Применение биоаналогов генно-инженерных биологических препаратов при ревматических заболеваниях
- Аннотация
- Статья
- Ссылки
- English
Для повышения доступности такого лечения необходимо расширять фармацевтический рынок за счет производства неоригинальных препаратов после истечения срока действия патента на оригинальные средства. В отношении ГИБП таковыми являются биоаналоги. Крупнейшие фармкомпании мира производят 20 биоаналогов ГИБП, зарегистрированных для применения при ревматических заболеваниях. В настоящее время три из них зарегистрированы в России, причем два (биоаналог ритуксимаба – препарат Ацеллбия® и биоаналог инфликсимаба) производятся российскими биофармацевтическими компаниями. В обзоре рассмотрены вопросы разработки и регистрации биоаналогов. Представлены результаты клинических исследований их эффективности при ревматических заболеваниях, проанализированы особенности назначения (выписка рецепта, перевод пациентов с оригинального препарата на неоригинальный и др.).
Для повышения доступности такого лечения необходимо расширять фармацевтический рынок за счет производства неоригинальных препаратов после истечения срока действия патента на оригинальные средства. В отношении ГИБП таковыми являются биоаналоги. Крупнейшие фармкомпании мира производят 20 биоаналогов ГИБП, зарегистрированных для применения при ревматических заболеваниях. В настоящее время три из них зарегистрированы в России, причем два (биоаналог ритуксимаба – препарат Ацеллбия® и биоаналог инфликсимаба) производятся российскими биофармацевтическими компаниями. В обзоре рассмотрены вопросы разработки и регистрации биоаналогов. Представлены результаты клинических исследований их эффективности при ревматических заболеваниях, проанализированы особенности назначения (выписка рецепта, перевод пациентов с оригинального препарата на неоригинальный и др.).
Введение
Генно-инженерные биологические препараты, такие как ингибиторы фактора некроза опухоли (ФНО), анти-В-клеточный препарат (ритуксимаб), блокатор активации Т-лимфоцитов (абатацепт), ингибитор интерлейкина (ИЛ) 6 (тоцилизумаб), позволяют достаточно эффективно контролировать активность иммуновоспалительных заболеваний у пациентов с недостаточным ответом на патогенетическую терапию первой линии. Речь, в частности, идет о больных ревматоидным артритом (РА), анкилозирующим спондилитом (АС), псориатическим артритом (ПсА), псориазом (Пс), ювенильным ревматоидным артритом (ЮРА), системными болезнями соединительной ткани, воспалительными заболеваниями кишечника (ВЗК).
Так, на фоне активной терапии метотрексатом (препарат первой линии) в дозе 25 мг/нед в сочетании с глюкокортикостероидами только 40–50% пациентов достигают ремиссии или низкой активности заболевания. В то же время последующее применение таргетной терапии ГИБП или ингибиторами янус-киназы в сочетании с метотрексатом позволяет добиться успеха у 75% пациентов [1].
Роль таргетных препаратов, в частности ГИБП, значительно возросла после принятия в качестве основной цели лечения РА и других хронических иммуновоспалительных заболеваний «достижение клинической ремиссии или как минимум низкой активности болезни» (Treat to Target – Т2Т) [2, 3], что нашло отражение в международных [4] и отечественных [5] клинических рекомендациях. Согласно данным Российского регистра больных ревматоидным артритом [6], частота назначения ГИБП составила 28,6% при средней продолжительности болезни 9,86 года.
В настоящее время вопрос о доступности ГИБП стоит очень остро. В первую очередь это связано с высокой стоимостью лечения. Так, в год на одного пациента, получающего ингибитор ФНО, тратится от 10 000 до 30 000 долл. США [7]. Это приводит к огромной нагрузке на бюджет системы здравоохранения в частности и страны в целом.
