺ–≤ –Ī–ł–ĺ–į–Ĺ–į–Ľ–ĺ–≥–į —ć—Ą—Ą–Ķ–ļ—ā–į–ľ –ĺ—Ä–ł–≥–ł–Ĺ–į–Ľ—Ć–Ĺ–ĺ–≥–ĺ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į.
–ü–ĺ–ī—Ö–ĺ–ī –ļ¬†–≤—č–Ī–ĺ—Ä—É –Ņ–Ķ—Ä–≤–ł—á–Ĺ–ĺ–Ļ –ļ–ĺ–Ĺ–Ķ—á–Ĺ–ĺ–Ļ —ā–ĺ—á–ļ–ł –≤¬†–ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź—Ö –Ī–ł–ĺ–į–Ĺ–į–Ľ–ĺ–≥–į –ĺ—ā–Ľ–ł—á–į–Ķ—ā—Ā—Ź –ĺ—ā¬†—ā–į–ļ–ĺ–≤–ĺ–≥–ĺ –≤¬†–ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź—Ö –ĺ—Ä–ł–≥–ł–Ĺ–į–Ľ—Ć–Ĺ–ĺ–≥–ĺ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į¬†‚Äď –≤¬†–ĺ–Ĺ–ļ–ĺ–Ľ–ĺ–≥–ł–ł –Ĺ–į–ł–Ī–ĺ–Ľ–Ķ–Ķ –Ņ—Ä–ł–Ķ–ľ–Ľ–Ķ–ľ–į —á–į—Ā—ā–ĺ—ā–į –ĺ—ā–≤–Ķ—ā–į. –Ę–ĺ—á–ļ–ł, –ļ–ĺ—ā–ĺ—Ä—č–Ķ –ł—Ā–Ņ–ĺ–Ľ—Ć–∑—É—é—ā—Ā—Ź –≤¬†–ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź—Ö –ĺ—Ä–ł–≥–ł–Ĺ–į–Ľ—Ć–Ĺ—č—Ö –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–ĺ–≤ (–ĺ–Ī—Č–į—Ź –≤—č–∂–ł–≤–į–Ķ–ľ–ĺ—Ā—ā—Ć, –≤—č–∂–ł–≤–į–Ķ–ľ–ĺ—Ā—ā—Ć, —Ā–≤–ĺ–Ī–ĺ–ī–Ĺ–į—Ź –ĺ—ā¬†–Ņ—Ä–ĺ–≥—Ä–Ķ—Ā—Ā–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź), –ľ–Ķ–Ĺ–Ķ–Ķ —á—É–≤—Ā—ā–≤–ł—ā–Ķ–Ľ—Ć–Ĺ—č –ī–Ľ—Ź –≤—č—Ź–≤–Ľ–Ķ–Ĺ–ł—Ź —Ä–į–∑–Ľ–ł—á–ł–Ļ –ľ–Ķ–∂–ī—É –Ī–ł–ĺ–į–Ĺ–į–Ľ–ĺ–≥–į–ľ–ł –ł¬†–ĺ—Ä–ł–≥–ł–Ĺ–į–Ľ—Ć–Ĺ—č–ľ–ł –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į–ľ–ł.
–í–į–∂–Ĺ—č–ľ –į—Ā–Ņ–Ķ–ļ—ā–ĺ–ľ –≤¬†–ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź—Ö –Ī–ł–ĺ–į–Ĺ–į–Ľ–ĺ–≥–ĺ–≤ —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź –ł–∑—É—á–Ķ–Ĺ–ł–Ķ –ł—Ö –Ņ—Ä–ĺ—Ą–ł–Ľ—Ź –Ī–Ķ–∑–ĺ–Ņ–į—Ā–Ĺ–ĺ—Ā—ā–ł, –ļ–ĺ—ā–ĺ—Ä—č–Ļ –ī–ĺ–Ľ–∂–Ķ–Ĺ —Ā–ĺ–ĺ—ā–≤–Ķ—ā—Ā—ā–≤–ĺ–≤–į—ā—Ć —ā–į–ļ–ĺ–≤–ĺ–ľ—É –ĺ—Ä–ł–≥–ł–Ĺ–į–Ľ—Ć–Ĺ–ĺ–≥–ĺ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į –ł¬†–ī–Ķ–ľ–ĺ–Ĺ—Ā—ā—Ä–ł—Ä–ĺ–≤–į—ā—Ć –ĺ—ā—Ā—É—ā—Ā—ā–≤–ł–Ķ –Ĺ–Ķ—Ö–į—Ä–į–ļ—ā–Ķ—Ä–Ĺ—č—Ö –ī–Ľ—Ź –ĺ—Ä–ł–≥–ł–Ĺ–į–Ľ—Ć–Ĺ–ĺ–≥–ĺ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į –Ņ–ĺ–Ī–ĺ—á–Ĺ—č—Ö —ć—Ą—Ą–Ķ–ļ—ā–ĺ–≤.
–í¬†–†–ĺ—Ā—Ā–ł–ł –ī–ĺ 2010¬†–≥.¬†–ĺ—ā—Ā—É—ā—Ā—ā–≤–ĺ–≤–į–Ľ–ł —á–Ķ—ā–ļ–ł–Ķ —ā—Ä–Ķ–Ī–ĺ–≤–į–Ĺ–ł—Ź –ļ¬†–ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ĺ–ľ—É –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—é –Ī–ł–ĺ–į–Ĺ–į–Ľ–ĺ–≥–ĺ–≤. –í¬†—Ä–Ķ–∑—É–Ľ—Ć—ā–į—ā–Ķ –Ĺ–į¬†–ĺ—ā–Ķ—á–Ķ—Ā—ā–≤–Ķ–Ĺ–Ĺ–ĺ–ľ —Ą–į—Ä–ľ–į—Ü–Ķ–≤—ā–ł—á–Ķ—Ā–ļ–ĺ–ľ —Ä—č–Ĺ–ļ–Ķ –Ņ–ĺ—Ź–≤–ł–Ľ–ł—Ā—Ć —ā–į–ļ –Ĺ–į–∑—č–≤–į–Ķ–ľ—č–Ķ copy biologics¬†‚Äď –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā—č, –ļ–ĺ—ā–ĺ—Ä—č–Ķ –Ĺ–Ķ–Ľ—Ć–∑—Ź –Ĺ–į–∑–≤–į—ā—Ć –Ī–ł–ĺ–į–Ĺ–į–Ľ–ĺ–≥–į–ľ–ł –ł–∑-–∑–į –ĺ—ā—Ā—É—ā—Ā—ā–≤–ł—Ź –ī–į–Ĺ–Ĺ—č—Ö —Ā—Ä–į–≤–Ĺ–ł—ā–Ķ–Ľ—Ć–Ĺ—č—Ö –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–Ļ. –°¬†—Ā–Ķ–Ĺ—ā—Ź–Ī—Ä—Ź 2010¬†–≥.¬†—Ä–ĺ—Ā—Ā–ł–Ļ—Ā–ļ–ł–ľ –∑–į–ļ–ĺ–Ĺ–ĺ–ī–į—ā–Ķ–Ľ—Ć—Ā—ā–≤–ĺ–ľ –∑–į–ļ—Ä–Ķ–Ņ–Ľ–Ķ–Ĺ–ĺ –ĺ–Ī—Ź–∑–į—ā–Ķ–Ľ—Ć–Ĺ–ĺ–Ķ –Ņ—Ä–ĺ–≤–Ķ–ī–Ķ–Ĺ–ł–Ķ —Ā—Ä–į–≤–Ĺ–ł—ā–Ķ–Ľ—Ć–Ĺ—č—Ö –ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ł—Ö –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–Ļ. –í¬†2014¬†–≥.¬†–≤—č—ą–Ľ–ĺ –≤¬†—Ā–≤–Ķ—ā –†—É–ļ–ĺ–≤–ĺ–ī—Ā—ā–≤–ĺ –Ņ–嬆—ć–ļ—Ā–Ņ–Ķ—Ä—ā–ł–∑–Ķ –Ľ–Ķ–ļ–į—Ä—Ā—ā–≤–Ķ–Ĺ–Ĺ—č—Ö —Ā—Ä–Ķ–ī—Ā—ā–≤ –§–ď–Ď–£ ¬ę–Ě–¶–≠–°–ú–ü¬Ľ –ú–ł–Ĺ–∑–ī—Ä–į–≤–į –†–ĺ—Ā—Ā–ł–ł, –≤¬†–ļ–ĺ—ā–ĺ—Ä–ĺ–ľ —ā—Ä–Ķ–Ī–ĺ–≤–į–Ĺ–ł—Ź –ļ¬†–ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź–ľ –Ī–ł–ĺ–į–Ĺ–į–Ľ–ĺ–≥–ĺ–≤ —Ā–ĺ–ĺ—ā–≤–Ķ—ā—Ā—ā–≤—É—é—ā —Ä–Ķ–ļ–ĺ–ľ–Ķ–Ĺ–ī–į—Ü–ł—Ź–ľ –í–ě–ó, EMA –ł¬†FDA. –°¬†1¬†—Ź–Ĺ–≤–į—Ä—Ź 2016¬†–≥.¬†–≤—Ā—ā—É–Ņ—Ź—ā –≤¬†—Ā–ł–Ľ—É –ü—Ä–į–≤–ł–Ľ–į –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–Ļ –Ī–ł–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ł—Ö –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–ĺ–≤ –ē–≤—Ä–į–∑–ł–Ļ—Ā–ļ–ĺ–≥–ĺ —ć–ļ–ĺ–Ĺ–ĺ–ľ–ł—á–Ķ—Ā–ļ–ĺ–≥–ĺ —Ā–ĺ—é–∑–į, –ļ–ĺ—ā–ĺ—Ä—č–Ķ –Ņ—Ä–Ķ–ī—Ā—ā–į–≤–Ľ—Ź—é—ā –į—É—ā–Ķ–Ĺ—ā–ł—á–Ĺ—č–Ļ –Ņ–Ķ—Ä–Ķ–≤–ĺ–ī –≤—Ā–Ķ—Ö —Ä–Ķ–ļ–ĺ–ľ–Ķ–Ĺ–ī–į—Ü–ł–Ļ –ē–≤—Ä–ĺ—Ā–ĺ—é–∑–į.
–ü–嬆—Ā–Ľ–ĺ–≤–į–ľ –†.–ź. –ė–≤–į–Ĺ–ĺ–≤–į, —Ą–į–ļ—ā–ł—á–Ķ—Ā–ļ–ł –∑–į–≤–Ķ—Ä—ą–Ķ–Ĺ–į —Ä–į–Ī–ĺ—ā–į –Ņ–嬆–Ņ—Ä–ł–≤–Ķ–ī–Ķ–Ĺ–ł—é —Ä–ĺ—Ā—Ā–ł–Ļ—Ā–ļ–ł—Ö —ā—Ä–Ķ–Ī–ĺ–≤–į–Ĺ–ł–Ļ –ļ¬†–ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź–ľ –Ī–ł–ĺ–į–Ĺ–į–Ľ–ĺ–≥–ĺ–≤ –≤¬†—Ā–ĺ–ĺ—ā–≤–Ķ—ā—Ā—ā–≤–ł–Ķ —Ā¬†–ľ–Ķ–∂–ī—É–Ĺ–į—Ä–ĺ–ī–Ĺ—č–ľ–ł —Ā—ā–į–Ĺ–ī–į—Ä—ā–į–ľ–ł.
–í¬†—Ā—ā—Ä–į–Ĺ–Ķ –∑–į–≤–Ķ—Ä—ą–ł–Ľ–į—Ā—Ć –ł¬†–Ī–ĺ–Ľ—Ć—ą–į—Ź —Ä–į–Ī–ĺ—ā–į –Ņ–嬆—Ā–ĺ–∑–ī–į–Ĺ–ł—é –ü—Ä–į–≤–ł–Ľ –ĺ—Ä–≥–į–Ĺ–ł–∑–į—Ü–ł–ł –Ņ—Ä–ĺ–ł–∑–≤–ĺ–ī—Ā—ā–≤–į –ł¬†–ļ–ĺ–Ĺ—ā—Ä–ĺ–Ľ—Ź –ļ–į—á–Ķ—Ā—ā–≤–į –Ľ–Ķ–ļ–į—Ä—Ā—ā–≤–Ķ–Ĺ–Ĺ—č—Ö —Ā—Ä–Ķ–ī—Ā—ā–≤, –ļ–ĺ—ā–ĺ—Ä—č–Ķ —Ā–ĺ–ĺ—ā–≤–Ķ—ā—Ā—ā–≤—É—é—ā –Ķ–≤—Ä–ĺ–Ņ–Ķ–Ļ—Ā–ļ–ł–ľ –Ņ—Ä–į–≤–ł–Ľ–į–ľ GMP. ¬ę–ö–ĺ–ľ–Ņ–į–Ĺ–ł—Ź BIOCAD –ĺ–ī–Ĺ–ĺ–Ļ –ł–∑¬†–Ņ–Ķ—Ä–≤—č—Ö –≤¬†—ć—ā–ĺ–ľ¬†–≥–ĺ–ī—É –Ņ–ĺ–Ľ—É—á–ł–Ľ–į –∑–į–ļ–Ľ—é—á–Ķ–Ĺ–ł–Ķ –嬆—Ā–ĺ–ĺ—ā–≤–Ķ—ā—Ā—ā–≤–ł–ł –Ņ—Ä–ĺ–ł–∑–≤–ĺ–ī–ł—ā–Ķ–Ľ—Ź –Ľ–Ķ–ļ–į—Ä—Ā—ā–≤–Ķ–Ĺ–Ĺ—č—Ö —Ā—Ä–Ķ–ī—Ā—ā–≤ —ā—Ä–Ķ–Ī–ĺ–≤–į–Ĺ–ł—Ź–ľ —Ā—ā–į–Ĺ–ī–į—Ä—ā–į –Ĺ–į–ī–Ľ–Ķ–∂–į—Č–Ķ–Ļ –Ņ—Ä–ĺ–ł–∑–≤–ĺ–ī—Ā—ā–≤–Ķ–Ĺ–Ĺ–ĺ–Ļ –Ņ—Ä–į–ļ—ā–ł–ļ–ł¬Ľ,¬†‚Äď –ļ–ĺ–Ĺ—Ā—ā–į—ā–ł—Ä–ĺ–≤–į–Ľ –†.–ź.¬†–ė–≤–į–Ĺ–ĺ–≤.
–ú–Ķ–∂–ī—É —ā–Ķ–ľ –≤–ĺ –ľ–Ĺ–ĺ–≥–ł—Ö —Ā—ā—Ä–į–Ĺ–į—Ö –Ņ–ĺ–ļ–į –Ņ–ĺ–Ľ–Ĺ–ĺ—Ā—ā—Ć—é –Ĺ–Ķ¬†—Ä–Ķ—ą–Ķ–Ĺ—č –≤–ĺ–Ņ—Ä–ĺ—Ā—č –≤–∑–į–ł–ľ–ĺ–∑–į–ľ–Ķ–Ĺ—Ź–Ķ–ľ–ĺ—Ā—ā–ł –ĺ—Ä–ł–≥–ł–Ĺ–į–Ľ—Ć–Ĺ—č—Ö –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–ĺ–≤ –ł¬†–Ī–ł–ĺ–į–Ĺ–į–Ľ–ĺ–≥–ĺ–≤. –í—Ā–Ķ —á–į—Č–Ķ –∑–≤—É—á–ł—ā –ľ–Ĺ–Ķ–Ĺ–ł–Ķ, —á—ā–ĺ, –Ķ—Ā–Ľ–ł –Ī–ł–ĺ–į–Ĺ–į–Ľ–ĺ–≥ –Ņ—Ä–ĺ—ą–Ķ–Ľ –≤—Ā–Ķ –Ĺ–Ķ–ĺ–Ī—Ö–ĺ–ī–ł–ľ—č–Ķ —Ā—Ä–į–≤–Ĺ–ł—ā–Ķ–Ľ—Ć–Ĺ—č–Ķ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź, –ĺ—Ä–ł–≥–ł–Ĺ–į–Ľ—Ć–Ĺ—č–Ļ –Ī–ł–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ł–Ļ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā –ľ–ĺ–∂–Ķ—ā –Ī—č—ā—Ć –∑–į–ľ–Ķ–Ĺ–Ķ–Ĺ –Ī–ł–ĺ–į–Ĺ–į–Ľ–ĺ–≥–ĺ–ľ.
–í¬†–Ĺ–į—Ā—ā–ĺ—Ź—Č–Ķ–Ķ –≤—Ä–Ķ–ľ—Ź –≤–ĺ –§—Ä–į–Ĺ—Ü–ł–ł –∑–į–ļ–ĺ–Ĺ–ĺ–ī–į—ā–Ķ–Ľ—Ć–Ĺ–ĺ –∑–į–ļ—Ä–Ķ–Ņ–Ľ–Ķ–Ĺ–į –≤–ĺ–∑–ľ–ĺ–∂–Ĺ–ĺ—Ā—ā—Ć –į–≤—ā–ĺ–ľ–į—ā–ł—á–Ķ—Ā–ļ–ĺ–Ļ –∑–į–ľ–Ķ–Ĺ—č –ĺ—Ä–ł–≥–ł–Ĺ–į–Ľ—Ć–Ĺ—č—Ö –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–ĺ–≤ –Ī–ł–ĺ–į–Ĺ–į–Ľ–ĺ–≥–į–ľ–ł —ɬ†–Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤, –Ĺ–į—á–ł–Ĺ–į—é—Č–ł—Ö –Ľ–Ķ—á–Ķ–Ĺ–ł–Ķ –Ī–ł–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ł–ľ–ł –Ľ–Ķ–ļ–į—Ä—Ā—ā–≤–Ķ–Ĺ–Ĺ—č–ľ–ł —Ā—Ä–Ķ–ī—Ā—ā–≤–į–ľ–ł. –ü–ĺ–ī–ĺ–Ī–Ĺ—č–Ļ –Ņ–ĺ—Ä—Ź–ī–ĺ–ļ –ī–Ķ–Ļ—Ā—ā–≤—É–Ķ—ā –ł¬†–≤ –Ī–ĺ–Ľ—Ć—ą–ł–Ĺ—Ā—ā–≤–Ķ —Ā–ļ–į–Ĺ–ī–ł–Ĺ–į–≤—Ā–ļ–ł—Ö —Ā—ā—Ä–į–Ĺ.
–í¬†—Ā–ł–Ľ—É —Ā–Ņ–Ķ—Ü–ł—Ą–ł—á–Ĺ–ĺ—Ā—ā–ł —Ā–ł—Ā—ā–Ķ–ľ—謆–≥–ĺ—Ā—É–ī–į—Ä—Ā—ā–≤–Ķ–Ĺ–Ĺ—č—Ö –∑–į–ļ—É–Ņ–ĺ–ļ –≤¬†–†–ĺ—Ā—Ā–ł–ł —ɬ†–ĺ—ā–Ķ—á–Ķ—Ā—ā–≤–Ķ–Ĺ–Ĺ—č—Ö –≤—Ä–į—á–Ķ–Ļ –ľ–Ķ–Ĺ—Ć—ą–Ķ —Ā–≤–ĺ–Ī–ĺ–ī—č –≤—č–Ī–ĺ—Ä–į –≤¬†–Ĺ–į–∑–Ĺ–į—á–Ķ–Ĺ–ł–ł –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į. –ú–ł–Ĺ–∑–ī—Ä–į–≤ –†–ĺ—Ā—Ā–ł–ł —Ä–į–∑—Ä–į–Ī–ĺ—ā–į–Ľ –Ņ—Ä–ĺ–Ķ–ļ—ā –Ņ–ĺ—Ā—ā–į–Ĺ–ĺ–≤–Ľ–Ķ–Ĺ–ł—Ź –嬆–Ņ—Ä–ĺ—Ü–Ķ–ī—É—Ä–Ķ —É—Ā—ā–į–Ĺ–ĺ–≤–Ľ–Ķ–Ĺ–ł—Ź –≤–∑–į–ł–ľ–ĺ–∑–į–ľ–Ķ–Ĺ—Ź–Ķ–ľ–ĺ—Ā—ā–ł –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–ĺ–≤. –Ē–ĺ–ļ—É–ľ–Ķ–Ĺ—ā –≤—Ā—ā—É–Ņ–ł—ā –≤¬†—Ā–ł–Ľ—É –≤¬†2016¬†–≥.¬†–í¬†–Ī–Ľ–ł–∂–į–Ļ—ą–ł–Ķ —ā—Ä–ł¬†–≥–ĺ–ī–į –≤—Ā–Ķ —Ä–į–Ĺ–Ķ–Ķ –∑–į—Ä–Ķ–≥–ł—Ā—ā—Ä–ł—Ä–ĺ–≤–į–Ĺ–Ĺ—č–Ķ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā—č –Ņ—Ä–ĺ–Ļ–ī—É—ā –Ņ—Ä–ĺ–≤–Ķ—Ä–ļ—É –Ĺ–į¬†—Ā–ĺ–ĺ—ā–≤–Ķ—ā—Ā—ā–≤–ł–Ķ –∂–Ķ—Ā—ā–ļ–ł–ľ —Ā–ĺ–≤—Ä–Ķ–ľ–Ķ–Ĺ–Ĺ—č–ľ —ā—Ä–Ķ–Ī–ĺ–≤–į–Ĺ–ł—Ź–ľ. –°¬†2018¬†–≥.¬†—ɬ†—É—á—Ä–Ķ–∂–ī–Ķ–Ĺ–ł–Ļ –∑–ī—Ä–į–≤–ĺ–ĺ—Ö—Ä–į–Ĺ–Ķ–Ĺ–ł—Ź –Ņ–ĺ—Ź–≤–ł—ā—Ā—Ź –≤–ĺ–∑–ľ–ĺ–∂–Ĺ–ĺ—Ā—ā—Ć –∑–į–ļ—É–Ņ–į—ā—Ć –Ľ–Ķ–ļ–į—Ä—Ā—ā–≤–Ķ–Ĺ–Ĺ—č–Ķ —Ā—Ä–Ķ–ī—Ā—ā–≤–į —ā–ĺ–Ľ—Ć–ļ–ĺ —Ā¬†–ī–ĺ–ļ–į–∑–į–Ĺ–Ĺ–ĺ–Ļ –≤–∑–į–ł–ľ–ĺ–∑–į–ľ–Ķ–Ĺ—Ź–Ķ–ľ–ĺ—Ā—ā—Ć—é —Ä–Ķ—Ą–Ķ—Ä–Ķ–Ĺ—ā–Ĺ–ĺ–ľ—É –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā—É.
–ü–ĺ—Ź–≤–Ľ–Ķ–Ĺ–ł–Ķ –Ĺ–į¬†–ĺ—ā–Ķ—á–Ķ—Ā—ā–≤–Ķ–Ĺ–Ĺ–ĺ–ľ —Ą–į—Ä–ľ–į—Ü–Ķ–≤—ā–ł—á–Ķ—Ā–ļ–ĺ–ľ —Ä—č–Ĺ–ļ–Ķ –ļ–į—á–Ķ—Ā—ā–≤–Ķ–Ĺ–Ĺ—č—Ö –ł¬†–Ī–Ķ–∑–ĺ–Ņ–į—Ā–Ĺ—č—Ö –Ī–ł–ĺ–į–Ĺ–į–Ľ–ĺ–≥–ĺ–≤¬†‚Äď –Ņ–ĺ–∑–ł—ā–ł–≤–Ĺ–į—Ź —ā–Ķ–Ĺ–ī–Ķ–Ĺ—Ü–ł—Ź. –Ě–į–Ņ—Ä–ł–ľ–Ķ—Ä, –Ņ–ĺ—Ā–Ľ–Ķ –Ņ–ĺ—Ź–≤–Ľ–Ķ–Ĺ–ł—Ź –Ņ–Ķ—Ä–≤–ĺ–≥–ĺ –Ī–ł–ĺ–į–Ĺ–į–Ľ–ĺ–≥–į¬†–≥—Ä–į–Ĺ—É–Ľ–ĺ—Ü–ł—ā–į—Ä–Ĺ–ĺ–≥–ĺ –ļ–ĺ–Ľ–ĺ–Ĺ–ł–Ķ—Ā—ā–ł–ľ—É–Ľ–ł—Ä—É—é—Č–Ķ–≥–ĺ —Ą–į–ļ—ā–ĺ—Ä–į (–Ņ—Ä–Ķ–Ņ–į—Ä–į—ā –õ–Ķ–Ļ–ļ–ĺ—Ā—ā–ł–ľ¬ģ –ļ–ĺ–ľ–Ņ–į–Ĺ–ł–ł BIOCAD) —Ā—Ä–Ķ–ī–Ĺ—Ź—Ź —Ā—ā–ĺ–ł–ľ–ĺ—Ā—ā—Ć –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–ĺ–≤ —ć—ā–ĺ–Ļ¬†–≥—Ä—É–Ņ–Ņ—č —Ā–Ĺ–ł–∑–ł–Ľ–į—Ā—Ć –Ī–ĺ–Ľ–Ķ–Ķ —á–Ķ–ľ –≤¬†–ī–≤–į —Ä–į–∑–į, –į¬†–Ņ–ĺ—ā—Ä–Ķ–Ī–Ľ–Ķ–Ĺ–ł–Ķ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–ĺ–≤ –ļ–ĺ–Ľ–ĺ–Ĺ–ł–Ķ—Ā—ā–ł–ľ—É–Ľ–ł—Ä—É—é—Č–Ķ–≥–ĺ —Ą–į–ļ—ā–ĺ—Ä–į –≤–ĺ–∑—Ä–ĺ—Ā–Ľ–ĺ –≤¬†—ā—Ä–ł —Ä–į–∑–į. –ʖ嬆–Ķ—Ā—ā—Ć –≤¬†—ā—Ä–ł —Ä–į–∑–į –Ī–ĺ–Ľ—Ć—ą–Ķ –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ –Ĺ–į—á–į–Ľ–ł –Ņ–ĺ–Ľ—É—á–į—ā—Ć —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ–Ķ –Ľ–Ķ—á–Ķ–Ĺ–ł–Ķ.
–Ē—Ä—É–≥–ĺ–Ļ –Ņ—Ä–ł–ľ–Ķ—Ĭ†‚Äď –Ī–ł–ĺ–į–Ĺ–į–Ľ–ĺ–≥ —Ä–ł—ā—É–ļ—Ā–ł–ľ–į–Ī–į¬†‚Äď –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā –ź—Ü–Ķ–Ľ–Ľ–Ī–ł—Ź (—Ä–ł—ā—É–ļ—Ā–ł–ľ–į–Ī), –Ņ—Ä–ĺ–ł–∑–≤–Ķ–ī–Ķ–Ĺ–Ĺ—č–Ļ –ļ–ĺ–ľ–Ņ–į–Ĺ–ł–Ķ–Ļ BIOCAD,¬†‚Äď –Ņ–Ķ—Ä–≤—č–Ļ —Ä–ĺ—Ā—Ā–ł–Ļ—Ā–ļ–ł–Ļ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā –ľ–ĺ–Ĺ–ĺ–ļ–Ľ–ĺ–Ĺ–į–Ľ—Ć–Ĺ—č—Ö –į–Ĺ—ā–ł—ā–Ķ–Ľ. –Ď–ĺ–Ľ–Ķ–Ķ –Ĺ–ł–∑–ļ–į—Ź —Ā—ā–ĺ–ł–ľ–ĺ—Ā—ā—Ć –ĺ—ā–Ķ—á–Ķ—Ā—ā–≤–Ķ–Ĺ–Ĺ–ĺ–≥–ĺ –Ī–ł–ĺ–į–Ĺ–į–Ľ–ĺ–≥–į –Ņ–嬆—Ā—Ä–į–≤–Ĺ–Ķ–Ĺ–ł—é —Ā¬†–ĺ—Ä–ł–≥–ł–Ĺ–į–Ľ—Ć–Ĺ—č–ľ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–ĺ–ľ (–Ĺ–į¬†23%) –Ņ–ĺ–∑–≤–ĺ–Ľ–ł–Ľ–į¬†–≥–ĺ—Ā—É–ī–į—Ä—Ā—ā–≤—É —Ā—ć–ļ–ĺ–Ĺ–ĺ–ľ–ł—ā—Ć —Ā–≤—č—ą–Ķ 30 –ľ–Ľ–Ĺ –ī–ĺ–Ľ–Ľ–į—Ä–ĺ–≤ –°–®–ź. –í¬†–Ĺ–į—Ā—ā–ĺ—Ź—Č–Ķ–Ķ –≤—Ä–Ķ–ľ—Ź –Ī–ĺ–Ľ–Ķ–Ķ 1000 –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ –Ņ–Ķ—Ä–Ķ—ą–Ľ–ł –Ĺ–į¬†–ł—Ā–Ņ–ĺ–Ľ—Ć–∑–ĺ–≤–į–Ĺ–ł–Ķ —Ä–ĺ—Ā—Ā–ł–Ļ—Ā–ļ–ĺ–≥–ĺ –Ī–ł–ĺ–į–Ĺ–į–Ľ–ĺ–≥–į —Ä–ł—ā—É–ļ—Ā–ł–ľ–į–Ī–į. –ü—Ä–ł —ć—ā–ĺ–ľ —Ā–ł–≥–Ĺ–į–Ľ–ĺ–≤ –嬆—Ä–į–∑–Ľ–ł—á–ł—Ź—Ö –≤¬†–Ņ—Ä–ĺ—Ą–ł–Ľ–Ķ –Ī–Ķ–∑–ĺ–Ņ–į—Ā–Ĺ–ĺ—Ā—ā–ł –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–ĺ–≤ –Ĺ–Ķ¬†–∑–į—Ä–Ķ–≥–ł—Ā—ā—Ä–ł—Ä–ĺ–≤–į–Ĺ–ĺ.
–í¬†–Ī–Ľ–ł–∂–į–Ļ—ą–Ķ–ľ –Ī—É–ī—É—Č–Ķ–ľ –Ĺ–į¬†—Ä–ĺ—Ā—Ā–ł–Ļ—Ā–ļ–ĺ–ľ —Ą–į—Ä–ľ–į—Ü–Ķ–≤—ā–ł—á–Ķ—Ā–ļ–ĺ–ľ —Ä—č–Ĺ–ļ–Ķ –Ņ–ĺ—Ź–≤–ł—ā—Ā—Ź —Ü–Ķ–Ľ—č–Ļ —Ä—Ź–ī –Ņ—Ä–ĺ–ł–∑–≤–Ķ–ī–Ķ–Ĺ–Ĺ—č—Ö –ļ–ĺ–ľ–Ņ–į–Ĺ–ł–Ķ–Ļ BIOCAD –Ī–ł–ĺ–į–Ĺ–į–Ľ–ĺ–≥–ĺ–≤ (—Ä–ł—Ā.¬†1).