В 2017 г. в нашей стране ГИБП и таргетных синтетических препаратов было закуплено на 10 643,39 млн руб. Большая часть указанной суммы израсходована на приобретение инфликсимаба (ИНФ), адалимумаба (АДА), ритуксимаба (РТМ), этанерцепта (ЭЦ) [8]. Учитывая тенденцию к повышению распространенности иммуновоспалительных заболеваний, в первую очередь за счет РА и спондилоартритов, в ближайшем будущем можно прогнозировать увеличение потребности в ГИБП [9].
Высокая стоимость биологических препаратов стала существенным препятствием к получению эффективного лечения в экономически нестабильных и развивающихся странах [10]. Например, в Венгрии доля пациентов с РА, которые лечились биологическими препаратами, в два раза ниже, чем в странах Евросоюза [10, 11].
Одним из способов снижения стоимости лечения является производство неоригинальных препаратов (генерики, или дженерики), что возможно только по истечении срока действия патентов на оригинальные лекарственные средства. Необходимо отметить, что благодаря генерикам стоимость лечения нередко уменьшается в несколько раз.
Согласно международному стандарту, генерик – это лекарственный продукт с доказанной фармацевтической, биологической и терапевтической эквивалентностью оригинальному препарату [12]. Данный термин применяется к относительно низкомолекулярным веществам, которые производятся путем традиционного химического синтеза и для которых может быть доказана идентичность основного действующего вещества (субстанции), а также биоэквивалентность фармакокинетических характеристик. К сожалению, даже в этом случае, особенно при регистрации генериков по упрощенной схеме, существует вероятность поставки на фармацевтический рынок некачественных препаратов [12, 13].
Ситуация с аналогами ГИБП значительно сложнее. В отношении неоригинальных ГИБП применяется англоязычный термин «biosimilars», то есть биологически подобные. В отечественной литературе для их обозначения используют термины «биоаналоги», «биосимиляры», «биоподобные средства», «воспроизведенные биологические препараты», «биофармацевтические препараты» [14].
Федеральным законом от 22.12.2014 № 429-ФЗ [15] введены следующие термины:
- биотехнологические лекарственные препараты – лекарственные препараты, производство которых осуществляется с использованием биотехнологических процессов и методов (в том числе ДНК-рекомбинантной технологии, технологии контролируемой экспрессии генов, кодирующих биологически активные белки в прокариотах и эукариотах, включая измененные клетки млекопитающих), гибридомного метода и метода моноклональных антител*;
- биоаналоговый (биоподобный) лекарственный препарат (биоаналог) – биологический лекарственный препарат, схожий по параметрам качества, эффективности и безопасности с референтным биологическим лекарственным препаратом в такой же лекарственной форме и имеющий идентичный способ введения.
Таким образом, согласно указанному закону аналогом термина «biosimilars» является термин «биоаналог».
ГИБП, биоаналог и биотерапевтический продукт
ГИБП относятся к белковым биофармацевтическим препаратам, которые существенно отличаются от других крупных молекул. Это вещества, получаемые не химическим, а биологическим методом с использованием специальных клеточных линий, которые культивируют в строго контролируемых условиях. Посттрансляционные модификации белковых молекул ГИБП (например, гликозилирование и образование молекулярной складчатой конформации) также должны строго контролироваться. Правильная структура белка гарантирует достижение необходимой биологической активности [16].