–ó–į–≤–Ķ—Ä—ą–į–Ķ—ā—Ā—Ź –Ņ—Ä–ĺ—Ü–Ķ–ī—É—Ä–į —Ä–Ķ–≥–ł—Ā—ā—Ä–į—Ü–ł–ł –Ī–ł–ĺ–į–Ĺ–į–Ľ–ĺ–≥–į –Ī–Ķ–≤–į—Ü–ł–∑—É–ľ–į–Ī–į, –Ņ—Ä–ĺ—Ö–ĺ–ī–ł—ā –Ņ—Ä–ĺ—Ü–Ķ–ī—É—Ä—É —Ä–Ķ–≥–ł—Ā—ā—Ä–į—Ü–ł–ł –Ī–ł–ĺ–į–Ĺ–į–Ľ–ĺ–≥ —ā—Ä–į—Ā—ā—É–∑—É–ľ–į–Ī–į, –ĺ—Ā—É—Č–Ķ—Ā—ā–≤–Ľ—Ź—é—ā—Ā—Ź –ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ł–Ķ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź —Ä–į–∑–Ĺ—č—Ö —Ą–į–∑ –Ī–ł–ĺ–į–Ĺ–į–Ľ–ĺ–≥–ĺ–≤ –ł–Ĺ—ā–Ķ—Ä—Ą–Ķ—Ä–ĺ–Ĺ–į –Ī–Ķ—ā–į-1–į, –Ņ—ć–≥–ł–Ĺ—ā–Ķ—Ä—Ą–Ķ—Ä–ĺ–Ĺ–į –į–Ľ—Ć—Ą–į-2–į, –ī–į—Ä–Ī—ć–Ņ–ĺ—ć—ā–ł–Ĺ–į, –ł–Ĺ—Ą–Ľ–ł–ļ—Ā–ł–ľ–į–Ī–į, –į–ī–į–Ľ–ł–ľ—É–ľ–į–Ī–į.
–í–ľ–Ķ—Ā—ā–Ķ —Ā¬†—ā–Ķ–ľ, –ļ–į–ļ –ĺ—ā–ľ–Ķ—ā–ł–Ľ –†.–ź. –ė–≤–į–Ĺ–ĺ–≤, –Ņ—Ä–ĺ–ł–∑–≤–ĺ–ī—Ā—ā–≤–ĺ –Ī–ł–ĺ–į–Ĺ–į–Ľ–ĺ–≥–ĺ–≤¬†‚Äď —ā–ĺ–Ľ—Ć–ļ–ĺ –Ņ–Ķ—Ä–≤—č–Ļ —ć—ā–į–Ņ —Ä–į–∑–≤–ł—ā–ł—Ź –ļ–ĺ–ľ–Ņ–į–Ĺ–ł–ł BIOCAD. –ē–Ķ –Ī—É–ī—É—Č–Ķ–Ķ —Ā–≤—Ź–∑–į–Ĺ–ĺ —Ā¬†—Ä–į–∑—Ä–į–Ī–ĺ—ā–ļ–ĺ–Ļ –ĺ—Ä–ł–≥–ł–Ĺ–į–Ľ—Ć–Ĺ—č—Ö –Ľ–Ķ–ļ–į—Ä—Ā—ā–≤–Ķ–Ĺ–Ĺ—č—Ö —Ā—Ä–Ķ–ī—Ā—ā–≤. –°–Ķ–Ļ—á–į—Ā –≤¬†—Ä–į–∑—Ä–į–Ī–ĺ—ā–ļ–Ķ –ł¬†–Ĺ–į —Ä–į–∑–Ĺ—č—Ö —Ā—ā–į–ī–ł—Ź—Ö –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–Ļ –Ĺ–į—Ö–ĺ–ī—Ź—ā—Ā—Ź 11 –ł–Ĺ–Ĺ–ĺ–≤–į—Ü–ł–ĺ–Ĺ–Ĺ—č—Ö –Ľ–Ķ–ļ–į—Ä—Ā—ā–≤–Ķ–Ĺ–Ĺ—č—Ö —Ā—Ä–Ķ–ī—Ā—ā–≤ (—Ä–ł—Ā.¬†2).
¬ę–í¬†–Ĺ–į—Ā—ā–ĺ—Ź—Č–Ķ–Ķ –≤—Ä–Ķ–ľ—Ź –ľ—č –∑–į–≤–Ķ—Ä—ą–ł–Ľ–ł —Ä–į–∑—Ä–į–Ī–ĺ—ā–ļ—É –į–Ĺ—ā–ł—ā–Ķ–Ľ–į –ļ¬†PD-1, –ļ–ĺ—ā–ĺ—Ä–ĺ–Ķ –Ņ–嬆—Ą—É–Ĺ–ļ—Ü–ł–ĺ–Ĺ–į–Ľ—Ć–Ĺ–ĺ–Ļ –į–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā–ł –Ĺ–Ķ¬†—É—Ā—ā—É–Ņ–į–Ķ—ā –Ĺ–ł–≤–ĺ–Ľ—É–ľ–į–Ī—É. –í–ĺ –≤—ā–ĺ—Ä–ĺ–ľ –ļ–≤–į—Ä—ā–į–Ľ–Ķ 2016¬†–≥.¬†–Ĺ–į—á–Ĺ–Ķ—ā—Ā—Ź –Ķ–≥–ĺ –ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ĺ–Ķ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–Ķ. –Ě–į¬†–Ī–ĺ–Ľ–Ķ–Ķ —Ä–į–Ĺ–Ĺ–ł—Ö —Ā—ā–į–ī–ł—Ź—Ö –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–Ļ –Ĺ–į—Ö–ĺ–ī—Ź—ā—Ā—Ź –Ķ—Č–Ķ –Ĺ–Ķ—Ā–ļ–ĺ–Ľ—Ć–ļ–ĺ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–ĺ–≤, –ļ–ĺ—ā–ĺ—Ä—č–Ķ –Ī—É–ī—É—ā –Ņ—Ä–ł–ľ–Ķ–Ĺ—Ź—ā—Ć—Ā—Ź –≤¬†–ļ–ĺ–ľ–Ī–ł–Ĺ–į—Ü–ł–ł –≤¬†–ĺ–Ī–Ľ–į—Ā—ā–ł –ł–ľ–ľ—É–Ĺ–ĺ–ĺ–Ĺ–ļ–ĺ–Ľ–ĺ–≥–ł–ł¬Ľ,¬†‚Äď –Ņ–ĺ—Ź—Ā–Ĺ–ł–Ľ –ī–ĺ–ļ–Ľ–į–ī—á–ł–ļ.
–í¬†2016¬†–≥.¬†–ļ–ĺ–ľ–Ņ–į–Ĺ–ł—Ź –Ņ–Ľ–į–Ĺ–ł—Ä—É–Ķ—ā –Ņ—Ä–ĺ–≤–Ķ–ī–Ķ–Ĺ–ł–Ķ –ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ł—Ö –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–Ļ —ā–į–ļ–ł—Ö –ł–Ĺ–Ĺ–ĺ–≤–į—Ü–ł–ĺ–Ĺ–Ĺ—č—Ö –Ľ–Ķ–ļ–į—Ä—Ā—ā–≤–Ķ–Ĺ–Ĺ—č—Ö —Ā—Ä–Ķ–ī—Ā—ā–≤, –ļ–į–ļ –Ī–ł—Ā–Ņ–Ķ—Ü–ł—Ą–ł—á–Ķ—Ā–ļ–ł–Ķ –į–Ĺ—ā–ł—ā–Ķ–Ľ–į –ļ¬†HER2/HER3 –ł¬†EGFR/HER3. –Ē–≤–ĺ–Ļ–Ĺ–ĺ–Ķ –Ī–Ľ–ĺ–ļ–ł—Ä–ĺ–≤–į–Ĺ–ł–Ķ —ć—ā–ł—Ö –ľ–ł—ą–Ķ–Ĺ–Ķ–Ļ –Ņ–ĺ–∑–≤–ĺ–Ľ–ł—ā —É–≤–Ķ–Ľ–ł—á–ł—ā—Ć —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć —ā–Ķ—Ä–į–Ņ–ł–ł —Ä–į–ļ–į –ľ–ĺ–Ľ–ĺ—á–Ĺ–ĺ–Ļ –∂–Ķ–Ľ–Ķ–∑—č –ł¬†–ļ–ĺ–Ľ–ĺ—Ä–Ķ–ļ—ā–į–Ľ—Ć–Ĺ–ĺ–≥–ĺ —Ä–į–ļ–į —Ā–ĺ–ĺ—ā–≤–Ķ—ā—Ā—ā–≤–Ķ–Ĺ–Ĺ–ĺ. –í¬†–Ĺ–į—á–į–Ľ–Ķ 2016¬†–≥.¬†–Ņ—Ä–Ķ–ī–Ņ–ĺ–Ľ–į–≥–į–Ķ—ā—Ā—Ź —Ā—ā–į—Ä—ā –ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ł—Ö –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–Ļ –ł–Ĺ–≥–ł–Ī–ł—ā–ĺ—Ä–į CDK8/19, –ļ–ĺ—ā–ĺ—Ä—č–Ļ –ĺ–Ī–Ľ–į–ī–į–Ķ—ā –ĺ–≥—Ä–ĺ–ľ–Ĺ—č–ľ –Ņ–ĺ—ā–Ķ–Ĺ—Ü–ł–į–Ľ–ĺ–ľ –ļ–į–ļ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā –Ņ–Ķ—Ä–≤–ĺ–Ļ –Ľ–ł–Ĺ–ł–ł —ā–Ķ—Ä–į–Ņ–ł–ł –ļ–į—Ā—ā—Ä–į—Ü–ł–ĺ–Ĺ–Ĺ–ĺ-—Ä–Ķ–∑–ł—Ā—ā–Ķ–Ĺ—ā–Ĺ–ĺ–≥–ĺ —Ä–į–ļ–į –Ņ—Ä–Ķ–ī—Ā—ā–į—ā–Ķ–Ľ—Ć–Ĺ–ĺ–Ļ –∂–Ķ–Ľ–Ķ–∑—č –ł¬†ER-–Ņ–ĺ–∑–ł—ā–ł–≤–Ĺ–ĺ–≥–ĺ —Ä–į–ļ–į –ľ–ĺ–Ľ–ĺ—á–Ĺ–ĺ–Ļ –∂–Ķ–Ľ–Ķ–∑—č.
–ó–į–≤–Ķ—Ä—ą–į—Ź –≤—č—Ā—ā—É–Ņ–Ľ–Ķ–Ĺ–ł–Ķ, –†.–ź.¬†–ė–≤–į–Ĺ–ĺ–≤ –Ņ–ĺ–ĺ–Ī–Ķ—Č–į–Ľ —á–Ķ—Ä–Ķ–∑¬†–≥–ĺ–ī –Ņ—Ä–Ķ–ī—Ā—ā–į–≤–ł—ā—Ć –Ņ–Ķ—Ä–≤—č–Ķ —Ä–Ķ–∑—É–Ľ—Ć—ā–į—ā—č –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–Ļ –Ĺ–ĺ–≤—č—Ö –ĺ—Ä–ł–≥–ł–Ĺ–į–Ľ—Ć–Ĺ—č—Ö –Ľ–Ķ–ļ–į—Ä—Ā—ā–≤–Ķ–Ĺ–Ĺ—č—Ö –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–ĺ–≤, —Ä–į–∑—Ä–į–Ī–ĺ—ā–į–Ĺ–Ĺ—č—Ö –ļ–ĺ–ľ–Ņ–į–Ĺ–ł–Ķ–Ļ BIOCAD.
–ė—ā–ĺ–≥–ł —ā—Ä–Ķ—ā—Ć–Ķ–Ļ —Ą–į–∑—č –ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ł—Ö –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–Ļ –Ī–ł–ĺ–į–Ĺ–į–Ľ–ĺ–≥–į —ā—Ä–į—Ā—ā—É–∑—É–ľ–į–Ī–į –ļ–ĺ–ľ–Ņ–į–Ĺ–ł–ł BIOCAD
–í–ĺ –≤—ā–ĺ—Ä–ĺ–ľ –ļ–≤–į—Ä—ā–į–Ľ–Ķ 2015¬†–≥.¬†–Ī—č–Ľ –∑–į–≤–Ķ—Ä—ą–Ķ–Ĺ –į–Ĺ–į–Ľ–ł–∑ —Ä–Ķ–∑—É–Ľ—Ć—ā–į—ā–ĺ–≤ –ľ–Ķ–∂–ī—É–Ĺ–į—Ä–ĺ–ī–Ĺ–ĺ–≥–ĺ —Ā—Ä–į–≤–Ĺ–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ–≥–ĺ –ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ĺ–≥–ĺ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź —Ą–į–∑—č III –Ņ–Ķ—Ä–≤–ĺ–≥–ĺ –ĺ—ā–Ķ—á–Ķ—Ā—ā–≤–Ķ–Ĺ–Ĺ–ĺ–≥–ĺ –Ī–ł–ĺ–į–Ĺ–į–Ľ–ĺ–≥–į —ā—Ä–į—Ā—ā—É–∑—É–ľ–į–Ī–į¬†‚Äď –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į BCD-022 –ł¬†–ĺ—Ä–ł–≥–ł–Ĺ–į–Ľ—Ć–Ĺ–ĺ–≥–ĺ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į —ā—Ä–į—Ā—ā—É–∑—É–ľ–į–Ī–į –ď–Ķ—Ä—Ü–Ķ–Ņ—ā–ł–Ŭģ. –Ę—Ä–į—Ā—ā—É–∑—É–ľ–į–Ī¬†‚Äď –ļ–Ľ—é—á–Ķ–≤–ĺ–Ļ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā —ā–į—Ä–≥–Ķ—ā–Ĺ–ĺ–Ļ —ā–Ķ—Ä–į–Ņ–ł–ł HER2-–Ņ–ĺ–Ľ–ĺ–∂–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ–≥–ĺ —Ä–į–ļ–į –ľ–ĺ–Ľ–ĺ—á–Ĺ–ĺ–Ļ –∂–Ķ–Ľ–Ķ–∑—č (–†–ú–Ė), –ĺ–ī–Ĺ–į–ļ–ĺ –Ķ–≥–ĺ –Ņ—Ä–ł–ľ–Ķ–Ĺ–Ķ–Ĺ–ł–Ķ –≤¬†—Ä—É—ā–ł–Ĺ–Ĺ–ĺ–Ļ –ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ĺ–Ļ –Ņ—Ä–į–ļ—ā–ł–ļ–Ķ –Ņ–ĺ–ļ–į –ĺ–≥—Ä–į–Ĺ–ł—á–Ķ–Ĺ–ĺ –≤—č—Ā–ĺ–ļ–ĺ–Ļ —Ā—ā–ĺ–ł–ľ–ĺ—Ā—ā—Ć—é. –í–Ĺ–Ķ–ī—Ä–Ķ–Ĺ–ł–Ķ –≤¬†–ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ—É—é –Ņ—Ä–į–ļ—ā–ł–ļ—É –≤–ĺ—Ā–Ņ—Ä–ĺ–ł–∑–≤–Ķ–ī–Ķ–Ĺ–Ĺ–ĺ–≥–ĺ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į —ā—Ä–į—Ā—ā—É–∑—É–ľ–į–Ī–į –Ī—É–ī–Ķ—ā —Ā–Ņ–ĺ—Ā–ĺ–Ī—Ā—ā–≤–ĺ–≤–į—ā—Ć –∑–Ĺ–į—á–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ–ľ—É –Ņ–ĺ–≤—č—ą–Ķ–Ĺ–ł—é –ī–ĺ—Ā—ā—É–Ņ–Ĺ–ĺ—Ā—ā–ł —Ā–ĺ–≤—Ä–Ķ–ľ–Ķ–Ĺ–Ĺ–ĺ–Ļ –≤—č—Ā–ĺ–ļ–ĺ—ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ–Ļ —ā–Ķ—Ä–į–Ņ–ł–ł –ī–Ľ—Ź –Ī–ĺ–Ľ—Ć–Ĺ—č—Ö –†–ú–Ė.
–Ě–į—É—á–Ĺ—č–Ļ —Ā–ĺ—ā—Ä—É–ī–Ĺ–ł–ļ –ĺ—ā–ī–Ķ–Ľ–Ķ–Ĺ–ł—Ź –ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ĺ–Ļ —Ą–į—Ä–ľ–į–ļ–ĺ–Ľ–ĺ–≥–ł–ł –ł¬†—Ö–ł–ľ–ł–ĺ—ā–Ķ—Ä–į–Ņ–ł–ł –†–ĺ—Ā—Ā–ł–Ļ—Ā–ļ–ĺ–≥–ĺ –ĺ–Ĺ–ļ–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ĺ–≥–ĺ –Ĺ–į—É—á–Ĺ–ĺ–≥–ĺ —Ü–Ķ–Ĺ—ā—Ä–į –ł–ľ. –Ě.–Ě. –Ď–Ľ–ĺ—Ö–ł–Ĺ–į, –ļ.–ľ.–Ĺ. –ē–ļ–į—ā–Ķ—Ä–ł–Ĺ–į –ě–Ľ–Ķ–≥–ĺ–≤–Ĺ–į –ė–ď–Ě–ź–Ę–ě–í–ź –ĺ–∑–Ĺ–į–ļ–ĺ–ľ–ł–Ľ–į —É—á–į—Ā—ā–Ĺ–ł–ļ–ĺ–≤ —Ā–ł–ľ–Ņ–ĺ–∑–ł—É–ľ–į —Ā¬†—Ä–Ķ–∑—É–Ľ—Ć—ā–į—ā–į–ľ–ł –ľ–Ķ–∂–ī—É–Ĺ–į—Ä–ĺ–ī–Ĺ–ĺ–≥–ĺ –ľ–Ĺ–ĺ–≥–ĺ—Ü–Ķ–Ĺ—ā—Ä–ĺ–≤–ĺ–≥–ĺ –ī–≤–ĺ–Ļ–Ĺ–ĺ–≥–ĺ —Ā–Ľ–Ķ–Ņ–ĺ–≥–ĺ —Ä–į–Ĺ–ī–ĺ–ľ–ł–∑–ł—Ä–ĺ–≤–į–Ĺ–Ĺ–ĺ–≥–ĺ –ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ĺ–≥–ĺ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź —Ą–į–∑—謆III, –ļ–ĺ—ā–ĺ—Ä–ĺ–Ķ –Ņ—Ä–ĺ–≤–ĺ–ī–ł–Ľ–ĺ—Ā—Ć –≤¬†–Ī–ĺ–Ľ–Ķ–Ķ —á–Ķ–ľ 30 –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į—ā–Ķ–Ľ—Ć—Ā–ļ–ł—Ö —Ü–Ķ–Ĺ—ā—Ä–į—Ö –Ĺ–į¬†—ā–Ķ—Ä—Ä–ł—ā–ĺ—Ä–ł–ł –†–§, –Ī–Ľ–ł–∂–Ĺ–Ķ–≥–ĺ –ł¬†–ī–į–Ľ—Ć–Ĺ–Ķ–≥–ĺ –∑–į—Ä—É–Ī–Ķ–∂—Ć—Ź.
–í¬†–ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–ł –Ņ—Ä–ł–Ĺ—Ź–Ľ–ł —É—á–į—Ā—ā–ł–Ķ –≤¬†–ĺ–Ī—Č–Ķ–Ļ —Ā–Ľ–ĺ–∂–Ĺ–ĺ—Ā—ā–ł 126¬†–Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–ļ —Ā¬†HER2-–Ņ–ĺ–Ľ–ĺ–∂–ł—ā–Ķ–Ľ—Ć–Ĺ—č–ľ –ľ–Ķ—ā–į—Ā—ā–į—ā–ł—á–Ķ—Ā–ļ–ł–ľ –†–ú–Ė. –í—Ā–Ķ –Ī–ĺ–Ľ—Ć–Ĺ—č–Ķ –Ī—č–Ľ–ł —Ā—ā—Ä–į—ā–ł—Ą–ł—Ü–ł—Ä–ĺ–≤–į–Ĺ—č —Ā–ĺ–≥–Ľ–į—Ā–Ĺ–ĺ –≤–ĺ–∑—Ä–į—Ā—ā—É, –Ņ—Ä–Ķ–ī—ą–Ķ—Ā—ā–≤—É—é—Č–Ķ–Ļ —ā–Ķ—Ä–į–Ņ–ł–ł, —Ā—ā–į—ā—É—Ā—ɬ†–≥–ĺ—Ä–ľ–ĺ–Ĺ–į–Ľ—Ć–Ĺ—č—Ö —Ä–Ķ—Ü–Ķ–Ņ—ā–ĺ—Ä–ĺ–≤ –ł¬†—Ä–į–Ĺ–ī–ĺ–ľ–ł–∑–ł—Ä–ĺ–≤–į–Ĺ—č –Ĺ–į¬†–ī–≤–Ķ¬†–≥—Ä—É–Ņ–Ņ—č –≤¬†—Ā–ĺ–ĺ—ā–Ĺ–ĺ—ą–Ķ–Ĺ–ł–ł 1:1. –ü–į—Ü–ł–Ķ–Ĺ—ā–ļ–ł –Ņ–Ķ—Ä–≤–ĺ–Ļ¬†–≥—Ä—É–Ņ–Ņ—č –Ņ–ĺ–Ľ—É—á–į–Ľ–ł BCD-022 –≤¬†–Ĺ–į–≥—Ä—É–∑–ĺ—á–Ĺ–ĺ–Ļ –ī–ĺ–∑–Ķ 8 –ľ–≥/–ļ–≥ –ĺ–ī–Ĺ–ĺ–ļ—Ä–į—ā–Ĺ–ĺ (–ĺ–ī–ł–Ĺ —Ü–ł–ļ–Ľ) —Ā¬†–Ņ–Ķ—Ä–Ķ—Ö–ĺ–ī–ĺ–ľ –Ĺ–į¬†–Ņ–ĺ–ī–ī–Ķ—Ä–∂–ł–≤–į—é—Č—É—é –ī–ĺ–∑—É 6 –ľ–≥/–ļ–≥ –ļ–į–∂–ī—č–Ķ —ā—Ä–ł –Ĺ–Ķ–ī–Ķ–Ľ–ł (–Ņ—Ź—ā—Ć —Ü–ł–ļ–Ľ–ĺ–≤). –ü–į—Ü–ł–Ķ–Ĺ—ā–ļ–ł –≤—ā–ĺ—Ä–ĺ–Ļ¬†–≥—Ä—É–Ņ–Ņ—č –Ņ–ĺ–Ľ—É—á–į–Ľ–ł –ď–Ķ—Ä—Ü–Ķ–Ņ—ā–ł–Ŭģ –Ņ–嬆–į–Ĺ–į–Ľ–ĺ–≥–ł—á–Ĺ–ĺ–Ļ —Ā—Ö–Ķ–ľ–Ķ. –ö—Ä–ĺ–ľ–Ķ —ā–ĺ–≥–ĺ, –≤¬†–ĺ–Ī–Ķ–ł—Ö¬†–≥—Ä—É–Ņ–Ņ–į—Ö –≤–≤–ĺ–ī–ł–Ľ—Ā—Ź –Ņ–į–ļ–Ľ–ł—ā–į–ļ—Ā–Ķ–Ľ 175 –ľ–≥/–ľ2 –≤–Ĺ—É—ā—Ä–ł–≤–Ķ–Ĺ–Ĺ–ĺ –≤¬†—ā–Ķ—á–Ķ–Ĺ–ł–Ķ —ā—Ä–Ķ—Ö —á–į—Ā–ĺ–≤ –≤¬†–Ņ–Ķ—Ä–≤—č–Ļ –ī–Ķ–Ĺ—Ć –ļ–į–∂–ī–ĺ–≥–ĺ —ā—Ä–Ķ—Ö–Ĺ–Ķ–ī–Ķ–Ľ—Ć–Ĺ–ĺ–≥–ĺ —Ü–ł–ļ–Ľ–į. –õ–Ķ—á–Ķ–Ĺ–ł–Ķ –Ņ—Ä–ĺ–ī–ĺ–Ľ–∂–į–Ľ–ĺ—Ā—Ć –ī–ĺ —ą–Ķ—Ā—ā–ł —Ü–ł–ļ–Ľ–ĺ–≤ –ł–Ľ–ł –ī–ĺ –Ņ—Ä–ĺ–≥—Ä–Ķ—Ā—Ā–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł—Ź –Ľ–ł–Ī–ĺ –ī–ĺ –Ĺ–į—Ā—ā—É–Ņ–Ľ–Ķ–Ĺ–ł—Ź –Ĺ–Ķ–Ņ–Ķ—Ä–Ķ–Ĺ–ĺ—Ā–ł–ľ–ĺ–Ļ —ā–ĺ–ļ—Ā–ł—á–Ĺ–ĺ—Ā—ā–ł. –ü–嬆–ĺ–ļ–ĺ–Ĺ—á–į–Ĺ–ł–ł —ą–Ķ—Ā—ā–ł —Ü–ł–ļ–Ľ–ĺ–≤ —ā–Ķ—Ä–į–Ņ–ł–ł –Ī–ĺ–Ľ—Ć–Ĺ—č–Ķ —Ā¬†–Ņ–ĺ–Ľ–Ĺ—č–ľ –Ľ–ł–Ī–ĺ —á–į—Ā—ā–ł—á–Ĺ—č–ľ –ĺ—ā–≤–Ķ—ā–ĺ–ľ –ł–Ľ–ł —Ā—ā–į–Ī–ł–Ľ–ł–∑–į—Ü–ł–Ķ–Ļ –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł—Ź –Ņ–嬆—Ä–Ķ—ą–Ķ–Ĺ–ł—é –≤—Ä–į—á–į-–ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į—ā–Ķ–Ľ—Ź –Ņ–Ķ—Ä–Ķ–≤–ĺ–ī–ł–Ľ–ł—Ā—Ć –≤¬†–Ņ–Ķ—Ä–ł–ĺ–ī –Ņ–ĺ–ī–ī–Ķ—Ä–∂–ł–≤–į—é—Č–Ķ–≥–ĺ –Ľ–Ķ—á–Ķ–Ĺ–ł—Ź –ł¬†–Ĺ–į–Ī–Ľ—é–ī–Ķ–Ĺ–ł—Ź.
–ě–Ī–Ķ¬†–≥—Ä—É–Ņ–Ņ—č –Ī—č–Ľ–ł —Ā–ĺ–Ņ–ĺ—Ā—ā–į–≤–ł–ľ—č –Ņ–嬆—Ö–į—Ä–į–ļ—ā–Ķ—Ä–ł—Ā—ā–ł–ļ–į–ľ –ĺ—Ā–Ĺ–ĺ–≤–Ĺ–ĺ–≥–ĺ –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł—Ź –ł¬†–ī–Ķ–ľ–ĺ–≥—Ä–į—Ą–ł—á–Ķ—Ā–ļ–ł–ľ –Ņ–į—Ä–į–ľ–Ķ—ā—Ä–į–ľ. –Ď–ĺ–Ľ—Ć—ą–ł–Ĺ—Ā—ā–≤–ĺ –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–ļ –ł–ľ–Ķ–Ľ–ł –ł–Ĺ–≤–į–∑–ł–≤–Ĺ—č–Ļ —Ä–į–ļ –Ĺ–Ķ—Ā–Ņ–Ķ—Ü–ł—Ą–ł—á–Ķ—Ā–ļ–ĺ–≥–ĺ —ā–ł–Ņ–į —Ā¬†–≥–ł–Ņ–Ķ—Ä—ć–ļ—Ā–Ņ—Ä–Ķ—Ā—Ā–ł–Ķ–Ļ HER2¬†3+, –Ī–ĺ–Ľ–Ķ–Ķ —á–Ķ–ľ —ɬ†50% –Ī–ĺ–Ľ—Ć–Ĺ—č—Ö –ĺ—ā—Ā—É—ā—Ā—ā–≤–ĺ–≤–į–Ľ–į —ć–ļ—Ā–Ņ—Ä–Ķ—Ā—Ā–ł—Ź¬†–≥–ĺ—Ä–ľ–ĺ–Ĺ–į–Ľ—Ć–Ĺ—č—Ö —Ä–Ķ—Ü–Ķ–Ņ—ā–ĺ—Ä–ĺ–≤. –ú–Ķ—ā–į—Ā—ā–į–∑—č –≤¬†–ī–≤—É—Ö –ł¬†–Ī–ĺ–Ľ–Ķ–Ķ –ĺ—Ä–≥–į–Ĺ–į—Ö –Ī—č–Ľ–ł –∑–į—Ä–Ķ–≥–ł—Ā—ā—Ä–ł—Ä–ĺ–≤–į–Ĺ—č –Ņ—Ä–ł–ľ–Ķ—Ä–Ĺ–ĺ —É¬†–Ņ–ĺ–Ľ–ĺ–≤–ł–Ĺ—č –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–ļ –≤¬†–ĺ–Ī–Ķ–ł—Ö¬†–≥—Ä—É–Ņ–Ņ–į—Ö. –ě—Ā–Ĺ–ĺ–≤–Ĺ—č–ľ–ł –ľ–Ķ—Ā—ā–į–ľ–ł –Ľ–ĺ–ļ–į–Ľ–ł–∑–į—Ü–ł–ł –ĺ—ā–ī–į–Ľ–Ķ–Ĺ–Ĺ—č—Ö –ľ–Ķ—ā–į—Ā—ā–į–∑–ĺ–≤ –Ī—č–Ľ–ł –Ņ–Ķ—á–Ķ–Ĺ—Ć, –Ľ–Ķ–≥–ļ–ł–Ķ, –Ľ–ł–ľ—Ą–į—ā–ł—á–Ķ—Ā–ļ–ł–Ķ —É–∑–Ľ—č, –ļ–ĺ—Ā—ā–ł. –Ď–ĺ–Ľ—Ć—ą–ł–Ĺ—Ā—ā–≤—É –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–ļ –Ņ—Ä–ĺ–≤–ĺ–ī–ł–Ľ–ł —Ö–ł—Ä—É—Ä–≥–ł—á–Ķ—Ā–ļ–ĺ–Ķ –Ľ–Ķ—á–Ķ–Ĺ–ł–Ķ –ł¬†–Ľ—É—á–Ķ–≤—É—é —ā–Ķ—Ä–į–Ņ–ł—é. –ú–Ķ–Ĺ–Ķ–Ķ –Ņ–ĺ–Ľ–ĺ–≤–ł–Ĺ—č –ł–∑¬†–Ĺ–ł—Ö –Ņ–ĺ–Ľ—É—á–į–Ľ–ł –į–ī—ä—é–≤–į–Ĺ—ā–Ĺ—É—é –ł–Ľ–ł –Ĺ–Ķ–ĺ–į–ī—ä—é–≤–į–Ĺ—ā–Ĺ—É—é —Ö–ł–ľ–ł–ĺ—ā–Ķ—Ä–į–Ņ–ł—é.
–≠—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć —ā–Ķ—Ä–į–Ņ–ł–ł –ĺ—Ü–Ķ–Ĺ–ł–≤–į–Ľ–į—Ā—Ć –Ņ–嬆–ī–į–Ĺ–Ĺ—č–ľ –ļ–ĺ–ľ–Ņ—Ć—é—ā–Ķ—Ä–Ĺ–ĺ–Ļ —ā–ĺ–ľ–ĺ–≥—Ä–į—Ą–ł–ł (–ö–Ę) —Ā¬†–ł—Ā–Ņ–ĺ–Ľ—Ć–∑–ĺ–≤–į–Ĺ–ł–Ķ–ľ –ļ—Ä–ł—ā–Ķ—Ä–ł–Ķ–≤ RECIST 1.1. –ö–Ę –Ņ—Ä–ĺ–≤–ĺ–ī–ł–Ľ–ł –Ĺ–į¬†—Ā–ļ—Ä–ł–Ĺ–ł–Ĺ–≥–Ķ, –Ņ–ĺ—Ā–Ľ–Ķ —ā—Ä–Ķ—Ö –ł¬†—ą–Ķ—Ā—ā–ł —Ü–ł–ļ–Ľ–ĺ–≤ —ā–Ķ—Ä–į–Ņ–ł–ł. –ē—Ā–Ľ–ł –Ņ—Ä–ł –ö–Ę –Ņ–ĺ—Ā–Ľ–Ķ —ā—Ä–Ķ—ā—Ć–Ķ–≥–ĺ –ł–Ľ–ł —ą–Ķ—Ā—ā–ĺ–≥–ĺ —Ü–ł–ļ–Ľ–į —ā–Ķ—Ä–į–Ņ–ł–ł –Ī—č–Ľ –≤—č—Ź–≤–Ľ–Ķ–Ĺ –Ņ–ĺ–Ľ–Ĺ—č–Ļ –ł–Ľ–ł —á–į—Ā—ā–ł—á–Ĺ—č–Ļ –ĺ—ā–≤–Ķ—ā, –≤—č–Ņ–ĺ–Ľ–Ĺ—Ź–Ľ–ł –ö–Ę —á–Ķ—Ä–Ķ–∑ —á–Ķ—ā—č—Ä–Ķ –Ĺ–Ķ–ī–Ķ–Ľ–ł –ī–Ľ—Ź –Ņ–ĺ–ī—ā–≤–Ķ—Ä–∂–ī–Ķ–Ĺ–ł—Ź –ī–ĺ—Ā—ā–ł–≥–Ĺ—É—ā–ĺ–≥–ĺ –ĺ—ā–≤–Ķ—ā–į. –≠—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć —ā–Ķ—Ä–į–Ņ–ł–ł (–Ľ—É—á—ą–ł–Ļ –ĺ—ā–≤–Ķ—ā) –ĺ—Ü–Ķ–Ĺ–ł–≤–į–Ľ–į—Ā—Ć –Ņ–嬆—Ä–Ķ–∑—É–Ľ—Ć—ā–į—ā–į–ľ –ö–Ę –Ĺ–Ķ–∑–į–≤–ł—Ā–ł–ľ—č–ľ —Ā–Ņ–Ķ—Ü–ł–į–Ľ–ł—Ā—ā–ĺ–ľ, –∑–į—Ā–Ľ–Ķ–Ņ–Ľ–Ķ–Ĺ–Ĺ—č–ľ –≤¬†–ĺ—ā–Ĺ–ĺ—ą–Ķ–Ĺ–ł–ł –Ņ—Ä–ĺ–≤–ĺ–ī–ł–ľ–ĺ–≥–ĺ –Ī–ĺ–Ľ—Ć–Ĺ—č–ľ –Ľ–Ķ—á–Ķ–Ĺ–ł—Ź.
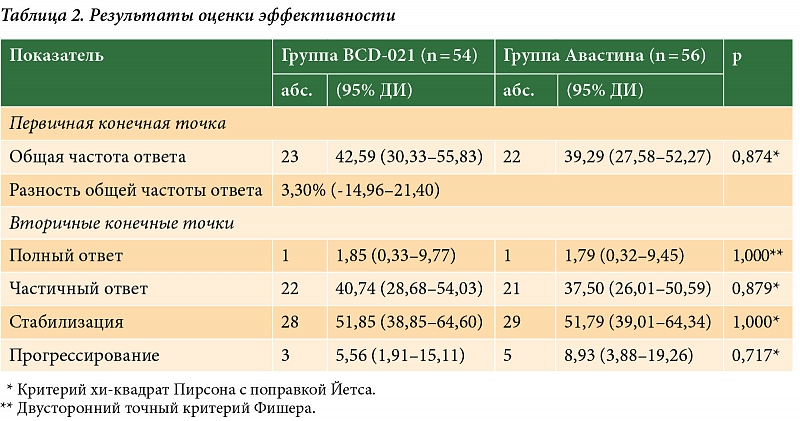
–ü–Ķ—Ä–≤–ł—á–Ĺ–ĺ–Ļ –ļ–ĺ–Ĺ–Ķ—á–Ĺ–ĺ–Ļ —ā–ĺ—á–ļ–ĺ–Ļ –į–Ĺ–į–Ľ–ł–∑–į —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā–ł –Ī—č–Ľ–į –ĺ–Ī—Č–į—Ź —á–į—Ā—ā–ĺ—ā–į –ĺ—ā–≤–Ķ—ā–ĺ–≤ (–Ņ–ĺ–Ľ–Ĺ—č–Ķ –ĺ—ā–≤–Ķ—ā—č + —á–į—Ā—ā–ł—á–Ĺ—č–Ķ –ĺ—ā–≤–Ķ—ā—č), –≤—ā–ĺ—Ä–ł—á–Ĺ—č–ľ–ł¬†‚Äď —á–į—Ā—ā–ĺ—ā–į –Ņ–ĺ–Ľ–Ĺ—č—Ö –ł¬†—á–į—Ā—ā–ł—á–Ĺ—č—Ö –ĺ—ā–≤–Ķ—ā–ĺ–≤, —á–į—Ā—ā–ĺ—ā–į —Ā—ā–į–Ī–ł–Ľ–ł–∑–į—Ü–ł–ł –ĺ–Ņ—É—Ö–ĺ–Ľ–Ķ–≤–ĺ–≥–ĺ –ĺ—ā–≤–Ķ—ā–į –ł¬†—á–į—Ā—ā–ĺ—ā–į –Ņ—Ä–ĺ–≥—Ä–Ķ—Ā—Ā–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł—Ź. –í¬†–į–Ĺ–į–Ľ–ł–∑ —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā–ł —Ā—É–ľ–ľ–į—Ä–Ĺ–ĺ –≤–ĺ—ą–Ľ–ł 110¬†–Ī–ĺ–Ľ—Ć–Ĺ—č—Ö: 56¬†–ł–∑¬†–≥—Ä—É–Ņ–Ņ—č BCD-022, 54¬†–ł–∑¬†–≥—Ä—É–Ņ–Ņ—č –ď–Ķ—Ä—Ü–Ķ–Ņ—ā–ł–Ĺ–į.
–ě–Ī—Č–į—Ź —á–į—Ā—ā–ĺ—ā–į –ĺ—ā–≤–Ķ—ā–į (–ě–ß–ě) –≤¬†–≥—Ä—É–Ņ–Ņ–Ķ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į BCD-022 —Ā–ĺ—Ā—ā–į–≤–ł–Ľ–į 53,57% (95%-–Ĺ—č–Ļ –ī–ĺ–≤–Ķ—Ä–ł—ā–Ķ–Ľ—Ć–Ĺ—č–Ļ –ł–Ĺ—ā–Ķ—Ä–≤–į–Ľ¬†(–Ē–ė) 40,70‚Äď65,98%), –į¬†–≤¬†–≥—Ä—É–Ņ–Ņ–Ķ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į –ď–Ķ—Ä—Ü–Ķ–Ņ—ā–ł–Ŭģ¬†‚Äď 53,70% (95% –Ē–ė 40,60‚Äď66,31%) —Ā–ĺ–ĺ—ā–≤–Ķ—ā—Ā—ā–≤–Ķ–Ĺ–Ĺ–ĺ. –†–į–∑–Ĺ–ł—Ü–į –ě–ß–ě –≤¬†–≥—Ä—É–Ņ–Ņ–Ķ –ł—Ā—Ā–Ľ–Ķ–ī—É–Ķ–ľ–ĺ–Ļ —ā–Ķ—Ä–į–Ņ–ł–ł –ł¬†–≤¬†–≥—Ä—É–Ņ–Ņ–Ķ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į —Ā—Ä–į–≤–Ĺ–Ķ–Ĺ–ł—Ź —Ā–ĺ—Ā—ā–į–≤–ł–Ľ–į -0,13% (95% –Ē–ė -19,83‚Äď18,35%) (—Ä = 0,862, –ļ—Ä–ł—ā–Ķ—Ä–ł–Ļ —Ö–ł-–ļ–≤–į–ī—Ä–į—ā –ü–ł—Ä—Ā–ĺ–Ĺ–į —Ā¬†–Ņ–ĺ–Ņ—Ä–į–≤–ļ–ĺ–Ļ –ô–Ķ—ā—Ā–į). –Ě–ł–∂–Ĺ—Ź—Ź¬†–≥—Ä–į–Ĺ–ł—Ü–į —Ä–į—Ā—Ā—á–ł—ā–į–Ĺ–Ĺ–ĺ–≥–ĺ 95% –Ē–ė (-19,83%) –Ņ—Ä–Ķ–≤—č—ą–į–Ľ–į —É—Ā—ā–į–Ĺ–ĺ–≤–Ľ–Ķ–Ŗŗɗ鬆–≥—Ä–į–Ĺ–ł—Ü—É –Ĺ–Ķ¬†–ľ–Ķ–Ĺ—Ć—ą–Ķ–Ļ —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā–ł, —á—ā–ĺ –Ņ–ĺ–∑–≤–ĺ–Ľ–ł–Ľ–ĺ —Ā–ī–Ķ–Ľ–į—ā—Ć –≤—č–≤–ĺ–ī –嬆–Ĺ–Ķ –ľ–Ķ–Ĺ—Ć—ą–Ķ–Ļ —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā–ł –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į BCD-022 –Ņ–嬆—Ā—Ä–į–≤–Ĺ–Ķ–Ĺ–ł—é —Ā¬†–Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–ĺ–ľ –ď–Ķ—Ä—Ü–Ķ–Ņ—ā–ł–Ŭģ. –ě—ā–Ĺ–ĺ—ą–Ķ–Ĺ–ł–Ķ —ą–į–Ĺ—Ā–ĺ–≤ –ī–Ľ—Ź –ě–ß–ě —Ā–ĺ—Ā—ā–į–≤–ł–Ľ–ĺ 0,9947 (95% –Ē–ė 0,470‚Äď2,105), —á—ā–ĺ —ā–į–ļ–∂–Ķ —É–ļ–į–∑—č–≤–į–Ľ–ĺ –Ĺ–į¬†–ĺ—ā—Ā—É—ā—Ā—ā–≤–ł–Ķ —Ā—ā–į—ā–ł—Ā—ā–ł—á–Ķ—Ā–ļ–ł –∑–Ĺ–į—á–ł–ľ—č—Ö —Ä–į–∑–Ľ–ł—á–ł–Ļ –≤¬†—ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā–ł –ľ–Ķ–∂–ī—ɬ†–≥—Ä—É–Ņ–Ņ–į–ľ–ł —ā–Ķ—Ä–į–Ņ–ł–ł. –°—ā–į—ā–ł—Ā—ā–ł—á–Ķ—Ā–ļ–ł –∑–Ĺ–į—á–ł–ľ—č—Ö —Ä–į–∑–Ľ–ł—á–ł–Ļ –Ĺ–Ķ¬†–Ī—č–Ľ–ĺ –ł¬†–Ņ—Ä–ł —Ā—Ä–į–≤–Ĺ–Ķ–Ĺ–ł–ł –Ņ—Ä–ĺ—á–ł—Ö –Ņ–į—Ä–į–ľ–Ķ—ā—Ä–ĺ–≤ –ĺ—Ü–Ķ–Ĺ–ļ–ł —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā–ł.
–ź–Ĺ–į–Ľ–ł–∑ –Ī–Ķ–∑–ĺ–Ņ–į—Ā–Ĺ–ĺ—Ā—ā–ł –Ņ—Ä–ĺ–≤–ĺ–ī–ł–Ľ–ł —Ā—ā–į–Ĺ–ī–į—Ä—ā–Ĺ—č–ľ–ł –ľ–Ķ—ā–ĺ–ī–į–ľ–ł¬†‚Äď –Ĺ–į¬†–ĺ—Ā–Ĺ–ĺ–≤–į–Ĺ–ł–ł –ī–į–Ĺ–Ĺ—č—Ö –嬆—Ä–Ķ–≥–ł—Ā—ā—Ä–į—Ü–ł–ł –Ĺ–Ķ–∂–Ķ–Ľ–į—ā–Ķ–Ľ—Ć–Ĺ—č—Ö —Ź–≤–Ľ–Ķ–Ĺ–ł–Ļ (–Ě–Į) –ł¬†—Ā–Ķ—Ä—Ć–Ķ–∑–Ĺ—č—Ö –Ĺ–Ķ–∂–Ķ–Ľ–į—ā–Ķ–Ľ—Ć–Ĺ—č—Ö —Ź–≤–Ľ–Ķ–Ĺ–ł–Ļ (–°–Ě–Į), —Ä–Ķ–∑—É–Ľ—Ć—ā–į—ā–ĺ–≤ –ļ–Ľ–ł–Ĺ–ł–ļ–ĺ-–Ľ–į–Ī–ĺ—Ä–į—ā–ĺ—Ä–Ĺ—č—Ö –į–Ĺ–į–Ľ–ł–∑–ĺ–≤, –ł–Ĺ—Ā—ā—Ä—É–ľ–Ķ–Ĺ—ā–į–Ľ—Ć–Ĺ—č—Ö –ĺ–Ī—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–Ļ –ł¬†–ī–į–Ĺ–Ĺ—č—Ö —Ą–ł–∑–ł–ļ–į–Ľ—Ć–Ĺ–ĺ–≥–ĺ –ĺ—Ā–ľ–ĺ—ā—Ä–į, –ļ–ĺ—ā–ĺ—Ä—č–Ķ –ĺ—Ā—É—Č–Ķ—Ā—ā–≤–Ľ—Ź–Ľ–ł—Ā—Ć —Ā–ĺ–≥–Ľ–į—Ā–Ŗ嬆–≥—Ä–į—Ą–ł–ļ—É –Ņ—Ä–ĺ—Ü–Ķ–ī—É—Ä. –í¬†–į–Ĺ–į–Ľ–ł–∑ –Ī–Ķ–∑–ĺ–Ņ–į—Ā–Ĺ–ĺ—Ā—ā–ł –≤–ĺ—ą–Ľ–ł –≤—Ā–Ķ –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ļ–ł, –Ņ–ĺ–Ľ—É—á–ł–≤—ą–ł–Ķ —Ö–ĺ—ā—Ź –Ī—č –ĺ–ī–Ĺ–ĺ –≤–≤–Ķ–ī–Ķ–Ĺ–ł–Ķ –ł—Ā—Ā–Ľ–Ķ–ī—É–Ķ–ľ–ĺ–≥–ĺ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į –ł–Ľ–ł –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į —Ā—Ä–į–≤–Ĺ–Ķ–Ĺ–ł—Ź. –°—É–ľ–ľ–į—Ä–Ĺ–ĺ 124¬†–Ņ–į—Ü–ł–Ķ–Ĺ—ā–ļ–ł: 63¬†–ł–∑¬†–≥—Ä—É–Ņ–Ņ—č BCD-022, 61¬†–ł–∑¬†–≥—Ä—É–Ņ–Ņ—č –ď–Ķ—Ä—Ü–Ķ–Ņ—ā–ł–Ĺ–į. –í¬†—Ü–Ķ–Ľ–ĺ–ľ –Ĺ–į¬†–Ņ—Ä–ĺ—ā—Ź–∂–Ķ–Ĺ–ł–ł –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź –ļ–į–ļ–ł–Ķ-–Ľ–ł–Ī–ĺ –Ě–Į –Ī—č–Ľ–ł –∑–į—Ä–Ķ–≥–ł—Ā—ā—Ä–ł—Ä–ĺ–≤–į–Ĺ—č —É¬†62 (98,41%) –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–ļ¬†–≥—Ä—É–Ņ–Ņ—č –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į BCD-022 –ł¬†—É 60 (98,36%) –Ī–ĺ–Ľ—Ć–Ĺ—č—Ö¬†–≥—Ä—É–Ņ–Ņ—č –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į –ď–Ķ—Ä—Ü–Ķ–Ņ—ā–ł–Ŭģ. –°–Ě–Į –Ī—č–Ľ–ł –≤—č—Ź–≤–Ľ–Ķ–Ĺ—č –≤¬†–ĺ–Ī—Č–Ķ–Ļ —Ā–Ľ–ĺ–∂–Ĺ–ĺ—Ā—ā–ł —ɬ†–ī–Ķ—Ā—Ź—ā–ł –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–ļ: —ɬ†—á–Ķ—ā—č—Ä–Ķ—Ö (6,35%) –ł–∑¬†–≥—Ä—É–Ņ–Ņ—č BCD-022 –ł¬†—ą–Ķ—Ā—ā–ł (9,84%) –ł–∑¬†–≥—Ä—É–Ņ–Ņ—č –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į —Ā—Ä–į–≤–Ĺ–Ķ–Ĺ–ł—Ź (–Ņ—Ä–ł—á–Ķ–ľ —ɬ†–ĺ–ī–Ĺ–ĺ–Ļ –Ī–ĺ–Ľ—Ć–Ĺ–ĺ–Ļ –∑–į—Ä–Ķ–≥–ł—Ā—ā—Ä–ł—Ä–ĺ–≤–į–Ĺ–ĺ –ī–≤–į –°–Ě–Į). –Ę–į–ļ–ł–ľ –ĺ–Ī—Ä–į–∑–ĺ–ľ, –≤¬†–≥—Ä—É–Ņ–Ņ–Ķ —Ā—Ä–į–≤–Ĺ–Ķ–Ĺ–ł—Ź –Ĺ–į—Ā—á–ł—ā—č–≤–į–Ľ–ĺ—Ā—Ć —Ā–Ķ–ľ—Ć (11,48%) –°–Ě–Į. –í¬†–Ī–ĺ–Ľ—Ć—ą–ł–Ĺ—Ā—ā–≤–Ķ —Ā–Ľ—É—á–į–Ķ–≤ –°–Ě–Į –Ī—č–Ľ–ł –ĺ–Ī—É—Ā–Ľ–ĺ–≤–Ľ–Ķ–Ĺ—č –Ĺ–į–Ľ–ł—á–ł–Ķ–ľ —Ā–ĺ–Ņ—É—ā—Ā—ā–≤—É—é—Č–Ķ–Ļ –Ņ–į—ā–ĺ–Ľ–ĺ–≥–ł–ł, –≤–ĺ–∑–ī–Ķ–Ļ—Ā—ā–≤–ł–Ķ–ľ —Ö–ł–ľ–ł–ĺ–Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–ĺ–≤, –Ņ—Ä–ł–ľ–Ķ–Ĺ—Ź–Ķ–ľ—č—Ö –≤¬†—Ä–į–ľ–ļ–į—Ö –ļ–ĺ–ľ–Ī–ł–Ĺ–ł—Ä–ĺ–≤–į–Ĺ–Ĺ–ĺ–Ļ —Ö–ł–ľ–ł–ĺ—ā–Ķ—Ä–į–Ņ–ł–ł, –Ľ–ł–Ī–ĺ –ī—Ä—É–≥–ł–ľ–ł –Ĺ–Ķ¬†—Ā–≤—Ź–∑–į–Ĺ–Ĺ—č–ľ–ł —Ā¬†–ł—Ā—Ā–Ľ–Ķ–ī—É–Ķ–ľ–ĺ–Ļ —ā–Ķ—Ä–į–Ņ–ł–Ķ–Ļ –Ņ—Ä–ł—á–ł–Ĺ–į–ľ–ł (—ā–į–Ī–Ľ.¬†1). –ü–嬆–ľ–Ĺ–Ķ–Ĺ–ł—é –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į—ā–Ķ–Ľ–Ķ–Ļ, —ā–ĺ–Ľ—Ć–ļ–ĺ —É¬†–ĺ–ī–Ĺ–ĺ–Ļ (1,59%) –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ļ–ł –ł–∑¬†–≥—Ä—É–Ņ–Ņ—č BCD-022 –ł¬†—á–Ķ—ā—č—Ä–Ķ—Ö (6,56%) –ł–∑¬†–≥—Ä—É–Ņ–Ņ—č –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į —Ā—Ä–į–≤–Ĺ–Ķ–Ĺ–ł—Ź —Ā–≤—Ź–∑—Ć —Ā¬†–ł—Ā—Ā–Ľ–Ķ–ī—É–Ķ–ľ–ĺ–Ļ —ā–Ķ—Ä–į–Ņ–ł–Ķ–Ļ –Ī—č–Ľ–į —Ä–į—Ā—Ü–Ķ–Ĺ–Ķ–Ĺ–į –ļ–į–ļ ¬ę–≤–Ķ—Ä–ĺ—Ź—ā–Ĺ–į—Ź¬Ľ –ł–Ľ–ł ¬ę–≤–ĺ–∑–ľ–ĺ–∂–Ĺ–į—Ź¬Ľ.
–°—Ä–Ķ–ī–ł –Ě–Į –Ĺ–į–ł–Ī–ĺ–Ľ–Ķ–Ķ —á–į—Ā—ā–ĺ –≤—Ā—ā—Ä–Ķ—á–į–Ľ–ł—Ā—Ć —Ź–≤–Ľ–Ķ–Ĺ–ł—Ź¬†–≥–Ķ–ľ–į—ā–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ĺ–Ļ —ā–ĺ–ļ—Ā–ł—á–Ĺ–ĺ—Ā—ā–ł, –≤–ļ–Ľ—é—á–į–≤—ą–ł–Ķ –Ĺ–Ķ–Ļ—ā—Ä–ĺ–Ņ–Ķ–Ĺ–ł—é, –Ľ–Ķ–Ļ–ļ–ĺ–Ņ–Ķ–Ĺ–ł—é, –Ľ–ł–ľ—Ą–ĺ–Ņ–Ķ–Ĺ–ł—é, –į–Ĺ–Ķ–ľ–ł—é –ł¬†—ā—Ä–ĺ–ľ–Ī–ĺ—Ü–ł—ā–ĺ–Ņ–Ķ–Ĺ–ł—é. –Ě–Ķ—Ā–ļ–ĺ–Ľ—Ć–ļ–ĺ —Ä–Ķ–∂–Ķ —Ä–Ķ–≥–ł—Ā—ā—Ä–ł—Ä–ĺ–≤–į–Ľ–ł—Ā—Ć –ĺ—ā–ļ–Ľ–ĺ–Ĺ–Ķ–Ĺ–ł—Ź —Ä—Ź–ī–į –Ī–ł–ĺ—Ö–ł–ľ–ł—á–Ķ—Ā–ļ–ł—Ö –Ņ–ĺ–ļ–į–∑–į—ā–Ķ–Ľ–Ķ–Ļ:¬†–≥–ł–Ņ–Ķ—Ä–≥–Ľ–ł–ļ–Ķ–ľ–ł—Ź,¬†–≥–ł–Ņ–Ķ—Ä—É—Ä–ł–ļ–Ķ–ľ–ł—Ź, –Ņ–ĺ–≤—č—ą–Ķ–Ĺ–ł–Ķ —É—Ä–ĺ–≤–Ĺ–Ķ–Ļ –Ľ–į–ļ—ā–į—ā–ī–Ķ–≥–ł–ī—Ä–ĺ–≥–Ķ–Ĺ–į–∑—č (–õ–Ē–ď), –į—Ā–Ņ–į—Ä—ā–į—ā–į–ľ–ł–Ĺ–ĺ—ā—Ä–į–Ĺ—Ā—Ą–Ķ—Ä–į–∑—č (–ź–°–Ę), –į–Ľ–į–Ĺ–ł–Ĺ–į–ľ–ł–Ĺ–ĺ—ā—Ä–į–Ĺ—Ā—Ą–Ķ—Ä–į–∑—č (–ź–õ–Ę), —Č–Ķ–Ľ–ĺ—á–Ĺ–ĺ–Ļ —Ą–ĺ—Ā—Ą–į—ā–į–∑—č (–©–§), –ľ–ĺ—á–Ķ–≤–ł–Ĺ—č. –°—Ä–Ķ–ī–ł –Ņ—Ä–ĺ—á–ł—Ö –Ě–Į –Ĺ–į–ł–Ī–ĺ–Ľ–Ķ–Ķ —Ä–į—Ā–Ņ—Ä–ĺ—Ā—ā—Ä–į–Ĺ–Ķ–Ĺ–Ĺ—č–ľ–ł –Ī—č–Ľ–ł –į–Ľ–ĺ–Ņ–Ķ—Ü–ł–ł, –Ĺ–į—Ä—É—ą–Ķ–Ĺ–ł—Ź —Ā–嬆—Ā—ā–ĺ—Ä–ĺ–Ĺ—č —Ā–ļ–Ķ–Ľ–Ķ—ā–Ĺ–ĺ-–ľ—č—ą–Ķ—á–Ĺ–ĺ–Ļ —Ā–ł—Ā—ā–Ķ–ľ—č (–į—Ä—ā—Ä–į–Ľ–≥–ł–ł, –ľ–ł–į–Ľ–≥–ł–ł, –ĺ—Ā—Ā–į–Ľ–≥–ł–ł, –Ī–ĺ–Ľ—Ć –≤¬†—Ā–Ņ–ł–Ĺ–Ķ), —Ā–嬆—Ā—ā–ĺ—Ä–ĺ–Ĺ—č –Ņ–ł—Č–Ķ–≤–į—Ä–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ–Ļ —Ā–ł—Ā—ā–Ķ–ľ—č (—ā–ĺ—ą–Ĺ–ĺ—ā–į, —Ä–≤–ĺ—ā–į, –ī–ł–į—Ä–Ķ—Ź, —Ā—ā–ĺ–ľ–į—ā–ł—ā), —Ā–嬆—Ā—ā–ĺ—Ä–ĺ–Ĺ—č —Ā–Ķ—Ä–ī–Ķ—á–Ĺ–ĺ-—Ā–ĺ—Ā—É–ī–ł—Ā—ā–ĺ–Ļ —Ā–ł—Ā—ā–Ķ–ľ—č (—ā–į—Ö–ł–ļ–į—Ä–ī–ł—Ź, –Ņ–į—Ä–ĺ–ļ—Ā–ł–∑–ľ–į–Ľ—Ć–Ĺ–į—Ź —Ą–ł–Ī—Ä–ł–Ľ–Ľ—Ź—Ü–ł—Ź –Ņ—Ä–Ķ–ī—Ā–Ķ—Ä–ī–ł–Ļ, –∂–Ķ–Ľ—É–ī–ĺ—á–ļ–ĺ–≤–į—Ź —ć–ļ—Ā—ā—Ä–į—Ā–ł—Ā—ā–ĺ–Ľ–ł—Ź), —Ā–Ľ–į–Ī–ĺ—Ā—ā—Ć,¬†–≥–ĺ–Ľ–ĺ–≤–Ĺ–į—Ź –Ī–ĺ–Ľ—Ć, —Ā–嬆—Ā—ā–ĺ—Ä–ĺ–Ĺ—č –ĺ—Ä–≥–į–Ĺ–ĺ–≤ –ī—č—Ö–į–Ĺ–ł—Ź¬†‚Äď –ĺ—Ā—ā—Ä–į—Ź –Ņ–Ĺ–Ķ–≤–ľ–ĺ–Ĺ–ł—Ź, –ĺ—Ā—ā—Ä—č–Ķ —Ä–Ķ—Ā–Ņ–ł—Ä–į—ā–ĺ—Ä–Ĺ—č–Ķ –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł—Ź. –°—ā–į—ā–ł—Ā—ā–ł—á–Ķ—Ā–ļ–ł –∑–Ĺ–į—á–ł–ľ—č—Ö —Ä–į–∑–Ľ–ł—á–ł–Ļ –ľ–Ķ–∂–ī—ɬ†–≥—Ä—É–Ņ–Ņ–į–ľ–ł –Ĺ–Ķ¬†–≤—č—Ź–≤–Ľ–Ķ–Ĺ–ĺ –Ĺ–ł¬†–Ņ–ĺ –ĺ–ī–Ĺ–ĺ–ľ—É –ł–∑¬†–∑–į—Ä–Ķ–≥–ł—Ā—ā—Ä–ł—Ä–ĺ–≤–į–Ĺ–Ĺ—č—Ö –Ě–Į.
–í–į–∂–Ĺ—č–ľ –Ņ–į—Ä–į–ľ–Ķ—ā—Ä–ĺ–ľ –Ī–Ķ–∑–ĺ–Ņ–į—Ā–Ĺ–ĺ—Ā—ā–ł –Ņ—Ä–ł –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–ł –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–ĺ–≤ –ľ–ĺ–Ĺ–ĺ–ļ–Ľ–ĺ–Ĺ–į–Ľ—Ć–Ĺ—č—Ö –į–Ĺ—ā–ł—ā–Ķ–Ľ —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź –ł–ľ–ľ—É–Ĺ–ĺ–≥–Ķ–Ĺ–Ĺ–ĺ—Ā—ā—Ć. –í¬†–ī–į–Ĺ–Ĺ–ĺ–ľ –ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ĺ–ľ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–ł –ĺ—Ü–Ķ–Ĺ–ł–≤–į–Ľ–ł —á–į—Ā—ā–ĺ—ā—É –ĺ–Ī—Ä–į–∑–ĺ–≤–į–Ĺ–ł—Ź –ł¬†—ā–ł—ā—Ä —Ā–≤—Ź–∑—č–≤–į—é—Č–ł—Ö –ł¬†–Ĺ–Ķ–Ļ—ā—Ä–į–Ľ–ł–∑—É—é—Č–ł—Ö –į–Ĺ—ā–ł—ā–Ķ–Ľ –ļ¬†—ā—Ä–į—Ā—ā—É–∑—É–ľ–į–Ī—É. –ė—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–Ķ –ł–ľ–ľ—É–Ĺ–ĺ–≥–Ķ–Ĺ–Ĺ–ĺ—Ā—ā–ł –≤—č—Ź–≤–ł–Ľ–ĺ —ā—Ä–ł (4,76%) —Ā–Ľ—É—á–į—Ź –Ņ–ĺ—Ź–≤–Ľ–Ķ–Ĺ–ł—Ź –į–Ĺ—ā–ł—ā–Ķ–Ľ –ļ¬†—ā—Ä–į—Ā—ā—É–∑—É–ľ–į–Ī—É –≤¬†–≥—Ä—É–Ņ–Ņ–Ķ –ł—Ā—Ā–Ľ–Ķ–ī—É–Ķ–ľ–ĺ–≥–ĺ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į –ł¬†–ĺ–ī–ł–Ĺ (1,64%) —Ā–Ľ—É—á–į–Ļ –≤¬†–≥—Ä—É–Ņ–Ņ–Ķ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į —Ā—Ä–į–≤–Ĺ–Ķ–Ĺ–ł—Ź (—Ä = 0,619). –ė–∑¬†–Ĺ–ł—Ö –Ĺ–Ķ–Ļ—ā—Ä–į–Ľ–ł–∑—É—é—Č–į—Ź –į–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć –Ī—č–Ľ–į –≤—č—Ź–≤–Ľ–Ķ–Ĺ–į —ɬ†–ĺ–ī–Ĺ–ĺ–Ļ –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ļ–ł –ł–∑¬†–≥—Ä—É–Ņ–Ņ—č –ł—Ā—Ā–Ľ–Ķ–ī—É–Ķ–ľ–ĺ–≥–ĺ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į –ł¬†–ĺ–ī–Ĺ–ĺ–Ļ –Ī–ĺ–Ľ—Ć–Ĺ–ĺ–Ļ –ł–∑¬†–≥—Ä—É–Ņ–Ņ—č —Ā—Ä–į–≤–Ĺ–Ķ–Ĺ–ł—Ź (—Ä = 1,0). –Ę–į–ļ–ł–ľ –ĺ–Ī—Ä–į–∑–ĺ–ľ, —Ā—ā–į—ā–ł—Ā—ā–ł—á–Ķ—Ā–ļ–ł –∑–Ĺ–į—á–ł–ľ—č—Ö —Ä–į–∑–Ľ–ł—á–ł–Ļ –ľ–Ķ–∂–ī—ɬ†–≥—Ä—É–Ņ–Ņ–į–ľ–ł –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į BCD-022 –ł¬†–Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į –ď–Ķ—Ä—Ü–Ķ–Ņ—ā–ł–Ŭģ –Ņ–嬆—á–į—Ā—ā–ĺ—ā–Ķ –ĺ–Ī—Ä–į–∑–ĺ–≤–į–Ĺ–ł—Ź –ļ–į–ļ —Ā–≤—Ź–∑—č–≤–į—é—Č–ł—Ö, —ā–į–ļ –ł¬†–Ĺ–Ķ–Ļ—ā—Ä–į–Ľ–ł–∑—É—é—Č–ł—Ö –į–Ĺ—ā–ł—ā–Ķ–Ľ –Ĺ–Ķ¬†–Ī—č–Ľ–ĺ. –ě–Ī–į –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į —Ö–į—Ä–į–ļ—ā–Ķ—Ä–ł–∑–ĺ–≤–į–Ľ–ł—Ā—Ć –Ĺ–ł–∑–ļ–ĺ–Ļ —á–į—Ā—ā–ĺ—ā–ĺ–Ļ –≤—č—Ź–≤–Ľ–Ķ–Ĺ–ł—Ź –į–Ĺ—ā–ł—ā–Ķ–Ľ –ļ¬†—ā—Ä–į—Ā—ā—É–∑—É–ľ–į–Ī—É.