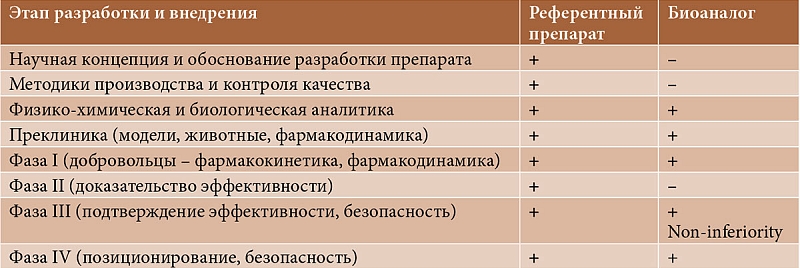
Молекулы ГИБП – моноклональные антитела (например, АДА) и гибридные молекулы, несущие растворимые рецепторы (например, ЭЦ). Их клинический профиль зависит от таких факторов, как конформационная структура, иммуногенность и взаимодействие с целевыми антигенами. Кроме того, он очень чувствителен к изменениям не только последовательности аминокислот, но и к молекулярным посттрансляционным изменениям, а также к наличию возможных примесей. Поскольку биоаналог не производится так же, как референтный препарат (используются другие клеточные линии, другие условия производства, может отличаться технология очистки и пр.), незначительные молекулярные различия в огромных молекулах иммуноглобулинов почти неизбежны [10]. С течением времени аналитические характеристики оригинальных ГИБП также могут существенно меняться. Это может быть обусловлено различными объективными факторами, в том числе масштабированием производства, оптимизацией процесса очистки белка [14], особенно при изменении технологии производства [17]. Поэтому процесс разработки биоаналогов значительно ближе к процессу разработки оригинального препарата, чем процесс разработки генерика (табл. 1).
Если сравнивать биоаналоги и генерики по их соотношению с оригинальным продуктом, то различие будет отмечаться не только в дизайне, но и в процессе производства, композиции и рецептуре [18]. В связи с невозможностью произвести абсолютный аналог референтного ГИБП биоаналогом является биотерапевтический продукт, который должен быть сопоставим с ранее лицензированным биотерапевтическим продуктом по определенным критериям [7, 15, 19–21]. Подобно генерикам биоаналоги лицензируются только после истечения срока действия патента на референтный препарат.
Генерики в отличие от биоаналогов могут быть одобрены без предоставления существенных клинических данных. Для биоаналогов проведение предрегистрационных клинических исследований фазы III обязательно [15]. При этом исследования должны быть рандомизированные, предполагать прямое сравнение референтного препарата и биоаналога, соответствовать требованию non-inferiority (препарат не должен уступать референтному препарату по параметрам эффективности и безопасности) (см. табл. 1). Уровень, свидетельствующий об отсутствии значимых различий, устанавливает регулирующий орган каждой страны в отдельности, поскольку на данный момент времени единый критерий отсутствует. В некоторых случаях этот уровень может быть связан со степенью снижения эффективности, которую готовы принять пациенты [10]. Различие результатов лечения по ключевым параметрам не должно отклоняться более чем на 15% ни в худшую, ни в лучшую сторону. Воспроизведенный препарат теоретически может оказаться достоверно эффективнее оригинального. Для таких препаратов используется термин «biobetter» (биологически улучшенный). В этом случае препарат должен рассматриваться как новый, оригинальный.
Несмотря на требования к сопоставимости, небольшие различия молекул биоаналогов и оригинальных препаратов неизбежны, в частности в отношении гликозилирования и конформации [7, 22]. Это потенциально может влиять на фармакодинамику, фармакокинетику и иммуногенность лекарственного средства.
Иммуногенность является важной проблемой всех биотерапевтических средств, и, по-видимому, именно незначительные молекулярные различия приводят к клинически важным различиям в иммуногенности. Белковая природа ГИБП обусловливает образование в организме пациента антител. При этом могут вырабатываться как связывающие, так и нейтрализующие антитела. Первые преимущественно повышают клиренс препарата, вторые – существенно снижают его эффективность. Антитела к препарату также могут быть причиной инфузионных осложнений и других иммунных реакций [23–25].
Биоаналоги следует отличать от биотерапевтических продуктов, для которых сопоставимость не доказывается. Для их обозначения используют термины «biomimics» («биоподражатели») или «intended copies» (предполагаемые копии) [10, 26]. Как правило, несопоставимыми являются препараты, которые не подвергались строгим оценкам и не удовлетворяют нормативным требованиям для биоаналогов [10]. Эти продукты обладают ограниченными аналитическими данными клинических испытаний, что затрудняет обоснованное сравнение их профилей безопасности и эффективности с таковыми референтных средств. В некоторых странах с менее строгими нормативными требованиями для регистрации лекарственных средств несопоставимые препараты выпускают на фармацевтический рынок без предварительного проведения клинических исследований или раскрытия степени их биоподобия [10]. Применение таких препаратов в Российской Федерации не должно допускаться.
Современные биоаналоги
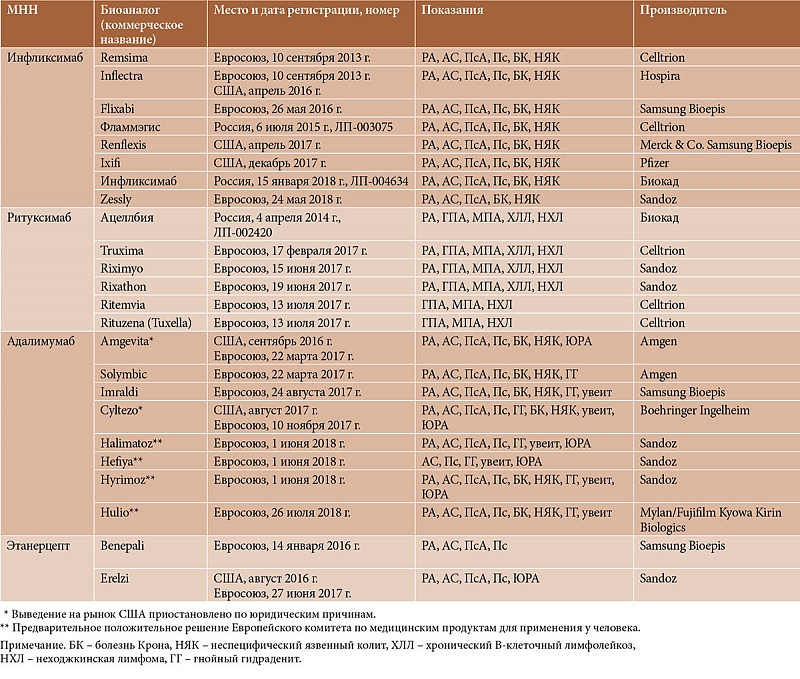
В связи с тем что срок действия значительного числа патентов на биологические агенты уже истек или истечет в ближайшие годы, количество биоаналогов постоянно будет увеличиваться. В таблице 2 приведены последние официальные данные о биоаналогах, зарегистрированных для лечения ревматических заболеваний в России, Евросоюзе и США [27–30].
В настоящее время крупнейшие фармацевтические компании мира производят восемь биоаналогов ИНФ, шесть – РТМ, четыре – АДА (еще четыре предварительно одобрены для регистрации в Евросоюзе), два – ЭЦ. В отношении всех этих препаратов получены достаточно объемные данные, подтверждающие их сопоставимость с референтными продуктами. Так, для первого из зарегистрированных в мире биоаналогов из класса моноклональных антител – ИНФ CT-P13 (в России зарегистрирован под торговым наименованием Фламмэгис®) проведена серия рандомизированных клинических исследований при РА и АС [31–36], в которых в том числе была продемонстрирована безопасность перевода пациентов с оригинального препарата на неоригинальный.
Перевод пациентов с оригинального препарата на биоаналог
Вопрос о возможности и целесообразности перевода пациентов с оригинального препарата на биоаналог является одним из наиболее болезненных и широко обсуждаемых. Взаимозаменяемость является стандартом для генериков. Однако в отношении биоаналогов ГИБП данную ситуацию следует рассматривать как перевод с одного препарата группы на другой препарат этой же группы [10]. Это может иметь клиническое значение, например, у пациентов, получающих ингибиторы ФНО. Если один препарат неэффективен, второй может быть эффективным, но если лечение вторым препаратом неэффективно, любой другой ингибитор ФНО с большой вероятностью будет неэффективным. В этом случае биоаналог может считаться вторым ингибитором ФНО, и при неудаче возможность переключения на следующий ингибитор ФНО практически исключена [7].