–§–į—Ä–ľ–į–ļ–ĺ–ļ–ł–Ĺ–Ķ—ā–ł–ļ—É –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–ĺ–≤ –ĺ—Ü–Ķ–Ĺ–ł–≤–į–Ľ–ł –Ņ–嬆—Ä–Ķ–∑—É–Ľ—Ć—ā–į—ā–į–ľ –ĺ–Ņ—Ä–Ķ–ī–Ķ–Ľ–Ķ–Ĺ–ł—Ź —ā—Ä–į—Ā—ā—É–∑—É–ľ–į–Ī–į –≤¬†—Ā—č–≤–ĺ—Ä–ĺ—ā–ļ–Ķ –ļ—Ä–ĺ–≤–ł –Ī–ĺ–Ľ—Ć–Ĺ—č—Ö –≤¬†–ļ–ĺ–Ĺ–ļ—Ä–Ķ—ā–Ĺ—č–Ķ –≤—Ä–Ķ–ľ–Ķ–Ĺ–Ĺ—č–Ķ —ā–ĺ—á–ļ–ł. –ü—Ä–ĺ–≤–ĺ–ī–ł–Ľ–ł –į–Ĺ–į–Ľ–ł–∑ —Ā—ā–į–Ĺ–ī–į—Ä—ā–Ĺ—č—Ö —Ą–į—Ä–ľ–į–ļ–ĺ–ļ–ł–Ĺ–Ķ—ā–ł—á–Ķ—Ā–ļ–ł—Ö –Ņ–į—Ä–į–ľ–Ķ—ā—Ä–ĺ–≤ –Ņ–ĺ—Ā–Ľ–Ķ –Ņ–Ķ—Ä–≤–ĺ–≥–ĺ –ł¬†—ą–Ķ—Ā—ā–ĺ–≥–ĺ —Ü–ł–ļ–Ľ–ĺ–≤ —ā–Ķ—Ä–į–Ņ–ł–ł, –į¬†—ā–į–ļ–∂–Ķ –ĺ–Ņ—Ä–Ķ–ī–Ķ–Ľ—Ź–Ľ–ł –ļ–ĺ–Ĺ—Ü–Ķ–Ĺ—ā—Ä–į—Ü–ł–ł —ā—Ä–į—Ā—ā—É–∑—É–ľ–į–Ī–į –Ņ–Ķ—Ä–Ķ–ī –ļ–į–∂–ī—č–ľ –≤–≤–Ķ–ī–Ķ–Ĺ–ł–Ķ–ľ —Ā¬†–Ņ–ĺ—Ā–Ľ–Ķ–ī—É—é—Č–ł–ľ —Ä–į—Ā—á–Ķ—ā–ĺ–ľ –°trough. –ü—Ä–ĺ–≤–Ķ–ī–Ķ–Ĺ–Ĺ—č–Ļ –į–Ĺ–į–Ľ–ł–∑ –Ņ–ĺ–ļ–į–∑–į–Ľ, —á—ā–ĺ –ļ–į–ļ –Ņ–ĺ—Ā–Ľ–Ķ –ĺ–ī–Ĺ–ĺ–ļ—Ä–į—ā–Ĺ–ĺ–≥–ĺ, —ā–į–ļ –ł¬†–Ņ–ĺ—Ā–Ľ–Ķ –ľ–Ĺ–ĺ–≥–ĺ–ļ—Ä–į—ā–Ĺ–ĺ–≥–ĺ –≤–≤–Ķ–ī–Ķ–Ĺ–ł—Ź –ł—Ā—Ā–Ľ–Ķ–ī—É–Ķ–ľ–ĺ–≥–ĺ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į –ł¬†–Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į —Ā—Ä–į–≤–Ĺ–Ķ–Ĺ–ł—Ź –ļ–ĺ–Ĺ—Ü–Ķ–Ĺ—ā—Ä–į—Ü–ł–ł —ā—Ä–į—Ā—ā—É–∑—É–ľ–į–Ī–į –≤¬†–ļ—Ä–ĺ–≤–ł –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–ļ –ł–∑–ľ–Ķ–Ĺ—Ź–Ľ–ł—Ā—Ć –į–Ĺ–į–Ľ–ĺ–≥–ł—á–Ĺ—č–ľ –ĺ–Ī—Ä–į–∑–ĺ–ľ.
–Ě–į—Ā—ā–ĺ—Ź—Č–Ķ–Ķ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–Ķ –≤—č–Ņ–ĺ–Ľ–Ĺ–Ķ–Ĺ–ĺ —Ā¬†—É—á–Ķ—ā–ĺ–ľ –Ķ–≤—Ä–ĺ–Ņ–Ķ–Ļ—Ā–ļ–ł—Ö —Ä–Ķ–ļ–ĺ–ľ–Ķ–Ĺ–ī–į—Ü–ł–Ļ –Ņ–嬆–ĺ—Ü–Ķ–Ĺ–ļ–Ķ –Ī–ł–ĺ—ć–ļ–≤–ł–≤–į–Ľ–Ķ–Ĺ—ā–Ĺ–ĺ—Ā—ā–ł, –į¬†—ā–į–ļ–∂–Ķ —Ä—É–ļ–ĺ–≤–ĺ–ī—Ā—ā–≤–į –Ņ–嬆–ł–∑—É—á–Ķ–Ĺ–ł—é –Ī–ł–ĺ–į–Ĺ–į–Ľ–ĺ–≥–ĺ–≤ –ľ–ĺ–Ĺ–ĺ–ļ–Ľ–ĺ–Ĺ–į–Ľ—Ć–Ĺ—č—Ö –į–Ĺ—ā–ł—ā–Ķ–Ľ. –°—ā–į—ā–ł—Ā—ā–ł—á–Ķ—Ā–ļ–ĺ–Ķ —Ā—Ä–į–≤–Ĺ–Ķ–Ĺ–ł–Ķ –Ņ–嬆–Ņ–Ķ—Ä–≤–ł—á–Ĺ–ĺ–Ļ —Ą–į—Ä–ľ–į–ļ–ĺ–ļ–ł–Ĺ–Ķ—ā–ł—á–Ķ—Ā–ļ–ĺ–Ļ –ļ–ĺ–Ĺ–Ķ—á–Ĺ–ĺ–Ļ —ā–ĺ—á–ļ–Ķ (AUC(0‚Äď504)) –Ņ–ĺ–∑–≤–ĺ–Ľ–ł–Ľ–ĺ –Ņ–ĺ–Ľ—É—á–ł—ā—Ć 90% –Ē–ė –ī–Ľ—Ź –ĺ—ā–Ĺ–ĺ—ą–Ķ–Ĺ–ł—Ź —Ā—Ä–Ķ–ī–Ĺ–ł—Ö¬†–≥–Ķ–ĺ–ľ–Ķ—ā—Ä–ł—á–Ķ—Ā–ļ–ł—Ö AUC(0‚Äď504)¬†‚Äď 83,31‚Äď113,55%. –Ē–į–Ĺ–Ĺ—č–Ļ –ł–Ĺ—ā–Ķ—Ä–≤–į–Ľ —Ā–ĺ–ĺ—ā–≤–Ķ—ā—Ā—ā–≤—É–Ķ—ā —É—Ā—ā–į–Ĺ–ĺ–≤–Ľ–Ķ–Ĺ–Ĺ—č–ľ –Ņ—Ä–Ķ–ī–Ķ–Ľ–į–ľ —ć–ļ–≤–ł–≤–į–Ľ–Ķ–Ĺ—ā–Ĺ–ĺ—Ā—ā–ł —Ą–į—Ä–ľ–į–ļ–ĺ–ļ–ł–Ĺ–Ķ—ā–ł—á–Ķ—Ā–ļ–ł—Ö –Ņ–ĺ–ļ–į–∑–į—ā–Ķ–Ľ–Ķ–Ļ 80‚Äď125%. 90% –Ē–ė –ī–Ľ—Ź –ĺ—ā–Ĺ–ĺ—ą–Ķ–Ĺ–ł—Ź —Ā—Ä–Ķ–ī–Ĺ–ł—Ö¬†–≥–Ķ–ĺ–ľ–Ķ—ā—Ä–ł—á–Ķ—Ā–ļ–ł—Ö –°m–į—Ö –ł—Ā—Ā–Ľ–Ķ–ī—É–Ķ–ľ–ĺ–≥–ĺ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į –ł¬†–Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į —Ā—Ä–į–≤–Ĺ–Ķ–Ĺ–ł—Ź —Ā–ĺ—Ā—ā–į–≤–ł–Ľ 88,33‚Äď111,14%, —á—ā–ĺ —ā–į–ļ–∂–Ķ —Ā–ĺ–ĺ—ā–≤–Ķ—ā—Ā—ā–≤—É–Ķ—ā –ł–Ĺ—ā–Ķ—Ä–≤–į–Ľ—É 80‚Äď125%. –°—Ä–į–≤–Ĺ–Ķ–Ĺ–ł–Ķ –ł—Ā–Ņ—č—ā—É–Ķ–ľ—č—Ö¬†–≥—Ä—É–Ņ–Ņ –Ņ–嬆–Ņ—Ä–ĺ—á–ł–ľ –≤—ā–ĺ—Ä–ł—á–Ĺ—č–ľ –ļ–ĺ–Ĺ–Ķ—á–Ĺ—č–ľ —ā–ĺ—á–ļ–į–ľ (–Ęmax, T1/2) —Ā¬†–Ņ–ĺ–ľ–ĺ—Č—Ć—é —Ā—ā–į–Ĺ–ī–į—Ä—ā–Ĺ—č—Ö —Ā—ā–į—ā–ł—Ā—ā–ł—á–Ķ—Ā–ļ–ł—Ö –ļ—Ä–ł—ā–Ķ—Ä–ł–Ķ–≤ —ā–į–ļ–∂–Ķ –Ņ–ĺ–ļ–į–∑–į–Ľ–ĺ –ĺ—ā—Ā—É—ā—Ā—ā–≤–ł–Ķ —Ā—ā–į—ā–ł—Ā—ā–ł—á–Ķ—Ā–ļ–ł –∑–Ĺ–į—á–ł–ľ—č—Ö —Ä–į–∑–Ľ–ł—á–ł–Ļ. –ü—Ä–ł –į–Ĺ–į–Ľ–ł–∑–Ķ –Ĺ–į¬†—ą–Ķ—Ā—ā–ĺ–ľ —Ü–ł–ļ–Ľ–Ķ —Ā—ā–į—ā–ł—Ā—ā–ł—á–Ķ—Ā–ļ–ĺ–Ķ —Ā—Ä–į–≤–Ĺ–Ķ–Ĺ–ł–Ķ –Ņ–嬆–≤—Ā–Ķ–ľ –ĺ—Ā–Ĺ–ĺ–≤–Ĺ—č–ľ —Ą–į—Ä–ľ–į–ļ–ĺ–ļ–ł–Ĺ–Ķ—ā–ł—á–Ķ—Ā–ļ–ł–ľ –Ņ–į—Ä–į–ľ–Ķ—ā—Ä–į–ľ (AUC(0-504), –°m–į—Ö, –Ęmax, T1/2) —Ā¬†–Ņ–ĺ–ľ–ĺ—Č—Ć—é —Ā—ā–į–Ĺ–ī–į—Ä—ā–Ĺ—č—Ö —Ā—ā–į—ā–ł—Ā—ā–ł—á–Ķ—Ā–ļ–ł—Ö –ļ—Ä–ł—ā–Ķ—Ä–ł–Ķ–≤ –Ņ—Ä–ĺ–ī–Ķ–ľ–ĺ–Ĺ—Ā—ā—Ä–ł—Ä–ĺ–≤–į–Ľ–ĺ –ĺ—ā—Ā—É—ā—Ā—ā–≤–ł–Ķ —Ā—ā–į—ā–ł—Ā—ā–ł—á–Ķ—Ā–ļ–ł –∑–Ĺ–į—á–ł–ľ—č—Ö —Ä–į–∑–Ľ–ł—á–ł–Ļ –ľ–Ķ–∂–ī—ɬ†–≥—Ä—É–Ņ–Ņ–į–ľ–ł. –°—ā–į—ā–ł—Ā—ā–ł—á–Ķ—Ā–ļ–ł –∑–Ĺ–į—á–ł–ľ—č—Ö —Ä–į–∑–Ľ–ł—á–ł–Ļ –Ņ–嬆–Ņ–ĺ–ļ–į–∑–į—ā–Ķ–Ľ—é Ctrough –ľ–Ķ–∂–ī—ɬ†–≥—Ä—É–Ņ–Ņ–į–ľ–ł –Ĺ–Ķ¬†–∑–į—Ä–Ķ–≥–ł—Ā—ā—Ä–ł—Ä–ĺ–≤–į–Ĺ–ĺ –Ĺ–ł¬†–≤¬†–ĺ–ī–Ĺ–ĺ–ľ –ł–∑¬†—Ü–ł–ļ–Ľ–ĺ–≤. –°–Ľ–Ķ–ī–ĺ–≤–į—ā–Ķ–Ľ—Ć–Ĺ–ĺ, –ľ–ĺ–∂–Ĺ–ĺ —Ā–ī–Ķ–Ľ–į—ā—Ć –≤—č–≤–ĺ–ī –ĺ–Ī¬†—ć–ļ–≤–ł–≤–į–Ľ–Ķ–Ĺ—ā–Ĺ–ĺ—Ā—ā–ł —Ą–į—Ä–ľ–į–ļ–ĺ–ļ–ł–Ĺ–Ķ—ā–ł—á–Ķ—Ā–ļ–ł—Ö —Ā–≤–ĺ–Ļ—Ā—ā–≤ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–ĺ–≤ BCD-022 –ł¬†–ď–Ķ—Ä—Ü–Ķ–Ņ—ā–ł–Ŭģ –Ņ—Ä–ł –≤–Ĺ—É—ā—Ä–ł–≤–Ķ–Ĺ–Ĺ–ĺ–ľ –≤–≤–Ķ–ī–Ķ–Ĺ–ł–ł.
–†–Ķ–∑—é–ľ–ł—Ä—É—Ź –ł–Ĺ—Ą–ĺ—Ä–ľ–į—Ü–ł—é, –Ņ–ĺ–Ľ—É—á–Ķ–Ĺ–Ĺ—É—é –≤¬†—Ä–į–ľ–ļ–į—Ö –ļ—Ä—É–Ņ–Ĺ–ĺ–ľ–į—Ā—ą—ā–į–Ī–Ĺ–ĺ–≥–ĺ –ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ĺ–≥–ĺ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź –Ī–ł–ĺ–į–Ĺ–į–Ľ–ĺ–≥–į —ā—Ä–į—Ā—ā—É–∑—É–ľ–į–Ī–į, –ī–ĺ–ļ–Ľ–į–ī—á–ł–ļ, –ļ–ĺ—ā–ĺ—Ä–į—Ź —ā–į–ļ–∂–Ķ —Ź–≤–Ľ—Ź–Ľ–į—Ā—Ć –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į—ā–Ķ–Ľ–Ķ–ľ –≤¬†–ĺ–ī–Ĺ–ĺ–ľ –ł–∑¬†–≤–Ķ–ī—É—Č–ł—Ö –ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ł—Ö —Ü–Ķ–Ĺ—ā—Ä–ĺ–≤, –ĺ—ā–ľ–Ķ—ā–ł–Ľ–į, —á—ā–ĺ –ī–į–Ĺ–Ĺ—č–Ķ —Ā–≤–ł–ī–Ķ—ā–Ķ–Ľ—Ć—Ā—ā–≤—É—é—ā –嬆—ā–Ķ—Ä–į–Ņ–Ķ–≤—ā–ł—á–Ķ—Ā–ļ–ĺ–Ļ —ć–ļ–≤–ł–≤–į–Ľ–Ķ–Ĺ—ā–Ĺ–ĺ—Ā—ā–ł –ł—Ā—Ā–Ľ–Ķ–ī—É–Ķ–ľ–ĺ–≥–ĺ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į BCD-022 –ł¬†–Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į —Ā—Ä–į–≤–Ĺ–Ķ–Ĺ–ł—Ź –ď–Ķ—Ä—Ü–Ķ–Ņ—ā–ł–Ŭģ.
–ė—ā–ĺ–≥–ł –ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ĺ–≥–ĺ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź —Ą–į–∑—č III –Ī–ł–ĺ–į–Ĺ–į–Ľ–ĺ–≥–į –Ī–Ķ–≤–į—Ü–ł–∑—É–ľ–į–Ī–į –≤¬†–Ņ–ĺ–Ņ—É–Ľ—Ź—Ü–ł–ł –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ —Ā¬†–Ĺ–Ķ–ľ–Ķ–Ľ–ļ–ĺ–ļ–Ľ–Ķ—ā–ĺ—á–Ĺ—č–ľ —Ä–į–ļ–ĺ–ľ –Ľ–Ķ–≥–ļ–ĺ–≥–ĺ
–í–Ķ–ī—É—Č–ł–Ļ –Ĺ–į—É—á–Ĺ—č–Ļ —Ā–ĺ—ā—Ä—É–ī–Ĺ–ł–ļ –ĺ—ā–ī–Ķ–Ľ–į —Ö–ł—Ä—É—Ä–≥–ł—á–Ķ—Ā–ļ–ĺ–Ļ –Ņ—É–Ľ—Ć–ľ–ĺ–Ĺ–ĺ–Ľ–ĺ–≥–ł–ł –°–į–Ĺ–ļ—ā-–ü–Ķ—ā–Ķ—Ä–Ī—É—Ä–≥—Ā–ļ–ĺ–≥–嬆–≥–ĺ—Ā—É–ī–į—Ä—Ā—ā–≤–Ķ–Ĺ–Ĺ–ĺ–≥–ĺ –ľ–Ķ–ī–ł—Ü–ł–Ĺ—Ā–ļ–ĺ–≥–ĺ —É–Ĺ–ł–≤–Ķ—Ä—Ā–ł—ā–Ķ—ā–į –ł–ľ.¬†–į–ļ–į–ī.¬†–ė.–ü.¬†–ü–į–≤–Ľ–ĺ–≤–į, –ī.–ľ.–Ĺ. –°–Ķ—Ä–≥–Ķ–Ļ –í–Ľ–į–ī–ł–ľ–ł—Ä–ĺ–≤–ł—á –ě–†–õ–ě–í –Ĺ–į—á–į–Ľ —Ā–≤–ĺ–Ķ –≤—č—Ā—ā—É–Ņ–Ľ–Ķ–Ĺ–ł–Ķ —Ā¬†–ļ—Ä–į—ā–ļ–ĺ–≥–ĺ –Ĺ–į–Ņ–ĺ–ľ–ł–Ĺ–į–Ĺ–ł—Ź –嬆—ā–ĺ–ľ, –ļ–į–ļ–ĺ–Ļ —Ā–Ķ—Ä—Ć–Ķ–∑–Ĺ–ĺ–Ļ –Ņ—Ä–ĺ–Ī–Ľ–Ķ–ľ–ĺ–Ļ –ī–Ľ—Ź —Ā–ĺ–≤—Ä–Ķ–ľ–Ķ–Ĺ–Ĺ–ĺ–Ļ –ĺ–Ĺ–ļ–ĺ–Ľ–ĺ–≥–ł–ł —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź –Ľ–Ķ—á–Ķ–Ĺ–ł–Ķ —Ä–į–ļ–į –Ľ–Ķ–≥–ļ–ĺ–≥–ĺ, –嬆–ī–ĺ—Ā—ā–ł–∂–Ķ–Ĺ–ł—Ź—Ö –≤¬†—ā–Ķ—Ä–į–Ņ–ł–ł —ć—ā–ĺ–≥–ĺ –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł—Ź –ł¬†–ľ–Ķ—Ā—ā–Ķ –Ī–Ķ–≤–į—Ü–ł–∑—É–ľ–į–Ī–į –≤¬†—Ā—Ö–Ķ–ľ–Ķ –Ľ–Ķ—á–Ķ–Ĺ–ł—Ź.
–†–į–ļ –Ľ–Ķ–≥–ļ–ĺ–≥–嬆‚Äď –ĺ–ī–Ĺ–ĺ –ł–∑¬†—Ā–į–ľ—č—Ö —Ä–į—Ā–Ņ—Ä–ĺ—Ā—ā—Ä–į–Ĺ–Ķ–Ĺ–Ĺ—č—Ö –∑–Ľ–ĺ–ļ–į—á–Ķ—Ā—ā–≤–Ķ–Ĺ–Ĺ—č—Ö –Ĺ–ĺ–≤–ĺ–ĺ–Ī—Ä–į–∑–ĺ–≤–į–Ĺ–ł–Ļ –≤¬†–ľ–ł—Ä–Ķ. –†–į–ļ –Ľ–Ķ–≥–ļ–ĺ–≥–ĺ –∑–į–Ĺ–ł–ľ–į–Ķ—ā —ā—Ä–Ķ—ā—Ć–Ķ –ľ–Ķ—Ā—ā–ĺ –Ņ–嬆—á–ł—Ā–Ľ—É –Ķ–∂–Ķ–≥–ĺ–ī–Ĺ–ĺ –≤—č—Ź–≤–Ľ—Ź–Ķ–ľ—č—Ö —Ā–Ľ—É—á–į–Ķ–≤ –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ķ–ľ–ĺ—Ā—ā–ł –ł¬†–Ņ–Ķ—Ä–≤–ĺ–Ķ –Ņ–嬆–Ķ–∂–Ķ–≥–ĺ–ī–Ĺ–ĺ–ľ—É —á–ł—Ā–Ľ—É —Ā–ľ–Ķ—Ä—ā–Ķ–Ļ —Ā—Ä–Ķ–ī–ł –≤—Ā–Ķ—Ö –∑–Ľ–ĺ–ļ–į—á–Ķ—Ā—ā–≤–Ķ–Ĺ–Ĺ—č—Ö –ĺ–Ņ—É—Ö–ĺ–Ľ–Ķ–Ļ. –ü—Ä–ł —ć—ā–ĺ–ľ –Ĺ–į¬†–ī–ĺ–Ľ—é –Ĺ–Ķ–ľ–Ķ–Ľ–ļ–ĺ–ļ–Ľ–Ķ—ā–ĺ—á–Ĺ–ĺ–≥–ĺ —Ä–į–ļ–į –Ľ–Ķ–≥–ļ–ĺ–≥–ĺ (–Ě–ú–†–õ) –≤¬†—Ā—ā—Ä—É–ļ—ā—É—Ä–Ķ –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ķ–ľ–ĺ—Ā—ā–ł —Ä–į–ļ–ĺ–ľ –Ľ–Ķ–≥–ļ–ĺ–≥–ĺ –Ņ—Ä–ł—Ö–ĺ–ī–ł—ā—Ā—Ź 80%. –ó–į –Ņ–ĺ—Ā–Ľ–Ķ–ī–Ĺ–ł–Ķ –ī–Ķ—Ā—Ź—ā–ł–Ľ–Ķ—ā–ł—Ź –Ī–Ľ–į–≥–ĺ–ī–į—Ä—Ź —Ä–į–∑–≤–ł—ā–ł—é –∑–Ĺ–į–Ĺ–ł–Ļ –嬆–ľ–ĺ–Ľ–Ķ–ļ—É–Ľ—Ź—Ä–Ĺ—č—Ö –ľ–Ķ—Ö–į–Ĺ–ł–∑–ľ–į—Ö –Ņ–į—ā–ĺ–≥–Ķ–Ĺ–Ķ–∑–į –Ě–ú–†–õ –ī–ĺ—Ā—ā–ł–≥–Ĺ—É—ā—č –∑–į–ľ–Ķ—ā–Ĺ—č–Ķ —É—Ā–Ņ–Ķ—Ö–ł –≤¬†—Ä–į–∑—Ä–į–Ī–ĺ—ā–ļ–Ķ –Ņ–ĺ–ī—Ö–ĺ–ī–ĺ–≤ –ļ¬†–Ľ–Ķ—á–Ķ–Ĺ–ł—é —ć—ā–ĺ–≥–ĺ –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł—Ź, –≤¬†–Ņ–Ķ—Ä–≤—É—é –ĺ—á–Ķ—Ä–Ķ–ī—Ć –Ĺ–į–Ņ—Ä–į–≤–Ľ–Ķ–Ĺ–Ĺ–ĺ–Ļ, —ā–į—Ä–≥–Ķ—ā–Ĺ–ĺ–Ļ —ā–Ķ—Ä–į–Ņ–ł–ł. –í–Ĺ–Ķ–ī—Ä–Ķ–Ĺ—č –≤¬†–ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ—É—é –Ņ—Ä–į–ļ—ā–ł–ļ—É –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā—č, –Ī–Ľ–ĺ–ļ–ł—Ä—É—é—Č–ł–Ķ —Ą–į–ļ—ā–ĺ—Ä —Ä–ĺ—Ā—ā–į —ć–Ĺ–ī–ĺ—ā–Ķ–Ľ–ł—Ź —Ā–ĺ—Ā—É–ī–ĺ–≤ (VEGF), —Ä–Ķ—Ü–Ķ–Ņ—ā–ĺ—Ä —ć–Ņ–ł–ī–Ķ—Ä–ľ–į–Ľ—Ć–Ĺ–ĺ–≥–ĺ —Ą–į–ļ—ā–ĺ—Ä–į —Ä–ĺ—Ā—ā–į (EGFR), –ļ–ł–Ĺ–į–∑—É –į–Ĺ–į–Ņ–Ľ–į—Ā—ā–ł—á–Ķ—Ā–ļ–ĺ–Ļ –Ľ–ł–ľ—Ą–ĺ–ľ—č (ALK).
–ě–ī–ł–Ĺ –ł–∑¬†–Ņ–Ķ—Ä–≤—č—Ö –ł¬†–Ĺ–į–ł–Ī–ĺ–Ľ–Ķ–Ķ –ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ł –ł–∑—É—á–Ķ–Ĺ–Ĺ—č—Ö —ā–į—Ä–≥–Ķ—ā–Ĺ—č—Ö –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–ĺ–≤ –≤¬†–ĺ–Ĺ–ļ–ĺ–Ľ–ĺ–≥–ł–ł¬†‚Äď –Ī–Ķ–≤–į—Ü–ł–∑—É–ľ–į–Ī,¬†–≥—É–ľ–į–Ĺ–ł–∑–ł—Ä–ĺ–≤–į–Ĺ–Ĺ–ĺ–Ķ –ľ–ĺ–Ĺ–ĺ–ļ–Ľ–ĺ–Ĺ–į–Ľ—Ć–Ĺ–ĺ–Ķ –į–Ĺ—ā–ł—ā–Ķ–Ľ–ĺ –ļ¬†VEGF. –ü—Ä–Ķ–Ņ–į—Ä–į—ā –ĺ–Ī–Ľ–į–ī–į–Ķ—ā –ī–ĺ–ļ–į–∑–į–Ĺ–Ĺ–ĺ–Ļ –≤¬†–ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ł—Ö –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź—Ö —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć—é –≤¬†—ā–Ķ—Ä–į–Ņ–ł–ł –Ņ–ĺ–∑–ī–Ĺ–ł—Ö —Ā—ā–į–ī–ł–Ļ –Ĺ–Ķ–ľ–Ķ–Ľ–ļ–ĺ–ļ–Ľ–Ķ—ā–ĺ—á–Ĺ–ĺ–≥–ĺ —Ä–į–ļ–į –Ľ–Ķ–≥–ļ–ĺ–≥–ĺ; –Ķ–≥–ĺ –ł—Ā–Ņ–ĺ–Ľ—Ć–∑–ĺ–≤–į–Ĺ–ł–Ķ –Ĺ–Ķ¬†—ā–ĺ–Ľ—Ć–ļ–ĺ —É–≤–Ķ–Ľ–ł—á–ł–≤–į–Ķ—ā —á–į—Ā—ā–ĺ—ā—É –ĺ–Ņ—É—Ö–ĺ–Ľ–Ķ–≤–ĺ–≥–ĺ –ĺ—ā–≤–Ķ—ā–į –Ĺ–į¬†—Ö–ł–ľ–ł–ĺ—ā–Ķ—Ä–į–Ņ–ł—é –ł¬†–≤—č–∂–ł–≤–į–Ķ–ľ–ĺ—Ā—ā—Ć –Ī–Ķ–∑ –Ņ—Ä–ĺ–≥—Ä–Ķ—Ā—Ā–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł—Ź, –Ŗ嬆–ł¬†–Ņ—Ä–ĺ–ī–Ľ–Ķ–≤–į–Ķ—ā –∂–ł–∑–Ĺ—Ć –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤. –°–ĺ–≥–Ľ–į—Ā–Ĺ–ĺ –į–ļ—ā—É–į–Ľ—Ć–Ĺ—č–ľ –ľ–Ķ–∂–ī—É–Ĺ–į—Ä–ĺ–ī–Ĺ—č–ľ —Ä–Ķ–ļ–ĺ–ľ–Ķ–Ĺ–ī–į—Ü–ł—Ź–ľ, –Ņ—Ä–ł–ľ–Ķ–Ĺ–Ķ–Ĺ–ł–Ķ –Ī–Ķ–≤–į—Ü–ł–∑—É–ľ–į–Ī–į –Ņ–ĺ–ļ–į–∑–į–Ĺ–ĺ –≤¬†–Ņ–Ķ—Ä–≤–ĺ–Ļ –Ľ–ł–Ĺ–ł–ł –Ľ–Ķ—á–Ķ–Ĺ–ł—Ź —Ä–į—Ā–Ņ—Ä–ĺ—Ā—ā—Ä–į–Ĺ–Ķ–Ĺ–Ĺ–ĺ–≥–ĺ –Ĺ–Ķ–Ņ–Ľ–ĺ—Ā–ļ–ĺ–ļ–Ľ–Ķ—ā–ĺ—á–Ĺ–ĺ–≥–ĺ –Ě–ú–†–õ. –ö¬†—Ā–ĺ–∂–į–Ľ–Ķ–Ĺ–ł—é, –≤–ĺ–∑–ľ–ĺ–∂–Ĺ–ĺ—Ā—ā–ł –ł—Ā–Ņ–ĺ–Ľ—Ć–∑–ĺ–≤–į–Ĺ–ł—Ź –Ī–Ķ–≤–į—Ü–ł–∑—É–ľ–į–Ī–į –≤¬†–ļ–Ľ–ł–Ĺ–ł–ļ–Ķ –∑–Ĺ–į—á–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ –ĺ–≥—Ä–į–Ĺ–ł—á–ł–≤–į—é—ā—Ā—Ź –≤—č—Ā–ĺ–ļ–ĺ–Ļ —Ā—ā–ĺ–ł–ľ–ĺ—Ā—ā—Ć—é –ĺ—Ä–ł–≥–ł–Ĺ–į–Ľ—Ć–Ĺ–ĺ–≥–ĺ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į.