Вопросы взаимозаменяемости
Согласно предварительным рекомендациям Ассоциации ревматологов России [14], при назначении оригинальных препаратов и биоаналогов следует указывать их торговое наименование, а не международное непатентованное название (МНН). Это необходимо для того, чтобы избежать автоматической (или случайной) замены референтного препарата на биоаналог в аптечной сети, отслеживать эффективность и безопасность того или иного лекарственного препарата в рамках фармаконадзора.
В медицинской документации помимо МНН следует указать торговое наименование ГИБП, а также факт перевода с оригинального препарата на биоаналог [37, 38]. Несмотря на очевидные экономические преимущества, автоматически заменять оригинальные ГИБП на их биоаналоги не рекомендуется.
Взаимозаменяемость препаратов устанавливается на основании полученной доказательной базы в отношении эквивалентности эффективности, безопасности, иммуногенности.
Без сомнений, биоаналоги, характеризующиеся высокой сопоставимостью с референтными препаратами, могут быть взаимозаменяемыми, как это зачастую происходит в реальной практике. В то же время необходимо учитывать три важных аспекта:
- если референтный препарат и биоаналог признаются взаимозаменяемыми в одном случае, то при регистрации в будущем другого биоаналога другого производителя он также должен быть признан взаимозаменяемым;
- аналогичным образом должна быть допущена взаимозаменяемость биоаналогов внутри одного МНН. Например, переход с одного биоаналога инфликсимаба на другой (другого производителя), что может иметь место в настоящее время в нашей стране (см. табл. 2);
- замена референтного препарата на биоаналог и переключение с одного биоаналога на другой ассоциируется с большей ответственностью врача, чем в случае низкомолекулярных генериков. Замена производится не на абсолютно аналогичный.
Авторы полагают, что врач должен иметь право выписывать рецепт как по торговому названию, так и по МНН, поскольку это отражает реальную практику. При первичном назначении ГИБП по согласованию с пациентом из соображений повышения доступности препарат может быть выписан по МНН, поскольку в этой ситуации ни один из вариантов препарата (оригинальный либо один из биоаналогов) не имеет преимуществ. В случае продолжения терапии возможен перевод пациента с одного препарата на другой (в том числе с биоаналога на оригинальный) одного класса. Однако он может быть нецелесообразен по организационным соображениям. В этом случае врач должен иметь право выписать рецепт по торговому названию.
Конечно, необходимо учитывать, что с юридической точки зрения аспекты, связанные с выпиской рецептов по торговому наименованию, противоречат текущим нормативным актам Российской Федерации. Очевидно, что в данном случае правовая база деятельности врача требует обсуждения и доработки.
Таким образом, вопросы, связанные с заменой оригинальных ГИБП на биоаналоги и переключением с одного биоаналога на другой, требуют дополнительного рассмотрения и достижения консенсуса специалистов.
Согласно рекомендациям экспертов Международной федерации фармацевтических производителей и ассоциаций, замена оригинального препарата на биоаналог допустима при выполнении следующих условий [37]:
- врач, назначающий лечение, должен иметь возможность установить, какой конкретный препарат будет выдан пациенту, и его решение должно исходить в первую очередь из медицинских соображений;
- врач должен принимать решение о переводе с одного биологического препарата на другой только после обсуждения этого вопроса с пациентом, при этом в процессе лечения необходимо обеспечить адекватный мониторинг состояния больного.
В некоторых ситуациях замена оригинального препарата на биоаналог не рекомендуется [37]. К таковым относятся потеря эффективности, непереносимость (в этом случае лучше перейти на препарат другой группы), риск дискредитировать препараты данной группы в глазах пациента.