–ü–Ķ—Ä—Ā–Ņ–Ķ–ļ—ā–ł–≤–Ĺ—č–Ļ –Ņ—É—ā—Ć –Ņ–ĺ–≤—č—ą–Ķ–Ĺ–ł—Ź –ī–ĺ—Ā—ā—É–Ņ–Ĺ–ĺ—Ā—ā–ł —Ā–ĺ–≤—Ä–Ķ–ľ–Ķ–Ĺ–Ĺ–ĺ–Ļ —ā–Ķ—Ä–į–Ņ–ł–ł, –į–ļ—ā–ł–≤–Ĺ–ĺ —Ä–į–∑–≤–ł–≤–į—é—Č–ł–Ļ—Ā—Ź –≤¬†–Ĺ–į—ą–ł –ī–Ĺ–ł –≤–ĺ –≤—Ā–Ķ–ľ¬†–ľ–ł—Ä–Ķ, –≤¬†—ā–ĺ–ľ —á–ł—Ā–Ľ–Ķ –≤¬†–°–®–ź –ł¬†–ē–≤—Ä–ĺ–Ņ–Ķ–Ļ—Ā–ļ–ĺ–ľ –°–ĺ—é–∑–Ķ,¬†‚Äď –≤–Ĺ–Ķ–ī—Ä–Ķ–Ĺ–ł–Ķ –≤¬†–ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ—É—é –Ņ—Ä–į–ļ—ā–ł–ļ—É –Ī–ł–ĺ–į–Ĺ–į–Ľ–ĺ–≥–ĺ–≤. –ö–ĺ–ľ–Ņ–į–Ĺ–ł–Ķ–Ļ BIOCAD —Ä–į–∑—Ä–į–Ī–ĺ—ā–į–Ĺ –Ņ–Ķ—Ä–≤—č–Ļ –≤¬†–†–ĺ—Ā—Ā–ł–ł –Ī–ł–ĺ–į–Ĺ–į–Ľ–ĺ–≥ –Ī–Ķ–≤–į—Ü–ł–∑—É–ľ–į–Ī–į. –°.–í. –ě—Ä–Ľ–ĺ–≤ —Ä–į—Ā—Ā–ļ–į–∑–į–Ľ –嬆–Ķ–≥–ĺ —Ä–į–∑—Ä–į–Ī–ĺ—ā–ļ–Ķ, –Ņ–ĺ–ī—Ä–ĺ–Ī–Ĺ–ĺ –ĺ—Ā—ā–į–Ĺ–ĺ–≤–ł–≤—ą–ł—Ā—Ć –Ĺ–į¬†—Ä–Ķ–∑—É–Ľ—Ć—ā–į—ā–į—Ö –Ņ—Ä–ĺ–≤–Ķ–ī–Ķ–Ĺ–Ĺ–ĺ–≥–ĺ –ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ĺ–≥–ĺ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź —Ą–į–∑—č III, –ł¬†–Ņ–ĺ–ī–Ķ–Ľ–ł–Ľ—Ā—Ź –Ľ–ł—á–Ĺ—č–ľ –ĺ–Ņ—č—ā–ĺ–ľ –Ņ—Ä–ł–ľ–Ķ–Ĺ–Ķ–Ĺ–ł—Ź –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į –≤¬†–ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ĺ–Ļ –Ņ—Ä–į–ļ—ā–ł–ļ–Ķ.
–Ě–į¬†–ī–ĺ–ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ĺ–ľ —ć—ā–į–Ņ–Ķ —Ä–į–∑—Ä–į–Ī–ĺ—ā–ļ–ł –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į –Ī—č–Ľ –Ņ—Ä–ĺ–≤–Ķ–ī–Ķ–Ĺ –≤–Ķ—Ā—Ć –Ĺ–Ķ–ĺ–Ī—Ö–ĺ–ī–ł–ľ—č–Ļ –ĺ–Ī—ä–Ķ–ľ —Ā—Ä–į–≤–Ĺ–ł—ā–Ķ–Ľ—Ć–Ĺ—č—Ö –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–Ļ —Ā¬†–ĺ—Ä–ł–≥–ł–Ĺ–į–Ľ—Ć–Ĺ—č–ľ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–ĺ–ľ –Ī–Ķ–≤–į—Ü–ł–∑—É–ľ–į–Ī–į –ź–≤–į—Ā—ā–ł–Ŭģ, –≤¬†—Ö–ĺ–ī–Ķ –ļ–ĺ—ā–ĺ—Ä—č—Ö –ĺ—Ü–Ķ–Ĺ–ł–≤–į–Ľ–ł —Ā—ā—Ä—É–ļ—ā—É—Ä–Ĺ—č–Ķ, —Ą–ł–∑–ł–ļ–ĺ-—Ö–ł–ľ–ł—á–Ķ—Ā–ļ–ł–Ķ –ł¬†–Ī–ł–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ł–Ķ —Ā–≤–ĺ–Ļ—Ā—ā–≤–į, —Ā–Ņ–Ķ—Ü–ł—Ą–ł—á–Ķ—Ā–ļ—É—é –į–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć, —ā–ĺ–ļ—Ā–ł–ļ–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ł–Ķ —Ö–į—Ä–į–ļ—ā–Ķ—Ä–ł—Ā—ā–ł–ļ–ł, —Ą–į—Ä–ľ–į–ļ–ĺ–ļ–ł–Ĺ–Ķ—ā–ł–ļ—É. –ü—Ä–ĺ–≤–Ķ–ī–Ķ–Ĺ–Ĺ—č–Ķ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź –Ņ–ĺ–ļ–į–∑–į–Ľ–ł –ĺ—ā—Ā—É—ā—Ā—ā–≤–ł–Ķ –∑–Ĺ–į—á–ł–ľ—č—Ö —Ä–į–∑–Ľ–ł—á–ł–Ļ –ľ–Ķ–∂–ī—É –Ī–ł–ĺ–į–Ĺ–į–Ľ–ĺ–≥–ĺ–ľ –Ī–Ķ–≤–į—Ü–ł–∑—É–ľ–į–Ī–į –ł¬†–ĺ—Ä–ł–≥–ł–Ĺ–į–Ľ—Ć–Ĺ—č–ľ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–ĺ–ľ –Ī–Ķ–≤–į—Ü–ł–∑—É–ľ–į–Ī–į –Ņ–嬆–≤—Ā–Ķ–ľ –ł–∑—É—á–į–≤—ą–ł–ľ—Ā—Ź —Ö–į—Ä–į–ļ—ā–Ķ—Ä–ł—Ā—ā–ł–ļ–į–ľ, —á—ā–ĺ –ĺ–Ņ—Ä–Ķ–ī–Ķ–Ľ–ł–Ľ–ĺ –≤–ĺ–∑–ľ–ĺ–∂–Ĺ–ĺ—Ā—ā—Ć –Ņ—Ä–ĺ–ī–ĺ–Ľ–∂–Ķ–Ĺ–ł—Ź –ł–∑—É—á–Ķ–Ĺ–ł—Ź –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į –≤¬†–ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ł—Ö –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź—Ö. –í¬†–ļ–ĺ–Ĺ—Ü–Ķ 2014¬†–≥.¬†–Ī—č–Ľ–ł –Ņ–ĺ–ī–≤–Ķ–ī–Ķ–Ĺ—č –ł—ā–ĺ–≥–ł —Ā—Ä–į–≤–Ĺ–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ–≥–ĺ –ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ĺ–≥–ĺ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź —Ą–į–∑—č III –Ī–ł–ĺ–į–Ĺ–į–Ľ–ĺ–≥–į –Ī–Ķ–≤–į—Ü–ł–∑—É–ľ–į–Ī–į –Ņ—Ä–ĺ–ł–∑–≤–ĺ–ī—Ā—ā–≤–į –ļ–ĺ–ľ–Ņ–į–Ĺ–ł–ł BIOCAD (BCD-021) –ł¬†–ĺ—Ä–ł–≥–ł–Ĺ–į–Ľ—Ć–Ĺ–ĺ–≥–ĺ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į –ź–≤–į—Ā—ā–ł–Ŭģ, –Ņ—Ä–ĺ–≤–ĺ–ī–ł–ľ–ĺ–≥–ĺ –≤¬†26 –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į—ā–Ķ–Ľ—Ć—Ā–ļ–ł—Ö —Ü–Ķ–Ĺ—ā—Ä–į—Ö –Ĺ–į¬†—ā–Ķ—Ä—Ä–ł—ā–ĺ—Ä–ł–ł –†–ĺ—Ā—Ā–ł–ł –ł¬†—Ā—ā—Ä–į–Ĺ –Ī–Ľ–ł–∂–Ĺ–Ķ–≥–ĺ –∑–į—Ä—É–Ī–Ķ–∂—Ć—Ź. –†–Ķ–∑—É–Ľ—Ć—ā–į—ā—č –ī–į–Ĺ–Ĺ–ĺ–≥–ĺ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź –Ī—č–Ľ–ł –Ņ—Ä–Ķ–ī—Ā—ā–į–≤–Ľ–Ķ–Ĺ—č –≤¬†–ł—é–Ĺ–Ķ 2015¬†–≥.¬†–≤ —Ä–į–ľ–ļ–į—Ö –Ņ–ĺ—Ā—ā–Ķ—Ä–Ĺ–ĺ–Ļ —Ā–Ķ—Ā—Ā–ł–ł –Ķ–∂–Ķ–≥–ĺ–ī–Ĺ–ĺ–≥–ĺ —Ā–ĺ–Ī—Ä–į–Ĺ–ł—Ź –ź–ľ–Ķ—Ä–ł–ļ–į–Ĺ—Ā–ļ–ĺ–≥–ĺ –ĺ–Ī—Č–Ķ—Ā—ā–≤–į –ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ĺ–Ļ –ĺ–Ĺ–ļ–ĺ–Ľ–ĺ–≥–ł–ł (American Society of Clinical Oncology¬†‚Äď ASCO).
–í¬†–ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–ł –Ņ—Ä–ł–Ĺ—Ź–Ľ–ł —É—á–į—Ā—ā–ł–Ķ 138¬†–Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ —Ā¬†–≤–Ņ–Ķ—Ä–≤—č–Ķ –≤—č—Ź–≤–Ľ–Ķ–Ĺ–Ĺ—č–ľ –Ĺ–Ķ–ĺ–Ņ–Ķ—Ä–į–Ī–Ķ–Ľ—Ć–Ĺ—č–ľ –ł–Ľ–ł –ľ–Ķ—ā–į—Ā—ā–į—ā–ł—á–Ķ—Ā–ļ–ł–ľ –Ĺ–Ķ–Ņ–Ľ–ĺ—Ā–ļ–ĺ–ļ–Ľ–Ķ—ā–ĺ—á–Ĺ—č–ľ –Ě–ú–†–õ, –ļ–ĺ—ā–ĺ—Ä—č–Ķ –Ī—č–Ľ–ł —Ä–į–Ĺ–ī–ĺ–ľ–ł–∑–ł—Ä–ĺ–≤–į–Ĺ—č –≤¬†—Ā–ĺ–ĺ—ā–Ĺ–ĺ—ą–Ķ–Ĺ–ł–ł¬†1:1 –≤¬†–≥—Ä—É–Ņ–Ņ—É –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į BCD-021 –ł¬†–≥—Ä—É–Ņ–Ņ—É –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į –ź–≤–į—Ā—ā–ł–Ŭģ. –°—Ö–Ķ–ľ–į –Ľ–Ķ—á–Ķ–Ĺ–ł—Ź –Ī—č–Ľ–į —Ā–Ľ–Ķ–ī—É—é—Č–Ķ–Ļ: –≤¬†–ĺ–Ī–Ķ–ł—Ö¬†–≥—Ä—É–Ņ–Ņ–į—Ö –Ņ–į—Ü–ł–Ķ–Ĺ—ā—č –Ņ–ĺ–Ľ—É—á–į–Ľ–ł –Ņ–į–ļ–Ľ–ł—ā–į–ļ—Ā–Ķ–Ľ –≤¬†–ī–ĺ–∑–Ķ 175¬†–ľ–≥/–ľ2 –≤–Ĺ—É—ā—Ä–ł–≤–Ķ–Ĺ–Ĺ–ĺ –≤¬†—ā–Ķ—á–Ķ–Ĺ–ł–Ķ —ā—Ä–Ķ—Ö —á–į—Ā–ĺ–≤ –ł¬†–ļ–į—Ä–Ī–ĺ–Ņ–Ľ–į—ā–ł–Ĺ –≤¬†–ī–ĺ–∑–Ķ, –ī–ĺ—Ā—ā–į—ā–ĺ—á–Ĺ–ĺ–Ļ –ī–Ľ—Ź –ī–ĺ—Ā—ā–ł–∂–Ķ–Ĺ–ł—Ź AUC 6 –ľ–≥/–ľ–Ľ √ó –ľ–ł–Ĺ, –≤–Ĺ—É—ā—Ä–ł–≤–Ķ–Ĺ–Ĺ–ĺ –≤¬†—ā–Ķ—á–Ķ–Ĺ–ł–Ķ 15‚Äď30¬†–ľ–ł–Ĺ—É—ā, —Ā—Ä–į–∑—É –Ņ–ĺ—Ā–Ľ–Ķ –Ņ–į–ļ–Ľ–ł—ā–į–ļ—Ā–Ķ–Ľ–į. –°—Ä–į–∑—É –Ņ–ĺ—Ā–Ľ–Ķ –ļ–į—Ä–Ī–ĺ–Ņ–Ľ–į—ā–ł–Ĺ–į –≤¬†–∑–į–≤–ł—Ā–ł–ľ–ĺ—Ā—ā–ł –ĺ—ā¬†–Ņ—Ä–ł—Ā–≤–ĺ–Ķ–Ĺ–Ĺ–ĺ–Ļ¬†–≥—Ä—É–Ņ–Ņ—č –Ņ–į—Ü–ł–Ķ–Ĺ—ā—č –Ņ–ĺ–Ľ—É—á–į–Ľ–ł BCD-021 –ł–Ľ–ł –ź–≤–į—Ā—ā–ł–Ŭģ –≤¬†–ī–ĺ–∑–Ķ 15¬†–ľ–≥/–ļ–≥ –≤–Ĺ—É—ā—Ä–ł–≤–Ķ–Ĺ–Ĺ–ĺ –≤¬†–≤–ł–ī–Ķ 90-–ľ–ł–Ĺ—É—ā–Ĺ–ĺ–Ļ –ł–Ĺ—Ą—É–∑–ł–ł. –í—Ā–Ķ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā—č –≤–≤–ĺ–ī–ł–Ľ–ł—Ā—Ć –≤¬†–Ņ–Ķ—Ä–≤—č–Ļ –ī–Ķ–Ĺ—Ć –ļ–į–∂–ī–ĺ–≥–ĺ —ā—Ä–Ķ—Ö–Ĺ–Ķ–ī–Ķ–Ľ—Ć–Ĺ–ĺ–≥–ĺ —Ü–ł–ļ–Ľ–į. –õ–Ķ—á–Ķ–Ĺ–ł–Ķ –Ņ—Ä–ĺ–ī–ĺ–Ľ–∂–į–Ľ–ĺ—Ā—Ć –ī–ĺ —ą–Ķ—Ā—ā–ł —Ü–ł–ļ–Ľ–ĺ–≤ –ł–Ľ–ł –ī–ĺ –Ņ—Ä–ĺ–≥—Ä–Ķ—Ā—Ā–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł—Ź –Ľ–ł–Ī–ĺ –ī–ĺ –Ĺ–į—Ā—ā—É–Ņ–Ľ–Ķ–Ĺ–ł—Ź –Ĺ–Ķ–Ņ–Ķ—Ä–Ķ–Ĺ–ĺ—Ā–ł–ľ—č—Ö —Ź–≤–Ľ–Ķ–Ĺ–ł–Ļ —ā–ĺ–ļ—Ā–ł—á–Ĺ–ĺ—Ā—ā–ł.
–ü—Ä–ł –ĺ—Ü–Ķ–Ĺ–ļ–Ķ —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā–ł –Ņ–Ķ—Ä–≤–ł—á–Ĺ–ĺ–Ļ –ļ–ĺ–Ĺ–Ķ—á–Ĺ–ĺ–Ļ —ā–ĺ—á–ļ–ĺ–Ļ –Ī—č–Ľ–į –ĺ–Ņ—Ä–Ķ–ī–Ķ–Ľ–Ķ–Ĺ–į –ĺ–Ī—Č–į—Ź —á–į—Ā—ā–ĺ—ā–į –ĺ—ā–≤–Ķ—ā–ĺ–≤ (—á–į—Ā—ā–ł—á–Ĺ—č–Ļ –ĺ—ā–≤–Ķ—ā + –Ņ–ĺ–Ľ–Ĺ—č–Ļ –ĺ—ā–≤–Ķ—ā) –Ņ–ĺ—Ā–Ľ–Ķ –Ņ–ĺ–Ľ—É—á–Ķ–Ĺ–ł—Ź –ī–ĺ —ą–Ķ—Ā—ā–ł –ļ—É—Ä—Ā–ĺ–≤ —ā–Ķ—Ä–į–Ņ–ł–ł, –ĺ—Ü–Ķ–Ĺ–ł–≤–į–≤—ą–į—Ź—Ā—Ź –Ņ–嬆–ī–į–Ĺ–Ĺ—č–ľ –ļ–ĺ–ľ–Ņ—Ć—é—ā–Ķ—Ä–Ĺ–ĺ–Ļ —ā–ĺ–ľ–ĺ¬≠–≥—Ä–į—Ą–ł–ł —Ā¬†–ł—Ā–Ņ–ĺ–Ľ—Ć–∑–ĺ–≤–į–Ĺ–ł–Ķ–ľ –ļ—Ä–ł—ā–Ķ—Ä–ł–Ķ–≤ RECIST¬†1.1. –Ē–Ľ—Ź –ĺ—Ü–Ķ–Ĺ–ļ–ł —Ą–į—Ä–ľ–į–ļ–ĺ–ļ–ł–Ĺ–Ķ—ā–ł–ļ–ł –≤¬†—Ö–ĺ–ī–Ķ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź –ĺ–Ņ—Ä–Ķ–ī–Ķ–Ľ—Ź–Ľ–ł —Ā—č–≤–ĺ—Ä–ĺ—ā–ĺ—á–Ĺ—č–Ķ –ļ–ĺ–Ĺ—Ü–Ķ–Ĺ—ā—Ä–į—Ü–ł–ł –Ī–Ķ–≤–į—Ü–ł–∑—É–ľ–į–Ī–į —ɬ†–≤—Ā–Ķ—Ö –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ –Ņ–Ķ—Ä–Ķ–ī –ļ–į–∂–ī—č–ľ –≤–≤–Ķ–ī–Ķ–Ĺ–ł–Ķ–ľ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–ĺ–≤, –į¬†—ā–į–ļ–∂–Ķ –≤¬†–ļ–ĺ–Ĺ–ļ—Ä–Ķ—ā–Ĺ—č—Ö –≤—Ä–Ķ–ľ–Ķ–Ĺ–Ĺ—č—Ö —ā–ĺ—á–ļ–į—Ö –Ĺ–į¬†–Ņ–Ķ—Ä–≤–ĺ–ľ –ł¬†—ą–Ķ—Ā—ā–ĺ–ľ —Ü–ł–ļ–Ľ–į—Ö —Ö–ł–ľ–ł–ĺ—ā–Ķ—Ä–į–Ņ–ł–ł. –ü—Ä–ł –ĺ—Ü–Ķ–Ĺ–ļ–Ķ –Ī–Ķ–∑–ĺ–Ņ–į—Ā–Ĺ–ĺ—Ā—ā–ł –į–Ĺ–į–Ľ–ł–∑–ł—Ä–ĺ–≤–į–Ľ–ł —á–į—Ā—ā–ĺ—ā—É –Ě–Į, –°–Ě–Į, —á–į—Ā—ā–ĺ—ā—É –ĺ—ā–ľ–Ķ–Ĺ—č –Ľ–Ķ—á–Ķ–Ĺ–ł—Ź –≤—Ā–Ľ–Ķ–ī—Ā—ā–≤–ł–Ķ –Ě–Į. –Ē–ĺ–Ņ–ĺ–Ľ–Ĺ–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ľ–ł –ł–ľ–ľ—É–Ĺ–ĺ–≥–Ķ–Ĺ–Ĺ–ĺ—Ā—ā—Ć —ā–Ķ—Ä–į–Ņ–ł–ł.
–ď—Ä—É–Ņ–Ņ—č –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ –Ī—č–Ľ–ł —Ā–ĺ–Ņ–ĺ—Ā—ā–į–≤–ł–ľ—č –Ņ–嬆–ł—Ā—Ö–ĺ–ī–Ĺ—č–ľ –ī–Ķ–ľ–ĺ–≥—Ä–į—Ą–ł—á–Ķ—Ā–ļ–ł–ľ –Ņ–į—Ä–į–ľ–Ķ—ā—Ä–į–ľ (–≤–ĺ–∑—Ä–į—Ā—ā, –≤–Ķ—Ā, —Ä–ĺ—Ā—ā, –Ņ–ĺ–Ľ), –ĺ–Ī—Č–Ķ–ľ—É —Ā–ĺ—Ā—ā–ĺ—Ź–Ĺ–ł—é –Ņ–嬆—ą–ļ–į–Ľ–Ķ ECOG –ł¬†—Ö–į—Ä–į–ļ—ā–Ķ—Ä–ł—Ā—ā–ł–ļ–į–ľ –ĺ—Ā–Ĺ–ĺ–≤–Ĺ–ĺ–≥–ĺ –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł—Ź (–Ņ—Ä–ĺ–ī–ĺ–Ľ–∂–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ—Ā—ā—Ć –≤—Ä–Ķ–ľ–Ķ–Ĺ–ł —Ā¬†–ľ–ĺ–ľ–Ķ–Ĺ—ā–į –Ņ–ĺ—Ā—ā–į–Ĺ–ĺ–≤–ļ–ł –ī–ł–į–≥–Ĺ–ĺ–∑–į, –Ņ—Ä–Ķ–ī—ą–Ķ—Ā—ā–≤—É—é—Č–į—Ź —ā–Ķ—Ä–į–Ņ–ł—Ź, –ľ–ĺ—Ä—Ą–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–į—Ź —Ö–į—Ä–į–ļ—ā–Ķ—Ä–ł—Ā—ā–ł–ļ–į –ĺ–Ņ—É—Ö–ĺ–Ľ–ł, —á–ł—Ā–Ľ–ĺ –ł¬†–Ľ–ĺ–ļ–į–Ľ–ł–∑–į—Ü–ł—Ź –ĺ—ā–ī–į–Ľ–Ķ–Ĺ–Ĺ—č—Ö –ľ–Ķ—ā–į—Ā—ā–į–∑–ĺ–≤). –£¬†–Ī–ĺ–Ľ—Ć—ą–ł–Ĺ—Ā—ā–≤–į –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ –Ī—č–Ľ–į –ī–ł–į–≥–Ĺ–ĺ—Ā—ā–ł—Ä–ĺ–≤–į–Ĺ–į —Ā—ā–į–ī–ł—Ź¬†IV –Ě–ú–†–õ (49 (90,74%) –ł¬†46 (82,14%) –Ī–ĺ–Ľ—Ć–Ĺ—č—Ö –≤¬†–≥—Ä—É–Ņ–Ņ–į—Ö BCD-021 –ł¬†–ź–≤–į—Ā—ā–ł–Ĺ–į —Ā–ĺ–ĺ—ā–≤–Ķ—ā—Ā—ā–≤–Ķ–Ĺ–Ĺ–ĺ) (—Ä > 0,05).
–í¬†–į–Ĺ–į–Ľ–ł–∑ —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā–ł –Ī—č–Ľ–ł –≤–ļ–Ľ—é—á–Ķ–Ĺ—č –≤—Ā–Ķ –Ī–ĺ–Ľ—Ć–Ĺ—č–Ķ, –ļ–ĺ—ā–ĺ—Ä—č–Ķ –Ņ–ĺ–Ľ—É—á–ł–Ľ–ł —Ö–ĺ—ā—Ź –Ī—č –ĺ–ī–Ĺ–ĺ –≤–≤–Ķ–ī–Ķ–Ĺ–ł–Ķ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į BCD-021 –ł–Ľ–ł –ź–≤–į—Ā—ā–ł–Ĺ–į –ł¬†—É –ļ–ĺ—ā–ĺ—Ä—č—Ö –ľ–ĺ–∂–Ĺ–ĺ –Ī—č–Ľ–ĺ –ĺ—Ü–Ķ–Ĺ–ł—ā—Ć –ī–ĺ—Ā—ā–ł–≥–Ĺ—É—ā—č–Ļ –ĺ—ā–≤–Ķ—ā (n = 110, 54 (78,26%) –Ņ–į—Ü–ł–Ķ–Ĺ—ā–į¬†–≥—Ä—É–Ņ–Ņ—č BCD-021 –ł¬†56 (81,20%) –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤¬†–≥—Ä—É–Ņ–Ņ—č –ź–≤–į—Ā—ā–ł–Ĺ–į).
–Ě–į¬†–ĺ—Ā–Ĺ–ĺ–≤–į–Ĺ–ł–ł –ī–į–Ĺ–Ĺ—č—Ö –ö–Ę –ĺ–Ī—Č–į—Ź —á–į—Ā—ā–ĺ—ā–į –ĺ—ā–≤–Ķ—ā–į –≤¬†–≥—Ä—É–Ņ–Ņ–Ķ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į BCD-021 —Ā–ĺ—Ā—ā–į–≤–ł–Ľ–į 42,59% (95% –Ē–ė 30,33‚Äď55,83%), –į¬†–≤¬†–≥—Ä—É–Ņ–Ņ–Ķ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į –ź–≤–į—Ā—ā–ł–Ŭģ¬†‚Äď 39,29% (95% –Ē–ė 27,58‚Äď52,27%).
–†–į–∑–Ĺ–ł—Ü–į –Ņ–ĺ–ļ–į–∑–į—ā–Ķ–Ľ—Ź –≤¬†–≥—Ä—É–Ņ–Ņ–Ķ –ł—Ā—Ā–Ľ–Ķ–ī—É–Ķ–ľ–ĺ–Ļ —ā–Ķ—Ä–į–Ņ–ł–ł –ł¬†–≤¬†–≥—Ä—É–Ņ–Ņ–Ķ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į —Ā—Ä–į–≤–Ĺ–Ķ–Ĺ–ł—Ź —Ā–ĺ—Ā—ā–į–≤–ł–Ľ–į 3,30% —Ā¬†95% –Ē–ė -14,96‚Äď21,40% (—Ä = 0,874, –ļ—Ä–ł—ā–Ķ—Ä–ł–Ļ —Ö–ł-–ļ–≤–į–ī—Ä–į—ā –ü–ł—Ä—Ā–ĺ–Ĺ–į —Ā¬†–Ņ–ĺ–Ņ—Ä–į–≤–ļ–ĺ–Ļ –ô–Ķ—ā—Ā–į). –Ě–ł–∂–Ĺ—Ź—Ź¬†–≥—Ä–į–Ĺ–ł—Ü–į —Ä–į—Ā—Ā—á–ł—ā–į–Ĺ–Ĺ–ĺ–≥–ĺ 95% –Ē–ė (-14,96%) –Ņ—Ä–Ķ–≤—č—Ā–ł–Ľ–į —É—Ā—ā–į–Ĺ–ĺ–≤–Ľ–Ķ–Ŗŗɗ鬆–≥—Ä–į–Ĺ–ł—Ü—É –Ĺ–Ķ¬†–ľ–Ķ–Ĺ—Ć—ą–Ķ–Ļ —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā–ł, –į¬†—Ā–Ľ–Ķ–ī–ĺ–≤–į—ā–Ķ–Ľ—Ć–Ĺ–ĺ, –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā BCD-021 –Ņ—Ä–ĺ–ī–Ķ–ľ–ĺ–Ĺ—Ā—ā—Ä–ł—Ä–ĺ–≤–į–Ľ –Ĺ–Ķ¬†–ľ–Ķ–Ĺ—Ć—ą—É—é —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć –Ņ–嬆—Ā—Ä–į–≤–Ĺ–Ķ–Ĺ–ł—é —Ā¬†–Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–ĺ–ľ –ź–≤–į—Ā—ā–ł–Ŭģ. –ě—ā–Ĺ–ĺ—ą–Ķ–Ĺ–ł–Ķ —ą–į–Ĺ—Ā–ĺ–≤ –ī–Ľ—Ź –ě–ß–ě —Ā–ĺ—Ā—ā–į–≤–ł–Ľ–ĺ 1,145 (95% –Ē–ė 0,500‚Äď2,629), —á—ā–ĺ —ā–į–ļ–∂–Ķ —É–ļ–į–∑—č–≤–į–Ľ–ĺ –Ĺ–į¬†–ĺ—ā—Ā—É—ā—Ā—ā–≤–ł–Ķ —Ā—ā–į—ā–ł—Ā—ā–ł—á–Ķ—Ā–ļ–ł –∑–Ĺ–į—á–ł–ľ—č—Ö —Ä–į–∑–Ľ–ł—á–ł–Ļ –≤¬†—ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā–ł –ľ–Ķ–∂–ī—ɬ†–≥—Ä—É–Ņ–Ņ–į–ľ–ł —ā–Ķ—Ä–į–Ņ–ł–ł. –ü—Ä–ł —Ā—Ä–į–≤–Ĺ–Ķ–Ĺ–ł–ł –ī–ĺ–Ņ–ĺ–Ľ–Ĺ–ł—ā–Ķ–Ľ—Ć–Ĺ—č—Ö –Ņ–į—Ä–į–ľ–Ķ—ā—Ä–ĺ–≤ –ĺ—Ü–Ķ–Ĺ–ļ–ł —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā–ł —ā–į–ļ–∂–Ķ –Ĺ–Ķ¬†–≤—č—Ź–≤–Ľ–Ķ–Ĺ–ĺ —Ā—ā–į—ā–ł—Ā—ā–ł—á–Ķ—Ā–ļ–ł –∑–Ĺ–į—á–ł–ľ—č—Ö —Ä–į–∑–Ľ–ł—á–ł–Ļ (—ā–į–Ī–Ľ.¬†2).