Необходимо отметить, что все вопросы, связанные с заменой референтного препарата на биоаналог, относятся исключительно к официально зарегистрированным по соответствующим показаниям и доказавшим свою сопоставимость препаратам (собственно биоаналогам) и ни в коей мере не относятся к биомимикам (intended copies). Последние не могут считаться сопоставимыми и взаимозаменяемыми. Как уже было сказано выше, по мнению авторов, клиническое применение таких препаратов в Российской Федерации не должно допускаться.
Экстраполяция показаний
Еще один важный вопрос, связанный с применением оригинальных и неоригинальных препаратов, – экстраполяция показаний. Если референтный продукт имеет более одного показания к применению, эффективность и безопасность биоаналога должны быть подтверждены в отдельности для каждого из заявленного показания. В то же время на практике возможны ситуации, когда биоаналог зарегистрирован по всем показаниям, имеющимся у референтного средства, но сопоставимость первого и второго доказана только в отношении одной-двух нозологий.
Экстраполяция показаний является важным преимуществом препарата и в отношении стоимости лечения [38].
Необходимость экстраполяции показаний поддержана экспертами Всемирной организации здравоохранения. Для ее подтверждения должны быть выполнены следующие условия [30]:
- для определения потенциальных различий между продуктами используется чувствительная модель клинических испытаний;
- механизмы действия при изучаемой патологии и экстраполированной одинаковы;
- безопасность и иммуногенность биоаналога достаточно охарактеризованы, и нет никаких уникальных/дополнительных проблем с безопасностью в случае экстраполяции;
- наличие убедительных аргументов в пользу того, что эффективность при изучаемой патологии может быть экстраполирована на другие заболевания.
К дополнительным факторам, которые следует учитывать для обоснования экстраполяции, относятся клинический опыт использования референтного продукта, различие профиля безопасности/иммуногенности между терапевтическими показаниями (с одной стороны, сопутствующие заболевания, терапия и иммунологический статус пациента, с другой – реакции, связанные с клетками-мишенями (например, лизис опухолевых клеток)), степень, в которой функциональные фрагменты молекулы могут характеризоваться и сравниваться [39]. В этом случае экстраполяция показаний рациональна, поскольку позволяет избежать ненужной траты времени и средств на проведение дополнительных исследований.
Российские биоаналоги
В настоящее время в России доступны три биоаналога ГИБП (см. табл. 2). Первым биоаналогом, зарегистрированным в нашей стране для лечения ревматических заболеваний, стал ИНФ CT-P13 (Фламмэгис®). На момент регистрации были опубликованы результаты рандомизированных исследований о его применении у пациентов с РА и АС [31–36]. В отношении других показаний (Пс, ПсА, ВЗК) проведена экстраполяция.
Последние годы ряд крупных фармацевтических компаний нашей страны начали развивать направление биофармации («Фармстандарт», «Р-Фарм», «Биокад»). В разработке биоаналогов наиболее продвинулась компания «Биокад», производящая РТМ и ИНФ.
Предрегистрационные исследования российских биоаналогов полностью соответствовали международным стандартам. Созданные биоаналоги прошли аналитический этап, этап доклинических испытаний и клинических исследований на чувствительной и гомогенной популяции пациентов. Кроме того, уже имеется опыт клинического применения BCD-020 (РТМ) по гематологическим показаниям [40]. Необходимо отметить, что автоматическая экстраполяция ревматологических показаний с оригинального препарата была неправомочна по причине различия мишеней: CD20+-клетки опухоли при лимфопролиферативных заболеваниях и определенный пул интактных В-лимфоцитов при ревматических нозологиях.
Для оценки эффективности, безопасности и иммуногенности российского биоаналога РТМ при лечении РА были проведены многоцентровые клинические исследования фазы III – BIORA и ALTERRA [41, 42]. Согласно результатам исследования BIORA профиль безопасности оригинального препарата РТМ (Мабтера®, компания «Рош-Москва») и биоаналога (Ацеллбия®, компания «Биокад»), используемых в качестве второй линии биологической терапии у пациентов с РА, оказался сопоставимым, иммуногенность препаратов оставалась низкой. Полученные результаты свидетельствуют о биологическом сходстве препаратов Мабтера® и Ацеллбия®. Перевод пациентов с оригинального препарата на биоаналог и наоборот не влиял отрицательно на показатели эффективности, безопасности и иммуногенности [41].