–í¬†–į–Ĺ–į–Ľ–ł–∑ –Ī–Ķ–∑–ĺ–Ņ–į—Ā–Ĺ–ĺ—Ā—ā–ł –Ī—č–Ľ–ł –≤–ļ–Ľ—é—á–Ķ–Ĺ—č –≤—Ā–Ķ –Ī–ĺ–Ľ—Ć–Ĺ—č–Ķ, –Ņ–ĺ–Ľ—É—á–ł–≤—ą–ł–Ķ —Ö–ĺ—ā—Ź –Ī—č –ĺ–ī–Ĺ–ĺ –≤–≤–Ķ–ī–Ķ–Ĺ–ł–Ķ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į BCD-021 –ł–Ľ–ł –ź–≤–į—Ā—ā–ł–Ĺ–į (n = 134). –ě–Ī–į –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į –Ņ—Ä–ĺ–ī–Ķ–ľ–ĺ–Ĺ—Ā—ā—Ä–ł—Ä–ĺ–≤–į–Ľ–ł –Ņ—Ä–ł–Ķ–ľ–Ľ–Ķ–ľ—č–Ļ –Ņ—Ä–ĺ—Ą–ł–Ľ—Ć —ā–ĺ–ļ—Ā–ł—á–Ĺ–ĺ—Ā—ā–ł –ł¬†–Ņ–Ķ—Ä–Ķ–Ĺ–ĺ—Ā–ł–ľ–ĺ—Ā—ā–ł. –Ě–Ķ¬†–≤—č—Ź–≤–Ľ–Ķ–Ĺ–ĺ –∑–Ĺ–į—á–ł–ľ—č—Ö —Ä–į–∑–Ľ–ł—á–ł–Ļ –ľ–Ķ–∂–ī—ɬ†–≥—Ä—É–Ņ–Ņ–į–ľ–ł –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–ĺ–≤ –ļ–į–ļ –Ņ–嬆–ĺ–Ī—Č–Ķ–Ļ —á–į—Ā—ā–ĺ—ā–Ķ –Ě–Į, —ā–į–ļ –ł¬†–Ņ–ĺ —á–į—Ā—ā–ĺ—ā–Ķ –ļ–į–∂–ī–ĺ–≥–ĺ –ł–∑¬†–∑–į—Ä–Ķ–≥–ł—Ā—ā—Ä–ł—Ä–ĺ–≤–į–Ĺ–Ĺ—č—Ö –Ě–Į. –°—Ä–Ķ–ī–ł –Ě–Į –Ĺ–į–ł–Ī–ĺ–Ľ–Ķ–Ķ —á–į—Ā—ā–ĺ –≤—Ā—ā—Ä–Ķ—á–į–Ľ–ł—Ā—Ć —Ź–≤–Ľ–Ķ–Ĺ–ł—Ź¬†–≥–Ķ–ľ–į—ā–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ĺ–Ļ —ā–ĺ–ļ—Ā–ł—á–Ĺ–ĺ—Ā—ā–ł, –≤–ļ–Ľ—é—á–į–≤—ą–ł–Ķ –Ĺ–Ķ–Ļ—ā—Ä–ĺ–Ņ–Ķ–Ĺ–ł—é, –Ľ–Ķ–Ļ–ļ–ĺ–Ņ–Ķ–Ĺ–ł—é, –Ľ–ł–ľ—Ą–ĺ–Ņ–Ķ–Ĺ–ł—é, –į–Ĺ–Ķ–ľ–ł—é –ł¬†—ā—Ä–ĺ–ľ–Ī–ĺ—Ü–ł—ā–ĺ–Ņ–Ķ–Ĺ–ł—é. –Ě–Ķ—Ā–ļ–ĺ–Ľ—Ć–ļ–ĺ —Ä–Ķ–∂–Ķ —Ä–Ķ–≥–ł—Ā—ā—Ä–ł—Ä–ĺ–≤–į–Ľ–ł—Ā—Ć –ĺ—ā–ļ–Ľ–ĺ–Ĺ–Ķ–Ĺ–ł—Ź —Ä—Ź–ī–į –Ī–ł–ĺ—Ö–ł–ľ–ł—á–Ķ—Ā–ļ–ł—Ö –Ņ–ĺ–ļ–į–∑–į—ā–Ķ–Ľ–Ķ–Ļ –ļ—Ä–ĺ–≤–ł:¬†–≥–ł–Ņ–Ķ—Ä–≥–Ľ–ł–ļ–Ķ–ľ–ł—Ź,¬†–≥–ł–Ņ–Ķ—Ä—É—Ä–ł–ļ–Ķ–ľ–ł—Ź, —É–≤–Ķ–Ľ–ł—á–Ķ–Ĺ–ł–Ķ —É—Ä–ĺ–≤–Ĺ—Ź –ľ–ĺ—á–Ķ–≤–ł–Ĺ—č, –Ņ–ĺ–≤—č—ą–Ķ–Ĺ–ł–Ķ –į–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā–ł –õ–Ē–ď, –ź–°–Ę, –ź–õ–Ę, –©–§. –°—Ä–Ķ–ī–ł –Ņ—Ä–ĺ—á–ł—Ö –Ě–Į –Ĺ–į–ł–Ī–ĺ–Ľ–Ķ–Ķ —á–į—Ā—ā–ĺ –Ĺ–į–Ī–Ľ—é–ī–į–Ľ–ł—Ā—Ć –į–Ľ–ĺ–Ņ–Ķ—Ü–ł–ł, –Ĺ–į—Ä—É—ą–Ķ–Ĺ–ł—Ź —Ā–嬆—Ā—ā–ĺ—Ä–ĺ–Ĺ—č —Ā–ļ–Ķ–Ľ–Ķ—ā–Ĺ–ĺ-–ľ—č—ą–Ķ—á–Ĺ–ĺ–Ļ —Ā–ł—Ā—ā–Ķ–ľ—č (–į—Ä—ā—Ä–į–Ľ–≥–ł–ł, –ľ–ł–į–Ľ–≥–ł–ł, –ĺ—Ā—Ā–į–Ľ–≥–ł–ł, –Ī–ĺ–Ľ—Ć –≤¬†—Ā–Ņ–ł–Ĺ–Ķ), —Ā–嬆—Ā—ā–ĺ—Ä–ĺ–Ĺ—č –Ņ–ł—Č–Ķ–≤–į—Ä–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ–Ļ —Ā–ł—Ā—ā–Ķ–ľ—č (—ā–ĺ—ą–Ĺ–ĺ—ā–į, —Ä–≤–ĺ—ā–į, –ī–ł–į—Ä–Ķ—Ź, —Ā—ā–ĺ–ľ–į—ā–ł—ā), —Ā–嬆—Ā—ā–ĺ—Ä–ĺ–Ĺ—č —Ā–Ķ—Ä–ī–Ķ—á–Ĺ–ĺ-—Ā–ĺ—Ā—É–ī–ł—Ā—ā–ĺ–Ļ —Ā–ł—Ā—ā–Ķ–ľ—č (—ā–į—Ö–ł–ļ–į—Ä–ī–ł—Ź), —Ā–Ľ–į–Ī–ĺ—Ā—ā—Ć,¬†–≥–ĺ–Ľ–ĺ–≤–Ĺ–į—Ź –Ī–ĺ–Ľ—Ć, –į¬†—ā–į–ļ–∂–Ķ –Ĺ–į—Ä—É—ą–Ķ–Ĺ–ł—Ź —Ā–嬆—Ā—ā–ĺ—Ä–ĺ–Ĺ—č –ĺ—Ä–≥–į–Ĺ–ĺ–≤ –ī—č—Ö–į–Ĺ–ł—Ź (–Ľ–Ķ–≥–ĺ—á–Ĺ–ĺ–Ķ –ļ—Ä–ĺ–≤–ĺ—ā–Ķ—á–Ķ–Ĺ–ł–Ķ, –ļ—Ä–ĺ–≤–ĺ—Ö–į—Ä–ļ–į–Ĺ—Ć–Ķ, –ĺ–ī—č—ą–ļ–į, –ļ–į—ą–Ķ–Ľ—Ć).
–°–Ě–Į –≤—č—Ź–≤–Ľ–Ķ–Ĺ—č –≤¬†–ĺ–Ī—Č–Ķ–Ļ —Ā–Ľ–ĺ–∂–Ĺ–ĺ—Ā—ā–ł —ɬ†22¬†–Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤: —ɬ†14 (20,59%)¬† –ł–∑¬†–≥—Ä—É–Ņ–Ņ—č BCD-021 (—ɬ†–ī–≤—É—Ö –ł–∑¬†–Ĺ–ł—Ö –∑–į—Ä–Ķ–≥–ł—Ā—ā—Ä–ł—Ä–ĺ–≤–į–Ĺ–ĺ –Ņ–嬆–ī–≤–į –°–Ě–Į) –ł¬†–≤–ĺ—Ā—Ć–ľ–ł (12,12%) –ł–∑¬†–≥—Ä—É–Ņ–Ņ—č —Ā—Ä–į–≤–Ĺ–Ķ–Ĺ–ł—Ź. –í¬†–Ī–ĺ–Ľ—Ć—ą–ł–Ĺ—Ā—ā–≤–Ķ —Ā–Ľ—É—á–į–Ķ–≤ –°–Ě–Į –Ī—č–Ľ–ł –ĺ–Ī—É—Ā–Ľ–ĺ–≤–Ľ–Ķ–Ĺ—č –Ĺ–į–Ľ–ł—á–ł–Ķ–ľ —Ā–ĺ–Ņ—É—ā—Ā—ā–≤—É—é—Č–Ķ–Ļ –Ņ–į—ā–ĺ–Ľ–ĺ–≥–ł–ł, –≤–ĺ–∑–ī–Ķ–Ļ—Ā—ā–≤–ł–Ķ–ľ —Ö–ł–ľ–ł–ĺ–Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–ĺ–≤, –Ņ—Ä–ł–ľ–Ķ–Ĺ—Ź–Ķ–ľ—č—Ö –≤¬†—Ä–į–ľ–ļ–į—Ö –ļ–ĺ–ľ–Ī–ł–Ĺ–ł—Ä–ĺ–≤–į–Ĺ–Ĺ–ĺ–Ļ —Ö–ł–ľ–ł–ĺ—ā–Ķ—Ä–į–Ņ–ł–ł, –Ľ–ł–Ī–ĺ –ī—Ä—É–≥–ł–ľ–ł –Ņ—Ä–ł—á–ł–Ĺ–į–ľ–ł, –Ĺ–Ķ¬†—Ā–≤—Ź–∑–į–Ĺ–Ĺ—č–ľ–ł —Ā¬†–ł—Ā—Ā–Ľ–Ķ–ī—É–Ķ–ľ–ĺ–Ļ —ā–Ķ—Ä–į–Ņ–ł–Ķ–Ļ. –Ě–Ķ¬†—É—Ā—ā–į–Ĺ–ĺ–≤–Ľ–Ķ–Ĺ–ĺ –∑–Ĺ–į—á–ł–ľ—č—Ö —Ä–į–∑–Ľ–ł—á–ł–Ļ –ľ–Ķ–∂–ī—ɬ†–≥—Ä—É–Ņ–Ņ–į–ľ–ł –Ņ–嬆—á–į—Ā—ā–ĺ—ā–Ķ –≤—Ā—ā—Ä–Ķ—á–į–Ķ–ľ–ĺ—Ā—ā–ł –Ľ—é–Ī—č—Ö –°–Ě–Į (—Ä > 0,05).
–°–ĺ–≥–Ľ–į—Ā–Ĺ–ĺ –ľ–Ķ–∂–ī—É–Ĺ–į—Ä–ĺ–ī–Ĺ—č–ľ —Ä–Ķ–ļ–ĺ–ľ–Ķ–Ĺ–ī–į—Ü–ł—Ź–ľ –Ņ–嬆–ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—é –≤–ĺ—Ā–Ņ—Ä–ĺ–ł–∑–≤–Ķ–ī–Ķ–Ĺ–Ĺ—č—Ö –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–ĺ–≤ –ľ–ĺ–Ĺ–ĺ–ļ–Ľ–ĺ–Ĺ–į–Ľ—Ć–Ĺ—č—Ö –į–Ĺ—ā–ł—ā–Ķ–Ľ, –ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ł–Ķ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā–ł –ł¬†–Ī–Ķ–∑–ĺ–Ņ–į—Ā–Ĺ–ĺ—Ā—ā–ł –ī–ĺ–Ľ–∂–Ĺ—č –≤–ļ–Ľ—é—á–į—ā—Ć –ĺ—Ü–Ķ–Ĺ–ļ—É –ł–ľ–ľ—É–Ĺ–ĺ–≥–Ķ–Ĺ–Ĺ–ĺ—Ā—ā–ł –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–ĺ–≤. –ü–嬆—Ä–Ķ–∑—É–Ľ—Ć—ā–į—ā–į–ľ –ĺ–Ī—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ —É—Ā—ā–į–Ĺ–ĺ–≤–Ľ–Ķ–Ĺ–ĺ –Ņ–嬆–ĺ–ī–Ĺ–ĺ–ľ—É —Ā–Ľ—É—á–į—é –Ņ–ĺ—Ź–≤–Ľ–Ķ–Ĺ–ł—Ź —Ā–≤—Ź–∑—č–≤–į—é—Č–ł—Ö –į–Ĺ—ā–ł—ā–Ķ–Ľ (—Ā¬†–Ĺ–Ķ–Ļ—ā—Ä–į–Ľ–ł–∑—É—é—Č–Ķ–Ļ –į–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć—é) –ļ¬†–Ī–Ķ–≤–į—Ü–ł–∑—É–ľ–į–Ī—É –≤¬†–ļ–į–∂–ī–ĺ–Ļ¬†–≥—Ä—É–Ņ–Ņ–Ķ. –Ę–į–ļ–ł–ľ –ĺ–Ī—Ä–į–∑–ĺ–ľ, –ĺ–Ī–į –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į —Ö–į—Ä–į–ļ—ā–Ķ—Ä–ł–∑–ĺ–≤–į–Ľ–ł—Ā—Ć –Ĺ–ł–∑–ļ–ĺ–Ļ –ł–ľ–ľ—É–Ĺ–ĺ–≥–Ķ–Ĺ–Ĺ–ĺ—Ā—ā—Ć—é.
–í¬†—Ü–Ķ–Ľ–ĺ–ľ –į–Ĺ–į–Ľ–ł–∑ –Ī–Ķ–∑–ĺ–Ņ–į—Ā–Ĺ–ĺ—Ā—ā–ł –Ņ—Ä–ĺ–ī–Ķ–ľ–ĺ–Ĺ—Ā—ā—Ä–ł—Ä–ĺ–≤–į–Ľ –ĺ—ā—Ā—É—ā—Ā—ā–≤–ł–Ķ –∑–Ĺ–į—á–ł–ľ—č—Ö —Ä–į–∑–Ľ–ł—á–ł–Ļ –ľ–Ķ–∂–ī—ɬ†–≥—Ä—É–Ņ–Ņ–į–ľ–ł –Ņ–嬆–ĺ—Ā–Ĺ–ĺ–≤–Ĺ—č–ľ –Ņ–į—Ä–į–ľ–Ķ—ā—Ä–į–ľ –Ī–Ķ–∑–ĺ–Ņ–į—Ā–Ĺ–ĺ—Ā—ā–ł.
–ź–Ĺ–į–Ľ–ł–∑ –Ņ–ĺ–ļ–į–∑–į—ā–Ķ–Ľ–Ķ–Ļ —Ą–į—Ä–ľ–į–ļ–ĺ–ļ–ł–Ĺ–Ķ—ā–ł–ļ–ł –Ņ—Ä–ĺ–≤–ĺ–ī–ł–Ľ–ł –≤¬†—Ā–ĺ–ĺ—ā–≤–Ķ—ā—Ā—ā–≤–ł–ł —Ā¬†–ľ–Ķ–∂–ī—É–Ĺ–į—Ä–ĺ–ī–Ĺ—č–ľ–ł —Ä–Ķ–ļ–ĺ–ľ–Ķ–Ĺ–ī–į—Ü–ł—Ź–ľ–ł –Ņ–嬆–ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—é –Ī–ł–ĺ—ć–ļ–≤–ł–≤–į–Ľ–Ķ–Ĺ—ā–Ĺ–ĺ—Ā—ā–ł. –†–Ķ–∑—É–Ľ—Ć—ā–į—ā—č –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź –Ņ–ĺ–ļ–į–∑–į–Ľ–ł, —á—ā–ĺ –ļ–į–ļ –Ņ–ĺ—Ā–Ľ–Ķ –ĺ–ī–Ĺ–ĺ–ļ—Ä–į—ā–Ĺ–ĺ–≥–ĺ, —ā–į–ļ –ł¬†–Ņ–ĺ—Ā–Ľ–Ķ –ľ–Ĺ–ĺ–≥–ĺ–ļ—Ä–į—ā–Ĺ–ĺ–≥–ĺ –≤–≤–Ķ–ī–Ķ–Ĺ–ł—Ź –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–ĺ–≤ BCD-021 –ł¬†–ź–≤–į—Ā—ā–ł–Ŭģ –ļ–ĺ–Ĺ—Ü–Ķ–Ĺ—ā—Ä–į—Ü–ł–ł –Ī–Ķ–≤–į—Ü–ł–∑—É–ľ–į–Ī–į –≤¬†–ļ—Ä–ĺ–≤–ł –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ –ł–∑–ľ–Ķ–Ĺ—Ź–Ľ–ł—Ā—Ć –į–Ĺ–į–Ľ–ĺ–≥–ł—á–Ĺ—č–ľ –ĺ–Ī—Ä–į–∑–ĺ–ľ. –°—ā–į—ā–ł—Ā—ā–ł—á–Ķ—Ā–ļ–ĺ–Ķ —Ā—Ä–į–≤–Ĺ–Ķ–Ĺ–ł–Ķ –Ņ–嬆–Ņ–Ķ—Ä–≤–ł—á–Ĺ–ĺ–Ļ —Ą–į—Ä–ľ–į–ļ–ĺ–ļ–ł–Ĺ–Ķ—ā–ł—á–Ķ—Ā–ļ–ĺ–Ļ –ļ–ĺ–Ĺ–Ķ—á–Ĺ–ĺ–Ļ —ā–ĺ—á–ļ–Ķ (AUC(0‚Äď504)) –Ņ–ĺ–∑–≤–ĺ–Ľ–ł–Ľ–ĺ –Ņ–ĺ–Ľ—É—á–ł—ā—Ć 90% –Ē–ė –ī–Ľ—Ź –ĺ—ā–Ĺ–ĺ—ą–Ķ–Ĺ–ł—Ź —Ā—Ä–Ķ–ī–Ĺ–ł—Ö¬†–≥–Ķ–ĺ–ľ–Ķ—ā—Ä–ł—á–Ķ—Ā–ļ–ł—Ö AUC(0‚Äď504)¬†‚Äď 80,67‚Äď109,69%. –Ē–į–Ĺ–Ĺ—č–Ļ –ł–Ĺ—ā–Ķ—Ä–≤–į–Ľ —Ā–ĺ–ĺ—ā–≤–Ķ—ā—Ā—ā–≤—É–Ķ—ā —É—Ā—ā–į–Ĺ–ĺ–≤–Ľ–Ķ–Ĺ–Ĺ—č–ľ –Ņ—Ä–Ķ–ī–Ķ–Ľ–į–ľ —ć–ļ–≤–ł–≤–į–Ľ–Ķ–Ĺ—ā–Ĺ–ĺ—Ā—ā–ł —Ą–į—Ä–ľ–į–ļ–ĺ–ļ–ł–Ĺ–Ķ—ā–ł—á–Ķ—Ā–ļ–ł—Ö –Ņ–ĺ–ļ–į–∑–į—ā–Ķ–Ľ–Ķ–Ļ 80‚Äď125%. 90% –Ē–ė –ī–Ľ—Ź –ĺ—ā–Ĺ–ĺ—ą–Ķ–Ĺ–ł—Ź —Ā—Ä–Ķ–ī–Ĺ–ł—Ö¬†–≥–Ķ–ĺ–ľ–Ķ—ā—Ä–ł—á–Ķ—Ā–ļ–ł—Ö –°m–į—Ö –ł—Ā—Ā–Ľ–Ķ–ī—É–Ķ–ľ–ĺ–≥–ĺ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į –ł¬†–Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į —Ā—Ä–į–≤–Ĺ–Ķ–Ĺ–ł—Ź —Ā–ĺ—Ā—ā–į–≤–ł–Ľ 89,12‚Äď111,35%, —á—ā–ĺ —ā–į–ļ–∂–Ķ —Ā–ĺ–ĺ—ā–≤–Ķ—ā—Ā—ā–≤—É–Ķ—ā —É—Ā—ā–į–Ĺ–ĺ–≤–Ľ–Ķ–Ĺ–Ĺ–ĺ–ľ—É –ł–Ĺ—ā–Ķ—Ä–≤–į–Ľ—É 80‚Äď125%. –ü—Ä–ł —Ā—Ä–į–≤–Ĺ–Ķ–Ĺ–ł–ł –ł—Ā–Ņ—č—ā—É–Ķ–ľ—č—Ö¬†–≥—Ä—É–Ņ–Ņ –Ņ–嬆–Ņ—Ä–ĺ—á–ł–ľ –≤—ā–ĺ—Ä–ł—á–Ĺ—č–ľ –ļ–ĺ–Ĺ–Ķ—á–Ĺ—č–ľ —ā–ĺ—á–ļ–į–ľ (–Ęmax, T1/2) —Ā¬†–Ņ–ĺ–ľ–ĺ—Č—Ć—é —Ā—ā–į–Ĺ–ī–į—Ä—ā–Ĺ—č—Ö —Ā—ā–į—ā–ł—Ā—ā–ł—á–Ķ—Ā–ļ–ł—Ö –ļ—Ä–ł—ā–Ķ—Ä–ł–Ķ–≤ —Ā—ā–į—ā–ł—Ā—ā–ł—á–Ķ—Ā–ļ–ł –∑–Ĺ–į—á–ł–ľ—č–Ķ —Ä–į–∑–Ľ–ł—á–ł—Ź –ľ–Ķ–∂–ī—ɬ†–≥—Ä—É–Ņ–Ņ–į–ľ–ł —ā–į–ļ–∂–Ķ –ĺ—ā—Ā—É—ā—Ā—ā–≤–ĺ–≤–į–Ľ–ł. –ü—Ä–ł –į–Ĺ–į–Ľ–ł–∑–Ķ –Ĺ–į¬†—ą–Ķ—Ā—ā–ĺ–ľ —Ü–ł–ļ–Ľ–Ķ —Ā—ā–į—ā–ł—Ā—ā–ł—á–Ķ—Ā–ļ–ĺ–Ķ —Ā—Ä–į–≤–Ĺ–Ķ–Ĺ–ł–Ķ –Ņ–嬆–≤—Ā–Ķ–ľ –ĺ—Ā–Ĺ–ĺ–≤–Ĺ—č–ľ —Ą–į—Ä–ľ–į–ļ–ĺ–ļ–ł–Ĺ–Ķ—ā–ł—á–Ķ—Ā–ļ–ł–ľ –Ņ–į—Ä–į–ľ–Ķ—ā—Ä–į–ľ (AUC(0‚Äď504), –°m–į—Ö, –Ęmax, T1/2) —Ā¬†–Ņ–ĺ–ľ–ĺ—Č—Ć—é —Ā—ā–į–Ĺ–ī–į—Ä—ā–Ĺ—č—Ö —Ā—ā–į—ā–ł—Ā—ā–ł—á–Ķ—Ā–ļ–ł—Ö –ļ—Ä–ł—ā–Ķ—Ä–ł–Ķ–≤ –Ņ–ĺ–ļ–į–∑–į–Ľ–ĺ –ĺ—ā—Ā—É—ā—Ā—ā–≤–ł–Ķ —Ā—ā–į—ā–ł—Ā—ā–ł—á–Ķ—Ā–ļ–ł –∑–Ĺ–į—á–ł–ľ—č—Ö —Ä–į–∑–Ľ–ł—á–ł–Ļ –ľ–Ķ–∂–ī—ɬ†–≥—Ä—É–Ņ–Ņ–į–ľ–ł. –°—ā–į—ā–ł—Ā—ā–ł—á–Ķ—Ā–ļ–ł –∑–Ĺ–į—á–ł–ľ—č—Ö —Ä–į–∑–Ľ–ł—á–ł–Ļ –Ņ–嬆–Ņ–ĺ–ļ–į–∑–į—ā–Ķ–Ľ—é Ctrough –ľ–Ķ–∂–ī—ɬ†–≥—Ä—É–Ņ–Ņ–į–ľ–ł –Ĺ–Ķ¬†–∑–į—Ä–Ķ–≥–ł—Ā—ā—Ä–ł—Ä–ĺ–≤–į–Ĺ–ĺ –Ĺ–ł¬†–≤¬†–ĺ–ī–Ĺ–ĺ–ľ –ł–∑¬†—Ü–ł–ļ–Ľ–ĺ–≤. –°–Ľ–Ķ–ī–ĺ–≤–į—ā–Ķ–Ľ—Ć–Ĺ–ĺ, –ľ–ĺ–∂–Ŗ嬆–≥–ĺ–≤–ĺ—Ä–ł—ā—Ć –ĺ–Ī¬†—ć–ļ–≤–ł–≤–į–Ľ–Ķ–Ĺ—ā–Ĺ–ĺ—Ā—ā–ł —Ą–į—Ä–ľ–į–ļ–ĺ–ļ–ł–Ĺ–Ķ—ā–ł—á–Ķ—Ā–ļ–ł—Ö —Ā–≤–ĺ–Ļ—Ā—ā–≤ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–ĺ–≤ BCD-021 –ł¬†–ź–≤–į—Ā—ā–ł–Ŭģ –Ņ—Ä–ł –≤–Ĺ—É—ā—Ä–ł–≤–Ķ–Ĺ–Ĺ–ĺ–ľ –≤–≤–Ķ–ī–Ķ–Ĺ–ł–ł.
–†–Ķ–∑—é–ľ–ł—Ä—É—Ź —Ā–ļ–į–∑–į–Ĺ–Ĺ–ĺ–Ķ, –°.–í.¬†–ě—Ä–Ľ–ĺ–≤ –Ņ–ĺ–ī—á–Ķ—Ä–ļ–Ĺ—É–Ľ, —á—ā–ĺ –į–Ĺ–į–Ľ–ł–∑ –ī–į–Ĺ–Ĺ—č—Ö –ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ĺ–≥–ĺ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź –Ņ–ĺ–ī—ā–≤–Ķ—Ä–ī–ł–Ľ –ĺ—ā—Ā—É—ā—Ā—ā–≤–ł–Ķ –∑–Ĺ–į—á–ł–ľ—č—Ö —Ä–į–∑–Ľ–ł—á–ł–Ļ –ľ–Ķ–∂–ī—É –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į–ľ–ł BCD-021 –ł¬†–ź–≤–į—Ā—ā–ł–Ŭģ –Ņ–嬆–≤—Ā–Ķ–ľ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–≤—ą–ł–ľ—Ā—Ź –Ņ–ĺ–ļ–į–∑–į—ā–Ķ–Ľ—Ź–ľ. –ü—Ä–Ķ–Ņ–į—Ä–į—ā BCD-021 –ľ–ĺ–∂–Ķ—ā –Ī—č—ā—Ć —Ä–Ķ–ļ–ĺ–ľ–Ķ–Ĺ–ī–ĺ–≤–į–Ĺ –ļ¬†–≤–Ĺ–Ķ–ī—Ä–Ķ–Ĺ–ł—é –≤¬†–ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ—É—é –Ņ—Ä–į–ļ—ā–ł–ļ—É, —á—ā–ĺ –Ņ–ĺ–∑–≤–ĺ–Ľ–ł—ā –Ņ–ĺ–≤—č—Ā–ł—ā—Ć –ī–ĺ—Ā—ā—É–Ņ–Ĺ–ĺ—Ā—ā—Ć —Ā–ĺ–≤—Ä–Ķ–ľ–Ķ–Ĺ–Ĺ–ĺ–Ļ —ā–Ķ—Ä–į–Ņ–ł–ł –ī–Ľ—Ź –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ —Ā–嬆–ľ–Ĺ–ĺ–≥–ł–ľ–ł –∑–Ľ–ĺ–ļ–į—á–Ķ—Ā—ā–≤–Ķ–Ĺ–Ĺ—č–ľ–ł –ĺ–Ņ—É—Ö–ĺ–Ľ—Ź–ľ–ł –ł¬†–∑–Ĺ–į—á–ł–ľ–ĺ —É–Ľ—É—á—ą–ł—ā—Ć —Ä–Ķ–∑—É–Ľ—Ć—ā–į—ā—č —ā–Ķ—Ä–į–Ņ–ł–ł, –ĺ–ī–Ĺ–ĺ–≤—Ä–Ķ–ľ–Ķ–Ĺ–Ĺ–ĺ —É–ľ–Ķ–Ĺ—Ć—ą–ł–≤ –∑–į—ā—Ä–į—ā—č –Ĺ–į¬†–Ľ–Ķ—á–Ķ–Ĺ–ł–Ķ.
–ě—Ä–ł–≥–ł–Ĺ–į–Ľ—Ć–Ĺ—č–Ļ –ĺ—ā–Ķ—á–Ķ—Ā—ā–≤–Ķ–Ĺ–Ĺ—č–Ļ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā –ď-–ö–°–§ –Ņ—Ä–ĺ–Ľ–ĺ–Ĺ–≥–ł—Ä–ĺ–≤–į–Ĺ–Ĺ–ĺ–≥–ĺ –ī–Ķ–Ļ—Ā—ā–≤–ł—Ź —ć–ľ–Ņ—ć–≥—Ą–ł–Ľ–≥—Ä–į—Ā—ā–ł–ľ: –į–Ĺ–į–Ľ–ł–∑ —Ä–Ķ–∑—É–Ľ—Ć—ā–į—ā–ĺ–≤ —Ä–Ķ–≥–ł—Ā—ā—Ä–į—Ü–ł–ĺ–Ĺ–Ĺ–ĺ–≥–ĺ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź
–ö–ĺ–ľ–Ņ–į–Ĺ–ł–Ķ–Ļ BIOCAD —Ä–į–∑—Ä–į–Ī–ĺ—ā–į–Ĺ –Ĺ–ĺ–≤—č–Ļ –ĺ—Ä–ł–≥–ł–Ĺ–į–Ľ—Ć–Ĺ—č–Ļ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā –Ņ–Ķ–≥–ł–Ľ–ł—Ä–ĺ–≤–į–Ĺ–Ĺ–ĺ–≥–ĺ —Ą–ł–Ľ–≥—Ä–į—Ā—ā–ł–ľ–į¬†‚Äď —ć–ľ–Ņ—ć–≥—Ą–ł–Ľ¬≠–≥—Ä–į—Ā—ā–ł–ľ –Ņ—Ä–ĺ–Ľ–ĺ–Ĺ–≥–ł—Ä–ĺ–≤–į–Ĺ–Ĺ–ĺ–≥–ĺ –ī–Ķ–Ļ—Ā—ā–≤–ł—Ź –ī–Ľ—Ź –Ņ—Ä–ĺ—Ą–ł–Ľ–į–ļ—ā–ł–ļ–ł —ā–į–ļ–ł—Ö —ā—Ź–∂–Ķ–Ľ—č—Ö –ĺ—Ā–Ľ–ĺ–∂–Ĺ–Ķ–Ĺ–ł–Ļ —Ö–ł–ľ–ł–ĺ—ā–Ķ—Ä–į–Ņ–ł–ł, –ļ–į–ļ –Ĺ–Ķ–Ļ—ā—Ä–ĺ–Ņ–Ķ–Ĺ–ł—Ź. –ü—Ä–Ķ–Ņ–į—Ä–į—ā —É—Ā–Ņ–Ķ—ą–Ĺ–ĺ –Ņ—Ä–ĺ—ą–Ķ–Ľ –ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ł–Ķ –ł—Ā–Ņ—č—ā–į–Ĺ–ł—Ź. –í—Ä–į—á-–ĺ–Ĺ–ļ–ĺ–Ľ–ĺ–≥ –ź—Ä—Ö–į–Ĺ–≥–Ķ–Ľ—Ć—Ā–ļ–ĺ–≥–ĺ –ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ĺ–≥–ĺ –ĺ–Ĺ–ļ–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ĺ–≥–ĺ –ī–ł—Ā–Ņ–į–Ĺ—Ā–Ķ—Ä–į –ú–į—Ä–ł–Ĺ–į –Ě–ł–ļ–ĺ–Ľ–į–Ķ–≤–Ĺ–į –Ě–ē–ß–ź–ē–í–ź —Ä–į—Ā—Ā–ļ–į–∑–į–Ľ–į –嬆—Ö–ĺ–ī–Ķ –ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ĺ–Ļ —Ä–į–∑—Ä–į–Ī–ĺ—ā–ļ–ł –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į –ł¬†–Ķ–≥–ĺ —É–Ĺ–ł–ļ–į–Ľ—Ć–Ĺ—č—Ö –ĺ—Ā–ĺ–Ī–Ķ–Ĺ–Ĺ–ĺ—Ā—ā—Ź—Ö.
–§–ł–∑–ł–ļ–ĺ-—Ö–ł–ľ–ł—á–Ķ—Ā–ļ–ł–Ķ —Ā–≤–ĺ–Ļ—Ā—ā–≤–į –ľ–ĺ–Ľ–Ķ–ļ—É–Ľ—č –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į —ć–ľ–Ņ—ć–≥—Ą–ł–Ľ–≥—Ä–į—Ā—ā–ł–ľ –ł–ľ–Ķ—é—ā –ĺ—ā–Ľ–ł—á–ł—Ź –ĺ—ā¬†–Ņ–Ķ—Ä–≤–ĺ–≥–ĺ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į –Ņ–Ķ–≥–ł–Ľ–ł—Ä–ĺ–≤–į–Ĺ–Ĺ–ĺ–≥–ĺ —Ą–ł–Ľ–≥—Ä–į—Ā—ā–ł–ľ–į –Ĺ–į¬†—Ä—č–Ĺ–ļ–Ķ¬†‚Äď –Ě–Ķ—É–Ľ–į—Ā—ā–ł–ľ–į. –ú–ĺ–Ľ–Ķ–ļ—É–Ľ—Ź—Ä–Ĺ–į—Ź –ľ–į—Ā—Ā–į –ĺ—Ā—ā–į—ā–ļ–į –Ņ–ĺ–Ľ–ł—ć—ā–ł–Ľ–Ķ–Ĺ–≥–Ľ–ł–ļ–ĺ–Ľ—Ź –≤¬†—Ā–ĺ—Ā—ā–į–≤–Ķ —ć–ľ–Ņ—ć–≥—Ą–ł–Ľ–≥—Ä–į—Ā—ā–ł–ľ–į —Ā–ĺ—Ā—ā–į–≤–Ľ—Ź–Ķ—ā 30 –ļ–Ē–į, –≤¬†—ā–ĺ –≤—Ä–Ķ–ľ—Ź –ļ–į–ļ –≤¬†—Ā–ĺ—Ā—ā–į–≤–Ķ –Ě–Ķ—É–Ľ–į—Ā—ā–ł–ľ–į —Ā–ĺ–ī–Ķ—Ä–∂–ł—ā—Ā—Ź –Ņ–ĺ–Ľ–ł—ć—ā–ł–Ľ–Ķ–Ĺ–≥–Ľ–ł–ļ–ĺ–Ľ—Ć –ľ–į—Ā—Ā–ĺ–Ļ 20 –ļ–Ē–į. –£–≤–Ķ–Ľ–ł—á–Ķ–Ĺ–ł–Ķ –ľ–į—Ā—Ā—č –Ņ–ĺ–Ľ–ł—ć—ā–ł–Ľ–Ķ–Ĺ–≥–Ľ–ł–ļ–ĺ–Ľ—Ź –Ņ—Ä–ł–≤–ĺ–ī–ł—ā –ļ¬†—ā–ĺ–ľ—É, —á—ā–ĺ –ĺ–Ĺ —Ā—ā–Ķ—Ä–ł—á–Ķ—Ā–ļ–ł –ī–į–Ľ—Ć—ą–Ķ —Ä–į—Ā–Ņ–ĺ–Ľ–į–≥–į–Ķ—ā—Ā—Ź –ĺ—ā¬†–į–ļ—ā–ł–≤–Ĺ–ĺ–Ļ —á–į—Ā—ā–ł –ľ–ĺ–Ľ–Ķ–ļ—É–Ľ—č –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į¬†‚Äď —Ą–ł–Ľ–≥—Ä–į—Ā—ā–ł–ľ–į. –Ď–Ľ–į–≥–ĺ–ī–į—Ä—Ź —ć—ā–ĺ–ľ—É —Ā–Ņ–Ķ—Ü–ł—Ą–ł—á–Ķ—Ā–ļ–į—Ź –į–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į —ć–ľ–Ņ—ć–≥—Ą–ł–Ľ¬≠–≥—Ä–į—Ā—ā–ł–ľ –Ņ—Ä–Ķ–≤—č—ą–į–Ķ—ā —ā–į–ļ–ĺ–≤—É—é –Ě–Ķ—É–Ľ–į—Ā—ā–ł–ľ–į¬†‚Äď 84‚Äď94 –ł¬†68% —Ā–ĺ–ĺ—ā–≤–Ķ—ā—Ā—ā–≤–Ķ–Ĺ–Ĺ–ĺ.
–Ě–į¬†—Ā–Ķ–≥–ĺ–ī–Ĺ—Ź—ą–Ĺ–ł–Ļ –ī–Ķ–Ĺ—Ć –≤—č–Ņ–ĺ–Ľ–Ĺ–Ķ–Ĺ –≤–Ķ—Ā—Ć –ĺ–Ī—ä–Ķ–ľ —Ā—Ä–į–≤–Ĺ–ł—ā–Ķ–Ľ—Ć–Ĺ—č—Ö —Ą–ł–∑–ł–ļ–ĺ-—Ö–ł–ľ–ł—á–Ķ—Ā–ļ–ł—Ö, –ī–ĺ–ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ł—Ö –ł¬†–ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ł—Ö –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–Ļ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į —ć–ľ–Ņ—ć–≥—Ą–ł–Ľ–≥—Ä–į—Ā—ā–ł–ľ. –í¬†–ļ–ĺ–Ĺ—Ü–Ķ 2014¬†–≥.¬†–∑–į–≤–Ķ—Ä—ą–Ķ–Ĺ –į–Ĺ–į–Ľ–ł–∑ –ī–į–Ĺ–Ĺ—č—Ö –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź —Ą–į–∑—č III, –≤¬†–ļ–ĺ—ā–ĺ—Ä–ĺ–ľ –Ĺ–į—Ä—Ź–ī—É —Ā¬†–ī—Ä—É–≥–ł–ľ–ł –ľ–Ķ–ī–ł—Ü–ł–Ĺ—Ā–ļ–ł–ľ–ł —Ü–Ķ–Ĺ—ā—Ä–į–ľ–ł —É—á–į—Ā—ā–≤–ĺ–≤–į–Ľ–ł –ł¬†—Ā–Ņ–Ķ—Ü–ł–į–Ľ–ł—Ā—ā—č –ź—Ä—Ö–į–Ĺ–≥–Ķ–Ľ—Ć—Ā–ļ–ĺ–≥–ĺ –ĺ–Ĺ–ļ–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ĺ–≥–ĺ –ī–ł—Ā–Ņ–į–Ĺ—Ā–Ķ—Ä–į. –ú.–Ě. –Ě–Ķ—á–į–Ķ–≤–į –Ņ—Ä–Ķ–ī—Ā—ā–į–≤–ł–Ľ–į —É—á–į—Ā—ā–Ĺ–ł–ļ–į–ľ —Ā–ł–ľ–Ņ–ĺ–∑–ł—É–ľ–į –ĺ—Ā–Ĺ–ĺ–≤–Ĺ—č–Ķ —Ä–Ķ–∑—É–Ľ—Ć—ā–į—ā—č –∑–į–≤–Ķ—Ä—ą–Ķ–Ĺ–Ĺ–ĺ–≥–ĺ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź.
–í¬†–ľ–Ķ–∂–ī—É–Ĺ–į—Ä–ĺ–ī–Ĺ–ĺ–ľ –ľ–Ĺ–ĺ–≥–ĺ—Ü–Ķ–Ĺ—ā—Ä–ĺ–≤–ĺ–ľ –ī–≤–ĺ–Ļ–Ĺ–ĺ–ľ —Ā–Ľ–Ķ–Ņ–ĺ–ľ —Ä–į–Ĺ–ī–ĺ–ľ–ł–∑–ł—Ä–ĺ–≤–į–Ĺ–Ĺ–ĺ–ľ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–ł —Ā¬†–ī–≤–ĺ–Ļ–Ĺ—č–ľ –ľ–į—Ā–ļ–ł—Ä–ĺ–≤–į–Ĺ–ł–Ķ–ľ —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć –Ĺ–ĺ–≤–ĺ–≥–ĺ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į BCD-017 (—ć–ľ–Ņ—ć–≥—Ą–ł–Ľ¬≠–≥—Ä–į—Ā—ā–ł–ľ) –ĺ—Ü–Ķ–Ĺ–ł–≤–į–Ľ–ł –Ĺ–į¬†–Ņ—Ä–ĺ—ā—Ź–∂–Ķ–Ĺ–ł–ł —á–Ķ—ā—č—Ä–Ķ—Ö —Ü–ł–ļ–Ľ–ĺ–≤ —Ö–ł–ľ–ł–ĺ—ā–Ķ—Ä–į–Ņ–ł–ł —ɬ†–Ī–ĺ–Ľ—Ć–Ĺ—č—Ö –†–ú–Ė. –ü—Ä–Ķ–Ņ–į—Ä–į—ā–ĺ–ľ —Ā—Ä–į–≤–Ĺ–Ķ–Ĺ–ł—Ź –Ī—č–Ľ –Ĺ–Ķ–Ņ–Ķ–≥–ł–Ľ–ł—Ä–ĺ–≤–į–Ĺ–Ĺ—č–Ļ —Ą–ł–Ľ–≥—Ä–į—Ā—ā–ł–ľ (–õ–Ķ–Ļ–ļ–ĺ—Ā—ā–ł–ľ¬ģ), –Ņ—Ä–ĺ–ł–∑–≤–ĺ–ī–ł–ľ—č–Ļ —ā–į–ļ–∂–Ķ –ļ–ĺ–ľ–Ņ–į–Ĺ–ł–Ķ–Ļ BIOCAD. –í¬†–ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–ł —É—á–į—Ā—ā–≤–ĺ–≤–į–Ľ–ł 135¬†–Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–ļ —Ā¬†–†–ú–Ė —Ā—ā–į–ī–ł–Ļ II‚ÄďIV, —Ä–į–Ĺ–ī–ĺ–ľ–ł–∑–ł—Ä–ĺ–≤–į–Ĺ–Ĺ—č—Ö –≤¬†—ā—Ä–ł¬†–≥—Ä—É–Ņ–Ņ—č –≤¬†—Ā–ĺ–ĺ—ā–Ĺ–ĺ—ą–Ķ–Ĺ–ł–ł 1:1:1 (–Ņ–į—Ü–ł–Ķ–Ĺ—ā–ļ–ł –≤¬†–ī–≤—É—Ö¬†–≥—Ä—É–Ņ–Ņ–į—Ö –Ņ–ĺ–Ľ—É—á–į–Ľ–ł –ī–≤–Ķ —Ä–į–∑–Ĺ—č—Ö –ī–ĺ–∑–ł—Ä–ĺ–≤–ļ–ł —ć–ľ–Ņ—ć–≥—Ą–ł–Ľ–≥—Ä–į—Ā—ā–ł–ľ–į, –≤¬†—ā—Ä–Ķ—ā—Ć–Ķ–Ļ¬†–≥—Ä—É–Ņ–Ņ–Ķ¬†‚Äď –Ĺ–Ķ–Ņ–Ķ–≥–ł–Ľ–ł—Ä–ĺ–≤–į–Ĺ–Ĺ—č–Ļ —Ą–ł–Ľ–≥—Ä–į—Ā—ā–ł–ľ). –í—Ā–Ķ —É—á–į—Ā—ā–Ĺ–ł—Ü—č –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź –Ņ–ĺ–Ľ—É—á–į–Ľ–ł —Ö–ł–ľ–ł–ĺ—ā–Ķ—Ä–į–Ņ–ł—é –Ņ–嬆—Ā—Ö–Ķ–ľ–Ķ ¬ę–ī–ĺ—Ü–Ķ—ā–į–ļ—Ā–Ķ–Ľ 75¬†–ľ–≥/–ľ2 + –ī–ĺ–ļ—Ā–ĺ—Ä—É–Ī–ł—Ü–ł–Ĺ 50¬†–ľ–≥/–ľ2¬Ľ. –Ě–į¬†–≤—ā–ĺ—Ä–ĺ–Ļ –ī–Ķ–Ĺ—Ć, –Ĺ–Ķ¬†–ľ–Ķ–Ĺ–Ķ–Ķ —á–Ķ–ľ —á–Ķ—Ä–Ķ–∑ 24¬†—á–į—Ā–į –Ņ–ĺ—Ā–Ľ–Ķ –≤–≤–Ķ–ī–Ķ–Ĺ–ł—Ź —Ö–ł–ľ–ł–ĺ—ā–Ķ—Ä–į–Ņ–ł–ł, –Ĺ–į—á–ł–Ĺ–į–Ľ–ĺ—Ā—Ć –≤–≤–Ķ–ī–Ķ–Ĺ–ł–Ķ –ł—Ā—Ā–Ľ–Ķ–ī—É–Ķ–ľ–ĺ–Ļ —ā–Ķ—Ä–į–Ņ–ł–ł.
–ü–į—Ü–ł–Ķ–Ĺ—ā–ļ–į–ľ –Ņ–Ķ—Ä–≤–ĺ–Ļ¬†–≥—Ä—É–Ņ–Ņ—č –ĺ–ī–Ĺ–ĺ–ļ—Ä–į—ā–Ĺ–ĺ –Ņ–ĺ–ī–ļ–ĺ–∂–Ĺ–ĺ –≤–≤–ĺ–ī–ł–Ľ–ł –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā BCD-017 –≤¬†–ī–ĺ–∑–Ķ 6¬†–ľ–≥ —Ā¬†–Ņ–ĺ—Ā–Ľ–Ķ–ī—É—é—Č–ł–ľ –Ķ–∂–Ķ–ī–Ĺ–Ķ–≤–Ĺ—č–ľ –≤–≤–Ķ–ī–Ķ–Ĺ–ł–Ķ–ľ –Ņ–Ľ–į—Ü–Ķ–Ī–ĺ (0,0083¬†–ľ–Ľ/–ļ–≥). –ü–į—Ü–ł–Ķ–Ĺ—ā–ļ–į–ľ –≤—ā–ĺ—Ä–ĺ–Ļ¬†–≥—Ä—É–Ņ–Ņ—č –Ĺ–į¬†–≤—ā–ĺ—Ä–ĺ–Ļ –ī–Ķ–Ĺ—Ć –ĺ–ī–Ĺ–ĺ–ļ—Ä–į—ā–Ĺ–ĺ –Ņ–ĺ–ī–ļ–ĺ–∂–Ĺ–ĺ –≤–≤–ĺ–ī–ł–Ľ–ł –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā BCD-017 7,5¬†–ľ–≥ —Ā¬†–Ņ–ĺ—Ā–Ľ–Ķ–ī—É—é—Č–ł–ľ –Ķ–∂–Ķ–ī–Ĺ–Ķ–≤–Ĺ—č–ľ –≤–≤–Ķ–ī–Ķ–Ĺ–ł–Ķ–ľ –Ņ–Ľ–į—Ü–Ķ–Ī–ĺ (0,0083¬†–ľ–Ľ/–ļ–≥). –ü–Ľ–į—Ü–Ķ–Ī–ĺ –≤¬†–Ņ–Ķ—Ä–≤—č—Ö –ī–≤—É—Ö¬†–≥—Ä—É–Ņ–Ņ–į—Ö –≤–≤–ĺ–ī–ł–Ľ–ł –ī–ĺ –ī–ĺ—Ā—ā–ł–∂–Ķ–Ĺ–ł—Ź –į–Ī—Ā–ĺ–Ľ—é—ā–Ĺ–ĺ–≥–ĺ —á–ł—Ā–Ľ–į –Ĺ–Ķ–Ļ—ā—Ä–ĺ—Ą–ł–Ľ–ĺ–≤ (–ź–ß–Ě) 10 √ó 109/–Ľ, –Ŗ嬆–Ĺ–Ķ –Ī–ĺ–Ľ–Ķ–Ķ 14¬†–ī–Ĺ–Ķ–Ļ. –ü–į—Ü–ł–Ķ–Ĺ—ā–ļ–į–ľ¬†–≥—Ä—É–Ņ–Ņ—č —Ā—Ä–į–≤–Ĺ–Ķ–Ĺ–ł—Ź –Ņ–Ľ–į—Ü–Ķ–Ī–ĺ –≤–≤–ĺ–ī–ł–Ľ–ĺ—Ā—Ć –ĺ–ī–Ĺ–ĺ–ļ—Ä–į—ā–Ĺ–ĺ, –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā —Ā—Ä–į–≤–Ĺ–Ķ–Ĺ–ł—Ź –≤–≤–ĺ–ī–ł–Ľ—Ā—Ź –≤¬†–ī–ĺ–∑–Ķ 5¬†–ľ–≥/–ļ–≥ –ī–ĺ —ā–Ķ—Ö –Ņ–ĺ—Ä, –Ņ–ĺ–ļ–į –ź–ß–Ě –Ĺ–Ķ¬†–ī–ĺ—Ā—ā–ł–≥–į–Ľ–ĺ 10 √ó 109/–Ľ, –Ŗ嬆–Ĺ–Ķ –Ī–ĺ–Ľ–Ķ–Ķ 14¬†–ī–Ĺ–Ķ–Ļ. –ė—Ā–Ņ–ĺ–Ľ—Ć–∑–ĺ–≤–į–Ĺ–ł–Ķ –ī–≤–ĺ–Ļ–Ĺ–ĺ–≥–ĺ –ľ–į—Ā–ļ–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź –Ņ–ĺ–ī—Ä–į–∑—É–ľ–Ķ–≤–į–Ľ–ĺ –Ņ—Ä–ł–ľ–Ķ–Ĺ–Ķ–Ĺ–ł–Ķ –ī–≤—É—Ö –≤–ł–ī–ĺ–≤ –Ņ–Ľ–į—Ü–Ķ–Ī–嬆‚Äď –Ņ–Ķ—Ä–≤—č–Ļ –≤–ł–ī, –≤–Ĺ–Ķ—ą–Ĺ–Ķ –Ĺ–Ķ¬†–ĺ—ā–Ľ–ł—á–ł–ľ—č–Ļ –ĺ—ā¬†–Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į —Ā—Ä–į–≤–Ĺ–Ķ–Ĺ–ł—Ź,¬†‚Äď –≤¬†–≥—Ä—É–Ņ–Ņ–į—Ö –ł—Ā—Ā–Ľ–Ķ–ī—É–Ķ–ľ–ĺ–≥–ĺ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į, –≤—ā–ĺ—Ä–ĺ–Ļ, –Ĺ–Ķ¬†–ĺ—ā–Ľ–ł—á–ł–ľ—č–Ļ –ĺ—ā¬†–ł—Ā—Ā–Ľ–Ķ–ī—É–Ķ–ľ–ĺ–≥–ĺ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į,¬†‚Äď –≤¬†–≥—Ä—É–Ņ–Ņ–Ķ —Ā—Ä–į–≤–Ĺ–Ķ–Ĺ–ł—Ź.
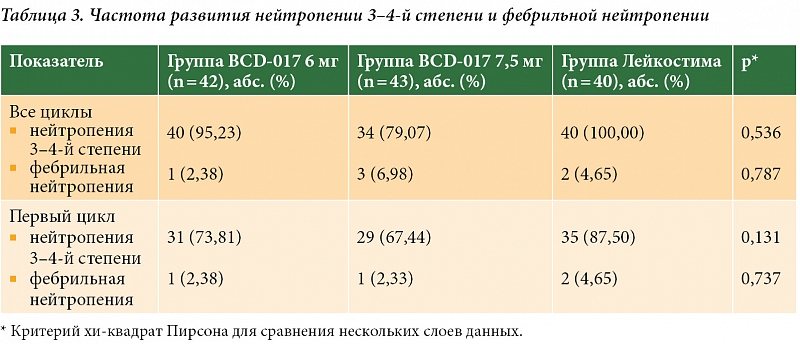
–ě—Ā–Ĺ–ĺ–≤–Ĺ—č–ľ –Ņ–ĺ–ļ–į–∑–į—ā–Ķ–Ľ–Ķ–ľ, –Ņ–嬆–ļ–ĺ—ā–ĺ—Ä–ĺ–ľ—É –ĺ—Ü–Ķ–Ĺ–ł–≤–į–Ľ–ł —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć —ā–Ķ—Ä–į–Ņ–ł–ł, –≤¬†–ī–į–Ĺ–Ĺ–ĺ–ľ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–ł –Ī—č–Ľ–į –Ņ—Ä–ĺ–ī–ĺ–Ľ–∂–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ—Ā—ā—Ć –Ĺ–Ķ–Ļ—ā—Ä–ĺ–Ņ–Ķ–Ĺ–ł–ł 4-–Ļ¬†—Ā—ā–Ķ–Ņ–Ķ–Ĺ–ł —ā—Ź–∂–Ķ—Ā—ā–ł –Ĺ–į¬†–Ņ–Ķ—Ä–≤–ĺ–ľ —Ü–ł–ļ–Ľ–Ķ —Ö–ł–ľ–ł–ĺ—ā–Ķ—Ä–į–Ņ–ł–ł. –ú–į–ļ—Ā–ł–ľ–į–Ľ—Ć–Ĺ—č–Ķ –Ņ–ĺ–ļ–į–∑–į—ā–Ķ–Ľ–ł –Ņ—Ä–ĺ–ī–ĺ–Ľ–∂–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ—Ā—ā–ł –Ĺ–Ķ–Ļ—ā—Ä–ĺ–Ņ–Ķ–Ĺ–ł–ł 4-–Ļ¬†—Ā—ā–Ķ–Ņ–Ķ–Ĺ–ł –ĺ—ā–ľ–Ķ—á–į–Ľ–ł—Ā—Ć –≤¬†–≥—Ä—É–Ņ–Ņ–Ķ —Ą–ł–Ľ–≥—Ä–į—Ā—ā–ł–ľ–į, —Ä–į–∑–Ľ–ł—á–ł—Ź —Ā¬†–Ņ–ĺ–ļ–į–∑–į—ā–Ķ–Ľ—Ź–ľ–ł –≤¬†–≥—Ä—É–Ņ–Ņ–į—Ö –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į BCD-017 –Ĺ–ĺ—Ā–ł–Ľ–ł —Ā—ā–į—ā–ł—Ā—ā–ł—á–Ķ—Ā–ļ–ł –∑–Ĺ–į—á–ł–ľ—č–Ļ —Ö–į—Ä–į–ļ—ā–Ķ—Ä. –ö—Ä–ĺ–ľ–Ķ —ā–ĺ–≥–ĺ, —Ā–ĺ–≥–Ľ–į—Ā–Ĺ–ĺ –ī–į–Ĺ–Ĺ—č–ľ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź, –Ĺ–į¬†–Ņ—Ä–ĺ—ā—Ź–∂–Ķ–Ĺ–ł–ł –≤—Ā–Ķ—Ö —á–Ķ—ā—č—Ä–Ķ—Ö —Ü–ł–ļ–Ľ–ĺ–≤ —ā—Ź–∂–Ķ–Ľ–į—Ź –Ĺ–Ķ–Ļ—ā—Ä–ĺ–Ņ–Ķ–Ĺ–ł—Ź (3‚Äď4-–Ļ¬†—Ā—ā–Ķ–Ņ–Ĺ–ł) —á–į—Č–Ķ –ł–ľ–Ķ–Ľ–į –ľ–Ķ—Ā—ā–ĺ –≤¬†–≥—Ä—É–Ņ–Ņ–Ķ —Ą–ł–Ľ–≥—Ä–į—Ā—ā–ł–ľ–į, –Ķ–Ķ —á–į—Ā—ā–ĺ—ā–į —Ā–ĺ—Ā—ā–į–≤–Ľ—Ź–Ľ–į 100%. –Ē–Ľ—Ź —Ā—Ä–į–≤–Ĺ–Ķ–Ĺ–ł—Ź: –≤¬†–≥—Ä—É–Ņ–Ņ–Ķ, –Ņ–ĺ–Ľ—É—á–į–≤—ą–Ķ–Ļ BCD-017 7,5¬†–ľ–≥, —á–į—Ā—ā–ĺ—ā–į —ā—Ź–∂–Ķ–Ľ–ĺ–Ļ –Ĺ–Ķ–Ļ—ā—Ä–ĺ–Ņ–Ķ–Ĺ–ł–ł –Ī—č–Ľ–į –Ĺ–ł–∂–Ķ¬†‚Äď 79,07%. –Ę–Ķ–ľ –Ĺ–Ķ¬†–ľ–Ķ–Ĺ–Ķ–Ķ –ī–į–Ĺ–Ĺ—č–Ķ —Ä–į–∑–Ľ–ł—á–ł—Ź –Ĺ–Ķ¬†–ī–ĺ—Ā—ā–ł–≥–į–Ľ–ł —Ā—ā–į—ā–ł—Ā—ā–ł—á–Ķ—Ā–ļ–ĺ–Ļ –∑–Ĺ–į—á–ł–ľ–ĺ—Ā—ā–ł. –§–Ķ–Ī—Ä–ł–Ľ—Ć–Ĺ–į—Ź –Ĺ–Ķ–Ļ—ā—Ä–ĺ–Ņ–Ķ–Ĺ–ł—Ź –≤¬†—Ö–ĺ–ī–Ķ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź –≤—Ā—ā—Ä–Ķ—á–į–Ľ–į—Ā—Ć —Ä–Ķ–ī–ļ–ĺ, –Ķ–Ķ —á–į—Ā—ā–ĺ—ā–į –Ĺ–Ķ¬†–ĺ—ā–Ľ–ł—á–į–Ľ–į—Ā—Ć –≤¬†–≥—Ä—É–Ņ–Ņ–į—Ö (—ā–į–Ī–Ľ.¬†3).
–í¬†—ā–Ķ—á–Ķ–Ĺ–ł–Ķ –Ņ–Ķ—Ä–≤–ĺ–≥–ĺ —Ü–ł–ļ–Ľ–į —ā–Ķ—Ä–į–Ņ–ł–ł —ā—Ź–∂–Ķ–Ľ—č–Ķ –ł–Ĺ—Ą–Ķ–ļ—Ü–ł–ł –Ĺ–Ķ¬†—Ä–Ķ–≥–ł—Ā—ā—Ä–ł—Ä–ĺ–≤–į–Ľ–ł—Ā—Ć –Ĺ–ł¬†–≤¬†–ĺ–ī–Ĺ–ĺ–Ļ¬†–≥—Ä—É–Ņ–Ņ–Ķ, –į¬†—Ā–ĺ –≤—ā–ĺ—Ä–ĺ–≥–ĺ –Ņ–嬆—á–Ķ—ā–≤–Ķ—Ä—ā—č–Ļ —Ü–ł–ļ–Ľ¬†‚Äď —ā–ĺ–Ľ—Ć–ļ–ĺ –≤¬†–≥—Ä—É–Ņ–Ņ–Ķ –õ–Ķ–Ļ–ļ–ĺ—Ā—ā–ł–ľ–į (10%). –ö¬†—Ā–Ľ—É—á–į—Ź–ľ —ā—Ź–∂–Ķ–Ľ–ĺ–Ļ –ł–Ĺ—Ą–Ķ–ļ—Ü–ł–ł –≤¬†—Ö–ĺ–ī–Ķ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź –ĺ—ā–Ĺ–ĺ—Ā–ł–Ľ–ł—Ā—Ć –Ņ–嬆–ĺ–ī–Ĺ–ĺ–ľ—É —Ā–Ľ—É—á–į—é –Ņ–Ĺ–Ķ–≤–ľ–ĺ–Ĺ–ł–ł, —Ä–ĺ–∂–ł—Ā—ā–ĺ–≥–ĺ –≤–ĺ—Ā–Ņ–į–Ľ–Ķ–Ĺ–ł—Ź –Ņ—Ä–į–≤–ĺ–≥–ĺ –Ņ—Ä–Ķ–ī–Ņ–Ľ–Ķ—á—Ć—Ź, –≤–ĺ—Ā–Ņ–į–Ľ–Ķ–Ĺ–ł—Ź –į—ā–Ķ—Ä–ĺ–ľ—č –Ņ—Ä–Ķ–ī–Ņ–Ľ–Ķ—á—Ć—Ź, —Ą—É—Ä—É–Ĺ–ļ—É–Ľ–į –ĺ–Ī–Ľ–į—Ā—ā–ł –Ĺ–ĺ—Ā–į.