Результаты исследования ALTERRA свидетельствуют о более высокой эффективности в качестве первой линии терапии РА препарата Ацеллбия® в сочетании с метотрексатом по сравнению с применением только метотрексата [42]. В целом препарат продемонстрировал достаточную сопоставимость с референтным препаратом и в настоящее время активно применяется в ревматологической практике по зарегистрированным показаниям – РА, гранулематоз с полиангиитом (ГПА), или болезнь Вегенера, микроскопический полиангиит (МПА) [43].
В 2018 г. зарегистрирован российский биоаналог ИНФ BCD-055 (компания «Биокад»). Изучение BCD-055 проводилось в рамках доклинических испытаний и клинических исследований (ASART-1, ASART-2, LIRA). Данный препарат сравнивали с референтным препаратом Ремикейд® (компания «МСД Фармасьютикалс»).
Фармакокинетика, эффективность и безопасность BCD-055 изучались в многоцентровых рандомизированных двойных слепых клинических исследованиях фаз I и III. Участниками исследований стали пациенты с активным АС [44]. В исследовании ASART-1 (фаза I) оценивали фармакокинетическую эквивалентность и безопасность препаратов BCD-055 и Ремикейд®, в исследовании ASART-2 (фаза III) – их эффективность и безопасность. Пациенты были рандомизированы (в исследовании ASART-1 в соотношении 1:1, в исследовании ASART-2 – 2:1) на две группы: получавшие BCD-055 или оригинальный препарат в дозе 5 мг/кг на нулевой, второй и шестой неделях, затем каждую восьмую неделю.
В анализ результатов по фармакокинетике (ASART-1) включен 81 пациент, в анализ эффективности и безопасности (ASART-2) – 199. Параметры фармакокинетики BCD-055 были эквивалентны таковым оригинального ИНФ. Ответа ASAS 20 на 30-й неделе достигли 81,30 и 67,74% пациентов групп BCD-055 и Ремикейда (р = 0,061). При анализе дополнительных конечных точек (индексы BASDAI, BASMI, BASFI, MASES, показатели качества жизни, экскурсия грудной клетки, счет патологически измененных суставов) достоверных различий между группами не выявлено. В период наблюдения нежелательные реакции были зарегистрированы у 48,48 и 58,21% пациентов в группах BCD-055 и Ремикейда (различия недостоверны).
В обобщенный анализ результатов [45] исследований ASART-1, ASART-2, LIRA были включены пациенты с активным АС и РА: 91, 198 и 195 соответственно (сравнительное исследование, проведенное в России и Белоруссии). BCD-055 не уступал оригинальному ИНФ как у пациентов с РА, так и у больных АС: ответ ACR 20 к 14-й неделе был достигнут у 75,83% пациентов в группе BCD-055 и у 74,19% больных в группе Ремикейда (p = 0,951), ответ ASAS 20 к 30-й неделе – у 81,30 и 67,74% соответственно (p = 0,061).
Результаты исследования ASART-2 продемонстрировали достижение ASAS 20/ASAS 40 на 14-й, 30-й и 54-й неделях у сопоставимого числа пациентов (р > 0,05) [47]. На 54-й неделе доля пациентов, получавших BCD-055 и Ремикейд® и достигших ASAS 20, в популяции ITT (Intention to Treat) составила 67,42 и 52,24% (р = 0,053), в популяции РР (Per Protocol) – 80,91 и 68,63% (р = 0,128). Достижение ASAS 40 в группах BCD-055 и Ремикейда наблюдалось у 53,03 и 38,81% пациентов в популяции ITT (р = 0,081), у 63,64 и 50,98% – в популяции РР (р = 0,177).