–ü—Ä–ł –ĺ—Ü–Ķ–Ĺ–ļ–Ķ –Ĺ–į–ī–ł—Ä–į —É—Ä–ĺ–≤–Ĺ—Ź –Ĺ–Ķ–Ļ—ā—Ä–ĺ—Ą–ł–Ľ–ĺ–≤ –Ī—č–Ľ–ĺ –≤—č—Ź–≤–Ľ–Ķ–Ĺ–ĺ, —á—ā–ĺ –≤¬†–≥—Ä—É–Ņ–Ņ–Ķ —Ą–ł–Ľ–≥—Ä–į—Ā—ā–ł–ľ–į –ĺ–Ĺ –Ī—č–Ľ –ī–ĺ—Ā—ā–ĺ–≤–Ķ—Ä–Ĺ–ĺ –Ĺ–ł–∂–Ķ, —á–Ķ–ľ –≤¬†–≥—Ä—É–Ņ–Ņ–į—Ö BCD-017¬†6 –ł¬†7,5¬†–ľ–≥. –ü—Ä–ł —ć—ā–ĺ–ľ –≤¬†–≥—Ä—É–Ņ–Ņ–Ķ BCD-017 7,5¬†–ľ–≥ –Ĺ–į¬†–Ņ–Ķ—Ä–≤–ĺ–ľ —Ü–ł–ļ–Ľ–Ķ —ā–Ķ—Ä–į–Ņ–ł–ł –Ĺ–į–ī–ł—Ä –Ĺ–Ķ–Ļ—ā—Ä–ĺ—Ą–ł–Ľ–ĺ–≤ –Ņ–ĺ—á—ā–ł –≤¬†–ī–≤–į —Ä–į–∑–į –Ņ—Ä–Ķ–≤—č—ą–į–Ľ –Ņ–ĺ–ļ–į–∑–į—ā–Ķ–Ľ–ł –≤¬†–≥—Ä—É–Ņ–Ņ–Ķ —Ą–ł–Ľ–≥—Ä–į—Ā—ā–ł–ľ–į –ł¬†–Ī—č–Ľ –Ī–Ľ–ł–∑–ĺ–ļ –ļ¬†–Ĺ–ł–∂–Ĺ–Ķ–Ļ¬†–≥—Ä–į–Ĺ–ł—Ü–Ķ –Ĺ–ĺ—Ä–ľ—č, –≤¬†—ā–ĺ –≤—Ä–Ķ–ľ—Ź –ļ–į–ļ –≤¬†–≥—Ä—É–Ņ–Ņ–Ķ —Ą–ł–Ľ–≥—Ä–į—Ā—ā–ł–ľ–į —Ā–ĺ–ĺ—ā–≤–Ķ—ā—Ā—ā–≤–ĺ–≤–į–Ľ –Ĺ–Ķ–Ļ—ā—Ä–ĺ–Ņ–Ķ–Ĺ–ł–ł 4-–Ļ¬†—Ā—ā–Ķ–Ņ–Ķ–Ĺ–ł.
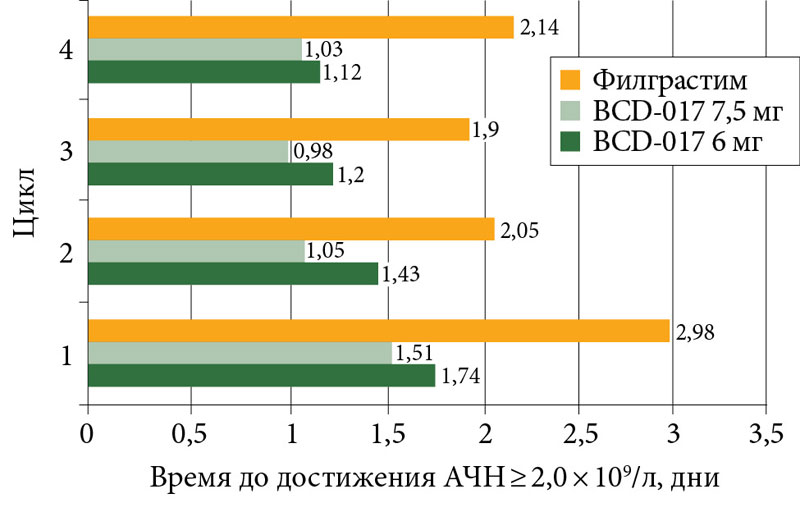
–í—Ä–Ķ–ľ—Ź –ī–ĺ –≤–ĺ—Ā—Ā—ā–į–Ĺ–ĺ–≤–Ľ–Ķ–Ĺ–ł—Ź –Ĺ–ĺ—Ä–ľ–į–Ľ—Ć–Ĺ–ĺ–≥–ĺ –ź–ß–Ě –Ņ–ĺ—Ā—ā–Ķ–Ņ–Ķ–Ĺ–Ĺ–ĺ —É–ľ–Ķ–Ĺ—Ć—ą–į–Ľ–ĺ—Ā—Ć —Ā¬†–Ņ–Ķ—Ä–≤–ĺ–≥–ĺ –Ņ–嬆—á–Ķ—ā–≤–Ķ—Ä—ā—č–Ļ —Ü–ł–ļ–Ľ (—Ä–ł—Ā.¬†3).
–Ě–į–ł–Ī–ĺ–Ľ–Ķ–Ķ –ī–Ľ–ł—ā–Ķ–Ľ—Ć–Ĺ—č–ľ –Ņ—Ä–ĺ—Ü–Ķ—Ā—Ā –≤–ĺ—Ā—Ā—ā–į–Ĺ–ĺ–≤–Ľ–Ķ–Ĺ–ł—Ź –Ĺ–Ķ–Ļ—ā—Ä–ĺ—Ą–ł–Ľ–ĺ–≤ –≤¬†–ļ—Ä–ĺ–≤–ł –ī–ĺ –Ĺ–ĺ—Ä–ľ–į–Ľ—Ć–Ĺ–ĺ–≥–ĺ —É—Ä–ĺ–≤–Ĺ—Ź –Ī—č–Ľ –≤¬†–≥—Ä—É–Ņ–Ņ–Ķ –õ–Ķ–Ļ–ļ–ĺ—Ā—ā–ł–ľ–į. –Ę–į–ļ, –≤–ĺ –≤—Ä–Ķ–ľ—Ź —á–Ķ—ā–≤–Ķ—Ä—ā–ĺ–≥–ĺ —Ü–ł–ļ–Ľ–į –ī–ĺ –ī–ĺ—Ā—ā–ł–∂–Ķ–Ĺ–ł—Ź –Ĺ–ĺ—Ä–ľ–į–Ľ—Ć–Ĺ–ĺ–≥–ĺ —á–ł—Ā–Ľ–į –Ĺ–Ķ–Ļ—ā—Ä–ĺ—Ą–ł–Ľ–ĺ–≤ –≤¬†–≥—Ä—É–Ņ–Ņ–Ķ –õ–Ķ–Ļ–ļ–ĺ—Ā—ā–ł–ľ–į –Ņ–ĺ—ā—Ä–Ķ–Ī–ĺ–≤–į–Ľ–ĺ—Ā—Ć 2,14 –ī–Ĺ—Ź. –í¬†–≥—Ä—É–Ņ–Ņ–į—Ö BCD-017¬†6¬†–ľ–≥ –ł¬†BCD-017¬†7,5¬†–ľ–≥ —ć—ā–ł –Ņ–ĺ–ļ–į–∑–į—ā–Ķ–Ľ–ł —Ā–ĺ—Ā—ā–į–≤–ł–Ľ–ł 1,12 –ł¬†1,03¬†–ī–Ĺ—Ź —Ā–ĺ–ĺ—ā–≤–Ķ—ā—Ā—ā–≤–Ķ–Ĺ–Ĺ–ĺ.
–Ě–Į –≤¬†—Ö–ĺ–ī–Ķ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź –Ĺ–į–Ī–Ľ—é–ī–į–Ľ–ł—Ā—Ć –Ņ—Ä–į–ļ—ā–ł—á–Ķ—Ā–ļ–ł —ɬ†–≤—Ā–Ķ—Ö –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–ļ, –Ŗ嬆–≤¬†–Ī–ĺ–Ľ—Ć—ą–ł–Ĺ—Ā—ā–≤–Ķ —Ā–Ľ—É—á–į–Ķ–≤ –Ī—č–Ľ–ł —Ā–≤—Ź–∑–į–Ĺ—č —Ā¬†–Ņ—Ä–ł–ľ–Ķ–Ĺ—Ź–≤—ą–Ķ–Ļ—Ā—Ź —Ā—Ö–Ķ–ľ–ĺ–Ļ —Ö–ł–ľ–ł–ĺ—ā–Ķ—Ä–į–Ņ–ł–ł. –ß–į—Ā—ā–ĺ—ā–į —Ź–≤–Ľ–Ķ–Ĺ–ł–Ļ¬†–≥–Ķ–ľ–į—ā–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ĺ–Ļ —ā–ĺ–ļ—Ā–ł—á–Ĺ–ĺ—Ā—ā–ł –Ĺ–Ķ¬†–ł–ľ–Ķ–Ľ–į –∑–Ĺ–į—á–ł–ľ—č—Ö —Ä–į–∑–Ľ–ł—á–ł–Ļ –≤–ĺ –≤—Ā–Ķ—Ö¬†–≥—Ä—É–Ņ–Ņ–į—Ö –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź, –∑–į –ł—Ā–ļ–Ľ—é—á–Ķ–Ĺ–ł–Ķ–ľ –Ľ–Ķ–Ļ–ļ–ĺ–Ņ–Ķ–Ĺ–ł–ł 4-–Ļ¬†—Ā—ā–Ķ–Ņ–Ķ–Ĺ–ł, —á–į—Č–Ķ –Ĺ–į–Ī–Ľ—é–ī–į–≤—ą–Ķ–Ļ—Ā—Ź –≤¬†–≥—Ä—É–Ņ–Ņ–Ķ —Ą–ł–Ľ–≥—Ä–į—Ā—ā–ł–ľ–į, —á—ā–ĺ –ļ–ĺ—Ā–≤–Ķ–ŖŖ嬆–≥–ĺ–≤–ĺ—Ä–ł–Ľ–ĺ –≤¬†–Ņ–ĺ–Ľ—Ć–∑—É –Ī–ĺ–Ľ—Ć—ą–Ķ–Ļ —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā–ł –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į BCD-017. –Ě–Ķ¬†–≤—č—Ź–≤–Ľ–Ķ–Ĺ–ĺ –ī–ĺ—Ā—ā–ĺ–≤–Ķ—Ä–Ĺ—č—Ö —Ä–į–∑–Ľ–ł—á–ł–Ļ –ľ–Ķ–∂–ī—ɬ†–≥—Ä—É–Ņ–Ņ–į–ľ–ł –Ņ–嬆–Ĺ–Ķ–≥–Ķ–ľ–į—ā–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ł–ľ —Ź–≤–Ľ–Ķ–Ĺ–ł—Ź–ľ –ł¬†–Ě–Į, –≤¬†—ā–ĺ–ľ —á–ł—Ā–Ľ–Ķ —Ā–≤–ĺ–Ļ—Ā—ā–≤–Ķ–Ĺ–Ĺ—č–ľ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į–ľ¬†–≥—Ä–į–Ĺ—É–Ľ–ĺ—Ü–ł—ā–į—Ä–Ĺ–ĺ–≥–ĺ –ļ–ĺ–Ľ–ĺ–Ĺ–ł–Ķ—Ā—ā–ł–ľ—É–Ľ–ł—Ä—É—é—Č–Ķ–≥–ĺ —Ą–į–ļ—ā–ĺ—Ä–į (–ď-–ö–°–§) –ł¬†–ľ–Ķ—Ā—ā–Ĺ—č–ľ —Ä–Ķ–į–ļ—Ü–ł—Ź–ľ. –°–Ě–Į —á–į—Č–Ķ –ĺ—ā–ľ–Ķ—á–į–Ľ–ł—Ā—Ć –≤¬†–≥—Ä—É–Ņ–Ņ–Ķ –õ–Ķ–Ļ–ļ–ĺ—Ā—ā–ł–ľ–į (11,63%), –Ŗ嬆—Ä–į–∑–Ľ–ł—á–ł—Ź –ľ–Ķ–∂–ī—ɬ†–≥—Ä—É–Ņ–Ņ–į–ľ–ł –Ī—č–Ľ–ł –Ĺ–Ķ–ī–ĺ—Ā—ā–ĺ–≤–Ķ—Ä–Ĺ—č.
–Ę–į–ļ–ł–ľ –ĺ–Ī—Ä–į–∑–ĺ–ľ, –ł—ā–ĺ–≥–ł –ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ĺ–≥–ĺ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź —Ą–į–∑—č III –Ņ—Ä–ĺ–ī–Ķ–ľ–ĺ–Ĺ—Ā—ā—Ä–ł—Ä–ĺ–≤–į–Ľ–ł –Ĺ–Ķ¬†–ľ–Ķ–Ĺ—Ć—ą—É—é —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć –Ĺ–ĺ–≤–ĺ–≥–ĺ –Ņ–Ķ–≥–ł–Ľ–ł—Ä–ĺ–≤–į–Ĺ–Ĺ–ĺ–≥–ĺ —Ą–ł–Ľ–≥—Ä–į—Ā—ā–ł–ľ–į¬†‚Äď –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į —ć–ľ–Ņ—ć–≥—Ą–ł–Ľ–≥—Ä–į—Ā—ā–ł–ľ –Ņ–嬆—Ā—Ä–į–≤–Ĺ–Ķ–Ĺ–ł—é —Ā¬†–Ĺ–Ķ–Ņ–Ķ–≥–ł–Ľ–ł—Ä–ĺ–≤–į–Ĺ–Ĺ—č–ľ —Ą–ł–Ľ–≥—Ä–į—Ā—ā–ł–ľ–ĺ–ľ¬†‚Äď –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–ĺ–ľ –õ–Ķ–Ļ–ļ–ĺ—Ā—ā–ł–ľ¬ģ. –ü—Ä–ł —ć—ā–ĺ–ľ –Ņ—Ä–ĺ—Ą–ł–Ľ—Ć –Ī–Ķ–∑–ĺ–Ņ–į—Ā–Ĺ–ĺ—Ā—ā–ł –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į —ć–ľ–Ņ—ć–≥—Ą–ł–Ľ–≥—Ä–į—Ā—ā–ł–ľ –Ī—č–Ľ –į–Ĺ–į–Ľ–ĺ–≥–ł—á–Ķ–Ĺ —ā–į–ļ–ĺ–≤–ĺ–ľ—É –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į –õ–Ķ–Ļ–ļ–ĺ—Ā—ā–ł–ľ¬ģ.
–Ě–į¬†–ĺ—Ā–Ĺ–ĺ–≤–į–Ĺ–ł–ł –į–Ĺ–į–Ľ–ł–∑–į –ī–į–Ĺ–Ĺ—č—Ö –ĺ–Ī¬†—ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā–ł, –Ī–Ķ–∑–ĺ–Ņ–į—Ā–Ĺ–ĺ—Ā—ā–ł –ł¬†—Ą–į—Ä–ľ–į–ļ–ĺ–ļ–ł–Ĺ–Ķ—ā–ł–ļ–Ķ –ī–ĺ–∑–į –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į —ć–ľ–Ņ—ć–≥—Ą–ł–Ľ–≥—Ä–į—Ā—ā–ł–ľ 7,5¬†–ľ–≥ –Ī—č–Ľ–į –≤—č–Ī—Ä–į–Ĺ–į –ļ–į–ļ –Ĺ–į–ł–Ī–ĺ–Ľ–Ķ–Ķ –Ņ—Ä–Ķ–ī–Ņ–ĺ—á—ā–ł—ā–Ķ–Ľ—Ć–Ĺ–į—Ź –ī–Ľ—Ź –ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ĺ–≥–ĺ –Ņ—Ä–ł–ľ–Ķ–Ĺ–Ķ–Ĺ–ł—Ź, –Ņ–ĺ—Ā–ļ–ĺ–Ľ—Ć–ļ—É –ĺ–Ī–Ķ—Ā–Ņ–Ķ—á–ł–≤–į–Ľ–į –Ĺ–į–ł–Ī–ĺ–Ľ—Ć—ą—É—é —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć –Ī–Ķ–∑ –Ņ–ĺ–≤—č—ą–Ķ–Ĺ–ł—Ź —ā–ĺ–ļ—Ā–ł—á–Ĺ–ĺ—Ā—ā–ł.
–í¬†–∑–į–ļ–Ľ—é—á–Ķ–Ĺ–ł–Ķ –ú.–Ě.¬†–Ě–Ķ—á–į–Ķ–≤–į —Ä–į—Ā—Ā–ļ–į–∑–į–Ľ–į –ĺ–Ī¬†–ĺ–Ņ—č—ā–Ķ –Ņ—Ä–ł–ľ–Ķ–Ĺ–Ķ–Ĺ–ł—Ź –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į BCD-017 –≤¬†—Ä–į–ľ–ļ–į—Ö –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź —Ą–į–∑—謆III –≤¬†–ź—Ä—Ö–į–Ĺ–≥–Ķ–Ľ—Ć—Ā–ļ–ĺ–ľ –ĺ–Ī–Ľ–į—Ā—ā–Ĺ–ĺ–ľ –ĺ–Ĺ–ļ–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ĺ–ľ –ī–ł—Ā–Ņ–į–Ĺ—Ā–Ķ—Ä–Ķ. –í¬†–ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–Ķ –Ī—č–Ľ–ĺ –≤–ļ–Ľ—é—á–Ķ–Ĺ–ĺ 36¬†–Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–ļ. –Ē–į–Ĺ–Ĺ—č–Ķ, –Ņ–ĺ–Ľ—É—á–Ķ–Ĺ–Ĺ—č–Ķ –≤¬†—ć—ā–ĺ–ľ –ī–ł—Ā–Ņ–į–Ĺ—Ā–Ķ—Ä–Ķ, —Ā–ĺ–ĺ—ā–≤–Ķ—ā—Ā—ā–≤–ĺ–≤–į–Ľ–ł –ĺ–Ī—Č–ł–ľ —Ä–Ķ–∑—É–Ľ—Ć—ā–į—ā–į–ľ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź. –ü–嬆–ľ–Ĺ–Ķ–Ĺ–ł—é –ú.–Ě.¬†–Ě–Ķ—á–į–Ķ–≤–ĺ–Ļ, –≤–Ĺ–Ķ–ī—Ä–Ķ–Ĺ–ł–Ķ —ć–ľ–Ņ—ć–≥—Ą–ł–Ľ–≥—Ä–į—Ā—ā–ł–ľ–į –≤¬†–ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ—É—é –Ņ—Ä–į–ļ—ā–ł–ļ—É –Ī—É–ī–Ķ—ā —Ā–Ņ–ĺ—Ā–ĺ–Ī—Ā—ā–≤–ĺ–≤–į—ā—Ć –Ņ–ĺ–≤—č—ą–Ķ–Ĺ–ł—é –Ī–Ķ–∑–ĺ–Ņ–į—Ā–Ĺ–ĺ—Ā—ā–ł —ā–Ķ—Ä–į–Ņ–ł–ł –ł¬†—É–Ľ—É—á—ą–ł—ā —Ä–Ķ–∑—É–Ľ—Ć—ā–į—ā—č –Ľ–Ķ—á–Ķ–Ĺ–ł—Ź –Ī–ĺ–Ľ—Ć–Ĺ—č—Ö, –Ņ–ĺ–Ľ—É—á–į—é—Č–ł—Ö –ľ–ł–Ķ–Ľ–ĺ—Ā—É–Ņ—Ä–Ķ—Ā—Ā–ł–≤–Ĺ—É—é —Ö–ł–ľ–ł–ĺ—ā–Ķ—Ä–į–Ņ–ł—é –Ņ–嬆–Ņ–ĺ–≤–ĺ–ī—É –∑–Ľ–ĺ–ļ–į—á–Ķ—Ā—ā–≤–Ķ–Ĺ–Ĺ—č—Ö –Ĺ–ĺ–≤–ĺ–ĺ–Ī—Ä–į–∑–ĺ–≤–į–Ĺ–ł–Ļ.
–ó–į–ļ–Ľ—é—á–Ķ–Ĺ–ł–Ķ
–ü—Ä–Ķ–ī—Ā—ā–į–≤–Ľ–Ķ–Ĺ–Ĺ—č–Ķ –Ĺ–į¬†—Ā–ł–ľ–Ņ–ĺ–∑–ł—É–ľ–Ķ —Ä–Ķ–∑—É–Ľ—Ć—ā–į—ā—č –ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ł—Ö –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–Ļ –Ņ–ĺ–ī—ā–≤–Ķ—Ä–∂–ī–į—é—ā –∑–į–Ĺ–ł–ľ–į–Ķ–ľ—č–Ķ –ļ–ĺ–ľ–Ņ–į–Ĺ–ł–Ķ–Ļ BIOCAD –≤–Ķ–ī—É—Č–ł–Ķ –Ņ–ĺ–∑–ł—Ü–ł–ł —Ā—Ä–Ķ–ī–ł –ĺ—ā–Ķ—á–Ķ—Ā—ā–≤–Ķ–Ĺ–Ĺ—č—Ö —Ą–į—Ä–ľ–į—Ü–Ķ–≤—ā–ł—á–Ķ—Ā–ļ–ł—Ö –ļ–ĺ–ľ–Ņ–į–Ĺ–ł–Ļ. –ė—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź —Ą–į–∑—謆III —Ā¬†—É—á–į—Ā—ā–ł–Ķ–ľ –Ī–ĺ–Ľ—Ć–Ĺ—č—Ö –†–ú–Ė –ł¬†–Ě–ú–†–õ —É–Ī–Ķ–ī–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ –ī–ĺ–ļ–į–∑–į–Ľ–ł —ā–Ķ—Ä–į–Ņ–Ķ–≤—ā–ł—á–Ķ—Ā–ļ—É—é —ć–ļ–≤–ł–≤–į–Ľ–Ķ–Ĺ—ā–Ĺ–ĺ—Ā—ā—Ć –Ī–ł–ĺ–į–Ĺ–į–Ľ–ĺ–≥–į —ā—Ä–į—Ā—ā—É–∑—É–ľ–į–Ī–į –ł¬†–Ī–ł–ĺ–į–Ĺ–į–Ľ–ĺ–≥–į –Ī–Ķ–≤–į—Ü–ł–∑—É–ľ–į–Ī–į –Ņ—Ä–ĺ–ł–∑–≤–ĺ–ī—Ā—ā–≤–į –ļ–ĺ–ľ–Ņ–į–Ĺ–ł–ł BIOCAD –ĺ—Ä–ł–≥–ł–Ĺ–į–Ľ—Ć–Ĺ—č–ľ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į–ľ. –í–Ĺ–Ķ–ī—Ä–Ķ–Ĺ–ł–Ķ —ć—ā–ł—Ö –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–ĺ–≤ –≤¬†–ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ—É—é –Ņ—Ä–į–ļ—ā–ł–ļ—É –ľ–ĺ–∂–Ķ—ā –Ņ–ĺ–≤—č—Ā–ł—ā—Ć –ī–ĺ—Ā—ā—É–Ņ–Ĺ–ĺ—Ā—ā—Ć —Ā–ĺ–≤—Ä–Ķ–ľ–Ķ–Ĺ–Ĺ–ĺ–Ļ –Ņ—Ä–ĺ—ā–ł–≤–ĺ–ĺ–Ņ—É—Ö–ĺ–Ľ–Ķ–≤–ĺ–Ļ —ā–Ķ—Ä–į–Ņ–ł–ł –ī–Ľ—Ź –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ —Ā¬†—Ā–į–ľ—č–ľ–ł —Ä–į—Ā–Ņ—Ä–ĺ—Ā—ā—Ä–į–Ĺ–Ķ–Ĺ–Ĺ—č–ľ–ł –≤¬†–†–ĺ—Ā—Ā–ł–ł –∑–Ľ–ĺ–ļ–į—á–Ķ—Ā—ā–≤–Ķ–Ĺ–Ĺ—č–ľ–ł –Ĺ–ĺ–≤–ĺ–ĺ–Ī—Ä–į–∑–ĺ–≤–į–Ĺ–ł—Ź–ľ–ł –ł¬†—Ā–Ĺ–ł–∑–ł—ā—Ć –∑–į—ā—Ä–į—ā—č –Ĺ–į¬†—ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ–Ķ –Ľ–Ķ—á–Ķ–Ĺ–ł–Ķ.
–ü–Ķ—Ä–≤—č–Ļ –ĺ—Ä–ł–≥–ł–Ĺ–į–Ľ—Ć–Ĺ—č–Ļ –ĺ—ā–Ķ—á–Ķ—Ā—ā–≤–Ķ–Ĺ–Ĺ—č–Ļ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā –ď-–ö–°–§ –Ņ—Ä–ĺ–Ľ–ĺ–Ĺ–≥–ł—Ä–ĺ–≤–į–Ĺ–Ĺ–ĺ–≥–ĺ –ī–Ķ–Ļ—Ā—ā–≤–ł—Ź, —Ä–į–∑—Ä–į–Ī–ĺ—ā–į–Ĺ–Ĺ—č–Ļ –ļ–ĺ–ľ–Ņ–į–Ĺ–ł–Ķ–Ļ BIOCAD, –ī–ĺ–ļ–į–∑–į–Ľ —Ā–≤–ĺ—é —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć –ł¬†–Ī–Ķ–∑–ĺ–Ņ–į—Ā–Ĺ–ĺ—Ā—ā—Ć –≤¬†—Ā—Ä–į–≤–Ĺ–Ķ–Ĺ–ł–ł —Ā¬†–∑–ĺ–Ľ–ĺ—ā—č–ľ —Ā—ā–į–Ĺ–ī–į—Ä—ā–ĺ–ľ –Ņ—Ä–ĺ—Ą–ł–Ľ–į–ļ—ā–ł–ļ–ł –Ĺ–Ķ–Ļ—ā—Ä–ĺ–Ņ–Ķ–Ĺ–ł–ł —ɬ†–Ī–ĺ–Ľ—Ć–Ĺ—č—Ö, –Ņ–ĺ–Ľ—É—á–į—é—Č–ł—Ö —Ö–ł–ľ–ł–ĺ—ā–Ķ—Ä–į–Ņ–ł—鬆–Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–ĺ–ľ —Ą–ł–Ľ–≥—Ä–į—Ā—ā–ł–ľ. –£–ī–ĺ–Ī–Ĺ—č–Ļ —Ä–Ķ–∂–ł–ľ –≤–≤–Ķ–ī–Ķ–Ĺ–ł—Ź –ł¬†–≤—č—Ā–ĺ–ļ–į—Ź —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į —ć–ľ–Ņ—ć–≥—Ą–ł–Ľ–≥—Ä–į—Ā—ā–ł–ľ –Ņ–ĺ–∑–≤–ĺ–Ľ—Ź—é—ā —Ä–į—Ā—Ā–ľ–į—ā—Ä–ł–≤–į—ā—Ć –Ķ–≥–ĺ –Ņ—Ä–ł–ľ–Ķ–Ĺ–Ķ–Ĺ–ł–Ķ –ļ–į–ļ –≤–Ķ—Ā—Ć–ľ–į –Ņ–Ķ—Ä—Ā–Ņ–Ķ–ļ—ā–ł–≤–Ĺ—č–Ļ –ľ–Ķ—ā–ĺ–ī –Ņ—Ä–ĺ—Ą–ł–Ľ–į–ļ—ā–ł–ļ–ł —ā–į–ļ–ł—Ö —ā—Ź–∂–Ķ–Ľ—č—Ö –ĺ—Ā–Ľ–ĺ–∂–Ĺ–Ķ–Ĺ–ł–Ļ —Ö–ł–ľ–ł–ĺ—ā–Ķ—Ä–į–Ņ–Ķ–≤—ā–ł—á–Ķ—Ā–ļ–ĺ–≥–ĺ –Ľ–Ķ—á–Ķ–Ĺ–ł—Ź, –ļ–į–ļ —Ą–Ķ–Ī—Ä–ł–Ľ—Ć–Ĺ–į—Ź –Ĺ–Ķ–Ļ—ā—Ä–ĺ–Ņ–Ķ–Ĺ–ł—Ź –ł¬†–Ĺ–Ķ–Ļ—ā—Ä–ĺ–Ņ–Ķ–Ĺ–ł—á–Ķ—Ā–ļ–ł–Ķ –ł–Ĺ—Ą–Ķ–ļ—Ü–ł–ł, –≤¬†–ī–į–Ĺ–Ĺ–ĺ–Ļ –Ņ–ĺ–Ņ—É–Ľ—Ź—Ü–ł–ł –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤.
–£–≤–į–∂–į–Ķ–ľ—č–Ļ –Ņ–ĺ—Ā–Ķ—ā–ł—ā–Ķ–Ľ—Ć uMEDp!
–£–≤–Ķ–ī–ĺ–ľ–Ľ—Ź–Ķ–ľ –í–į—Ā –ĺ —ā–ĺ–ľ, —á—ā–ĺ –∑–ī–Ķ—Ā—Ć —Ā–ĺ–ī–Ķ—Ä–∂–ł—ā—Ā—Ź –ł–Ĺ—Ą–ĺ—Ä–ľ–į—Ü–ł—Ź, –Ņ—Ä–Ķ–ī–Ĺ–į–∑–Ĺ–į—á–Ķ–Ĺ–Ĺ–į—Ź –ł—Ā–ļ–Ľ—é—á–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ –ī–Ľ—Ź —Ā–Ņ–Ķ—Ü–ł–į–Ľ–ł—Ā—ā–ĺ–≤ –∑–ī—Ä–į–≤–ĺ–ĺ—Ö—Ä–į–Ĺ–Ķ–Ĺ–ł—Ź.
–ē—Ā–Ľ–ł –í—č –Ĺ–Ķ —Ź–≤–Ľ—Ź–Ķ—ā–Ķ—Ā—Ć —Ā–Ņ–Ķ—Ü–ł–į–Ľ–ł—Ā—ā–ĺ–ľ –∑–ī—Ä–į–≤–ĺ–ĺ—Ö—Ä–į–Ĺ–Ķ–Ĺ–ł—Ź, –į–ī–ľ–ł–Ĺ–ł—Ā—ā—Ä–į—Ü–ł—Ź –Ĺ–Ķ –Ĺ–Ķ—Ā–Ķ—ā –ĺ—ā–≤–Ķ—ā—Ā—ā–≤–Ķ–Ĺ–Ĺ–ĺ—Ā—ā–ł –∑–į –≤–ĺ–∑–ľ–ĺ–∂–Ĺ—č–Ķ –ĺ—ā—Ä–ł—Ü–į—ā–Ķ–Ľ—Ć–Ĺ—č–Ķ –Ņ–ĺ—Ā–Ľ–Ķ–ī—Ā—ā–≤–ł—Ź, –≤–ĺ–∑–Ĺ–ł–ļ—ą–ł–Ķ –≤ —Ä–Ķ–∑—É–Ľ—Ć—ā–į—ā–Ķ —Ā–į–ľ–ĺ—Ā—ā–ĺ—Ź—ā–Ķ–Ľ—Ć–Ĺ–ĺ–≥–ĺ –ł—Ā–Ņ–ĺ–Ľ—Ć–∑–ĺ–≤–į–Ĺ–ł—Ź –í–į–ľ–ł –ł–Ĺ—Ą–ĺ—Ä–ľ–į—Ü–ł–ł —Ā –Ņ–ĺ—Ä—ā–į–Ľ–į –Ī–Ķ–∑ –Ņ—Ä–Ķ–ī–≤–į—Ä–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ–Ļ –ļ–ĺ–Ĺ—Ā—É–Ľ—Ć—ā–į—Ü–ł–ł —Ā –≤—Ä–į—á–ĺ–ľ.
–Ě–į–∂–ł–ľ–į—Ź –Ĺ–į –ļ–Ĺ–ĺ–Ņ–ļ—É ¬ę–í–ĺ–Ļ—ā–ł¬Ľ, –í—č –Ņ–ĺ–ī—ā–≤–Ķ—Ä–∂–ī–į–Ķ—ā–Ķ, —á—ā–ĺ —Ź–≤–Ľ—Ź–Ķ—ā–Ķ—Ā—Ć –≤—Ä–į—á–ĺ–ľ –ł–Ľ–ł —Ā—ā—É–ī–Ķ–Ĺ—ā–ĺ–ľ –ľ–Ķ–ī–ł—Ü–ł–Ĺ—Ā–ļ–ĺ–≥–ĺ –≤—É–∑–į.