По количеству пациентов со связывающими и нейтрализующими антителами к препарату группы также были сопоставимы: в группе BCD-055 – 21,26 и 3,15% (р = 0,920), в группе Ремикейда – 20,63 и 6,35% (р = 0,443) соответственно.
В ходе исследования зафиксирован один летальный исход в группе исследуемого препарата. Необходимо отметить, что он не был связан с проводившимся лечением.
Частота выявления и профиль иммуногенности в обеих группах не различались, так же как частота выбывания пациентов из исследования по причине развития нежелательных реакций. Большинство зафиксированных побочных явлений характеризовались как легкие и средние.
Российский биоаналог ИНФ зарегистрирован для лечения РА, АС, ПсА, Пс, ВЗК, то есть по всем показаниям, имеющимся у референтного препарата.
Таким образом, биоаналоги РТМ и ИНФ, применяющиеся в Российской Федерации, обладают высокой сопоставимостью с оригинальными препаратами, их эффективность и безопасность доказаны.
Заключение
Несомненно, введение в клиническую практику биоаналогов позволит снизить стоимость лечения и сделать его доступным для многих пациентов. Подтверждением тому служит опыт Евросоюза. Так, в странах Евросоюза различие цен производителей биоаналогов и оригинальных препаратов (этанерцепта, ритуксимаба и инфликсимаба) составило 31–39%, розничных цен – 11–143% [46]. Проведенный в России анализ [8] показал, что использование одного только биоаналога РТМ (препарат Ацеллбия®, компания «Биокад») для новых пациентов и пациентов, у которых терапия ГИБП оказалась недостаточно эффективной, позволяет существенно снизить затраты государства на лечение. Уменьшение нагрузки на бюджет в трехлетней перспективе составит от 6% в системе общего медицинского страхования до 13% по региональной льготе (с учетом сохранения цены биоаналога на среднем уровне 2017 г.). Это позволит высвободить более 1,5 млрд руб.
Таким образом, разработка биоаналогов – это магистральный путь развития мировой биофармацевтической индустрии. Внедрение в практику препаратов, полный цикл производства которых осуществляется на территории Российской Федерациии и которые в полной мере отвечают высоким стандартам, а также соответствуют зарубежным референтным медикаментозным средствам, является чрезвычайно важным для повышения качества оказания медицинской помощи больным тяжелыми иммуновоспалительными ревматическими заболеваниями.
D.E. Karateev, E.L. Luchikhina
Moscow Regional Research and Clinical Institute
Contact person: Dmitry Evgenyevich Karateev, dekar@inbox.ru
The role of targeted therapy for chronic immuno-inflammatory diseases, including biological drugs (biologics), has increased significantly over the past few years. One of the most important tools in terms of increasing the availability of new treatment options is to expand the market by producing non-original drugs after the expiration of the patent protection of original medications. Regarding the biologics, these are biosimilars (in Russian equivalent is the term “bioanalogs”). The world's largest pharmaceutical companies currently produce 20 biosimilars registered for use in rheumatic diseases, of which 3 biosimilars are approved in Russia, 2 of them (rituximab biosimilar and Acellbia®, infliximab biosimilar) are produced by the Russian biopharmaceutical company. The review describes the main features of the development and registration of biosimilars, the results of clinical studies in rheumatic diseases, and the features of application in the practice of a rheumatologist (prescription, switching, etc.).
Уважаемый посетитель uMEDp!
Уведомляем Вас о том, что здесь содержится информация, предназначенная исключительно для специалистов здравоохранения.
Если Вы не являетесь специалистом здравоохранения, администрация не несет ответственности за возможные отрицательные последствия, возникшие в результате самостоятельного использования Вами информации с портала без предварительной консультации с врачом.
Нажимая на кнопку «Войти», Вы подтверждаете, что являетесь врачом или студентом медицинского вуза.